Drugs, Health Technologies, Health Systems
Reimbursement Review
Omaveloxolone (Skyclarys)
Sponsor: Biogen Canada Inc.
Therapeutic area: Friedreich’s ataxia
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Ethics Review
Clinical Review
Abbreviations
9-HPT
9-hole peg test
ADL
activities of daily living
AE
adverse event
ALT
alanine aminotransferase
AST
aspartate aminotransferase
CDA-AMC
Canada’s Drug Agency
CGIC
Clinical Global Impression of Change
CI
confidence interval
DSMB
Data Safety Monitoring Board
FA
Friedreich’s ataxia
FACOMS
Friedreich’s Ataxia Clinical Outcome Measures Study
FAS
full analysis set
FARA
Friedreich’s Ataxia Research Alliance
FARS
Friedreich’s Ataxia Rating Scale
FARS-ADL
Friedreich’s Ataxia Rating Scale Activities of Daily Living
GAA
guanine-adenine-adenine
GAA1
guanine-adenine-adenine 1
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
IWRS
interactive web response system
LDL
low-density lipoprotein
MAR
missing at random
MDC
Muscular Dystrophy Canada
mFARS
modified Friedreich’s Ataxia Rating Scale
MMRM
mixed model for repeated measures
NAF
National Ataxia Foundation
OLE
open-label extension
PGIC
Patient Global Impression of Change
PD
pharmacodynamic
PK
pharmacokinetic
RCT
randomized controlled trial
RWE
real-world evidence
SAE
serious adverse event
SD
standard deviation
SE
standard error
ULN
upper limit of normal
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information on the Application Submitted for Review
Item | Description |
|---|---|
Drug product | Omaveloxolone (Skyclarys), administered at a recommended dose of 150 mg orally once daily (3 capsules of 50 mg each) |
Sponsor | Biogen Canada Inc. |
Indication | For the treatment of Friedreich’s ataxia in patients aged 16 years or older |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | March 13, 2025 |
Recommended dose | 150 mg (3 capsules of 50 mg each) taken orally once daily |
NOC = Notice of Compliance.
Introduction
Friedreich’s ataxia (FA) is a rare autosomal recessive ataxia caused by loss-of-function mutations from trinucleotide repeat expansions in the FXN gene located on chromosome 9q13.1,2 FA accounts for up to one-half of all hereditary ataxia cases and affects approximately 1 in 30,000 to 1 in 50,000 individuals.2 In Canada, estimates suggest that 300 to 1,000 patients are affected.3-5 FA typically presents in childhood or adolescence with a complex neurologic phenotype characterized by progressive gait ataxia. Additional cerebellar signs including dysarthria and dysphagia; peripheral motor and sensory neuropathy combined with pyramidal signs; and, in advanced disease, visual and hearing impairment.6-8 Most affected individuals develop hypertrophic cardiomyopathy, which in some cases may precede the onset of ataxia. Diabetes mellitus and skeletal abnormalities, such as pes cavus and scoliosis, are also common. The disease shortens life expectancy considerably because of cardiac complications, with a mean age at death of 37 years. Currently, there are no approved disease-modifying therapies available in Canada for FA. Management focuses on supportive care, rehabilitation, and symptomatic treatment of complications.9,10
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of omaveloxolone, administered at a recommended dose of 150 mg orally once daily (as 3 capsules of 50 mg each), in the treatment of FA in adults and adolescents aged 16 years and older.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of the input provided by the patient and clinician groups who responded to our call for input and from clinical experts consulted for the purpose of this review.
Patient Input
Four patient groups — Muscular Dystrophy Canada (MDC), the National Ataxia Foundation (NAF), the Ataxia Canada – Claude St-Jean Foundation, and Friedreich’s Ataxia Research Alliance (FARA) — provided input. MDC identified and contacted adults living with FA and parents of children aged 16 years and over living with FA to participate in a health care experience survey and semistructured virtual interviews. The organization gathered insights from 85 individuals (aged 16 years to 70 years) with a confirmed FA diagnosis on diagnostics delays, gaps in treatment, emotional and social effects, and access to care and support systems. NAF conducted a survey of its community members with FA living in different provinces across Canada and received 14 responses (from 9 people with FA and 5 caregivers). Ataxia Canada gathered the experiences of patients and caregivers in Canada through a combination of interviews and a survey and received 85 responses. Feedback from FARA regarding the disease experience was drawn from 2 sources: a white paper that included views from parents of children with FA living in the US, highlighting the importance of pediatric inclusion in clinical trials, and a patient-focused drug development meeting that included 145 patients and caregivers, during which participants were polled about their current disease state, experience with different symptoms, and perspectives on future treatments.
Ataxia Canada and MDC highlighted that patients with FA experience significant impacts with respect to coordinating and/or maintaining balance; mobility and scoliosis; productivity at home and work; independence and social participation; and mental health. Additionally, FARA noted that patients and caregivers indicated experiencing neurologic symptoms (e.g., regular falls and trouble with balance, walking, and coordinating hands and/or arms) and fatigue. NAF, Ataxia Canada, and FARA indicated that people with FA and their caregivers spend hours engaging in symptom-based therapies (such as occupational therapy, speech therapy, and physiotherapy), visiting medical specialists, and obtaining mobility devices, which can be challenging, expensive, and time-consuming.
The patient groups agreed that there is a significant need for a treatment that is a cure for FA or can reverse its effects. Patients are also seeking a treatment that slows and/or stops disease progression, promotes better symptom management, helps them regain and/or preserve mobility, increases energy levels, improves health-related quality of life (HRQoL) and independence, prevents complications (such as scoliosis or diabetes), and reduces the physical and emotional burden on families and caregivers. Currently, individuals require genetic testing to confirm the presence of biallelic mutations of the FXN gene. MDC and NAF reported that the testing process was easy and provided clarity about the diagnosis but was often emotionally challenging.
Four respondents (1 each from MDC and NAF and 2 from Ataxia Canada) shared their experiences with accessing omaveloxolone through clinical trials. All 4 respondents (2 of whom were already wheelchair users) highlighted that being on the drug slowed their disease progression. The viewpoints gathered by FARA from individuals with FA and parents of individuals with FA who participated in the MOXIe trial included slowed progression, longer retention of motor function (i.e., improved endurance, speech, and ability to walk, stay upright, and eat), better ability to cope with fatigue, and minimal side effects (i.e., transient elevation of liver enzymes, cholesterol, headache, nausea, and diarrhea).
Clinician Input
Input From Clinical Experts Consulted for This Review
The information in this section is based on the input received from a panel of 4 clinical specialists consulted by Canada’s Drug Agency (CDA-AMC) for the purpose of this review.
The clinical experts emphasized that there are currently no approved disease-modifying treatments for FA in Canada; thus, there is a critical unmet need. Current management relies on supportive care and symptom management, which do not alter the underlying progressive disease course.
The experts indicated that omaveloxolone would represent the first disease-modifying therapy for FA and be positioned as a first-line option for eligible patients aged 16 years and older with genetically confirmed disease. They noted that it would be used alongside existing supportive care measures rather than replacing them.
Regarding patient selection, the experts suggested that while all patients with confirmed FA could theoretically benefit, those at earlier disease stages may show more discernible stabilization before irreversible neurologic damage occurs. However, they emphasized that nonambulatory patients should not be excluded, given that preserving upper limb and/or bulbar function could still provide meaningful benefit.
With respect to assessing treatment response, the experts acknowledged challenges in translating the clinical trial measures to routine practice. While the modified Friedreich’s Ataxia Rating Scale (mFARS) was used in trials and natural history studies, it is not commonly employed in clinical settings. Furthermore, the experts suggested monitoring for 2 years to establish efficacy but noted that even partial slowing of disease progression may be valuable. According to the experts, treatment discontinuation should be based primarily on safety and tolerability rather than on lack of improvement alone, given the progressive nature of FA, the variability of the disease, its rate of progression, and the absence of alternative disease-modifying options.
The experts emphasized that diagnosis and treatment should be guided by specialists experienced in FA management, such as neuromuscular or movement disorder neurologists. They also noted that, while regular monitoring is important, requiring intensive specialized outcome measures (such as standardized scales designed for clinical trials that require trained users for reliable administration) could limit equitable access to treatment.
Clinician Group Input
A single clinician group input was received from the Neuromuscular Disease Network for Canada. Input from 4 clinicians familiar with clinical trials of FA treatments, specifically omaveloxolone, was gathered through 1-to-1 submissions and group discussions. The group noted that the primary therapeutic goals are to slow disease progression, preserve or enhance function, extend survival, and improve well-being.
The clinician group indicated that omaveloxolone is poised to be incorporated into the current treatment paradigm. However, the group also highlighted that there is not enough evidence to establish which patients are most likely to respond to omaveloxolone. Regarding the outcomes used to determine a patient’s response, the clinician group indicated standardized tests used in neurologic exams (i.e., mFARS) and functional assessments (such as the Friedreich’s Ataxia Rating Scale – Activities of Daily Living [FARS-ADL]). Measurements every 6 months in the first year and then annually were noted as reasonable and practical. In terms of a clinically meaningful response to treatment, the group noted that there should be an improvement in patient function and well-being. For example, this could be reflected by just a 1-point improvement in upright stability score compared to expected progression, indicating preserved balance in the short term and predicting delayed loss of ambulation.
The clinician group noted that omaveloxolone may be discontinued due to lack of efficacy (determined after 1 year, based on Clinical Global Impression of Change [CGIC] and Patient Global Impression of Change [PGIC]) or side effects (i.e., evidence of organ dysfunction). Additionally, it was recommended that a statin be prescribed to manage the cardiovascular risk factors associated with increased low-density lipoprotein (LDL) (a common side effect of omaveloxolone); this was deemed a better approach than discontinuing omaveloxolone. Lastly, the clinician group highlighted that people with FA must be treated at specialized centres that offer comprehensive interdisciplinary care, regardless of omaveloxolone treatment. For patients without easy access to such centres, care should be managed by a neurologist who is knowledgeable about the disease and its management.
Drug Program Input
Drug plans submitted questions concerning comparators as well as the initiation, renewal, discontinuation, and generalizability of omaveloxolone. The clinical expert panel convened by CDA-AMC provided advice on the potential implementation issues raised by the drug program. Refer to Table 4 for more details.
Clinical Evidence
Systematic Review
Description of Studies
One pivotal, phase II, randomized controlled trial (RCT) (the MOXIe Part 2 trial; N = 103) was included in the review to evaluate whether omaveloxolone 150 mg once daily improved modified mFARS scores compared to placebo after 48 weeks of treatment in patients aged 16 years to 40 years with an mFARS score of 20 to 80 and genetically confirmed FA. The trial included secondary end points assessing changes in activities of daily living (ADLs), upper limb function (9-hole peg test [9-HPT]), mobility (25-foot timed walk test), and frequency of falls.
Despite randomization, there were imbalances in baseline characteristics between the treatment groups. Compared to the placebo group, the omaveloxolone group had a higher proportion of males (60% males versus 33% females), more patients with cardiomyopathy (48% versus 29%), higher baseline mFARS scores (40.94 versus 38.77), and more patients with guanine-adenine-adenine 1 (GAA1) ( i.e., the shorter expanded allele) repeat lengths of greater than or equal to 675 (███ ██ ██%). The mean age was similar between groups (approximately 24 years), and the majority of patients in both groups were ambulatory (93%), ██████ █████ ████ ████████ █████████ ███████. Other disease characteristics were generally balanced, including the mean age of FA onset (15 years), disease duration (4.8 years), and prevalence of conditions like scoliosis (74%) and sensory neuropathy (49%).
Efficacy Results
Change in mFARS Score at Week 48
The mFARS measures neurologic function across 4 domains: bulbar function, upper limb coordination, lower limb coordination, and upright stability. Scores range from 0 to 93, with higher scores indicating greater impairment. Assessments were conducted at baseline and week 48.
In the prespecified primary analysis population (full analysis set [FAS]), patients receiving omaveloxolone had a mean baseline mFARS score of 40.94 points (standard deviation [SD] = 10.39 points) and showed a mean improvement (decrease) from baseline of █████ ██████ ███ █████) at week 48. The placebo group had a mean baseline score of 38.77 points (SD = 11.03 points) and showed a mean worsening (increase) from baseline of █████ ██████ ███ █████). The mixed model for repeated measures (MMRM) estimate of mean difference in change from baseline between the omaveloxolone and placebo groups was −2.40 points (95% confidence interval [CI], −4.31 to −0.50 points; P = 0.0141).
In the all-randomized population, which included patients with severe pes cavus, omaveloxolone treatment improved mFARS scores by an estimated mean difference of −1.93 points relative to placebo (95% CI, −3.70 to −0.15 points; P = 0.0342). The mean baseline scores were █████ ███ █████) for omaveloxolone ███ █████ ███ █████) for placebo, with mean changes from baseline ██ █████ ███ █████) and ████ ███ █████), respectively.
Change in Performance on the 9-HPT
The 9-HPT measures upper-extremity function based on the time taken to place and remove 9 pegs in a pegboard. The test was performed at baseline and week 48, with faster times indicating better function. Results are reported as the reciprocal of average time (1 per second) for the dominant hand.
In the FAS population, patients receiving omaveloxolone had a mean reciprocal of average time (1 per second) baseline value of ██████ ███ ███████) and showed little to no change from baseline (mean change ████████ ██ █████) at week 48. The placebo group had a mean reciprocal of average time (1 per second) baseline value of ██████ ███ ███████) and showed little to no change (mean change ████████ ██ ███████). The MMRM estimated mean difference between groups was ██████ ████ ███ ███████ ██ ███████ | | ██████).
Change in Performance on a 25-Foot Timed Walk Test
The 25-foot timed walk test measures mobility based on the time taken to walk 25 feet. Assessments were conducted at baseline and week 48, with results reported as the reciprocal of average walk time (1 per second). Higher values indicate better function.
In the FAS population, patients receiving omaveloxolone had a mean baseline score of ██████ ███ ███████) and showed a decline from baseline of –██████ ███ ███████) at week 48. The placebo group had a mean baseline score of ██████ ███ ███████) and showed a decline of –██████ ███ ███████). The MMRM estimated mean difference between groups was 0.0058 (95% CI, –0.0099 to 0.0214; P = 0.4635).
Frequency of Falls at Week 48
Falls were recorded daily by patients in a study diary throughout the 48-week treatment period. A fall was defined as “the patient unintentionally coming to rest on the ground or at a lower level.”
In the FAS population, patients receiving omaveloxolone reported a mean of ████ █████ ███ █████) during treatment compared to ████ █████ ███ █████) in the placebo group. The Poisson estimated difference in the incidence rate of falls between the omaveloxolone and placebo groups was █████ ████ ███ █████ ██ █████ | | ██████).
ADLs at Week 48
The ADL assessment included 9 questions evaluating speech, swallowing, cutting food and/or handling utensils, dressing, performing personal hygiene activities, falling, and walking, as well as bladder function and quality of sitting position. The total scores range from 0 to 36, with higher scores indicating greater impairment. Assessments were conducted at baseline and week 48.
In the FAS population, patients receiving omaveloxolone had a mean baseline ADL score of 10.738 (SD = 4.7663) and showed an improvement (decrease) from baseline of ██████ ██████ ███ ██████) at week 48. The placebo group had a mean baseline score of 9.869 (SD = 4.8339) and showed a worsening (increase) of ██████ ██████ ███ ██████). The MMRM estimated mean difference between groups was –1.30 points (95% CI, █████ ██ ██████ P = 0.0420).
Harms Results
All patients in both treatment groups experienced at least 1 adverse event (AE) during the 48-week trial. Common AEs occurring more frequently with omaveloxolone than with placebo included nausea (33.3% versus 13.5%), abdominal pain (21.6% versus 5.8%), diarrhea (19.6% versus 9.6%), fatigue (21.6% versus 13.5%), and increased liver enzymes (alanine aminotransferase [ALT] = 37.3% versus 1.9%; aspartate aminotransferase [AST] = 21.6% versus 1.9%). Serious adverse events (SAEs) occurred in 9.8% of patients receiving omaveloxolone versus 5.8% of patients receiving placebo. Treatment discontinuations due to AEs were more frequent in the group receiving omaveloxolone (7.8% versus 3.8%). Notable harms of special interest included liver enzyme elevations, which occurred more often in the group receiving omaveloxolone. No deaths were reported during the study period.
Critical Appraisal
Overall, the MOXIe trial demonstrated acceptable internal validity, benefiting from its randomized, double-blind, placebo-controlled design, proper allocation concealment, validated primary outcome measure, and appropriate statistical methods. Key limitations affecting internal validity included baseline imbalances between treatment groups despite randomization, with the omaveloxolone group having characteristics suggesting more advanced disease (i.e., higher mFARS scores, longer GAA1 repeat lengths, and a greater proportion of patients with cardiomyopathy). Furthermore, the imbalance in the male-to-female ratio between the groups receiving omaveloxolone and placebo may also bias the results against omaveloxolone, as observed in subgroup analyses showing better responses among males. The impact of some of these imbalances was explored in post hoc analyses that suggested a potential underestimation of treatment effect in the primary end point. Additionally, a higher discontinuation rate in the group receiving omaveloxolone versus placebo (13.7% versus 3.8%), primarily due to AEs, raises concerns about potential bias from missing data. The absence of an established minimum clinically important difference for mFARS also creates uncertainty in interpreting the clinical significance of the observed treatment effect. All outcomes other than the primary outcome either include the null or are outside the statistical testing hierarchy.
