Drugs, Health Technologies, Health Systems
Reimbursement Review
Daridorexant (Quviviq)
Sponsor: Idorsia Pharmaceuticals Canada Ltd.
Therapeutic area: Insomnia
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AASM
American Academy of Sleep Medicine
AE
adverse event
AESI
adverse event of special interest
BWSQ
Benzodiazepine Withdrawal Symptom Questionnaire
CBT
cognitive behavioural therapy
CBT-I
cognitive behavioural therapy for insomnia
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CID
chronic insomnia disorder
CNS
central nervous system
C-SSRS
Columbia Suicide Severity Rating Scale
CYP3A4
cytochrome P450 3A4
DORA
dual orexin receptor antagonist
DSM-5
Diagnostic and Statistical Manual of Mental Disorders (Fifth Edition)
GABA
gamma-aminobutyric acid
GRADE
Grading of Recommendations Assessment, Development and Evaluation
IDSIQ
Insomnia Daytime Symptoms and Impacts Questionnaire
ISI
Insomnia Severity Index
LPS
latency to persistent sleep
LSM
least squares mean
LTE
long-term extension
MDSC
Mood Disorders Society of Canada
MID
minimal important difference
PSG
polysomnography
RCT
randomized controlled trial
REM
rapid eye movement
S1
stage 1 sleep
S2
stage 2 sleep
SAE
serious adverse event
SD
standard deviation
SDQ
Sleep Diary Questionnaire
sLSO
subjective latency to sleep onset
sTST
subjective total sleep time
sWASO
subjective wake after sleep onset
SWS
slow-wave sleep
TEAE
treatment-emergent adverse event
TST
total sleep time
VAS
visual analogue scale
WASO
wake after sleep onset
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Daridorexant (Quviviq), 25 mg oral tablet, 50 mg oral tablet |
Sponsor | Idorsia Pharmaceuticals Canada Ltd. |
Indication | Quviviq (daridorexant) is indicated for the management of adult patients with insomnia, characterized by difficulties with sleep onset and/or sleep maintenance |
Reimbursement request | For moderate to severe CID, when CBT-I is inappropriate, unavailable, or has failed, characterized by difficulty initiating or maintaining sleep, early awakenings; occurring at least 3 times weekly; lasting a minimum of 3 months; and an ISI score of ≥ 15 |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | April 28, 2023 |
Recommended dose | The recommended dose of daridorexant for adults is 1 tablet of 50 mg once per night, taken orally in the evening within 30 minutes before going to bed, with at least 7 hours remaining before planned awakening Dosage adjustments: The recommended dose of daridorexant is 25 mg when used with moderate CYP3A4 inhibitors. The recommended dose in patients with moderate hepatic impairment is 25 mg once per night |
CBT-I = cognitive behavioural therapy for insomnia; CID = chronic insomnia disorder; CYP3A4 = cytochrome P450 3A4; ISI = Insomnia Severity Index; NOC = Notice of Compliance.
Introduction
Insomnia is a sleep disorder characterized by persistent difficulties falling asleep, maintaining sleep, or waking up too early, despite having adequate opportunities to sleep.1 For a diagnosis of chronic insomnia disorder (CID), symptoms must occur at least 3 times per week for at least 3 months and be present despite adequate conditions for sleep, per the Diagnostic and Statistical Manual of Mental Disorders (Fifth Edition) (DSM-5).1 It must also be associated with significant distress or impairment of daytime functioning, and impairment in social, occupational, academic, behavioural, or other important areas of functioning. The consequences of untreated insomnia include decreased work productivity, increased health care use, and a higher risk of accidents. Insomnia is more prevalent in older adults, females, and those with a family history of insomnia or coexisting psychiatric and medical conditions.2 Both CID and decreased total sleep time are independent risk factors for suicidal ideation and behaviour.2
Cognitive behavioural therapy for insomnia (CBT-I) is considered a first-line treatment for insomnia;3 however, it is not effective in all patients and has limitations related to adherence and accessibility in Canada.4-8 Pharmacological treatments, such as benzodiazepines, nonbenzodiazepine gamma-aminobutyric acid (GABA) agonists, and other hypnotics, are frequently used but are associated with adverse effects, including next-day sedation, cognitive impairment, increased risk of falls and accidents, and the potential for tolerance, dependence, and withdrawal symptoms.9-11 Consensus recommendations for the management of chronic insomnia in Canada, published in October 2024, note that there is a lack of long-term data on the efficacy of these pharmacotherapies; their use is generally recommended for short-term management. The consensus recommendations also note that dual orexin receptor antagonists (DORAs) may have benefits that outweigh their risks for long-term use;12 however, no DORAs are publicly funded in Canada at the time of this review. A 2023 RAND Europe report estimated that the global prevalence of CID in the general adult population ranges from 6.0% to 14.8% based on data from 7 studies comprising 6 countries in Europe, the UK, and the US.13 The report estimated that the prevalence of CID in the general adult population in Canada at the time was 8.8% (95% confidence interval [CI], 5.0% to 12.0%).13
Daridorexant (Quviviq) is a DORA that acts on both orexin 1 receptors and orexin 2 receptors and is equipotent on both. The terminal half-life of daridorexant is approximately 8 hours. Tablets of daridorexant are available in 25 mg and 50 mg formats. The recommended dose of daridorexant for adults is 50 mg orally once per night, taken in the evening in the 30 minutes before bedtime, when at least 7 hours remain before planned awakening. For patients with moderate hepatic impairment or who use moderate cytochrome P450 3A4 (CYP3A4) inhibitors, the recommended dose of daridorexant is 25 mg.14 Daridorexant received a Health Canada Notice of Compliance on April 28, 2023, for the management of adult patients with insomnia that is characterized by difficulties with sleep onset and/or sleep maintenance. The Health Canada indication is broader than the sponsor’s reimbursement request, which is for the treatment of insomnia in adults who have received a diagnosis of CID based on the most recent version of the DSM.15
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of daridorexant, 25 mg and 50 mg oral tablets, used in the treatment of patients with moderate to severe CID when CBT-I is inappropriate, unavailable, or has failed. CID is characterized by difficulty initiating or maintaining sleep, early awakenings; at least 3 times weekly, lasting a minimum duration of 3 months, and an Insomnia Severity Index (ISI) score of at least 15.
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of input provided to Canada’s Drug Agency (CDA-AMC) by the patient and clinician groups that responded to our call for input and from clinical experts consulted by for the purpose of this review.
Patient Input
CDA-AMC received patient input from 4 groups, including the Gastrointestinal Society, which provided information from meetings with health care experts and researchers, as well as results from surveys conducted on digestive and liver diseases and disorders; Menopause Chicks, which surveyed women aged 45 years to 64 years who were experiencing sleep disruptions; a group of 8 adults in Canada living with CID who represent a range of patient groups; and a joint submission from Mood Disorders Society of Canada (MDSC) and Migraine Canada, which surveyed and interviewed patients with insomnia, including 1 patient with experience with the treatment under review.
The input from the Gastrointestinal Society noted that CID is an independent condition that is also closely linked to a range of comorbidities, including cardiovascular disease, diabetes, obesity, cancer, and gastrointestinal diseases. In the survey conducted by Menopause Chicks, more than 85% of patients surveyed believe the underlying cause of their insomnia to be hormone changes. The input from adults in Canada living with CID stated that the negative effects of CID are often underestimated; impacts on the daily life of those affected are significant, with symptoms including persistent fatigue, difficulty concentrating, physical discomfort (such as joint pain and muscle soreness), a pervasive lack of energy that impedes self-care and routines (i.e., exercise, maintaining household responsibilities, childcare), emotional strain (feelings of frustration and anxiety), and social isolation due to exhaustion. Patients expressed that those around them often fail to understand their struggles, adding to feelings of isolation. They also noted that sleep difficulties exacerbate other health conditions, like migraines and depression, and they often miss time at work, school, or volunteering because of their symptoms. The Gastrointestinal Society noted that beyond the workplace, insufficient sleep can affect an individual’s emotional well-being, behaviour, and interactions, contributing to memory lapses, accidents, injuries, and mood disturbances. In the MDSC and Migraine Canada input, patients reported concerns about the long-term impacts of CID on their mental and physical health. Those patients also noted that partners and family members often endure sleepless nights alongside their loved ones, leading to stress, frustration, and relationship strain.
Pharmacological treatments that patients have used include sedative-hypnotics, GABA agonists, antidepressants, antipsychotics, DORAs (lemborexant), and cannabinoids. Input from Menopause Chicks noted that most patients surveyed were prescribed selective serotonin reuptake inhibitors as a sleep aid. The inputs from adults in Canada living with CID and MDSC and Migraine Canada highlighted that patients also try nonpharmaceutical options, including lifestyle adjustments, strict sleep routines, meditation, and exercise. Because of high unmet needs, some patients also take over-the-counter supplements and drugs, such as antihistamines, melatonin, magnesium, L-theanine, herbal products (e.g., chamomile, lavender, valerian root), and antihistamines (diphenhydramine). Patients reported that these treatment options provide short-term relief, relaxation, and minor sleep support; however, drawbacks include grogginess, impaired functioning, inconsistent results with continued persistent daily fatigue, and high costs. Across the patient group inputs, many patients expressed fear of dependency on medicated sleep aids and concerns about long-term side effects, including potential cognitive impacts, and many patients noted that currently available treatment options do not address the core issue of achieving deep, restorative sleep.
As such, the patient groups emphasized the need for treatments that offer consistent and restorative sleep, reduced or eliminated grogginess, and a low risk of dependency and cognitive side effects. Patients noted that addressing these unmet needs could reduce physical, emotional, and financial strains, as well as offer a better quality of life, through improved sleep and daily functioning. For the 1 patient who reported experience with daridorexant in the MDSC and Migraine Canada input, caregivers reported an improvement in the patient’s ability to fall asleep and stay asleep without any outward signs of next-day side effects.
Clinician Input
Input From Clinical Experts Consulted for This Review
The clinical experts consulted by CDA-AMC for this review noted that, because of the multifactorial nature of CID and its frequent comorbidities, it is important to have a variety of pharmacological and nonpharmacological approaches in addition to CBT-I. The experts agreed that, to improve compliance, there is a need for targeted therapies to address the multiple factors contributing to insomnia (e.g., physiologic, biologic, circadian, psychiatric) that are associated with long-term efficacy, improved tolerance, and fewer adverse effects.
The experts noted that when CBT-I is not helpful or not possible, daridorexant may be a first-line pharmacological option for insomnia disorder. However, the experts noted that deprescribing the traditional sedative-hypnotics poses a challenge, as patients often prefer current treatment options because of their rapid onset of action. As such, transitioning to daridorexant may be difficult, as DORAs may take 4 weeks to 8 weeks to demonstrate results.
Per the clinical experts, patients with CID, especially those who may have significant functional impairment, would benefit the most from new treatments such as daridorexant, whereas other drugs might be more appropriate for patients with comorbid conditions such as chronic pain or depression. The experts suggested using DORAs preferentially in patients for whom minimizing adverse effects is a priority, given their favourable side-effect profile. Narcolepsy would be an absolute contraindication for DORAs.
The clinical experts mentioned that response to treatment is usually assessed with the patient’s impression of improvement in sleep quality, sleep quantity, daytime functioning, and other symptoms. They noted that patients will likely have their own subjective standard of what they would define as an adequate benefit from treatment. The clinical experts also noted that optimal treatment response to daridorexant may not be observed until 8 weeks of treatment. As such, patients may opt to discontinue treatment because of lack of efficacy. Adverse effects (e.g., hallucinations; vivid, disturbing dreams; suicidality) that outweigh any potential benefits are also potential reasons for the discontinuation of treatment.
Clinician Group Input
Twelve clinician submissions provided input for this review, including from the Mood Disorders Research and Treatment Service, the Family Physician Airways Group of Canada, the Canadian Consortium of Sleep and Sleep Interested Physicians, the Synergy Medical Clinic, and MedSleep, as well as from psychiatrists in British Columbia and Quebec, family physicians, a neurologist, and an inpatient mental health pharmacist. All contributors had experience working with patients diagnosed with CID. The input stated that current treatment options for insomnia include nonpharmacological therapies, such as sleep hygiene and CBT-I, which can be costly and have limited availability in Canada. The input stated that pharmacological options, particularly GABA agonists, are only prescribed for short-term use, are limited by poor efficacy and next-day sedation, often require dose escalation, can lead to dependence or nonresponse, can lead to withdrawal symptoms upon discontinuation, and are associated with cognitive side effects. In addition, there are limited or no clinical data supporting their use for patients with CID. As such, to improve patient outcomes, the clinician submissions noted that there is a need for treatments that improve daytime functioning and are tolerable, affordable, and safe.
The clinician input highlighted the unique mechanism of action of daridorexant, which reduces wakefulness without causing sedation. Daridorexant would be used in the first-line setting for CID, after CBT-I or when CBT-I is not suitable, according to the input. With regard to assessing response to treatment, the clinician groups noted that, given the prevalence of insomnia in Canada and limited resources, frequent polysomnograms are not practical for use in clinical practice. Some of the clinician groups highlighted the use of the ISI screening tool and other patient-reported outcomes used in research to assess treatment response; however, the clinical experts consulted for this review noted that the ISI tool may not be used commonly in Canadian clinical practice. The clinician groups noted that a clinically meaningful response to treatment includes improved next-day functioning, improved duration and quality of sleep, and a reduction in ISI score. Potential reasons for discontinuing treatment include side effects such as reduced daytime functioning, nightmares, and sleep paralysis. Various clinicians agreed that daridorexant would be particularly suitable for patients at risk for drug dependence or abuse and for older patients, given the unsuitability of many other drugs because of the potential for cognitive impairment and falls. The clinicians agreed with the clinical experts consulted for this review that daridorexant would likely be prescribed by primary care physicians in outpatient settings, and that no specific dependence or rebound issues would need to be addressed for patients discontinuing daridorexant.
Drug Program Input
The drug programs identified the following jurisdictional implementation issues: relevant comparators, initiation of therapy, continuation or renewal of therapy, discontinuation of therapy, prescribing of therapy, and care provision issues. Refer to Table 4 for details.
Clinical Evidence
Systematic Review
Description of Studies
Two phase III, double-blind, placebo-controlled, randomized trials — Study 301 (N = 930) and Study 302 (N = 617) — were included in this review. Both studies were designed to assess the efficacy and safety of daridorexant (50 mg and 25 mg in Study 301, and 25 mg in Study 302) in adult patients with CID after month 1 and month 3 of treatment. The primary outcomes in the included trials were change from baseline in sleep maintenance (wakefulness after sleep onset [WASO]) and sleep onset (latency to persistent sleep [LPS]), measured objectively with polysomnography (PSG). Other outcomes relevant to the current review were change from baseline in sleep duration (subjective total sleep time [sTST]), sleep quality, daytime functioning, ISI score, total sleep time (TST) for each sleep stage, and safety.
Both studies enrolled adult patients with moderate to severe CID, based on DSM-5 criteria. Patients with specific comorbid conditions were excluded, such as those with acute or unstable psychiatric conditions, suicidal ideation with intent, alcohol or drug abuse, any lifetime history of suicide attempt, sleep-related breathing disorders, or narcolepsy. Patients who were unable or unwilling to discontinue concomitant use of moderate CYP3A4 inhibitors were also excluded from the trials. The baseline characteristics of the patients enrolled in Study 301 and Study 302 were generally similar across treatment groups, with a mean age of 55.1 years (standard deviation [SD] = 15.4 years) to 56.7 years (SD = 14.1 years) in the 2 studies. Most patients identified as white (86.7% to 92.6%), 6.0% to 9.7% identified as Black or African American, and 0.6% to 3.6% identified as Asian. In both studies, CBT-I was rarely used; only 3 patients in Study 301 received CBT-I at screening.
Efficacy Results
Sleep Maintenance
Sleep maintenance, reported as change from baseline in WASO, was 1 of the primary outcomes in Study 301 and Study 302. In Study 301, the mean WASO at baseline was 95.48 minutes (SD = 37.81 minutes) in the daridorexant 50 mg arm, 97.87 minutes (SD = 38.77 minutes) in the daridorexant 25 mg arm, and 102.51 minutes (SD = 40.77 minutes) in the placebo arm. At month 1, the least squares mean (LSM) change from baseline in WASO was –28.98 minutes (95% CI, –32.67 to –25.30 minutes), –18.40 minutes (95% CI, –22.13 to –14.67 minutes), and –6.20 minutes (95% CI, –9.93 to –2.48 minutes) in the daridorexant 50 mg, daridorexant 25 mg, and placebo groups, respectively. The LSM difference in change from baseline in WASO at 1 month compared to placebo was –22.78 minutes (97.5% CI, –28.75 to –16.82 minutes) in favour of daridorexant 50 mg and was –12.20 minutes (97.5% CI, –18.19 to –6.21 minutes) in favour of daridorexant 25 mg. At month 3, the LSM change from baseline was –29.41 minutes (95% CI, –33.40 to –25.43 minutes) in the daridorexant 50 mg arm, –22.97 minutes (95% CI, –26.96 to –18.99 minutes) in the daridorexant 25 mg arm, and –11.11 minutes (95% CI, –15.13 to –7.09 minutes) in the placebo arm. The LSM difference in change from baseline in WASO compared to placebo was –18.3 minutes (97.5% CI, –24.76 to –11.85 minutes) in favour of daridorexant 50 mg and was –11.86 minutes (97.5% CI, –18.30 to –5.42 minutes) in favour of daridorexant 25 mg.
In Study 302, the mean WASO at baseline was 106.31 minutes (SD = 49.10) in the daridorexant 25 mg arm and 108.07 minutes (SD = 48.71 minutes) in the placebo arm. At month 1, the LSM change from baseline was –24.19 minutes (95% CI, –28.47 to –19.91 minutes) for daridorexant 25 mg and –12.57 minutes (95% CI, –16.82 to –8.32 minutes) for placebo, corresponding to an LSM difference of –11.62 minutes (95% CI, –17.60 to –5.63 minutes) in favour of daridorexant 25 mg. Results of the subgroup analysis showed that in male patients, there was no difference between groups (mean difference = 3.94; 95% CI, –13.58 to 5.69). At month 3, the LSM change from baseline in the daridorexant 25 mg arm was –24.25 minutes (95% CI, –29.02 to –19.47 minutes) and in the placebo arm was –10.25 minutes (95% CI, –16.95 to –3.55 minutes), representing an LSM difference of –14.00 minutes (97.5% CI, –18.76 to –9.24 minutes) in favour of daridorexant 25 mg.
Sleep Onset
Sleep onset, reported as change from baseline in LPS, was 1 of the primary outcomes in Study 301 and Study 302. In Study 301, the mean LPS at baseline was 63.62 minutes (SD = 37.39 minutes) in the daridorexant 50 mg arm, 67.27 minutes (SD = 38.56 minutes) in the daridorexant 25 mg arm, and 66.54 minutes (SD = 39.77 minutes) in the placebo arm. At month 1, the LSM change from baseline in LPS was –31.20 minutes (95% CI, –34.51 to –27.90 minutes) in the daridorexant 50 mg arm, –28.17 minutes (95% CI, –31.51 to –24.83 minutes) in daridorexant 25 mg group, and –19.85 minutes (95% CI, –23.18 to –16.52 minutes) in the placebo group. The LSM difference in change from baseline in LPS at 1 month compared to placebo was –11.35 minutes (97.5% CI, –16.694 to –6.015 minutes) in favour of daridorexant 50 mg and –8.32 minutes (97.5% CI, –13.69 to –2.96 minutes) in favour of daridorexant 25 mg. At month 3, the LSM change from baseline in LPS was –34.80 minutes (95% CI, –38.12, to –31.49 minutes) in the daridorexant 50 mg arm, –30.73 minutes (95% CI, –34.04 to –27.41 minutes) in the daridorexant 25 mg arm, and –23.13 minutes (95% CI, –26.46 to –19.80 minutes) in the placebo arm. The LSM difference in change from baseline in LPS at 3 months compared to placebo was –11.67 minutes (97.5% CI, –17.03 to –6.32 minutes) in favour of daridorexant 50 mg and was –7.59 minutes (97.5% CI, –12.94 to –2.25 minutes) in favour of daridorexant 25 mg.
In Study 302, the mean LPS at baseline was 68.88 minutes (SD = 40.55 minutes) in the daridorexant 25 mg arm and 71.82 minutes (SD = 46.09 minutes) in the placebo arm. At month 1, the LSM change from baseline was –26.46 minutes (95% CI, –30.63 to –22.29 minutes) in the daridorexant 25 mg arm and –20.01 minutes (95% CI, –24.15 to –15.88 minutes) in the placebo arm, corresponding to an LSM difference of –6.45 minutes (95% CI, 12.28, to –0.61 minutes) in favour of daridorexant 25 mg. At month 3, the LSM change from baseline was –28.91 minutes (95% CI, –33.41 to –24.40 minutes) in the daridorexant 25 mg arm and –19.89 minutes (95% CI, –24.38 to –15.41 minutes) in the placebo arm, representing an LSM difference of –9.01 minutes (97.5% CI, –15.34 to –2.68 minutes) in favour of daridorexant 25 mg.
Sleep Duration
Sleep duration, measured subjectively, was reported as change from baseline in sTST was a secondary outcome in Study 301 and Study 302.
In Study 301, the mean sTST at baseline was 313.18 minutes (SD = 57.60 minutes) in the daridorexant 50 mg arm, 309.85 minutes (SD = 60.11 minutes) in the daridorexant 25 mg arm, and 315.89 minutes (SD = 53.14 minutes) in the placebo arm. At month 1, the LSM change from baseline in sTST was 43.62 minutes (95% CI, 38.17 to 49.06 minutes) in the daridorexant 50 mg arm, 34.18 minutes (95% CI, 28.72 to 39.65 minutes) in the daridorexant 25 mg arm, and 21.56 minutes (95% CI, 16.10 to 27.02 minutes) in the placebo arm. The LSM difference in change from baseline in sTST at 1 month compared to placebo was 22.06 minutes (97.5% CI, 13.30 to 30.18 minutes) in favour of daridorexant 50 mg and 12.62 minutes (97.5% CI, 3.85 to 21.39 minutes) in favour of daridorexant 25 mg. At month 3, the LSM change from baseline was 57.67 minutes (95% CI, 51.17 to 64.17 minutes) in the daridorexant 50 mg arm, 47.83 minutes (95% CI, 41.33 to 54.33 minutes) in the daridorexant 25 mg arm, and 37.90 minutes (95% CI, 31.39 to 44.40) in the placebo arm, representing an LSM difference of 19.77 minutes (97.5% CI, 9.30 to 30.24 minutes) in favour of daridorexant 50 mg and 9.93 minutes (97.5% CI, –0.54 to 20.40 minutes) in favour of daridorexant 25 mg.
In Study 302, the mean sTST at baseline was 308.49 minutes (SD = 52.85 minutes) in the daridorexant 25 mg arm and 307.57 minutes (SD = 51.52 minutes) in the placebo arm. At month 1, the LSM change from baseline in sTST was 43.77 minutes (95% CI, 38.14 to 49.41) in the daridorexant 25 mg arm and 27.64 minutes (95% CI, 22.02 to 33.27 minutes) in the placebo arm. The mean change from baseline in sTST was 16.13 minutes longer with daridorexant 25 mg than with placebo (95% CI, 8.22 to 24.04 minutes). At month 3, the LSM change from baseline in sTST was 56.18 minutes (95% CI, 49.81 to 62.55 minutes) in the daridorexant 25 mg arm and 37.12 minutes (95% CI, 30.78 to 43.46 minutes) in the placebo arm, representing an LSM difference of 19.06 minutes (95% CI, 10.13 to 27.99 minutes) in favour of daridorexant 25 mg.
Sleep Quality
Sleep quality was determined with the Sleep Diary Questionnaire (SDQ) and was considered an exploratory outcome in the included studies. In Study 301, the mean visual analogue scale (VAS) score for sleep quality at baseline was 36.23 (SD = 17.03) in the daridorexant 50 mg arm, 35.56 (SD = 17.77) in the daridorexant 25 mg arm, and 35.60 (SD = 17.78) in the placebo arm. At month 1, the mean change from baseline was 14.21 points (SD = 18.95 points) in the daridorexant 50 mg arm, 11.35 points (SD = 15.65 points) in the daridorexant 25 mg arm, and 7.04 points (SD = 13.74 points) in the placebo arm. At month 3, the mean change from baseline was 20.21 points (SD = 22.15 points) in the daridorexant 50 mg arm, 18.20 (SD = 19.10 points) in the daridorexant 25 mg arm, and 13.95 points (SD = 18.85 points) in the placebo arm.
In Study 302, the mean VAS sleep quality score at baseline was 37.94 (SD = 15.02) in the daridorexant 25 mg arm and 36.91 (SD = 14.77) in the placebo arm. At month 1, the mean change from baseline was 11.20 points (SD = 15.55 points) in the daridorexant 25 mg arm and 9.41 points (SD = 14.44 points) in the placebo arm. At month 3, the mean change from baseline was 17.77 points (SD = 18.55 points) in the daridorexant 25 mg arm and 13.18 points (SD = 17.33 points) in the placebo arm.
Change From Baseline in IDSIQ Total Score
In Study 301, the mean Insomnia Daytime Symptoms and Impacts Questionnaire (IDSIQ) total score at baseline was 74.52 points (SD = 25.16 points) in the daridorexant 50 mg arm, 73.06 points (SD = 24.55 points) in the daridorexant 25 mg arm, and 73.55 points (SD = 24.64 points) in the placebo arm. At month 1, the LSM difference in change from baseline in IDSIQ total score compared to placebo was –7.24 points (95% CI, –9.785 to –4.698 points) in favour of daridorexant 50 mg and –2.94 points (95% CI, –5.487 to –0.385 points) in favour of daridorexant 25 mg. At month 3, the LSM difference between groups compared to placebo was –7.20 points (95% CI, –10.544 to –3.862 points) in favour of daridorexant 50 mg and –3.46 points (95% CI, –6.809 to –0.113 points) in favour of daridorexant 25 mg.
In Study 302, the mean IDSIQ total score at baseline was 73.14 points (SD = 21.21 points) in the daridorexant 25 mg arm and 74.48 points (SD = 20.26 points) in the placebo arm. At month 1, the mean difference from baseline was 3.11 points (95% CI, –5.807 to –0.412 points) lower in the daridorexant arm than in the placebo arm. At month 3, the IDSIQ total score was 4.23 points (95% CI, –7.477 to –0.986 points) lower in the daridorexant 25 mg arm than in the placebo arm.
Change From Baseline in ISI Score
The change from baseline in ISI score was an exploratory outcome in the included trials. In Study 301, the mean ISI score at baseline was 19.3 points (SD = 4.0 points) in the daridorexant 50 mg arm, 19.0 points (SD = 4.3 points) in the daridorexant 25 mg arm, and 19.2 points (SD = 4.0 points) in the placebo arm. At month 1, the LSM change from baseline was –4.81 points (95% CI, –5.36 to –4.26 points) in the daridorexant 50 mg arm, –4.07 points (95% CI, –4.63 to –3.51 points) in the daridorexant 25 mg arm, and –3.05 points (95% CI, –3.60 to –2.50 points) in the placebo arm. Compared to placebo, there was a decrease in ISI score of –1.76 points (95% CI, –2.54 to –0.99 points) in the daridorexant 50 mg arm and –1.02 points (95% CI, –1.80 to –0.24 points) in the daridorexant 25 mg arm. At month 3, the LSM change from baseline was –7.17 points (95% CI, –7.84 to –6.50 points) in the daridorexant 50 mg arm, –6.02 points (95% CI, –6.69 to –5.35 points) in the daridorexant 25 mg arm, and –5.19 points (95% CI, –5.86 to –4.52 points) in the placebo arm. Compared to placebo, there was a decrease in ISI score of –1.98 points (95% CI, –2.92 to –1.04 points) in the daridorexant 50 mg arm and –0.83 points (95% CI, –1.78 to 0.11 points) in the daridorexant 25 mg arm.
In Study 302, the mean ISI score at baseline was 19.5 points (SD = 4.0 points) in the daridorexant 25 mg arm and 19.6 points (SD = 4.1 points) in the placebo arm. At month 1, the mean change in ISI score from baseline was –5.1 points (SD = 5.2 points) in the daridorexant 25 mg arm and –3.8 points (SD = 4.6 points) in the placebo arm. At month 3, the mean change from baseline in ISI score was –6.9 points (SD = 6.0 points) in the daridorexant 25 mg arm and –5.4 points (SD = 5.5 points) in the placebo arm.
Change From Baseline in Duration of TST in Each Sleep Stage
The change from baseline in duration of TST in each sleep stage (i.e., stage 1 [S1], stage 2 [S2], slow-wave sleep [SWS], and rapid eye movement [REM]) was an exploratory end point in the included trials.
In Study 301, for S1 at month 1, the change from baseline was 5.41 minutes in the daridorexant 50 mg arm, 4.26 minutes in the daridorexant 25 mg arm, and 2.59 minutes in the placebo arm. For S2 at month 1, the change from baseline was 34.74 minutes in the daridorexant 50 mg arm, 29.79 minutes in the daridorexant 25 mg arm, and 16.10 minutes in the placebo arm. For SWS at month 1, the change from baseline was 2.16 minutes in the daridorexant 50 mg arm, 0.71 minutes in the daridorexant 25 mg arm, and1.60 minutes in the placebo arm. For REM at month 1, the change from baseline was 15.51 minutes in the daridorexant 50 mg arm, 13.50 minutes in the daridorexant 25 mg arm, and 9.22 minutes in the placebo arm. For S1 at month 3, the change from baseline was 6.89 minutes in the daridorexant 50 mg arm, 5.67 minutes in the daridorexant 25 mg arm, and 4.51 minutes in the placebo arm. For S2 at month 3, the change from baseline was 37.69 minutes in the daridorexant 50 mg arm, 36.19 minutes in the daridorexant 25 mg arm, and 25.07 minutes in the placebo arm. For SWS at month 3, the change from baseline was –0.20 minutes in the daridorexant 50 mg arm, 0.20 minutes in the daridorexant 25 mg arm, and –1.54 minutes in the placebo arm. For REM at month 3, the change from baseline was 16.21 minutes in the daridorexant 50 mg arm, 12.55 minutes in the daridorexant 2,550 mg arm, and 11.65 minutes in the placebo arm.
In Study 302, for S1 at month 1, the change from baseline was 3.64 minutes in the daridorexant 25 mg arm and 1.81 minutes in the placebo arm. For S2 at month 1, the change from baseline was 31.54 minutes in the daridorexant 25 mg and 24.09 minutes in the placebo arm. For SWS at month 1, the change from baseline was 0.45 minutes in the daridorexant 25 mg arm and 0.90 minutes in the placebo arm. For REM at month 1, the change from baseline was 14.19 minutes in the daridorexant 25 mg arm and 7.31 minutes in the placebo arm. For S1 at month 3, the change from baseline was 5.46 minutes in the daridorexant 25 mg arm and 2.10 minutes in the placebo arm. For S2 at month 3, the change from baseline was 31.36 minutes in the daridorexant 25 mg arm and 25.45 minutes in the placebo arm. For SWS at month 3, the change from baseline was –0.91 minutes in the daridorexant 25 mg arm and 0.65 minutes in the placebo arm. For REM at month 3, the change from baseline was 13.86 minutes in the daridorexant 25 mg arm and 6.94 minutes in the placebo arm.
Harms Results
The overall incidence of treatment-emergent adverse events (TEAEs) in Study 301 was generally similar across groups, affecting 41.3% of patients in the daridorexant 25 mg arm, 39.3% in the daridorexant 50 mg arm, and 37.2% in the placebo arm. The most common TEAEs in the daridorexant 25 mg, daridorexant 50 mg, and placebo arms were nasopharyngitis (9.0% versus 7.8% versus 7.8%) and headache (5.5% versus 6.5 versus 3.9%). Serious adverse events (SAEs) were experienced by 2 (0.6%) patients in the daridorexant 25 mg arm, 3 (1.0%) patients in the daridorexant 50 mg arm, and 7 (2.3%) patients in the placebo arm. Adverse events (AEs) leading to withdrawals were reported for 7 patients (2.3%) in the daridorexant 25 mg arm, 3 patients (1.0%) in the daridorexant 50 mg arm, and 10 patients (3.2%) in the placebo arm. One patient died from a myocardial infarction, although it was not considered to be related to treatment.
In Study 302, the overall incidence of TEAEs was similar in the daridorexant 25 mg arm and the placebo arm (41.2% versus 36.3%). The most common TEAEs in the daridorexant 25 mg arm and the placebo arm were nasopharyngitis (4.2% versus 6.5%) and headache (5.2% versus 3.6%). SAEs were reported in 3 patients (1%) in the daridorexant 25 mg arm and 4 patients (1.3%) in the placebo arm. Four patients (1.3%) in the daridorexant 25 mg arm and 7 patients (2.3%) in the placebo arm stopped treatment because of AEs. No deaths were reported in Study 302.
Critical Appraisal
Study 301 and Study 302 were multicentre, double-blind, phase III, randomized controlled trials (RCTs). The randomization and allocation concealment processes used in the studies were judged to be appropriate. Treatment allocation was stratified by age (< 65 years or ≥ 65 years), which, according to the clinical experts consulted for this review, is a clinically important variable, particularly with regard to harms (i.e., morning sedation and night dizziness) and the risk of injury from a fall. Overall, baseline characteristics were generally balanced and similar in the treatment groups in both studies. Treatment discontinuations were relatively infrequent (7.1% to 9.0% in Study 301 and 5.8% to 7.4% in Study 302), and most of the patients in Study 301 and Study 302 completed the double-blinded treatment period; thus, the risk of attrition bias was considered low. In both studies, the overall rate of missing data was low (< 10% missing) for all relevant outcomes. Low overall missingness and balanced discontinuation rates across groups reduced concerns related to this potential overestimation, but do not completely rule out bias from informative missingness. Sensitivity analyses using multiple imputation under missing-not-at-random assumptions further supported the robustness of results, as statistical significance was maintained even under conservative imputations. Therefore, any impact of bias because of missing data on the study results was likely low. Multiplicity adjustment using alpha-splitting was conducted for primary and secondary outcomes to control for type I error. Other outcomes (e.g., other efficacy outcomes and exploratory outcomes) were not adjusted for multiplicity, so there is an increased risk of false-positive conclusions in statistically significant results. The primary outcomes in both studies were objectively measured using PSG during sleep studies, and subjectively measured outcomes were captured using patient self-assessment of sleep or validated questionnaires. Although PSG provides objective data on sleep parameters and is less prone to reporting bias, results may not fully capture real-world sleep patterns. Sleep disturbances related to the sleep-study setting may alter sleep architecture and limit the accuracy and generalizability of PSG results. According to the clinical experts consulted for this review, self-reporting of a patient’s sleep may not align with the more objective measures. The experts noted, for example, that patients with insomnia have been shown to overestimate the time taken to fall asleep (sleep onset latency) and underestimate total sleeping time. The clinical experts emphasized that insomnia is largely a subjective condition and that sleep complaints are very individualized; therefore, self-reported sleep quality and perceived changes in a patient’s sleep may be more clinically meaningful than PSG-derived metrics. However, subjective measures may be more prone to bias, including recall bias and placebo effects, that cannot be easily measured or accounted for, making it difficult to determine the true magnitude and certainty of treatment effects. Thresholds of clinical significance for all relevant outcomes, except for sleep quality on VAS, were provided by the sponsor. According to the clinical experts consulted for this review, these provided thresholds were clinically relevant and acceptable.
There were several limitations in the included studies that could affect the generalizability of results to the clinical setting in Canada. In both studies, most patients who were screened for the trials failed (72.4% in Study 301 and 74.9% in Study 302). The most common reason for screening failure was not meeting the inclusion or exclusion criteria, although the submission did not provide any details on the exact criteria that were failed during screening. Patients who did not meet specific sleep parameters on PSG and those with comorbidities (e.g., acute or unstable psychiatric conditions) were excluded. According to the clinical experts, PSG or other sleep studies are not used to diagnose CID and are not routinely performed in clinical settings for patients with CID. Also, patients who were excluded because of comorbidities could have otherwise been potential candidates for treatment with daridorexant. Thus, the screening process may have led to a study population that does not reflect the broader population with CID in Canada, thereby limiting the generalizability of the results. Although mean age, duration of disease, and distribution of sex in the study populations were consistent with Canadian settings, the clinical experts noted that the proportion of Asian patients were considerably lower in the 2 studies (especially Study 301) than in Canadian settings. It is unclear whether this could affect the study results beyond cultural differences related to sleep among different racial groups. According to the Health Canada product monograph, daridorexant is contraindicated for patients who use strong CYP3A4 inhibitors, although daridorexant 25 mg is indicated for patients receiving moderate CYP3A4 inhibitors (or those with moderate hepatic impairment). However, the use of moderate or strong CYP3A4 inhibitors was prohibited during the studies, and patients unwilling or unable to discontinue those medications were excluded from both studies. Therefore, any interpretation of efficacy or harms results in the daridorexant 25 mg treatment group presents a challenge. Several medications, including concomitant pharmacological treatments for insomnia or other central nervous system (CNS)-related medications, were prohibited in the studies. According to the clinical experts, patients with CID seen in Canadian clinical settings would often be taking 1 or more CNS-related medications prohibited in the studies. Although only a minority of patients in Canada receive CBT-I as first-line therapy, the clinical experts noted that the proportion would likely be higher than that observed in the trials (n = 3 [0.3%] in Study 301 and n = 0 in Study 302). As previously noted, sleep studies are not used in clinical settings to diagnose or assess insomnia; instead, patients are usually interviewed about their sleep and how they feel. Additionally, subjective measures used in the studies, such as IDSIQ, ISI, and SDQ, are not routinely used in clinical practice, per the clinical experts consulted for this review.
GRADE Summary of Findings and Certainty of the Evidence
The selection of outcomes for Grading of Recommendations Assessment, Development and Evaluation (GRADE) was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
efficacy outcomes (sleep maintenance, sleep onset, sleep duration, sleep quality)
harms outcomes (SAEs).
Table 2: Summary of Findings for Daridorexant 50 mg vs. Placebo for Patients With CID
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Daridorexant 50 mg (95% CI) | Placebo (95% CI) | Difference (97.5% CI) | |||||
Sleep maintenance | |||||||
LSM change from baseline in WASO at 3 months, minutes Follow-up: 3 months | 620 (1 RCT) | NA | –29.41 (–33.40 to –25.43) | –11.11 (–15.13 to –7.09) | –18.30 (–24.76 to –11.85)a | Lowb,c | Daridorexant may result in a reduction (improvement) in sleep maintenance (the time spent awake after onset of persistent sleep) at 3 months compared with placebo. The clinical importance of the reduction is unclear. |
Sleep onset | |||||||
LSM change from baseline in LPS, minutes Follow-up: 3 months | 620 (1 RCT) | NA | –34.80 (–38.118 to –31.490) | –23.13 (–26.464 to –19.803) | –11.67 (–17.027 to –6.320)a | Lowb,d | Daridorexant may result in a reduction (improvement) in sleep onset (LPS is the time from wakefulness to 10 consecutive minutes of sleep) at 3 months compared with placebo. The clinical importance of the reduction is unclear. |
Sleep duration | |||||||
LSM change from baseline in sTST, minutes Follow-up: 3 months | 620 (1 RCT) | NA | 57.67 (51.17 to 64.17) | 37.90 (31.39 to 44.40) | 19.77 (9.30 to 30.24)a | Lowe,f | Daridorexant may result in an increase in subjective total sleep duration at 3 months compared with placebo. The clinical importance of the reduction is unclear. |
Sleep quality | |||||||
Mean change from baseline in SDQ VAS Follow-up: 3 months | 620 (1 RCT) | NA | Daridorexant 50 mg: 20.21 (SD = 22.15) Placebo: 13.95 (SD = 18.85) Difference: NR | Very lowg,h | The evidence is very uncertain about the effect of daridorexant on change from baseline in SDQ VAS compared with placebo. | ||
Harms | |||||||
Proportion of patients with 1 or more SAEs Follow-up: end of study | 620 (1 RCT) | NA | 10 per 1,000 | 23 per 1,000 | NR | Moderatei,j | Daridorexant 50 mg likely results in little to no difference in the occurrence of SAEs compared with placebo. |
CI = confidence interval; CID = chronic insomnia disorder; LPS = latency to persistent sleep; LSM = least squares mean; NA = not applicable; NR = not reported; RCT = randomized controlled trial; SAE = serious adverse event; SD = standard deviation; sTST = subjective total sleep time; VAS = visual analogue scale; vs. = versus; WASO = wake after sleep onset.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aAfter hypothesis-testing adjustment for multiplicity, the alpha or threshold of significance was estimated to be 0.025. Thus, 97.5% CIs are reported for these between-group differences.
bRated down once for serious indirectness. The outcomes were measured using polysomnography (PSG). According to the clinical experts, PSG is not used in clinical settings to assess the efficacy of treatment. The external validity of the trial is limited by the large number of patients excluded at screening.
cRated down once for serious imprecision. Based on a minimal important difference (MID) threshold of 20 minutes, the point estimate of the effect and the 97.5% CI includes the possibility of a trivial effect as well as a nontrivial effect.
dRated down once for serious imprecision. Based on an MID threshold of 10 minutes, the point estimate of the effect is larger than the threshold, but the 97.5% CI includes the possibility of a trivial effect as well as a nontrivial effect.
eRated down once for serious indirectness. The outcome was self-reported. Clinical experts confirmed that patients are likely to underestimate one’s own duration of sleep. The external validity of the trial is also limited by the large number of patients excluded at screening.
fRated down once for serious imprecision. Based on an MID threshold of 30 minutes, the point estimate of the effect, as well as the 97.5% CI, includes the possibility of a trivial effect as well as a nontrivial effect.
gAnalysis of this outcome was not adjusted for multiplicity; the results are considered to be supportive evidence.
hRated down once for serious indirectness. Rated down twice for very serious imprecision. The absolute difference between the groups, with CIs, was unavailable. The MID threshold for this outcome is unclear.
iEven though the external validity of the trial is limited by the large number of patients excluded at screening, not rated down for indirectness.
jRated down once for serious imprecision. The number of events did not meet the optimal information size.
Sources: Details included in the table are from the sponsor’s Summary of Clinical Evidence, Clinical Study Reports for Study 30116 and Study 302,17 and additional information provided by the sponsor.
Long-Term Extension Study
Description of Study
Study 303 was a 40-week, long-term extension (LTE) study (N = 804) that assessed the safety and tolerability of daridorexant in adult and older patients with CID. It was a multicentre, double-blind, placebo-controlled trial conducted at 94 sites in 14 countries, including 7 in Canada. Patients who completed Study 301 or Study 302 were eligible, and patients who had previously received placebo were rerandomized to continue placebo or to receive daridorexant 25 mg. In total, there were 137 patients receiving daridorexant 50 mg (all from Study 301), 270 receiving daridorexant 25 mg (132 from Study 301 and 138 from Study 302), and 255 patients receiving placebo, who were rerandomized in a 1:1 ratio to either daridorexant 25 mg (N = 127 [66 from Study 301 and 61 from Study 302], termed the ex-placebo to daridorexant 25 mg arm) or placebo (N = 128). Study 303 also included a daridorexant 10 mg arm (N = 142), though daridorexant 10 mg is not a Health Canada–approved dose and was not summarized within this report. A 30-day safety follow-up period assessed AEs and medications. Of the 662 patients in the 4 relevant treatment groups, 459 (69.3%) completed the study. Demographic and most baseline characteristics were balanced across treatment groups, consistent with prior studies.
Efficacy Results
Change From Baseline in sWASO
Throughout the extension study, mean subjective wake after sleep onset (sWASO) reductions from baseline were maintained across treatment groups. At week 36, LSM differences between the treatment and placebo groups in the change in sWASO from the confirmatory study baseline was –2.01 minutes (95% CI, –14.71 to 10.68 minutes; P = 0.7554) in the daridorexant 50 mg group and –1.51 minutes (95% CI, –12.65 to 9.62 minutes; P = 0.5148) in the daridorexant 25 mg group.
Change From Baseline in sLSO
In the extension study, mean reductions in subjective latency to sleep onset (sLSO) from baseline were observed in all treatment groups, with numerically greater improvements in the active treatment groups than in the placebo groups. At week 36, compared to placebo, the LSM treatment difference in sLSO from confirmatory study baseline was –9.19 minutes (95% CI, –18.45 to 0.07 minutes; P = 0.0517) in the daridorexant 25 mg group and –8.76 minutes (95% CI, –19.34 to 1.82 minutes; P = 0.1044) in the 50 mg group.
Change From Baseline in sTST
In the extension study, changes in sTST from confirmatory study baseline were numerically greater in the daridorexant groups than in the placebo group, with the most pronounced change in the 50 mg group. At week 36, compared to placebo, the LSM treatment difference in sTST was 17.77 minutes (95% CI, –0.35 to 35.90 minutes; P = 0.0546) in the daridorexant 50 mg group and 5.26 minutes (95% CI, –10.59 to 21.11 minutes; P = 0.5148) in the daridorexant 25 mg group.