Overall, the external validity of the MOXIe Part 2 trial was limited by eligibility restrictions, the exclusion of patients with significant cardiac issues, and the capping of the number of patients with severe pes cavus. The sponsor justified the decision to cap the number of patients with severe pes cavus based on evidence from the MOXIe Part 1 trial suggesting that these patients may represent a different subtype of FA and also because their severe pes cavus would likely interfere with their ability to perform assessments that required standing or pedalling. Clinical experts involved in this review suggested that all patients with FA have some degree of pes cavus, which has not been shown to be clinically prognostic. The trial excluded patients aged younger than 16 years despite most patients being diagnosed at around 11 years of age; this exclusion created uncertainty about treatment effects in pediatric populations. However, the current Health Canada indication restricts the population to patients aged 16 years or older. This limitation to external validity is of limited impact if the drug is prescribed according to the indication. The restriction to patients with mFARS scores ranging from 20 to 80 and the exclusion of those with significant cardiac issues limits generalizability to patients with more severe disease. Furthermore, the outcomes used in the trial, particularly mFARS scores, are not routinely implemented in clinical practice, a situation that creates challenges when it comes to translating trial results to real-world assessment of treatment response. While the absence of Canadian sites was noted, clinical experts did not consider this a major limitation, given the similarity of the patient population and treatment approaches across countries.
GRADE Summary of Findings and Certainty of the Evidence
The selection of outcomes for the Grading of Recommendations Assessment, Development and Evaluation (GRADE) assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
change in mFARS score
change in 9-HTP performance
change in 25-foot timed walk test results
change in frequency of falls
change in ADLs.
Table 2: Summary of Findings for Omaveloxolone vs. Placebo for Patients With FA
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Omaveloxolone | Difference | |||||
Change in mFARS at week 48 | |||||||
Change in mFARS score (less is better) Follow-up: 48 weeks | 82 (1 RCT) | NA | ████ ██████ | █████ ██████ | ████ █████ ██████ █████ █████ ██ ████ ██████ | Moderatea | Compared to placebo, omaveloxolone likely results in slower progression of the mFARS score in patients with FA. |
Change in performance on 9-HPT at week 48 | |||||||
Change in 9-HTP performance (reciprocal of average walk time [per second], where more time is better) Follow-up: 48 weeks | 82 (1 RCT) | NA | ███████ ████████ | ███████ ████████ | ██████ ████ ████████ ███████ █████ ██ ██████ █████ | Lowb | Omaveloxolone may result in little to no difference in the change in 9-HTP compared to placebo in patients with FA. |
Change in performance on a 25-foot timed walk test at week 48 | |||||||
Change in 25-foot timed walk test (reciprocal of average walk time [per second], where more time is better) Follow-up: 48 weeks | 82 (1 RCT) | NA | ███████ 1 per second | ███████ 1 per second | 0.0058 more 1 per second (██████ █████ ██ ██████ █████ | Lowb | Compared to placebo, omaveloxolone may result in little to no difference in in 25-foot timed walk test results in patients with FA. |
Change in frequency of falls at week 48 | |||||||
Change in frequency of falls (less is better) Follow-up: 48 weeks | 82 (1 RCT) | NA | ████ █████ █████ ██ █████████ | ████ █████ █████ ██ █████████ | ████ █████ █████████ ████ ██ █████ █████ ██████ ████ █████ | Lowb | Compared to placebo, omaveloxolone may result in little to no difference in the frequency of falls in patients with FA. |
Change in ADLs at week 48 | |||||||
Changes in ADLs (less is better) Follow-up: 48 weeks | 82 (1 RCT) | NA | █████ ██████ | ██████ ██████ | ████ █████ ██████ █████ █████ ██ ████ ██████ | Moderatec | Omaveloxolone likely results in a decrease in ADL scores in patients with FA. |
9-HPT = 9-hole peg test; ADL = activities of daily living; CDA-AMC = Canada’s Drug Agency; CI = confidence interval; FA = Friedreich's ataxia; mFARS = modified Friedreich’s Ataxia Rating Scale; MID = minimal important difference; NA = not applicable; RCT = randomized controlled trial; vs. = versus.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aNo published between-group MID was identified. Clinical experts consulted by CDA-AMC identified 2 points as the potential clinically meaningful threshold. Rated down 1 level down for imprecision because the upper bound of the 95% CI suggests no clinically meaningful difference, while the lower bound of the 95% CI suggests benefit.
bNo published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects; therefore, the null was used. Rated down 2 levels for very serious imprecision because the lower CI suggests harm while the upper CI suggests benefit of little to no difference.
cNo published between-group MID was identified, and the clinical experts consulted by CDA-AMC identified 1 point as the potential clinically relevance threshold. Rated down 1 level for imprecision because the upper bound of the CI suggests no clinically meaningful difference, while the lower bound of the CI suggests clinically meaningful difference. Not rated down for imprecision; there is a between-group difference of less than the null and a CI that excludes the null.
Source: Sponsor’s Summary of Clinical Evidence.11
Long-Term Extension Study
Description of Study
One ongoing, open-label extension (OLE) study was included to assess the long-term safety and tolerability of omaveloxolone in patients with FA following the completion of Part 1 or Part 2 of the MOXIe trial. The study enrolled 149 patients from the MOXIe Part 1 and MOXIe Part 2 trials, including 106 patients who were omaveloxolone-naive (referred to as the placebo-omaveloxolone group) and 43 who had previously received omaveloxolone (referred to as the omaveloxolone-omaveloxolone group). All patients received omaveloxolone 150 mg daily, with interim analysis data available up to 144 weeks. Baseline characteristics were generally balanced between groups; however, the placebo-omaveloxolone group had a higher proportion of males than females (55.7% versus 37.2%) and patients with pes cavus (█████ ██ ████%).
Efficacy Results
At 144 weeks, the mean changes from baseline in mFARS score were 3.37 points (SD = 4.939 points) in the placebo-omaveloxolone group and 2.28 points (SD = 5.896 points) in the omaveloxolone-omaveloxolone group. ADL scores showed mean increases (indicating worsening) of 1.873 points in the placebo-omaveloxolone group and 1.286 points in the omaveloxolone-omaveloxolone group at week 144. Outcomes for additional functional measures, including the 9-HPT and 25-foot timed walk test, showed similar patterns.
Harms Results
The safety profile in the OLE was consistent with that of the controlled trial. Common AEs included coronavirus infection (18.8%), increased ALT (18.8%), headache (18.1%), upper respiratory tract infection (16.8%), nausea (16.1%), and fatigue (13.4%). SAEs occurred in 8.7% of patients (7.5% in the placebo-omaveloxolone group, 11.6% in the omaveloxolone-omaveloxolone group); ████ discontinued due to AEs (███% in the placebo-omaveloxolone group, ███% in the omaveloxolone-omaveloxolone group). Liver enzyme elevations remained a notable AE of special interest, but appeared to be manageable with monitoring. No deaths were reported.
Critical Appraisal
The main limitations of the long-term extension study are the lack of a control group, the open-label design with subjective outcomes, and potential selection bias (given that the study enrolled only patients who had completed the original trials). There is also a risk of attrition bias because the number of patients contributing to the analyses declined steadily over time; the final outcome measures are based on less than half the number of originally enrolled patients. In addition, the COVID-19 pandemic affected study visits and treatment continuity, with 14.8% of patients experiencing treatment interruptions. The use of historical controls for contextualizing progression rates, while informative, has inherent limitations due to potential differences in patient populations and assessment methods.
The clinical experts consulted for this review suggested that, aside from the exclusion of pediatric patients, the eligibility criteria of the OLE resulted in a study population comparable to patients in Canada. However, the trial’s strict inclusion and exclusion criteria, including constraints around cardiac involvement, may have led to a cohort that was healthier than what is typically encountered in routine clinical practice involving the treatment of FA in Canada. Furthermore, the study did not include any sites in Canada, which may reduce the generalizability and applicability of the results to Canadian practice.
Indirect Comparisons
No indirect treatment comparisons were submitted for this review.
Studies Addressing Gaps in the Evidence From the Systematic Review
Description of Studies
A propensity score–matched analysis compared long-term outcomes between patients in the MOXIe trial extension (N = 136) and matched controls from the Friedreich’s Ataxia Clinical Outcome Measures Study (FACOMS) natural history database (N = 136). Patients were matched on key characteristics, including age, sex, baseline mFARS score, age at FA onset, and baseline gait score, with a mean follow-up of approximately 2.5 years in both cohorts.
Efficacy Results
The estimated 3-year change from baseline in mFARS score was 6.61 points (standard error [SE] = 0.65 points) in matched FACOMS patients compared to 3.00 points (SE = 0.66 points) in patients in the MOXIe trial extension, representing a statistically significant difference of −3.61 points (95% CI, −1.79 to −5.43 points) favouring omaveloxolone. This suggests that patients in the MOXIe OLE experienced slower increases in mFARS scores than patients included from the matched FACOMS group (indicative of slowed disease progression).
Harms Results
Safety outcomes were not assessed in this analysis.
Critical Appraisal
The choice of study design was considered appropriate, given the constraints of the rare disease. The real-world evidence (RWE) used comparative evidence to describe treatment efficacy in a treatment-naive population; however, the choice of baseline in the FACOMS group could introduce indication bias due to unmeasured confounding factors. While the timing of treatment initiation was not an issue, the primary analysis cohort — patients who had completed 48 weeks of follow-up — may have differed systematically from patients in the FACOMS group. Limited bias due to exposure or outcome misclassification was noted; however, measurement error at year 3 could slightly favour omaveloxolone. Propensity score–matching variables were sufficient, and diagnostic results showed comparability between the FACOMS and MOXIe cohorts. However, the estimation of progression at 3 years relied on a missing at random (MAR) assumption, with missing outcome analyses not provided, raising concerns about dropout due to AEs.
Conclusions
Evidence from 1 phase II RCT suggests that, compared to placebo, omaveloxolone likely results in slower decline in neurologic function, as measured by mFARS scores over 48 weeks in patients with FA aged 16 years and older. It likely results in improvements in ADL measures, such as activities involving upper limb function and mobility, and may reduce the frequency of falls. While this is suggestive of functional relevance, with the clinical experts suggesting 2-point and 1-point potentially meaningful thresholds for mFARS and ADL, respectively, the actual clinical significance of these observations is uncertain due to the lack of an established minimal clinically important difference in mFARS and ADL scores. Other functional outcomes, including upper limb function, mobility, and frequency of falls, did not show significant improvements with treatment.
Comparison with natural history controls suggest that the treatment benefit of omaveloxolone may be maintained for up to 3 years. However, these studies do not support clear conclusions to be drawn about whether disease progression will continue to be slower among patients treated with omaveloxolone than among matched controls. A long-term extension study suggests that benefit may be observed within the first year, given that treatment-naive patients were observed to have better responses. However, these findings are limited by the open-label design and potential selection bias, and the magnitude of these group differences is uncertain. The safety profile appears manageable, with liver enzyme elevations being the primary concern requiring monitoring. Common adverse effects, including gastrointestinal symptoms and fatigue, were generally mild to moderate in severity.
Important evidence gaps remain, particularly regarding efficacy in pediatric patients (who account for a significant proportion of newly diagnosed cases), efficacy in patients with more advanced disease or significant cardiac involvement, and long-term comparative efficacy (i.e., beyond 3 years). Additionally, while the mFARS captures aspects of neurologic function, there is limited evidence for outcomes identified as important by patients, such as HRQoL, fatigue, and ability to maintain independence. Given that FA is a progressive, debilitating condition with no approved disease-modifying treatment, even a modest slowing of progression could be meaningful to patients. However, uncertainty remains about the magnitude and durability of benefit.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of omaveloxolone, administered at a recommended dose of 150 mg orally once daily (as 3 capsules of 50 mg each), for the treatment of FA in adults and adolescents aged 16 years and older.
Disease Background
The contents of this section have been informed by materials submitted by the sponsor and by clinical expert input. The following information has been summarized and validated by the review team.
FA is a rare autosomal recessive ataxia caused by loss-of-function mutations from trinucleotide repeat expansions in the FXN gene located on chromosome 9q13.1,2 The majority of patients have an expanded guanine-adenine-adenine (GAA) trinucleotide repeat in intron 1 of both alleles of the FXN gene.2 A longer GAA trinucleotide repeat translates to more FXN silencing, leading to an early onset of disease and rapid disease progression.1,12 FA accounts for up to one-half of all hereditary ataxia cases.2 It occurs in approximately 1 in 30,000 to 1 in 50,000 individuals,2 and the GAA triplet repeat expansion that causes FA is found only in individuals of European, North African, Middle Eastern, or Indian origin.2 Estimates based on 2010 Canadian data suggest that there are 300 to 750 patients with FA in Canada. More recent consultations with Ataxia Canada suggest that the number may be as high as 1,000.5
Common neurological symptoms to present at disease onset include gait instability (i.e., ataxia), which may manifest as clumsiness and/or increased falls, or dysmetria, which may manifest as problems with hand skills and fine motor tasks. While less common, patients may also present with sensory loss or dysarthria.6 Patients may also present with non-neurologic symptoms (i.e., cardiac involvement, diabetes mellitus, and skeletal abnormalities); however, this presentation is much less common, accounting for fewer than 10% of cases.6,7 Onset of FA typically occurs in childhood or adolescence, generally appearing between 5 years and 20 years of age,8 with a mean age at onset of 10 years to 15 years. Age at disease onset is inversely correlated with the number of GAA repeats1 and is an important predictor of overall disease severity and speed of progression.2
Diagnosis of FA typically occurs during childhood or adolescence.7,8 It is based on clinical suspicion of symptoms (the majority of which are neurologic) and confirmed by genetic testing.9 Other suggestive clinical findings include musculoskeletal features, such as scoliosis or pes cavus; hypertrophic nonobstructive cardiomyopathy; optic atrophy and/or deafness; and endocrinological features, such as glucose intolerance or diabetes mellitus.13 Testing options include single-gene testing or a multigene panel.13 Single-gene testing is targeted at identifying abnormal GAA expansion in intron 1 of FXN; however, if only 1 expanded allele is detected (which happens in approximately 4% of cases), sequence analysis of FXN is also performed.8,13 Multigene testing, in which other genes of interest are assessed in addition to FXN, is not recommended as a first-line strategy in typical cases. However, it may be helpful for atypical presentations.13 Genetic testing is readily available in Canada. Patients require a referral for a genetic test from a neurologist or geneticist.
Upon presentation of symptoms, a diagnosis of FA is typically confirmed within 2 years to 3 years.6 For patients who present with non-neurologic symptoms, the time to diagnosis may be longer because this is an uncommon presentation. In a European prospective, observational, natural history study of more than 600 patients with FA conducted by the European Friedreich's Ataxia Consortium for Translational Studies, the median time to diagnosis was 5 years for patients with non-neurologic onset of symptoms.6 Even after controlling for the effect of age at examination, age at onset, and presentation of symptoms before or after 1996 (when the FXN gene was discovered), a diagnostic delay of approximately 7 years was observed among patients.6 Another natural history analysis of patients with FA found that those with early onset declined 50% more quickly than patients with a more typical onset and twice as quickly as those with an intermediate onset.14 Results from a 4-year cohort analysis in the consortium study found that patients experienced progressively worse functioning and reduced ability to perform ADLs. Loss of ambulation was also identified as an important driver of disease progression, particularly contributing to declines in speech and upper limb function.15
FA considerably shortens life expectancy; patients with FA have a mean age of death of 37 years. Those with late-onset disease tend to have a longer life expectancy, with many living until the 40 years to 50 years.9,10 Common causes of death due to FA include cardiac complications (60% of deaths), pneumonia, aspiration, diabetic coma, stroke, and trauma sequelae.9,10,16 FA can result in significant morbidity and mortality. Because patients are at greater risk of developing comorbidities (e.g., cardiomyopathy and diabetes) and physical disabilities (e.g., scoliosis), they face greater limitations in their ability to engage in social and physical activities. They also experience more fatigue, pain, unsteadiness, emotional issues, and eating and drinking difficulties. They may feel an overall lack of control and experience losses of independence and autonomy, resulting in substantial impairments to their quality of life.