ISI Score
At week 40, the mean change in ISI score was –9.8 in the daridorexant 50 mg group, –8.5 in the daridorexant 25 mg group, –4.3 in the ex-placebo to daridorexant 25 mg group, and –7.5 in the placebo group. A 6-point or greater decrease in ISI score was achieved by 74.7% of patients in the 50 mg group, 66.1% in the 25 mg group, 35.8% in the ex-placebo group, and 53.9% in the placebo group.
Change From Baseline in SDQ VAS Scores
Improvements from baseline in quality of sleep (VAS) were maintained in all treatment groups throughout the extension study. At week 36, the mean change from baseline was 27.4 (SD = 23.6) in the daridorexant 50 mg group, 22.4 (SD = 21.6) in the daridorexant 25 mg group, 9.7 (SD = 16.3) in the ex-placebo to daridorexant 25 mg group, and 21.9 (SD = 19.2) in the placebo group. Similar results were reported for all other VAS domain end points.
Change From Baseline in IDISQ Total Score
Mean reductions in IDSIQ total score from baseline were maintained throughout the extension study among patients in the daridorexant 50 mg and 25 mg groups. At week 36, the LSM treatment difference in IDSIQ total score, compared to placebo, was –9.12 (95% CI, –15.59 to –2.66; P = 0.0058) in the daridorexant 50 mg group and –4.52 (95% CI, –10.15 to 1.12, P = 0.11161) in the daridorexant 25 mg group.
Harms Results
In Study 303, from the double-blind treatment period until up to 30 days after the double-blind study treatment end date, the proportion of patients experiencing TEAEs was 40.1% in the daridorexant 50 mg group, 38.4% in the daridorexant 25 mg group, 38.1% in the ex-placebo to daridorexant 25 mg group, and 35.2% in the placebo group. Most TEAEs were mild or moderate, with nasopharyngitis being the most common (4.7% to 8.7%). The incidence of SAEs was less than 5.2% in all treatment groups. Most SAEs were reported by a single patient and were not considered to be related to the study medication, except for 1 case of orthostatic intolerance (in the daridorexant 25 mg group) and 1 instance of depression and/or suicidal ideation (in the placebo group). One death occurred in the daridorexant 25 mg group, but it was unrelated to treatment.
In terms of adverse events of special interest (AESIs), less than 3% of patients in each group experienced falls or other events; hallucinations and/or sleep paralysis, narcolepsy-like symptoms, and suicide and/or self-harm were reported by 1 patient each. No patients reported suicidal ideation or suicidal behaviour during the double-blind portion of the study or during the placebo run-out period. Mean Benzodiazepine Withdrawal Symptom Questionnaire (BWSQ) scores were low and similar across treatment groups, with minor changes from the last double-blind assessment to the placebo run-out period. There was no indication of rebound insomnia, assessed with mean sTST, in any treatment group during the placebo run-out period. Next-morning residual effects, assessed with mean VAS morning sleepiness score, improved from baseline throughout the study for all treatment groups.
Critical Appraisal
In the extension study, baseline characteristics were balanced across groups, consistent with the confirmatory studies. However, only patients who completed Study 301 or Study 302 were included, which could bias the results in favour of treatment. Furthermore, patients in the placebo arm of Study 303 were already receiving placebo in their confirmatory study (Study 301 or Study 302). Their continued participation suggests that they were likely good responders to placebo, which may explain their improved sleep-related outcomes (i.e., sTST, sWASO, and sLSO) reported in the extension study. The study lacked multiplicity adjustments, had many tests, and had no statistical sample size considerations, raising the risk of type I error. High dropout rates (28.5% to 35.2%) could have overrepresented patients more likely to benefit from treatment.
Of the 662 patients in the 4 treatment groups assessed in this review, 7 (1.1%) identified as Asian, 38 (5.7%) as Black or African American, 2 (0.3%) as another race, and 421 (63.6%) as white, which doesn't reflect the ethnic diversity seen in patients with CID , according to the clinical experts consulted. Additionally, the exclusion of individuals with acute and unstable mental health conditions and the low percentage of patients (7.3%) with psychiatric disorders in the extension study limits the generalizability of results to this patient population. The clinical experts also noted that tools like ISI and IDSIQ are not commonly used in clinical practice, further limiting generalizability.
Indirect Comparisons
The sponsor determined that it was infeasible and inappropriate to conduct an indirect treatment comparison for daridorexant; thus, no indirect evidence was submitted for this review.
Studies Addressing Gaps in the Evidence From the Systematic Review
No studies addressing gaps in the evidence were submitted by the sponsor for this review.
Conclusions
CID is a common condition with a significant clinical burden on daily life, including impaired daytime functioning and severe negative impacts on mental and physical health and on quality of life. The limited effective or well-tolerated, long-term treatment options available highlights an unmet clinical need for novel treatments that address the multiple facets of insomnia.
Evidence for this review came from 2 double-blind, placebo-controlled trials (Study 301 and Study 302), which evaluated the efficacy and safety of daridorexant 50 mg and daridorexant 25 mg in adults with CID, defined according to DSM-5 criteria, and 1 LTE study (Study 303). Outcomes evaluated in the studies were clinically relevant and aimed to address the needs identified by patients and clinicians, including sleep maintenance, onset, duration, and quality. Study 301 demonstrated that at 3 months, compared to placebo, treatment with daridorexant 50 mg resulted in a statistically significant improvement (reduction) in sleep maintenance measured by WASO (LSM difference, –18.3 minutes; 97.5% CI, –24.76 to –11.85 minutes) and in sleep onset measured by LPS (LSM difference, –11.67 minutes; 97.5% CI, –17.03 to –6.32 minutes), and an increase in sleep duration (LSM difference, 19.77 minutes; 97.5% CI, 9.30 to 30.24 minutes). However, it is uncertain whether the results for these outcomes were clinically meaningful, given the effect sizes and 97.5% CIs, which contained the possibility of benefit as well as the possibility of no benefit. Results for change from baseline in insomnia symptoms (measured with ISI) and daytime functioning (measured with IDSIQ) appeared to improve, but given the subjective nature of the outcomes, are uncertain. It is also unclear whether the results were clinically meaningful compared to placebo, although the pharmacokinetic profile of daridorexant is theoretically beneficial for daytime symptoms. DORAs are known to have an acceptable safety profile, and the harms observed in the pivotal trials were considered manageable by the clinical experts consulted for this review. Additionally, no safety signals were identified for rebound insomnia, next-morning residual effects, or suicidality. The findings were generally consistent in the LTE study. There is no direct or indirect comparative evidence between daridorexant and relevant treatments for patients with CID; thus, the comparative efficacy and safety of daridorexant remain unknown.
Both Study 301 and Study 302 evaluated the 25 mg dose of daridorexant, and results were consistent with the 50 mg dose, although the magnitude of the results was not as high. As such, there was evidence of a dose-response relationship with respect to efficacy outcomes; however, this was not observed for harms. Because the population of patients meeting the indication for daridorexant 25 mg (i.e., patients receiving moderate CYP3A4 inhibitors or with moderate hepatic impairment) was excluded from the trials, limited conclusions can be drawn about the efficacy and safety of the daridorexant 25 mg dose. Because CID frequently occurs alongside other psychiatric or medical conditions, the exclusion of patients with comorbid psychiatric disorders and those using certain medications (e.g., antidepressants or antipsychotics) from the pivotal studies limits the generalizability of the findings to real-world clinical practice.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of daridorexant, 25 mg oral tablet and 50 mg oral tablet, in the treatment adult patients with CID.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
Insomnia is characterized by persistent sleep difficulty, despite adequate sleep opportunity, and is associated daytime dysfunction.1 Patients with insomnia experience dissatisfaction with the quality or duration of their sleep because of difficulties initiating or maintaining sleep or waking up too early. For a diagnosis of CID — defined in the DSM-5 as an insomnia disorder — symptoms must occur at least 3 times per week for at least 3 months and be present despite adequate conditions for sleep.1 It must also be associated with significant distress, the impairment of daytime functioning, or the impairment of social, occupational, academic, behavioural, or other important areas of functioning. The etiology of insomnia includes a combination of predisposing factors that increase the risk for CID (e.g., chronic mental health or neurologic conditions); precipitating events that lead to sleep disruption (e.g., severe accident leading to physical injury, death of a close family member, or change in occupation); and perpetuating factors that consist of behavioural and cognitive compensatory responses to ѕlеерlеѕsոеѕs that may contribute to persistent physiologic arousal and to the continuation of insomnia (e.g., worry about sleep loss or clock-watching in bed).18
A range of risk factors has been implicated in the development of insomnia, including older age, family history of insomnia, and female sex.2 Insomnia often coexists with psychiatric disorders (e.g., anxiety, depression, bipolar disorder), medical conditions (e.g., pulmonary disease, diabetes, cancer), and neurologic conditions (e.g., epilepsy, dementias, multiple sclerosis).2 CID has a negative impact on the daytime functioning, safety, and quality of life of patients. Patients with CID report increased fatigue, confusion, tension, anxiety, and depression. Although patients tend to overestimate the magnitude of their performance deficits, as well as the magnitude of their sleep deficits,2 insomnia has been associated with decreased work productivity, increased health care use, and an increased risk of accidents. Patients with CID frequently seek over-the-counter remedies and have an increased risk of developing substance use disorders. When untreated, the symptoms of insomnia can exacerbate existing health issues and lead to new physical and mental health complications over time, including cardiovascular, psychiatric, and neurologic conditions. Both CID and decreased total sleep time are independent risk factors for suicidal ideation and behaviour.2
There is a wide range of prevalence estimates for insomnia2 because of variances in case definitions, assessment procedures, sample characteristics, and lengths of assessment.19 In general, approximately 30% of adults report experiencing insomnia symptoms of any severity at some point in their lives, with around 10% to 15% also experiencing daytime consequences, such as fatigue.19 When the more stringent diagnostic criteria of the DSM-5 or the International Classification of Sleep Disorders are used, CID prevalence rates tend to cluster between 6% and 10%.19 A 2023 RAND Europe report estimated that the global prevalence of CID in the general adult population to range from 6.0% to 14.8%, based on data from 7 studies comprising 6 countries in Europe, the UK, and the US.13 The report estimated the prevalence of CID in the general adult population in Canada at the time to be 8.8% (95% CI, 5.6% to 12.0%).13
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
The goal of treatment for patients with CID is to reduce nighttime and daytime symptoms (i.e., improving daytime functioning over a long period of time because of the chronicity of the disease). Consensus recommendations for the management of CID in Canada, published in October 2024, highlight the importance of specifically treating insomnia, even in the presence of comorbidities.12 This consensus aligns with international guidelines, including the European Sleep Research Society 2023 update. The consensus recommendations, the European guidelines, and the clinical experts consulted for this review recommend CBT-I as the first-line treatment for chronic insomnia.12,20 The clinical experts noted that CBT-I is effective for many but not all patients, and in Canada, given the limited provider awareness, limited access to CBT-I, and financial constraints, pharmacological treatment remains necessary.4-8
There are currently no approved or recommended pharmacological treatments indicated for CID on Canadian public drug plans. The clinical experts consulted for this review noted that trazadone, an atypical antidepressant, is commonly used off-label in Canada because of its hypnotic effects at low doses. Other currently used pharmacotherapeutic options in Canada include hypnotics (e.g., Z-drugs [zolpidem and zopiclone]), off-label benzodiazepines, and GABA agonists. The clinical experts explained that the efficacy of pharmacotherapeutic options, including benzodiazepines and Z-drugs, has been confirmed in multiple short-term studies (up to 4 weeks), but long-term data (3 months to 12 months) are limited. There is also the potential for misuse with these drugs as long-term therapy in the absence of suitable alternatives. Moreover, these therapies are associated with potentially concerning adverse effects, including daytime drowsiness, risk of falls, risk of addiction and abuse, and rebound insomnia after discontinuation.9-11 The consensus recommendations for the management of chronic insomnia in Canada, published in October 2024, note that there is a lack of long-term data on the efficacy of these pharmacotherapies, and their use is generally recommended for short-term management. Although the consensus recommendations note that DORAs may have benefits that outweigh risks for long-term use,12 no DORAs are publicly funded in Canada as of this review. The risks associated with these pharmacological drugs are particularly pronounced in older, more frail adults, which is a population with a high prevalence of CID.21-23 Thus, these options should be limited to the short-term management of insomnia. There is also a lack of evidence supporting the benefit of off-label antidepressants and antipsychotics in patients with CID, as well as concerns regarding their safety profile. The clinical experts highlighted the need to reduce the use of medications with limited evidence or low risk-benefit ratios as newly approved pharmacotherapies are integrated.12
Drug Under Review
The key characteristics of daridorexant and other treatments available for CID are summarized in Table 3. Health Canada approval for daridorexant was granted in April 2023.14
Daridorexant is a DORA that acts on both orexin 1 receptors and orexin 2 receptors equipotently. Daridorexant antagonizes the activation of orexin receptors through orexin neuropeptides, which, consequently, decreases the wake drive, allowing sleep to occur. The terminal half-life of daridorexant is approximately 8 hours. Tablets of daridorexant are available in 25 mg and 50 mg doses. The recommended dose of daridorexant for adults is 1 tablet of 50 mg once per night, taken orally in the evening in the 30 minutes before bedtime, when at least 7 hours remain before planned awakening. For patients with moderate hepatic impairment or who use moderate CYP3A4 inhibitors, the recommended dose of daridorexant is 25 mg.14
Daridorexant is indicated for the management of adult patients with insomnia characterized by difficulties with sleep onset and/or sleep maintenance. The sponsor’s request for reimbursement is for a more targeted indication than that specified in the product monograph. Specifically, the request is for patients with moderate to severe CID in whom CBT-I is inappropriate, unavailable, or has failed, and whose CID is characterized by difficulty initiating or maintaining sleep, early awakenings; occurring at least 3 times weekly, lasting a minimum duration of 3 months, and an ISI score of at least 15.15
Table 3: Key Characteristics of Daridorexant, Lemborexant, Z-Drugs, Benzodiazepines, Doxepin, Antidepressants, and Antipsychotics
Drug name | Mechanism of action | Indicationa | Route of administration | Recommended dosage | Serious adverse effects or safety issues | Other |
|---|---|---|---|---|---|---|
Daridorexant | Dual orexin receptor antagonist that acts on both orexin 1 receptors and orexin 2 receptors equipotently. Daridorexant antagonizes the activation of orexin receptors through orexin neuropeptides (orexin A and orexin B), which, consequently, decreases the wake drive, allowing sleep to occur. | For the management of adult patients with insomnia, characterized by difficulties with sleep onset and/or sleep maintenance. | Oral | The recommended dose of daridorexant for adults is 1 tablet of 50 mg once per night, taken orally in the 30 minutes before bed, when at least 7 hours remain before planned awakening. The recommended dose is 25 mg when used with moderate CYP3A4 inhibitors. | Contraindicated for patients with narcolepsy. Not recommended for patients with severe hepatic impairment. Concomitant use with strong CYP3A inhibitors should be avoided. | NA |
Lemborexant | Competitive antagonist of OX1R and OX2R. Blocking the binding of the wake-promoting neuropeptides orexin A and orexin B to receptors (OX1R and OX2R) is thought to suppress the wake drive. | For the treatment of insomnia, characterized by difficulties with sleep onset and/or sleep maintenance. | Oral | 5 mg once per night in the few minutes before bed when at least 7 hours remain before planned awakening. Dose may be increased to 10 mg, depending on response and tolerability. | Contraindicated for patients with narcolepsy. Not recommended for patients with severe hepatic impairment. Concomitant use with other CNS depressants, CYP3A inhibitors, or CYP3A inducers should be avoided. Increased risk of daytime impairment if taken when less than 7 hours of sleep remain or if a higher-than-recommended dose taken. May cause drowsiness and increase the risk of falls. | NA |
Z-drugs | GABA A receptor positive modulators are presumed to exert therapeutic effects by binding to the benzodiazepine site of the alpha-1 subunit that contains GABA A receptors, increasing the frequency of chloride channel opening and resulting in the inhibition of neuronal excitation. | Short-term use (usually not exceeding 7 to 10 days) for the following: treatment and symptomatic relief of insomnia characterized by difficulty falling asleep frequent nocturnal awakenings and/or early morning awakenings disturbed sleep that results in impaired daytime functioning. | Oral, sublingual. | Varies by drug | Contraindicated for patients with complex sleep behaviours (e.g., night eating, somnambulism) and no recollection of such activities; personal or family history of sleepwalking; severe hepatic impairment. Can lead to abuse, misuse, addiction, physical dependence, and withdrawal reactions, which can result in overdose or death, especially when combined with opioids, alcohol, or illicit drugs. Concomitant use with opioids may result in profound sedation, respiratory depression, coma, and death. Can produce severe or life-threatening withdrawal symptoms. | Drugs: zolpidem, zopiclone |
Benzodiazepines | Depressants of the CNS, believed to enhance or facilitate the effects of the inhibitory neurotransmitter GABA and act as agonists at the benzodiazepine receptors sites. | Temazepam: for the symptomatic relief of transient and short-term insomnia characterized by difficulty falling asleep, frequent nocturnal awakenings, and/or early morning awakenings. Treatment should usually not exceed 7 to 10 consecutive days. | Oral | Varies by drug | Dose-dependent ataxia or dizziness; dependence and/or withdrawal symptoms. Additive sedation with CNS depressants (e.g., alcohol). Rebound insomnia may occur on withdrawal. Risk of withdrawal symptoms after abrupt discontinuation. Risk of fall, risk of development of tolerance, risk of dependence. Risk of injuries while driving. | Drugs: temazepam, lorazepam, clonazepam, others |
Doxepin | Doxepin binds with high affinity to the histamine H1 receptor, where it functions as an antagonist. The exact mechanism by which doxepin exerts its sleep maintenance effect is unknown, but is believed to be due to its antagonism of the H1 receptor. | For the treatment and symptomatic relief of insomnia characterized by frequent nocturnal awakening and/or early morning awakenings. | Oral | 6 mg once daily, although 3 mg once daily may be appropriate for some patients. For older adult patients, 3 mg once daily, which can be increased to 6 mg if clinically indicated. | Contraindicated for patients with hypersensitivity to other dibenzoxepin compounds, untreated narrow angle glaucoma, or severe urinary retention. Serious side effects and death have been reported following the concomitant use of certain drugs with MAOIs. Contraindicated in patients taking MAOIs or who have used MAOIs in the previous 2 weeks. Associated with complex sleep-related behaviours. Should not be consumed with alcohol. | NA |
Antidepressants | Unclear in humans | None for insomnia | Oral | Varies by drug | Drowsiness, orthostatic hypotension, nausea, vomiting, headache, dry mouth, priapism (rare). Toxicity may be increased by inhibitors of CYP3A4. Effectiveness may be decreased by inducers of CYP3A4, CYP2D6, or CYP1A2, depending on the drug. | Drugs: trazodone, mirtazapine, TCAs |
Antipsychotics | Interact with a broad range of neurotransmitter receptors, with direct and indirect effects | None for insomnia | Oral | Varies by drug | Sedation, dizziness, weight gain, orthostatic hypotension, hepatic aminotransferase elevation, headache, anticholinergic effects, increased risk of diabetes and dyslipidemia, movement disorders; may lower thyroid hormone levels; may induce modest QTc prolongation. Additive sedation with CNS depressants; may potentiate antihypertensive drug effects; inhibitors of CYP3A4 may increase quetiapine levels; inducers of CYP3A4 may decrease quetiapine levels. Use with caution in conjunction with drugs known to prolong the QTc interval. | Drugs: quetiapine, olanzapine, many others |
CNS = central nervous system; CYP3A4 = cytochrome P450 3A4; GABA = gamma-aminobutyric acid; MAOI = monoamine oxidase inhibitor; NA = not applicable; OX1R = orexin receptor 1; OX2R = orexin receptor 2; TCA = tricyclic antidepressants.
aHealth Canada–approved indication.
Sources: Product monographs for daridorexant,14 Dayvigo (lemborexant),24 Sublinox (zolpidem),25 Restoril (temazepam),26 Silenor (doxepin),27trazodone,28 and Apo-Quetiapine XR (quetiapine fumarate extended-release).29
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
CDA-AMC received patient input from 4 groups, including the Gastrointestinal Society, which provided information from meetings with health care experts and researchers, as well as results from surveys conducted on digestive and liver diseases and disorders; Menopause Chicks, which surveyed women aged 45 years to 64 years who were experiencing sleep disruptions; a group of 8 adults in Canada living with CID who represent a range of patient groups; and a joint submission from MDSC and Migraine Canada, which surveyed and interviewed patients with insomnia, including 1 patient who had experience with the treatment under review.
The input from the Gastrointestinal Society noted that CID is an independent condition that is closely linked to a range of comorbidities, including cardiovascular disease, diabetes, obesity, cancer, and gastrointestinal diseases. In the survey conducted by Menopause Chicks, more than 85% of patients said they believe the underlying cause of their insomnia is hormone changes. The group of adults in Canada living with CID stated that the negative effects of CID are often underestimated, and explained that symptoms can have significant impacts on their daily life, including persistent fatigue, difficulty concentrating, physical discomfort (such as joint pain and muscle soreness), a pervasive lack of energy that impedes self-care and routines (i.e., exercise, maintaining household responsibilities, childcare), emotional strain (feelings of frustration and anxiety), and social isolation due to exhaustion. The patients explained that those around them often fail to understand their struggles, adding to feelings of isolation. The group’s input noted that sleep difficulties exacerbate other health conditions, like migraines and depression, and that patients often miss time at work, school, or volunteering due to their symptoms. The Gastrointestinal Society noted that beyond the workplace, insufficient sleep can affect an individual’s emotional well-being, behaviour, and interactions, contributing to memory lapses, accidents, injuries, and mood disturbances. The input from MDSC and Migraine Canada described patients’ concerns about the long-term impacts of CID on their mental and physical health. The input also noted that partners and family members often endure sleepless nights alongside their loved ones, leading to stress, frustration, and relationship strain.
Pharmacological treatments that patients have used include sedatives-hypnotics, GABA agonists, antidepressants, antipsychotics, DORAs (lemborexant), and cannabinoids. Input from Menopause Chicks noted that most patients surveyed were prescribed selective serotonin reuptake inhibitors as a sleep aid. Inputs from the group of adults in Canada living with CID and from MDSC and Migraine Canada highlighted that often patients try nonpharmaceutical options, including lifestyle adjustments, strict sleep routines, meditation, and exercise. Because of high unmet needs, some patients also take over-the-counter supplements and drugs such as antihistamines, melatonin, magnesium, L-theanine, herbal products (e.g., chamomile, lavender, valerian root), and antihistamines (diphenhydramine). Patients reported that these treatment options provide short-term relief, relaxation, and minor sleep support. However, drawbacks include grogginess, impaired functioning, inconsistent results with continued persistent daily fatigue, and high costs. Across inputs, many patients expressed a fear of dependency on medicated sleep aids and concerns about long-term side effects, including potential cognitive impacts; however, patients across submissions noted that currently available treatment options do not address the core issue of achieving deep, restorative sleep.
As such, the patient groups emphasized the need for treatments that offer consistent and restorative sleep, reduced or eliminated grogginess, and a low risk of dependency or cognitive side effects. Patients noted that addressing these unmet needs could reduce physical, emotional, and financial strains, as well as offer a better quality of life through improved sleep and daily functioning. For the 1 patient who had experience with daridorexant in the MDSC and Migraine Canada input, caregivers reported an improvement in the patient’s ability to fall asleep and stay asleep, without any outward signs of next-day side effects.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of CID.
Unmet Needs
Given the multifactorial nature of CID and its frequent comorbidities, the clinical experts noted that it is important to have a variety of pharmacological and nonpharmacological approaches, in addition to CBT-I. While CBT-I is a known and effective treatment for insomnia disorder, issues related to access to CBT-I, such as financial barriers and provider awareness, limit the use of this intervention in clinical practice. The experts also highlighted that up to a third of patients may not respond to CBT-I. In terms of pharmacological options, the experts agreed that there is a need for targeted therapies to address the multiple factors that contribute to insomnia (e.g., physiologic, biologic, circadian, psychiatric) and are associated with long-term efficacy, improved tolerance, and a reduction in adverse effects, improving compliance.
Place in Therapy
The clinical experts indicated that daridorexant may be a first-line pharmacological therapy for insomnia disorder, if CBT-I is not effective or not possible. The experts stated that deprescribing traditional sedative-hypnotics poses a challenge, as patients often prefer current treatment options due to their rapid onset of action. As such, they noted that transitioning to daridorexant may be difficult as DORAs may take 4 weeks to 8 weeks to demonstrate results. The experts also noted the utility of DORAs used in combination with other pharmacologic drugs; this is specifically reserved for patients with complex insomnia difficulties.
Patient Population
According to the clinical experts, applying the generally agreed upon symptomatic diagnosis of CID is appropriate to identify patients suitable for treatment with daridorexant. Individuals with significant functional impairment (e.g., social, occupational, physical) are most in need of intervention. They noted that other drugs might be more appropriate for patients with comorbid conditions, such as chronic pain or depression. The experts suggested using DORAs preferentially for patients in whom minimizing adverse effects is a priority, given their favourable side-effect profile. Narcolepsy would be an absolute contraindication for DORAs.
Because CID is mostly symptom guided, the clinical experts mentioned that, generally, diagnostic tests or sleep studies (e.g., PSG) are not needed unless there is evidence of treatment resistance or other disorders (e.g., comorbid obstructive sleep apnea and insomnia or periodic limb movement disorder).
Assessing the Response Treatment
The clinical experts mentioned that response to treatment would initially be assessed after about 8 weeks of treatment and would be determined by a patient’s impression of improvement in sleep quality, quantity, and daytime functioning, and other symptoms. They noted that every patient will likely have their own subjective standard of what they would define as adequate benefit from treatment and that it is presently unclear which patients would best respond to treatment. Formal questionnaires or objectively measured end points are likely not used in clinical settings.
Discontinuing Treatment
According to the clinical experts, lack of efficacy after a reasonable trial at an optimal dose and/or possible adverse effects (e.g., hallucinations, vivid disturbing dreams, suicidality) that outweigh any potential benefits are potential reasons to discontinue treatment. The experts also noted that the optimal time to determine treatment efficacy is after around 8 weeks of treatment. The clinical experts also flagged that patients may opt to discontinue treatment sooner, citing a lack of efficacy because they are used to the immediate hypnotic effects of Z-drugs and benzodiazepines.
Prescribing Considerations
The clinical experts mentioned that any physician or nurse practitioner can diagnose and treat CID with daridorexant as a first-line therapy. They also noted that a specialist is not required for diagnosis or treatment, and should generally be reserved for resistant insomnia or multiple concomitant sleep disorders. The experts noted that the dosing schedule is straightforward as indicated and that the side-effect profile of daridorexant is generally manageable and easy to discuss with the patients.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
Twelve clinician submissions provided input for this review, including from the Mood Disorders Research and Treatment Service, the Family Physician Airways Group of Canada, the Canadian Consortium of Sleep and Sleep Interested Physicians, the Synergy Medical Clinic, and MedSleep, as well as from psychiatrists in British Columbia and Quebec, family physicians, a neurologist, and an inpatient mental health pharmacist. All contributors had experience working with patients diagnosed with CID. The input stated that current treatment options for insomnia include nonpharmacological therapies, such as sleep hygiene and CBT-I, which can be costly and has limited availability in Canada. The input stated that pharmacological options, particularly GABA agonists, are only prescribed for short-term use, are limited by poor efficacy and next-day sedation, often require dose escalation, can lead to dependence or nonresponse, can lead to withdrawal symptoms upon discontinuation, and are associated with cognitive side effects. In addition, there are limited or no clinical data supporting their use for patients with CID. As such, to improve patient outcomes, the clinician submissions noted that there is a need for treatments that improve daytime function, are tolerable, affordable, and safe.
The clinician input highlighted the unique mechanism of action of daridorexant, which reduces wakefulness without causing sedation. Daridorexant would be used in the first-line setting for CID, after CBT-I or when CBT-I is not suitable, according to the input. With regard to assessing response to treatment, the clinician submissions noted that, given the prevalence of insomnia in Canada and limited resources, frequent polysomnograms are not practical for use in clinical practice. Some of the clinician submissions highlighted the use of the ISI screening tool and other patient-reported outcomes used in research to assess treatment response; however, the clinical experts consulted for this review noted the ISI tool may be less commonly used in Canadian clinical practice. The clinician submissions noted that a clinically meaningful response to treatment includes improved next-day functioning, improved duration and quality of sleep, and a reduction in ISI score. Potential reasons for discontinuing treatment include side effects, such as reduced daytime functioning, nightmares, and sleep paralysis. Various clinicians agreed that daridorexant would be particularly suitable for patients at risk for drug dependence or abuse and for older patients, given the unsuitability of many other drugs because of the potential for cognitive impairment and falls. The clinicians agreed with the clinical experts consulted for this review that daridorexant would likely be prescribed by primary care physicians in outpatient settings, and no specific dependence or rebound issues would need to be addressed for patients discontinuing daridorexant.
Drug Program Input
The drug programs provide input on each drug being reviewed through the Reimbursement Review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
What is the current standard of care for CID? Please include sleep hygiene, CBT-I, and pharmacotherapy used off-label, such as melatonin (for adults 18 years to 64 years and for older adults ≥ 65 years). | CBT-I is the standard of care for patients with CID, if it is available and if the patients are willing to do it. Sleep hygiene by itself is not indicated in CID, as it is not effective for chronic insomnia. There are 4 drugs approved by Health Canada for insomnia (i.e., DORAs, Z-drugs [zolpidem, zopiclone]), and doxepin [for short-term use]). Trazadone is used frequently in clinical settings as an off-label therapy. Several drugs, including antidepressants and serotonin dopamine antagonists (previously known as atypical antipsychotics), are used off-label. Clinicians often prescribe medications based on experience and the failure of previous treatment. |
What proportion of patients 65 years and older experience CID in Canada? Do you have any concerns about patients 65 years and older who are prescribed daridorexant, given the degree of polypharmacy seen in this cohort and the risk of related adverse drug events, including falls and hospitalization? | In Canada, 15% to 20% of patients 65 years and older experience CID. The clinical experts did not identify any additional risk with daridorexant in this population. |
Can you comment on the efficacy of provider-initiated CBT-I vs. mobile CBT-I apps in CID? | Current data suggest that mobile CBT-I can be as effective as provider-initiated CBT-I in the proper context. However, the effectiveness, to some extent, depends on the ability of patients to use the app. |
What is the place in therapy for daridorexant in CID? | The clinical experts indicated that daridorexant may be used as a first-line pharmacological therapy for insomnia disorder, if CBT-I is not effective or not possible. The experts also noted that DORAs may be used in combination with other pharmacologic drugs that are specifically reserved for patients with complex insomnia difficulties. |
Lemborexant received a do not reimburse recommendation from CDEC because of its unclear clinical impact related to variability and data issues, the lack of comparative efficacy relative to short-term pharmacological options, and the questionable improvements on outcomes important to patients. As such, lemborexant is currently not funded by jurisdictions in Canada. | This is a comment from the drug plans to inform CDEC deliberations. The sponsor noted that the population considered in the review of lemborexant is broader than that described in the reimbursement request for daridorexant. |
Considerations for initiation of therapy | |
Should the initiation criteria for daridorexant reflect the inclusion criteria for Study 301? If daridorexant is initiated, how long would clinicians need to follow patients to monitor efficacy and/or safety? In practice, is the ISI tool used by all practitioners to screen for moderate to severe insomnia? How and when is the ISI tool used in practice? Are there other validated tools used to screen for moderate to severe insomnia? | The clinical experts noted that the inclusion criteria for the studies evaluating daridorexant excluded patients with comorbid psychiatric or other disorders, which would not be the case in clinical practice. The clinical experts stated that in the primary care setting, if patients have insomnia and are older than 18 years, daridorexant could be initiated. The experts also noted that once initiated, 2 months would be a reasonable time to monitor for treatment response and follow a patient for efficacy. When patients have safety concerns, they are asked to discontinue treatment and discuss them with the clinician during their next visit. The clinical experts noted that the ISI tool is not routinely used by practitioners in Canada. However, tools like the Pittsburgh Sleep Quality Index or the ISI may be used in select settings or for research purposes. Insomnia is routinely screened and monitored during clinical assessment, which usually consists of patient-reported sleep history (i.e., difficulties initiating and/or maintaining sleep, daytime function, frequency and severity of symptoms). |
Are patients with stable coexisting psychiatric illnesses (e.g., patients with substance use disorder who are taking opioid agonist treatment plus ADHD, stimulants plus depression, or antidepressants) or medical conditions (e.g., diabetes plus heart disease) eligible for daridorexant? | The clinical experts said they consider these patients eligible for treatment with daridorexant. The only absolute contraindication to daridorexant is narcolepsy. |
In practice, what happens when a patient presents with CID and a BMI less than 18.5 kg/m2 or greater than 40.0 kg/m2, or when a patient presents with CID and an MMSE score less than 25? Is a benefit-risk assessment of individual patients performed before daridorexant is prescribed? | The clinical experts do not consider BMI to be a concern, and noted that patients are generally not screened for cognitive impairment. A benefit-risk assessment would not be performed for individual patients before daridorexant is prescribed. |
Should all patients with CID be offered CBT-I before the initiation of daridorexant? How long after the initiation of CBT-I should the patient be assessed for response? And when would daridorexant be initiated in this context? | Yes, all patients with CID should be offered CBT-I, but it is not always available. If patients receive CBT-I, they could be assessed after 2 or 3 months (once the course is finished) to determine if any pharmaceutical drug would be of further benefit. |
Considerations for continuation or renewal of therapy | |
What tools are used in clinical practice to assess and monitor the therapeutic response of daridorexant in patients with CID? | Clinical assessment is the primary way to assess and monitor therapeutic response to daridorexant. No tools are routinely used in clinical practice. |
In patients with CID on daridorexant, is there a definition of full vs. partial responder? If so, how are partial responders defined and managed? | Generally, there is no standard clinical definition of what a partial response is in patients with CID. |
Considerations for discontinuation of therapy | |
What parameters define loss of response, absence of clinical benefit, or worsening of CID? | The patient’s assessment of sleep, as well as daytime functioning, could define loss of response, absence of clinical benefit, or worsening of CID. |
Considerations for prescribing of therapy | |
Which discipline would diagnose, assess, and treat patients with CID in Canada? | Any and all disciplines could diagnose, assess, and treat patients with CID. Examples include family physicians, psychiatrists, internists, and neurologists. |
Care provision issues | |
Drugs used to treat patients with CID must meet a wide range of requirements, including no development of tolerance, no risk of dependence, and no withdrawal symptoms. Does daridorexant meet these requirements? | According to the evidence included in the pivotal studies, daridorexant is not associated with dependence or withdrawal symptoms. |
ADHD = attention-deficit/hyperactivity disorder; BMI = body mass index; CBT-I = cognitive behavioural therapy for insomnia; CID = chronic insomnia disorder; CDEC = Canadian Drug Expert Committee; DORA = dual orexin receptor antagonist; ISI = Insomnia Severity Index; MMSE = Mini Mental State Examination; vs. = versus.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of daridorexant 25 mg and 50 mg oral tablets in the treatment of moderate to severe CID when CBT-I is inappropriate, unavailable, or has failed. CID is characterized by difficulty initiating or maintaining sleep, early awakenings, at least 3 occurrences weekly, a minimum duration of 3 months, and an ISI score of at least 15. The focus will be placed on comparing daridorexant to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of daridorexant is presented in 2 sections, with a CDA-AMC critical appraisal of the evidence included at the end of each section. The first section, the Systematic Review, includes pivotal studies and RCTs that were selected in accordance with the sponsor’s systematic review protocol, plus additional evidence that was identified as relevant to the indication under review. The CDA-AMC assessment of the certainty of the evidence in the first section, using the GRADE approach, follows the critical appraisal of the evidence. The second section includes a sponsor-submitted LTE study. No indirect evidence or additional studies were included.
Included Studies
Clinical evidence from the following are included in the review and appraised in this document:
2 pivotal trials identified in the systematic review
1 LTE study.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5. Two phase III, double-blind, parallel-group, placebo-controlled, randomized trials were included in this review. Both studies were conducted between 2018 and 2020, with database locks on March 31, 2020 (Study 301), and June 18, 2020 (Study 302). Study 301 was conducted across 10 countries in Europe and North America, including 4 sites in Canada. Study 302 was conducted across 11 countries in Europe, North America, and Asia, including 5 sites in Canada.
Study 301 and Study 302 were both designed to assess efficacy and safety of daridorexant in patients with insomnia disorder. Study 301 compared daridorexant 50 mg and daridorexant 25 mg to placebo, whereas Study 302 compared daridorexant 10 mg and daridorexant 25 mg to placebo. Because the 10 mg dosage is not approved by Health Canada, data are not presented for that group.
Table 5: Details of Studies Included in the Systematic Review — Study 301 and Study 302
Detail | Study 301 | Study 302 |
|---|---|---|
Designs and populations | ||
Study design | Phase III, multicentre, double-blind, placebo-controlled, parallel-group, randomized trial. | Phase III, multicentre, double-blind, placebo-controlled, parallel-group, randomized trial. |
Locations | The study was conducted at 75 sites in 10 countries (Australia, Canada, Denmark, Germany, Italy, Poland, Serbia, Spain, Switzerland, and the US) | The study was conducted at 81 sites in 11 countries (Belgium, Bulgaria, Canada, Czech Republic, Finland, France, Germany, Hungary, Republic of South Korea, Sweden, and the US) |
Patient enrolment dates | Start date: June 4, 2018 End date: February 25, 2020 | Start date: May 29, 2018 End date: May 14, 2020 |
Randomized (N) | Total: N = 930
| Total: N = 924
|
Inclusion criteria |
| |
Exclusion criteria |
| |
Drugs | ||
Intervention |
|
|
Comparators | Placebo film-coated tablets, with 1 tablet taken in the evening, orally, during the single-blind placebo run-in period (13 to 24 days); during the randomized, double-blind treatment period (84 ± 2 days); and during the single-blind placebo run-out period (7 + 2 days) | Placebo film-coated tablets, with 1 tablet taken in the evening, orally, during the single-blind placebo run-in period (13 to 24 days); during the randomized, double-blind treatment period (84 ± 2 days); and during the single-blind placebo run-out period (7 + 2 days) |
Study duration | ||
Screening phase | 7 to 18 days (visit 1 to visit 2) | |
Run-in phase | 13 to 24 days (visit 2 to visit 4) | |
Treatment phase | From randomization (day 1, visit 4) until EODBT (morning of day 85, visit 8) | |
Follow-up phase | From EODBT (evening of day 85, visit 9) until EOS (i.e., optional enrolment in Study 303 or follow-up telephone call 30 days after the last dose of double-blind treatment for patients who did not enter the extension study) | |
Outcomes | ||
Primary end point |
| |
Secondary and exploratory end points | Secondary end points:
Other efficacy end points:
Other exploratory end points
| |
Publication status | ||
Publications | Clinicaltrial.gov (NCT03545191)30 Mignot et al.31 Fietze et al.32 Phillips-Beyer et al. (2023)33 Clinical Study Report for Study 30116 | Clinicaltrial.gov (NCT03575104)34 Mignot et al.31 Clinical Study Report for Study 30217 |
BMI = body mass index; CBT = cognitive behavioural therapy; CYP3A4 = cytochrome P450 3A4; DSM-5 = Diagnostic and Statistical Manual of Mental Disorders (Fifth Edition); EODBT = end of double-blind treatment; EOS = end of study; IDSIQ = Insomnia Daytime Symptoms and Impacts Questionnaire; ISI = Insomnia Severity Index; LPS = latency to persistent sleep; PGA-S = Patient Global Assessment of Disease Severity; PGI-C = Patient Global Impression of Change; PGI-S = Patient Global Impression of Severity; PSG = polysomnography; REM = rapid eye movement; SDQ = Sleep Disorders Questionnaire; sLSO = subjective latency to sleep onset; sTST = subjective total sleep time; SWS = slow-wave sleep; S1 = stage 1 sleep; S2 = stage 2 sleep; TST = total sleep time; VAS = visual analogue scale; WASO = wake after sleep onset.
aAs reported in the SDQ, completed at home between visit 2 and visit 3.
Sources: Clinical Study Report for Study 301,16 Mignot et al.,31 Clinical Study Report for Study 302.17 Details included in the table are from the sponsor’s Summary of Clinical Evidence and Clinical Study Reports.
Figure 1 shows the design of Study 301 and Study 302. Both studies consisted of a screening phase, a double-blind treatment phase, and a safety follow-up phase. Patients who completed study treatment (double-blind treatment and placebo run-out treatment) in the core study were eligible to enter an extension study (Study 303).
The screening period lasted from 7 days to 18 days (from visit 1 to visit 2) and consisted of an initial verification of eligibility, followed by a one-night PSG assessment and a minimum of 7 daily SDQ entries. The screening phase also included a placebo run-in period that lasted from 13 days to 24 days (from visit 2 to visit 4) and consisted of confirmation of eligibility, single-blind placebo treatment once daily, and 2 PSG nights (visit 3) performed after patients had completed at least 7 daily SDQ entries.
The double-blind treatment phase ran from randomization (day 1, visit 4) until the end of double-blind treatment (morning of day 85, visit 8). Randomization was performed with an Interactive Response Technology system, and treatment allocation was stratified by age into 2 categories (< 65 years and ≥ 65 years). In Study 301, patients were randomized in a 1:1:1 ratio to daridorexant 25 mg, daridorexant 50 mg, or placebo; in Study 302, patients were randomized to daridorexant 25 mg or placebo. Double-blind study treatment was given once daily from day 1 to day 84.
The safety follow-up phase consisted of 2 periods. The first period was a placebo run-out period, from the end of double-blind treatment (evening of day 85, visit 9) until the end of treatment (day 92, visit 10), which included 7 days of single-blind placebo treatment once daily (day 85 to day 91) and 1 PSG night (day 85 to day 86). The second period involved a safety follow-up period, from the end of treatment (day 92, visit 10) until the end of the study, which was either the enrolment date in Study 303 or the date of the 30-day follow-up telephone call (day 115, visit 11) for patients who did not enter the extension study. During this period, information on AEs, SAEs, and concomitant medications was collected.
Figure 1: Study Design of Study 301 and Study 302
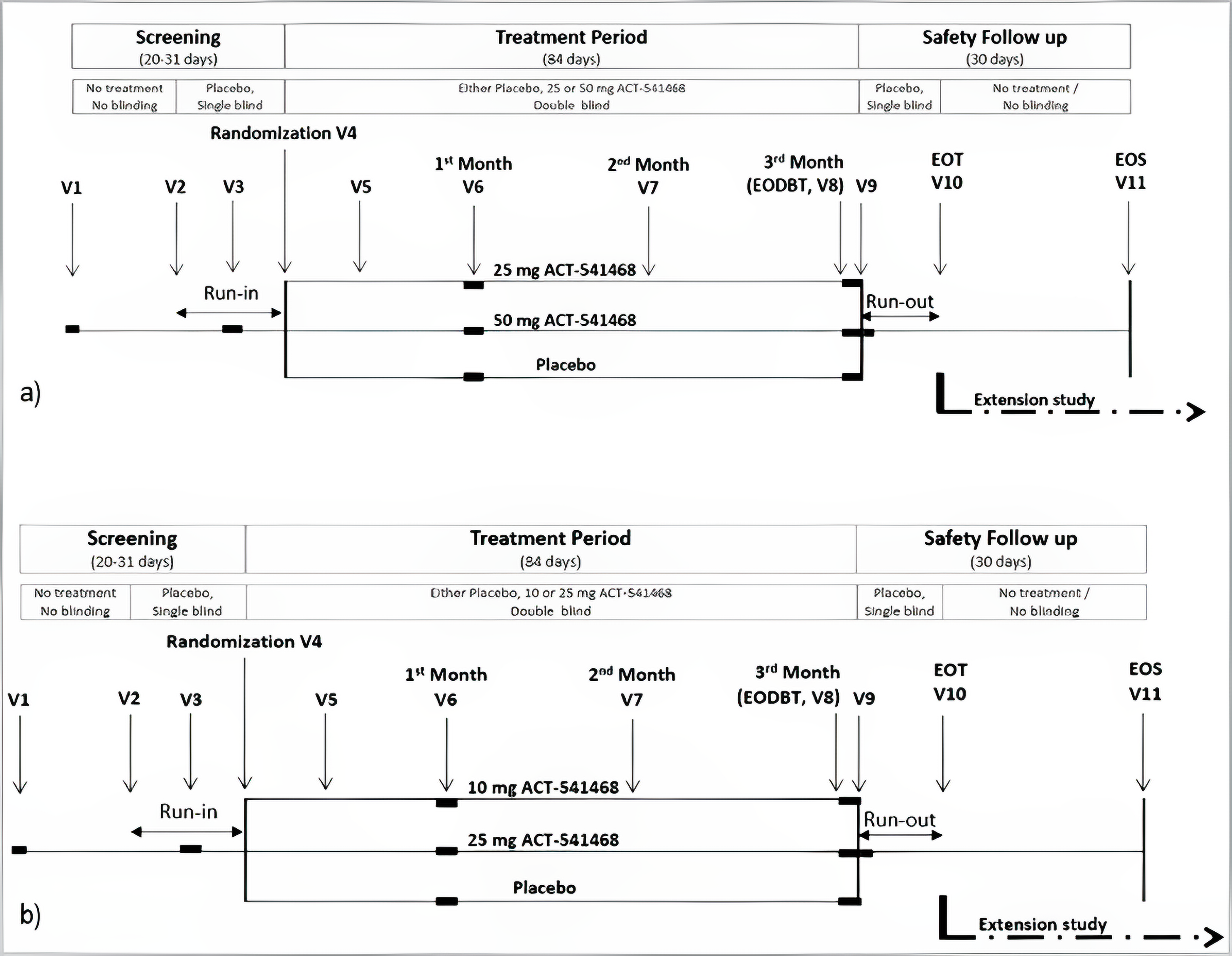
ACT-541468 = daridorexant; EODBT = end of double-blind treatment; EOT = end of treatment; EOS = end of study; V = visit.