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
According to clinicians consulted by CDA-AMC for this review, there are currently no approved disease-modifying therapies available in Canada for the treatment of FA. Therefore, management of FA focuses on supportive, rehabilitative, and symptomatic measures. The latest set of FA treatment guidelines was published in 2022.17,18 At the time of publication, these guidelines stated that there was no approved pharmacological treatment for FA, but that research into potential therapeutic drugs had advanced considerably in the past 2 decades.18 The guidelines also stated that, despite significant progress in the search for disease-modifying drugs, the chronic, progressive course of FA cannot yet be significantly slowed;18 these guidelines were published before omaveloxolone became available. Management of FA has historically focused on symptomatic and supportive care.8,9,19 As FA progresses, affected individuals experience worsening gait and limb ataxia, motor weakness, reflex and sensory loss, impairments in speech and swallowing, hearing loss, reduced visual acuity, and bladder dysfunction.7 Currently, given that there are no pharmacological treatments for weakness or ataxia,8 management approaches focus on orthotics, adaptive equipment, walking aids, wheelchairs, and physical therapy.19 Some neurologic features can be managed through selective pharmacological therapies. For example, spasticity can be managed with baclofen, tizanidine, or botulinum toxin; neuropathic pain can be managed with gabapentin or pregabalin; and urinary urgency can be managed with anticholinergic drugs, such as oxybutynin.8
Non-neurologic features of FA can include cardiac complications, diabetes mellitus, and skeletal abnormalities (including scoliosis and foot abnormalities).7 There are no FA-specific medications to prevent or treat cardiac disease progression; therefore, cardiomyopathy is typically managed according to general cardiology guidelines,8 including with antiarrhythmic drugs, anti–cardiac failure medication, anticoagulants, and pacemaker insertion.13 Diabetes mellitus in individuals with FA may be treated with diet changes and, if necessary, oral hypoglycemic medications or insulin.7,13
Patients with FA may experience curvature of the spine and abnormalities of the feet. Nonsurgical interventions, such as physical therapy and bracing to stabilize the spine during growth, can be used to address these complications; however, in severe cases, surgery may be used.8,19 Orthopedic surgeons, neurologists, physiatrists, or rehabilitation specialists should be consulted when making decisions to address such complications. Vision and hearing problems can be treated with corrective devices.19 Speech therapy can help patients maximize their verbal communication skills.19
Input from clinicians consulted by CDA-AMC for the purpose of this review suggests that nonpharmacological interventions form the foundation of FA management. These include physiotherapy, occupational therapy, speech and language therapy, orthotic supports for gait and upper limb function, and adaptive equipment to help maintain mobility and independence. Routine cardiac evaluations and interventions (e.g., medications for cardiomyopathy, heart rhythm management) are an integral component of care, given the high prevalence of cardiac involvement in FA. Psychological support and genetic counselling should also be offered to patients and their families. Pharmacological options in current Canadian practice are limited to symptomatic and adjunctive treatments. The use of coenzyme Q10 and vitamin E, while not strongly evidence-based, is common due to their theoretical antioxidant benefits and relatively benign safety profiles. As per the clinical experts consulted by CDA-AMC, these are generally well-tolerated, over-the-counter interventions thought to potentially support mitochondrial function or reduce oxidative stress; however, high-quality evidence demonstrating meaningful clinical impact is lacking. No other drugs are routinely used in Canada specifically to slow or halt disease progression in FA. Any off-label use of drugs without a Health Canada indication is done on a case-by-case basis, typically informed by limited and inconclusive research data; this may sometimes be facilitated through special access processes.
Drug Under Review
The key characteristics of omaveloxolone for the treatment of FA are summarized in Table 3.
FA is associated with the inhibition of nuclear factor erythroid 2-related factor 2 (Nrf2), which is involved in cellular response and oxidative stress.20 The suppression of Nrf2 in FA results in oxidative damage, leading to cell death and tissue degradation. Activation of Nrf2 by omaveloxolone has been shown to restore Nrf2 levels, increase Nrf2 activity, rescue mitochondrial dysfunction, and restore redox balance.20 The precise mechanism by which omaveloxolone exerts therapeutic effects in patients with FA is unknown.20
The recommended dose of omaveloxolone is 150 mg (3 capsules of 50 mg each) taken orally once daily. The Health Canada–approved indication is for the treatment of FA in patients aged 16 years and older. The sponsor’s reimbursement request aligns with the Health Canada indication. Omaveloxolone has been approved by regulatory agencies in the US, European Union, and Switzerland. It is currently being reviewed by health technology assessment agencies in England (National Institute for Health and Care Excellence) and Australia (Pharmaceutical Benefits Advisory Committee).
Table 3: Key Characteristics of Omaveloxolone
Characteristic | Omaveloxolone |
|---|---|
Mechanism of action | An orally bioavailable triterpenoid analogue and potent activator of Nrf2. The drug selectively and reversibly binds to KEAP1, allowing for nuclear translocation of Nrf2 and transcription of its target genes. The precise mechanism by which the drug exerts therapeutic effects in patients with Friedreich’s ataxia is unknown. |
Indication | For the treatment of Friedreich’s ataxia in patients aged 16 years or older |
Route of administration | Oral |
Recommended dose | 150 mg (3 capsules of 50 mg each) |
Serious adverse effects or safety issues | Elevation of BNP, lipid abnormalities, increases in ALT and/or AST, headache, fatigue, nausea, diarrhea, oropharyngeal pain, back pain, muscle spasms, influenza, and decreased appetite |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; BNP = B-type natriuretic peptide; Nrf2 = nuclear factor erythroid 2-related factor 2.
Source: Draft product monograph for Skyclarys.20
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
CDA-AMC received 4 patient group submissions: 1 each from MDC, NAF, Ataxia Canada – Claude-St-Jean Foundation, and FARA. MDC supports people affected by muscular dystrophies and related muscle diseases in Canada. Its mission is to enhance the lives of those affected by neuromuscular disorders by providing support through all stages of disease progression so individuals have the appropriate tools to navigate the challenges. Its programs and services help patients and caregivers navigate the system. The organization also shares educational materials and evidence-based information on new treatments, provides financial assistance, and works toward improving the overall well-being and quality of life of patients and their family members. NAF was established specifically to help people with ataxia and their families, with a mission to accelerate the development of treatments by partnering with pharmaceutical companies, researchers, and clinicians in search of new treatments or a cure while providing vital services for families affected by ataxia. Ataxia Canada – Claude-St-Jean Foundation is a charity and community of women, men, adults, teenagers, and children with various forms of ataxia across the country. Its mission is to improve the well-being of people with familial ataxia and support research initiatives. FARA is a national, public, not-for profit organization dedicated to supporting scientific research in search of treatments and a cure for FA. It promotes public awareness and aligns with clinicians, patients, scientists, government agencies, pharmaceutical companies, and other organizations committed to finding a cure.
Neuromuscular service support staff at MDC identified and contacted adults living with FA and parents of children aged 16 years or older with FA to participate in a health care experience survey and semistructured virtual interviews. The survey was shared through e-blasts, personalized invites, and patient online groups in Canada. It gathered insights into diagnosis delays, gaps in treatment, emotional and social effects, and access to care and support systems. MDC received data from a total of 85 individuals (40 males and 45 females) who had received a confirmed diagnosis of FA between the ages of 16 years and 70 years from all provinces in Canada. NAF conducted a survey of its community members with FA in provinces across Canada. A total of 14 responses were received, including from 9 people with FA (living in British Columbia, Manitoba, and Ontario) and 5 caregivers (living in New Brunswick, Ontario, and Quebec). Ataxia Canada gathered the experiences of patients and caregivers in Canada through a combination of interviews and a survey. Of the 85 responders (who represented every Canadian province, including the Northwest Territories), most patients (72%) had experienced disease onset before the age of 15 years, and 50% had been diagnosed between the ages of 8 years and 15 years. Feedback from FARA regarding disease experience was drawn from 2 sources: a white paper that included views from parents of children with FA living in the US, highlighting the importance of pediatric inclusion in clinical trials; and a patient-focused drug development meeting that included 145 patients and caregivers (most of whom resided in the US, with 3% living in Canada) who were asked about their current disease state, experience with different symptoms, and perspectives on future treatments. Views regarding patients’ experiences with omaveloxolone were collected by FARA in January 2021 as part of the FA Community Response Letter, which was shared through the organization’s website, email lists, and social media channels. The letter was signed by 74,070 individuals from 118 countries; of these, 3,157 were from Canada. Written testimony was provided by individuals with varying durations of experience with FA (from very recent diagnoses to diagnoses 15 or more years ago). Respondents included individuals living with FA (n = 1,924); parents of children with FA (n = 688); those who had experience taking omaveloxolone through clinical trials (MOXIe trial, n = 70); and family members who had a loved one with FA who had participated in the MOXIe trials (n = 148).
FA is a recessive neurodegenerative disease caused by biallelic mutations in the FXN gene. It is a progressive, debilitating, life-shortening condition. People with the condition progressively lose ambulation and the ability to accomplish ADLs 2 decades to 3 decades following initial symptom onset. Ataxia Canada and MDC highlighted that patients with FA have significant challenges and/or impairments in coordination and/or balance, mobility and scoliosis, productivity at home and work, independence and social participation, and mental health. Additionally, FARA noted that patients and caregivers report fatigue and neurological symptoms (i.e., trouble with balancing, walking, and coordinating hands and/or arms and having regular falls) as having the most impact on daily quality of life. Currently, there is no approved medication for FA in Canada. NAF, Ataxia Canada, and FARA indicated that people with FA and their caregivers spend hours engaging in symptom-based therapies (such as occupational therapy, speech therapy, and physiotherapy), visiting medical specialists, and obtaining mobility devices, all of which can be challenging, expensive, and time-consuming. Ataxia Canada noted that individuals with FA use anticoagulation treatments for permanent, persistent, or paroxysmal atrial fibrillation as well as muscle relaxants (i.e., baclofen, tizanidine, benzodiazepines, dantrolene sodium, and/or intramuscular botulinum toxin injection) to manage symptoms. Current management strategies do not slow progression; this was noted as a major limitation to their effectiveness. MDC noted that the majority of the patients (n = 61) had no prior experience with treatments for FA; however, many reported using coenzyme Q10 and creatinine to manage symptoms or said they had been prescribed beta blockers to support heart function. Very few respondents (n = 3) had access to targeted treatments for FA through participation in clinical trials. MDC and NAF noted that health disparities for people with FA and their caregivers can arise because some do not have access to specialist clinics, live far away from ataxia centres or movement disorder clinics, and/or may find it difficult to travel as their disease progresses.
Four respondents (1 each from MDC and NAF, 2 from Ataxia Canada) shared their experiences with accessing omaveloxolone through clinical trials. All highlighted that being on the drug had slowed their disease progression (2 of these respondents were already wheelchair-dependent). In addition, 1 respondent highlighted feeling a slight reversal or progression of neurological symptoms such as maintaining balance, writing, swallowing, talking, and stamina. Mild side effects were noted at the beginning of treatment, such as rapid heart rate (infrequently) for 6 weeks and occasional diarrhea for 6 months. The viewpoints gathered by FARA of individuals with FA and parents of individuals who participated in the MOXIe trial indicated that patients who received omaveloxolone through the trial experienced slowing progression, longer retention of motor function (i.e., improved endurance, speech, and ability to walk, stay upright, and eat), and better ability to cope with fatigue. They reported minimal side effects; these included headache, nausea, diarrhea, and transient elevation of liver enzymes and cholesterol.
Currently, individuals require genetic testing to confirm the presence of biallelic mutations of the FXN gene. Ataxia Canada, NAF, and MDC highlighted that the majority of their respondents had undergone genetic testing. MDC and NAF highlighted that the process was easy, especially for people with family members who had been diagnosed with FA. While the process was often emotionally challenging, it provided clarity about the diagnosis. Additionally, MDC noted that some respondents found certain tests, such as nerve conduction studies or shock therapy, to be uncomfortable. Ataxia Canada noted the use of the mFARS scale to evaluate a patient’s status (i.e., assessment of their bulbar function, upper limb coordination, lower limb coordination, and ability to stand and walk). However, the organization highlighted that only a few clinicians may be familiar with using the scale and that it becomes less sensitive to change as patients transfer to wheelchair use. Therefore, use of the scale should be limited in such cases for determining treatment eligibility.
The patient groups agreed that there is a significant need for a treatment that can cure FA or reverse its effects. Patients are seeking a treatment that slows and/or stops disease progression, offers better symptom management, helps them regain and/or preserve their mobility, increases their energy levels, and improves their HRQoL and independence while reducing the physical and emotional burden on families and caregivers and preventing complications like scoliosis and diabetes. MDC noted that patients were concerned about the cost of the treatment (if it would not be covered), which could add to their existing financial burden (i.e., the costs involved in travelling to specialist centres and managing symptoms through various therapies). Furthermore, NAF highlighted those respondents with FA had a moderate to major impact on their and their family members’ quality of life.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). In this review, a total of 3 clinical experts were consulted (2 pediatric specialists and 1 who treats adults). In addition, as part of the review of omaveloxolone, a panel of 4 clinical experts from across Canada (3 pediatric specialists and 1 who treats adults) was convened to characterize patients’ unmet therapeutic needs, assist in identifying and communicating situations in which there are gaps in the evidence that could be addressed through the collection of additional data, promote the early identification of potential implementation challenges, gain further insight into the clinical management of patients living with the condition, and explore the potential place in therapy of the drug (e.g., potential reimbursement conditions). A summary of these discussions follows.
Unmet Needs
The experts emphasized that currently, there are no approved treatments in Canada that can halt, slow, or reverse the underlying neurodegenerative process in FA. The standard of care consists of nonpharmacological interventions (e.g., physiotherapy, occupational therapy, speech therapy, nutritional support, and adaptive devices) and symptomatic management (e.g., through coenzyme Q10 and vitamin E, management of cardiomyopathies, and treatments for spasticity); none of these offer a confirmed disease-modifying effect. These strategies do not alter the disease trajectory or prevent progressive loss of independence. Despite multidisciplinary involvement and comprehensive supportive care, patients and families continue to face the certainty of ongoing neurologic decline.
Consequently, the largest gap in the current therapeutic landscape for FA is the lack of any proven disease-modifying drug. A drug that could provide durable stabilization or slow the progression of the disease would represent a paradigm shift and meet a critical, longstanding unmet need in the management of FA.
Place in Therapy
Omaveloxolone would represent the first therapy in Canada to specifically target the underlying pathophysiology of FA rather than to solely manage its symptoms. This would mark a significant departure from the current treatment landscape, which is limited to supportive and symptomatic measures. As such, the introduction of omaveloxolone would be expected to fill a critical gap. This would position it as a first-line disease-modifying option for patients who meet the indicated age threshold (≥ 16 years) and have a confirmed genetic diagnosis of FA. Patients would not be required to try other treatments first, given that there are no alternatives that modify disease progression; omaveloxolone would likely be offered as soon as a patient is eligible. It would be used in conjunction with existing supportive strategies — not replacing them, but potentially providing the disease-targeted component. This also applies broadly to adults with FA who continue to experience progressive disability.
Patient Population
FA is a rare but genetically definable disorder. Diagnosis involves genetic testing for GAA repeats in the FXN gene and is usually straightforward. While rare ataxic mimics exist, misdiagnosis is uncommon. Patients are typically identified in childhood (often around the age of 11 years and sometime as early as 8 years, according to the pediatric clinical experts); by age 16 years, many have advanced manifestations. Although pediatric age of onset is the most common, a sizable proportion of patients are diagnosed in their twenties, thirties; diagnosis beyond these age groups is rare. Earlier onset tends to predict a more severe and rapidly progressive course, but there is wide heterogeneity. The age of disease onset is the most influential factor in determining the prognosis of patients. Patients who experience adult onset typically show slower progression, fewer cardiac complications, and longer survival. While trial data have focused on patients aged 16 years to 40 years, experts believe that all patients with genetically confirmed FA could, in theory, benefit, with those at the earlier stages (i.e., with lower disease burden) potentially showing more discernible stabilization. However, the experts acknowledge that there is no clinical evidence to substantiate the theoretical benefit in the pediatric population or in patients aged older than 40 years. Thus, the most suitable patients may be those who still retain considerable function (i.e., before there has been irreversible neurologic damage).
The clinical expert specializing in adult patients observed that those with advanced or late-onset disease may meaningfully benefit from slowed progression or preservation of upper limb or bulbar function. Nonambulatory patients should not be excluded; preserving upper limb or bulbar function could still be meaningful and translate into better communication and preserved autonomy that can be measured through some domains of the ADL tool (i.e., eating and personal hygiene). Although the magnitude of measurable benefit might be greatest in those with milder disease severity, all patients with FA technically need disease-modifying interventions.
Assessing the Response to Treatment
According to clinical experts, it is challenging to harmonize how treatment response is measured in clinical trials and in clinical practice. The clinical trial used the mFARS as a primary outcome. While this is a valid research tool, it is time-consuming and not routinely employed in standard clinic visits. Other trial measures (e.g., the 25-foot timed walk test and 9-HPT) are not standard in routine care and can be impractical or risky (e.g., timed walking tests might increase fall risk).
The clinical experts reported that in practice, clinicians rely on a combination of observed clinical stability (e.g., in gait, upper limb coordination, and bulbar function), reports from patients and/or their families, and annual or semiannual neurologic assessments. A clinically meaningful response might simply be a slowing of progression or a stabilization over 12 months rather than the improvement of any specific metric. “Buying time” is meaningful. The experts expressed concerns about relying on patient-reported outcomes alone, given the high unmet need in this patient population. Tools that are objective and standardized, but practical, are preferred.