Sources: Clinical Study Reports for Study 30116 and Study 302.17
Populations
Inclusion and Exclusion Criteria
Study 301 and Study 302 enrolled adult patients, aged 18 years or older, who were diagnosed with insomnia disorder, according to the DSM-5 criteria. Patients with moderate to severe insomnia per the ISI (a score of 15 or more) at screening were deemed eligible for enrolment. Other key inclusion criteria included self-reported insufficient sleep quantity for at least 3 nights a week for at least 3 months before the screening visit, and for at least 3 of 7 nights on the SDQ completed during the placebo run-in period before the run-in PSG nights. Patients also had to meet sleep parameters on the PSG nights at visit 3, such as average TST less than 420 minutes, WASO for 30 minutes or more, or a sleep onset time of 20 minutes or more. Key exclusion criteria included self-reported daytime napping of at least 1 hour per day on at least 3 days per week. Patients with specific conditions were also excluded, such as those with acute or unstable psychiatric conditions, suicidal ideation with intent or any lifetime history of suicide attempt, alcohol or drug abuse, sleep-related breathing disorders, periodic limb movement disorder, restless legs syndrome, circadian rhythm disorder, REM behaviour disorder, narcolepsy, or apnea and/or hypopnea.
Interventions
In both trials, the study phases alternated between a single-blind placebo period, a double-blind treatment period, and another single-blind placebo period. The placebo periods included a run-in period (13 days to 24 days) and a run-out period (7 + 2 days), during which patients were administered placebo tablets that matched the daridorexant tablets.
During the double-blind treatment period, patients received the following interventions, based on their assigned treatment arm.
Study 301:
Daridorexant film-coated tablets, 25 mg, with 1 tablet taken in the evening, orally, during the randomized double-blind treatment period (84 ± 2 days)
Daridorexant film-coated tablets, 50 mg, with 1 tablet taken in the evening, orally, during the randomized double-blind treatment period (84 ± 2 days)
Placebo film-coated tablets, with 1 tablet taken in the evening, orally, during the single-blind placebo run-in period (13 days to 24 days), the randomized double-blind treatment period (84 ± 2 days), and the single-blind placebo run-out period (7 + 2 days).
Study 302:
Daridorexant film-coated tablets, 25 mg, with 1 tablet taken in the evening, orally, during the randomized double-blind treatment period (84 ± 2 days)
Placebo film-coated tablets, with 1 tablet taken in the evening, orally, during the single-blind placebo run-in period (13 days to 24 days), the randomized double-blind treatment period (84 ± 2 days), and the single-blind placebo run-out period (7 + 2 days).
The investigational treatment and its matching placebo were indistinguishable, and all treatment wallets were packaged in the same way. At the site, 1 tablet of study treatment was taken orally at least 2 hours after the last meal and 30 ± 5 minutes before lights off on the evening of each PSG night. At home, the study treatment was taken orally at bedtime. If 1 or more doses were missed, the next dose had to be taken on the evening of the following day.
Concomitant pharmacological treatments for insomnia or other CNS-related medications were prohibited during the study to optimize the evaluation of the safety and efficacy profile of daridorexant. Cognitive behaviour therapy (CBT) for any indication was only allowed if the treatment started at least 1 month before visit 3 and the patient agreed to continue CBT throughout the study.
Outcomes
A list of efficacy and safety end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence, as well as any outcomes identified as important to this review by the clinical experts consulted for this review and in input from patient and clinician groups and public drug plans. Using the same considerations, CDA-AMC selected end points that were considered to be most relevant to expert committee deliberations and finalized this list of end points in consultation with members of the expert committee.
Select summarized efficacy end points were assessed using GRADE. They included sleep maintenance, sleep onset, sleep duration, and sleep quality. For the safety end points, the incidence of SAEs was assessed using GRADE.
Objective Outcomes
Objective outcomes were measured in both studies using PSG.
Sleep Maintenance (WASO)
One of the primary efficacy end points in Study 301 and Study 302 was change from baseline in WASO, defined as the time spent awake after the onset of persistent sleep (beginning of the first continuous 20 epochs [i.e., 10 minutes]) scored as nonawake. Improved sleep is indicated by a decrease in WASO duration.
PSG recordings were performed by technologists during sleep-study visits over the course of the study. The sponsors reported that the construct validity of the end point was demonstrated with a positive and statistically significant correlation (Spearman rank correlation, 0.61; P value < 0.0001) between the actigraphy-derived measure, which is another objective measure of sleep disorders, and the PSG-derived measure of WASO.35 The mean of multiple WASO measurements over 3 nights demonstrated substantial test-retest reliability, with an intraclass correlation coefficient of 0.64 (95% CI, 0.52 to 0.73) in patients with insomnia.36 According to American Academy of Sleep Medicine (AASM) expert consensus, the threshold for a clinically meaningful difference from placebo in group-level results is a mean change of at least 20 minutes between the treated and placebo arms.37 The clinical experts consulted for this review confirmed that this minimal important difference (MID) is clinically meaningful.
Table 6: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | Study 301 | Study 302 |
|---|---|---|---|
Change from baselinea in WASO (sleep maintenance)b | Month 1c and month 3c | Primary | Primary |
Change from baselinea in LPS (sleep onset)b | Month 1c and month 3c | Primary | Primary |
Change from baselinea in sTSTb | Month 1c and month 3c | Secondary | Secondary |
Change from baselined in sleep quality, determined by SDQ VAS scores | Month 1e and month 3e | Exploratory | Exploratory |
Change from baseline in ISI score (total score) | Month 1 and month 3 | Exploratory | Exploratory |
Change from baseline in IDSIQ total score | Month 1f and month 3f | Other efficacy end points | Other efficacy end points |
Change from baseline in duration of TST in each sleep stage (S1, S2, SWS, and REM) | Month 1 and month 3 | Exploratory | Exploratory |
TEAEs, SAEs, AESIs (i.e., narcolepsy-like symptoms, suicide and/or self-injury)g | Up to 30 days after double-blind study treatment discontinuation or until enrolment in the extension study | Safety | Safety |
Occurrence of suicidal ideation and/or behaviour on double-blind study treatment, based on C-SSRS | During double-blind study treatment During the placebo run-out period | Safety | Safety |
Rebound insomnia, assessed based on WASO, LPS, and sTST | During the placebo run-out period,h compared to baselined | Safety | Safety |
Next-morning residual effect (referred to in the protocol as next-day residual effect), assessed based on changes from baselined in morning sleepiness score on the SDQ VAS | Month 1f and month 3f | Safety | Safety |
AESI = adverse event of special interest; C-SSRS = Columbia Suicide Severity Rating Scale; IDSIQ = Insomnia Daytime Symptoms and Impacts Questionnaire; ISI = Insomnia Severity Index; LPS = latency to persistent sleep; REM = rapid eye movement; SAE = serious adverse event; SDQ = Sleep Diary Questionnaire; sTST = subjective total sleep time; SWS = slow-wave sleep; TEAE = treatment-emergent adverse event; TST= total sleep time; VAS = visual analogue scale; WASO = wake after sleep onset.
aBaseline: mean of the 2 polysomnography (PSG) nights at visit 3.
bTo account for the concurrent evaluation (multiple comparison) of 2 distinct end point categories (sleep maintenance and sleep onset), a Bonferroni correction was applied. The remaining hypotheses were tested using the gatekeeping strategy.
cMonth 1 and month 3: mean of the 2 PSG nights at visit 6 and visit 8, respectively.
dBaseline: mean value based on the screening SDQ or IDSIQ entries performed at home in the 7 days immediately preceding the first PSG at visit 3.
eMonth 1 and month 3: mean value based on the SDQ or IDSIQ entries performed at home in the 7 days immediately preceding the first PSG at visit 6 and visit 8, respectively.
fMonth 3: visit 8 or, if that is missing, week 12 of the questionnaire.
gAn Independent Safety Board reviewed blinded data on selected adverse events and safety assessment results and adjudicated whether the submitted cases were AESIs.
hPlacebo run-out period: mean value based on the SDQ entries performed in the 7 days immediately after the PSG night at visit 9.
Sources: Clinical Study Reports for Study 30116 and Study 30217. Details included in the table are from the sponsor’s Summary of Clinical Evidence and the Clinical Study Reports.
Sleep Onset (LPS)
The other primary end point in Study 301 and Study 302 was the change from baseline in LPS (i.e., sleep onset). LPS was defined as the time from the start of recording to the beginning of the first continuous 20 epochs (i.e., 10 minute) scored as nonawake (i.e., epochs scored as S1, S2, SWS, or REM), on PSG. An improved sleep is indicated by a decrease in LPS duration. According to AASM expert consensus, the suggested threshold for a clinically meaningful difference from placebo in group-level results is defined as a mean change of at least 10 minutes between the treated and placebo arms.37 The clinical experts consulted for this review confirmed that this MID is clinically meaningful.
Subjective Outcomes
Patient-reported outcome instruments were used in Study 301 and Study 302 to evaluate patients' perceptions of their sleep issues and how these problems affect their daytime activities. They included quality of sleep measured on a VAS of SDQ, ISI, and IDSIQ. Table 7 presents the descriptions of the outcome measures and a summary of the interpretation and validity of these instruments.
Sleep Duration (sTST)
The secondary end point was the change from baseline in sTST. sTST provides an overall assessment of sleep times, incorporating time to sleep onset and wake time after sleep onset. According to the sponsor, sTST data permitted the translation of the PSG results to a benefit felt by patients. sTST was defined as the time reported by the patient in response to the SDQ morning questionnaire question: In total, how long did you sleep last night? In addition, rebound insomnia, an exploratory end point, was evaluated by comparing the sTST value obtained during the placebo run-out period to the baseline sTST value. In terms of end point validity, sTST has demonstrated a moderate to strong convergent validity with total ISI score, actigraphy-derived TST, and PSG-derived TST.38-40 Following an assessment of data from an open-label, phase II trial of zolpidem and pooled data from a phase III, placebo-controlled trial of daridorexant in patients with moderate to severe insomnia, it was concluded that an improvement of 55 minutes from baseline in sTST is considered a clinically meaningful improvement of this outcome.41,42 Of note, according to AASM expert consensus, the suggested threshold for a clinically meaningful difference from placebo in group-level results is defined as a mean change of at least 30 minutes between the treated and placebo arms.37
Sleep Quality
Change from baseline in sleep quality was an exploratory end point of the included trials, and was measured using the SDQ, which is described in Table 7. The SDQ was developed by the sponsor, based on the Consensus Sleep Diary, morning administration, questionnaire and adapted for use in clinical trials. The SDQ was completed by the patient, without study staff input or interference, and included a morning and evening questionnaire and a VAS. The morning and evening questionnaire collected information on self-reported sleep characteristics (sleep induction and maintenance), habitual napping, bedtime, and timing of study treatment intake. The VAS collected information on quality of sleep, depth of sleep, and morning sleepiness (all assessed in the morning), and daytime alertness and daily ability to function (both assessed in the evening) by asking patients to report their feelings by placing a mark on a VAS.
Daytime Functioning
Daytime functioning was measured using IDSIQ total score, which is described in Table 7. The IDSIQ is a patient-reported outcome measure structured in 3 domains (alert/cognition, mood, sleepiness). The IDSIQ sleepiness domain score, based on patient responses to 4 items, could range from 0 to 40 (whole numbers only), with higher scores indicating a greater burden of illness during the daytime.
Insomnia Symptoms
The assessment of global insomnia symptom severity was conducted using the ISI, which is described in Table 7. Eligible patients had to have self-reported insomnia of at least moderate severity (ISI score ≥ 15) at screening. The ISI is a 7-item questionnaire that assesses insomnia symptoms and their impact on the severity of sleep onset and sleep maintenance difficulties, and any insomnia-related interference with daytime functioning. The assessment is based on a 5-point Likert scale (range, 0 to 4), where the composite score was obtained by summing the 7 rated dimensions that measure patients’ perceptions of their insomnia. A score of 15 to 21 indicates a moderate level of insomnia, and a score of 22 to 28 indicates severe insomnia.
Safety End Points
Safety end points included AEs reported by the investigators (i.e., TEAEs, SAEs, AEs leading to premature discontinuation of study treatment), independent safety board–adjudicated AESIs (i.e., narcolepsy-like symptoms and suicide and/or self-injury). Other safety end points relevant to the current review include next-morning residual effect, rebound insomnia, and the occurrence of suicidal ideation and/or behaviour.
Next-Morning Residual Effect
The next-morning residual effect was a safety end point assessed using changes from baseline in the morning sleepiness score on the VAS of the SDQ. For the quality of sleep end point, a positive change from baseline indicates a better outcome, meaning a decrease in morning sleepiness.
Rebound Insomnia
Rebound insomnia, characterized by the return or exacerbation of sleep onset or sleep maintenance difficulties upon discontinuation of the treatment, was a safety outcome of interest in this review. It was assessed using WASO, LPS at the start of the placebo run-out period (visit 9) compared to baseline (visit 3), and the subjective sleep parameter sTST from the placebo run-out period compared to baseline.
Occurrence of Suicidal Ideation and/or Behaviour
The Columbia Suicide Severity Rating Scale (C-SSRS) questionnaire was used to identify the presence and severity of suicidal ideation and suicidal behaviours, which were safety end points. The C-SSRS also captures information on the intensity of ideation (frequency, duration, controllability, and deterrents) and reasons for the most severe types of ideations. The C-SSRS contains 10 outcome categories, each of which can be responded to with a binary response (yes or no). A trained health care professional, in accordance with local requirements and local clinical practice, was required to complete the questionnaire together with the patient on a tablet device and assess the findings. Moreover, good convergent and divergent validity was demonstrated between the C-SSRS and other multi-informant suicidal ideation and behavioural scales.43
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
WASO | PSG-derived measure that refers to periods of wakefulness that occur after defined sleep onset. It measures time spent awake (in minutes) after initial sleep onset until final awakening.44-46 A shorter duration indicates improved sleep. | Validity: Moderate correlation between PSG-derived and actigraphy-derived WASO (r = 0.61).35 ROC analysis showed that PSG-derived WASO does not accurately distinguish between those with insomnia and those without (AUC = 0.55).47 Reliability: Moderate test-retest reliability with multiple LPS measurements over 3 nights (ICC = 0.69; 95% CI, 0.59 to 0.77) in patients with insomnia.36 Responsiveness: No data were found. | In AASM guidelines (2017) and an AASM systematic review (2021) based on expert consensus, the suggested threshold for a clinically important difference relative to placebo (between-group change) is a mean change of ≥ 20 minutes.37,48 |
LPS | PSG-derived measure that estimates the time (in minutes) from the start of recording to the start of the first of 20 consecutive epochs of nonwakefulness.36,46,49 A shorter duration indicates improved sleep. | Validity: ROC analysis showed that PSG-derived LPS does not accurately distinguish between those with insomnia and those without (AUC = 0.63).47 Reliability: Substantial test-retest reliability with multiple LPS measurements over 3 nights (ICC = 0.80; 95% CI, 0.73 to 0.85) in patients with insomnia.36 Responsiveness: No data were found. | In AASM guidelines (2017) based on expert consensus, the suggested threshold for a clinically important difference relative to placebo (between-group change) is a mean change of ≥ 10 minutes.37 |
sTST | Self-reported measure derived from a sleep diary that estimates the duration of sleep (in minutes) from sleep onset to sleep offset.44-46 A longer sTST indicates improved sleep. | Validity: Convergent validity was supported by a strong correlation between the total ISI score and sTST (r = –0.54).38 Strong correlation between sTST and actigraphy-derived TST (r = 0.64).40 Discriminant validity has been demonstrated between patients with insomnia and those in the control group (AUC = 0.87; 95% CI, 0.80 to 0.93).50 Reliability and responsiveness: No data were found. | In AASM guidelines (2017) based on expert consensus, the suggested threshold for a clinically important difference relative to placebo (between-group change) is a mean change of ≥ 30 minutes.37 In a study evaluating adults with insomnia disorder using DSM-5 criteria, the mean change was ≥ 55 minutes from baseline to day 15 (within-group change).42 |
ISI | Self-reported 7-item instrument used to assess the severity of insomnia by scoring the severity of sleep onset and sleep maintenance difficulties and any insomnia-related interference with daytime functioning. Each item is rated on a 5-point Likert scale, with the total score ranging from 0 to 28.38 Higher scores indicate more severe insomnia. | Validity: Strong correlations between ISI items and subjective sleep measures: ISI total score correlated with PSQI (r = 0.80) and with the SF-12 mental component (r = –0.51).38 Moderate to strong correlations with sleep diary measures (r = 0.32 to 0.91).44 Reliability: High internal consistency (Cronbach alpha = 0.90 to 0.92).38,51 Responsiveness: Sensitive to changes in perceived sleep difficulties related to treatment.38,51 | In a study evaluating adult patients with primary insomnia, a 6-point reduction was thought to represent a clinically meaningful improvement (within-group change).52 In a study evaluating patients with insomnia complaints, insomnia diagnosis, and no insomnia, there was a 7-point reduction in moderate improvements and a 9-point reduction in marked improvements (within-group change).38 In a study assessing RCTs for insomnia interventions, 6 studies used a relative decrease from 2.8 to 4.0 points (between-group change) as the MID.53 |
IDSIQ | Self-reported 14-item instrument used to evaluate daytime functioning in people with insomnia. Consists of 3 main domains: alert/cognition, mood, and sleepiness. A lower score indicates improvement in daytime functioning.54 | Validity: Moderate to strong correlations with the PROMIS Sleep T score in patients with and without insomnia. Weak to moderate correlations with ISI total score in patients with and without insomnia.54 Reliability: High internal consistency (alpha = 0.917). Good test-retest reliability (ICC = 0.856 to 0.911).54 Responsiveness: Decreases in mean total scores among patients with insomnia who reported decreases or no change in disease severity on PGI-C, PGA-S, or ISI scales.54 | In a study evaluating adults with insomnia disorder using DSM-5 criteria, there was a ≥ 17-point reduction in the total score, a 9-point reduction in the alert/cognition domain, and a 4-point reduction in the mood and sleepiness domains (within-group change).33 In a study evaluating adults with and without insomnia disorder using DSM-5 criteria, the suggested threshold for a clinically meaningful important difference is a ≥ 20-point reduction in the total score (within-group change).54 |
SDQ VAS | Self-reported instrument; patients report their feelings by placing a mark on a VAS indicating their quality of sleep, depth of sleep, and morning sleepiness (assessed in the morning), and daytime alertness and daily ability to function (assessed in the evening).16 | Validity: Strong correlation between the IDSIQ total score and both VAS quality of sleep and VAS morning sleepiness.55 Reliability and responsiveness: No data were found. | No data were found. |
AASM = American Academy of Sleep Medicine; AUC = area under the curve; CI = confidence interval; DSM-5 = Diagnostic and Statistical Manual of Mental Disorders (Fifth Edition); ICC = intraclass correlation coefficient; IDSIQ = Insomnia Daytime Symptoms and Impacts Questionnaire; ISI = Insomnia Severity Index; LPS = latency to persistent sleep; MID = minimal important difference; PGA-S: Patient Global Assessment of Disease Severity; PGI-C = Patient Global Impression of Change; PROMIS = Patient-Reported Outcomes Measurement Information System; PSG = polysomnography; PSQI = Pittsburgh Sleep Quality Index; RCT = randomized controlled trial; ROC = receiver operating characteristic; SDQ = Sleep Diary Questionnaire; SF-12 = 12-Item Short Form Survey; sTST = subjective total sleep time; TST = total sleep time; VAS = visual analogue scale; WASO = wake after sleep onset.
Statistical Analysis
Sample Size and Power Calculation
Study 301 and Study 302 used similar assumptions and calculations to estimate sample size. The difference compared to placebo in the mean change from baseline to month 1 and to month 3 (± the assumed SD for each treatment group) for WASO, LPS, and sTST was assumed to be 15 minutes (± 40 minutes), 15 minutes (± 40 minutes), and 20 minutes (± 54 minutes), respectively (based on phase II data from 2 studies: AC-078A201 and AC-078A202).56,57 The corresponding effect size (mean/SD) was approximately 0.37.
In Study 301, based on a two-sample z-test, a sample size of at least 900 patients randomized to daridorexant 25 mg, daridorexant 50 mg, or placebo in a 1:1:1 ratio (i.e., 300 per group) would provide 98.9% power to detect an effect size of 0.37 for a single hypothesis test. Similarly, in Study 302, a sample size of at least 900 patients randomized to daridorexant 10 mg, daridorexant 25 mg, or placebo would provide that power to detect an effect size of 0.37. In both studies, this accounts for the Bonferroni correction, in which the significance level (alpha) is halved and set to a 2-sided 2.5% significance level. However, as the number of null hypotheses (end points) to test increases, the power decreases. The power calculation assumed that all null hypotheses were independent, which is a conservative assumption for power calculations. Consequently, 900 subjects were needed to provide at least 90% power to detect an effect size of 0.37 when 9 independent null hypotheses were tested.
Efficacy End Points
In both studies, the analysis of the primary and key secondary end points, involving change from baseline in WASO, LPS, and sTST, employed a longitudinal data analysis method (specifically, a linear mixed-effect model). They are outlined in Table 8.
The analysis models were adjusted for multiple values, such as the baseline value of the relevant response variable (WASO, LPS, or sTST), age group (< 65 years, ≥ 65 years), treatment (higher dose, lower dose, placebo), visit (month 1, month 3), and the interaction of treatment by visit and baseline by visit. Study-wise type I error control was implemented across the 2 primary and 2 secondary end points, as well as across 2 time points of outcome assessments (month 1 and month 3) and 2 dose-level comparisons versus placebo. Thus, overall, 16 independent null hypotheses were tested in the study, with appropriate type I error control for each of the included studies.
As a way to evaluate the efficacy hypotheses, appropriate contrasts were computed to test the treatment differences of interest. Difference of interest was the difference in LSM change from baseline between higher-dose daridorexant and placebo and between lower-dose daridorexant and placebo, both at month 1 and month 3.
The analysis of the other efficacy end points, specifically concerning the IDSIQ total score, was performed using the same model as the main analysis, which is the linear mixed-effects model. The LSM for each treatment group at each time point was displayed along with associated standard errors and 95% CIs.
For all end points, observed values at month 1 and month 3, along with baseline data, are reported as mean (SD). The analysis of the exploratory end points (change from baseline to month 1 and to month 3 of the respective variables), as well as the safety end points, was summarized descriptively using the observed values.
Partially missing data were handled with implicit imputation, which means that missing data points are given the same value as the mean of the nonmissing data points of that same time point or week. WASO and LPS values were handled as follows: if 1 of the 2 values was missing for baseline, month 1, or month 3, the single value available was used as the mean for that time point. If both values were missing for a time point, then the mean value was considered missing for that time point. The same approach was used for the Sheehan Disability Scale. For sTST and IDSIQ sleepiness domain score values, patients had to have at least 2 days of data during each week to calculate a weekly mean. Otherwise, the mean value was considered missing for that week. The same approach was used for the IDSIQ total score and VAS scores.
Statistical Testing
The type I error rate was controlled for the testing of multiple null hypotheses associated with the primary and secondary end points assessed at month 1 and month 3 of treatment and the 2 dose levels (i.e., 25 mg and 50 mg in Study 301 and 10 mg and 25 mg in Study 302) included in the studies. For each of the primary end points (change from baseline in WASO [sleep maintenance] and LPS [sleep onset]) and secondary end points (change from baseline in sTST [sleep quantity], and IDSIQ sleepiness domain score [daytime function]), 4 null hypotheses were defined as follows:
Hypothesis 1: Higher dose – placebo = 0 at month 1
Hypothesis 2: Higher dose – placebo = 0 at month 3
Hypothesis 3: Lower dose – placebo = 0 at month 1
Hypothesis 4: Lower dose – placebo = 0 at month 3
In these 4 hypotheses, higher dose (i.e., 50 mg in Study 301, 25 mg in Study 302), lower dose (i.e., 25 mg in Study 301, 10 mg in Study 302), and placebo represent the mean change from baseline for the given end point (WASO, LPS, sTST, or IDSIQ sleepiness domain score) and time point (month 1 or month 3).
Each null hypothesis was tested against the alternative hypothesis that daridorexant improved the respective end point at the given dose and time point, compared to placebo.
The order of testing and the alpha level applied to each null hypothesis was based on the Bonferroni-based gatekeeping procedure that controlled the study-wise type I error at a two-sided 5% significance level.58 To account for the concurrent evaluation (multiple comparison) of 2 distinct end point categories (i.e., sleep maintenance and sleep onset), a Bonferroni correction was applied. Both end point categories were tested at half of the two-sided 5% significance level.
The remaining hypotheses were tested using the gatekeeping strategy, moving from month 1 to month 3 for higher-dose daridorexant versus placebo and then from month 1 to month 3 for lower-dose daridorexant versus placebo. The prespecified proportion of alpha (weight) that was distributed once a given null hypothesis (node) was rejected is shown on the arrow in Figure 2 (Study 301) and Figure 3 (Study 302). If a certain null hypothesis could not be rejected, then the alpha level used for that test was absorbed at that node and not distributed further.
For the other secondary end point, change from baseline in IDSIQ total score, nominal two-sided P values (versus placebo) were not adjusted under type I error rate control (i.e., not adjusted for multiplicity).
For the primary and secondary end points, the linear mixed-effects model was run using an unstructured covariance matrix, shared across treatment groups, to model the correlation among repeated measurements. A restricted maximum likelihood approach was used to derive (unbiased) estimates of variance components. The Kenward-Roger approximation was used to compute the denominator degrees of freedom and adjust standard errors.59
Subgroup Analyses
Subgroup analyses for the primary and secondary efficacy end points, as outlined in the statistical analysis plan and included as a relevant subgroup in the systematic literature review, were conducted in both studies. The subgroups were based on age (≥ 65 years, < 65 years) and sex (male, female) in the studies.
Various efficacy end points were analyzed, including the change from baseline in WASO, LPS, sTST, and ISI score. The next-morning residual effect was assessed as a safety end point. Regarding primary and secondary end points, the same model as for the main analysis (i.e., linear mixed-effects model) was used, except that age was not used as covariate in the model. Treatment effect estimates (LSM difference between daridorexant 50 mg and placebo, along with 95% CIs) are depicted in forest plots for primary and secondary efficacy end points. All results presented are descriptive, as these secondary analyses were not controlled for type I error rate or specifically powered. For the same reasons, tests for interactions between the treatment groups and age were not conducted.
Figure 2: Hypothesis and Overall Testing Strategy — Study 301
H = hypothesis; IDSIQ = Insomnia Daytime Symptoms and Impacts Questionnaire; LPS = latency to persistent sleep; sTST = subjective total sleep time; WASO = wake after sleep onset.
Source: Clinical Study Report for Study 301.16
Figure 3: Hypothesis and Overall Testing Strategy — Study 302
H = hypothesis; IDSIQ = Insomnia Daytime Symptoms and Impacts Questionnaire; LPS = latency to persistent sleep; sTST = subjective total sleep time; WASO = wake after sleep onset.
Source: Clinical Study Report for Study 302.17
Sensitivity Analyses
Regarding sensitivity analyses, models based on the multiple imputation method were performed to assess the robustness of the conclusions of the main analysis and departures from the missing-at-random assumption, and the bias that could result when the outcomes for patients who discontinued the study differed from those who completed the study.60 For this matter, 2 models that adopted missing-not-at-random assumptions were used: a control-based imputation method called jump to reference; and a delta-adjusted imputation method. In addition, to provide a reference for the sensitivity analyses under MNAR, the main analyses were repeated using a multiple imputation method under the missing-at-random assumption. For all multiple imputation analyses, a preliminary step was performed to obtain a monotone missing data pattern. Any nonmonotone (or intermediate) missing data were imputed using the Markov chain Monte Carlo method under missing-at-random assumptions within each treatment group. Imputation for monotone missing data was performed sequentially, 1 visit at a time, using the linear regression method.
Table 8: Statistical Analysis of Efficacy End Points — Study 301 and Study 302
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Primary end points | ||||
Change from baseline in WASO (sleep maintenance) at month 1 and month 3 and change from baseline in LPS (sleep onset) at month 1 and month 3 | Linear mixed-effects model with unstructured covariance matrix | Baseline value of the variable, age group (< 65 years, ≥ 65 years), treatment (higher dose, lower dose, placebo), visit (month 1, month 3), and the interaction of treatment by visit and baseline by visit | Implicit imputation: If 1 of the 2 values was missing for baseline, month 1, or month 3, the single value available was used as the mean for that time point. If both values were missing for a time point, then the mean value was considered missing for that time point | MI method: Jump to reference and a delta-adjusted imputation method under MNAR MI method: Jump to reference and a delta-adjusted imputation method under MAR (performed to provide a reference) |
Secondary end points | ||||
Change from baseline in sTST at month 1 and month 3 | Linear mixed-effects model with unstructured covariance matrix | Baseline value of the variable, age group (< 65 years, ≥ 65 years), treatment (higher dose, lower dose, placebo), visit (month 1, month 3), and the interaction of treatment by visit and baseline by visit | Implicit imputation: Patients had to have at least 2 days of data during each week to calculate a weekly mean; otherwise, the mean value was considered missing for that week | MI methodology: Jump to reference and a delta-adjusted imputation method under MNAR MI methodology: Jump to reference and a delta-adjusted imputation method under MAR (performed to provide a reference) |
Other secondary and exploratory end points | ||||
Change from baseline in IDSIQ total score | Linear mixed-effects model with unstructured covariance matrix | Baseline value of the variable, age group (< 65 years, ≥ 65 years), treatment (higher dose, lower dose, placebo), visit (month 1, month 3), and the interaction of treatment by visit and baseline by visit | Implicit imputation: Patients had to have at least 2 days of data during each week to calculate a weekly mean; otherwise, the mean value was considered missing for that week | NA |
Change from baseline in sleep quality (SDQ VAS score) | Descriptive statistics | NA | Implicit imputation: Patients had to have at least 2 days of data during each week to calculate a weekly mean; otherwise, the mean value was considered missing for that week | NA |
Change from baseline in ISI scores | Descriptive statistics | NA | No method to impute missing values was applied (i.e., unimputed missing data) | NA |
Change from baseline in duration of TST in each sleep stage (S1, S2, SWS, and REM) | Descriptive statistics | NA | No method to impute missing values was applied | NA |
IDSIQ = Insomnia Daytime Symptoms and Impacts Questionnaire; ISI = Insomnia Severity Index; LPS = latency to persistent sleep; MAR = missing at random; MI = multiple imputation; MNAR = missing not at random; NA = not applicable; REM = rapid eye movement; S1 = stage 1 sleep; S2 = stage 2 sleep; SDQ = Sleep Diary Questionnaire; sTST = subjective total sleep time; SWS = slow-wave sleep; VAS = visual analogue scale; WASO = wake after sleep onset.
Sources: Clinical Study Reports for Study 30116 and Study 302,17 statistical analysis plan.60 Details included in the table are from the sponsor’s Summary of Clinical Evidence and the Clinical Study Reports.
Analysis Populations
Table 9 summarizes the analysis populations defined in Study 301 and Study 302.
Table 9: Analysis Populations — Study 301 and Study 302
Population | Definition | Application |
|---|---|---|
Screened analysis set | Comprised all patients who entered screening and received a subject identification number | Disposition of patients, previous and concomitant medications |
Full analysis set | Comprised all patients assigned (i.e., randomized) to a double-blind study treatment | Demographic characteristics, baseline characteristics, and efficacy end points |
Safety set | Comprised all patients who received at least 1 dose of a double-blind study treatment | Disposition of patients, previous and concomitant medications, exposure to interventions, and safety end points |
Treatment withdrawal set | Comprised all patients in the safety set who received at least 1 dose of a single-blind placebo treatment in the placebo run-out period | End points assessing withdrawal symptoms (i.e., BWSQ total score) and rebound insomnia based on sTST |
BWSQ = Benzodiazepine Withdrawal Symptom Questionnaire; sTST = subjective total sleep time.
Sources: Clinical Study Reports for Study 30116 and Study 302.17 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results
Patient Disposition
Patient disposition in Study 301 and Study 302 is shown in Table 10. In Study 301, a total of 3,326 patients were screened. Most patients (n = 2,396 [72%]) failed screening, either at the screening stage or after the single-blind run-in period. Not meeting the inclusion or exclusion criteria was the most common reason for screening failure, although no information was provided on which of the criteria were failed during screening. Overall, 930 patients were randomized in a 1:1:1 ratio to daridorexant 25 mg (N = 310), daridorexant 50 mg (N = 310), or placebo (N = 310). In total, 91.2% of randomized patients completed Study 301. Treatment was discontinued by 24 patients (7.7%) in the daridorexant 25 mg arm, 22 patients (7.1%) in the daridorexant 50 mg arm, and 28 patients (9.0%) in the placebo arm, primarily due to patient withdrawal (2.6% to 2.9%) and AEs (1.0% to 3.2%).
In Study 302, a total of 3,683 patients were screened. Most patients (n = 2,759 [74.9%]) failed screening, either at the screening stage or after the single-blind run-in period. Not meeting the inclusion or exclusion criteria was the most common reason for screening failure, although no information was provided on which of the criteria were failed during screening. Overall, 924 patients were randomized in a 1:1:1 ratio to daridorexant 10 mg (N = 307; data not shown), daridorexant 25 mg (N = 309), or placebo (N = 308). In total, 91.9% of randomized patients completed Study 302. Treatment was discontinued by 23 patients (7.4%) in the daridorexant 25 mg arm and 18 patients (5.8%) in the placebo arm, primarily due to patient withdrawal (4.2% versus 1.3%) and AEs (1.3% versus 2.3%).
Table 10: Summary of Patient Disposition — Study 301 and Study 302
Patient disposition | Study 301 | Study 302 | |||
|---|---|---|---|---|---|
Daridorexant 25 mg (N = 310) | Daridorexant 50 mg (N = 310) | Placebo (N = 310) | Daridorexant 25 mg (N = 309) | Placebo (N = 308) | |
Screened, N | 3,326 | 3,683 | |||
Screen failure, N (%) | |||||
Not eligible, per inclusion or exclusion criteria | 1,164 (89.3) | 1,351 (91.2) | |||
Withdrawal by patient | 121 (9.3) | 116 (7.8) | |||
Lost to follow-up | 10 (0.8) | 11 (0.7) | |||
Other | 9 (0.7) | 4 (0.3) | |||
Entered SB run-in period, N | 2,022 | 2,201 | |||
Discontinued SB run-in period, n (%) | 1,092 (32.8) | 1,277 (34.7) | |||
Not eligible, per inclusion or exclusion criteria | 1,004 (91.9) | 1,184 (92.7) | |||
Adverse events | 4 (0.4) | 1 (0.1) | |||
Withdrawal by patient | 75 (6.9) | 66 (5.2) | |||
Lost to follow-up | 3 (0.3) | 4 (0.3) | |||
Other | 6 (0.5) | 22 (1.7) | |||
Randomized, N | 310 | 310 | 310 | 309 | 308 |
Treated, N (%) | 310 (100.0) | 308 (99.4) | 309 (99.7) | 308 (99.7) | 306 (99.4) |
Discontinued treatment, n (%) | 24 (7.7) | 22 (7.1) | 28 (9.0) | 23 (7.4) | 18 (5.8) |
Reason for treatment discontinuation, n (%) | |||||
Death | 1 (0.3) | 0 | 0 | 0 | 0 |
Adverse events | 7 (2.3) | 3 (1.0) | 10 (3.2) | 4 (1.3) | 7 (2.3) |
Withdrawal by patient | 8 (2.6) | 8 (2.6) | 9 (2.9) | 13 (4.2) | 4 (1.3) |
Lost to follow-up | 1 (0.3) | 1 (0.3) | 3 (1.0) | 2 (0.6) | 0 |
Lack of efficacy | 5 (1.6) | 6 (1.9) | 4 (1.3) | 4 (1.3) | 5 (1.6) |
Other | 2 (0.6) | 4 (1.3) | 2 (0.6) | 0 | 2 (0.6) |
Completed DB treatment period | 286 (92.3) | 286 (92.3) | 281 (90.6) | 285 (92.2) | 288 (93.5) |
Discontinued from study, n (%) | 22 (7.1) | 25 (8.1) | 30 (9.7) | 29 (9.4) | 22 (7.1) |
Reason for study discontinuation, n (%) | |||||
Death | 1 (0.3) | 0 | 0 | 0 | 0 |
Adverse events | 4 (1.3) | 2 (0.6) | 7 (2.3) | 3 (1.0) | 4 (1.3) |
Withdrawal by patient | 13 (4.2) | 10 (3.2) | 12 (3.9) | 16 (5.2) | 11 (3.6) |
Lost to follow-up | 1 (0.3) | 2 (0.6) | 3 (1.0) | 2 (0.6) | 1 (0.3) |
Other | 3 (1.0) | 11 (3.5) | 8 (2.6) | 8 (2.6) | 6 (1.9) |
Completed study | 288 (92.9) | 285 (91.9) | 280 (90.3) | 280 (90.6) | 286 (92.9) |
Analysis sets | |||||
Full analysis set, N | 310 | 310 | 310 | 309 | 308 |
Safety, N | 310 | 308 | 309 | 308 | 306 |
DB = double-blind; SB = single-blind.
Sources: Clinical Study Reports for Study 30116 and Study 302.17 Details included in the table are from the sponsor’s Summary of Clinical Evidence and the Clinical Study Reports.
Baseline Characteristics
The baseline characteristics outlined in Table 11 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
The baseline characteristics of the patients enrolled were similar between groups within studies, and generally comparable across the trials. The mean age per treatment group ranged from 55.1 years (SD = 15.4 years) to 56.7 years (SD = 14.1 years) across the studies. There was roughly an equal proportion of patients aged 65 years or older in the 2 studies. More than two-thirds of participants in both studies were female, and the male-female distribution was generally similar across treatment groups and studies. Most patients identified as white (86.7% to 92.6%), but 6.0% to 9.7% identified as Black or African American, and 0.6% to 3.6% identified as Asian. People of other races made up a smaller proportion of those enrolled. Of note, there was a higher proportion of patients who identified as Asian in Study 302 than in Study 301, likely because some study sites for Study 302 were located in South Korea. On average, the patients enrolled in the studies had been diagnosed with CID for 10 or more years, and the mean ISI score at baseline ranged from 19 (SD = 4.3) to 19.6 (SD = 4.1).
Table 11: Summary of Baseline Characteristics — Study 301 and Study 302
Characteristic | Study 301 | Study 302 | |||
|---|---|---|---|---|---|
Daridorexant 25 mg (N = 310) | Daridorexant 50 mg (N = 310) | Placebo (N = 310) | Daridorexant 25 mg (N = 309) | Placebo (N = 308) | |
Age at screening, years | |||||
Mean age (SD) | 55.8 (15.3) | 55.5 (15.3) | 55.1 (15.4) | 56.3 (14.4) | 56.7 (14.1) |
< 65, n (%) | 189 (61.0) | 189 (61.0) | 188 (60.6) | 188 (60.8) | 187 (60.7) |
≥ 65, n (%) | 121 (39.0) | 121 (39.0) | 122 (39.4) | 121 (39.2) | 121 (39.3) |
Sex, n (%) | |||||
Male | 95 (30.6) | 111 (35.8) | 100 (32.3) | 91 (29.4) | 103 (33.4) |
Female | 215 (69.4) | 199 (64.2) | 210 (67.7) | 218 (70.6) | 205 (66.6) |
Race, n (%) | |||||
American Indian or Alaska Native | 0 | 1 (0.3) | 0 | 0 | 0 |
Asian | 3 (1.0) | 4 (1.3) | 2 (0.6) | 11 (3.6) | 10 (3.2) |
Black or African American | 19 (6.1) | 30 (9.7) | 28 (9.0) | 26 (8.4) | 29 (9.4) |
Native Hawaiian or other Pacific Islander | 1 (0.3) | 1 (0.3) | 0 | 1 (0.3) | 1 (0.3) |
Other | 0 | 0 | 2 (0.6) | 0 | 0 |
White or Caucasian | 287 (92.6) | 274 (88.4) | 278 (89.7) | 271 (87.7) | 267 (86.7) |
Body mass index at screening, kg/m2 | |||||
Mean (SD) | 26.65 (4.36) | 26.28 (4.28) | 26.43 (4.12) | 26.11 (4.32) | 26.23 (4.32) |
Time since diagnosis, years | |||||
Mean (SD) | 10.17 (10.1) | 10.73 (10.7) | 10.96 (10.5) | 11.73 (11.86) | 10.54 (10.52) |
WASO, minutes | |||||
N | 310 | 309 | 309 | 309 | 308 |
Mean (SD) | 97.9 (38.8) | 95.5 (37.8) | 102.5 (40.8) | 106.031 (49.10) | 108.073 (48.71) |
LPS, minutes | |||||
N | 310 | 309 | 309 | 309 | 308 |
Mean (SD) | 67.273 (38.56) | 63.619 (37.39) | 66.535 (39.77) | 68.877 (40.55) | 71.815 (46.09) |
sTST, minutes | |||||
N | 310 | 309 | 309 | 309 | 308 |
Mean (SD) | 309.848 (60.11) | 313.178 (57.60) | 315.886 (53.14) | 308.489 (52.85) | 307.570 (51.52) |
IDSIQ sleepiness domain score | |||||
N | 308 | 309 | 308 | 308 | 307 |
Mean (SD) | 22.119 (6.88) | 22.479 (7.21) | 22.260 (6.95) | 22.242 (6.20) | 22.571 (5.76) |
ISI score | |||||
N | 310 | 308 | 309 | 308 | 306 |
Mean (SD) | 19.0 (4.3) | 19.3 (4.0) | 19.2 (4.0) | 19.5 (4.0) | 19.6 (4.1) |
IDSIQ = Insomnia Daytime Symptoms and Impacts Questionnaire; ISI = Insomnia Severity Index; LPS = latency to persistent sleep; SD = standard deviation; sTST = subjective total sleep time; WASO = wake after sleep onset.
Sources: Clinical Study Report for Study 30116 and Study 302.17 Details included in the table are from the sponsor’s Summary of Clinical Evidence and the Clinical Study Reports.
Exposure to Study Treatments
Exposure to study medications is summarized in Table 12. In both studies, treatment adherence was measured by the amount of study drug returned at each study visit. Patients also recorded their adherence in the SDQ every morning. Overall, there was high adherence to the interventions in both studies (more than 98%).
Table 12: Summary of Patient Exposure From the Studies Included in the Systematic Review
Exposure | Study 301 | Study 302 | |||
|---|---|---|---|---|---|
Daridorexant 25 mg (N = 310) | Daridorexant 50 mg (N = 310) | Placebo (N = 310) | Daridorexant 25 mg (N = 309) | Placebo (N = 308) | |
Safety set (N) | 310 | 308 | 309 | 307 | 306 |
Duration of double-blind treatment, days | |||||
n | 310 | 308 | 309 | 308 | 306 |
Mean (SD) | 80.3 (15.5) | 82.1 (11.3) | 81.9 (13.1) | 81.0 (14.5) | 81.8 (13.1) |
Median (range) | 84 (2 to 122) | 84 (15 to 117) | 84 (7 to 145) | 84 (3 to 117) | 84 (2 to 101) |
Exposure to double-blind treatment | |||||
Patient-years | 68.1 | 69.2 | 69.3 | 68.3 | 68.6 |
Adherence, % | |||||
n | 299 | 304 | 301 | 295 | 296 |
Mean (SD) | 98.63 (5.24) | 99.21 (4.11) | 98.37 (6.85) | 99.49 (4.22) | 98.75 (4.81) |
Median (range) | 100.0 (61.8 to 113.4) | 100.0 (73.5 to 128.0) | 100.0 (45.2 to 129.5) | 100.0 (74.1 to 133.3) | 100.0 (70.2 to 117.6) |
Treatment withdrawal seta (N) | 286 | 286 | 280 | 284 | 285 |
Duration of placebo run-out treatment, days | |||||
n | 286 | 286 | 280 | 284 | 283 |
Mean (SD) | 8.5 (3.4) | 8.6 (3.8) | 8.3 (2.9) | 8.4 (3.8) | 8.6 (5.0) |
Median (range) | 8 (2 to 34) | 8 (1 to 35) | 7 (4 to 27) | 8 (1 to 35) | 7 (3 to 49) |
SD = standard deviation.
aComprised all patients in the safety set who received at least 1 dose of the single-blind placebo treatment in the placebo run-out period.