Furthermore, given that no validated minimal clinically important difference for these scales exists in real-world practice, and that natural progression is heterogeneous, objective definitions of response may be elusive. However, clinical experts believe that a reduction or halting of expected annual declines (e.g., of approximately 2 mFARS points per year) could be considered a meaningful success. For patients who are nonambulatory, changes in ADL scores (e.g., ability to perform personal care independently or speak more clearly) may be more relevant. However, similarly, the experts were not aware of an established minimal important difference for ADL; they suggested that a change of 1 point could be clinically informative.
Discontinuing Treatment
According to clinical experts, the decision to stop therapy should be guided primarily by safety and tolerability rather than by a lack of improvement or disease progression alone. This perspective arises from the recognition that even a partial slowing of progression is valuable, and that discontinuing the only available treatment that could provide some stabilizing effect may be detrimental to the patient’s long-term function. AEs — particularly persistent or severe liver function abnormalities (i.e., ALT and/or AST elevations) — or severe allergic reactions might prompt discontinuation. However, absent severe toxicity, it would be hard to justify stopping therapy in a disease with no alternatives. The experts favoured individualized clinical judgment over rigid stopping rules.
However, while the experts did not necessarily view discontinuation based on efficacy outcomes as a favourable approach, they noted the need to objectively monitor the progress of patients. Lack of response could be determined after 2 years of treatment. Data on disease progression in individuals who discontinued omaveloxolone after prolonged use are not yet published, but they will start to become available, mostly from US-based cohorts. These data will be important in developing future guidance about omaveloxolone discontinuation.
Prescribing Considerations
According to clinical experts, diagnosis and treatment should be guided by specialists experienced in FA, such as neuromuscular or movement disorder neurologists. While no official companion diagnostic beyond genetic testing is necessary, confirmed genetic diagnosis is integral to ensuring appropriate patient selection. This therapy would likely be prescribed and monitored in specialized clinics capable of assessing neurologic function and coordinating care, rather than in general community settings.
Experts underscored the importance of regular liver function monitoring (e.g., due to elevated transaminases observed in the clinical trials). Similarly, the noted increases in LDL cholesterol may require additional monitoring or management (e.g., statins). Patients with cardiac issues (e.g., advanced cardiomyopathy) were largely excluded from the trials; however, the experts do not view such patients as necessarily ineligible. Rather, additional monitoring by a cardiologist may be required.
Requiring intensive or frequent specialized outcome measures for reimbursement (e.g., mFARS assessments) could limit equitable access, favouring patients who live closer to large centres with more resources. Experts acknowledged the need for quantifiable measures to monitor progression, but suggested annual intervals would be sufficient to establish a quantifiable measure of disease progression.
Additional Considerations
The experts noted that the efficacy of omaveloxolone beyond the study duration remains uncertain due to lack of robust evidence on long-term efficacy. In terms of evaluating outcomes important in clinical trials, the experts recognized that the mFARS and other ataxia scales are not routinely used in clinical practice. Frequency of falls, upper limb function, bulbar function, and stabilization of disease are more meaningful in practice; these are at least partially captured in the FARS-ADL scale. This scale tightly correlates with the mFARS, supporting the meaningfulness of mFARS and providing an additional useful standardized assessment tool. Patient quality of life measures, while potentially informative, may be less reliable because of bias and high patient expectations.
Experts highlighted strong patient demand: many individuals with FA may seek immediate access to omaveloxolone, given the historical absence of any disease-modifying option. The experts also noted that a number of therapeutics within the development pipeline may be approved in the coming years, potentially creating opportunities for combination or sequential therapy.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
A single submission was received from the Neuromuscular Disease Network for Canada. The network was launched in January 2000, bringing together Canada’s leading clinical, scientific, technical, and patient expertise to improve care, research, and collaboration in neuromuscular disease. Its mission is to improve the treatment of all neuromuscular diseases for all people in Canada. Input from 4 clinicians familiar with clinical trials of omaveloxolone for FA was gathered from 1-to-1 submissions and group discussions.
Treatment strategies for FA remain complex, requiring interdisciplinary management by a team of medical and health professionals. FA management addresses not only motor impairment, but also associated challenges, such as vision and hearing loss, psychological and cognitive issues, cardiomyopathy, diabetes, and skeletal abnormalities. Current treatment focuses on rehabilitation and managing complications. The primary therapeutic goals are to slow disease progression, preserve or enhance function, extend survival, and improve patient well-being.
The clinician group indicated that omaveloxolone is poised to be incorporated into the current treatment paradigm. The experts highlighted that all patients with FA would benefit from an intervention to slow disease progression, but that people with milder symptoms and in earlier stages of FA were most likely to benefit. In addition, the need applies to ambulatory patients, given that treatment may extend their ability to walk, and to nonambulatory patients, given that treatment could help them maintain upper limb function, speech for a longer period. Currently, there is not enough evidence to establish which patients are most likely to respond to omaveloxolone.
Regarding the outcomes used to determine a patient’s response, the clinician group indicated that standardized tests are used for neurologic exams (i.e., the mFARS) and functional assessments (i.e., the FARS-ADL). The use of simple clinician- and patient-related outcome measures like the CGIC and PGIC was also recommended. Measurements every 6 months in the first year and then annually were noted as reasonable and practical. In terms of a clinically meaningful response to treatment, the group noted that there should be an improvement in patient function and well-being (for example, this could be reflected by just a 1-point improvement in upright stability score, indicating better balance in the short-term and predicting delayed loss of ambulation).
The clinician group highlighted that omaveloxolone may be discontinued due to lack of efficacy (to be determined after a year, based on CGIC and PGIC) or side effects (i.e., evidence of organ dysfunction). Transient increases in aminotransferases without bilirubin changes and transient increases in NT-ProBNP without evidence of heart failure do not require discontinuation. Additionally, in the context of increased LDL (a common side effect of omaveloxolone), it was recommended that a statin be prescribed to manage cardiovascular risk factors rather than discontinuing omaveloxolone. Lastly, the clinician group highlighted that people with FA must be treated at specialized centres that offer comprehensive interdisciplinary care, regardless of omaveloxolone treatment. For patients without easy access to such centres, care should be managed by a neurologist knowledgeable about the disease and its management. Equipping providers with the necessary educational tools for prescribing and monitoring the drug will be crucial for ensuring effective treatment.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Are there other medications in the pipeline that show potential to slow the progression of FA? | There are no currently approved disease-modifying therapies for FA, and no other medications are well-established to slow FA progression. Experts noted that some therapeutics are currently in the development pipeline (e.g., vatiquinone) and could present future opportunities; however, they were unaware of any imminent pipeline drugs with clear evidence of disease modification. While research is ongoing globally, omaveloxolone would be the first of its kind in this space, if approved. |
Considerations for initiation of therapy | |
In the lifespan of a person with FA, when is genetic testing usually done, and who pays for this test? | The clinical experts noted that genetic testing is typically done at the time of suspected diagnosis, usually in childhood or adolescence. It is covered by provincial health care plans in Canada; however, this may vary by jurisdiction. |
Overall, should the initiation criteria for omaveloxolone reflect the inclusion criteria for MOXIe Part 2 trial? | The clinical experts noted that strict adherence to the MOXIe trial criteria may not be fully necessary. While genetic confirmation and age thresholds, as per Health Canada’s indication, should be respected, excluding certain subgroups (e.g., those with more severe disease) may not be desirable in clinical practice. The clinical experts noted the lack of clinical evidence of efficacy and safety for patients aged younger than 16 years, or 40 years and older. However, they also noted that theoretically, patients aged younger than 16 years or aged 40 years and older could also benefit from omaveloxolone although evidence of such theoretical benefit is lacking. The study only included patients with a baseline mFARS score between 20 points to 80 points. The experts noted that the lower bound (20 points) ensures only that patients are symptomatic and may not be a useful criterion for initiation. However, measuring progress in patients with a baseline mFARS score of more than 80 points could be challenging. |
In practice, what is the proportion of patients who are > 40 years of age? What proportion of these patients are diagnosed with FA > 40 years? | The clinical experts noted that late diagnosis in patients aged 40 years and older is rare. Most patients are identified well before this age. The proportion of patients aged 40 years and older living with FA exists but is relatively small, as diagnosis typically occurs in childhood or early adolescence. |
In practice, what is the proportion of patients who are younger than 16 years with a diagnosis of FA? | The clinical experts noted that the majority of patients diagnosed with FA are aged younger than 16 years, often around 10 years to 15 years of age. Pediatric populations comprise an important portion of patients under clinical care. |
Should the initiation criteria for omaveloxolone reflect the age thresholds in the MOXIe Part 2 trial? | While the trial included patients aged ≥ 16 years, the experts acknowledge that there will be pressure to consider use in younger patients. Given the limited data, it may be prudent to initially follow the age threshold from the trial (≥ 16 years); clinical trial evidence over time may support earlier use. |
If a patient with FA discontinues treatment (e.g., due to side effects), are they eligible for re-treatment? | The clinical experts indicated that there is no known contraindication to rechallenging after side effect resolution. If the reason for stopping (e.g., transaminase elevations) resolves and the patient and/or their family wishes to try again, reinitiation at the physician’s discretion may be reasonable. |
Considerations for continuation or renewal of therapy | |
In patients with FA, is there a definition of a full versus partial responder? How would you monitor continuous response? | No standardized definition of “full” or “partial” responder exists. Response may be interpreted as stabilization or slower disease progression. Monitoring would be through clinical judgment, patient function (gait, upper limb coordination, bulbar function), and possibly adapted objective measures every 6 months to 12 months rather than through the strict use of trial scales. |
Considerations for discontinuation of therapy | |
The recommendation by the sponsor is to assess patients for progression annually after starting treatment. What parameters would inform the discontinuation of omaveloxolone? | Persistent severe adverse events (e.g., significant liver enzyme elevations), severe allergic reactions, or patient preference might warrant discontinuation. Disease progression alone may not be a reason to stop, given that even partial stabilization is considered beneficial. |
If a patient is achieving therapeutic response on omaveloxolone at 150 mg/day, is a “drug holiday” a consideration? | There is no established rationale for a drug holiday if the patient is stable and tolerating therapy. Experts suggested that discontinuation could risk losing whatever benefit was gained, and reintroducing therapy without data on off-on effects is uncertain. |
Considerations for prescribing of therapy | |
Omaveloxolone is dosed at 150 mg/day. Apart from hepatic impairment — a condition for which there are lower dosing recommendations, according to the product monograph — are there any instances or circumstances in which a lower dose of omaveloxolone may be appropriate? | The experts did not identify other clear scenarios. The 150 mg/day dose resulted from the initial, dose-seeking part of the MOXIe trial, during which lower and higher doses were tested. Although numbers were low, the results clearly pointed to 150 mg/day as the optimal dose. Standard dosing is 150 mg/day unless hepatic impairment necessitates adjustments. Pediatric dosing or off-label modifications are not supported by current evidence. |
Given that patients with FA require a multidisciplinary approach, can you comment on where patients will be receiving care across Canada? | Patients are typically followed in specialty neuromuscular or movement disorder clinics, which are often affiliated with tertiary care centres. Care is multidisciplinary, involving neurologists, cardiologists, physiotherapists, occupational therapists, and genetic counsellors. This care model ensures comprehensive management and appropriate monitoring if omaveloxolone is introduced. Ideally, establishing additional sites in Canada of the Friedreich’s Ataxia Global Clinical Consortium would facilitate proper care and participation in clinical research of patients with FA. |
Generalizability | |
With the extensive exclusion criteria of the MOXIe Part 2 trial, what subgroups of patients with FA will this medication not be indicated (e.g., patients with active substance use or with concomitant medications that cause significant drug-drug interactions)? | While no subgroup is explicitly excluded in practice, those with severe cardiac disease or other major comorbidities were underrepresented in the trial. Clinicians would exercise caution, but no absolute exclusion is anticipated. Substance use or complex drug interactions would need individual assessment. |
If a patient is deemed “ambulatory” or “nonambulatory,” as per the definitions within the trial, should it affect access to omaveloxolone? | The experts do not support restricting treatment based on ambulatory status. Patients who are nonambulatory may still benefit (e.g., preserved upper limb or bulbar function). |
System and economic issues | |
As it is a rare drug, access will be variable, given jurisdictions may have a specific department managing requests for rare drugs. | For CDEC consideration. |
CDEC = Canadian Drug Expert Committee; FA = Friedreich’s ataxia; mFARS = modified Friedreich’s Ataxia Rating Scale.
Clinical Evidence
The objective of this clinical review is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of omaveloxolone 150 mg orally once daily for the treatment of FA in adults and adolescents aged 16 years and older. The focus will be on comparing omaveloxolone to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of omaveloxolone is presented in 4 sections, with our critical appraisal of the evidence included at the end of each. The first section, the Systematic Review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The review team’s assessment of the certainty of the evidence in this first section (using the GRADE approach) follows the critical appraisal of the evidence. The second section includes 1 sponsor-submitted, long-term extension study. The third section includes indirect evidence from the sponsor. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following are included in the review and appraised in this document:
1 pivotal study (an RCT) identified through a systematic review
1 long-term extension study
1 additional study addressing gaps in evidence.
Systematic Review
The contents in this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5.
Table 5: Details of Studies Included in the Systematic Review
Detail | MOXle Part 2 trial |
|---|---|
Designs and populations | |
Study design | Randomized, international, placebo-controlled, double-blind, parallel-group, phase II study |
Locations | 1 site in Australia, 1 site in Austria, 1 site in Italy, 1 site in the UK, and 7 sites in the US |
Patient enrolment dates | First patient enrolled: October 20, 2017 Last patient enrolled: October 31, 2019 |
Randomized (N) | N = 103 n = 51 in the omaveloxolone group n = 52 in the placebo group |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Omaveloxolone, 150 mg, administered orally once daily |
Comparator | Placebo administered orally once daily |
Study duration | |
Screening phase | Up to 60 days |
Treatment phase | 48 weeks |
Follow-up phase | 4 weeks |
Long-term extension phase | Currently ongoing. The interim analysis summarized in Section 3 represents data up to 144 weeks (with a DBL on March 24, 2022). The final analysis was planned for Q4 2024. |
Outcomes | |
Primary end points |
|
Secondary and exploratory end points | Key secondary:
Other secondary:
Exploratory:
|
Publication status | |
Publications | Lynch et al. (2020)21 Lynch et al. (2023)22 Hendrix et al. (2023)23 Study identifiers NCT02255435 RTA 408-C-1402 2015 to 002762 to 23 |
9-HPT = 9-hole peg test; ALT = alanine aminotransferase; AST = aspartate aminotransferase; CGIC = Clinical Global Impression of Change; CoQ10 = coenzyme Q10; DBL = database lock; eGFR = estimated glomerular filtration rate; FA = Friedreich’s ataxia; FA-ADL = Friedreich’s ataxia – Activities of Daily Living; mFARS = modified Friedreich’s Ataxia Rating Scale; PGIC = Patient Global Impression of Change; SF-36 = Short Form (36) Health Survey; ULN = upper limit of normal.
Source: MOXIe Part 2 trial Clinical Study Report.24 Details are from the sponsor’s Summary of Clinical Evidence.11
The MOXIe trial was conducted in 2 parts:
Part 1 was a randomized, placebo-controlled, double-blind, dose-ranging study to evaluate the safety, efficacy, pharmacokinetic (PK) activity, and pharmacodynamic (PD) activity of omaveloxolone at various doses (2.5 mg, 5 mg, 10 mg, 20 mg, 40 mg, 80 mg, 160 mg, and 300 mg) in patients with FA. Part 1 enrolled a total of 69 patients with a mean age of 26 years and a relatively equal proportion of female (54%) and male (46%) patients. The study concluded that a 160 mg dose of omaveloxolone was preferable for safety, efficacy, and PKs. Omaveloxolone is available as 2.5 mg, 10 mg, and 50 mg capsules. A dose of 150 mg was selected for further investigation in Part 2 because it was expected to provide a similar pharmacologic response to 160 mg while also reducing the number of capsules patients are required to take each day.25,26 A detailed summary of Part 1 of the MOXIe trial is not provided for this report because the direct comparative evidence for omaveloxolone 150 mg versus placebo is informed through Part 2.
Part 2 was a multicentre, randomized, placebo-controlled, double-blind, parallel-group study to evaluate the safety and efficacy of omaveloxolone 150 mg in patients with FA.
The key evidence informing the efficacy and safety of omaveloxolone is based on Part 2 of the MOXIe trial. The MOXIe trial was conducted at 11 sites across 5 countries. The trial consisted of a 48-week treatment phase followed by a 4-week follow-up phase after patients received their last doses; thus, patients’ safety follow-up visits occurred at 52 weeks. Following randomization, patients self-administered omaveloxolone orally once daily for 48 weeks. The 150 mg dose of omaveloxolone was determined from Part 1 of the trial based on reviews of efficacy, safety, PK, and PD data by a Data Safety Monitoring Board (DSMB) and the sponsor. A total of 103 patients were randomized into Part 2 of the MOXIe trial, with 51 patients in the omaveloxolone group and 52 patients in the placebo group. Patients were randomized 1 to 1 through a centralized interactive web response system (IWRS) to receive omaveloxolone 150 mg or placebo. Randomization was stratified by pes cavus status (severe pes cavus versus no severe pes cavus, determined using a flashlight test developed by the sponsor). Patients in the severe pes cavus stratum were not to comprise more than 20% of patients enrolled in the MOXIe Part 2 trial.
The number of patients with severe pes cavus was capped based on observations made during the MOXIe Part 1 trial. This decision was based on the observation that omaveloxolone did not lead to significant improvements in key end points, including mFARS and exercise testing, among patients with severe pes cavus. The lack of improvement was attributed to the requirement of these end points to achieve balance and coordination, which may be difficult for patients who have less surface area contact of their foot with the floor. The clinical diagnosis of pes cavus was defined as a loss of lateral support. Loss of lateral support was determined by having a patient stand barefoot and bear weight. If light from a flashlight could be seen under the patient’s arch, then the patient was diagnosed as having pes cavus, per the study’s definition.27
Patients who were enrolled into Part 1 and Part 2 of the MOXIe trial were eligible to roll into an OLE study to assess the long-term safety and tolerability of omaveloxolone in patients with FA.
Populations
Inclusion and Exclusion Criteria
Key inclusion criteria mandated that participants have genetically confirmed FA, exhibit a moderate level of functional disability (i.e., mFARS score from 20 to 80), and maintain stable exercise regimens. Additionally, eligible patients had to demonstrate adequate cardiac and renal function (i.e., left ventricular ejection fraction of at least 40% and estimated glomerular filtration rate of at least 60 mL/min/1.73 m2). Exclusion criteria included clinically significant cardiac disease (i.e., beyond the mild to moderate cardiomyopathy that is typically associated with FA). Furthermore, patients could be excluded if they were using antioxidant supplements or medications that could interfere with cytochrome P450 enzymes or if they had recently participated in other interventional studies.
Interventions
Patients randomized into the MOXIe Part 2 trial received either omaveloxolone 150 mg or placebo. Omaveloxolone was self-administered by patients and taken orally as 3 capsules of 50 mg once daily in the morning on an empty stomach. Patients tracked their doses using diaries and were instructed to record the date and time of each dose.
Because the study was double-blinded, all patients, investigators, site personnel, and laboratories with direct involvement in the conduct of the study (or their designees) were blinded to treatment assignments. The only individuals with access to treatment assignments for the MOXIe Part 2 trial included individuals who maintained the IWRS or DSMB, members of the unblinded statistical team providing data to and analyzing data for the DSMB, and safety personnel without direct involvement in the conduct of the study who were assigned to report unblinded data to regulatory authorities as required.
A list of permitted and excluded concomitant medications was prespecified. Permitted medications included antibiotics, daily multivitamins, pain medications, other medications for the management of comorbidities, and contraceptives. Excluded medications included other investigational therapies, anticoagulants (except baby Aspirin up to 81 mg), specified medications within 14 days of study day 1 (i.e., antioxidant supplements and antispasticity drugs), and specified medications within 7 days of study day 1 (i.e., herbal preparations, sensitivity substrates for cytochrome P450 2C8 or 3A4, moderate or strong inducers of cytochrome P450 3A4, and substrates for p-glycoprotein transporter). Patients requiring chronic medications were instructed to maintain their doses and dose schedules throughout the MOXIe trial period, as medically feasible. Those requiring intermittent medications were advised to avoid taking them on days when PK samples were collected, when medically feasible.
Outcomes
A list of efficacy end points assessed in this Clinical Review report is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on the outcomes included in the sponsor’s Summary of Clinical Evidence as well as on any outcomes identified as important to this review, according to the clinical experts consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, the selected end points were considered those most relevant to inform expert committee deliberations and the list of end points was finalized in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE. The available clinical trial assessing the efficacy and safety of omaveloxolone had a predefined time point for reported end points at week 48. Consequently, this time point was prioritized as the most clinically relevant. Outcomes that were determined to be the most clinically relevant and had potential applicability to clinical practice were prioritized. Prioritization of outcomes was based on input from clinicians, patients, sponsors, and health economics experts. Key secondary outcomes in the trial (PGIC at week 48 and CGIC at week 48) are presented in Appendix 1, but are not part of the outcomes that have undergone the GRADE process.
Table 6: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | MOXle Part 2 trial |
|---|---|---|
Change in mFARS | Week 48 | Primarya |
Change in performance on 9-HPT | Week 48 | Other secondary |
Change in performance on a 25-foot timed walk test | Week 48 | Other secondary |
Frequency of falls | Week 48 | Other secondary |
Change in FA-ADL | Week 48 | Other secondary |
Harms | Week 48 | Safety |
9-HPT = 9-hole peg test; FA-ADL = Friedreich’s ataxia – Activities of Daily Living; mFARS = modified Friedreich’s Ataxia Rating Scale.
aStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
Source: MOXIe Part 2 trial Clinical Study Report.24 Details are from the sponsor’s Summary of Clinical Evidence.11
Friedreich’s Ataxia Rating Scale
The mFARS includes 4 domains from the Friedreich’s Ataxia Rating Scale (FARS) examination (i.e., bulbar function, upper limb coordination, lower limb coordination, and upright stability),but excludes the neurologic section. The mFARS is calculated as the full neurologic FARS assessment minus section D (peripheral nervous system). A higher score on the FARS or mFARS examination signifies more severe physical impairment, and a reduction in FARS or mFARS score signifies improvement in functioning. The change in mFARS score from baseline (relative to placebo) was the primary end point of the MOXIe Part 2 trial. Previous studies assessing the natural history of FA suggest that upright stability is a key measure correlated to the progression of patient’s disease and ultimate loss of ambulation, while upper limb coordination is associated with progression among nonambulatory patients.14,28
9-Hole Peg Test
The 9-HPT is a brief, standardized, quantitative test of upper-extremity (arm and hand) function. Both the dominant and nondominant hands were tested twice (with 2 consecutive trials of the dominant hand followed immediately by 2 consecutive trials of the nondominant hand). Longer test times reflect more impairment in the patient’s upper-extremity function; thus, a decrease from baseline suggests improvement.
25-Foot Timed Walk Test
The 25-foot timed walk test is a quantitative mobility and leg function performance test based on the time (in seconds) needed to complete a 25-foot walk. Longer test times reflect more impairment in the patient’s ability to walk; decreases in time from baseline suggest improvement. If a patient used an assistive device for the 25-foot timed walk test on day 1, the same assistive device was used for all 25-foot walk assessments.
Falls Diary
Throughout Part 2 of the study, patients were instructed to record any instances of falls in a study diary. A fall was defined as “the patient unintentionally coming to rest on the ground or at a lower level.” Items tracked in the falls diary included, but were not limited to, the date and time of each fall, the location of the fall, the activity that preceded the fall, the perceived cause of the fall, and whether an injury was sustained during the fall. Patients were provided the fall diary at screening and were to record fall incidents from screening to week 48.
Activities of Daily Living
Patients were instructed to answer 9 questions on the ADL survey to assess 9 concepts: speech, swallowing, cutting food and handling utensils, dressing, personal hygiene, falling, walking, quality of sitting position, and bladder function. The total score can range from 0 (no ADL impairments) to 36 (most severe ADL impairments).
Harms
AEs, SAEs, withdrawal due to AEs, deaths, and AEs of special interest were reported.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
mFARS | The mFARS includes 4 domains from the FARS examination (bulbar function, upper limb coordination, lower limb coordination, and upright stability) but excludes the neurologic section that is not considered clinically meaningful (i.e., peripheral nervous system assessment). The mFARS is scored out of 93, with higher scores indicating greater impairment. | The overall validity and reliability of the mFARS were supported by Rummey et al. (2019),29 Rummey et al. 2020),30 and Tai et al. (2021).31 Validity Among a sample of 1,011 participants with FA, each item within the subscale (e.g., cough and modified bulbar function, sitting posture, and upright stability) correlated with the overall subscale (Pearson correlations [r] ranged from 0.32 to 0.90).29 The overall subscale intercorrelation ranged from 0.57 to 0.76 (modified bulbar and upper limb r = 0.63; modified bulbar and lower limb r = 0.57; modified bulbar and upright stability r = 0.55; upper limb and lower limb r = 0.68; upper limb and upright stability r = 0.66; lower limb and upright stability r = 0.76).29 Reliability A study consisting of 172 patients with FA measuring test-retest reliability observed an ICC of 0.95 (95% CI, 0.94 to 0.96).30 In another study, mFARS assessments were conducted by video with 19 individuals. The authors observed similar test-retest reliability in lower limb coordination (ICC = 0.96; 95% CI, 0.90 to 0.98), upright stability (ICC = 0.97; 95% CI, 0.93 to 0.99), and total mFARS (ICC = 0.97; 95% CI, 0.95 to 0.99). In addition, they evaluated a lower test-retest reliability for upper limb coordination (ICC = 0.86; 95% CI, 0.63 to 0.94) and bulbar function (ICC = 0.38; 95% CI, −0.57 to 0.76).31 Responsiveness No relevant information was found from the submitted references | |
9-HPT | 9-HPT is a brief, standardized, quantitative test of upper-extremity (arm and hand) function. Both the dominant and nondominant hands were tested twice (2 consecutive trials of the dominant hand followed immediately by 2 consecutive trials of the nondominant hand). Longer test times reflect more impairment on the patient’s upper-extremity function; thus, a decrease from baseline suggests an improvement. | No information was available for validity, reliability, or responsiveness. | |
25-foot timed walk test | The 25-foot timed walk test is a quantitative mobility and leg function performance test based on the time (in seconds) needed to complete a 25-foot walk. Longer test times reflect more impairment in the patient’s ability to walk; decreases in time from baseline suggest an improvement. If a patient used an assistive device for the 25-foot timed walk test on day 1, the same assistive device was used for all 25-foot walk assessments. | Validity In 20 individuals with FA, the 25-foot timed walk test had moderate correlation with the average daily step count (r = 0.585; P < 0.01).32 Reliability and responsiveness No data are available from the submitted references for patients with FA. | |
FA-ADL | 9 questions on the ADL survey assessed 9 concepts: speech, swallowing, cutting food and handling utensils, dressing, personal hygiene, falling, walking, quality of sitting position, and bladder function. The total score can range from 0 (no ADL impairments) to 36 (most severe ADL impairments). | Validity In a sample of 298 patients with ataxia across a representative spectrum of degenerative ataxias (FA [n = 57], RFC1 [n = 44], and SCA3 [n = 28]), FARS-ADL was correlated with FARS stage (spearman correlation: rho = 0.78; n = 259; P < 0.001), clinical disease severity (rhoSARA = 0.80), and patient-reported impairment (rhoPROM-ataxia = 0.69; rhoEQ5D-VAS = –0.37).6 FARS-ADL and SARA showed several item cross-correlations as well.33 Reliability No relevant information was found in the submitted references. Responsiveness The FA-ADL was sensitive to change across different ataxia genotypes within a trial-relevant time scale of only 1 year. The FA-ADL showed significant and specific progression only in patients with worsening PGIC (0.11 to 1.78; P = 0.027; n = 39) and stability in patients with stable PGIC (–0.80 to 1.06; P = 0.782; n = 39).33 | The minimal important change was 1.1 points, based on intraindividual variability among patients with stable PGIC.33 |
9-HPT = 9-hole peg test; ADL = activities of daily living; CI = confidence interval; FA = Friedreich’s ataxia; FA-ADL = Friedreich’s ataxia – Activities of Daily Living; FARS = Friedreich’s Ataxia Rating Scale; FARS-ADL = Friedreich’s Ataxia Rating Scale – Activities of Daily Living; ICC = intraclass correlation coefficient; MCID = minimal clinically important difference; MID = minimal important difference; mFARS = modified Friedreich’s Ataxia Rating Scale; PGIC = Patient Global Impression of Change; PROM = patient-reported outcome measure; SARA = Scale for the Assessment and Rating of Ataxia; SCA3 = spinocerebellar ataxia type 3; RFC1 = replication factor C subunit 1.
Statistical Analysis
Sample-Size and Power Calculation
Patients Without Severe Pes Cavus
The protocol sample-size calculations were based on 100 patients (i.e., the total planned enrolment), 80 of whom would be without severe pes cavus. A mixed model with repeated measures based on the 80 patients without severe pes cavus had approximately 85% power to detect a 2.0-point difference between omaveloxolone and placebo in change from baseline in mFARS at week 48 under the following model specifications:
████████ ████████████ ██████ ██ ███ ███ ███ ███ ███ ███ ██████ ████████ ████████ ██████████ █████████ ████ ███████████ █ ███████ ████████████ ██ ███ ████ ███████ ██ ███ Two-sided type I error rate of 0.05
a difference of 2.0 points between the change in mFARS in the omaveloxolone and placebo groups
an SD of change in mFARS of 3.5 points.
Patients With Severe Pes Cavus
███ ███████ ████ ██████ ███ █████ ████ ██ ████████ ███ █████████████ ███ █████ ██ ██████ █ ██████████ ██ ███ ██████ ██ ██████ ████ ████████ ██ ██████ █████ ███ ███████████ ███ ███ ███████ ██████ ███ █████ █████ ██████ ███████ ███████ ███████ ███ █████ ██ ████ ███████ ███ ████████ ██ ██ ███ ███ ██ ████████ ██████ █ ██████████ ███████ ███████████ ███ ████ ████ ████ ███████ ████ ██████████ ██████████████ ██ ███ █████ █████ ████ ██ ███ ████████ █████████ ██ ██████ ███ █████ ███ ███████ ██ ██████ █ ████ ██ ███████ ████████ ████ ██ ███████ ███████ ███ ██████████ ██ ██████ █ ███████ █████ ████████ ███ ████ ███████ ██ █████ ████ █ ██████████ █████ ██ ████ █████ ███ █████████ █████ ████ ███ ███████ ███ █████████ ██ ██████ ███ █████ ███ ███ ███████ ██████████.27
Statistical Testing
The primary objective of the trial was to assess change in mFARS score at week 48. Key secondary and secondary end points were tested hierarchically to maintain the family-wise overall type I error rate of 0.05 upon observation of statistical significance for the primary end point. Formal testing of the hierarchy continued as long as statistical significance was observed for each tested end point. If end points did not demonstrate statistical significance, formal testing of the subsequent end points was not to occur. Key secondary and secondary end points were tested in the following order:
PGIC at week 48 (key secondary)
CGIC at week 48 (key secondary)
change in 9-HPT at week 48
change in 25-foot timed walk test at week 48
frequency of falls over 48 weeks
change in peak work during maximal exercise testing at week 48
change in ADL score at week 48.
All other end points were exploratory and presented with nominal significance levels.
Post Hoc Analysis
It was observed in the study that, while the distribution of many baseline characteristics was similar between treatment groups, some imbalances occurred despite randomization. The cohort receiving omaveloxolone had characteristics consistent with slightly more advanced disease than the cohort receiving placebo: higher mFARS scores (a mean score of 40.9 points versus 38.8 points), longer GAA1 repeat lengths (a mean of 739.2 versus 693.8), and, most importantly, a greater proportion of patients with a history of cardiomyopathy (48% versus 29%). To assess the impacts of these imbalances on the primary end point, mFARS pos-hoc analyses included MMRM evaluations with covariates, analyses of covariance at week 48 with and without imputation, and tipping point analysis. Correlations with ALT and AST were explored for the FAS and all-randomized populations. Additionally, ADL post hoc analyses included distributions of total scores and changes from baseline to weeks 24, 36, and 48 for FAS and all-randomized populations.
Subgroup Analyses
Subgroup analyses were conducted for the primary efficacy end point: change in mFARS score at week 48. The analyses were conducted among subgroups that had a sufficient number of patients to warrant these analyses:
age: younger than 18 years, 18 years or older
sex: female, male
geographic location: US, other
ethnicity: non-Hispanic or Latino, Hispanic or Latino
race: white, nonwhite [wording from original source]
GAA1 repeat length greater than or equal to 675: yes, no.
Subgroup analyses of geographic location, ethnicity, and race were not conducted because the subgroups were not sufficiently large. An additional subgroup analysis of ambulatory status (i.e., ambulatory or nonambulatory) was conducted post hoc. Subgroup analyses were not adjusted for multiplicity.
Table 8: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Primary | ||||
Change in mFARS | MMRM with treatment group, time, interaction between treatment and time, and interaction between baseline mFARS and time | Study site, baseline mFARS | No imputation; model estimates are valid under a MAR assumption | Tipping point with multiple imputation, treatment-based multiple imputation, control-based multiple imputation, and the MMRM fit to the PP population |
Secondary | ||||
Change in performance on 9-HPT | MMRM | Study site, baseline 9-HPT | MAR | None |
Change in performance on 25-foot timed walk test | MMRM | Study site, baseline 25-foot timed walk test | MAR | None |
Frequency of falls | Poisson model to compute incidence rate of falls with a natural logarithm of time on study (days) included as an offset term | None | MAR | None |
9-HPT = 9-hole peg test; MAR = missing at random; MMRM = mixed model for repeated measures; mFARS = modified Friedreich’s Ataxia Rating Scale; PP = per protocol.