Sources: Clinical Study Reports for Study 30116 and Study 302.17 Details included in the table are from the sponsor’s Summary of Clinical Evidence and the Clinical Study Reports.
Table 13: Concomitant Medications Used During the Double-Blind Treatment Phase (Safety Set) — Study 301 and Study 302
Concomitant treatment | Study 301 | Study 302 | |||
|---|---|---|---|---|---|
Daridorexant 25 mg (N = 310) | Daridorexant 50 mg (N = 308) | Placebo (N = 309) | Daridorexant 25 mg (N = 308) | Placebo (N = 309) | |
CBT at screening, n (%) | 1 (0.3) | 1 (0.3) | 1 (0.3) | 0 | 0 |
Patients with at least 1 medication, n (%) | 205 (66.1) | 190 (61.7) | 188 (60.8) | 211 (68.5) | 208 (68.0) |
Most commonly taken, n (%) | |||||
Propionic acid derivatives | 43 (13.9) | 42 (13.6) | 45 (14.6) | 38 (12.3) | 28 (9.2) |
HMG-CoA reductase inhibitors | 37 (11.9) | 24 (7.8) | 34 (11.0) | 24 (7.8) | 36 (11.8) |
ACE inhibitors, plain | 21 (6.8) | 20 (6.5) | 30 (9.7) | 20 (6.5) | 37 (12.1) |
Thyroid hormones | 30 (9.7) | 24 (7.8) | 27 (8.7) | 35 (11.4) | 40 (13.1) |
Beta blocking drugs, selective | 34 (11.0) | 18 (5.8) | 24 (7.8) | 28 (9.1) | 26 (8.5) |
ACE = angiotensin-converting enzyme; CBT = cognitive behavioural therapy; HMG-CoA = beta-hydroxy-beta-methylglutaryl coenzyme A.
Sources: Clinical Study Reports for Study 30116 and Study 302.17 Details included in the table are from the sponsor’s Summary of Clinical Evidence and the Clinical Study Reports.
Efficacy
Sleep Maintenance
Sleep maintenance reported as change from baseline in WASO was 1 of the primary outcomes in Study 301 and Study 302. Results for change from baseline in WASO at month 1 and month 3 are presented in Table 14.
Study 301
In Study 301, the mean WASO at baseline was 95.48 minutes (SD = 37.81 minutes) in the daridorexant 50 mg arm, 97.87 minutes (SD = 38.77 minutes) in the daridorexant 25 mg arm, and 102.51 minutes (SD = 40.77 minutes) in the placebo arm.
At month 1, the LSM change from baseline in WASO was –28.98 minutes (95% CI, –32.67 to –25.30 minutes) in the daridorexant 50 mg arm, –18.40 minutes (95% CI, –22.13 to –14.67 minutes) in the daridorexant 25 mg arm, and –6.20 minutes (95% CI, –9.93 to –2.48 minutes) in the placebo arm. The LSM difference in change from baseline in WASO at 1 month compared to placebo was –22.78 minutes (97.5% CI, –28.75 to –16.82 minutes) in favour of daridorexant 50 mg and –12.20 minutes (97.5% CI, –18.19 to –6.21 minutes) in favour of daridorexant 25 mg.
At month 3, the LSM change from baseline was –29.41 minutes (95% CI, –33.40 to –25.43 minutes) in the daridorexant 50 mg arm, –22.97 minutes (95% CI, –26.96 to –18.99 minutes) in the daridorexant 25 mg arm, and –11.11 minutes (95% CI, –15.13 to –7.09 minutes) in the placebo arm. The LSM difference in change from baseline in WASO compared to placebo was –18.3 minutes (97.5% CI, –24.76 to –11.85 minutes) in favour of daridorexant 50 mg and –11.86 minutes (97.5% CI, –18.30 to –5.42 minutes) in favour of daridorexant 25 mg.
Results of the subgroup analysis based on age (< 65 years or ≥ 65 years) and sex (male or female) followed the same general pattern as the overall population, as shown in Appendix 1.
Study 302
In Study 302, the mean WASO at baseline was 106.31 minutes (SD = 49.10) in the daridorexant 25 mg arm and 108.07 minutes (SD = 48.71) in the placebo arm.
At month 1, the LSM change from baseline was –24.19 minutes (95% CI, –28.47 to –19.91 minutes) in the daridorexant 25 mg arm and –12.57 minutes (95% CI, –16.82 to –8.32 minutes) in the placebo arm, corresponding to an LSM difference of –11.62 minutes (95% CI, –17.60 to –5.63 minutes) in favour of daridorexant 25 mg. Results of the subgroup analysis showed that in male patients, there was no difference between groups (mean difference = 3.94; 95% CI, –13.58 to 5.69). Findings from the rest of the subgroups were consistent with the main results, as shown in Appendix 1.
At month 3, the LSM change from baseline was –24.25 minutes (95% CI, –29.02 to –19.47) in the daridorexant 25 mg arm and –14.00 minutes (95% CI, –18.76 to –9.24 minutes) in the placebo arm, representing an LSM difference of –10.25 minutes (95% CI, –16.95 to –3.55 minutes) in favour of daridorexant 25 mg. Results of the subgroup analysis were generally consistent with the main results, as shown in Appendix 1.
Sleep Onset
Sleep onset, reported as change from baseline in LPS, was 1 of the primary outcomes in Study 301 and Study 302. Table 14 presents the results at month 1 and month 3.
Study 301
In Study 301, the mean LPS at baseline was 63.62 minutes (SD = 37.39 minutes) in the daridorexant 50 mg arm, 67.27 minutes (SD = 38.56 minutes) in the daridorexant 25 mg arm, and 66.54 minutes (SD = 39.77 minutes) in the placebo arm.
At month 1, the LSM change from baseline in LPS was –31.20 minutes (95% CI, –34.51 to –27.90 minutes) in the daridorexant 50 mg arm, –28.17 (95% CI, –31.51 to –24.83 minutes) in the daridorexant 25 mg arm, and –19.85 minutes (95% CI, –23.18 to –16.52 minutes) in the placebo arm. The LSM difference in change from baseline in LPS at 1 month compared to placebo was –11.35 minutes (97.5% CI, –16.694 to –6.015 minutes) in favour of daridorexant 50 mg and –8.32 minutes (97.5% CI, –13.69 to –2.96 minutes) in favour of daridorexant 25 mg.
At month 3, the LSM change from baseline in LPS was –34.80 minutes (95% CI, –38.12, to –31.49 minutes) in the daridorexant 50 mg arm, –30.73 minutes (95% CI, –34.04 to –27.41 minutes minutes) in the daridorexant 25 mg arm, and –23.13 minutes (95% CI, –26.46 to –19.80 minutes) in the placebo arm. The LSM difference in change from baseline in LPS at 3 months compared to placebo was –11.67 minutes (97.5% CI, –17.03 to –6.32 minutes) in favour of daridorexant 50 mg and–7.59 minutes (97.5% CI, –12.94 to –2.25 minutes) in favour of daridorexant 25 mg.
Results of the subgroup analysis based on age (< 65 years or ≥ 65 years) and sex (male or female) followed the same general pattern as the overall population as shown in Appendix 1.
Table 14: Change From Baseline to Month 1 and Month 3 in WASO and LPS — Study 301 and Study 302 (Full Analysis Set)
Variable | Study 301 | Study 302 | |||
|---|---|---|---|---|---|
Daridorexant 25 mg (N = 310) | Daridorexant 50 mg (N = 310) | Placebo (N = 310) | Daridorexant 25 mg (N = 309) | Placebo (N = 308) | |
Change from baseline to month 1 and to month 3 in WASO (sleep maintenance), minutesa,b | |||||
N at baseline | 310 | 309 | 309 | 309 | 308 |
Baseline WASO, mean (SD) | 97.87 (38.77) | 95.48 (37.81) | 102.51 (40.77) | 106.03 (49.10) | 108.07 (48.71) |
Month 1 | |||||
N | 298 | 305 | 299 | 295 | 300 |
Observed value, mean (SD) | 76.90 (42.40) | 65.34 (34.98) | 91.72 (42.43) | 80.00 (44.38) | 92.90 (49.74) |
Change from baseline in WASO, mean (SD) | –20.49 (37.80) | –29.92 (36.96) | –10.63 (41.05) | –26.03 (43.82) | –15.26 (41.31) |
Change from baseline in WASO, LSM (95% CI) | –18.40 (–22.13 to –14.67) | –28.98 (–32.67 to –25.30) | –6.20 (–9.93 to –2.48) | –24.19 (–28.47 to –19.91) | –12.57 (–16.82 to –8.32) |
LSM difference vs. placebo (97.5% CI) | –12.20 (–18.19 to –6.21)c | –22.78 (–28.75 to –16.82) | NA | –11.62 (–17.60 to –5.63)d | NA |
P value | < 0.0001 | < 0.0001 | NA | 0.0001 | NA |
Month 3 | |||||
N | 289 | 287 | 283 | 281 | 283 |
Observed value, mean (SD) | 72.54 (39.61) | 65.24 (38.86) | 86.50 (43.15) | 79.57 (48.83) | 90.58 (46.64) |
Change from baseline in WASO, mean (SD) | –25.28 (43.21) | –29.99 (39.98) | –16.39 (44.18) | –26.06 (45.20) | –17.09 (51.71) |
Change from baseline in WASO, LSM (95% CI) | –22.97 (–26.96 to –18.99) | –29.41 (–33.40 to –25.43) | –11.11 (–15.13 to –7.09) | –24.25 (–29.02 to –19.47) | –14.00 (–18.76 to –9.24) |
LSM difference vs. placebo (97.5% CI) | –11.86 (–18.30 to –5.42) | –18.30 (–24.76 to –11.85) | NA | –10.25 (–16.95 to –3.55)d | NA |
P value | < 0.0001 | < 0.0001 | NA | 0.0028 | NA |
Change from baseline to month 1 and to month 3 in LPS (sleep onset), minutesa,c | |||||
N at baseline | 310 | 309 | 309 | 309 | 308 |
Baseline, mean (SD) | 67.27 (38.56) | 63.62 (37.39) | 66.54 (39.77) | 68.88 (40.55) | 71.82 (46.09) |
Month 1 | |||||
N | 298 | 305 | 299 | 295 | 300 |
Observed value, mean (SD) | 38.27 (32.43) | 33.81 (26.69) | 46.29 (36.16) | 42.16 (39.41) | 50.03 (40.01) |
Change from baseline in LPS, mean (SD) | –29.40 (36.55) | –29.67 (39.28) | –20.47 (40.25) | –26.60 (39.27) | –21.93 (41.11) |
Change from baseline in LPS, LSM (95% CL) | –28.17 (–31.51 to –24.83) | –31.20 (–34.51 to –27.90) | –19.85 (–23.18 to –16.52) | –26.46 (–30.63 to –22.29) | –20.01 (–24.15 to –15.88) |
LSM difference vs. placebo (97.5% CI) | –8.32 (–13.69 to –2.96) | –11.35 (–16.69 to –6.02) | NA | –6.45 (–12.28 to –0.61)d | NA |
P value | 0.0005 | < 0.0001 | NA | 0.0303 | NA |
Month 3 | |||||
N | 289 | 287 | 283 | 281 | 283 |
Observed value, mean (SD) | 35.66 (33.93) | 30.28 (23.41) | 42.92 (34.29) | 38.89 (36.96) | 49.23 (45.76) |
Change from baseline in LPS, mean (SD) | –31.95 (39.57) | –32.84 (34.55) | –24.53 (41.07) | −28.06 (41.84) | −21.83 (47.58) |
Change from baseline in LPS, LSM (95% CL) | –30.73 (–34.04 to –27.41) | –34.80 (–38.12 to –31.49) | –23.13 (–26.46 to –19.80) | –28.91 (–33.41 to –24.40) | –19.89 (–24.38 to –15.41) |
LSM difference vs. placebo (97.5% CI) | –7.59 (–12.94 to –2.25) | –11.67 (–17.03 to –6.32) | NA | –9.01 (–15.34 to −2.68)d | NA |
P value | 0.0015 | < 0.0001 | NA | 0.0053 | NA |
CI = confidence interval; CL = confidence limit; LPS = latency to persistent sleep; LSM = least squares mean; NA = not applicable; SD = standard deviation; vs. = versus; WASO = wake after sleep onset.
aThe P value has been adjusted for multiple testing. Note that alpha levels were based on hypothesis testing. For WASO: daridorexant 50 mg at month 1, alpha = 0.025; daridorexant 50 mg at month 3, alpha = 0.025; daridorexant 25 mg at month 1, alpha = 0.025; daridorexant 25 mg at month 3, alpha = 0.01875. For LPS: daridorexant 50 mg at month 1, alpha = 0.025; daridorexant 50mg at month 3, alpha = 0.025; daridorexant 25 mg at month 1, alpha = 0.025; daridorexant 25 mg at month 3, alpha = 0.01875.
bMixed models for repeated measures (MMRM) for change from baseline in WASO = baseline WASO + age group (< 65 years or ≥ 65 years) + treatment + visit + treatment × visit + baseline × visit.
cMMRM for change from baseline in LPS = baseline LPS + age group (< 65 years or ≥ 65 years) + treatment + visit + treatment × visit + baseline × visit.
dValues from Study 302 are 95% CI.
Sources: Clinical Study Reports for Study 30116 and Study 302.17
Study 302
In Study 302, the mean LPS at baseline was 68.88 minutes (SD = 40.55 minutes) in the daridorexant 25 mg arm and 71.82 minutes (SD = 46.09 minutes) in the placebo arm.
At month 1, the LSM change from baseline was –26.46 minutes (95% CI, –30.63 to –22.29 minutes) for daridorexant 25 mg and –20.01 minutes (95% CI, –24.15 to –15.88 minutes) for placebo, corresponding to an LSM difference of –6.45 minutes (95% CI, 12.28, to –0.61 minutes) in favour of daridorexant 25 mg. Findings from the subgroups were consistent with the main results, as shown in Appendix 1.
At month 3, the LSM change from baseline was –28.91 minutes (95% CI, –33.41 to –24.40 minutes) in the daridorexant 25 mg arm and –19.89 minutes (95% CI, –24.38 to –15.41 minutes) in the placebo arm, representing an LSM difference of –9.01 minutes (–15.34 to –2.68 minutes) in favour of daridorexant 25 mg. Results of the subgroup analysis were generally consistent with the main results, as shown in Appendix 1.
Sleep Duration
Sleep duration, measured subjectively, was reported as change from baseline in sTST and was a secondary outcome in Study 301 and Study 302. Table 15 presents the results at month 1 and month 3.
Study 301
In Study 301, the mean sTST at baseline was 313.18 minutes (SD = 57.60 minutes) in the daridorexant 50 mg arm, 309.85 minutes (SD = 60.11 minutes) in the daridorexant 25 mg arm, and 315.89 minutes (SD = 53.14 minutes) in the placebo arm.
At month 1, the LSM change from baseline in sTST was 43.62 minutes (95% CI, 38.17 to 49.06 minutes) in the daridorexant 50 mg arm, 34.18 minutes (95% CI, 28.72 to 39.65 minutes) in the daridorexant 25 mg arm, and 21.56 minutes (95% CI, 16.10 to 27.02 minutes) in the placebo arm. The LSM difference in change from baseline in sTST at 1 month compared to placebo was 22.06 minutes (97.5% CI, 13.30 to 30.18 minutes) in favour of daridorexant 50 mg and 12.62 minutes (97.5% CI, 3.85 to 21.39 minutes) in favour of daridorexant 25 mg.
At month 3, the LSM change from baseline was 57.67 minutes (95% CI, 51.17 to 64.17 minutes) in the daridorexant 50 mg arm, 47.83 minutes (95% CI, 41.33 to 54.33 minutes) in the daridorexant 25 mg arm, and 37.90 minutes (95% CI, 31.39 to 44.40 minutes) in the placebo arm. The LSM difference in change from baseline in sTST compared to placebo was 19.77 minutes (97.5% CI, 9.30 to 30.24 minutes) in favour of daridorexant 50 mg and 9.93 minutes (97.5% CI, –0.54 to 20.40 minutes) in favour of daridorexant 25 mg.
Study 302
In Study 302, the mean sTST at baseline was 308.49 minutes (SD = 52.85 minutes) in the daridorexant 25 mg arm and 307.57 minutes (SD = 51.52 minutes) in the placebo arm.
At month 1, the LSM change from baseline in sTST was 43.77 minutes (95% CI, 38.14 to 49.41 minutes) in the daridorexant 25 mg arm and 27.64 minutes (95% CI, 22.02 to 33.27 minutes) in the placebo arm. The mean change from baseline in sTST was 16.13 minutes longer with daridorexant 25 mg than with placebo (95% CI, 8.22 to 24.04 minutes).
At month 3, the LSM change from baseline in sTST was 56.18 minutes (95% CI, 49.81 to 62.55 minutes) in the daridorexant 25 mg arm and 37.12 minutes (95% CI, 30.78 to 43.46 minutes) in the placebo arm. The LSM difference compared to placebo was 19.06 minutes (95% CI, 10.13 to 27.99 minutes) in favour of daridorexant 25 mg.
Table 15: Change From Baseline to Month 1 and Month 3 in sTST — Study 301 and Study 302 (Full Analysis Set)
Variable | Study 301 | Study 302 | |||
|---|---|---|---|---|---|
Daridorexant 25 mg (N = 310) | Daridorexant 50 mg (N = 310) | Placebo (N = 310) | Daridorexant 25 mg (N = 309) | Placebo (N = 308) | |
Change from baseline to month 1 and to month 3 in sTSTa,b | |||||
N at baseline | 310 | 309 | 309 | 309 | 308 |
Baseline sTST, minutes, mean (SD) | 309.85 (60.11) | 313.18 (57.60) | 315.89 (53.14) | 308.49 (52.85) | 307.57 (51.52) |
Month 1 | |||||
N | 303 | 304 | 302 | 297 | 297 |
Observed value, mean (SD) | 345.16 (65.64) | 357.76 (73.64) | 337.72 (64.56) | 353.27(66.74) | 336.16 (62.52) |
Change from baseline in sTST, minutes, mean (SD) | 35.45 (48.56) | 44.57 (53.44) | 21.70 (44.04) | 44.46 (50.67) | 28.29 (48.60) |
Change from baseline in sTST, LSM (95% CI) | 34.18 (28.72 to 39.65) | 43.62 (38.17 to 49.06) | 21.56 (16.10 to 27.02) | 43.77 (38.14 to 49.41) | 27.64 (22.02 to 33.27) |
LSM difference vs. placebo (97.5% CI) | 12.62 (3.85 to 21.39) | 22.06 (13.30 to 30.18) | NA | 16.13 (8.22 to 24.04)c | NA |
P value | 0.0013 | < 0.0001 | NA | < 0.0001 | NA |
Month 3 | |||||
Number of patients contributing to the analysis | 292 | 289 | 289 | 285 | 287 |
Observed value, mean (SD) | 358.39 (72.03) | 371.76 (78.89) | 354.35 (72.73) | 365.46 (69.73) | 347.02 (64.70) |
Change from baseline in sTST, minutes, mean (SD) | 49.07 (55.32) | 58.46 (61.41) | 38.48 (54.99) | 55.91 (56.62) | 38.04 (54.99) |
Change from baseline in sTST, LSM (95% CI) | 47.83 (41.33 to 54.33) | 57.67 (51.17 to 64.17) | 37.90 (31.39 to 44.40) | 56.18 (49.81 to 62.55) | 37.12 (30.78 to 43.46) |
LSM difference vs. placebo (97.5% CI) | 9.93 (–0.54 to 20.40) | 19.77 (9.30 to 30.24) | NA | 19.06 (10.13 to 27.99)c | NA |
P value | 0.0334 | < 0.0001 | NA | < 0.0001 | NA |
CI = confidence interval; LSM = least squares mean; NA = not applicable; SD = standard deviation; sTST = subjective total sleep time; vs. = versus.
aThe P value has been adjusted for multiple testing. Note that alpha levels were based on hypothesis testing. For sTST, daridorexant 50 mg at month 1, alpha = 0.025; daridorexant 50 mg at month 3, alpha = 0.025; daridorexant 25 mg at month 1, alpha = 0.025; daridorexant 25 mg at month 3, alpha = 0.0375
bMixed models for repeated measures for change from baseline in sTST = baseline sTST + age group (< 65 years or ≥ 65 years) + treatment + visit + treatment × visit + baseline × visit.
cValues from Study 302 are 95% CI.
Sources: Clinical Study Reports for Study 30116 and Study 302.17 Details included in the table are from the sponsor’s Summary of Clinical Evidence and the Clinical Study Reports.
Sleep Quality
Sleep quality was determined using the SDQ and was considered an exploratory outcome in the included studies. Table 16 presents the reported sleep quality in Study 301 and Study 302 at month 1 and month 3.
Study 301
The mean VAS score for sleep quality at baseline was 36.23 (SD = 17.03) in the daridorexant 50 mg arm, 35.56 (SD = 17.77) in the daridorexant 25 mg arm, and 35.60 (SD = 17.78) in the placebo arm. In Study 301, there were within-group improvements in sleep quality from baseline in all groups, with numerically greater improvements in the active treatment groups than in the placebo group.
At month 1, the mean change from baseline was 14.21 points (SD = 18.95 points) in the daridorexant 50 mg arm, 11.35 points (SD = 15.65 points) in the daridorexant 25 mg arm, and 7.04 points (SD = 13.74 points) in the placebo arm.
At month 3, the mean change from baseline was 20.21 points (SD = 22.15 points) in the daridorexant 50 mg arm, 18.20 (SD = 19.10 points) in the daridorexant 25 mg arm, and 13.95 points (SD = 18.85 points) in the placebo arm.
Study 302
The mean VAS sleep quality score at baseline was 37.94 (SD = 15.02) in the daridorexant 25 mg arm and 36.91 (SD = 14.77) in the placebo arm. In Study 302, there were within-group improvements in sleep quality from baseline in both groups, with numerically greater improvements in the active treatment group than in the placebo group.
At month 1, the mean change from baseline was 11.20 points (SD = 15.55 points) in the daridorexant 25 mg arm and 9.41 points (SD = 14.44 points) in the placebo arm.
At month 3, the mean change from baseline was 17.77 points (SD = 18.55 points) in the daridorexant 25 mg arm and 13.18 points (SD = 17.33 points) in the placebo arm.
Change From Baseline in IDSIQ Total Score
The change from baseline in the IDSIQ total score, which is the sum of 3 IDSIQ domain scores (i.e., alert/cognition, sleepiness, and mood), was another efficacy end point in the included trials. The results are presented in Table 16. Per the sponsor’s request, results for the IDSIQ sleepiness domain are provided in Table 31 of Appendix 1.
Study 301
The mean IDSIQ total score at baseline was 74.52 points (SD = 25.16 points) in the daridorexant 50 mg arm, 73.06 points (SD = 24.55 points) in the daridorexant 25 mg arm, and 73.55 points (SD = 24.64 points) in the in the placebo arm.
The LSM difference in change from baseline in IDSIQ total score compared to placebo at month 1 was –7.24 points (95% CI, –9.785 to –4.698 points) in favour of daridorexant 50 mg and –2.94 points (95% CI, –5.487 to –0.385 points) in favour of daridorexant 25 mg. The LSM difference between groups at month 3 compared to placebo was –7.20 points (95% CI, –10.544 to –3.862 points) in favour of daridorexant 50 mg and –3.46 points (95% CI, –6.809 to –0.113 points) in favour of daridorexant 25 mg.
Study 302
The mean IDSIQ total scores at baseline were 73.14 points (SD = 21.21) in the daridorexant 25 mg arm, and 74.48 points (SD = 20.26) in the placebo arm.
At month 1, the mean difference from baseline in IDSIQ total score was 3.11 points (95% CI, –5.807 to –0.412 points) lower in the daridorexant arm than in the placebo arm. At month 3, the IDSIQ total score was 4.23 points (95% CI, –7.477 to –0.986) lower in the daridorexant 25 mg arm than in the placebo arm.
Table 16: Change From Baseline to Month 1 and Month 3 in Sleep Quality and IDSIQ Total Score — Study 301 and Study 302 (Full Analysis Set)
Variable | Study 301 | Study 302 | |||
|---|---|---|---|---|---|
Daridorexant 25 mg (N = 310) | Daridorexant 50 mg (N = 310) | Placebo (N = 310) | Daridorexant 25 mg (N = 309) | Placebo (N = 308) | |
Change from baseline to month 1 and to month 3 in sleep quality (assessed by VAS) | |||||
N at baseline | 310 | 309 | 309 | 309 | 308 |
Baseline, mean (SD) | 35.56 (17.77) | 36.23 (17.03) | 35.60 (17.78) | 37.94 (15.02) | 36.91 (14.77) |
Month 1 | |||||
N | 303 | 304 | 302 | 297 | 297 |
Observed value, mean (SD) | 46.93 (20.86) | 50.54 (21.19) | 42.45 (19.10) | 49.38 (18.13) | 46.32 (17.46) |
Change from baseline, mean (SD) | 11.35 (15.65) | 14.21 (18.95) | 7.04 (13.74) | 11.20 (15.55) | 9.41 (14.44) |
Month 3 | |||||
N | 292 | 289 | 289 | 285 | 287 |
Observed value, mean (SD) | 53.74 (23.04) | 56.32 (22.31) | 49.28 (22.40) | 55.76 (20.05) | 50.34 (19.51) |
Change from baseline, mean (SD) | 18.20 (19.10) | 20.21 (22.15) | 13.95 (18.85) | 17.77 (18.55) | 13.18 (17.33) |
Change from baseline to month 1 and to month 3 in IDSIQ total score | |||||
N at baseline | 308 | 309 | 308 | 308 | 307 |
Baseline IDSIQ, mean (SD) | 73.056 (24.551) | 74.521 (25.156) | 73.550 (24.642) | 73.137 (21.208) | 74.484 (20.255) |
Month 1 | |||||
N | 301 | 304 | 301 | 297 | 297 |
Observed value, mean (SD) | 63.945 (25.159) | 60.693 (26.690) | 67.447 (25.327) | 61.126 (22.040) | 65.444 (21.216) |
Change from baseline, LSM (95% CI) | –9.17 (–10.992 to –7.351) | –13.48 (–15.286 to –11.667) | –6.24 (–8.052 to –4.419) | –11.90 (–13.824 to –9.971) | –8.79 (–10.708 to –6.867) |
Difference vs. placebo (95% CI) | –2.94 (–5.487 to –0.385) | –7.24 (–9.785 to –4.698) | NA | –3.11 (–5.807 to –0.412) | NA |
P value (two-sided) | 0.0241 | < 0.0001 | NA | 0.0239 | NA |
Month 3 | |||||
N | 290 | 291 | 288 | 283 | 289 |
Observed value, mean (SD) | 57.395 (26.456) | 54.367 (27.228) | 61.358 (27.579) | 55.729 (24.236) | 60.776 (22.137) |
Change from baseline, LSM (95% CI) | –15.56 (–17.937 to –13.179) | –19.30 (–21.667 to –16.931) | –12.10 (–14.475 to –9.718) | –17.30 (–19.620 to –14.985) | –13.07 (–15.370 to –10.773) |
Difference vs. placebo (95% CI) | –3.46 (–6.809 to –0.113) | –7.20 (–10.544 to –3.862) | NA | –4.23 (–7.477 to –0.986) | NA |
P value (two-sided) | 0.0428 | < 0.0001 | NA | 0.0107 | NA |
CI = confidence interval; IDSIQ = Insomnia Daytime Symptoms and Impacts Questionnaire; LSM = least squares mean; NA = not applicable; SD = standard deviation; VAS = visual analogue scale; vs. = versus.
Sources: Clinical Study Reports for Study 30116 and Study 302.17 Details included in the table are from the sponsor’s Summary of Clinical Evidence and the Clinical Study Reports.
Change From Baseline in ISI Score
The change from baseline in ISI score was an exploratory outcome in the included trials. The results are presented in Table 17.
Study 301
The mean ISI score at baseline was 19.3 points (SD = 4.0 points) in the daridorexant 50 mg arm, 19.0 points (SD = 4.3 points) in the daridorexant 25 mg arm, and 19.2 (SD = 4.0 points) in the placebo arm.
At month 1, the LSM change from baseline was –4.81 points (95% CI, –5.36 to –4.26 points) in the daridorexant 50 mg arm, –4.07 points (95% CI, –4.63 to –3.51 points) in the daridorexant 25 mg arm, and –3.05 points (95% CI, –3.60 to –2.50 points) in the placebo arm. Compared to placebo, there was a decrease in ISI score of –1.76 points (95% CI, –2.54 to –0.99) in the daridorexant 50 mg arm and of –1.02 points (95% CI, –1.80 to –0.24) in the daridorexant 25 mg arm.
At month 3, the LSM change from baseline was –7.17 points (95% CI, –7.84 to –6.50 points) in the daridorexant 50 mg arm, –6.02 points (95% CI, –6.69 to –5.35 points) in the daridorexant 25 mg arm, and –5.19 points (95% CI, –5.86 to –4.52 points) in the placebo arm. Compared to placebo, there was a decrease in ISI score of –1.98 points (95% CI, –2.92 to –1.04 points) in the daridorexant 50 mg arm and –0.83 points (95% CI, –1.78 to 0.11 points) in the daridorexant 25 mg arm.
Study 302
The mean ISI score at baseline was 19.5 points (SD = 4.0 points) in the daridorexant 25 mg arm and 19.6 points (SD = 4.1 points) in the placebo arm. At month 1, the mean change from baseline was –5.1 points (SD = 5.2 points) in the daridorexant 25 mg arm and –3.8 points (SD = 4.6 points) in the placebo arm. At month 3, the mean change from baseline was –6.9 points (SD = 6.0 points) in the daridorexant 25 mg arm and –5.4 points (SD = 5.5 points) in the placebo arm.
Table 17: Change From Baseline to Month 1 and Month 3 in ISI Total Score — Study 301 and Study 302 (Full Analysis Set)
Variable | Study 301 | Study 302 | |||
|---|---|---|---|---|---|
Daridorexant 25 mg (N = 310) | Daridorexant 50 mg (N = 310) | Placebo (N = 310) | Daridorexant 25 mg (N = 309) | Placebo (N = 308) | |
Change from baseline to month 1 and to month 3 in ISI score | |||||
N at baseline | 310 | 308 | 309 | 308 | 306 |
Baseline ISI score, mean (SD) | 19.0 (4.3) | 19.3 (4.0) | 19.2 (4.0) | 19.5 (4.0) | 19.6 (4.1) |
Month 1 | |||||
N | 292 | 299 | 297 | 287 | 294 |
Observed value, mean (SD) | 14.8 (5.9) | 14.3 (5.8) | 16.1 (5.2) | 14.4 (5.8) | 15.8 (5.4) |
Change from baseline, mean (SD) | –4.1 (4.7) | –4.9 (5.5) | –3.1 (4.7) | -–5.1 (5.2) | –3.8 (4.6) |
Change from baseline, LSM (95% CI) | –4.07 (–4.63 to –3.51) | –4.81 (–5.36 to –4.26) | –3.05 (–3.60 to –2.50) | NR | NR |
Difference vs. placebo (95% CI) | –1.02 (–1.80 to –0.24) | –1.76 (–2.54 to –0.99) | NA | NR | NR |
P value | 0.0104 | < 0.0001 | NA | NR | NR |
Month 3 | |||||
N | 286 | 283 | 281 | 280 | 277 |
Observed value, mean (SD) | 12.9 (6.6) | 11.9 (6.3) | 13.8 (6.0) | 12.5 (6.0) | 14.1 (5.9) |
Change from baseline, mean (SD) | –6.0 (5.8) | −7.2 (6.5) | −5.4 (5.7) | –6.9 (6.0) | –5.4 (5.5) |
Change from baseline, LSM (95% CI) | –6.02 (–6.69 to –5.35) | –7.17 (–7.84 to –6.50) | –5.19 (–5.86 to –4.52) | NR | NR |
Difference vs. placebo (95% CI) | –0.83 (–1.78 to 0.11) | –1.98 (–2.92 to –1.04) | NA | NR | NR |
P value | 0.0826 | < 0.0001 | NA | NR | NR |
CI = confidence interval; ISI = Insomnia Severity Index; LSM = least squares mean; NA = not applicable; NR = not reported; SD = standard deviation; vs. = versus.
Sources: Clinical Study Reports for Study 30116 and Study 302.17 Details included in the table are from the sponsor’s Summary of Clinical Evidence and the Clinical Study Reports.
Change From Baseline in Duration of TST in Each Sleep Stage
The change from baseline in the duration of TST in each sleep stage (i.e., S1, S2, SWS, and REM) was an exploratory end point in the included trials. The results are presented in Table 18.
Study 301
For S1, the change from baseline to 1 month was 5.41 minutes in the daridorexant 50 mg arm, 4.26 minutes in the daridorexant 25 mg, and 2.59 minutes in the placebo arm. For S2, the change from baseline to 1 month was 34.74 minutes in the daridorexant 50 mg arm, 29.79 minutes in the daridorexant 25 mg arm, and 16.10 minutes in the placebo arm. For SWS, the change from baseline to 1 month was 2.16 minutes in daridorexant 50 mg arm, 0.71 minutes in the daridorexant 25 mg arm, and 1.60 minutes in the placebo arm. For REM, the change from baseline to 1 month was 15.51 minutes in the daridorexant 50 mg arm, 13.50 minutes in the daridorexant 25 mg arm, and 9.22 minutes in the placebo arm.
For S1, the change from baseline to 3 months was 6.89 minutes in the daridorexant 50 mg arm, 5.67 minutes in the daridorexant 25 mg arm, and 4.51 minutes in the placebo arm. For S2, the change from baseline was 37.69 minutes in the daridorexant 50 mg arm, 36.19 minutes in the daridorexant 25 mg arm, and 25.07 minutes in the placebo arm. For SWS, the change from baseline was –0.20 minutes in the daridorexant 50 mg arm, 0.20 minutes in the daridorexant 25 mg arm, and –1.54 minutes in the placebo arm. For REM, the change from baseline was 16.21 minutes in the daridorexant 50 mg arm, 12.55 minutes in the daridorexant 25 mg arm, and 11.65 minutes in the placebo arm.
Study 302
For S1, the change from baseline to month 1 was 3.64 minutes in the daridorexant 25 mg arm and 1.81 minutes in the placebo arm. For S2, the change from baseline to 1 month was 31.54 minutes in the daridorexant 25 mg arm and 24.09 minutes in the placebo arm. For SWS, the change from baseline to 1 month was 0.45 minutes in the daridorexant 25 mg arm and 0.90 minutes in the placebo arm. For REM, the change from baseline to 1 month was 14.19 minutes in the daridorexant 25 mg arm and 7.31 minutes in the placebo arm.
For S1, the change from baseline to month 3 was 5.46 minutes in the daridorexant 25 mg arm and 2.10 minutes in the placebo arm. For S2, the change from baseline to month 3 was 31.36 minutes in the daridorexant 25 mg arm and 25.45 minutes in the placebo arm. For SWS, the change from baseline to month 3 was –0.91 minutes in the daridorexant 25 mg arm and 0.65 minutes in the placebo arm. For REM. and the change from baseline to month 3 was 13.86 minutes in the daridorexant 25 mg arm and 6.94 minutes in the placebo arm.
The pattern of distribution of changes was similar across the groups, as shown in Appendix 1.
Table 18: Change From Baseline to Month 1 and Month 3 in Duration of TST in Each Sleep Stage — Study 301 and Study 302 (Full Analysis Set)
Variable | Study 301 | Study 302 | ||||
|---|---|---|---|---|---|---|
Daridorexant 25 mg (N = 310) | Daridorexant 50 mg (N = 310) | Placebo (N = 310) | Daridorexant 25 mg (N = 309) | Placebo (N = 308) | ||
N at baseline | 310 | 309 | 309 | 309 | 308 | |
N at month 1 | 298 | 305 | 299 | 295 | 300 | |
N at month 3 | 289 | 287 | 283 | 281 | 283 | |
Stage 1 sleep duration | ||||||
Baseline, mean (SD) | 35.82 (17.50) | 35.73 (18.61) | 34.10 (17.94) | 35.50 (18.71) | 38.18 (20.16) | |
Month 1 | ||||||
Observed value, mean (SD) | 40.16 (19.16) | 41.22 (23.07) | 36.67 (21.17) | 39.26 (21.22) | 40.18 (21.39) | |
Change from baseline, mean (SD) | 4.26 (14.67) | 5.41 (15.76) | 2.59 (13.97) | 3.64 (14.42) | 1.81 (16.13) | |
Month 3 | ||||||
Observed value, mean (SD) | 41.55 (21.22) | 42.7 (23.61) | 38.64 (19.32) | 41.35 (21.43) | 40.43 (21.51) | |
Change from baseline, mean (SD) | 5.67 (16.00) | 6.89 (16.56) | 4.51 (14.15) | 5.46 (16.99) | 2.10 (16.54) | |
Stage 2 sleep duration | ||||||
Baseline, mean (SD) | 178.82 (42.84) | 184.16 (40.77) | 180.36 (42.73) | 177.21 (48.44) | 174.59 (51.42) | |
Month 1 | ||||||
Observed value, mean (SD) | 208.48 (46.95) | 218.90 (40.53) | 196.40 (45.63) | 208.36 (48.16) | 198.64 (50.35) | |
Change from baseline, mean (SD) | 29.79 (37.43) | 34.74 (40.76) | 16.10 (40.45) | 31.54 (41.18) | 24.09 (37.89) | |
Month 3 | ||||||
Observed value, mean (SD) | 214.77 (41.07) | 222.20 (41.19) | 204.54 (45.33) | 209.86 (46.38) | 200.67 (49.94) | |
Change from baseline, mean (SD) | 36.19 (40.65) | 37.69 (41.38) | 25.07 (41.50) | 31.36 (41.88) | 25.45 (42.61) | |
SWS duration | ||||||
Baseline, mean (SD) | 44.45 (29.54) | 43.17 (31.53) | 43.11 (29.17) | 39.04 (30.55) | 32.67 (26.30) | |
Month 1 | ||||||
Observed value, mean (SD) | 45.09 (30.93) | 45.59 (33.20) | 44.56 (33.50) | 40.01 (32.85) | 33.50 (29.18) | |
Change from baseline, mean (SD) | 0.713 (19.61) | 2.16 (20.68) | 1.60 (21.57) | 0.447 (23.14) | 0.90 (20.43) | |
Month 3 | ||||||
Observed value, mean (SD) | 44.47 (32.25) | 43.11 (31.17) | 41.40 (31.79) | 38.84 (30.89) | 33.38 (29.30) | |
Change from baseline, mean (SD) | 0.202 (21.56) | –0.20 (22.27) | –1.54 (22.06) | –0.91 (24.08) | 0.65 (21.37) | |
REM duration | ||||||
Baseline, mean (SD) | 63.37 (20.60) | 65.18 (20.23) | 61.05 (20.52) | 60.90 (24.28) | 62.0 (23.74) | |
Month 1 | ||||||
Observed value, mean (SD) | 77.11 (23.07) | 80.74 (24.19) | 70.34 (23.45) | 75.04 (24.74) | 69.06 (23.96) | |
Change from baseline, mean (SD) | 13.50 (21.58) | 15.51 (24.61) | 9.22 (20.75) | 14.19 (21.0) | 7.31 (20.29) | |
Month 3 | ||||||
Observed value, mean (SD) | 76.04 (22.99) | 81.49 (24.5) | 72.36 (23.26) | 74.83 (24.24) | 69.19 (25.0) | |
Change from baseline, mean (SD) | 12.55 (24.42) | 16.21 (23.29) | 11.65 (23.06) | 13.86 (20.42) | 6.94 (20.24) | |
REM = rapid eye movement; SD = standard deviation; SWS = slow-wave sleep; TST = total sleep time.
Sources: Clinical Study Reports for Study 30116 and Study 302.17 Details included in the table are from the sponsor’s Summary of Clinical Evidence and the Clinical Study Reports.
Harms
Detailed harms data are summarized in Table 19.
Adverse Events
The overall incidence of TEAEs in Study 301 was generally similar across the groups; 41.3% of patients in the daridorexant 25 mg arm experienced TEAEs, as did 39.3% in the daridorexant 50 mg and 37.2% in the placebo arm. Similarly, in Study 302, the overall incidence of TEAEs was 41.2% in the daridorexant 25 mg arm and 36.3% in the placebo arm.
In Study 301, the most common TEAEs in the daridorexant 25 mg, daridorexant 50 mg, and placebo arms were nasopharyngitis (9.0% versus 7.8% versus 7.8%) and headache (5.5% versus 6.5% versus 3.9%). Other notable TEAEs included accidental overdose (1.9% versus 2.6% versus 1.9%), fatigue (2.3% versus 2.6% versus 0.6%), and dizziness (1.9% versus 2.3% versus 0.6%). The incidence of somnolence was similar in all groups, including the placebo group (3.5% versus 1.9% versus 2.3%).
In Study 302, the most common TEAEs in the daridorexant 25 mg and placebo arms were nasopharyngitis (4.2% versus 6.5%) and headache (5.2% versus 3.6%). Other notable TEAEs included accidental overdose (1.6% versus 1.0%), fatigue (3.6% versus 0.7%), and dizziness (2.3% versus 1.6%). The incidence of somnolence was 3.2% in the daridorexant 25 mg arm and 1.3% in the placebo arm.
Serious Adverse Events
The incidence of SAEs was generally low in both studies. In Study 301, SAEs were reported in 2 patients (0.6%) in the daridorexant 25 mg arm, 3 patients (1.0%) in the daridorexant 50 mg arm, and 7 patients (2.3%) in the placebo arm. In Study 302, SAEs were reported in 3 patients (1%) in the daridorexant 25 mg arm and 4 patients (1.3%) in the placebo arm.
Withdrawals Due to Adverse Events
In Study 301, AEs leading to withdrawal were reported in 7 patients (2.3%) in the daridorexant 25 mg, 3 patients (1.0%) in the daridorexant 50 mg arm, and 10 patients (3.2%) in the placebo arm. In Study 302, 4 patients (1.3%) in the daridorexant 25 mg arm and 7 patients (2.3%) in the placebo arm stopped treatment due to AEs.
Mortality
In Study 301, there was 1 death due to myocardial infarction, although it was not considered to be related to treatment (Table 19). No deaths were reported in Study 302.
Notable Harms
Notable harms of interest to this review included next-morning residual effects, rebound insomnia, and suicidal ideation, which were evaluated as part of the safety outcomes in Study 301 and Study 302. Other AESIs to this review included hallucinations and narcolepsy-like symptoms.
Next-Morning Residual Effect
Next-morning residual effect was a safety outcome in both studies and was flagged as important by the clinical experts. The morning sleepiness scores on the SDQ VAS were similar in the daridorexant and placebo groups at baseline in both studies. At month 1 and month 3, the mean observed values for morning sleepiness were higher than at baseline in all groups. In Study 301, change from baseline values at 3 months improved by 14.85 points in the daridorexant 25 mg arm, 15.28 points in the daridorexant 50 mg arm, and 11.50 points in the placebo arm. In Study 302, change from baseline values at 3 months improved by 14.91 points in the daridorexant 25 mg arm and 11.40 points in the placebo arm. In both studies, the change from baseline was numerically higher in the daridorexant groups than in the placebo groups; however, no statistical comparisons were made. Results are presented in Appendix 1.
Rebound Insomnia
Clinical experts consulted for this review noted that rebound insomnia, characterized by the worsening of sleep parameters upon discontinuation of the medication, as an important safety outcome. In Study 301 and Study 302, during the placebo run-out period, WASO and LPS were numerically lower, and the mean self-reported total sleep time was higher than baseline values, indicating an absence of rebound insomnia in both the daridorexant and placebo groups (Table 32). However, no statistical comparisons were made. Results are presented in Appendix 1.
Suicidal Ideation
In Study 301, no patients reported any TEAEs related to suicide or self-injury. Based on the C-SSRS, no patient-reported suicidal ideation or behaviour occurred during the study treatment (double-blind and placebo run-out) periods (data not shown).
In Study 302, 1 patient in the daridorexant 25 mg arm was found to have an AESI related to suicide or self-injury. Based on the C-SSRS, no patient-reported suicidal ideation or behaviour occurred during the study treatment (double-blind and placebo run-out) periods (data not shown).
Other Harms of Interest
In Study 301 and Study 302, 1 patient each (both in the daridorexant 25 mg arms) reported having hallucinations. Narcolepsy-like symptoms related to complex sleep behaviour, including hallucinations and sleep paralysis, were reported in 3 patients in each of the 2 studies. The results are presented in Table 19.
Table 19: Summary of Harms Results — Study 301 and Study 302
Adverse events | Study 301 | Study 302 | |||
|---|---|---|---|---|---|
Daridorexant 25 mg (N = 310) | Daridorexant 50 mg (N = 308) | Placebo (N = 309) | Daridorexant 25 mg (N = 308) | Placebo (N = 306) | |
Most common adverse eventsa (≥ 2%), n (%) | |||||
Patients with ≥ 1 TEAEa | 128 (41.3) | 121 (39.3) | 115 (37.2) | 127 (41.2) | 111 (36.3) |
Nasopharyngitis | 28 (9.0) | 24 (7.8) | 24 (7.8) | 13 (4.2) | 20 (6.5) |
Headache | 17 (5.5) | 20 (6.5) | 12 (3.9) | 16 (5.2) | 11 (3.6) |
Accidental overdose | 6 (1.9) | 8 (2.6) | 6 (1.9) | 5 (1.6) | 3 (1.0) |
Fatigue | 7 (2.3) | 8 (2.6) | 2 (0.6) | 11 (3.6) | 2 (0.7) |
Dizziness | 6 (1.9) | 7 (2.3) | 2 (0.6) | 7 (2.3) | 5 (1.6) |
Nausea | 1 (0.3) | 8 (2.6) | 4 (1.3) | 4 (1.3) | 3 (1.0) |
Somnolence | 11 (3.5) | 6 (1.9) | 7 (2.3) | 10 (3.2) | 4 (1.3) |
Fall | 2 (0.6) | 1 (0.3) | 8 (2.6) | 3 (1.0) | 3 (1.0) |
Upper respiratory tract infection | 1 (0.3) | 1 (0.3) | 4 (1.3) | 3 (1.0) | 7 (2.3) |
Diarrhea | 8 (2.6) | 2 (0.6) | 4 (1.3) | 3 (1.0) | 4 (1.3) |
SAEs,a n (%) | |||||
Patients with ≥ 1 SAE | 2 (0.6) | 3 (1.0) | 7 (2.3) | 3 (1.0) | 4 (1.3) |
Syncope | 0 | 1 (0.3) | 2 (0.6) | — | — |
Adenocarcinoma of the colon | 0 | 1 (0.3) | 0 | — | — |
Decreased hemoglobin | 0 | 1 (0.3) | 0 | — | — |
Postprocedural hemorrhage | 0 | 1 (0.3) | 0 | — | — |
Renal colic | 0 | 1 (0.3) | 0 | — | — |
Depression | 0 | 0 | 2 (0.6) | — | — |
Anal abscess | 0 | 0 | 1 (0.3) | — | — |
Ankle fracture | 0 | 0 | 1 (0.3) | — | — |
Herpes zoster | 0 | 0 | 1 (0.3) | — | — |
Panic attack | 0 | 0 | 1 (0.3) | — | — |
Lumbar radiculopathy | — | — | — | 1 (0.3) | 0 |
Schizophrenia | — | — | — | 1 (0.3) | 0 |
Hemoptysis | — | — | — | 1 (0.3) | 0 |
Hypertensive crisis | — | — | — | 0 | 1 (0.3) |
Joint dislocation | — | — | — | 0 | 1 (0.3) |
Meniscus injury | — | — | — | 0 | 1 (0.3) |
Rotator cuff syndrome | — | — | — | 0 | 1 (0.3) |
Patients who stopped treatment due to adverse events, n (%) | |||||
Patients who stopped treatment | 7 (2.3) | 3 (1.0) | 10 (3.2) | 4 (1.3) | 7 (2.3) |
Hyperthyroidism | 0 | 1 (0.3) | 0 | — | — |
Renal impairment | 0 | 1 (0.3) | 0 | — | — |
Supraventricular extrasystoles | 0 | 1 (0.3) | 0 | — | — |
Sedation complication | 1 (0.3) | 0 | 1 (0.3) | — | — |
Syncope | 0 | 0 | 2 (0.6) | — | — |
Anal abscess | 0 | 0 | 1 (0.3) | — | — |
Ankle fracture | 0 | 0 | 1 (0.3) | — | — |
Atrial tachycardia | 0 | 0 | 1 (0.3) | — | — |
Insomnia | 0 | 0 | 1 (0.3) | — | — |
Migraine with aura | 0 | 0 | 1 (0.3) | — | — |
Panic attack | 0 | 0 | 1 (0.3) | — | — |
Tinnitus | 0 | 0 | 1 (0.3) | — | — |
Apathy | — | — | — | 1 (0.3) | 0 |
Hallucinations, mixed | — | — | — | 1 (0.3) | 0 |
Poor quality sleep | — | — | — | 1 (0.3) | 0 |
Schizophrenia | — | — | — | 1 (0.3) | 0 |
Amnesia | — | — | — | 0 | 1 (0.3) |
Depression | — | — | — | 0 | 1 (0.3) |
Discomfort | — | — | — | 0 | 1 (0.3) |
Hypervigilance | — | — | — | 0 | 1 (0.3) |
Irritability | — | — | — | 0 | 1 (0.3) |
Paresthesia | — | — | — | 0 | 1 (0.3) |
Sciatica | — | — | — | 0 | 1 (0.3) |
Deaths, n (%) | |||||
Patients who died | 1 (0.3) | 0 | 0 | 0 | 0 |
Adverse events of special interest, n (%)a,b | |||||
Narcolepsy-like symptoms related to excessive daytime sleepiness | 2 (0.6) | 1 (0.3) | 1 (0.3) | 4 (1.3) | 1 (0.3) |
Somnolence | 2 (0.6) | 1 (0.3) | 1 (0.3) | 2 (0.6) | 1 (0.3) |
Narcolepsy-like symptoms related to complex sleep behaviour, including hallucinations and/or sleep paralysis | 2 (0.6) | 1 (0.3) | 0 | 3 (1.0) | 0 |
Sleep paralysis | 2 (0.6) | 1 (0.3) | 0 | 2 (0.6) | 0 |
Confusional state | 1 (0.3) | 0 | 0 | ||
Hallucination, visual | 1 (0.3) | 0 | 0 | 1 (0.3) | 0 |
Narcolepsy-like symptoms related to cataplexy | 0 | 0 | 0 | 0 | 0 |
Suicide and/or self-injury | 0 | 0 | 0 | 1 (0.3) | 0 |
SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aOccurring from the start of double-blind study treatment up until 30 days after the end of double-blind study treatment or enrolment in the Study 303 extension.
bAn Independent Safety Board reviewed blinded clinical patient data on selected adverse events and safety assessment results and adjudicated whether the submitted cases were adverse events of special interest.
Sources: Clinical Study Reports for Study 30116 and Study 302.17 Details included in the table are from the sponsor’s Summary of Clinical Evidence and the Clinical Study Reports.
Critical Appraisal
Internal Validity
Study 301 and Study 302 were multicentre, double-blind, phase III, randomized placebo-controlled trials. Randomization processes (i.e., using Interactive Response Technology) and the allocation concealment process used in the studies were judged to be appropriate and ensuring an overall balanced distribution of patients to either the active treatment or placebo groups. Treatment allocation was stratified by age (< 65 years or ≥ 65 years). According to the clinical experts, age is a clinically important variable, particularly with regard to harms (i.e., morning sedation and night dizziness) and the risk of injury due to a fall, and the stratification conducted in the included studies was appropriate.
Treatment discontinuations were relatively infrequent (7.1% to 9.0% in Study 301 and 5.8% to 7.4% in Study 302), as were permanent study discontinuations (7.1% to 9.7% in Study 301 and 7.1% to 9.4% in Study 302), and most of the participants in Study 301 and Study 302 completed the double-blinded treatment period; thus, the risk of attrition bias was considered low. Additionally, treatment adherence was more than 98% in both studies. Overall, the harms observed in the study, and the similar discontinuation rates across arms and studies, suggest that unblinding was unlikely.
Multiplicity adjustment using alpha-splitting was conducted for the primary and secondary outcomes to control for type I error. Other outcomes (i.e., efficacy outcomes such as IDSIQ total score and exploratory outcomes such as sleep quality, ISI scores, and duration of TST during each sleep stage) were not adjusted for multiplicity, so there is an increased risk of false-positive conclusions for statistically significant results. Similarly, subgroup analyses were not adjusted for multiplicity. Missing data were evaluated using implicit imputation, which raised concerns about bias that would overestimate the effect of daridorexant. As noted, discontinuations were infrequent and the overall rate of missing data was low (< 10%) in both studies for all relevant outcomes. Low overall missingness and balanced discontinuation rates across groups reduced concerns related to this potential overestimation, but did not completely rule out bias from informative missingness. Sensitivity analyses using multiple imputation under missing-not-at-random assumptions further supported the robustness of the results, as statistical significance was maintained even under conservative imputations. Therefore, any impact of bias due to missing data on study results was likely low.
The primary outcomes in both studies were objectively measured using PSG during sleep studies, whereas subjectively measured outcomes were captured using self-assessment of patients’ sleep or validated questionnaires. Although PSG provides objective data on sleep parameters and is less prone to reporting bias, its results may not fully capture real-world sleep patterns. Sleep disturbances related to sleep-study settings — including factors such as a separate sleeping location and the presence of monitoring equipment (e.g., connected probes) — may alter sleep architecture and limit the accuracy and generalizability of PSG results. According to the clinical experts consulted for this review, patients’ self-reporting of sleep may not align with the more objective measures. They noted, for example, that patients with insomnia have been found to overestimate the time taken to fall asleep (sleep onset latency) and to underestimate total sleeping time. The clinical experts emphasized that insomnia is largely a subjective condition and that sleep complaints are very individualized; therefore, self-reported sleep quality and perceived changes in patients’ sleep may be more clinically meaningful than PSG-derived metrics. However, subjective measures may be more prone to bias, including recall bias and placebo effects, that cannot be easily measured or accounted for, making it difficult to determine the true magnitude and certainty of treatment effects. Thresholds of clinical significance for all relevant outcomes, except for sleep quality by VAS, were provided by the sponsor. According to the clinical experts consulted for this review, these thresholds were clinically relevant and acceptable. They noted that the most clinically useful measure of improved sleep is a patient’s own perception. However, it can be difficult to quantify an exact value for improved sleep.
External Validity
There were several limitations in the included studies that could affect the generalizability of results to Canadian clinical settings. Among the patients who were screened for the trials, most patients failed screening (72.4% in Study 301 and 74.9% in Study 302), either at the initial screening stage or after the single-blind run-in period. The most common reason for screening failure was not meeting the inclusion or exclusion criteria, although the submission did not provide any additional details on the exact criteria that were failed during screening. Based on the eligibility criteria for the trials, patients who did not meet specific sleep parameters on PSG and those with comorbidities (e.g., acute or unstable psychiatric conditions) were excluded. According to the clinical experts, CID is diagnosed based on clinical assessment and patient interview rather than PSG or other sleep studies, which are not performed in clinical settings. Also, patients who were excluded based on comorbidities could have otherwise been potential candidates for treatment with daridorexant. Thus, the screening process may have led to a study population that does not reflect the broader population with CID in Canada, limiting the generalizability of the results.
The clinical experts confirmed that the baseline characteristics of patients in Study 301 and Study 302 were generally similar to those of patients seen in Canadian clinical settings, with some notable limitations. Although mean age, duration of disease, and distributions of sex in the trial populations were consistent with those in settings in Canada, the clinical experts noted that the proportion of Asian patients was considerably lower in the studies (especially Study 301) than in Canadian settings. It is unclear whether this would affect the study results, beyond cultural differences related to sleep among different racial groups.
According to the Health Canada product monograph, daridorexant is contraindicated for patients who use strong CYP3A4 inhibitors, although daridorexant 25 mg is indicated for patients receiving moderate CYP3A4 inhibitors (or those with moderate hepatic impairment). However, the use of moderate or strong CYP3A4 inhibitors was prohibited during the studies, and those unwilling or unable to discontinue those medications were excluded from both studies. Therefore, any interpretation of efficacy or harms results in the daridorexant 25 mg treatment group was a challenge.
Several medications, including concomitant pharmacological treatments for insomnia or other CNS-related medications, were prohibited in the studies. According to the clinical experts, patients with CID seen in Canadian clinical settings would often be taking 1 or more of the CNS-related medications prohibited in the studies. One notable example of prohibited medications was antihistamines; many people in the real world take over-the-counter antihistamines to manage allergies. Patients in the included studies were allowed to continue CBT-I throughout the study. Although only a minority of patients receive CBT-I in the real world, the proportion would likely be higher than that observed in the trials (3 patients [0.3%] in Study 301, and no patients in Study 302), and the clinical experts noted that, generally, this would be considered a first-line treatment. In the included trials, the most common reason for not using CBT was patient unawareness that CBT was a treatment option. As noted by the clinical experts consulted for this review, physician and patient unawareness and limited accessibility are the most common reasons that patients in real-world settings do not receive CBT-I. Overall, concerns regarding concomitant medications could lower the generalizability of the results. However, it is worth noting that these exclusions were likely implemented to prevent concern about influence from other sleep-active drugs.
As previously noted, sleep studies are not used in clinical settings to diagnose or assess insomnia; instead, patients are usually interviewed about their sleep and how they feel. Additionally, subjective measures used in the studies, such as IDSIQ, ISI, and SDQ, are not routinely used in clinical practice, per the clinical experts consulted for this review.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:61,62
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for daridorexant 50 mg versus placebo. As daridorexant 25 mg is only used for patients receiving moderate CYP3A4 inhibitors or those with moderate hepatic impairment, this group was not included in the GRADE assessment. Further, there is a dose-response relationship associated with daridorexant 25 mg and daridorexant 50 mg; thus, only the results for the 50 mg dose were included in the GRADE assessment.
Long-Term Extension Studies
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
Description of Studies
One LTE study (Study 303; N = 804) has been summarized to provide evidence on the long-term safety and tolerability of daridorexant in adult and older patients with CID. Study 303 was a multicentre, double-blind, parallel-group, randomized, placebo-controlled, 40-week extension to Study 301 and Study 302. Study 303 was conducted at 94 sites in 14 countries, with 7 sites in Canada.
As shown in Figure 4, Study 303 included a 30-day safety follow-up phase after the end of double-blind treatment to assess AEs, SAEs, and concomitant medications. This phase included an initial 7-day single-blind run-out period, during which all patients received a daily dose of placebo, followed by a safety follow-up period until the end of the study (week 44).
Figure 4: Study Design of Study 303
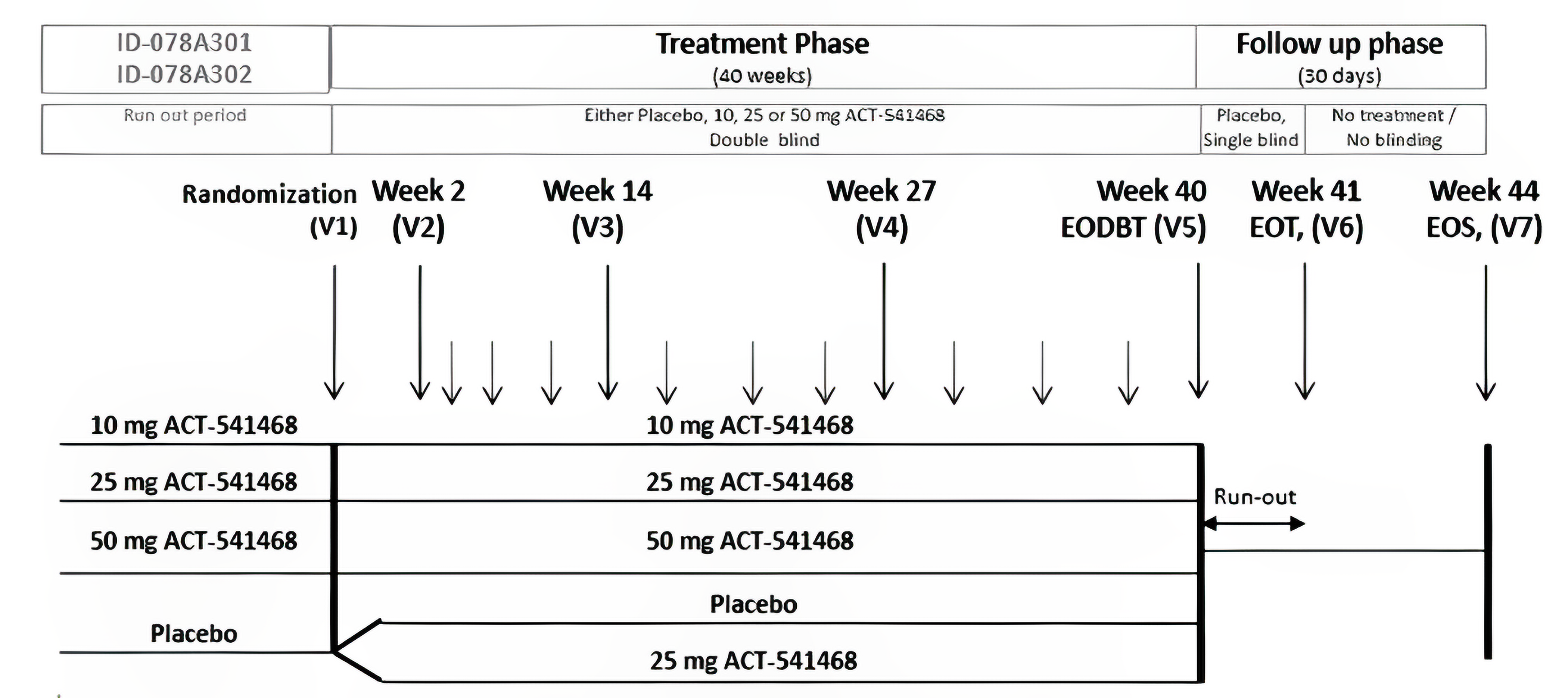
ACT-541468 = daridorexant; EODBT = end of double-blind treatment; EOS = end of study; EOT = end of treatment; ID-078A301 = Study 301; ID-078A302 = Study 302; V = visit.
Sources: Clinical Study Report for Study 303 and the sponsor’s Summary of Clinical Evidence.15,63 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Populations
For Study 303, eligible participants were required to have completed Study 301 or Study 302. Inclusion and exclusion criteria for Study 303 were generally in line with those in the pivotal trials, as identified in Table 5.
Interventions
Patients who received daridorexant in Study 301 or Study 302 maintained the same dose in Study 303, and patients who received placebo were rerandomized in a 1:1 ratio to continue to receive placebo or to receive daridorexant 25 mg. Because the Health Canada–recommended therapeutic daily dose of daridorexant for adults is 50 mg, or 25 mg for patients with moderate hepatic impairment, the results for the 10 mg dose were not included in this section. Prohibited concomitant therapies were in line with those in the pivotal trials.
Outcomes
Table 20 presents a list of relevant outcomes evaluated in Study 303. Unlike the pivotal trials, Study 303 assessed sWASO, which consisted of the self-reported time spent awake after sleep onset, reported in item 7 of the SDQ (In total, how long did these awakenings last?) and sLSO consisted of the self-reported time to fall asleep, reported in item 5 of the SDQ (How long did it take you to fall asleep?). Most efficacy results are presented in the table at week 12 and week 36 of the extension study, corresponding to month 6 and month 12 from the confirmatory study baseline.
Table 20: Key End Points in Study 303
Outcome measure | Time point | Study 303 |
|---|---|---|
Change from baseline in sWASO | Week 36a | Exploratory |
Change from baseline in sLSO | Week 36a | Exploratory |
Change from baseline in sTST | Week 36a | Exploratory |
Change from baseline in IDSIQ scores | Week 36a | Exploratory |
Change from baseline in SDQ VAS scores | Week 36a | Exploratory |
Change from baseline in ISI scores | Week 14,a week 27,a and week 40a | Exploratory |
TEAEs, SAEs, AESIs (i.e., narcolepsy-like symptoms, suicide and/or self-injury) | Up to 30 days after double-blind study treatment discontinuation | Safety |
Occurrence of suicidal ideation and/or behaviour based on C-SSRS | During double-blind study treatment and during the placebo run-out period | Safety |
Withdrawal effects (physical dependence) upon treatment discontinuation, assessed using changes in BWSQ total score | From the last assessment on double-blind treatment to the end of the placebo run-out period | Safety |
Rebound insomnia, assessed using the subjective sleep parameter of sTST | From baselinea to the end of the placebo run-out period | Safety |
Next-morning residual effect assessed using change from baseline over time in the morning sleepiness score on the SDQ VAS (mm) | Week 36a | Safety |
AESI = adverse event of special interest; BWSQ = Benzodiazepine Withdrawal Symptom Questionnaire; C-SSRS = Columbia Suicide Severity Rating Scale; IDSIQ = Insomnia Daytime Symptoms and Impacts Questionnaire; ISI = Insomnia Severity Index; SAE = serious adverse event; SDQ = Sleep Diary Questionnaire; sLSO = subjective latency to sleep onset; sTST = subjective total sleep time; sWASO = subjective wake after sleep onset; TEAE = treatment-emergent adverse event; VAS = visual analogue scale.
aBaseline refers to the last nonmissing assessment or value measured before or on the first day of double-blind study treatment in Study 301 or Study 302, unless otherwise noted in the report.
Sources: Clinical Study Report for Study 303 and the sponsor’s Summary of Clinical Evidence. 15,63 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Statistical Analysis
The analyses of efficacy end points were performed using the full analysis set. For all efficacy end points, change from baseline was analyzed using the confirmatory baseline, which was defined as the last nonmissing assessment or value measured before or on the first day of double-blind study treatment in Study 301 or Study 302. A linear mixed-effects model was used for the analyses of change from baseline in sTST, sWASO, sLSO, and in IDSIQ total score, sleepiness domain score, alert/cognition domain score, and mood domain score. The model was adjusted for the baseline value of the relevant response variable, age group per assigned strata, treatment, visit, the interaction of treatment by visit, and the interaction of baseline by visit. An unstructured covariance matrix was used to model the correlation among repeated measurements.
The treatment differences between daridorexant groups (25 mg and 50 mg) and placebo at week 12, week 24, and week 36 were based on the LSM change from confirmatory study baseline. For each daridorexant dose compared to placebo, the placebo-adjusted LSM, standard errors, 95% CI, and unadjusted two-sided P value were presented.
The end points for VAS scores, ISI, and the number of self-reported awakenings were summarized descriptively.
Unless otherwise noted, the safety set was used for tables and listings of safety data. For safety end points, TEAEs, SAEs, and AESIs were summarized descriptively. These events were collected from the start of double-blind treatment until up to 30 days after the end of double-blind treatment. Changes from baseline in safety outcomes were analyzed using the confirmatory study baseline, as previously defined.
No adjustment for multiple testing was implemented.
Handling of Missing Data
For sTST, sWASO, sLSO, IDSIQ scores, VAS scores, and the number of self-reported awakenings, at least 2 days of data for each week was required to calculate a weekly mean. Otherwise, the mean value was considered missing for that week.
Subgroups
The treatment effect of daridorexant versus placebo was evaluated across different subgroups. These included age at screening in Study 301 (< 65 years, 65 to 74 years, ≥ 75 years) for all end points, as well as sex, region, race, and body mass index at screening (< 30 kg/m2, ≥ 30 kg/m2) for sTST and IDSIQ scores. The analyses used the linear mixed-effects models previously described, applied separately to each subgroup.
Analysis Populations
The analysis sets used in Study 303 are described in Table 21.
Table 21: Analysis Populations in Study 303
Population | Definition | Application |
|---|---|---|
Full analysis set | All patients randomized to study treatment | Demographic characteristics, baseline characteristics, and efficacy end points |
Safety set | All patients who received at least 1 dose of the study treatment | Disposition of patients, previous and concomitant medications, exposure to interventions, and safety end points |
Treatment withdrawal set | All patients in the safety set who received at least 1 dose of the single-blind placebo treatment during the placebo run-out period | End points assessing withdrawal symptoms (i.e., BWSQ total score) and rebound insomnia based on sTST |
BWSQ = Benzodiazepine Withdrawal Symptom Questionnaire; sTST = subjective total sleep time.
Sources: Clinical Study Report for Study 303 and the sponsor’s Summary of Clinical Evidence.15,63 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results
Patient Disposition
A summary of patient disposition in Study 303 is presented in Table 22. In Study 303, all 137 patients in the daridorexant 50 mg group originated from the same dose group in Study 301. The daridorexant 25 mg group included 132 patients (48.9%) from Study 301 and 138 patients (51.1%) from Study 302, all from the corresponding dose group. Similarly, the placebo group comprised 57 patients (44.5%) from Study 301 and 71 patients (55.5%) from Study 302. Among the 127 patients who transitioned from placebo in the pivotal trials to daridorexant 25 mg in Study 303, 66 patients (52.0%) came from Study 301 and 61 patients (48.0%) from Study 302.
A total of 804 patients were enrolled in Study 303, with 662 patients enrolled into treatment arms of interest to this review. Across treatment arms of interest to this review, 208 patients (31.4%) prematurely discontinued treatment. The most frequent reason for treatment discontinuation was lack of efficacy, with the placebo group showing the highest rate (22.7%), followed by 10.7% the daridorexant 25 mg group, 9.5% in the daridorexant 50 mg group, and 7.9% in the ex-placebo to daridorexant 25 mg group. A total of 459 patients (69.3%) in these 4 treatment groups completed the study, and the remaining 203 patients (30.7%) discontinued prematurely, mainly due to patient withdrawal. The placebo group had the highest withdrawal rate (25.0%), followed by the ex-placebo to daridorexant 25 mg group (19.7%), the daridorexant 25 mg group (19.6%), and the daridorexant 50 mg group (15.3%).
Table 22: Patient Disposition in Study 303
Patient disposition | Daridorexant 50 mg (N = 137) | Daridorexant 25 mg (N = 270) | Ex-placebo to daridorexant 25 mg (N = 127) | Placebo (N = 128) |
|---|---|---|---|---|
Completed study, n (%) | 94 (68.6) | 193 (71.5) | 89 (70.1) | 83 (64.8) |
Discontinued treatment, n (%) | 44 (32.1) | 78 (28.9) | 36 (28.3) | 50 (39.1) |
Adverse events | 9 (6.6) | 10 (3.7) | 6 (4.7) | 6 (4.7) |
Withdrawal | 8 (5.8) | 23 (8.5) | 9 (7.1) | 8 (6.3) |
Lost to follow-up | 2 (1.5) | 4 (1.5) | 3 (2.4) | 0 |
Lack of efficacy | 13 (9.5) | 29 (10.7) | 29 (22.7) | 10 (7.9) |
Other | 12 (8.8) | 12 (4.4) | 8 (6.3) | 7 (5.5) |
Discontinued the study, n (%) | 43 (31.4) | 77 (28.5) | 38 (29.9) | 45 (35.2) |
Adverse event | 4 (2.9) | 5 (1.9) | 5 (3.9) | 5 (3.9) |
Withdrawal | 21 (15.3) | 53 (19.6) | 25 (19.7) | 32 (25.0) |
Lost to follow-up | 2 (1.5) | 4 (1.5) | 3 (2.4) | 0 |
Other | 16 (11.7) | 14 (5.2) | 5 (3.9) | 8 (6.3) |
Analysis sets | ||||
Full analysis set, N | 137 | 270 | 127 | 128 |
Safety,a N | 137 | 268 | 126 | 128 |
Treatment withdrawal set,b N | 93 | 79 | 38 | 78 |
aComprised patients who received at least 1 dose of the study treatment during the extension study.
bComprised patients in the safety set who received at least 1 dose of placebo run-out treatment.
Sources: Clinical Study Report for Study 303 and the sponsor’s Summary of Clinical Evidence.15,63 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Baseline Characteristics
The baseline characteristics of patients enrolled into treatment arms of interest to this review from Study 303 are summarized in Table 23. A total of 662 patients were enrolled into treatment arms of interest from Study 303, with 137 patients in the daridorexant 50 mg group, 270 in daridorexant 25 mg group, 127 in the ex-placebo to daridorexant 25 mg group, and 128 patients in the placebo group. Demographic characteristics and baseline efficacy characteristics were generally balanced across treatment groups, and were similar to those in the pivotal studies, with mean age ranging from 56.5 years to 59.2 years across treatment groups. Baseline values at the start of the pivotal trials for efficacy variables, such as sTST, IDSIQ scores, sLSO, and sWASO, were well balanced in the daridorexant 25 group, the daridorexant 50 mg group, and the placebo group. IDSIQ scores in the ex-placebo to daridorexant 25 mg group were lower (improved) at Study 303 baseline than scores in the other treatment groups at Study 301 and Study 302 baseline. However, IDSIQ scores in the ex-placebo to daridorexant 25 mg group were comparable to those in the placebo group at the extension study baseline; mean score on the sleepiness domain was 18.44 (SD = 7.38), on the mood domain was 14.64 (SD = 8.62), and on the alert/cognition domain was 24.95 (SD = 10.77), and IDSIQ total score was 57.32 (SD = 25.83).
Table 23: Summary of Baseline Characteristics in Study 303 (Full Analysis Set)
Characteristic | Daridorexant 50 mg (N = 137) | Daridorexant 25 mg (N = 270) | Ex-placebo to daridorexant 25 mg (N = 127) | Placebo (N = 128) |
|---|---|---|---|---|
Age, years, mean (SD) | 56.9 (13.6) | 57.6 (14.1) | 56.5 (15.5) | 59.2 (12.6) |
Sex, n (%) | ||||
Male | 39 (28.5) | 71 (26.3) | 44 (34.6) | 36 (28.1) |
Female | 98 (71.5) | 199 (73.7) | 83 (65.4) | 92 (71.9) |
Race, n (%) | ||||
Black or African American | 15 (10.9) | 19 (7.0) | 7 (5.5) | 8 (6.3) |
American Indian or Alaska Native | 1 (0.7) | 0 | 0 | 0 |
Native Hawaiian or other Pacific Islander | 0 | 0 | 0 | 1 (0.8) |
Asian | 0 | 8 (3.0) | 5 (3.9) | 2 (1.6) |
White | 121 (88.3) | 243 (90.0) | 115 (90.6) | 115 (89.8) |
Other | 0 | 0 | 0 | 2 (1.6) |
Patient characteristics | ||||
Medical history of psychiatric disorders, n (%) | 13 (9.5) | 22 (8.1) | 4 (3.1) | 5 (3.9) |
BMI, kg/m2, mean (SD) | 25.89 (4.24) | 26.57 (4.65) | 26.69 (4.32) | 25.90 (4.04) |
Patients taking ≥ 1 concomitant medication, n (%)a | 90 (65.7) | 195 (72.8) | 96 (76.2) | 91 (71.1) |
sTST, minutes, mean (SD) | 303.79 (65.08) | 299.52 (61.45) | 303.79 (65.08)c | 305.07 (56.51) |
ISI score,b mean (SD) | 19.9 (3.7) | 19.9 (4.2) | 15.0 (5.7)c | 19.4 (4.0) |
IDSIQ scores, mean (SD) | ||||
Sleepiness domain | 22.37 (6.56) | 22.54 (6.76) | 18.44 (7.38)c | 21.79 (6.56) |
Mood domain | 20.10 (8.01) | 19.43 (8.49) | 16.06 (8.74)c | 17.68 (8.01) |
Alert/cognition domain | 32.39 (1.00) | 32.24 (9.80) | 26.27 (10.95)c | 30.83 (9.14) |
Total score | 74.86 (23.52) | 74.21 (23.78) | 60.76 (25.79)c | 70.30 (22.13) |
BMI = body mass index; IDSIQ = Insomnia Daytime Symptoms and Impacts Questionnaire; ISI = Insomnia Severity Index; SD = standard deviation; sTST = subjective total sleep time.
Note: Baseline values from confirmatory Study 301 or Study 302 are reported unless otherwise noted.
aStudy concomitant therapies are any treatments that are either ongoing at the signing of informed consent for the extension study or initiated between the signing of informed consent and the end of the study.
bA score of 15 to 21 indicates moderate insomnia; a score of 22 to 28 indicates severe insomnia.
cValues are as reported at baseline of the extension study.
Sources: Clinical Study Report for Study 303 and the sponsor’s Summary of Clinical Evidence.15,63 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Exposure to Study Treatments
A summary of treatment exposure in Study 303 is presented in Table 24. Overall exposure (mean duration of double-blind treatment in the extension study) was 231 days in the daridorexant 25 mg group, 222 days in the daridorexant 50 mg group, 232 days in the ex-placebo to daridorexant 25 mg group, and 210 days in the placebo group. Compliance during the double-blind treatment phase was high, with a mean of more than 99% of patients in each group adhering to treatment, and was generally consistent between the groups; there were no treatment interruptions.
Table 24: Study Treatment Exposure and Adherence During the Double-Blind Treatment Period in Study 303 (Safety Set)
Exposure | Daridorexant 50 mg (N = 137) | Daridorexant 25 mg (N = 270) | Ex-placebo to daridorexant 25 mg (N = 127) | Placebo (N = 128) | |
|---|---|---|---|---|---|
Duration of double-blind treatment | |||||
Mean, days (SD) | 222.2 (93.2) | 231.4 (85.3) | 232.3 (88.6) | 209.7 (97.2) | |
Median, days (range) | 277 (1 to 308) | 277 (1 to 321) | 276 (9 to 359) | 274 (7 to 298) | |
Exposure to double-blind treatment, patient-years | 83.3 | 169.8 | 80.2 | 73.5 | |
Compliance | |||||
Mean, days treated (SD) | 99.78 (1.00) | 99.58 (2.88) | 99.67 (2.43) | 99.78 (1.59) | |
Median, days treated (range) | 100.0 (92.2 to 100.0) | 100.0 (59.6 to 100.0) | 100.0 (74.2 to 100.0) | 100.0 (83.5 to 100.0) | |
Duration of run-out treatmenta | |||||
Mean, days (SD) | 8.8 (5.1) | 8.4 (3.4) | 8.3 (3.2) | 7.9 (3.4) | |
Median, days (range) | 7 (4 to 35) | 7 (2 to 35) | 7 (3 to 23) | 7 (3 to 30) | |
SD = standard deviation.
aDuring the treatment withdrawal set.
Sources: Clinical Study Report for Study 303 and the sponsor’s Summary of Clinical Evidence.15,63 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Concomitant Medications and Cointerventions
A summary of concomitant medications in Study 303 is presented in Table 25. As in Study 301, therapies deemed necessary for a patient's well-being and not classified as prohibited concomitant medications were allowed. The use of concomitant therapies during double-blind treatment was defined as treatments ongoing at the start of the double-blind treatment or initiated during the double-blind period and continued until up to 1 day after the last double-blind dose. During the double-blind treatment phase, 65% of patients in the daridorexant 50 mg group, 71.6% in the daridorexant 25 mg group, 74.6% in the ex-placebo to daridorexant 25 mg group, and 70.3% in the placebo group reported using at least 1 concomitant medication. The most frequently used concomitant medications were propionic acid derivatives, thyroid hormones, beta-hydroxy-beta-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, angiotensin-converting-enzyme inhibitors, and proton pump inhibitors.
Table 25: Concomitant Medications Used During the Double-Blind Treatment Phase in Study 303 (Safety Set)
Concomitant medication | Daridorexant 50 mg (N = 137) | Daridorexant 25 mg (N = 270) | Ex-placebo to daridorexant 25 mg (N = 127) | Placebo (N = 128) |
|---|---|---|---|---|
Patients with at least 1 medication, n (%) | 89 (65.0) | 192 (71.6) | 94 (74.6) | 90 (70.3) |
Most common medications, n (%) | ||||
Propionic acid derivates | 18 (13.1) | 36 (13.4) | 16 (12.7) | 21 (16.4) |
Thyroid hormones | 12 (8.8) | 25 (9.3) | 12 (9.5) | 20 (15.6) |
Statins | 10 (7.3) | 36 (13.4) | 19 (15.1) | 16 (12.5) |
ACE inhibitors | 10 (7.3) | 26 (9.7) | 15 (11.9) | 16 (12.5) |
Proton pump inhibitors | 15 (10.9) | 20 (7.5) | 12 (9.5) | 9 (7.0) |
ACE = angiotensin-converting enzyme.
Sources: Clinical Study Report for Study 303 and sponsor’s Summary of Clinical Evidence.15,63 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Efficacy
For the daridorexant 50 mg, daridorexant 25 mg, and placebo groups, baseline refers to assessments conducted on or before the start of the double-blind study treatment in Study 301 or Study 302, representing the confirmatory study baseline. For the ex-placebo to daridorexant 25 mg group, baseline refers to assessments conducted on or before the start of the double-blind study treatment in the extension study. The exploratory efficacy analysis included the same subjective end points as Study 301.
The results for change from baseline in sWASO, sLSO, sTST, and IDSIQ total score in Study 303 are presented in Table 26.
Change From Baseline in sWASO
During the extension study, mean sWASO reductions from baseline were maintained across treatment groups. The LSM difference in the change in sWASO from the confirmatory study baseline to week 36 between the daridorexant 50 mg group and the placebo group was –2.01 minutes (95% CI, –14.71 to 10.68 minutes; P = 0.7554) and between the daridorexant 25 mg group and the placebo group was –1.51 minutes (95% CI, –12.65 to 9.62 minutes; P = 0.5148). For the ex-placebo to daridorexant 25 mg group, mean sWASO values in the extension study from week 4 onward were comparable to the other daridorexant groups. The LSM difference in from the extension study baseline to week 36 between the ex-placebo to daridorexant 25 mg group and the placebo group was –3.92 minutes (95% CI, –13.87 to 5.99 minutes; P = 0.4342).
Change From Baseline in sLSO
In the extension study, improvements were numerically greater in the daridorexant groups than in the placebo group for mean sLSO. The LSM treatment difference in sLSO from confirmatory study baseline to week 36 between the daridorexant 50 mg group and the placebo group was –8.76 minutes (95% CI, –19.34 to 1.82 minutes; P = 0.1044) and between the daridorexant 25 mg group and the placebo group was –9.19 minutes (95% CI, –18.45 to 0.07 minutes; P = 0.0517). The LSM difference in sLSO from extension study baseline to week 36 between the ex-placebo to daridorexant 25 mg group and the placebo group was –15.14 minutes (95% CI, –25.52 to –4.76 minutes; P = 0.0046).
Change From Baseline in sTST
In the extension study, changes in sTST from confirmatory study baseline were numerically greater in the daridorexant groups than in the placebo group, with the most pronounced change in the daridorexant 50 mg group. The LSM treatment difference in sTST from confirmatory study baseline to week 36 between the daridorexant 50 mg group and the placebo group was 17.77 minutes (95% CI, –0.35 to 35.90 minutes; P = 0.0546) and between the daridorexant 25 mg group and the placebo group was 5.26 minutes (95% CI, –10.59 to 21.11 minutes; P = 0.5148). In a post hoc analysis, the LSM treatment difference in sTST from extension study baseline to week 36 between the ex-placebo to daridorexant 25 mg group and the placebo group was 25.74 minutes (95% CI, 9.51 to 41.96; minutes P = 0.0021).
Change from Baseline in IDISQ Total Score
Mean reductions in IDSIQ total score from baseline were maintained throughout the extension study across all treatment groups. The LSM treatment difference in IDSIQ total score from confirmatory study baseline to week 36 between the daridorexant 50 mg group and the placebo group was –9.12 (95%CI, –15.59 to –2.66; P = 0.0058) and between the daridorexant 25 mg group and the placebo group was –4.52 (95% CI, –10.15 to 1.12, P = 0.11161). The mean IDSIQ total score in the ex-placebo to daridorexant 25 mg group was in a range similar to that in the other daridorexant treatment groups from week 4 onward in the extension study. The LSM difference in IDSIQ total score from extension study baseline to week 36 between the ex-placebo to daridorexant 25 mg group and the placebo group was –3.42 (95% CI, –7.95 to 1.11; P = 0.1377). Similar findings were observed for the IDSIQ sleepiness, mood, and alert/cognition domain scores.
Table 26: Change From Baseline in sTST, sWASO, sLSO, and IDSIQ Total Score in Study 303 (Full Analysis Set)
Variable | Daridorexant 50 mg (N = 137) | Daridorexant 25 mg (N = 270) | Ex-placebo to daridorexant 25 mg (N = 127)a | Placebo (N = 128) | |
|---|---|---|---|---|---|
sWASO, minutes | |||||
N at week 36 | 87 | 170 | 83 | 70 | |
Baseline, mean (SD) | 80.11 (57.33) | 90.47 (60.26) | 52.92 (44.81)b | 82.68 (52.39) | |
Change from baseline, LSM (95% CI) | –36.84 (–45.52 to –28.16) | –36.34 (–42.50 to –30.18) | –8.12 (–14.92 to –1.31)a | –34.83 (–44.14 to –25.52) | |
LSM difference vs. placebo (95% CI) | –2.01 (–14.71 to 10.68) | –1.51 (–12.65 to 9.62) | –3.92 (–13.87 to 5.99)a | NA | |
P value | 0.7554 | 0.7895 | 0.4342a | NA | |
sLSO, minutes | |||||
N at week 36 | 87 | 170 | 83 | 70 | |
Baseline, mean (SD) | 63.41 (40.30) | 66.44 (40.89) | 47.40 (33.94)b | 64.82 (39.95) | |
Change from baseline, LSM (95% CI) | –22.87 (–30.13 to –15.61) | –23.30 (–28.43 to –18.16) | –11.26 (–18.51 to –4.02)a | –14.11 (–21.84 to –6.38) | |
LSM difference vs. placebo (95% CI) | –8.76 (–19.34 to 1.82) | –9.19 (–18.45 to 0.07) | –15.14 (–25.52 to –4.76)a | NA | |
P value | 0.1044 | 0.0517 | 0.0046a | NA | |
sTST, minutes | |||||
N at week 36 | 87 | 170 | 83 | 70 | |
Baseline, mean (SD) | 303.79 (65.08) | 299.52 (61.45) | 351.65 (71.43)b | 305.07 (56.51) | |
Change from baseline, LSM (95% CI) | 75.16 (62.72 to 87.61) | 62.65 (53.85 to 71.46) | 34.25 (23.11 to 45.40)a | 57.39 (44.16 to 70.62) | |
LSM difference vs. placebo (95% CI) | 17.77 (–0.35 to 35.90) | 5.26 (–10.59 to 21.11) | 25.74 (9.51 to 41.96)a | NA | |
P value | 0.0546 | 0.5148 | 0.0021a | NA | |
IDSIQ total score | |||||
N at week 36 | 87 | 175 | 83 | 68 | |
Baseline, mean (SD) | 74.86 (23.52) | 74.21 (23.78) | 60.76 (25.79)b | 70.23 (22.13) | |
Change from baseline, LSM (95% CI) | –26.55 (–30.99 to –22.11) | –21.94 (–25.06 to –18.83) | –6.04 (–9.13 to –2.95)a | –17.43 (–22.14 to –12.71) | |
LSM difference vs. placebo (95% CI) | –9.12 (–15.59 to –2.66) | –4.5 (–10.15 to 1.12) | –3.42 (–7.95 to 1.11)a | NA | |
P value | 0.0058 | 0.1161 | 0.1377a | NA | |
CI = confidence interval; IDSIQ = Insomnia Daytime Symptoms and Impacts Questionnaire; LSM = least squares mean; NA = not applicable; SD = standard deviation; sLSO = subjective latency to sleep onset; sTST = subjective total sleep time; sWASO = subjective wake after sleep onset; vs. = versus.
Note: Baseline values from confirmatory Study 301 or Study 302 are reported unless otherwise noted.
aChange from extension study baseline between the placebo to daridorexant 25 mg group and the placebo group were conducted in a separate post hoc analysis.
bValues are as reported at baseline of the extension study.
Sources: Clinical Study Report for Study 303 and the sponsor’s Summary of Clinical Evidence.15,63 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Change From Baseline in SDQ VAS Scores
The results for change from baseline in SDQ VAS scores in the extension study are presented in Table 27. Improvements from baseline in quality of sleep were maintained in all treatment groups throughout the extension study. The mean change from confirmatory study baseline to week 36 was 27.4 (SD = 23.6) in the daridorexant 50 mg group, 22.4 (SD = 21.6) in the daridorexant 25 mg group, 9.7 (SD = 16.3) in the ex-placebo to daridorexant 25 mg group, and 21.9 (SD = 19.2) in the placebo group.
Similar results were reported for the VAS depth of sleep, daytime alertness, and ability to function end points.
Table 27: Change From Baseline in VAS Scores in Study 303 (Full Analysis Set)
Variable | Daridorexant 50 mg (N = 137) | Daridorexant 25 mg (N = 270) | Ex-placebo to daridorexant 25 mg (N = 127) | Placebo (N = 128) |
|---|---|---|---|---|
VAS quality of sleep | ||||
N at week 36 | 87 | 170 | 83 | 70 |
Baseline, mean (SD) | 34.0 (16.0) | 35.6 (17.0) | 49.8 (21.1)a | 38.1 (16.5) |
Change from baseline, mean (SD) | 27.4 (23.6) | 22.4 (21.6) | 9.7 (16.3)b | 21.9 (19.2) |
VAS depth of sleep | ||||
N at week 36 | 87 | 170 | 83 | 70 |
Baseline, mean (SD) | 33.9 (16.1) | 35.4 (16.8) | 50.0 (21.2)a | 37.9 (16.3) |
Change from baseline, mean (SD) | 26.7 (24.0) | 21.7 (21.9) | 9.0 (17.3)b | 20.6 (20.7) |
VAS daytime alertness | ||||
N at week 36 | 87 | 175 | 83 | 68 |
Baseline, mean (SD) | 41.0 (20.4) | 39.7 (19.6) | 52.4 (21.8)a | 41.0 (18.8) |
Change from baseline, mean (SD) | 21.3 (21.8) | 20.1 (23.3) | 7.2 (14.1)b | 18.2 (21.2) |
VAS Ability to function | ||||
N at week 36 | 87 | 175 | 83 | 68 |
Baseline, mean (SD) | 39.4 (19.2) | 40.2 (18.8) | 51.2 (22.4)a | 42.6 (19.0) |
Change from baseline, mean (SD) | 24.9 (24.9) | 19.1 (23.9) | 7.9 (18.2)b | 17.7 (22.0) |
SD = standard deviation; VAS = visual analogue scale.