Source: MOXIe Part 2 trial Clinical Study Report.24 Details are from the sponsor’s Summary of Clinical Evidence.11
Analysis Populations
A summary of the analysis populations used in the MOXIe Part 2 trial are included in Table 9. The primary efficacy analyses were conducted using the FAS, while safety analyses were conducted using the safety population. Other analyses sets were used for exploratory and sensitivity analyses.
Table 9: Analysis Populations in the MOXIe Part 2 Trial
Study | Population | Definition | Application |
|---|---|---|---|
MOXIe Part 2 trial | Full analysis set | The full analysis set included all patients enrolled without severe pes cavus who had at least 1 postbaseline measurement, categorized by their randomized treatment group (i.e., whether or not they received the study drug). | The primary efficacy analysis was based on patients without pes cavus. |
All-randomized populationa | The all-randomized set included all patients who were randomized, categorized by their randomized treatment group (i.e., whether or not they received the study drug). | Descriptive analyses of efficacy are presented using the all-randomized population. | |
Pes cavus population | The population of patients with pes cavus included all patients in the severe pes cavus stratum, categorized by their randomized treatment group (i.e., whether or not they received study drug). | Descriptive analyses of efficacy are presented using the population of patients with pes cavus. | |
Safety populationa | The safety population included all patients who received at least 1 dose of the randomized study drug and was used for the evaluation of safety variables. Patients who received at least 1 dose of omaveloxolone were classified in the omaveloxolone group. Patients who received at least 1 dose of placebo and no dose of omaveloxolone were classified in the placebo group. | Safety analyses were based on all enrolled patients. | |
Per-protocol population | The per-protocol population was defined as patients without severe pes cavus who received the study drug through week 48 and had no major protocol deviation that could potentially affect the efficacy assessments. | A sensitivity analysis exploring the robustness of the primary full analysis set findings was based on the patients in the per-protocol population. |
aThis analysis set included patients with and without severe pes cavus.
Source: MOXIe Part 2 trial Clinical Study Report.24 Details are from the sponsor’s Summary of Clinical Evidence.11
Results
Patient Disposition
A summary of patient disposition in the MOXIe Part 2 trial is provided in Table 10. A total of 156 patients were screened for eligibility; of these, 103 patients were randomized, including 51 patients in the group receiving omaveloxolone and 52 patients in the group receiving placebo. Of the 103 patients, 20 patients had pes cavus (n = 10 in each group).
Among all patients, 94 patients (91%) completed treatment through week 48 of the study, including 44 patients out of 51 patients (86.3%) randomized to the group receiving omaveloxolone and 50 patients out of 52 patients (96.2%) randomized to the group receiving placebo. Seven patients (13.7%) in the group receiving omaveloxolone and 2 patients (3.8%) in the group receiving placebo discontinued study treatment due to AEs (7.8% versus 3.8%, respectively) and withdrawal by patient (5.9% versus 0%, respectively). Most patients (93%) completed the study follow-up through week 52.
The majority of protocol deviations occurring during Part 2 of the MOXIe trial were minor. A total of 22 patients in the group receiving omaveloxolone and 19 patients in the group receiving placebo had at least 1 major protocol deviation. The most common reason for a major protocol deviation was deviation from the study procedure (n = 13 in the group receiving omaveloxolone and n = 7 in the group receiving placebo).34 Other protocol deviations included those related to the informed consent form (n = 5 versus n = 8 in the omaveloxolone and placebo groups, respectively), eligibility criteria (n = 3 versus n = 7), use of excluded concomitant medications (n = 3 versus n = 2), drug dispensing (n = 1 versus n = 0), visit window (n = 1 versus n = 0), and other (n = 1 versus n = 0).34
Table 10: Summary of Patient Disposition From Studies Included in the Systematic Review
Patient disposition | FAS N = 83 | All-randomized population N = 103 | ||
|---|---|---|---|---|
Omaveloxolone N = 41 | Placebo N = 42 | Omaveloxolone N = 51 | Placebo N = 52 | |
Screened, N | NA | NA | 156 | |
Reason for being screened out, N (%) | NA | NA | 53 | |
Not meeting inclusion criteria | NA | NA | 53 | |
Randomized, N | NA | NA | 51 | 52 |
Safety, N | 41 (100) | 42 (100) | 51 (100) | 52 (100) |
FAS, N | 40 (97.6) | 42 (100) | 40 (78.4) | 42 (80.8) |
PP, N | 29 (70.7) | 32 (76.2) | 29 (56.9) | 32 (61.5) |
Population with pes cavus, N | 0 | 0 | 10 (19.6) | 10 (19.2) |
Completed treatment through week 48, N (%) | 35 (85.4) | 40 (95.2) | 44 (86.3) | 50 (96.2) |
Discontinued study treatment, N (%) | | ██████ | | █████ | 7 (13.7) | 2 (3.8) |
Reason for discontinuation, N (%) | ||||
Adverse event | | █████ | | █████ | 4 (7.8) | 2 (3.8) |
Withdrawal by patient | | █████ | || | 3 (5.9) | 0 |
Completed study follow-up through week 52, N (%) | ██ ██████ | ██ █████ | 45 (88.2) | 51 (98.1) |
Completed study follow-up through week 52 and completed treatment through week 48 | ██ ██████ | ██ █████ | 44 (86.3) | 50 (96.2) |
Completed study follow-up through week 52, but did not complete treatment through week 48 | | █████ | | █████ | | █████ | | █████ |
Discontinued study | | ██████ | | █████ | 6 (11.8) | 1 (1.9) |
Administrative reasons | | █████ | || | | █████ | || |
Withdrawal by patient | | █████ | | █████ | | █████ | | █████ |
FAS = full analysis set; NA = not applicable; PP = per protocol.
Source: MOXIe trial Part 2 Clinical Study Report.24 Details are from the sponsor’s Summary of Clinical Evidence.11
Baseline Characteristics
A summary of baseline characteristics relevant to the review is provided in Table 11; baseline characteristics were summarized for the FAS and all-randomized population. In general, baseline characteristics in the FAS were similar between the groups receiving omaveloxolone versus placebo in the MOXIe Part 2 trial. Patients had a mean age of approximately 24 years and were mostly white (98%). Patients had a mean weight of 67 kg and a mean body mass index of ██ kg/m2. Patients had a mean peak work of 1.13 watt/kg, ADL of 10, age of onset of 15.5 years, were approximately 4.8 years since onset, and had a mean GAA1 repeat length of 717 ████ ████ █████ ████ ██ ████████ (██%), with a GAA1 repeat length greater than or equal to 675. The majority of patients (93%) had ambulatory status, ████████ ██████ ████ █████ ██ ███ ████████ ████████ ████ ████ ██ █████████ ██████. Most patients also had a history of █████████ ███%) and a history of scoliosis (74%). Approximately ██% of patients had a history of sensory neuropathy; ██% of patients had a history of cardiomyopathy; 30% had a history of swallowing difficulties; and 23% had a history of scoliosis surgery.
Although randomization was conducted using IWRS, there were some imbalances in patient baseline characteristics. While the mean age in each treatment group was similar, the proportions of patients aged younger than 18 years versus 18 years or older differed; a greater proportion of patients in the group receiving placebo versus omaveloxolone were younger than 18 years of age (31% versus 17.5%, respectively), and a greater proportion of patients in the group receiving omaveloxolone versus placebo were aged 18 years or older (82.5% versus 69.0%, respectively). There was also a greater proportion of females in the group receiving omaveloxolone versus placebo (60.0% versus 33.35, respectively). The group receiving omaveloxolone also had a higher proportion of patients with a GAA1 repeat length greater than or equal to 675 (████% versus ████%, respectively), a history of cardiomyopathy (47.5% versus 28.6%), and a history of scoliosis surgery (30.0% versus 16.7%). Despite the relatively balanced reporting of use of assistive devices across both treatment groups, there was a higher proportion of patients in the group receiving omaveloxolone who required assistance with a walker (█████ versus ████%). A slightly higher proportion of patients in the group receiving placebo had a history of areflexia (████% versus ████%, respectively).
In the FAS and all-randomized population, the majority of patients (> 90%) had a relevant medical history event recorded at baseline. In the all-randomized population, the most common medical history preferred terms (occurring in > 25% of patients in the omaveloxolone group) (in the omaveloxolone group and placebo group, respectively) were █████████ ██████ ███ ███████ ██████████ ██████ ███ ███████ ███████ ██████ ███ ███████ ███ ████████ ██████ ███ ███████ Overall, the high proportion of comorbidities observed at baseline in both treatment groups, but particularly in the omaveloxolone group, highlights the considerable morbidity experienced by patients with FA.
Table 11: Summary of Baseline Characteristics From Studies Included in the Systematic Review
Characteristic | FAS population | All-randomized population | ||
|---|---|---|---|---|
Omaveloxolone (N = 40) | Placebo (N = 42) | Omaveloxolone (N = 51) | Placebo (N = 52) | |
Mean age at screening (SD) | 24.2 (6.48) | 23.6 (7.79) | 23.4 (6.08) | 24.1 (7.85) |
Age at screening | ||||
< 18 years | 7 (17.5) | 13 (31.0) | 9 (17.6) | 15 (28.8) |
≥ 18 years | 33 (82.5) | 29 (69.0) | 42 (82.4) | 37 (71.2) |
Sex | ||||
Female | 24 (60.0) | 14 (33.3) | 31 (60.8) | 17 (32.7) |
Male | 16 (40.0) | 28 (66.7) | 20 (39.2) | 35 (67.3) |
Ethnicity | ||||
Hispanic or Latino | 1 (2.5) | 3 (7.1) | 2 (3.9) | 3 (5.8) |
Not Hispanic or Latino | 39 (97.5) | 39 (92.9) | 49 (96.1) | 49 (94.2) |
Race | ||||
White | 40 (100.0) | 40 (95.2) | 50 (98.0) | 50 (96.2) |
Other [wording from original source] | 0 | 2 (4.8) | 1 (2.0) | 2 (3.8) |
Mean weight, kg (SD) | 68.9 (18.4) | 66.3 (17.9) | ████ █████ | ████ ████ |
Mean BMI, kg/m2 (SD) | ████ █████ | ████ ████ | ████ █████ | ███ █████ |
Mean peak work, watt/kg (SD) | 1.08 (0.54) | 1.18 (0.61) | 1.09 (0.58) | 1.23 (0.62) |
Mean mFARS (SD) | 40.94 (10.39) | 38.77 (11.03) | 40.84 (10.15) | 37.94 (10.77) |
Mean ADL (SD) | 10.74 (4.77) | 9.87 (4.83) | 11.03 (4.49) | 9.85 (4.72) |
Mean age at FA onset, years (SD) | 15.9 (5.74) | 15.1 (5.34) | 14.8 (5.67) | 15.3 (5.31) |
Mean years since FA onset, years (SD) | ███ ██████ | ███ █████ | ███ ██████ | ███ █████ |
Mean GAA1 repeat length (SD) | 739.2 (214.88) | 693.8 (277.19) | 736.8 (206.80) | 676.2 (267.88) |
GAA1 repeat length ≥ 675, n (%) | ██ ██████ | ██ ██████ | ██ ██████ | ██ ██████ |
Ambulatory status, n (%) | 37 (92.5) | 39 (92.9) | ██ ██████ | ██ ██████ |
Ambulatory assistive devices, n (%) | ||||
Cane | | █████ | | ██████ | | █████ | | █████ |
Help of another person | || | | ██████ | || | | █████ |
Hold hand of caregiver | | █████ | || | | █████ | || |
Assistance from another person or trolley bag in balance-demanding situations | | █████ | || | | █████ | || |
Mobility scooter and wheelchair | || | | █████ | || | | █████ |
Rolling bag or rollator | || | | █████ | || | | █████ |
Uses scooter; also walker and wheelchair occasionally | | █████ | || | | █████ | || |
Walker | | ██████ | | ██████ | ██ ██████ | | ██████ |
Walking pole | || | | █████ | || | | █████ |
Wheelchair | | ██████ | | ██████ | ██ ██████ | | ██████ |
None | ██ ██████ | ██ ██████ | ██ ██████ | ██ ██████ |
History of cardiomyopathy, n (%) | 19 (47.5) | 12 (28.6) | 25 (49.0) | 15 (28.8) |
History of areflexia, n (%) | ██ ██████ | ██ ██████ | ██ ██████ | ██ ██████ |
History sensory neuropathy, n (%) | ██ ██████ | ██ ██████ | ██ ██████ | ██ ██████ |
History of swallowing difficulties, n (%) | ██ ██████ | ██ ██████ | ██ ██████ | ██ ██████ |
History of scoliosis, n (%) | 29 (72.5) | 32 (76.2) | 39 (76.5) | 37 (71.2) |
History of scoliosis surgery, n (%) | 12 (30.0) | 7 (16.7) | 16 (31.4) | 10 (19.2) |
ADL = activities of daily living; BMI = body mass index; FA = Friedreich's ataxia; FAS = full analysis set; GAA1 = guanosine-adenine-adenine 1; Max = maximum; mFARS = modified Friedreich’s Ataxia Rating Scale; Min = minimum; SD = standard deviation.
Source: MOXIe Part 2 trial Clinical Study Report.24 Details are from the sponsor’s Summary of Clinical Evidence.11
Exposure to Study Treatments
Study Treatments
█████ ██ ██████████ ███ ████████ ███ ██████████ ██ █████ █████████ ██████ ███ █████ █████ ████ ██ ████ ███ ███████ █████████ ███████ █ ██████████ ████ ██ ██████ ██ ██ █████████████ ███ █████████ ██ █ ████ ██ █████ █████ ██████ █ ████ ██ █████ █████ ██ ███ ███████ ██████ █ ████ ██ █████ █████ ████ ████████ ██ ████████ ████ █ ████ ██ █████ █████ ████████ █████ ███ ████ ██████████ ██ ████ █████████ ██████ █████ ████ █████ ██ ████████ ██ ███ █████████████ █████ ███ █████ ██ ████████ ██ ███ ███████ █████ ██████ ████ ███████████ ███ █████ ██████ ██ █████ █████████ ███ ███████ ██████ ████ █████████ ██████ ████ █ ████ ██ ███ ██ ███ █████████████ █████ ███ █████ ██ ███ ███████ ██████ ████ ████ ██ █████ ████ ████████ ██ ██████ █████████ █████ ██████ ███ ███████ █████████ ███████
Concomitant Medications and Cointerventions
███ ████████ ██ ████████ ██████ ██ ███ ████ ███ █████ ███████████ ███ ███ ██████ ██████████ ███ ██ █████ █ ███████████ ███████████ ██ ███ ██████ ███████████ ███ ████ ██████ ██████ ██ ████ ██ ████████ ██ ███ █████████████ ██████ ███████████ ███████████ ████████ ███ ███ █████████████ █████ ███ ███████ ██████ █████████████ ██████ ███ █████ ████ █████████ ██████ ███ ██████ ███ ███████████ ██████ ███ ███████
Table 12: Summary of Patient Exposure From Studies Included in the Systematic Review
Exposure | MOXle Part 2 trial | |
|---|---|---|
Omaveloxolone N = 51 | Placebo N = 52 | |
Cumulative dose dispensed (mg), mean (SD) | ██████ ████████ | || |
Number of doses received, mean (SD) | █████ ███████ | █████ ███████ |
Duration of treatment (days), mean (SD) | █████ ███████ | █████ ███████ |
Study drug compliance ≥ 80%, mean (%) | ██ ██████ | ██ ██████ |
Study drug compliance (%), mean (SD) | ██████ █████████ | █████ ███████ |
Total number of doses dispensed, mean (SD) | █████ ███████ | █████ ███████ |
Total number of doses returned, mean (SD) | ████ ███████ | ████ ███████ |
SD = standard deviation.
Source: MOXIe Part 2 trial Clinical Study Report.24 Details are from the sponsor’s Summary of Clinical Evidence.11
Efficacy
Change in mFARS Score at Week 48
In the primary analysis population (FAS), treatment with omaveloxolone improved mFARS score relative to placebo at week 48. Patients receiving omaveloxolone had a mean change from baseline of █████ ██████ ███ █████), while those receiving placebo had a mean change of █████ ██████ ███ █████). The MMRM estimate of mean difference in change from baseline between the groups receiving omaveloxolone and placebo was −2.40 points (95% CI, −4.31 to −0.50; P = 0.0141) favouring omaveloxolone. This was the primary outcome of the study and met the threshold for statistical significance.
In the all-randomized population, which included patients with severe pes cavus, omaveloxolone treatment improved mFARS scores by an estimated mean difference of −1.93 points relative to placebo (95% CI, −3.70 to −0.15; P = 0.0342).
Additional prespecified and post hoc analyses supported the primary analysis, demonstrating consistent benefits of omaveloxolone over placebo.
Change in Performance on 9-HPT
In the FAS population, patients receiving omaveloxolone had a mean change from baseline ██ ███████ ███ █ ███████), while those receiving placebo had a mean change ██ ███████ ███ █ ███████). The MMRM estimated mean difference in change from baseline between omaveloxolone and placebo was ██████ ████████████ ███ ███████ ██ ███████ | | ██████).