Note: Baseline values from confirmatory Study 301 or Study 302 are reported unless otherwise noted.
aValues are as reported at baseline of the extension study.
bChange from extension study baseline reported.
Sources: Clinical Study Report for Study 303 and the sponsor’s Summary of Clinical Evidence.15,63 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Change From Baseline in ISI Score
The results for change from baseline in ISI score in the extension study are presented in Table 28. In Study 303, the mean change from confirmatory study baseline to week 14, to week 27, and to week 40 in ISI score decreased in all treatment groups at each visit. At week 40, the mean change in ISI score was –9.8 (SD = 6.8) in the daridorexant 50 mg group, –8.5 (SD = 6.9) in the daridorexant 25 mg, –4.3 (SD = 5.7 in the ex-placebo to daridorexant 25 mg group, and –7.5 (SD = 7.0) in the placebo group. At week 40, a 6-point decrease in ISI score from confirmatory study baseline was achieved by 74.7% of patients in the daridorexant 50 mg group, in 66.1% of patients in the daridorexant 25 mg, in 35.8% of patients in the ex-placebo to daridorexant 25 mg group, and in 53.9% of patients in the placebo group.
Table 28: Change From Baseline in ISI Score in Study 303 (Full Analysis Set)
Variable | Daridorexant 50 mg (N = 137) | Daridorexant 25 mg (N = 270) | Ex-placebo to daridorexant 25 mg (N = 127) | Placebo (N = 128) |
|---|---|---|---|---|
N at baseline | 137 | 270 | 127a | 128 |
Baseline, mean (SD) | 19.9 (3.7) | 19.9 (4.2) | 15.0 (5.7)a | 19.4 (4.0) |
Week 14 | ||||
N at week 14 | 104 | 210 | 97 | 90 |
Observed value, mean (SD) | 11.3 (6.6) | 11.6 (6.6) | 10.6 (6.5) | 12.9 (6.4) |
Change from baseline, mean (SD) | –8.5 (7.2) | –8.1 (6.5) | –4.0 (5.9) | –6.4 (6.5) |
Week 27 | ||||
N at week 27 | 95 | 197 | 93 | 75 |
Observed value, mean (SD) | 10.7 (6.7) | 11.3 (6.8) | 10.0 (6.1) | 12.3 (6.4) |
Change from baseline, mean (SD) | –9.2 (7.0) | –8.6 (6.5) | –4.7 (5.4) | –7.0 (6.4) |
Week 40 | ||||
N at week 40 | 83 | 174 | 81 | 76 |
Observed value, mean (SD) | 10.2 (6.4) | 11.4 (7.0) | 10.5 (6.3) | 12.1 (6.8) |
Change from baseline, mean (SD) | –9.8 (6.8) | –8.5 (6.9) | –4.3 (5.7) | –7.5 (7.0) |
ISI = Insomnia Severity Index; SD = standard deviation.
Note: Baseline values from confirmatory Study 301 or Study 302 are reported unless otherwise noted.
aValues are as reported at baseline of the extension study.
Sources: Clinical Study Report for Study 303 and the sponsor’s Summary of Clinical Evidence.15,63 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Harms
During the double-blind treatment period of Study 303, from baseline until up to 30 days after the double-blind study treatment end date, the incidence of TEAEs was generally comparable across treatment groups. The overall percentage of patients experiencing TEAEs was 40.1% in the daridorexant 50 mg group, 38.4% in the daridorexant 25 mg group, 38.1% in the ex-placebo to daridorexant 25 mg group, and 35.2% in the placebo group. Most TEAEs were mild or moderate in intensity, and the most reported AE was nasopharyngitis, with frequencies ranging between 4.7% (placebo) and 8.8% (daridorexant 50 mg). Several TEAEs were reported only in the daridorexant groups, such as accidental overdose, somnolence, pneumonia, back pain, tonsillitis, and increased hepatic enzymes.
Overall, the incidence of treatment-emergent SAEs was reported for less than 5.2% of patients in any treatment group. SAEs reported in the daridorexant treatment groups, compared to none in the placebo group, included diverticulitis (2 patients in the 50 mg group; 1 patient in the 25 mg group) and confusional state (1 patient in the 50 mg group and 1 patient in the 25 mg groups). The rate of treatment discontinuation due to AEs was 6.6% in the daridorexant 50 mg group and 4.7% in the placebo group. One death occurred in the daridorexant 25 mg group due to a myocardial infarction and was not attributed to the treatment under review.
Few TEAEs of special interest occurred, with less than 3% of patients in each treatment group experiencing falls. Other events included hallucinations and/or sleep paralysis (1 patient in the daridorexant 50 mg group), narcolepsy-like symptoms related to excessive daytime sleepiness (1 patient in the daridorexant 25 mg group), and suicidal ideation (1 patient in the placebo group). A TEAE of suicidal ideation was reported in the daridorexant 50 mg group, but it was not classified as an AESI due to other contributing factors.
Table 29: Key Harms Data in Study 303 (Safety Set)
AE | Daridorexant 50 mg (N = 137) | Daridorexant 25 mg (N = 268) | Ex-placebo to daridorexant 25 mg (N = 126) | Placebo (N = 128) |
|---|---|---|---|---|
Most common AEs,a n (%) | ||||
Patients with ≥ 1 AE | 55 (40.1) | 103 (38.4) | 48 (38.1) | 45 (35.2) |
Nasopharyngitis | 12 (8.8) | 15 (5.6) | 11 (8.7) | 6 (4.7) |
Accidental overdose | 4 (2.9) | 3 (1.1) | 4 (3.2) | 0 |
Somnolence | 4 (2.9) | 2 (0.7) | 1 (0.8) | 0 |
Migraine | 1 (0.7) | 0 | 0 | 3 (2.3) |
Fall | 4 (2.9) | 6 (2.2) | 1 (0.8) | 2 (1.6) |
Headache | 3 (2.2) | 6 (2.2) | 2 (1.6) | 2 (1.6) |
Cough | 3 (2.2) | 2 (0.7) | 0 | 1 (0.8) |
Pneumonia | 3 (2.2) | 1 (0.4) | 1 (0.8) | 0 |
Back pain | 3 (2.2) | 7 (2.6) | 3 (2.4) | 0 |
Tonsillitis | 1 (0.7) | 1 (0.4) | 3 (2.4) | 0 |
Upper respiratory tract infection | 0 | 6 (2.2) | 0 | 2 (1.6) |
Sinusitis | 0 | 1 (0.4) | 3 (2.4) | 2 (1.6) |
Increased hepatic enzyme | 0 | 0 | 3 (2.4) | 0 |
SAEs,b n (%) | ||||
Patients with ≥ 1 SAE | 7 (5.1) | 12 (4.5) | 4 (3.2) | 2 (1.6) |
Diverticulitis | 1 (0.7) | 2 (0.7) | 0 | 0 |
Patients who stopped treatment due to AEs, n (%) | ||||
Patients who stopped | 9 (6.6) | 10 (3.7) | 6 (4.8) | 6 (4.7) |
Deaths, n (%) | ||||
Patients who died, n (%) | 0 | 1 (0.4) | 0 | 0 |
AESI,c n (%) | ||||
Patients with ≥ 1 AESI | 1 (0.7) | 1 (0.4) | 0 | 1 (0.8) |
Narcolepsy-like symptoms related to excessive daytime sleepiness | 0 | 1 (0.4) | 0 | 0 |
Narcolepsy-like symptoms related to complex sleep behaviour, including hallucinations and/or sleep paralysis | 1 (0.7) | 0 | 0 | 0 |
Narcolepsy-like symptoms related to cataplexy | 0 | 0 | 0 | 0 |
Suicide or self-injury | 0 | 0 | 0 | 1 (0.8) |
AE = adverse event; AESI = adverse event of special interest; SAE = serious adverse event.
Note: AEs and SAEs include those occurring from the start of the double-blind study treatment in Study 303 until up to 30 days after the end of the double-blind study treatment.
aOccurring in at least 2% of patients in any randomized group.
bOccurring in at least 2 patients in any randomized group.
cAESIs are as identified by the clinical experts consulted for this review.
Sources: Clinical Study Report for Study 303 and the sponsor’s Summary of Clinical Evidence.15,63 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Notable Harms
Notable harms of interest to this review included next-morning residual effects and rebound insomnia, which were evaluated as part of the efficacy outcomes in Study 303. Other harms of interest to this review are included in Table 30.
Next-Morning Residual Effect
Throughout the extension study, mean observed values for VAS morning sleepiness were higher than at confirmatory study baseline in all treatment groups. The mean change from confirmatory study baseline at week 36 was 21.0 (SD = 22.5) in the daridorexant 50 mg group and 17.5 (SD = 19.8) in the placebo group.
Rebound Insomnia
During the placebo run-out period, the mean observed values for sTST during the placebo run-out period were higher than at confirmatory study baseline in all treatment groups.
Suicidal Ideation and/or Behaviour
No patients reported suicidal ideation or behaviour, as assessed by the C-SSRS, during either the double-blind study period or the placebo run-out period.
BWSQ Total Scores Upon Treatment Discontinuation
Mean BWSQ total scores were low and similar across treatment groups. Changes from the last assessment on double-blind treatment to the placebo run-out period were minor, with no relevant differences between the daridorexant and placebo groups. No patient had a BWSQ score greater than 20 at the end of the run-out period.
Table 30: Notable Harms Data in Study 303
Variable | Daridorexant 50 mg (N = 137) | Daridorexant 25 mg (N = 270) | Ex-placebo to daridorexant 25 mg (N = 127) | Placebo (N = 128) |
|---|---|---|---|---|
Change from last double-blind assessment to end of the run-out period in BWSQ total scorea | ||||
N | 79 | 157 | 75 | 64 |
Last assessment on double-blind treatment (week 40), mean (SD) | 1.4 (2.3) | 2.0 (3.4) | 1.8 (3.2)b | 1.0 (1.7) |
Change from last assessment to end of the run-out period (week 41), mean (SD) | –0.4 (1.7) | –0.4 (2.4) | 0.1 (2.6)c | –0.4 (1.5) |
Change from baseline to end of the run-out period in sTSTa | ||||
N | 85 | 158 | 73 | 62 |
Baseline, mean (SD) | 309.15 (58.27) | 302.85 (58.85) | 352.69 (64.09)b | 306.44 (50.04) |
Change from baseline to end of the run-out period (week 41), mean (SD) | 59.33 (76.01) | 55.06 (62.67) | 16.09 (52.26)c | 55.66 (69.92) |
Change from baseline to week 36 in VAS morning sleepinessd | ||||
|---|---|---|---|---|
N | 87 | 170 | 83 | 70 |
Baseline, mean (SD) | 38.6 (19.9) | 37.7 (18.6) | 48.7 (20.7)b | 39.6 (18.1) |
Change from baseline (week 36), mean (SD) | 21.0 (22.5) | 19.4 (22.1) | 7.7 (13.9)c | 17.5 (19.8) |
BWSQ = Benzodiazepine Withdrawal Symptom Questionnaire; SD = standard deviation; sTST = subjective total sleep time; VAS = visual analogue scale.
Note: Baseline values from confirmatory Study 301 or Study 302 are reported unless otherwise noted.
aTreatment withdrawal set.
bValues are as reported at baseline of the extension study.
cChange from extension study baseline reported.
dSafety set.
Sources: Clinical Study Report for Study 303 and the sponsor’s Summary of Clinical Evidence.15,63 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
In the extension study, patients from the placebo arm of Study 301 and Study 302 were rerandomized in a 1:1 ratio to receive either placebo or daridorexant 25 mg. This ensured that treatment allocation was randomized within the extended study framework. The baseline characteristics of patients were well balanced across treatment groups, consistent with those seen in the confirmatory studies, supporting the comparability of the groups at baseline. Only patients who completed Study 301 or Study 302 were included in the extension study. This could bias the results in favour of daridorexant, as those who responded well to treatment were more likely to remain in the study. Patients in the placebo arm of Study 303 were already receiving placebo in their respective confirmatory study, and their continued participation suggests that they were likely good responders to placebo, which may explain the higher scores and improvements on sleep-related outcomes (i.e., sWASO, sLSO, and sTST) reported in the extension study. The LTE study did not apply multiplicity adjustments, conducted many tests, and lacked statistical sample size considerations, increasing the risk of a type I error; thus, the results should only be considered to be supportive of the pivotal studies and the overall effect of daridorexant. Many outcomes were assessed descriptively, which may limit the interpretability of certain findings. Furthermore, the high dropout rate in all treatment groups in the extension study (28.5% to 35.2%) could result in an overrepresentation of patients more likely to benefit from the treatment.
External Validity
Of the 662 patients in the 4 treatment groups assessed in Study 303, 7 (1.1%) identified as Asian, 38 (5.7%) as Black or African American, 2 (0.3%) as other race, and 421 (63.6%) as white, which the clinical experts agreed is not representative of the ethnic diversity seen among patients with CID in their clinical practices. The clinical experts consulted for this review highlighted that CID often coexists with psychiatric disorders. However, individuals with acute and unstable mental health conditions were excluded from the confirmatory studies, and therefore were also excluded in the LTE. In the extension study, only 7.3% of patients had a diagnosed psychiatric disorder. This exclusion may limit the generalizability of the findings and could influence treatment outcomes, as the impact of daridorexant on psychiatric disorders is unknown. The clinical experts also noted that assessment of response to treatment is largely based on a patient’s subjective experience. In addition, tools such as the ISI and the IDSIQ are not commonly used in clinical practice, which limits the generalizability of the results.
Indirect Evidence
The sponsor determined that it was infeasible and inappropriate to conduct an indirect treatment comparison for daridorexant, so no indirect evidence was submitted for this review.
Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in the systematic review evidence were submitted by the sponsor.
Discussion
Summary of Available Evidence
The evidence included in this review consisted of 2 pivotal studies (Study 301 and Study 302) and 1 LTE study (Study 303). No indirect treatment comparisons or studies addressing gaps in the evidence were included.
Study 301 and Study 302 were both phase III, double-blind, placebo-controlled, randomized trials designed to assess the efficacy and safety of daridorexant in adult patients with CID. Patients in Study 301 were randomized in a 1:1:1 ratio to 3-months of treatment with daridorexant 50 mg, daridorexant 25 mg, or placebo; patients in Study 302 were randomized in a 1:1:1 ratio to 3-months of treatment with daridorexant 25 mg, daridorexant 10 mg (data not shown), or placebo. Both studies enrolled patients with CID that met the DSM-5 criteria and without comorbidities, such as acute or unstable psychiatric conditions, alcohol or drug use disorder, suicidality, or sleep-related breathing disorder. The primary outcomes in the included trials were the change from baseline in sleep maintenance (measured by WASO) and in sleep onset (measured by LPS). Other outcomes relevant to the current review included the change from baseline in sleep duration (measured by sTST), sleep quality, daytime functioning (measured by IDSIQ), ISI score, and safety.
The patients in Study 301 (N = 930) and Study 302 (N = 617) had a mean age of approximately 55.5 years and 56.5 years, respectively, and 66% were female. Patients had been diagnosed with CID for more than 10 years, on average, and most of the patients had not received prior or concurrent CBT-I. At baseline, across the treatment groups in both studies, mean WASO ranged from 95.5 minutes to 108.07 minutes, mean LPS ranged from 63.62 minutes to 71.82 minutes, mean sTST ranged from 307.57 minutes to 315.89 minutes, and mean ISI scores ranged from 19.0 points to 19.6 points.
The LTE study (Study 303; N = 804; daridorexant 10 mg arm [N = 142] not included in this report) provided evidence on the long-term safety and tolerability of daridorexant in adult and older patients with CID. Study 303 was a double-blind, parallel-group, randomized, placebo-controlled, 40-week extension to Study 301 and Study 302. Participants in this study had a mean age of 56.5 years to 59.2 years across the treatment groups and were mostly female (at least 65% in each group). The outcomes were exploratory, and included change from baseline in ISI score, sleep duration (sTST), sleep quality, and IDSIQ scores. Safety outcomes and harms were also assessed.
Several known but not formally submitted studies have assessed the use of daridorexant in patients with CID and various comorbidities. Single-arm, real-world studies have reported ISI score improvements in patients with CID and psychiatric or neurologic comorbidities,64,65 whereas a randomized crossover study showed that is daridorexant safe in patients with mild-to moderate sleep apnea.66 These studies provide helpful context but are limited by their design (i.e., lack of a control group in single-arm studies), small sample sizes, and short duration.
Interpretation of Results
Efficacy
The pivotal trials addressed outcomes noted as important by both patients and clinicians. The patient groups indicated that consistent and restorative sleep without any next-day effects is important. According to the input provided in the clinician submissions, important outcomes include improved duration and quality of sleep, as well as improved next-day functioning. The clinical experts consulted for this review noted that Study 301 and Study 302 were the first clinical trials that attempted to evaluate next-day functioning, which is relevant because of the terminal 8-hour half-life of daridorexant.
Outcomes of the 2 pivotal trials used objective (i.e., PSG) and subjective measures, including validated patient-reported instruments. However, the clinical experts noted that PSG results cannot always fully capture real-world sleep patterns, partly because of suboptimal or altered sleep during PSG nights. This could limit the accuracy and generalizability of the results derived using PSG. For subjective measures, the literature states that patients with insomnia tend to overestimate time taken to fall asleep and underestimate total sleeping time,2 which was corroborated by the clinical experts consulted for this review. Considering the challenges associated with quantifying and evaluating sleep — which is individualized and subjective — there is uncertainty about the true magnitude of the results from the studies included in the review. Other key limitations of the pivotal trials include the large proportion of screening failures and the use of outcome measures not routinely used in clinical practice. Importantly, the trials excluded many common comorbid conditions and concurrent medications; therefore, the exclusion of patients with acute and unstable mental health disorders could reduce the generalizability of the results to Canadian clinical settings. Similar concerns were raised for the LTE study (Study 303).
Overall, evidence from the included studies suggests that sleep outcomes, including sleep maintenance, sleep onset, and sleep duration, improved in both the 25 mg and 50 mg daridorexant groups and in the placebo group. Daridorexant 50 mg resulted in a statistically significant improvement in sleep maintenance (LSM reduction in WASO at month 1 was 22.78 minutes [97.5% CI, –28.75 minutes to –16.82 minutes] and at month 3 was 18.30 minutes [97.5% CI, –24.76 minutes to –11.85 minutes]) compared to placebo. Based on an MID of 20 minutes, the difference was not considered clinically meaningful; however, the 97.5% CI did include the potential for a clinically meaningful benefit in WASO. Daridorexant 50 mg also resulted in a statistically significant improvement in sleep onset (LSM reduction in LPS at month 1 was 11.35 minutes [97.5 CI, –16.69 to –6.02] and at month 3 was 11.67 minutes [97.5% CI, –17.03 to –6.32]) compared to placebo. Based on an MID of 10 minutes, the difference was not considered clinically meaningful; however, the 97.5% CI included the potential for a clinically meaningful benefit. For both these outcomes, results from the daridorexant 25 mg group were consistent with those from the 50 mg dose, although the magnitude of benefit with 25 mg was not as substantial as that with 50 mg. Daridorexant demonstrated a dose-response relationship, with the 50 mg dose showing greater efficacy in sleep onset and maintenance than the 25 mg dose.
Sleep duration, assessed as patient self-reported sTST, was a secondary outcome. Compared to placebo, neither daridorexant group had mean changes from baseline that reached the suggested threshold for a clinically meaningful effect on sTST (30 minutes) at month 1 or month 3 in Study 301 or Study 302. The results of the objective and subjective outcomes were generally consistent. Results from the extension study, Study 303, showed that changes in sWASO, sLSO, and sTST were maintained over time for patients who remained on treatment with either 25 mg or 50 mg of daridorexant, with a greater effect than placebo for all outcomes at week 36. However, because the MIDs for sWASO and SLSO are unknown, it is unclear whether the results represent clinically meaningful improvements. It should be noted that patients who received placebo in the pivotal trials and continued treatment with placebo in Study 303 may have experienced a strong placebo effect, contributing to their positive response. This may explain the higher values for sWASO, sLSO, and sTST reported with placebo in the extension study than in the pivotal studies. Change from baseline in the duration of TST in each sleep stage (i.e., S1, S2, SWS, REM) in the pivotal trials was noted as a clinically important consideration by the clinical experts consulted for this review. Results from the included studies showed that there was generally an increase in the duration of each sleep stage in all treatment groups, which is consistent with the improved total sleep duration.
In both pivotal studies, daytime functioning, measured using the IDSIQ, was considered to be another efficacy end point. There was a numerical reduction (improvement) in IDSIQ scores from baseline in each treatment group (including placebo) at month 1 and month 3. A clinically meaningful reduction was denoted by a decrease in IDSIQ total score of 17 points or more. Improvements in both the daridorexant 25 mg group and daridorexant 50 mg group, compared to placebo, as well as between-group differences, at both time points were not considered clinically meaningful, based on this threshold. Improvements in IDSIQ total scores from baseline were consistently observed across all treatment groups throughout the extension study. Daridorexant acts by antagonizing the activation of orexin receptors by the orexin neuropeptides and, consequently, decreases the wake drive, allowing sleep to occur. With a terminal half-life of approximately 8 hours, it is expected that daridorexant will not affect daytime functioning and may contribute to reduced next-day residual effects, a key advantage over longer-acting hypnotics; however, the evidence for this is uncertain.
Sleep quality (determined with the SDQ) and qualitatively assessed sleep depth, morning sleepiness, and daytime functioning (assessed with a VAS) were exploratory outcomes in the trials. Results of the 2 pivotal trials suggest that daridorexant 50 mg and daridorexant 25 mg may result in a larger increase in sleep quality VAS at month 1 and month 3 than placebo. In the extension study, numerical improvements in the sleep quality VAS were consistently noted across all treatment groups. Nonetheless, the certainty of these results was diminished by study attrition, large standard deviations, and the lack of an MID, making it difficult to determine whether the differences are clinically meaningful.
Similar results were observed in the trials for the change from baseline in ISI scores, another exploratory outcome. At baseline, patients’ ISI scores were all above 19, indicating moderate insomnia (an ISI score of 15 to 21). After 3 months of treatment, ISI scores in the daridorexant groups were below 15 points in both studies, suggesting an improvement in insomnia symptom severity. For the daridorexant 50 mg group, the within-group change from baseline may be considered clinically important, but the difference compared to placebo was not considered clinically meaningful, based on the between-group MID of 2.8 points to 4.0 points. Results from the extension study demonstrated sustained reductions in mean ISI scores across all treatment groups over time. At week 40, the proportion of patients who experienced a clinically meaningful decrease in ISI scores (6 points) was higher in the daridorexant 25 mg group and the 50 mg daridorexant group (66.1% to 74.7%) than in the placebo group (53.9%). This may be because patients who enrolled and stayed in the extension study were better responders than those who discontinued before week 40. Overall, the changes in sleep quality and daytime functioning and the improvement in ISI scores were not controlled for multiplicity in any of the trials. Therefore, the potential for an inflated risk of type I error should be considered when interpretating these results, and they should only be considered supportive.
There is a lack of evidence evaluating the efficacy of daridorexant relative to other available nonpharmacological and pharmacological options for the treatment of CID, so the comparative efficacy and safety of daridorexant remains unknown.
Harms
As noted by the clinical experts consulted for this review, currently used pharmacotherapies, such as Z-drugs and off-label benzodiazepines, are often limited to short-term use for insomnia because of their adverse effects, which include daytime grogginess the next day, risk of withdrawal symptoms, abuse potential, and rebound insomnia upon discontinuation.12 Next-morning residual effect, measured as self-reported morning sleepiness on VAS, was numerically lower (improved) in the daridorexant groups than in the placebo group across all studies. In terms of risks related to withdrawal, the BWSQ total score was similar in all treatment groups during the placebo run-out period in the extension study, indicating no evidence of withdrawal symptoms upon treatment discontinuation. There was no indication of rebound insomnia during the placebo run-out period, measured with WASO and LPS in the pivotal trials. However, the clinical experts consulted on this review noted that measuring rebound insomnia may be complex and, ideally, should involve various sleep-related measures. As such, the extent to which these measures effectively capture the occurrence of rebound insomnia is unclear. No safety signals related to suicidal ideation or self-injury, measured with the C-SSRS, were observed in any of the studies. There were no AEs indicative of abuse potential, apart from accidental overdose, which occurred in no more than 2.6% of patients in any treatment group in the pivotal trials. Nasopharyngitis and headaches were the most common TEAEs in the pivotal trials. The proportion of patients who discontinued treatment because of AEs was low (< 2.5%) in both studies. Withdrawal due to TEAEs continued in the extension study, with 6.6% of patients in the 50 mg group and 3.7% of patients in the 25 mg group stopping daridorexant. The proportion of patients who experienced SAEs was slightly greater in the extension study, which may be due to the longer study duration. Few TEAEs of special interest occurred during the extension study, with less than 3% of patients in each treatment group experiencing falls; other events — road traffic accidents, hallucinations and/or sleep paralysis, narcolepsy-like symptoms related to excessive daytime sleepiness, and suicidal ideation — each occurred once across the various treatment groups. Thus, overall, daridorexant appears to be safe, with minimal adverse effects.
Conclusion
CID is a common condition with a significant clinical burden on daily life, including impaired daytime functioning, and severe negative impacts on mental and physical health and on quality of life. The availability of effective or well-tolerated long-term treatment options is limited, highlighting an unmet clinical need for novel treatments that address the multiple facets of insomnia.
Evidence for this review consisted of 2 double-blind, placebo-controlled trials (Study 301 and Study 302) and 1 LTE study (Study 303) that evaluated the efficacy and safety of daridorexant 50 mg and 25 mg in adults with CID that met DSM-5 criteria. Outcomes evaluated in the studies were clinically relevant and aimed to address the needs identified by patients and clinicians, including sleep maintenance, onset, duration, and quality. Study 301 demonstrated that at 3 months, compared to placebo, treatment with daridorexant 50 mg resulted in a statistically significant improvement (reduction) in sleep maintenance, measured as WASO (LSM difference, –18.3 minutes; 97.5% CI, –24.76 to –11.85 minutes), and sleep onset, measured as LPS (LSM difference, –11.67 minutes; 97.5% CI, –17.03 to –6.32 minutes), and an increase in sleep duration (LSM difference, 19.77 minutes; 97.5% CI, 9.30 to 30.24 minutes). However, it is uncertain whether the results for these outcomes are clinically meaningful, given the effect sizes and 97.5% CIs, which contained the possibility of benefit and the possibility of no benefit. Results for change from baseline in insomnia symptoms (ISI) and daytime functioning (measured as IDSIQ) appeared to improve; however, they are uncertain, given the subjective nature of the outcomes, and it is unclear whether the results are clinically meaningful compared to placebo, although the pharmacokinetic profile of daridorexant is theoretically beneficial for daytime symptoms. DORAs are known to have an acceptable safety profile, and the harms observed in the pivotal trials were considered manageable by the clinical experts consulted for this review. Additionally, no safety signals were identified regarding rebound insomnia, next-morning residual effects, or suicidality. Findings were generally consistent for the LTE study. There is no direct or indirect comparative evidence between daridorexant and relevant treatments for patients with CID; thus, the comparative efficacy and safety of daridorexant remains unknown.
Both Study 301 and Study 302 evaluated the 25 mg dose of daridorexant, and results were consistent with the 50 mg dose, although the magnitude of the results was not as high. As such, there was evidence of a dose-response relationship with respect to efficacy outcomes, but this was not observed for harms. Because the population of patients for which daridorexant 25 mg is indicated (i.e., patients on moderate CYP3A4 inhibitors or with moderate hepatic impairment) was excluded from the trials, minimal conclusions can be drawn about the efficacy and safety of the daridorexant 25 mg dose. As CID frequently occurs alongside other psychiatric or medical conditions, the exclusion of patients with comorbid psychiatric disorders and those using certain medications (e.g., antidepressants or antipsychotics) from the pivotal studies limits the generalizability of the findings to real-world clinical practice.
References
1.Neubauer DN. Evaluation and diagnosis of insomnia in adults. UpToDate; 2023. Accessed December 3, 2024. http://www.uptodate.com
2.Benca R. Risk factors, comorbidities, and consequences of insomnia in adults. UpToDate; 2024. Accessed December 3, 2024. http://www.uptodate.com
3.Huang K, Li S, He R, et al. Efficacy of cognitive behavioral therapy for insomnia (CBT-I) in older adults with insomnia: A systematic review and meta-analysis. Australas Psychiatry. 2022;30(5):592-597. doi: 10.1177/10398562221118516 PubMed
4.Morin CM, Benca R. Chronic insomnia. Lancet. 2012;379(9821):1129-41. doi: 10.1016/s0140-6736(11)60750-2 PubMed
5.Beaulieu-Bonneau S, Ivers H, Guay B, Morin CM. Long-Term Maintenance of Therapeutic Gains Associated With Cognitive-Behavioral Therapy for Insomnia Delivered Alone or Combined With Zolpidem. Sleep. 2017;40(3):zsx002. doi: 10.1093/sleep/zsx002 PubMed
6.Crain J, Raj M, Garland S, Jones S, Farah B. Current practice analysis: interventions for insomnia disorder. (CADTH technology review: no.7). CADTH; 2017. Accessed April 14, 2023. https://www.cadth.ca/sites/default/files/pdf/OP0527_Current_Practice_Analysis_Insomnia%20Disorder.pdf
7.Payne KA, Myhr G. Increasing Access to Cognitive-Behavioural Therapy (CBT) for the Treatment of Mental Illness in Canada: A Research Framework and Call for Action. Healthc Policy. 2010;5(3):e173-85. PubMed
8.Koffel E, Bramoweth AD, Ulmer CS. Increasing access to and utilization of cognitive behavioral therapy for insomnia (CBT-I): a narrative review. J Gen Intern Med. 2018;33(6):955-962. doi: 10.1007/s11606-018-4390-1 PubMed
9.Wilson S, Anderson K, Baldwin D, et al. British Association for Psychopharmacology consensus statement on evidence-based treatment of insomnia, parasomnias and circadian rhythm disorders: An update. J Psychopharmacol. 2019;33(8):923-947. doi: 10.1177/0269881119855343 PubMed
10.Fitzgerald T, Vietri J. Residual Effects of Sleep Medications Are Commonly Reported and Associated with Impaired Patient-Reported Outcomes among Insomnia Patients in the United States. Sleep Disord. 2015;2015:607148. doi: 10.1155/2015/607148 PubMed
11.Taylor S, Annand F, Burkinshaw P, et al. Dependence and withdrawal associated with some prescribed medicines: an evidence review. Public Health England; 2020. Accessed by sponsor, no date provided. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/940255/PHE_PMR_report_Dec2020.pdf
12.Morin CM, Khullar A, Robillard R, et al. Delphi consensus recommendations for the management of chronic insomnia in Canada. Sleep Med. 2024;124:598-605. doi: 10.1016/j.sleep.2024.09.038 PubMed
13.Hafner M, Romanelli RJ, Yerushalmi E, Troxel WM. The societal and economic burden of insomnia in adults: an international study. RAND Corporation; 2023. Accessed by sponsor, no date provided. https://www.rand.org/pubs/research_reports/RRA2166-1.html
14.Innomar Strategies Inc. Quviviq (as daridorexant hydrochloride): 25 mg and 50 mg, oral tablets [product monograph]. April 26, 2023. Updated November 8, 2024.
15.Idorsia Pharmaceuticals Canada Ltd. Quviviq (daridorexant) for the treatment of chronic insomnia disorder: clinical evidence template [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Quviviq (daridorexant), 25 mg and 50 mg, oral tablets. December 2, 2024.
16.Idorsia Pharmaceuticals Ltd. Clinical Study Report: ID-078A301. Multi-center, double-blind, randomized, placebo-controlled, parallel-group, polysomnography study to assess the efficacy and safety of ACT-541468 in adult and elderly subjects with insomnia disorder [internal sponsor's report]. October 15, 2020.
17.Idorsia Pharmaceuticals Ltd. Clinical Study Report: ID-078A302. Multi-center, double-blind, randomized, placebo-controlled, parallel-group, polysomnography study to assess the efficacy and safety of ACT-541468 in adult and elderly subjects with insomnia disorder [internal sponsor's report]. November 13, 2020.
18.Winkelman JW. Overview of the treatment of insomnia in adults. UpToDate; 2024. Accessed December 3, 2024. http://www.uptodate.com
19.Morin CM, Jarrin DC. Epidemiology of Insomnia: Prevalence, Course, Risk Factors, and Public Health Burden. Sleep Med Clin. 2022;17(2):173-191. doi: 10.1016/j.jsmc.2022.03.003 PubMed
20.Riemann D, Espie CA, Altena E, et al. The European Insomnia Guideline: An update on the diagnosis and treatment of insomnia 2023. J Sleep Res. 2023;32(6):e14035. doi: 10.1111/jsr.14035 PubMed
21.By the 2023 American Geriatrics Society Beers Criteria® Update Expert Panel. American Geriatrics Society 2023 updated AGS Beers Criteria® for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2023;71(7):2052-2081. doi: 10.1111/jgs.18372 PubMed
22.Dang-Vu TT, and Morin CM. Prise de position de la Société Québécoise de gériatrie (SQG) sur l'évaluation et la prise en charge de l'insomnie chez la personne âgée [Assessment and management of insomnia in the elderly]. Société Québécoise de gériatrie; 2019. Accessed by sponsor, no date provided. https://www.sqgeriatrie.org/dl2.php?file=2019-10_l39evaluation_et_la_prise_en_charge_de_l39insomnie_chez_la_perso.pdf
23.Hoang LUA, Gravel J. Gestion des hypnosédatifs en UCDG: annexe 7 du document, 3ième édition. Regroupement des Unités de Courte Durée Gériatriques et des services hospitaliers de gériatrie du Québec; 2021. Accessed by sponsor, no date provided. https://rushgq.org/wp-content/uploads/2021/10/Annexe_7_Fiche_RUSHGQ_Hypnosedatifs_6oct2021.pdf
24.CADTH. Reimbursement review: lemborexant (Dayvigo). Can J Health Technol. 2023;3(4). doi: 10.51731/cjht.2023.624
25.Bausch Health, Canada Inc. Sublinox (zolpidem tartrate): 5 mg and 10 mg, sublingual orally disintegrating tablets (ODT) [product monograph]. July 19, 2011. Updated March 17, 2022. Accessed February 21, 2025. https://pdf.hres.ca/dpd_pm/00065109.PDF
26.Apotex Inc. Temazepam (temazepam): 15 mg and 30 mg, capsules [product monograph]. 2020. Accessed February 21, 2025. https://pdf.hres.ca/dpd_pm/00059186.PDF
27.Paladin Labs Inc. Silenor (doxepin as doxepin hydrochloride): 3 and 6 mg, oral tablets [product monograph]. December 7, 2012. Accessed February 21, 2025. https://pdf.hres.ca/dpd_pm/00018598.PDF
28.PRO DOC LTÉE. Trazodone-50, Trazodone-100 (trazodone hydrochloride): 50 mg and 100 mg, tablets; Trazodone-150 D (trazodone hydrochloride): 150 mg, tablets [product monograph]. October 5, 2021. Accessed February 21, 2025. https://pdf.hres.ca/dpd_pm/00063106.PDF
29.Sanis Health Inc. Quetiapine Fumarate XR (quetiapine as quetiapine fumarate)): 50 mg, 150 mg, 200 mg, 300 mg and 400 mg, extended-release oral tablets [product monograph]. June 4, 2021. Updated April 6, 2022. Accessed February 21, 2025. https://pdf.hres.ca/dpd_pm/00065360.PDF
30.Idorsia Pharmaceuticals Ltd. NCT03545191: Study to Assess the Efficacy and Safety of ACT-541468 (Daridorexant) in Adult and Elderly Subjects With Insomnia Disorder. ClinicalTrials.gov. Updated March 25, 2022. Accessed by sponsor, no date provided. https://clinicaltrials.gov/study/NCT03545191
31.Mignot E, Mayleben D, Fietze I, et al. Safety and efficacy of daridorexant in patients with insomnia disorder: results from two multicentre, randomised, double-blind, placebo-controlled, phase 3 trials. Lancet Neurol. 2022;21(2):125-139. doi: 10.1016/s1474-4422(21)00436-1 PubMed
32.Fietze I, Bassetti CLA, Mayleben DW, Pain S, Seboek Kinter D, McCall WV. Efficacy and Safety of Daridorexant in Older and Younger Adults with Insomnia Disorder: A Secondary Analysis of a Randomised Placebo-Controlled Trial. Drugs Aging. 2022;39(10):795-810. doi: 10.1007/s40266-022-00977-4 PubMed
33.Phillips-Beyer A, Kawata AK, Kleinman L, Kinter DS. Meaningful Within-Patient Change on the Insomnia Daytime Symptoms and Impacts Questionnaire (IDSIQ): Analysis of Phase III Clinical Trial Data of Daridorexant. Pharmaceut Med. 2023;37(4):291-303. doi: 10.1007/s40290-023-00484-w PubMed
34.Idorsia Pharmaceuticals Ltd. NCT03575104: Study to Assess the Efficacy and Safety of ACT-541468 (Daridorexant) in Adult and Elderly Subjects With Insomnia Disorder. ClinicalTrials.gov. Updated March 25, 2022. Accessed February 21, 2025. https://clinicaltrials.gov/study/NCT03575104
35.Marino M, Li Y, Rueschman MN, et al. Measuring sleep: accuracy, sensitivity, and specificity of wrist actigraphy compared to polysomnography. Sleep. 2013;36(11):1747-55. doi: 10.5665/sleep.3142 PubMed
36.Gaines J, Vgontzas AN, Fernandez-Mendoza J, et al. Short- and Long-Term Sleep Stability in Insomniacs and Healthy Controls. Sleep. 2015;38(11):1727-34. doi: 10.5665/sleep.5152 PubMed
37.Sateia MJ, Buysse DJ, Krystal AD, Neubauer DN, Heald JL. Clinical Practice Guideline for the Pharmacologic Treatment of Chronic Insomnia in Adults: An American Academy of Sleep Medicine Clinical Practice Guideline. J Clin Sleep Med. 2017;13(2):307-349. doi: 10.5664/jcsm.6470 PubMed
38.Morin CM, Belleville G, Bélanger L, Ivers H. The Insomnia Severity Index: psychometric indicators to detect insomnia cases and evaluate treatment response. Sleep. 2011;34(5):601-8. doi: 10.1093/sleep/34.5.601 PubMed
39.Bianchi MT, Williams KL, McKinney S, Ellenbogen JM. The subjective-objective mismatch in sleep perception among those with insomnia and sleep apnea. J Sleep Res. 2013;22(5):557-68. doi: 10.1111/jsr.12046 PubMed
40.Jungquist CR, Pender JJ, Klingman KJ, Mund J. Validation of Capturing Sleep Diary Data via a Wrist-Worn Device. Sleep Disord. 2015;2015:758937. doi: 10.1155/2015/758937 PubMed
41.Phillips-Beyer A, Kawata AK, Kleinman L, Flamion B. The Sleep Diary Questionnaire: An Analysis of Meaningful Change in Subjective Total Sleep Time Using Phase 2 and Phase 3 Clinical Trial Data. Presented at: SLEEP, Associated Professional Sleep Societies, LLC (APSS); 4-8 June 2022; Charlotte (NC).
42.Phillips-Beyer A, Kawata AK, Kleinman L, Seboek Kinter D, Flamion B. Meaningful Within-Patient Change in Subjective Total Sleep Time in Patients with Insomnia Disorder: An Analysis of the Sleep Diary Questionnaire Using Data from Open-Label and Phase III Clinical Trials. Pharmaceut Med. 2024;38(2):133-144. doi: 10.1007/s40290-023-00512-9 PubMed
43.Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266-77. doi: 10.1176/appi.ajp.2011.10111704 PubMed
44.Bastien CH, Vallières A, Morin CM. Validation of the Insomnia Severity Index as an outcome measure for insomnia research. Sleep Med. 2001;2(4):297-307. doi: 10.1016/s1389-9457(00)00065-4 PubMed
45.Mallinson DC, Kamenetsky ME, Hagen EW, Peppard PE. Subjective sleep measurement: comparing sleep diary to questionnaire. Nat Sci Sleep. 2019;11:197-206. doi: 10.2147/NSS.S217867 PubMed
46.Sanchez-Ortuno MM, Edinger JD. Internight sleep variability: its clinical significance and responsiveness to treatment in primary and comorbid insomnia. J Sleep Res. 2012;21(5):527-34. doi: 10.1111/j.1365-2869.2012.01010.x PubMed
47.Edinger JD, Ulmer CS, Means MK. Sensitivity and specificity of polysomnographic criteria for defining insomnia. J Clin Sleep Med. 2013;9(5):481-91. doi: 10.5664/jcsm.2672 PubMed
48.Edinger JD, Arnedt JT, Bertisch SM, et al. Behavioral and psychological treatments for chronic insomnia disorder in adults: an American Academy of Sleep Medicine systematic review, meta-analysis, and GRADE assessment. J Clin Sleep Med. 2021;17(2):263-298. PubMed
49.Feige B, Al-Shajlawi A, Nissen C, et al. Does REM sleep contribute to subjective wake time in primary insomnia? A comparison of polysomnographic and subjective sleep in 100 patients. J Sleep Res. 2008;17(2):180-90. doi: 10.1111/j.1365-2869.2008.00651.x PubMed
50.Levenson JC, Troxel WM, Begley A, et al. A quantitative approach to distinguishing older adults with insomnia from good sleeper controls. J Clin Sleep Med. 2013;9(2):125-31. doi: 10.5664/jcsm.2404 PubMed
51.Gagnon C, Belanger L, Ivers H, Morin CM. Validation of the Insomnia Severity Index in primary care. J Am Board Fam Med. 2013;26(6):701-10. doi: 10.3122/jabfm.2013.06.130064 PubMed
52.Yang M, Morin CM, Schaefer K, Wallenstein GV. Interpreting score differences in the Insomnia Severity Index: using health-related outcomes to define the minimally important difference. Curr Med Res Opin. 2009;25(10):2487-94. doi: 10.1185/03007990903167415 PubMed
53.Qin Z, Zhu Y, Shi DD, Chen R, Li S, Wu J. The gap between statistical and clinical significance: time to pay attention to clinical relevance in patient-reported outcome measures of insomnia. BMC Med Res Methodol. 2024;24(1):177. doi: 10.1186/s12874-024-02297-0 PubMed
54.Hudgens S, Phillips-Beyer A, Newton L, Seboek Kinter D, Benes H. Development and Validation of the Insomnia Daytime Symptoms and Impacts Questionnaire (IDSIQ). Patient. 2021;14(2):249-268. doi: 10.1007/s40271-020-00474-z PubMed
55.Luyet P-P, Olivieri A, Braunstein G. Understanding daytime functioning in insomnia: responder and correlation analyses in patients treated with daridorexant. Sleep Sci Pract. 2023;7(1):7. doi: 10.1186/s41606-023-00089-x
56.Dauvilliers Y, Zammit G, Fietze I, et al. Daridorexant, a New Dual Orexin Receptor Antagonist to Treat Insomnia Disorder. Ann Neurol. 2020;87(3):347-356. doi: 10.1002/ana.25680 PubMed
57.Zammit G, Dauvilliers Y, Pain S, Sebök Kinter D, Mansour Y, Kunz D. Daridorexant, a new dual orexin receptor antagonist, in elderly subjects with insomnia disorder. Neurology. 2020;94(21):e2222-e2232. doi: 10.1212/wnl.0000000000009475 PubMed
58.Bretz F, Maurer W, Brannath W, Posch M. A graphical approach to sequentially rejective multiple test procedures. Stat Med. 2009;28(4):586-604. doi: 10.1002/sim.3495 PubMed
59.Kenward MG, Roger JH. Small sample inference for fixed effects from restricted maximum likelihood. Biometrics. 1997;53(3):983-97. PubMed
60.Idorsia Pharmaceuticals Ltd. Statistical Analysis Plan: ID-078A301, version 4. Multi-center, double-blind, randomized, placebo-controlled, parallel-group, polysomnography study to assess the efficacy and safety of ACT-541468 in adult and elderly subjects with insomnia disorder [internal sponsor's report]. March 25, 2020.
61.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi: 10.1016/j.jclinepi.2010.07.015 PubMed
62.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi: 10.1016/j.jclinepi.2019.10.014 PubMed
63.Idorsia Pharmaceuticals Ltd. Clinical Study Report: ID-078A303. Multi-center, double-blind, parallel-group, randomized, placebo-controlled, three doses, 40-week extension to studies ID-078A301 and ID-078A302 to assess the long-term safety and tolerability of ACT-541468 in adult and elderly subjects with insomnia disorder [internal sponsor's report]. September 21, 2021.
64.Palagini L, Alfi G, Gurrieri R, et al. Early experience with the new DORA daridorexant in patients with insomnia disorder and comorbid mental disturbances: Results of a naturalistic study with 3 months follow-up. J Sleep Res. 2024;33(6):e14196. doi: 10.1111/jsr.14196 PubMed
65.Fernandes M, Placidi F, Mercuri NB, Liguori C. Daridorexant treatment for chronic insomnia: a real-world retrospective single-center study. Neurol Sci. 2024;45(7):3443-3448. doi: 10.1007/s10072-024-07326-w PubMed
66.Boof ML, Ufer M, Fietze I, et al. Assessment of the effect of the dual orexin receptor antagonist daridorexant on various indices of disease severity in patients with mild to moderate obstructive sleep apnea. Sleep Med. 2022;92:4-11. doi: 10.1016/j.sleep.2021.11.015 PubMed
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Figure 5: Subgroup Analysis for Change From Baseline in WASO (in Minutes) at Month 1 and Month 3 — Study 301 (Full Analysis Set)
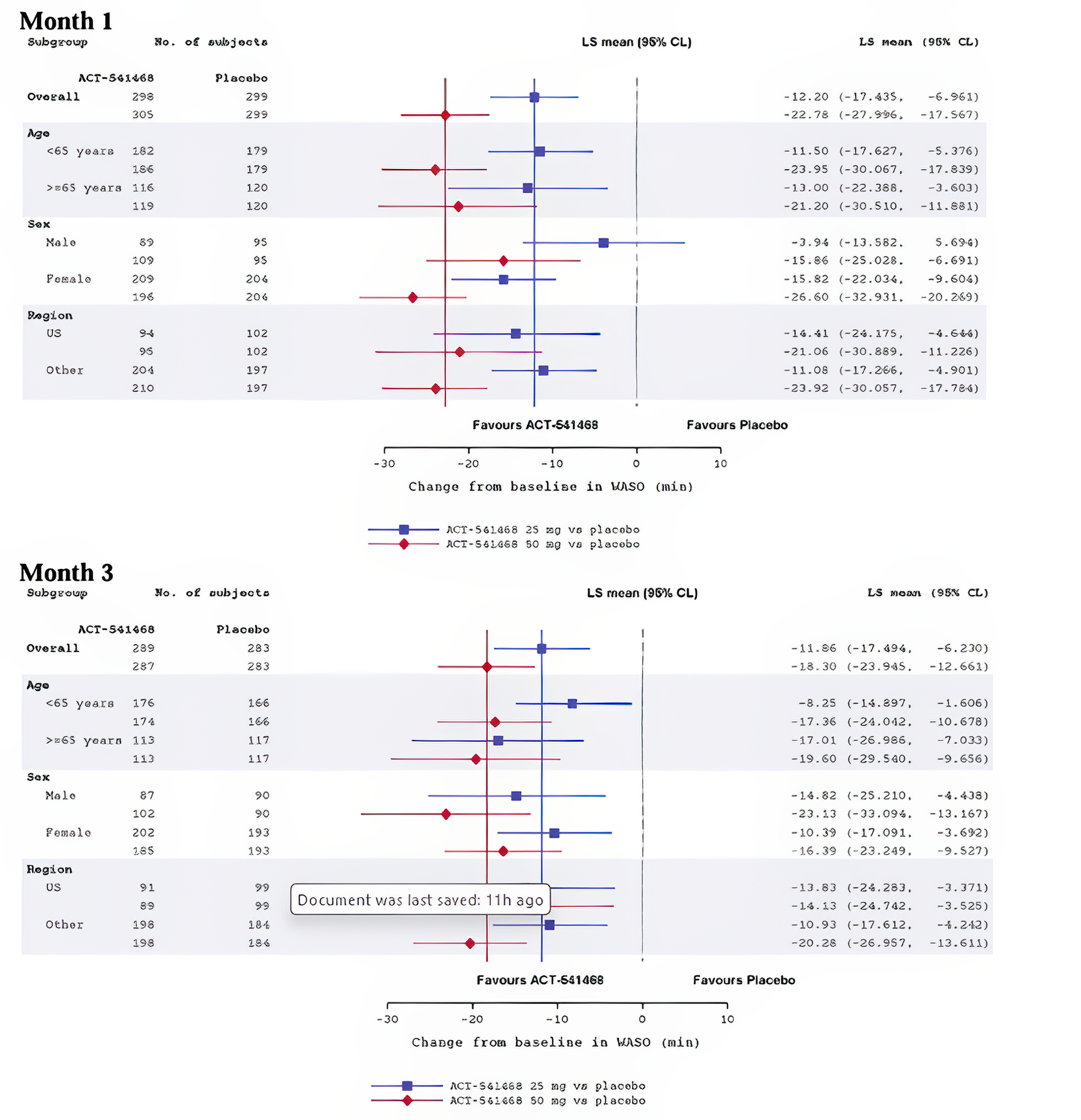
CL = confidence limit; LS Mean = least squares mean; WASO = wake after sleep onset.
Mixed-effects model for repeated measures: change from baseline in WASO = baseline WASO + age group (< 65; ≥ 65 years) + treatment + visit + treatment x visit + baseline x visit.
Age group is removed from the model for the subgroup analysis of age.
Solid vertical reference lines are the overall estimates.
Source: Clinical study report study 301.16
Figure 6: Subgroup Analysis for Change From Baseline in WASO (in Minutes) at Month 1 and Month 3 — Study 302 (Full Analysis Set)
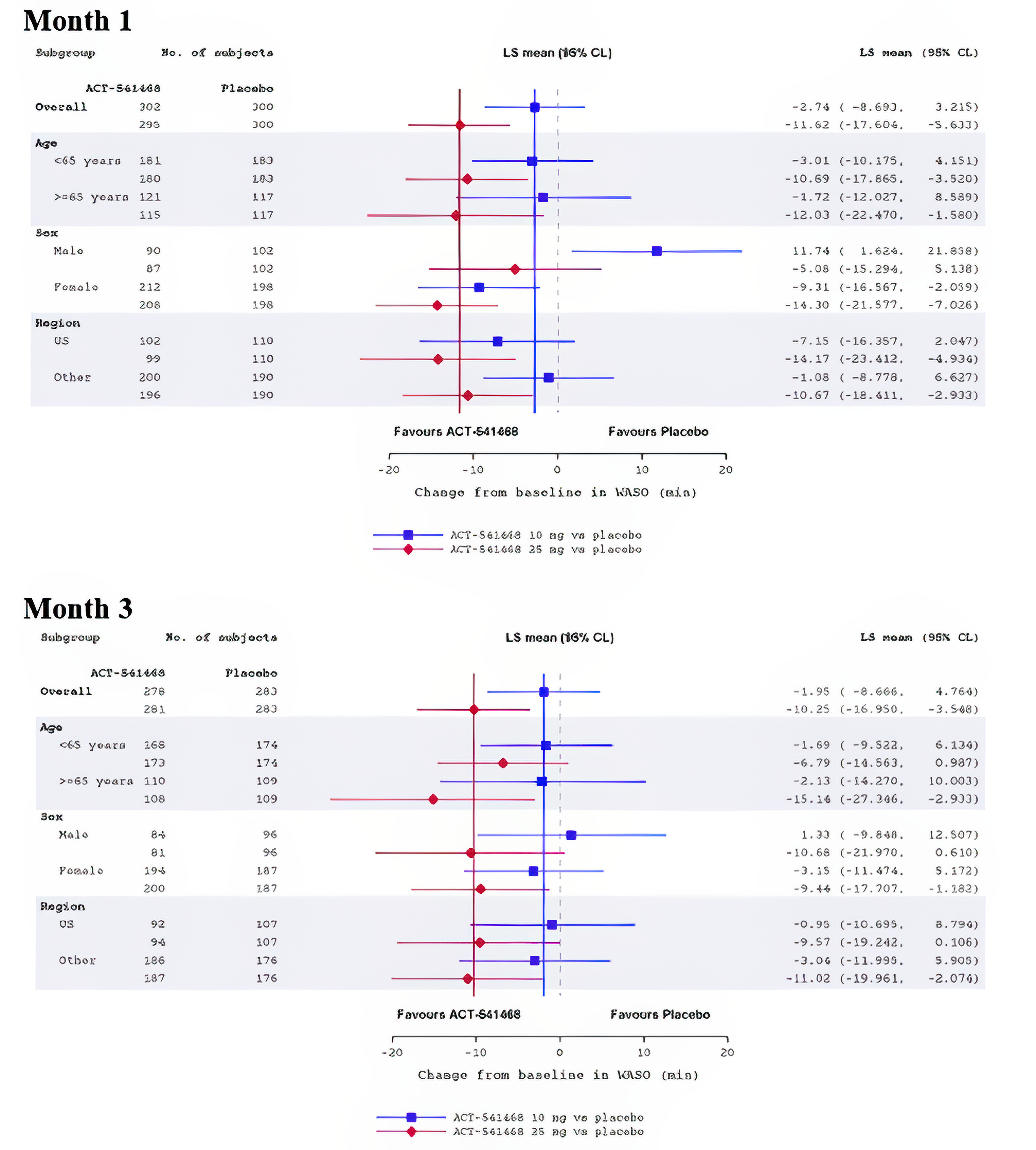
CL = confidence limit; LS Mean = least squares mean; WASO = wake after sleep onset.
Mixed-effects model for repeated measures: change from baseline in WASO = baseline WASO + age group (< 65; ≥ 65 years) + treatment + visit + treatment x visit + baseline x visit.
Age group is removed from the model for the subgroup analysis of age.
Solid vertical reference lines are the overall estimates.
Source: Clinical study report study 30217
Figure 7: Subgroup Analysis for Change From Baseline in LPS (in Minutes) at Month 1 and Month 3 — Study 301 (Full Analysis Set)
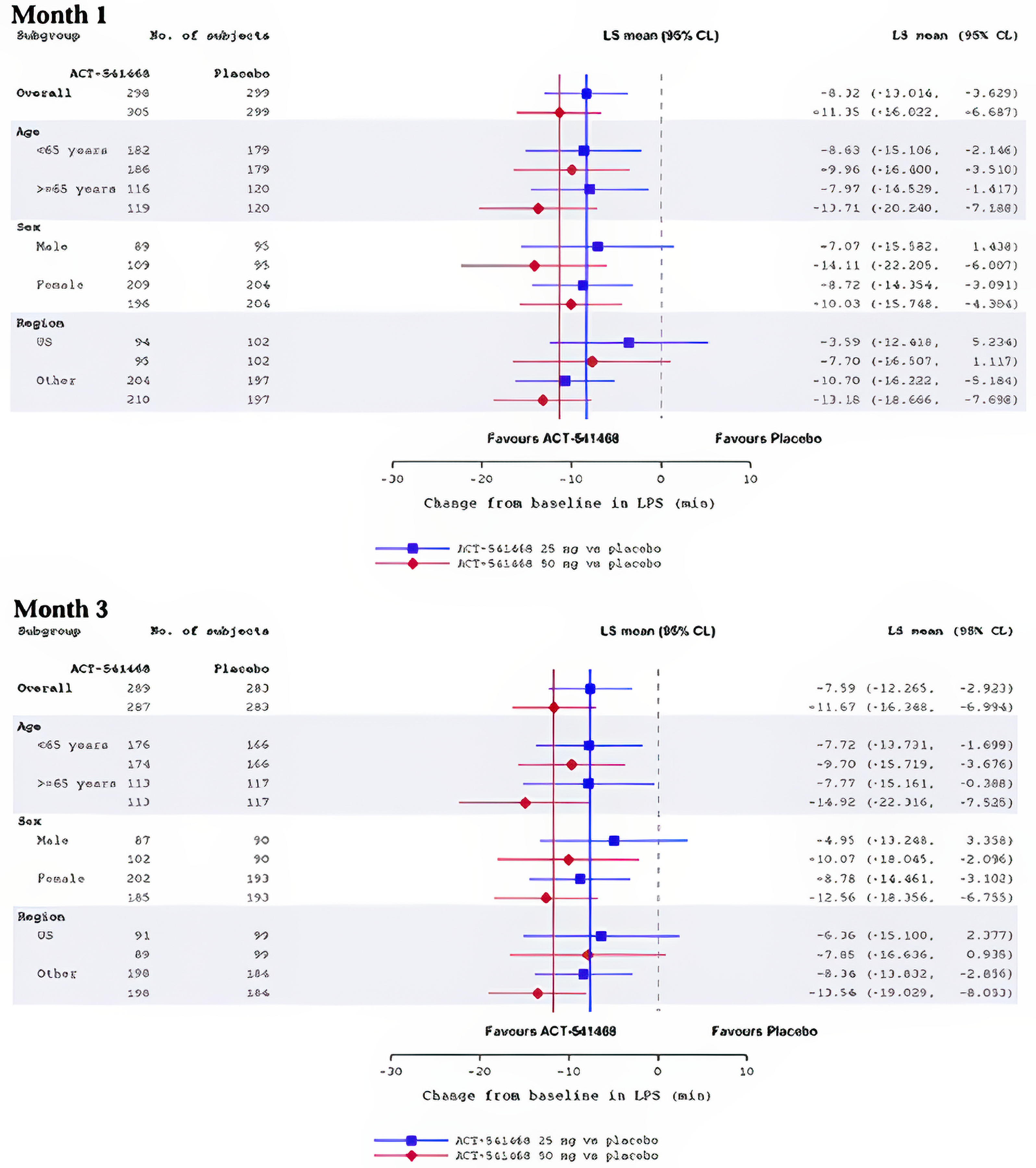
CL = confidence limit; LS Mean = least squares mean; LPS = latency to persistent sleep.
Mixed-effects model for repeated measures: change from baseline in LPS = baseline LPS + age group (< 65; ≥ 65 years) + treatment + visit + treatment x visit + baseline x visit.
Age group is removed from the model for the subgroup analysis of age.
Solid vertical reference lines are the overall estimates.
Source: Clinical study report study 301.16
Figure 8: Subgroup Analysis for Change From Baseline in LPS (in Minutes) at Month 1 and Month 3 — Study 302 (Full Analysis Set)
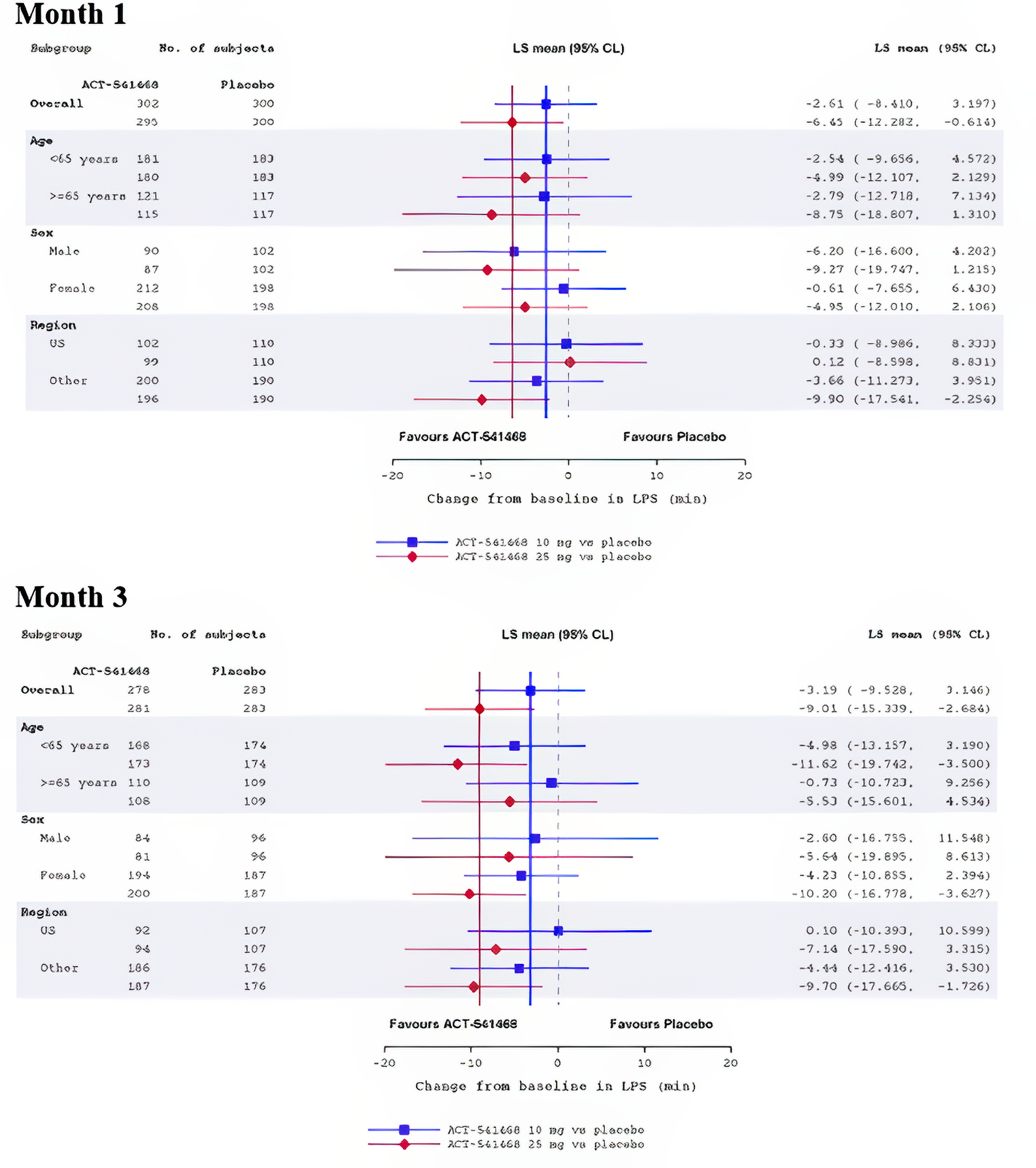
CL = confidence limit; LS Mean = least squares mean; LPS = latency to persistent sleep.
Mixed-effects model for repeated measures: change from baseline in LPS = baseline LPS + age group (< 65; ≥ 65 years) + treatment + visit + treatment x visit + baseline x visit.
Age group is removed from the model for the subgroup analysis of age.
Solid vertical reference lines are the overall estimates.
Source: Clinical study report study 302.17
Figure 9: Mean (± SE) Change From Baseline to Month 1 and Month 3 in the Duration of TST (in Minutes) in Each Sleep Stage Over the Whole Night — Study 301 (Full Analysis Set)
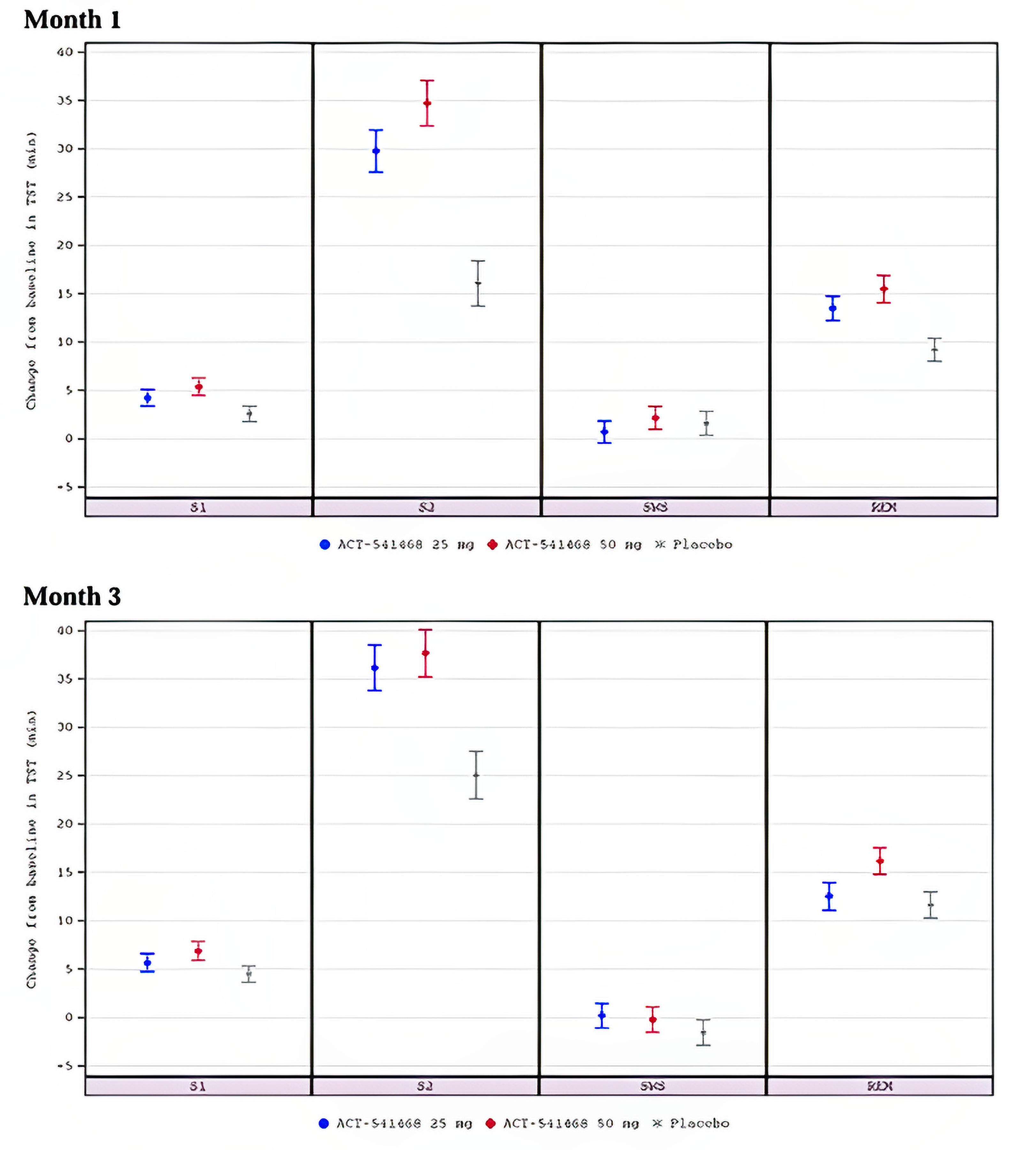
REM = rapid eye movement; S1 = sleep stage 1; S2 = sleep stage 2; SE = standard error; SWS = slow-wave sleep; TST = total sleep time.
Source: Clinical study report study 301.16
Figure 10: Mean (± SE) Change From Baseline to Month 1 and Month 3 in the Duration of TST (in Minutes) in Each Sleep Stage Over the Whole Night — Study 302 (Full Analysis Set)
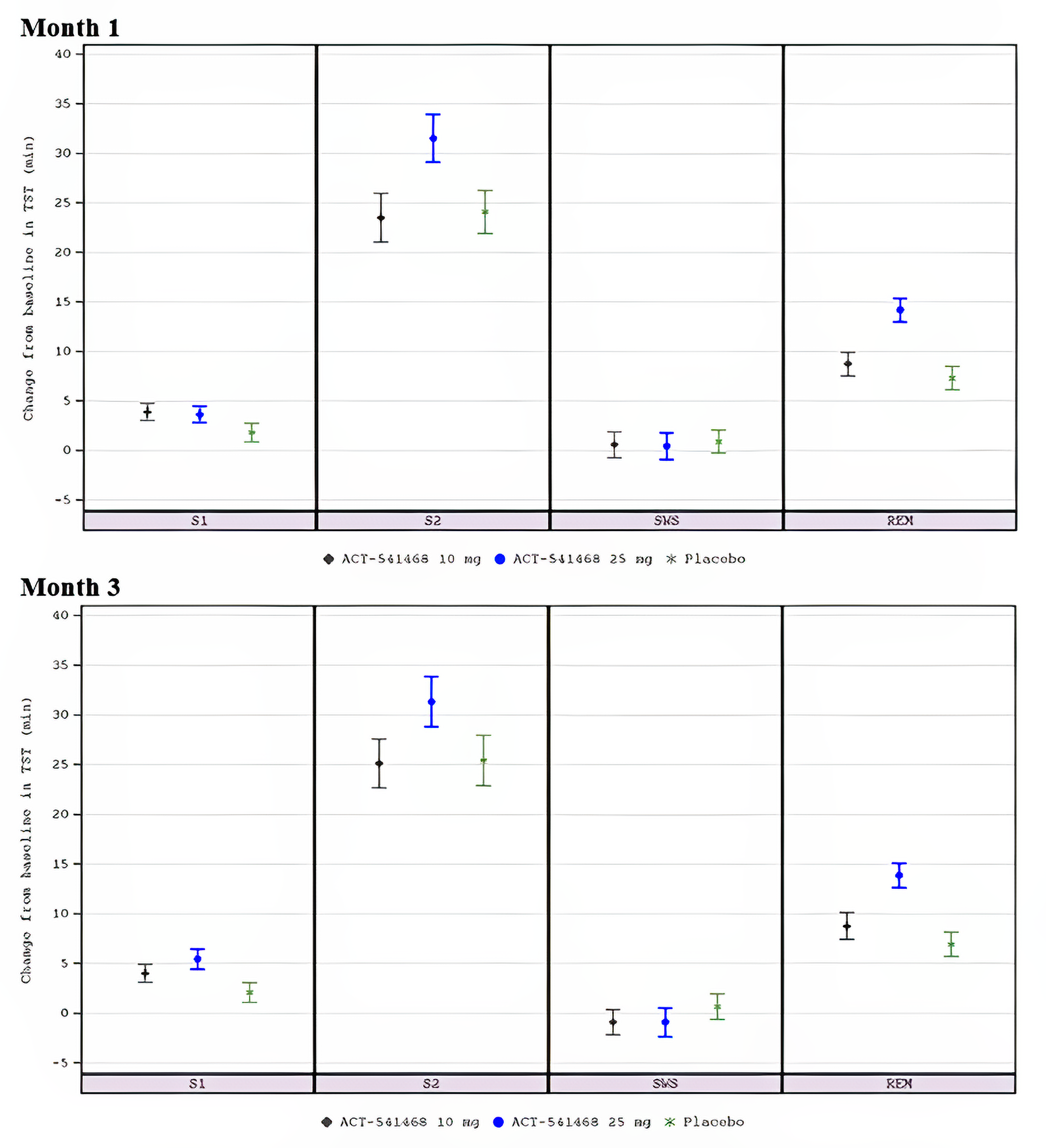
REM = rapid eye movement; S1 = sleep stage 1; S2 = sleep stage 2; SE = standard error; SWS = slow-wave sleep; TST = total sleep time.
Source: Clinical study report 302.17
Table 31: Change From Baseline to Month 1 and Month 3 in IDSIQ Sleepiness Domain Score — Study 301 and Study 302 (Full Analysis Set)
Variable | Study 301 | Study 302 | |||
|---|---|---|---|---|---|
Daridorexant 25mg (N = 310) | Daridorexant 50mg (N = 310) | Placebo (N = 310) | Daridorexant 25 mg (N = 309) | Placebo (N = 308) | |
Change from baseline to Month 1 and to Month 3 in IDSIQ sleepiness domain score a,b | |||||
N at baseline | 308 | 309 | 308 | 308 | 307 |
Baseline, mean (SD) | 22.12 (6.88) | 22.48 (7.21) | 22.26 (6.95) | 22.24 (6.19) | 22.57 (5.76) |
Month 1 | |||||
N | 301 | 304 | 301 | 297 | 297 |
Observed value, mean (SD) | 19.37 (7.12) | 18.62 (7.79) | 20.29(6.94) | 18.73 (6.45) | 19.77 (6.26) |
Change from baseline, mean (SD) | –2.73 (4.55) | –3.80 (5.61) | –2.01 (4.81) | –3.45 (5.61) | –2.83 (4.61) |
Change from baseline, LS Mean (95% CL) | –2.77 (–3.32, –2.23) | –3.77 (–4.31, –3.22) | –2.02 (–2.57, –1.48) | –3.51 (–4.10, –2.92) | –2.75 (–3.34, –2.16) |
Treatment group difference vs. placebo (98.75% CI) | –0.75 (–1.73, 0.23) | –1.75 (–2.72, –0.77) | NA | –0.75 (–1.58, 0.071)c | NA |
P value | 0.0547 | < 0.0001 | NA | 0.0733 | NA |
Month 3 | |||||
N | 290 | 291 | 288 | 283 | 289 |
Observed value, mean (SD) | 17.31 (7.63) | 16.55 (8.08) | 18.46 (7.79) | 16.97 (6.99) | 18.39 (6.59) |
Change from baseline, mean (SD) | –4.72 (6.26) | –5.85 (6.93) | –3.80 (6.62) | –5.29 (6.49) | –4.08 (5.97) |
Change from baseline, LS Mean (95% CL) | –4.78 (–5.49, –4.07) | –5.70 (–6.41, –4.99) | –3.79 (–4.50, –3.08) | –5.27 (–5.96, –4.57) | –4.01 (–4.71, –3.32) |
Treatment group difference vs. placebo (98.75% CI) | –0.99 (–2.27, 0.29) | –1.90 (–3.18, –0.63) | NA | –1.25 (–2.23, –0.28) c | NA |
P value | 0.0534 | 0.0002 | NA | 0.0120 | NA |
CI = confidence interval; CL = confidence limit; FAS = final analysis sample; IDSIQ = Insomnia Daytime Symptoms and Impacts Questionnaire; LS = least squares; NA = not applicable; SD = standard deviation
aThe P value has been adjusted for multiple testing. Note that alpha levels were based on hypothesis testing. For IDSIQ Sleepiness domain: daridorexant 50mg at month 1, alpha = 0.0125; daridorexant 50mg at month 3, alpha = 0.0125; daridorexant 25 mg at month 1, alpha = 0.0125; daridorexant 25 mg at month 3, alpha = 0.0375.
bMMRM for change from baseline in IDSIQ sleepiness domain score = baseline IDSIQ sleepiness domain score + age group (< 65; ≥ 65 years) + treatment + visit + treatment × visit + baseline × visit.
cValues from Study 302 are 95% CI.
Source: Clinical study report study 30116 and 302.17
Table 32: Change From Baseline to Placebo Run-Out Period in WASO, LPS, and sTST Rebound Insomnia — Study 301 and Study 302 (Treatment Withdrawal Set)
Key safety outcome | Study 301 | Study 302 | ||||
|---|---|---|---|---|---|---|
Daridorexant 25 mg (N = 286) | Daridorexant 50 mg (N = 286) | Placebo (N = 280) | Daridorexant 25 mg (N = 286) | Placebo (N = 280) | ||
WASO (minutes) | ||||||
N at baseline | 286 | 286 | 279 | 284 | 285 | |
Baseline, Mean (SD) | 98.26 (38.22) | 94.83 (37.68) | 103.48 (40.71) | 106.86 (48.80) | 108.33 (49.14) | |
Run-out | ||||||
N | 284 | 283 | 279 | 276 | 275 | |
Observed value, mean (SD) | 89.78 (53.57) | 92.23 (57.39) | 83.086 (45.37) | 99.36 (64.62) | 80.79 (55.06) | |
Change from baseline, mean (SD) | –8.64 (55.54) | –2.52 (52.36) | –20.392 (45.78) | –5.09 (57.9) | –26.17 (53.54) | |
LPS (minutes) | ||||||
N at baseline | 286 | 286 | 279 | 284 | 285 | |
Baseline, Mean (SD) | 67.44 (38.34) | 63.07(35.11) | 67.83 (40.85) | 67.52 (39.85) | 71.61 (44.50) | |
Run-out | ||||||
N | 285 | 284 | 279 | 277 | 276 | |
Observed value, mean (SD) | 49.93 (55.24) | 48.19 (49.57) | 40.01 (38.39) | 55.76 (68.56) | 52.84 (63.53) | |
Change from baseline, mean (SD) | –17.17 (56.68) | –15.04 (55.81) | –27.82 (47.20) | –10.26 (67.29) | –18.28 (63.79) | |
sTST (minutes) | ||||||
N at baseline | 286 | 286 | 279 | 284 | 285 | |
Baseline, Mean (SD) | 309.12 (60.71) | 313.19(58.01) | 317.07 (520.00) | 309.69 (51.57) | 308.75 (51.50) | |
Run-out | ||||||
N | 280 | 281 | 274 | 279 | 279 | |
Observed value, mean (SD) | 352.57 (71.05) | 356.89(73.46) | 359.38 (68.62) | 356.12 (66.63) | 351.57 (63.67) | |
Change from baseline, mean (SD) | 43.29 (53.82) | 42.94 (59.60) | 42.32 (52.70) | 46.75 (55.36) | 42.30 (53.79) | |
LPS = latency to persistent sleep; min = minute; mg = milligram; SD = standard deviation; sTST = subjective total sleep time; WASO = wake after sleep onset.
Source: Clinical study report study 30116 and 302.17 Details included in the table are from the sponsor’s Summary of Clinical Evidence and the clinical study reports.
Table 33: Change From Baseline in VAS Morning Sleepiness to Month 1 and Month 3, Next-Morning Residual Effect — Study 301 and Study 302 (Safety Set)
Key safety outcome | Study 301 | Study 302 | |||
|---|---|---|---|---|---|
Daridorexant 25 mg (N = 310) | Daridorexant 50 mg (N = 308) | Placebo (N = 309) | Daridorexant 25 mg (N = 308) | Placebo (N = 306) | |
N at baseline | 310 | 308 | 308 | 308 | 306 |
Baseline, Mean (SD) | 37.43 (18.79) | 38.02 (18.79) | 37.18 (18.95) | 38.60 (16.32) | 37.41 (15.66) |
Month 1 | |||||
N | 303 | 304 | 302 | 297 | 297 |
Observed value, mean (SD) | 45.71 (20.48) | 48.10 (21.38) | 41.84 (18.87) | 47.18 (18.22) | 44.48 (17.94) |
Change from baseline, mean (SD) | 8.20 (14.33) | 9.92 (15.41) | 4.93 (13.09) | 8.53 (14.87) | 7.13 (13.26) |
Month 3 | |||||
N | 292 | 289 | 289 | 285 | 287 |
Observed value, mean (SD) | 52.34 (23.00) | 53.30 (22.56) | 48.56 (21.28) | 53.50 (20.39) | 48.95 (20.27) |
Change from baseline, mean (SD) | 14.85 (19.41) | 15.28 (19.28) | 11.50 (17.67) | 14.91 (18.38) | 11.40 (16.47) |
mg = milligram; SD = standard deviation; VAS = visual analogue scale.
Source: Clinical study report study 30116 and 302.17 Details included in the table are from the sponsor’s Summary of Clinical Evidence and the clinical study reports.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
CBT-I
cognitive behavioural therapy for insomnia
CDA-AMC
Canada's Drug Agency
CI
confidence interval
CID
chronic insomnia disorder
DSM
Diagnostic and Statistical Manual of Mental Disorders
ICER
incremental cost-effectiveness ratio
ISI
Insomnia Severity Index
NHWS
National Health and Wellness Survey
QALY
quality-adjusted life-year
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Daridorexant (Quviviq), 25 mg and 50 mg oral tablets |
Indication | For the management of adult patients with insomnia, characterized by difficulties with sleep onset and/or sleep maintenance |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | April 28, 2023 |
Reimbursement request | For moderate to severe CID, when CBT-I is inappropriate, unavailable, or has failed, characterized by difficulty initiating or maintaining sleep, early awakenings, at least 3 occurrences weekly; a minimum duration of 3 months; and an ISI score ≥ 15 |
Sponsor | Idorsia Pharmaceuticals Canada Ltd. |
Submission history | No |
CBT-I = cognitive behavioural therapy for insomnia; CID = chronic insomnia disorder; ISI = Insomnia Severity Index; NOC = Notice of Compliance.
Table 2: Summary of the Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Decision tree |
Target population | Adults diagnosed according to the most recent version of the DSM referring to CID. |
Treatment | Daridorexant |
Dose regimen | 50 mg once per night |
Submitted price | $2.36 per 25 mg or 50 mg tablet |
Submitted treatment cost | $861.99 per year |
Comparator | No pharmacological treatment |
Perspective | Canadian publicly funded health care payer |
Outcome | QALYs |
Time horizon | 1 year |
Key data sources | One phase III, double-blind, placebo-controlled trial (Study 301) and a 40-week double-blind, placebo-control, long-term extension study (Study 303) |
Submitted results | ICER = $28,152 per QALY gained vs. no pharmacological treatment (incremental costs = $692; incremental QALYs = 0.025) |
Key limitations |
|
CDA-AMC reanalysis results |
|
CDA-AMC = Canada's Drug Agency; CID = chronic insomnia disorder; DSM = Diagnostic and Statistical Manual of Mental Disorders; ICER = incremental cost-effectiveness ratio; ISI = Insomnia Severity Index; QALY = quality-adjusted life-year; vs. = versus.
Conclusions
The sponsor-submitted evidence suggests that daridorexant (Quviviq) may result in a reduction in sleep maintenance and sleep onset and an increase in sleep duration, compared to placebo, at 3 months for patients with moderate to severe chronic insomnia disorder (CID) — when cognitive behavioural therapy for insomnia (CBT-I) is inappropriate, unavailable, or has failed — that is characterized by difficulty initiating or maintaining sleep, early awakenings, at least 3 occurrences weekly, a minimum duration of 3 months, and an ISI score of at least 15. However, it is uncertain whether the results for these outcomes are clinically meaningful, given the effect sizes and 97.5% confidence intervals (CIs), which contained the possibility of benefit as well as the possibility of no benefit. As noted in the Clinical Review Report, there was further considerable uncertainty associated with the outcomes due to challenges with the subjective and individualized nature of sleep evaluation. The Clinical Review Report concluded that daridorexant appeared to be generally safe, with a minimal difference in the occurrence of serious adverse events (AEs) compared to placebo. Because the sponsor determined it was unfeasible to generate indirect evidence, there was no direct or indirect evidence available to compare daridorexant to pharmacological treatments, so no conclusions can be drawn about the comparative effectiveness and safety of daridorexant compared to pharmacological treatments.
Input received by CDA-AMC for this review from patients and clinicians indicated that multiple alternative, off-label treatments are used to manage patients with CID in clinical practice, including benzodiazepines, Z-drugs (also known as nonbenzodiazepines), antidepressants, and antipsychotics. As such, the sponsor’s base case, which compared daridorexant to no treatment, is not informative for decision-making. The effectiveness and safety of daridorexant relative to these therapies for CID is unknown. Notably, these treatments are less costly than daridorexant at publicly available list prices. The cost of daridorexant is $2.36 nightly, whereas the range for currently used treatments is $0.30 to $1.74 nightly.
CDA-AMC identified several limitations of the sponsor’s pharmacoeconomic analysis, most of which stem from the uncertainty associated with the submitted clinical evidence (i.e., lack of comparative clinical evidence to relevant comparators, generalizability of the evidence). Although the sponsor estimated that daridorexant is associated with an incremental cost-effectiveness ratio (ICER) of $28,152 per quality-adjusted life-year (QALY) gained, compared to no pharmacological treatment, the incremental benefit is primarily derived from the mean change in Insomnia Severity Index (ISI) scores over time, whereas the incremental costs were driven by treatment acquisition costs. The results of the sponsor’s mapping algorithm may not meet face validity, as the clinical evidence did not suggest a meaningfully important between-group difference in ISI scores, yet the sponsor predicted an incremental QALY improvement of 0.025 (corresponding to approximately an additional 9 days of perfect health in a year for patients on daridorexant compared to no treatment). Of note, a majority (80%) of the incremental QALY gained occurred between 12 weeks and 52 weeks. The sponsor assumed that the ISI score at 12 weeks for patients in the no-pharmacological-treatment arm would be maintained throughout the modelled time horizon, but for daridorexant, ISI scores reflected the extension study. Together, the economic results are uncertain, as there is insufficient clinical evidence to conclude that daridorexant would provide a net clinical benefit compared to no pharmacological treatment.
Perspectives of Patients, Clinicians, and Drug Programs
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was provided by 4 groups: the Gastrointestinal Society, Mood Disorders Society of Canada, Migraine Canada, and Menopause Chicks. Surveys and interviews with patients and health care professionals indicated that insomnia negatively impacts patients’ physical and mental wellbeing by affecting their activities of daily living. Survey and interview respondents indicated considerable physical, mental, emotional, and social anguish, along with comorbidities such as cardiovascular disease, diabetes, obesity, cancer, and gastrointestinal diseases and disorders. The majority of patient respondents were receiving numerous different treatments, with varying levels of success, including cognitive behavioural therapy, life-style adjustments, and off-label medication, such as over-the-counter medication or supplements, benzodiazepines, Z-drugs, antidepressants, antipsychotics, and, in extreme circumstances, alcohol and recreational drugs. The patient groups indicated that current treatments carry with them risks of dependency and are for short-term use only, and noted that medications carry risks for abuse, misuse, withdrawal, next-day sedation, and other side effects. Patient input also indicated that the most important outcomes were the alleviation of symptoms (e.g., achievement of deep restorative sleep and daytime functioning) and an improvement in patients’ overall wellbeing without additional harm. Of those interviewed, a caregiver to a patient who had experience with daridorexant indicated that treatment improved sleep time with minimal side effects.
Twelve clinician groups provided input. Clinician input stated that current treatment pathways include nonpharmacological therapies, such as cognitive behavioural therapy, sleep hygiene, and off-label pharmacological medications, such as over-the-counter medication and supplements, benzodiazepines, Z-drugs, antidepressants, H1 antagonists, and anticonvulsants. Clinician input indicated that nonpharmacological therapies are often difficult to access and can be costly, whereas pharmacological medications can have cognitive side effects, can result in dependency, can cause refractory disease due to tolerance, and are not appropriate for long-term use. The clinician groups stated that current treatment goals are to improve sleep continuity and related daytime function; improvement in nocturnal symptoms only is not adequate. The clinicians stated that daridorexant is expected to be used for patients with chronic insomnia as early as in the first-line setting.
The drug plans questioned whether the provision of daridorexant should be limited to the inclusion criteria of Study 301 and to the place in therapy of daridorexant relative to other insomnia treatment options. The plans also sought to understand how disease progression is measured in clinical practice.
Several of these concerns were addressed in the sponsor’s model:
AEs were captured in the economic model in the form of a 1-time disutility and cost.
Utilities were measured using the EQ-5D-3L, which captured some of the patient-important outcomes identified, such as usual activities, pain and/or discomfort and anxiety and/or depression.
Impacts of sleep on daytime functioning and satisfaction with current sleep patterns were captured using the ISI.
CDA-AMC was unable to address the following concerns raised by input from the contributing groups:
The cost-effectiveness of daridorexant compared with both nonpharmacological and pharmacological alternatives is unknown because there is a lack of direct and indirect comparative clinical evidence.
Comorbidities were not captured in the sponsor’s model.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
Daridorexant is indicated for the management of adult patients with insomnia characterized by difficulties with sleep onset and/or sleep maintenance, whereas the reimbursement request is for the treatment of insomnia in adults diagnosed according to the criteria for CID in the most recent version of the Diagnostic and Statistical Manual of Mental Disorders (DSM).1 Canada’s Drug Agency (CDA-AMC) accepted a deviation request from the sponsor to focus the economic evaluation on the reimbursement-requested population, as follows: “for the treatment of insomnia in adults diagnosed according to the most recent version of the Diagnostic and Statistical Manual of Mental Disorders (DSM) referring to chronic insomnia disorder (CID),” and to exclude pharmacological comparators from the economic evaluation. The sponsor submitted a cost-utility analysis of daridorexant compared to no pharmacological treatment for the treatment of insomnia in adults who met the criteria for CID in the most recent version of the DSM.2 However, after receiving the clinical and pharmacoeconomic review reports, the sponsor revised the reimbursement-requested population for daridorexant patients with moderate to severe CID in whom CBT-I is inappropriate, unavailable, or has failed, which is characterized by difficulty initiating or maintaining sleep, early awakenings, at least 3 occurrences weekly, a minimum duration of 3 months, and an ISI score of at least 15. This revised reimbursement-requested population was noted to be more reflective of Study 301 and Study 303, but is narrower than in the Health Canada indication.
Daridorexant is available in 25 and 50 mg oral tablets.3 The recommended dose for daridorexant is a 50 mg oral tablet once per night, but should be reduced to 25 mg nightly for patients receiving moderate CYP3A4 inhibitors.3 At the submitted price of $2.36 for both the 25 mg and 50 mg tablets, the sponsor estimated that the annual treatment cost is $861.99 per patient, assuming 365.25 days per year.1 No treatment costs were assumed for the no-pharmacological-treatment arm.1
The analysis was conducted from a Canadian public health care payer perspective. The clinical outcome of interest was QALYs. The sponsor adopted a 1-year time horizon, and discounting was not applied.
Model Structure
The sponsor submitted a decision tree (Figure 1)1 in which patients with CID entered the model and were treated with daridorexant or no pharmacological treatment.1 ISI scores were assessed at each time point (i.e., 0 weeks, 4 weeks, 12 weeks, 26 weeks, 39 weeks, and 52 weeks).1 The model further applied a discontinuation rate for the daridorexant group only. Patients who discontinued were assumed to receive no pharmacological treatment1.