In the all-randomized population, which included additional patients, omaveloxolone treatment resulted in an estimated mean difference of ██████ 1 per second relative to placebo (95% CI, ███████ ██ ███████ | | ██████), indicating no significant difference between the groups.
Change in Performance on a 25-Foot Timed Walk Test
In the FAS population, omaveloxolone did not demonstrate a significant improvement in performance on the 25-foot timed walk test compared to placebo at week 48. Patients receiving omaveloxolone had a mean change from baseline in the reciprocal of average walk time of –██████ per second (SD ███████), whereas those receiving placebo had a mean change of –██████ per second (SD ███████). The MMRM estimated mean difference between omaveloxolone and placebo was 0.0058 per second (95% CI, ███████ ██ ██████; P = 0.4635).
In the all-randomized population, omaveloxolone treatment resulted in an estimated mean difference of ██████ per second relative to placebo (95% CI, ███████ ██ ███████ | | ██████), a nonsignificant difference between the treatment groups.
Frequency of Falls at Week 48
In the FAS population, treatment with omaveloxolone did not significantly reduce the frequency of falls compared to placebo at week 48. Patients receiving omaveloxolone reported a mean of ████ falls on treatment (SD █████), while those receiving placebo reported a mean of ████ falls (SD █████). The Poisson estimated difference in incidence rate of falls between omaveloxolone and placebo was █████ ████ ███ █████ ██ █████ | | ██████).
In the all-randomized population, omaveloxolone treatment led to an incidence rate difference of █████ compared to placebo (95% CI, █████ ██ █████ | | ██████), a nonsignificant difference in the frequency of falls between the groups.
ADLs at Week 48
In the FAS population, treatment with omaveloxolone improved ADL scores relative to placebo at week 48. Patients receiving omaveloxolone had a mean change from baseline of ██████ points (SD ██████), while those receiving placebo had a mean increase of █████ points (SD ██████). The MMRM estimated mean difference in change from baseline between omaveloxolone and placebo was –1.30 points (95% CI, █████ ██ █████; P = 0.0420) favouring omaveloxolone.
In the all-randomized population, which included patients with severe pes cavus, omaveloxolone treatment improved ADL scores by an estimated mean difference of █████ points relative to placebo (95% CI, –████ ██ █████ | | ██████).
Table 13: Summary of Key Efficacy Results From Studies Included in the Systematic Review
Variable | FAS population | All-randomized population | ||
|---|---|---|---|---|
Omaveloxolone (N = 40) | Placebo (N = 42) | Omaveloxolone (N = 51) | Placebo (N = 52) | |
Change in mFARS at week 48 | ||||
Number of patients contributing to the analysis, n (%) | 41 | 34 | ██ | ██ |
Baseline (points), mean (SD) | 40.94 (10.39) | 38.77 (11.03) | █████ ████ | ████ █████ |
Change from baseline (points), mean (SD) | ████ █████ | ███ ██████ | ████ █████ | ████ █████ |
Treatment group difference versus control, MMRM estimated means difference (95% CI) | −2.40 (−4.31 to −0.50) | Reference | ████ █████ ██ █████ | ███ |
P value | 0.0141 | Reference | ██████ | ███ |
Change in performance on 9-HPT at week 48a | ||||
Number of patients contributing to the analysis, n (%) | ██ | ██ | ██ | ██ |
Baseline (1/second), mean (SD) | ██████ █████████ | ██████ █████████ | ██████ █████████ | ██████ █████████ |
Change from baseline (1/second), mean (SD)b | ███████ █████████ | ███████ █████████ | ███████ █████████ | ███████ █████████ |
Treatment group difference versus control, MMRM estimated means difference (95% CI) | ██████ █████████ ███████ | Reference | ██████ █████████ ███████ | ███ |
P value | ██████ | Reference | ██████ | ███ |
Change in performance on a 25-foot timed walk test at week 48c | ||||
Number of patients contributing to the analysis, n (%) | ██ | ██ | ██ | ██ |
Baseline (reciprocal of average walk time [/sec]), mean (SD) | ██████ ████████) | ██████ █████████ | ██████ █████████ | ██████ █████████ |
Change from baseline (reciprocal of average walk time [/sec]), mean (SD)b | -██████ ████████) | -██████ █████████ | ███████ █████████ | ███████ █████████ |
Treatment group difference versus control, MMRM estimated means difference (95%CI) | 0.0058 (−███████ ██████) | Reference | ██████ █████████ ███████ | ███ |
P value | 0.4635 | Reference | ██████ | ███ |
Change in frequency of falls at week 48 | ||||
Number of patients contributing to the analysis, n (%) | ██ | ██ | ██ | ██ |
Number of falls while on treatment, mean (SD) | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
Treatment group difference versus control, incidence rate of falls, least square means difference (95%CI)d | ███ ███ ███ | ███ | ███ ███ ███ | ███ |
P value | ██████ | ███ | ██████ | ███ |
Change in ADLs at week 48e | ||||
Number of patients contributing to the analysis, n (%) | ██ | ██ | ██ | ██ |
Baseline (points), mean (SD) | 10.738 (4.7663) | 9.869 (4.8339) | ██████ ████████ | █████ ████████ |
Change from baseline (points), mean (SD) | ██████ ████████ | █████ ████████ | █████ █████ ███ | ██████ ████████ |
Treatment group difference versus control, MMRM estimated means difference (95% CI) | −1.30 ███████ █████) | Reference | ██ ███ ███ | ███ |
P value | 0.0420 | Reference | ██████ | ███ |
9-HPT = 9-hole peg test; ADL = activities of daily living; CI = confidence interval; FAS = full analysis set; mFARS = modified Friedreich’s Ataxia Rating Scale; MMRM = mixed model for repeated measures; SD = standard deviation.
aAnalysis based on reciprocal of average time, dominant hand.
bMean changes and P values estimated from MMRM analyses for 9-HPT, 25-foot timed walk test, peak work, and ADLs.
cAnalysis based on reciprocal of average walk time.
dComparison in the frequency of falls for the omaveloxolone versus placebo groups was estimated from the Poisson model, with the natural logarithm of time on study (days) included as an offset term.
eTotal ADL score.
Source: MOXIe Part 2 trial Clinical Study Report.24
Harms
Refer to Table 14 for harms data.
Adverse Events
Overall, all patients in the trial experienced at least 1 AE. The following AEs were reported in a greater proportion of patients in the omaveloxolone group than in the placebo group: nausea (33.3% versus 13.5%), abdominal pain (21.6% versus 5.8%), diarrhea (19.6% versus 9.6%), fatigue (21.6% versus 13.5%), ALT increase (37.3% versus 1.9%), AST increase (21.6% versus 1.9%), muscle spasm (15.7% versus 5.8%), back pain (13.7% versus 7.7%), and oropharyngeal pain (17.6% versus 5.8%). The following AE was reported in a greater proportion of patients in the placebo group than the omaveloxolone group: ligament sprain (15.4% versus 9.8%).
Serious Adverse Events
In the all safety population, SAEs were reported in 5 patients (9.8%) receiving omaveloxolone and in 3 patients (5.8%) receiving placebo. In the subgroup without pes cavus, SAEs occurred in | ████████ █████%) in the omaveloxolone group compared to | ████████ ████%) in the placebo group.
SAEs in the omaveloxolone group included anemia, various cardiac events (atrial fibrillation, palpitations, sinus tachycardia, ventricular tachycardia, and noncardiac chest pain), laryngitis, viral upper respiratory tract infection, and craniocerebral injury. In the placebo group, SAEs consisted of atrial fibrillation, gallbladder disorder, and ankle fracture.
Withdrawals Due to AEs
In the all safety population, treatment discontinuations due to AEs were more frequent in the group receiving omaveloxolone, with 4 patients (7.8%) in that group discontinuing treatment compared to 2 patients (3.8%) in the group receiving placebo. In the subgroup without pes cavus | | ████████ ████%) in the omaveloxolone group and | ████████ ████%) in the placebo group discontinued treatment due to AEs.
Mortality
No deaths were reported in either treatment group during the study.
Notable Harms
In the all safety population, elevations in liver enzymes were AEs of special interest associated with omaveloxolone treatment. Increases in ALT were reported in 19 patients (37.3%) receiving omaveloxolone compared to 1 patient (1.9%) on placebo. Similarly, AST elevations occurred in 11 patients (21.6%) in the omaveloxolone group versus 1 patient (1.9%) in the placebo group. In the subgroup without pes cavus, ALT increases were observed in ██ ████████ █████%) treated with omaveloxolone and | ███████ ████%) on placebo, while AST increases were reported in | ████████ █████%) receiving omaveloxolone compared to | ███████ ██████ on placebo. These findings indicate a higher incidence of liver enzyme elevations in patients treated with omaveloxolone compared to placebo.
Table 14: Summary of Harms Results From Studies Included in the Systematic Review
Adverse events | All safety population | Without pes cavus | ||
|---|---|---|---|---|
Omaveloxolone (N = 51) | Placebo (N = 52) | █████████ █ ██ | ██████████ █ ███ | |
Most common AEs, n (%)a | ||||
≥ 1 AE | 51 (100) | 52 (100) | ██ █████ | ██ █████ |
Gastrointestinal disorders | ||||
Nausea | 17 (33.3) | 7 (13.5) | ██ ██████ | | ██████ |
Abdominal pain | 11 (21.6) | 3 (5.8) | | ██████ | | █████ |
Diarrhea | 10 (19.6) | 5 (9.6) | | ██████ | | ██████ |
Vomiting | | ██████ | | ██████ | | ██████ | | █████ |
General disorders and administration-site conditions | ||||
Fatigue | 11 (21.6) | 7 (13.5) | | ██████ | | █████ |
Injuries, poisonings, and procedural complications | ||||
Contusion | 17 (33.3) | 19 (36.5) | ██ ██████ | ██ ██████ |
Excoriation | 13 (25.5) | 12 (23.1) | ██ ██████ | | ██████ |
Laceration | | ██████ | | ██████ | | ██████ | | ██████ |
Ligament sprain | | █████ | | ██████ | | █████ | | ██████ |
Investigations | ||||
ALT increase | 19 (37.3) | 1 (1.9) | ██ ██████ | | █████ |
AST increase | 11 (21.6) | 1 (1.9) | | ██████ | | █████ |
Musculoskeletal and connective tissue disorders | ||||
Arthralgia | | ██████ | ██ ██████ | | ██████ | | ██████ |
Muscle spasm | | ██████ | | █████ | | ██████ | | █████ |
Back pain | | ██████ | | █████ | | █████ | | █████ |
Nervous system disorders | ||||
Headache | 19 (37.3) | 13 (25.0) | ██ ██████ | ██ ██████ |
Dizziness | | █████ | | ██████ | | █████ | | █████ |
Respiratory, thoracic, and mediastinal disorders | ||||
Oropharyngeal pain | | ██████ | | █████ | | ██████ | | █████ |
Cough | | ██████ | | █████ | | █████ | | █████ |
Infections and infestations | ||||
Upper respiratory tract infection | 14 (27.5) | 15 (28.8) | ████ █████ | ██ ██████ |
Nasopharyngitis | | ██████ | | ██████ | | ██████ | | ██████ |
Metabolism and nutrition disorders | ||||
Decreased appetite | | ███████ | | █████ | | ██████ | | █████ |
SAEs, n (%)a | ||||
Patients with ≥ 1 SAE | 5 (9.8) | 3 (5.8) | | ██████ | | █████ |
Anemia | 1 (2.0) | 0 | | █████ | || |
Atrial fibrillation | 1 (2.0) | 1 (1.9) | | █████ | | █████ |
Palpitations | 1 (2.0) | 0 | | █████ | || |
Sinus tachycardia | 1 (2.0) | 0 | | █████ | || |
Ventricular tachycardia | 1 (2.0) | 0 | | █████ | || |
Noncardiac chest pain | 1 (2.0) | 0 | | █████ | || |
Gallbladder disorder | 0 | 1 (1.9) | || | | █████ |
Laryngitis | 1 (2.0) | 0 | | █████ | || |
Viral upper respiratory tract infection | 1 (2.0) | 0 | | █████ | || |
Ankle fracture | 0 | 1 (1.9) | || | || |
Craniocerebral injury | 1 (2.0) | 0 | | █████ | || |
Patients who stopped treatment due to AEs, n (%) | ||||
Patients who stopped | 4 (7.8) | 2 (3.8) | | █████ | | █████ |
Atrial fibrillation | || | | █████ | || | | █████ |
Erythrosis | || | | █████ | || | | █████ |
AST increase | | █████ | || | || | || |
ALT increase | | █████ | || | || | || |
Muscle spasms | | █████ | || | | █████ | || |
Ventricular tachycardia | | █████ | || | | █████ | || |
Rosacea | | █████ | || | | █████ | || |
Deaths, n (%) | ||||
Patients who died | || | || | || | || |
AEs of special interest, n (%) | ||||
ALT increase | 19 (37.3) | 1 (1.9) | ██ ██████ | | █████ |
AST increase | 11 (21.6) | 1 (1.9) | | ██████ | | █████ |
Liver injury | ██ | ██ | ██ | ██ |
AE = adverse event; ALT = alanine aminotransferase; AST = aspartate aminotransferase; SAE = serious adverse event; NR = not reported.
Note: Multiple events were counted once for each patient for each summary. AEs were coded using MedDRA Version 14.1.
aProportion greater than or equal to 10% in either treatment group in safety population.
Source: MOXIe Part 2 trial Clinical Study Report.24 Details are from the sponsor’s Summary of Clinical Evidence.11
Critical Appraisal
Internal Validity
The MOXIe Part 2 trial is a randomized, double-blind, placebo-controlled study appraised by the CDA-AMC review team as pivotal evidence to assess the use of omaveloxolone to treat FA exhibited several features that enhance its internal validity. The trial had an appropriate randomization design with clear inclusion and exclusion criteria; enrolled patients had genetic confirmation of FA. Central allocation through IWRS, coupled with a double-blind design and a placebo that matched the active treatment closely, ensured that patients, investigators, site personnel, and laboratory staff remained unaware of treatment assignments. It is possible that liver enzyme elevations in the group receiving omaveloxolone could have suggested treatment status, potentially leading to partial unblinding.
There were several imbalances in baseline characteristics across treatment groups in the trial. Specifically, the group receiving omaveloxolone had a higher proportion of males, a greater number of patients with a history of cardiomyopathy, higher baseline mFARS scores, and more individuals with GAA1 repeat lengths greater than or equal to 675. The imbalances in mFARS score, GAA1 repeat lengths, and cardiomyopathy history may suggest that patients in the group receiving omaveloxolone are at a more advanced stage of disease, potentially biasing efficacy against omaveloxolone. However, it is unclear if the higher proportion of males may create bias in favour of or against omaveloxolone.
Additional post hoc sensitivity analyses in the FAS population, performed to assess the impact of controlling for imbalances between the randomized cohorts in baseline disease characteristics (i.e., history of cardiomyopathy, GAA1 repeat length, and history of cardiomyopathy and GAA1 repeat length combined), showed a greater treatment effect than did the prespecified primary analysis. In addition, the prespecified subgroup analysis of mFARS scores based on sex suggests that male patients benefited more than female patients. This suggests that the imbalance in sex may bias the overall results against omaveloxolone.
Dropout rates were low overall, with 91% of participants completing the trial; reasons for discontinuation were clearly reported. However, discontinuations were more common in the group receiving omaveloxolone, generally due to AEs. This raises the possibility that missing data were not completely at random and that the MAR introduced bias in the results.
The use of mFARS as a primary end point was supported by its internal validity and reliability in measuring the physical performance of patients with FA; however, no minimal clinically important difference was established, nor is the measure routinely applied in clinical practice. The statistical analysis plan was prespecified and appropriate, employing MMRM methods and hierarchical testing procedures to control for multiple comparisons. Multiple sensitivity analyses supported the findings. The sample size was adequate to provide sufficient power for the primary outcome, based on the power calculation. However, it is unclear if that is the case for any outcomes beyond the primary outcome. The internal validity of secondary outcomes is further compromised by falling outside the statistical testing hierarchy. As such, all P values are considered nominal in nature.
External Validity
The clinical experts consulted during this review suggested that, aside from the exclusion of pediatric patients, the eligibility criteria of the MOXIe Part 2 trial were generally comparable to those applied to patients in Canada. However, the trial’s strict inclusion and exclusion criteria, including constraints around cardiac involvement, may have led to a cohort that was healthier than what is typically encountered in routine Canadian practice. Furthermore, and according to the pediatric clinical experts, most patients are diagnosed at an earlier age than 16 years (typically around 11 years). However, the clinical expert specializing in adult patients with FA expressed that the mean age of onset in the trial is representative of the population of patients in Canada. The exclusion of the pediatric population means there is no evidence of the safety and efficacy of omaveloxolone in that patient population. However, the approved indication is for patients aged 16 years and older. Finally, the eligibility criteria limited the patient population to those with mFARS scores ranging from 20 to 80. Therefore, considering the primary outcome, the generalizability of the results is limited to patients whose scores fall within this range.