Model Inputs
The baseline population characteristics used to inform the model were based on Study 301, a randomized, double-blind, placebo-controlled, 12-week, phase III trial (68.07% female; mean age = 56.08 years; ISI score = 19.25).4,5
The primary measure of efficacy in the model was the change in ISI scores based on a post hoc analysis, using a linear mixed-effects model to combine the observed ISI scores from Study 301 and Study 303.4-6 Study 303 was a double-blind, placebo-controlled, 40-week extension study of Study 301 in which patients continued daridorexant or, for those in the placebo group, were rerandomized to either continue with placebo or switch to daridorexant 25 mg. The post hoc analysis conducted to inform the economic model only included patients who continued on 50 mg daridorexant or placebo in the 40-week extension study. Because of rerandomization, the placebo sample size was reduced, effectively, from 128 to 57 patients.1,6 A half-cycle correction was applied to ISI scores.1 In the economic model, ISI scores for daridorexant were weighted based on the proportion of patients remaining or discontinuing treatment with their expected half-cycle–corrected ISI value.1 Treatment discontinuation was based on the observed discontinuation rates reported in Study 301 and Study 303 at each assessed time point, and discontinuation rates were applied to all patients, irrespective of their ISI scores (i.e., patients with lower ISI scores had the same likelihood of discontinuing as those with higher ISI scores).5,6 The sponsor assumed that patients receiving no pharmacological treatment would retain the ISI score they achieved at week 12 until the end of the 52-week time point assessments, whereas patients treated with daridorexant reflected the mixed-effects model results for ISI scores from Study 301 and Study 303.1
All-cause incidence rates for all grades of AEs were estimated from Study 301 and Study 303 clinical trial data for daridorexant and placebo, whereas the duration of acute AEs was sourced from the literature.7-9 These would have an impact on both utilities and costs.10
Utilities were derived using a mapping algorithm that transforms ISI scores to EQ-5D-3L scores, based on the National Health and Wellness Survey (NHWS) dataset, which contained both ISI assessments and EQ-5D scores for individuals with self-reported insomnia.11,12 The mapping algorithm was based on a generalized linear model that regressed ISI scores onto EQ-5D-3L utility values with a gamma distribution and log link.12,13 AE disutilities were sourced from literature and were applied as a 1-off decrement.10
Direct costs in the model included treatment acquisition, health care resources, (i.e., medical visits, emergency department visits, inpatient visits) and AE management. Drug costs for daridorexant were based on the sponsor’s submitted prices.2 Health care resource use rates were derived from a generalized linear model that linked health care resource use by ISI score, based on the NHWS dataset.14 Costs for each health care resource were sourced from Ontario’s Schedule of Benefits for Physician Services, and the Canadian Institute for Health Information patient cost estimator.15,16 AE management costs were informed by the Alberta Interactive Health Data Application and applied as a 1-time cost.1,17 All costs were reported in 2024 Canadian dollars and, when applicable, costs were inflated using the Bank of Canada’s inflation calculator.18
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (5,000 iterations). The deterministic and probabilistic results were similar to the probabilistic findings presented here. All results are based on publicly available list prices.
Base-Case Results
The sponsor’s probabilistic base case reported that daridorexant was associated with an expected cost of $2,106.78, and 0.71 QALYs over the 1-year time horizon.1 The ICER for daridorexant was $28,152 per QALY gained (incremental costs = $691.79; incremental QALY = 0.03) compared to no pharmacological treatment.1 Daridorexant had a 85% probability of being cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained.
The sponsor’s results were primarily driven by changes in ISI scores and drug-acquisition costs.1 The sponsor’s model predicted a decreasing ISI score over the assessment time points for patients receiving daridorexant and, as a result, an increasing QALY benefit for daridorexant compared to no pharmacological treatment.1 The majority of the cost difference was driven by treatment acquisition costs.1 Of note, mortality was not explicitly modelled, which means that daridorexant was assumed to not have any impact on extending life compared to no pharmacological treatment.1
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. no pharmacological treatment ($/QALY) |
|---|---|---|---|---|---|
No pharmacological treatment | 1,414.99 | Reference | 0.69 | Reference | Reference |
Daridorexant | 2,106.78 | 691.79 | 0.71 | 0.03 | 28,152 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Sensitivity and Scenario Analyses Results
The sponsor presented several deterministic scenario analyses, including the application of ISI scores based on different statistical models, the exclusion of treatment discontinuation, the exclusion of AEs, and the extrapolation of the time horizon to lifetime (51 years).1 In all scenario analyses, the ICER aligned with the sponsor’s base case; daridorexant remained cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained compared to no pharmacological treatment.
The sponsor conducted a scenario analysis from a societal perspective. This analysis included additional costs associated with lost productivity hours for a patient using 2 different approaches. In all analyses adopting a societal perspective, daridorexant dominated no pharmacological treatment (i.e., less costly, more effective).
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations of the sponsor’s analysis that have notable implications for the economic analysis:
Lack of comparative clinical evidence to relevant comparators limits the interpretation of the cost-effectiveness of daridorexant. The sponsor’s submitted economic model compared daridorexant to no treatment, based on the placebo arm of Study 301 and Study 303.1 However, according to the patient and clinician input gathered by CDA-AMC, patients have multiple different pharmacological treatment options (i.e., benzodiazepine receptor agonists, Z-drugs, and antidepressants). This was corroborated by clinician expert feedback obtained by CDA-AMC. These comparators were not included in the sponsor’s analysis, which is problematic, as it does not address the cost-effectiveness of daridorexant compared to all relevant treatment options funded publicly, albeit off-label. The sponsor’s choice of no treatment as the only relevant comparator has limited relevance to the decision problem.
CDA-AMC noted that the sponsor submitted a request for deviation, in which all pharmacological comparators would be excluded from the model and the analysis would be focused only on a comparison to sleep hygiene, explaining that an indirect treatment comparison was not feasible for the reimbursement-requested population of patients with chronic insomnia and that current pharmacological options are available for short-term use only. Although this request was approved by CDA-AMC, the sponsor was advised that the exclusion of relevant comparators would be considered a major limitation of its submission.19 The sponsor’s revised reimbursement request includes limiting the population to patients with moderate to severe CID in whom CBT-I is inappropriate, unavailable or has failed; however, current clinical practice guidelines recommend the adoption of pharmacological treatments in this population.20,21 The only comparator in the submitted economic model was no pharmacological treatment, informed by the efficacy and safety of placebo in Study 301 and Study 303, in which patients received placebo treatment only and in which concomitant medications were not permitted (refer to the next bullet: Limited generalizability of the trial population reduces the external validity of the economic model). However, as noted in the sponsor’s budget impact analysis (BIA), daridorexant is expected to displace the chronic use of off-label pharmacological treatments. This implies that pharmacological treatments are used in clinical practice to manage CID. As the sponsor determined that it was unfeasible to generate indirect evidence, CDA-AMC is unable to determine the cost-effectiveness of daridorexant against any pharmacological comparator.2
The sponsor’s base case comparing daridorexant to no treatment is not informative for decision-making. There are several other treatments used to manage patients with CID in clinical practice that, notably, at publicly available list price, are less costly than daridorexant (refer to Appendix 1).
Limited generalizability of the trial population reduces the external validity of the economic model. The sponsor’s submitted pharmacoeconomic evaluation was based on efficacy parameters obtained from Study 301 and Study 303.2,5,6 The clinical trial population may reflect the revised reimbursement-requested population (i.e., patients with moderate to severe CID in whom CBT-I is inappropriate, unavailable, or has failed, which is characterized by difficulty initiating or maintaining sleep, early awakenings, at least 3 occurrences weekly, a minimum duration of 3 months, and an ISI score of at least 15). But, as noted in the Clinical Review Report, of the patients screened for Study 301 who continued into Study 303, a majority failed screening because of inclusion criteria ineligibility. The trial inclusion criteria excluded patients receiving pharmacological treatments and those with comorbid conditions (e.g., acute or unstable psychiatric conditions), which may have excluded potential candidates for daridorexant treatment in clinical practice.2,5,6 Clinical expert feedback obtained by CDA-AMC indicated that the inclusion criteria for the trial population may not be generalizable to the broader population treated for chronic insomnia in Canada.
CDA-AMC was unable to address the limitation regarding the generalizability of the results from the trial population, which were used to inform the economic model.
Health utilities lack face validity. The sponsor’s submitted model incorporated published mapping algorithms, based on the NHWS dataset, to derive health utility values (i.e., EQ-5D-3L) from the ISI observations reported in Study 301 and Study 303.12 There is considerable uncertainty about the adoption of mapped utilities. Incorporating these mapped utilities resulted in an incremental QALY improvement of 0.025 (corresponding to approximately an additional 9 days of perfect health per year for patients on daridorexant compared to no treatment); however, as noted in the CDA-AMC Clinical Review Report, the results for ISI were uncertain, given the subjective nature of the outcomes. Although they appeared to improve, it was uncertain whether the differences reported between 50 mg of daridorexant and placebo were clinically meaningful. Because quality of life was not an outcome of interest in Study 301 or Study 303, there is no evidence to substantiate the quality of life improvements predicted in the sponsor’s economic model. Therefore, there is uncertainty about whether there would be a meaningful QALY benefit associated with daridorexant compared to no pharmacological treatment.
The sponsor further assumed that patients receiving no pharmacological treatment who continued into Study 303 (week 12 and onward) would retain the ISI score achieved at the end of Study 301.1 As a result, the QALY observed in the no-pharmacological-treatment health state does not change after week 12, inflating the incremental QALYs between daridorexant and no pharmacological treatment beyond this time point. Because the ISI score observed in the placebo arm at 12 weeks was maintained, 80% (0.020) of the incremental QALYs were observed between week 12 and week 52.
CDA-AMC was unable to address this limitation. The majority of incremental utility gains in the sponsor’s model occurred after the 12-week period; rather than being informed by the clinical evidence, the sponsor assumed that, for the rest of the model duration, patients receiving no pharmacological treatment would retain the ISI score they achieved at the end of week 12. As noted, the CDA-AMC Clinical Review Report does not report a quality of life improvement and the validity of the mapping exercise to derive health utility weights and inform the economic model remains uncertain.
The meaningfulness of the clinical outcomes for daridorexant versus no pharmacological treatment for the treatment of chronic insomnia is uncertain. As reported in the CDA-AMC Clinical Review Report, no conclusion could be drawn about whether there was a clinically meaningful improvement in ISI score between the 50 mg daridorexant arm and the placebo arm in Study 301. Additionally, other patient-important outcomes indicated in patient and clinician input, such as sleep quality and daytime functioning, were associated with uncertainty. Sleep quality and daytime functioning were measured with the Sleep Diary Questionnaire and the Insomnia Daytime Symptoms and Impacts Questionnaire, respectively. As noted in the Clinical Review Report, numerical increases in SDQ scores were observed in both the daridorexant and placebo arms; however, the interpretation of sleep quality outcomes was limited by study attrition, large standard deviations, and the lack of a defined minimally important difference. Insomnia Daytime Symptoms and Impacts Questionnaire scores appeared to be improved; however, it is uncertain whether the results were clinically meaningful, as meaningful between-group differences were not met. Together, this limited the certainty that daridorexant treatment provides a clinical benefit compared to placebo.
Given that Study 301 and Study 303 had no control for multiplicity, changes in ISI scores, sleep quality, and daytime functioning were associated with a low certainty of improvement compared to placebo. Thus, it is uncertain whether daridorexant provides a net benefit relative to no treatment.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Patients with acute insomnia were not included in the submitted economic model. | Clinical expert feedback obtained by CDA-AMC indicated that the use of daridorexant would be most appropriate in the chronic insomnia setting. It also noted that it would take 4 weeks to 8 weeks for daridorexant treatment to achieve full efficacy, so it would, therefore, be inappropriate for acute insomnia. Nonetheless, it was noted that there is a possibility that daridorexant would be inappropriately prescribed to manage acute insomnia if clinicians are not aware of its mechanism of action. |
The diagnosis of CID is based on DSM criteria. | Uncertain. The modelled population reflects the population assessed in Study 301, in which 1 of the inclusion criterion was the diagnosis of CID using DSM criteria. The clinician experts consulted by CDA-AMC indicated that such a diagnosis is conducted with clinician assessment and patient histories, and that the DSM is not stringently adhered to in clinical practice, which may limit the generalizability of the study results. |
The economic model only considers the 50 mg dose. | Acceptable. The clinical expert feedback obtained by CDA-AMC indicated that 50 mg of daridorexant would be the primary formulation used for treatment. The CDA-AMC Clinical Review Report noted that no conclusions were drawn about the efficacy and safety of the 25 mg dose. The 25 mg daridorexant dose is indicated for patients receiving moderate CYP3A4 inhibitors or those with moderate hepatic impairment.3 |
No mortality due to the time horizon. | Acceptable. Daridorexant is not expected to result in an increase in mortality, and applying a standard mortality ratio to a 1-year time horizon is not expected to impact results. |
One-year time horizon. | Inappropriate. Patients diagnosed with chronic insomnia are likely to experience long-term disease, with possible disease progression or regression for an extended time duration. Furthermore, patients prescribed daridorexant are likely to continue treatment beyond 1 year. |
CDA-AMC = Canada's Drug Agency; CID = chronic insomnia disorder; CYP3A4 = cytochrome P450 3A4; DSM = Diagnostic and Statistical Manual of Mental Disorders.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
CDA-AMC was unable to address several key limitations of the sponsor’s submission, including the uncertainty about the comparative clinical evidence and the uncertainty related to the mapping exercise to convert ISI scores to utility weights. As noted in the CDA-AMC Clinical Review Report, there is no comparative evidence, so no conclusions can be drawn about the magnitude and direction of the effects of daridorexant relative to relevant comparators. The economic model based the QALY benefit on the change in ISI score from baseline. As noted in the CDA-AMC Clinical Review Report, there was no clinically meaningful improvement in ISI score between daridorexant and placebo; any adjustments in ISI scores using alternative statistical analyses, such as linear mixed-effects model or the sponsor’s assumptions about ISI score changes after 12 weeks, are arbitrary in nature. These limitations prevented CDA-AMC from deriving a revised base-case estimate to inform the cost-effectiveness of daridorexant against relevant comparators for CID.
Although no conclusions can be drawn about the clinical effectiveness or safety of daridorexant compared to relevant off-label comparators, these treatments are less costly than daridorexant at publicly available list prices (daridorexant = $2.36 nightly; other treatments = $0.30 to $1.74 nightly) (Table 5 and Table 6).
Overall Conclusions
The sponsor-submitted evidence suggests that daridorexant may result in a reduction in sleep maintenance and sleep onset and an increase in sleep duration, compared to placebo, at 3 months for patients with moderate to severe CID in whom CBT-I is inappropriate, unavailable, or has failed, which is characterized by difficulty initiating or maintaining sleep, early awakenings, at least 3 occurrences weekly, a minimum duration of 3 months, and an ISI score of at least 15. However, it is uncertain whether the results for these outcomes are clinically meaningful, given the effect sizes and 97.5% CIs, which contained the possibility of benefit as well as the possibility of no benefit. As noted by the Clinical Review Report, there was considerable uncertainty associated with the outcomes, owing to challenges with the subjective and individualized nature of sleep evaluation. The Clinical Review Report concluded that daridorexant appears to be generally safe, with minimal differences in the occurrence of serious AEs compared to placebo. Because the sponsor determined that it was unfeasible to generate indirect evidence (there was no direct or indirect evidence available comparing daridorexant to relevant pharmacological treatments), no conclusions can be drawn about the effectiveness or safety of daridorexant compared to relevant pharmacological treatments.
Input received by CDA-AMC for this review from patients and clinicians indicated that multiple alternative, off-label treatments are used to manage patients with CID in clinical practice, including benzodiazepines, Z-drugs, antidepressants, and antipsychotics. As such, the sponsor’s base case, which compared daridorexant to no treatment, is not informative for decision-making. The effectiveness and safety of daridorexant relative to these therapies for CID is unknown. Notably, these treatments are less costly than daridorexant at publicly available list prices. The cost of daridorexant is $2.36 nightly, whereas the price range for currently used treatments is $0.30 to $1.74 nightly.
CDA-AMC identified several limitations of the sponsor’s pharmacoeconomic analysis, most of which stem from uncertainty associated with the submitted clinical evidence (i.e., lack of comparative clinical evidence to relevant comparators, generalizability of the evidence). Although the sponsor estimated daridorexant to be associated with an ICER of $28,152 per QALY gained compared to no pharmacological treatment, the incremental benefit is primarily derived from the mean change in ISI scores over time; in contrast, the incremental costs were driven by treatment acquisition costs. The results of the sponsor’s mapping algorithm may not meet face validity, as the clinical evidence did not suggest a meaningfully important between-group difference in ISI scores, yet the sponsor predicted an incremental QALY improvement of 0.025 (corresponding to approximately an additional 9 days of perfect health per year for patients receiving daridorexant compared to no treatment). Of note, a majority (80%) of the incremental QALY gained occurred between 12 weeks and 52 weeks. The sponsor assumed that the ISI score at 12 weeks for patients in the no-pharmacological-treatment arm would be maintained throughout the modelled time horizon, but for daridorexant, ISI scores reflected the extension study. Together, the economic results are uncertain, as there is insufficient clinical evidence to conclude that daridorexant would provide a net clinical benefit compared to no pharmacological treatment.
References
1.Idorsia Pharmaceuticals Canada Ltd. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Quviviq (daridorexant), 25 mg and 50 mg, oral tablets. December 2, 2024.
2.Idorsia Pharmaceuticals Canada Ltd. Drug Reimbursement Review sponsor submission: Quviviq (daridorexant), 25 mg and 50 mg, oral tablets [internal sponsor's package]. December 2, 2024.
3.Innomar Strategies Inc. Quviviq (daridorexant): 25 mg and 50 mg oral tablets [product monograph]. April 26, 2023. Updated November 8, 2024.
4.Mignot E, Mayleben D, Fietze I, et al. Safety and efficacy of daridorexant in patients with insomnia disorder: results from two multicentre, randomised, double-blind, placebo-controlled, phase 3 trials. Lancet Neurol. 2022;21(2):125-139. doi: 10.1016/s1474-4422(21)00436-1 PubMed
5.Idorsia Pharmaceuticals Ltd. Clinical Study Report: ID-078A301. Multi-center, double-blind, randomized, placebo-controlled, parallel-group, polysomnography study to assess the efficacy and safety of ACT-541468 in adult and elderly subjects with insomnia disorder [internal sponsor's report]. October 15, 2020.
6.Idorsia Pharmaceuticals Ltd. Clinical Study Report: ID-078A303. Multi-center, double-blind, parallel-group, randomized, placebo-controlled, three doses, 40-week extension to studies ID-078A301 and ID-078A302 to assess the long-term safety and tolerability of ACT-541468 in adult and elderly subjects with insomnia disorder [internal sponsor's report]. September 21, 2021.
7.DeGeorge KC, Ring DJ, Dalrymple SN. Treatment of the Common Cold. Am Fam Physician. 2019;100(5):281-289. PubMed
8.Waterer G. Recovery from community acquired pneumonia: the view from the top of the iceberg. Eur Respir J. 2017;49(6). doi: 10.1183/13993003.00571-2017 PubMed
9.Fabricant L, Ham B, Mullins R, Mayberry J. Prolonged pain and disability are common after rib fractures. Am J Surg. 2013;205(5):511-5; discussion 515-6. doi: 10.1016/j.amjsurg.2012.12.007
10.Falk Hvidberg M, Hernandez Alava M. Catalogues of EQ-5D-3L Health-Related Quality of Life Scores for 199 Chronic Conditions and Health Risks for Use in the UK and the USA. Pharmacoeconomics. 2023;41(10):1287-1388. doi: 10.1007/s40273-023-01285-4 PubMed
11.Chalet FX, Albanese E, Egea Santaolalla C, et al. Epidemiology and burden of chronic insomnia disorder in Europe: an analysis of the 2020 National Health and Wellness Survey. J Med Econ. 2024;27(1):1308-1319. doi: 10.1080/13696998.2024.2407631 PubMed
12.Chalet FX, Bujaroska T, Germeni E, et al. Mapping the Insomnia Severity Index Instrument to EQ-5D Health State Utilities: A United Kingdom Perspective. Pharmacoecon Open. 2023;7(1):149-161. doi: 10.1007/s41669-023-00388-0 PubMed
13.Wailoo AJ, Hernandez-Alava M, Manca A, et al. Mapping to Estimate Health-State Utility from Non-Preference-Based Outcome Measures: An ISPOR Good Practices for Outcomes Research Task Force Report. Value Health. 2017;20(1):18-27. doi: 10.1016/j.jval.2016.11.006 PubMed
14.Dauvilliers Y, Zammit G, Fietze I, et al. Daridorexant, a New Dual Orexin Receptor Antagonist to Treat Insomnia Disorder. Ann Neurol. 2020;87(3):347-356. doi: 10.1002/ana.25680 PubMed
15.Ontario Ministry of Health. Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 24, 2023)). 2023. Accessed February 20, 2024. https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf
16.Canadian Institute for Health Information. Patient cost estimator. 2024. Accessed by sponsor, no date provided. https://www.cihi.ca/en/patient-cost-estimator
17.Alberta Health Interactive Health Data Team. Hospital Inpatient Care Case Costs - CMG/Plex. 2015. Accessed October 29, 2024. https://open.alberta.ca/opendata/hospital-inpatient-care-case-costs-cmgplex
18.Bank of Canada. Inflation calculator. 2024. Accessed July 2, 2024. https://www.bankofcanada.ca/rates/related/inflation-calculator/
19.Canada's Drug Agency. Quviviq (daridorexant) – Presubmission Meeting Follow-up [internal document]. In: Drug Reimbursement Review sponsor submission: Quviviq (daridorexant), 25 mg and 50 mg, oral tablets. October 7, 2024.
20.Wilson S, Anderson K, Baldwin D, et al. British Association for Psychopharmacology consensus statement on evidence-based treatment of insomnia, parasomnias and circadian rhythm disorders: An update. J Psychopharmacol. 2019;33(8):923-947. doi: 10.1177/0269881119855343 PubMed
21.Riemann D, Espie CA, Altena E, et al. The European Insomnia Guideline: An update on the diagnosis and treatment of insomnia 2023. J Sleep Res. 2023;32(6):e14035. doi: 10.1111/jsr.14035 PubMed
22.AA Pharma Inc. Restoril (temazepam): 15 mg and 30 mg, oral capsules [product monograph; sponsor supplied reference]. December 31, 1980. Accessed November 16, 2021.
23.sanofi-aventis Canada Inc. Imovane (zopiclone): 5.0 mg and 7.5 mg, tablets [product monograph]. October 5, 2020.
24.Bonnet MH, Arand DL. The use of lorazepam TID for chronic insomnia. Int Clin Psychopharmacol. 1999;14(2):81-9. doi: 10.1097/00004850-199903000-00004 PubMed
25.Coe HV, Hong IS. Safety of low doses of quetiapine when used for insomnia. Ann Pharmacother. 2012;46(5):718-22. doi: 10.1345/aph.1Q697 PubMed
26.Jaffer KY, Chang T, Vanle B, et al. Trazodone for Insomnia: A Systematic Review. Innov Clin Neurosci. 2017;14(7-8):24-34. PubMed
27.Kamphuis J, Taxis K, Schuiling-Veninga CC, Bruggeman R, Lancel M. Off-Label Prescriptions of Low-Dose Quetiapine and Mirtazapine for Insomnia in The Netherlands. J Clin Psychopharmacol. 2015;35(4):468-70. doi: 10.1097/JCP.0000000000000338 PubMed
28.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2024. Accessed March 17, 2025. https://www.formulary.health.gov.on.ca/formulary/
29.B. C. Government. BC PharmaCare formulary search. 2024. Accessed March 17, 2025. https://pharmacareformularysearch.gov.bc.ca
30.Statistics Canada. Table: 17-10-0057-01 Projected population, by projection scenario, age and sex, as of July 1 (x 1,000). 2022. Accessed April 2023. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701
31.Ohayon MM, Reynolds CF, 3rd. Epidemiological and clinical relevance of insomnia diagnosis algorithms according to the DSM-IV and the International Classification of Sleep Disorders (ICSD). Sleep Med. 2009;10(9):952-60. doi: 10.1016/j.sleep.2009.07.008 PubMed
32.Hafner M, Romanelli RJ, Yerushalmi E, Troxel WM. The societal and economic burden of insomnia in adults: An international study. RAND Corporation; 2023. Accessed by sponsor, no date provided. https://www.rand.org/pubs/research_reports/RRA2166-1.html
33.Garland SN, Rowe H, Repa LM, Fowler K, Zhou ES, Grandner MA. A decade's difference: 10-year change in insomnia symptom prevalence in Canada depends on sociodemographics and health status. Sleep Health. 2018;4(2):160-165. doi: 10.1016/j.sleh.2018.01.003 PubMed
34.Towards Optimized Practice. Assessment to management of adult insomnia clinical practice guideline [sponsored supplied reference]. Alberta Medical Association; 2015. Accessed by sponsor, no date provided. https://actt.albertadoctors.org/media/1dqjq3fh/adult-insomnia-summary.pdf
35.Idorsia Pharmaceuticals Ltd. Advisory board survey results [sponsor supplied reference]. 2024.
36.The Conference Board of Canada. Understanding the Gap 2.0: A Pan-Canadian Analysis of Prescription Drug Insurance Coverage [sponsor supplied reference]. 2022.
37.Idorsia Pharmaceuticals Canada Ltd. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Quviviq (daridorexant), 25 mg and 50 mg, oral tablets. December 2, 2024.
38.IQVIA. MIDAS Global Edition – Monthly, Ex-factory, Full year [sponsor supplied reference]. 2022.
39.Crain J, Raj M, Garland S, Jones S, Farah B. Current practice analysis: interventions for insomnia disorder. (CADTH technology review: no.7). CADTH; 2017. Accessed April 14, 2023. https://www.cadth.ca/sites/default/files/pdf/OP0527_Current_Practice_Analysis_Insomnia%20Disorder.pdf
40.Centre for Effective Practice. Management of Chronic Insomnia. 2017. Accessed November 20, 2024. https://tools.cep.health/wp-content/uploads/2021/07/CEP_Management_of_Chronic_Insomnia_2017.pdf
Appendix 1: Cost Comparison Table
Please note that this appendix has not be copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 5: CDA-AMC Cost Comparison of Insomnia Medications
Treatment | Strength / concentration | Form | Price | Recommended dosagea | Nightly cost ($)a | Annual cost ($) |
|---|---|---|---|---|---|---|
Daridorexant (Quviviq) | 50 mg 25 mg | Tablet | $2.3600 | 50 mg nightly | $2.36 | $861 |
Table 6: CDA-AMC Cost Comparison of Insomnia Medications (Off-Label Treatments)
Treatment | Strength / concentration | Form | Price | Recommended dosagea | Nightly cost ($)a | Annual cost ($) |
|---|---|---|---|---|---|---|
Benzodiazepines | ||||||
Temazepam (generics) | 15 mg 30 mg | Tablet | $0.2461 $0.2980 | 30 mg nightly | $0.30 | $109 |
Lorazepam (generics) | 0.5 mg 1 mg 2 mg | Tablet | $0.0359 $0.0447 $0.0699 | 0.5 mg to 2 mg nightly | $0.04 to $0.07 | $13 to $26 |
Clonazepam (generic) | 0.5 mg 2 mg | Tablet | $0.0418 $0.0721 | 0.5 mg to 2 mg nightly | $0.04 to $0.07 | $15 to $26 |
Triazolam (generic) | 0.125 mg 0.25 mg | Tablet | $0.1496 $0.2551 | 0.125 mg to 0.25 mg nightly | $0.15 to $0.26 | $55 to $93 |
Flurazepam (generic) | 15 mg 30 mg | Tablet | $0.1166 $0.1364 | 15 to 30 mg nightly | $0.12 to $0.14 | $43 to $50 |
Z-drugs (also known as nonbenzodiazepines) | ||||||
Zopiclone (generics) | 5 mg 7.5 mg | Tablet | $0.2231 $0.4685 | 5 to 7.5 mg nightly | $0.22 to $0.47 | $81 to $171 |
Zolpidem tartrate (generic) | 5 mg 10 mg | Tablet | $1.1825 $1.1883 | 5 to 10 mg nightly | $1.18 to 1.19 | $432 to $434 |
Antidepressants | ||||||
Trazodone (generics) | 50 mg 100 mg 150 mg | Tablet | $0.0554 $0.0989 $0.1453 | 50 mg to 150 mg nightly | $0.06 to $0.15 | $20 to $53 |
Amitriptyline (generics) | 10 mg 25 mg 50 mg | Tablet | $0.0305 $0.0829 $0.1540 | 10 mg to 50 mg nightly | $0.03 to 0.15 | $11 to $56 |
Doxepin (Sinequan) | 10 mg 25 mg 50 mg 75 mg 100 mg | Tablet | $0.4075 $0.5000 $0.9274 $0.8711d $1.7388d | 10 mg to 100 mg nightly | $0.41 to $1.74 | $149 to $635 |
Mirtazapine (generics) | 15 mg 30 mg 45 mg | Orally Disintegrating Tablet | $0.4046 $0.8087 $1.2132 | 15 mg to 45 mg nightly | $0.40 to $1.21 | $148 to $443 |
Antipsychotics | ||||||
Quetiapine (generics) | 25 mg 100 mg 200 mg 300 mg | Immediate release tablet | $0.0494 $0.1318 $0.2647 $0.3863 | 25 mg to 300 mg nightly | $0.05 to $0.39 | $18 to $141 |
50 mg 150 mg 200 mg 300 mg 400 mg | Extended-release tablet | $0.2501 $0.4926 $0.6661 $0.9976 $1.3270 | 50 mg to 400 mg nightly | $0.25 to $1.33 | $91 to $485 | |
aAll recommended dosages were based on the product’s respective product monograph or published literature.3,22-27
bAll prices are from the Ontario Drug Benefit Formulary (accessed February 2025), unless otherwise indicated, and do not include dispensing fees28
cAnnual costs are calculated based on 365 days per year.
dCost sourced from BC Pharmacare Formulary (accessed March 2025).29
Appendix 2: Submission Quality
Please note that this appendix has not be copy-edited.
Description | Yes or No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to limitations about lack of relevant off-label comparators and external validity of the modelled population. |
Model has been adequately programmed and has sufficient face validity | No | Refer to limitations on model’s health utilities. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | The sponsor’s utility estimates were derived from a mapping algorithm in which the baseline characteristics (i.e., female proportion, age, baseline ISI score, and BMI) of the algorithm were not aligned with the population in Studies 301 and 303. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | For some model parameters, the sponsor arbitrarily incorporated uncertainty (i.e., change in ISI scores, adverse event probabilities, disutilities, medical costs, and adverse event costs) using a standard deviation equal to a 95 percentile in a normal distribution. This is unlikely to reflect the true uncertainty around the model’s parameters. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
BMI = body mass index; ISI = Insomnia Severity Index.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not be copy-edited.
AE = adverse event; HCRU = health care resource use; ISI = Insomnia Severity Index; QALY = quality-adjusted life year; Rx = prescription; SDS = Sheehan Disability Scale; WPAI = Work Productivity and Activity Index.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 8: Disaggregated Summary of the Sponsor’s Economic Evaluation Results (Probabilistic)
Parameter | Daridorexant | No pharmacological treatment |
|---|---|---|
QALYs | ||
Total | 0.71 | 0.69 |
QALYs | 0.72 | 0.69 |
Disutilities from AEs | −0.006 | −0.003 |
Costs ($) | ||
Total | $2,107 | $1,415 |
Treatment cost | $679 | $0 |
Medical costs | $1,267 | $1,315 |
AE costs | $161 | $100 |
AE = adverse event; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Submitted BIA and CDA-AMC Appraisal
Please note that this appendix has not be copy-edited.
Table 9: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
CID = chronic insomnia disorder; DSM = diagnostic and statistical manual of mental disorders.
Summary of Sponsor’s BIA
In the submitted BIA, the sponsor assessed the budget impact of reimbursing daridorexant for the management of adult patients with insomnia, characterized by difficulties with sleep onset and/or sleep maintenance (i.e., acute and chronic insomnia). A scenario analysis was further submitted that reflected the original reimbursement-requested population (i.e., adults diagnosed according to the most recent version of the DSM referring to CID). The BIA was conducted from the perspective of a Canadian public drug plan over a 3-year time horizon (2025 to 2028).
An epidemiological approach was taken to determine the number of patients eligible for daridorexant using data from literature and sponsor assumptions.30-36 The sponsor compared a reference scenario where patients are treated with benzodiazepines, antidepressants, and Z-drugs to a new drug scenario in which daridorexant would be reimbursed. Similar market shares were assumed in the reference scenario for patients with acute and chronic insomnia. In the new drug scenario, daridorexant was assumed to have a different market uptake for acute and chronic insomnia patients, informed by the sponsor’s internal projections. Drug costs were derived from the Ontario Drug Benefit and sponsor’s assumptions. Additionally, markups were included in the sponsor’s base-case results. Key inputs to the BIA are documented in Table 10.
Key assumptions to the BIA include:
The treatment duration for acute and chronic insomnia was assumed to be 30 and 253 days, respectively.
Drug utilization of different dosages of benzodiazepines, antidepressants, and Z-drugs were proportional to the number of doses available (e.g., if 2 dosages are available, each would take 50% of the drug’s utilization).
Table 10: Summary of Key Model Parameters
Parameter: target population | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) | |
|---|---|---|
Acute insomnia | Chronic insomnia | |
Statistics Canada population Prevalence of insomnia Annual increase in insomnia Proportion of patients prescribed with a pharmacological treatment | 26,410,468 / 26,674,474 / 26,937,210 15.20% 0.15% 20.00% | 26,410,468 / 26,674,474 / 26,937,210 8.80% 0.15% 50.00% |
Proportion of patients by age group | ||
Proportion of patients aged 18 to 64 Proportion of patients aged ≥ 65 | 76.81% 23.19% | 76.81% 23.19% |
Proportion of patients with public drug coverage | ||
Proportion of patients aged 18 to 64 with public coverage Proportion of patients aged ≥ 65 with public coverage | 24.74% 88.28% | 24.74% 88.28% |
Number of patients eligible for drug under review | 320,084 / 323,283 / 326,468 | 466,572 / 471,236 / 475,877 |
Market Uptake (3 years) | ||
Uptake (reference scenario) | ||
Acute insomnia Benzodiazepines Antidepressants Z-drugs (also known as nonbenzodiazepines) | 12.18% / 12.18% / 12.18% 44.66% / 44.66% / 44.66% 43.14% / 43.14% / 43.14% | |
Chronic insomnia Chronic use of acute insomnia treatments | 100% / 100% / 100% | |
Uptake (new drug scenario) | ||
Acute insomnia Daridorexant Benzodiazepines Antidepressants Z-drugs (also known as nonbenzodiazepines) | 0.50% / 1.00% / 1.50% 12.12% / 12.06% / 12.00% 44.44% / 44.21% / 43.99% 42.92% / 42.71% / 42.49% | |
Chronic insomnia Daridorexant Chronic use of acute insomnia treatments | 5.21% / 8.39% / 11.60% 94.80% / 91.62% / 88.40% | |
Cost of treatment (per patient, per day) | ||
Daridorexant Benzodiazepines Antidepressants Z-drugs | $2.36 $0.036 to $0.30 $0.044 to $0.08 $0.22 to $0.47 | |
Summary of the Sponsor’s BIA Results
The sponsor estimated that funding daridorexant for the management of adult patients with insomnia, characterized by difficulties with sleep onset and/or sleep maintenance (i.e., acute and chronic insomnia) would cost $86,506,575 across the 3-year time horizon (Year 1: $17,374,007; Year 2: $28,722,380; and Year 3: $40,410,188). When considering chronic insomnia patients only, the estimated total budget impact was $79,632,725 (Year 1: $16,243,936; Year: $26,438,867; Year 3: $36,949,922).37
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The population of the submitted BIA does not align with the reimbursement request. The revised reimbursement-requested population is for moderate to severe CID, when CBT-I is inappropriate, unavailable, or has failed, characterized by difficulty initiating or maintaining sleep; or early awakenings; occurring at least 3 times weekly; lasting a minimum of 3 months; and an ISI score ≥ 15. This population is narrower than the population captured in the sponsor’s BIA which reflected the Health Canada indication and the original reimbursement request as neither restricted the eligibility of daridorexant treatment by disease severity or specific sleep parameters.
CDA-AMC was unable to address this limitation. However, given that the sponsor’s revised reimbursement request is for a narrower population, the CDA-AMC reanalysis likely overestimates the budget impact of reimbursing daridorexant. The degree to which the budget impact may have been overestimated is unknown and is dependent on the extent to which the sponsor’s revised reimbursement population would narrow the number of patients eligible for daridorexant relative to their original reimbursement-requested population.
Relevant comparators and available doses were not included. The sponsor included benzodiazepines, antidepressants, and Z-drugs in their base-case analysis. However, clinician expert feedback elicited by CDA-AMC indicated that there were other relevant comparators used in clinical practice for chronic insomnia that were missing in the sponsor’s BIA, These included clonazepam, triazolam, flurazepam, zolpidem tartrate, doxepin, mirtazapine, and quetiapine. Additionally, even for the comparators included within the sponsor’s BIA, certain drug dosages were excluded (e.g., temazepam (15 mg), trazodone (100 mg, 150 mg), and amitriptyline (50 mg)). Clinician expert feedback sought by CDA-AMC noted that the listed doses are prescribed in Canada for CID. Clinical expert feedback further indicated that nitrazepam is not generally used for CID in clinical practice.
In reanalysis, CDA-AMC included the missing comparators and dosages. As a result, market shares were revised in line with the clinician expert feedback obtained by CDA-AMC (Figure 2). The sponsor reference scenario market shares were based on the IQVIA MIDAS Global database38 which incorporated manufacturer sales data of both public and private markets and included lemborexant which is not publicly reimbursed in Canada. Nonetheless, clinician expert feedback consulted by CDA indicated that the sponsor’s reference scenario market shares were somewhat aligned with their clinical practice as trazodone and zopiclone were perceived to capture the majority of the market share.
Market uptake of daridorexant is associated with uncertainty. The sponsor’s submitted base case assumed 5.21%, 8.39%, 11.60% of eligible patients with CID will receive daridorexant in Year 1, Year 2 and Year 3 respectively, based on the sponsor’s internal forecasts. BIA market share forecast was based on analogue product sales data (i.e., lemborexant), which as previously mentioned, is only funded through private insurance and out-of-pocket market. As the BIA is conducted in the public drug plan perspective, it remains uncertain how transferable the market uptake from nonpublic sources would be to public drug plans. Clinician expert opinions consulted by CDA-AMC indicated that market uptake is expected to be higher for chronic insomnia than the sponsor’s projections.
In reanalysis, CDA-AMC assumed 7.5%, 12.5%, and 15%, in line with the clinician expert feedback consulted by CDA-AMC.
Duration of treatment of chronic insomnia patients is associated with uncertainty and does not reflect clinical practice. The duration of treatment was assumed to be 253 days.2 The sponsor cited that patients with chronic insomnia will be treated for the same duration as Study 303.2 However, Study 303 was a 40-week extension of the 12-week Studies 301 and 302, implying that patients were intended to be treated for a total duration of a year.
Additionally, to reduce the risk of abuse and dependency associated with long-term use of comparators, clinical expert feedback indicated that comparators are typically prescribed in intervals; where patients will not be exposed to chronically to the comparator treatment.39,40 Due to the mechanism of action of daridorexant, there is a minimum period of 4 to 8 weeks before full efficacy is achieved in patients as the clinical trials demonstrated a gradual increase in effect size for sleep parameters.5,6 Thus, the assumption that there is a similar duration of treatment for daridorexant and chronic use of comparator treatments is unlikely to be appropriate and is likely to underestimate the budget impact. To demonstrate the impact of treatment duration, CDA-AMC revised duration of treatment for chronic insomnia to 365 days in reanalysis. However, CDA-AMC does note that this may overestimate the treatment cost for the comparator treatment given the expected differences in treatment duration due to the drugs’ pharmacodynamics.
Drug plan patient eligibility and enrolment is associated with uncertainty. In the sponsor’s BIA base case, the proportion of patients who were publicly eligible for public coverage was based on the enrolment rates of each jurisdiction with the exception of NIHB which was assumed to be 100% by the sponsor. Based on the sponsor’s submitted price, the annual cost of daridorexant does not exceed $900 and it may be unlikely that eligible patients would attempt to enrol for public coverage through their jurisdiction-specific drug plan if daridorexant were to become publicly reimbursed. However, there is uncertainty associated with this assumption.
To address the uncertainty associated with patient enrolment into public drug plans, CDA-AMC conducted a scenario analysis where the target population size was filtered based on proportion eligible rather than proportion enrolled.
CDA-AMC Reanalyses of the BIA
The CDA-AMC base-case reanalyses revised the reference scenario market shares, included relevant comparators, revised the market uptake of daridorexant for patients with chronic insomnia, and adjusted the duration of treatment for chronic insomnia.
Table 11: CDA-AMC Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Correctionsa to sponsor’s base case | ||
1. Markup and dispensing fees. | Included. | Removed. |
Changes to derive the CDA-AMC base case | ||
1. Added comparators | Excluded clonazepam, triazolam, flurazepam, zolpidem tartrate, doxepin, mirtazapine, and quetiapine as comparators. Market shares: Benzodiazepines: 12.18%, Z-drugs (also known as nonbenzodiazepines): 43.14% Antidepressants: 44.66% | Included clonazepam, triazolam, flurazepam, zolpidem tartrate, doxepin, mirtazapine, and quetiapine as comparators. Market shares: Benzodiazepines: 13.2% Z-drugs (also known as nonbenzodiazepines): 26.2%, Antidepressants and antipsychotics: 60.6% |
2. Daridorexant market uptake in chronic insomnia | 5.21%, 8.39%, and 11.6% from years 1 to 3 | 7.5%, 12.5% and 15% in years 1 to 3 |
3. Duration of treatment in chronic insomnia | 253 days. | 365 days. |
CDA-AMC base case | Reimbursement request population: 1 + 2 + 3 | |
BIA = budget impact analysis.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 12 and a more detailed breakdown is presented in Table 13. The CDA-AMC base-case reanalysis only presents the reimbursement-requested population given clinician expert feedback obtained by CDA-AMC indicated that chronic insomnia would be the expected place in therapy for daridorexant. The use of daridorexant for acute insomnia would be inappropriate given the drug’s noted mechanism of action as described here. CDA-AMC does not explicitly present any reanalysis on the full Health Canada population or the acute insomnia subgroup as clinical experts consulted by CDA-AMC were unable to validate the appropriateness of certain model inputs that informed the acute insomnia subpopulation (i.e., market size, market shares of treatment classes, and distribution of treatment classes).
Based on CDA-AMC reanalyses, the estimated budget impact of funding daridorexant for the management of adult patients diagnosed with CID according to the DSM is $26,558,223 in year 1, $44,717,188, in year 2, and $54,203,925 in year 3, with a 3-year budget impact of $125,479,336. All reanalyses were based on publicly available prices and the confidential prices of each treatment may impact the results of the CDA-AMC base case.
Table 12: Summary of the CDA-AMC Reanalyses of the BIA (Reimbursement-Requested Population)
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 79,632,725 |
CDA-AMC reanalysis 1 | 62,622,975 |
CDA-AMC reanalysis 2 | 91,050,134 |
CDA-AMC reanalysis 3 | 94,577,266 |
CDA-AMC base case | 125,479,336 |
BIA = budget impact analysis.
CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 13):
Increased the proportion of publicly covered patients to reflect the number of eligible patients in each jurisdiction.
Table 13: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA (Reimbursement-Requested Population)
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 24,101,756 | 24,752,195 | 25,006,853 | 25,261,029 | 75,020,077 |
New drug | 24,101,756 | 40,996,131 | 51,445,719 | 62,210,951 | 154,652,802 | |
Budget impact | 0 | 16,243,936 | 26,438,867 | 36,949,922 | 79,632,725 | |
Sponsor’s submitted base case corrected | Reference | 22,310,948 | 22,912,194 | 23,146,930 | 23,381,287 | 69,440,411 |
New drug | 22,310,948 | 36,286,364 | 44,912,794 | 53,797,549 | 134,996,708 | |
Budget impact | 0 | 13,374,170 | 21,765,864 | 30,416,262 | 65,556,297 | |
CDA-AMC base case | Reference | 48,339,318 | 49,641,988 | 50,150,572 | 50,658,334 | 150,450,894 |
New drug | 48,339,318 | 76,200,211 | 94,867,760 | 104,862,259 | 275,930,230 | |
Budget impact | 0 | 26,558,223 | 44,717,188 | 54,203,925 | 125,479,336 | |
CDA-AMC Scenario analysis: Public plan patient enrolment | Reference | 87,281,357 | 89,708,224 | 90,705,261 | 91,701,554 | 272,115,039 |
New drug | 87,281,357 | 137,701,687 | 171,583,385 | 189,821,327 | 499,106,400 | |
Budget impact | 0 | 47,993,464 | 80,878,124 | 98,119,773 | 226,991,360 |
BIA = budget impact analysis.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.