The clinical experts noted that baseline characteristics were reflective of the patient population aged 16 years of age and older. However, the pediatric clinical experts also communicated that patients are typically diagnosed around the age of 11 years, which is lower than the mean age of onset in the MOXIe Part 2 trial (i.e., 15.5 years) and may suggest that patients in Canada could be at a more advanced stage than patients in the trial. On the other hand, the clinical expert specializing in adult patients expressed that the age of onset is representative of patients in Canada. Given these differing perspectives, the effect of uncertainty regarding the comparability of age of onset between trial participants and patients in Canada remains undetermined. Furthermore, the study did not include any sites in Canada, reducing the generalizability and applicability of the results to the Canadian practice. However, the clinical experts did not feel that the lack of Canadian sites is a significant limitation. This is due to the similarity of the patient population and treatment paradigm with other countries that had representing sites.
The study capped the number of patients with severe pes cavus in the study and excluded these patients from the FAS. The sponsor justified this decision on evidence from the MOXIe Part 1 trial that suggested patients with severe pes cavus may represent a different subtype of FA that likely interferes with the ability to perform assessments that require standing or pedalling. Clinical experts involved in this review suggested that all patients with FA have a certain level of pes cavus, which has not been suggested as clinically prognostic of FA prognosis. Results from the all-randomized population suggest that the inclusion of patients with severe pes cavus may lead to a decrease in the observed benefit of omaveloxolone compared to placebo; however, this may be a limitation of the mFARS as a measure of FA progression.
While the duration of the study is reasonable, at 48 weeks, the clinical experts noted that due to the heterogeneity of the disease course, a longer duration with a comparative efficacy outcome assessed at 2 years would have been appropriate. While the sponsor provided OLE studies, several limitations related to the design of OLE studies (outlined in the relevant section) prevent us from understanding the comparative efficacy of omaveloxolone over a period longer than 48 weeks.
The outcomes used in the MOXIe Part 2 trial were appropriate for a clinical study setting, but are not commonly implemented in clinical practice. According to the clinical experts, none of the outcomes implemented in the study are used in practice. Specifically, the primary outcome, mFARS, is not used in clinical practice. This may represent challenges in clinical implementation and assessment of treatment response, and further limits the interpretation of the study to current practice. Limitation of the generalizability of outcomes is reinforced by the lack of a clear and formal minimal important difference. However, the clinical experts supported the use of a 2-point threshold as a meaningful change in the mFARS score, based on estimates from 1 natural history study.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for the outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:35,36
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of the effect. We describe evidence of very low certainty as “very uncertain.”
For RCTs: Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, or publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and its location relative to the threshold for a clinically important effect (when a threshold was available) or to the null. In the case of the outcomes presented in this report, a clinically important effect of 2 points was determined for mFARS, while the target of certainty was assessed based on the location of the point estimate relative to the null.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for omaveloxolone versus placebo.
Long-Term Extension Study
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Study
One OLE study (the Study 1402 extension) was included to assess the long-term safety and tolerability of omaveloxolone in patients with FA following the completion of Part 1 or Part 2 of the MOXIe trial. The OLE is ongoing; therefore, the preliminary results presented reflect an interim analysis based on the data cut-off date of March 24, 2022.
Part 1 of the MOXIe trial was a randomized, placebo-controlled, double-blind, dose-ranging study to evaluate the safety, efficacy, PK, and PD activity of omaveloxolone at various doses (2.5 mg, 5 mg, 10 mg, 20 mg, 40 mg, 80 mg, 160 mg, and 300 mg) in patients with FA. It included 69 patients, of whom 54% were female and 46% were male; the mean age was 26 years. The study concluded that a 160 mg dose of omaveloxolone was preferred for safety, efficacy, and PKs.
In the OLE, patients received omaveloxolone (150 mg) once daily until the drug became available through commercial channels or until patient withdrawal, whichever occurred first. Day 1 of the OLE was defined as the first day on which treatment was dispensed to a patient following completion of Part 1 or Part 2 of the MOXIe trial. All visits were enumerated with respect to this definition of day 1. Patients were to be scheduled for in-person assessments during treatment in the OLE on day 1, week 4, week 24, and every 24 weeks thereafter. Patients were also assessed by telephone contact on day 14. The end-of-study visit was scheduled 30 days after the last dose. The OLE was under way during the COVID-19 pandemic. Risk mitigation strategies were implemented to protect the health of patients while maintaining compliance with good clinical practice.
Given that patients in Part 2 of the MOXIe trial were followed continuously, their eligibility for the extension was assessed at the week 52 visit, and a separate screening visit was not required. For patients in Part 1, a screening visit to confirm eligibility for the extension was completed.
Populations
The inclusion criteria for the OLE consisted of previous enrolment in and completion of either the MOXIe Part 1 or Part 2 trials without any major protocol deviations. The exclusion criteria specified that patients should not have any uncontrolled comorbidities or clinically significant abnormality. Patients who discontinued treatment early in Part 1 or Part 2 of the MOXIe trial were not eligible to enrol in the OLE.
Patients were discontinued from treatment with omaveloxolone if they reported elevated transaminases (i.e., ALT or AST > 8 × the upper limit of normal [ULN]; ALT or AST > 5 × ULN for more than 2 weeks; ALT or AST > 3 × ULN and total bilirubin > 2 × ULN; or ALT or AST > 3 × ULN), with the appearance of fatigue, nausea, vomiting, right upper quadrant pain or tenderness, fever, rash, and/or eosinophilia (> 5%).
Interventions
All patients enrolled in the extension were assigned to receive oral omaveloxolone once daily at a dose of 150 mg (3 capsules of 50 mg each). Patients were treated with omaveloxolone until the drug became available through commercial channels or until patient withdrawal, whichever was sooner.
Patients were permitted to receive the following concomitant medications:
antioxidant supplements
antispasticity drugs.
Patients taking medication chronically were to be maintained on those same doses and dose schedules throughout the study period, as medically feasible.
Medications not permitted during the OLE included:
sensitive substrates for cytochrome P450 2C8 or 3A4 (e.g., repaglinide, midazolam, sildenafil)
moderate or strong inhibitors or inducers of cytochrome P450 3A4 (e.g., carbamazepine, phenytoin, ciprofloxacin, grapefruit juice)
substrates for p-glycoprotein transporter (e.g., ambrisentan, digoxin).
Outcomes
The primary objective of the OLE was to assess safety through AEs, SAEs, use of concomitant medications, laboratory test results, vital sign measurements, and weight. Assessment of long-term efficacy was a secondary objective. Efficacy outcomes assessed during the OLE included mFARS, PGIC, CGIC, 9-HPT, 25-foot timed walk test, and FARS-ADL.
A detailed description of these efficacy end points is provided in the Systematic Review section.
Statistical Analysis
Because Part 1 of the MOXIe trial was dose-ranging, patients who were treated with omaveloxolone received doses ranging from 2.5 mg to 300 mg. Based on the half-life of omaveloxolone 160 mg observed in Part 1 of the MOXIe trial (i.e., approximately 26 hours ± 13 hours), patients were not expected to have any lingering omaveloxolone drug effects more than 8 weeks after the date of their last dose. The last patient enrolled in Part 1 of the MOXIe trial completed their last visit on June 13, 2017. Patients in Part 1 of the MOXIe trial received treatment for a short period of time (i.e., 12 weeks) and were then off the study treatment for at least 12 months before enrolling in the OLE. Therefore, patients who were enrolled in the OLE from Part 1 of the MOXIe trial were considered treatment-naive and included in the placebo-omaveloxolone group.
Results of the March 24, 2022, interim analyses are presented using summary tables. Safety was monitored for both patient populations: those who participated in Part 1 of the study or who were initially randomized to placebo in Part 2 (known hereafter as the placebo-omaveloxolone group), and those who were initially randomized to omaveloxolone in Part 2 (known hereafter as the omaveloxolone-omaveloxolone group). Clinical outcomes were also summarized for all patients combined (known hereafter as the overall omaveloxolone group).
For patients initially randomized to placebo in Part 1 or Part 2 of the MOXIe trial, baseline values were defined as the last nonmissing assessment before the first study drug administration in the OLE, unless otherwise specified. If the first study drug administration in the OLE occurred after the date of drug dispensation in the extension, the last measurement before the first study drug administration was considered the day 1 measurement for the calculation of baseline. For patients in Part 2, extension day 1 (week 52) efficacy and safety results were pulled from the Part 2 analysis datasets if patients were not rescreened before the extension study.
Data are summarized using descriptive statistics. Continuous data are presented with the number of patients, mean, SD, median, minimum, and maximum (where applicable). Categorical data are summarized using frequency counts and percentages. If the mFARS assessment was not completed, there was no imputation; the imputation rules applied only to partially missing data. Data missing due to the COVID-19 pandemic were not imputed.
Results
Patient Disposition
A summary of the patient disposition during the OLE is reported in Table 15. A total of 149 patients were enrolled in the OLE, including ██ patients from Part 1 of the MOXIe trial and ██ patients from Part 2 of the MOXIe trial. All patients received at least 1 dose of omaveloxolone and were included in the safety population. All ██ patients enrolled from Part 1 of the MOXIe trial were considered treatment-naive upon enrolment in the OLE and were included in the placebo-omaveloxolone group. Of patients recruited from Part 2 of the trial, ██ patients had previously been randomized to placebo and were included in the placebo-omaveloxolone group, while 43 patients had previously been randomized to omaveloxolone and were included in the omaveloxolone-omaveloxolone group. More patients from the placebo-omaveloxolone group terminated the treatment early, either by patient choice or due to an AE.
Treatment Modifications Due to the COVID-19 Pandemic
█████████████ ███ ███████████ ███ ██ ███ ████████ █████████ █████████ ██ ████████ ███████ █████████ ██ ████████ ███████ ██ ███ ████████████ █████ ███ █ ████████ ███████ ██ ███ █████████ ██████ ███ ████ ██████ ██████ ███ █████████ ████████████ ███ ██████████████ ████ ███ ███████ ██ ███████ ███████ ██████ ██ ███ ████████████ █████ ███ █████ ██ ███ █████████ ███████ █████ ██████████ ███████ ███ █████████ ████████████ ███ ██ ███ ████████ ████████ ████████ ███████ ██████ █████ ██ ███ ████████████ █████ ███ █ ██ ███ █████████ ███████ ████████████ █████████ ██████ █████ ██████ ██████ ███ █████ █████ ██████ ██████ ███ ████ ████████ ████████ ██████ ███ ██ ████████ ████ ██ ████ ███ █████████ ██ ████████ ████████ ████████ ██ ████ ██ █████████ ██ ████████ ████████ ███ ████ ██ █████████ ██ ████████ ████████
Patient disposition | Placebo-omaveloxolone group N = 106 n (%) | Omaveloxolone-omaveloxolone group N = 43 n (%) | Overall omaveloxolone group N = 149 n (%) |
|---|---|---|---|
Safety population in extension | |||
From Part 1 | ██ ██████ | || | ██ ██████ |
From Part 2 | ██ ██████ | ██ █████ | ██ ██████ |
Continuing treatment | ██ ██████ | ██ ██████ | ███ ██████ |
Terminated treatment early | ██ ██████ | | █████ | ██ ██████ |
Reason for terminating treatment early | |||
Withdrawal by patient | ██ ██████ | | █████ | ██ ██████ |
Adverse event | | █████ | | █████ | ██ █████ |
Other | | █████ | || | | █████ |
OLE = open-label extension.
Notes: Based on the half-life of omaveloxolone 160 mg in Part 1 (i.e., approximately 26 hours ± 13 hours), patients were not expected to have lingering drug effects more than 8 weeks after the date of their last dose. For this reason, all patients who participated in Part 1 of the study (who were then off the drug for a period of time ranging from 21 months to 49 months) were treated as placebo-omaveloxolone (i.e., omaveloxolone-naive) in the extension study analysis.
The placebo-omaveloxolone group consisted of patients initially randomized to placebo in Part 2 or participating in Part 1 of the study. The omaveloxolone-omaveloxolone group consisted of patients initially randomized to omaveloxolone in Part 2.
Source: MOXIe OLE Clinical Study Report.37
Baseline Characteristics
Overall, patient characteristics were similar across treatment groups (Table 16). The mean age of patients was approximately 26 years; most were aged 18 years or older (92.6%). The mean mFARS was 42.66 points, and the mean ADL score was 12.50 points. The mean age at onset of FA was 15.2 years, and the mean length of time since onset of FA was 11.0 years. The majority of patients were without pes cavus (69.8%); 73 patients (49.0%) had a GAA1 repeat length greater than or equal to 675. However, more patients in the placebo-omaveloxolone group were male (55.7%), and more patients in the omaveloxolone-omaveloxolone group were female (62.8%).
Table 16: Baseline Characteristics of the Safety Population
Parameter (units) | Placebo-omaveloxolone group N = 106 | Omaveloxolone-omaveloxolone group N = 43 | Overall omaveloxolone group N = 149 |
|---|---|---|---|
Baseline age (years), mean (SD) | ████ ██████ | ████ ██████ | ████ ██████ |
Baseline age group, n (%) | |||
< 18 years | | █████ | | █████ | ██ █████ |
≥ 18 years | ██ ██████ | ██ ██████ | ███ ██████ |
Sex, n (%) | |||
Female | ██ ██████ | ██ ██████ | ██ ██████ |
Male | ██ ██████ | ██ ██████ | ██ ██████ |
Ethnicity, n (%) | |||
Hispanic or Latino | | █████ | | █████ | | █████ |
Non-Hispanic or Latino | ███ ██████ | ██ ██████ | ███ ██████ |
Race, n (%) | |||
White | ███ ██████ | ██ ██████ | ███ ██████ |
Nonwhite [wording from original source] | | █████ | | █████ | | █████ |
Weight (kg), mean (SD) | ██████ ███████ | █████ ██████ | █████ ██████ |
BMI (kg/m2) | |||
Mean (± SD) | ██████ ███████ | █████ █████ | █████ █████ |
Diastolic blood pressure (mm Hg) | |||
Mean (± SD) | ████ ██████ | ████ ██████ | ████ ██████ |
Systolic blood pressure (mm Hg) | |||
Mean (± SD) | █████ ███████ | █████ ██████ | ███ ██████ |
Heart rate (beats/min) | |||
Mean (± SD) | ████ ███████ | ████ ███████ | ████ ██████ |
mFARS | |||
Mean (± SD) | █████ ████████ | █████ ██████ | ████ ██████ |
ADL, mean (± SD) | █████ ███████ | █████ ██████ | █████ █████ |
Age at FA onset (years) | |||
Mean (± SD) | ████ ██████ | ████ ██████ | ████ ██████ |
Years since FA onset (years) | |||
Mean (± SD) | ████ ██████ | ███ ██████ | ████ ██████ |
GAA1 repeat length | ████ | ████ | █████ |
Mean (± SD) | █████ ████████ | █████ ██████ | █████ █████ |
GAA1 repeat length ≥ 675, n (%) | |||
No | ██ ██████ | ██ ██████ | ██ ██████ |
Yes | ██ ██████ | ██ ██████ | ██ ██████ |
Pes cavus, n (%) | |||
No | ██ ██████ | ██ ██████ | ███ ██████ |
Yes | ██ ██████ | | ██████ | ██ ██████ |
Ambulatory status, n (%) | |||
Ambulatory | ██ ██████ | ██ ██████ | ███ ██████ |
Nonambulatory | ██ █████ | | ██████ | ██ ██████ |
History of cardiomyopathy, n (%) | |||
No | ██ ██████ | ██ ██████ | ██ ██████ |
Yes | ██ ██████ | ██ ██████ | ██ ██████ |
ADL = activities of daily living; BMI = body mass index; FA = Friedreich’s ataxia; GAA1 = guanosine-adenine-adenine 1; mFARS = modified Friedreich’s Ataxia Rating Scale; OLE open-label extension; SD = standard deviation.
Note: The placebo-omaveloxolone group consisted of patients initially randomized to placebo in Part 2 or participating in Part 1 of the study. The omaveloxolone-omaveloxolone group consisted of patients initially randomized to omaveloxolone in Part 2.
aThe sponsor confirmed that the reported mean (SD) for GAA1 repeat length did not include all 149 patients, given that there was variation across the MOXIe Part 2 trial sites in genetic testing kits and their capabilities; this difference resulted in some sites being less able to determine the specific GAA1 repeat length.
Source: MOXIe OLE Clinical Study Report.37
Exposure to Study Treatments
██ ███ ███████ ████ ██████ ███ ████ ██████████ ████ █████████ █████ ███ ████████ ███ █████████ ██ ████ █ ████ ████████ ██ █████ ████ ██████ ████ █████ ██████████ ███ ████ █████ ██████ ███ ████ ████ ██ ████████████ █████ ███ █████████ █████ ███████ ███████ ████ ███ ██ ████████ ████████ █████████████ ██████ ███ ███ ██ ██ ████ ███ ███ █████ ██ ████████ ████████ █████████████ ██ ██ ████ ███ ████████ ████ ███ ████ ████████ ██ █████ ████ ██ treatment.
Efficacy
Modified FARS
Based on the natural history of FA progression, mFARS scores are expected to increase over time, indicating a patient’s worsening condition and neurologic function. Patients are expected to experience average increases of 1.91 points, 4.24 points, and 5.58 points in mFARS after 1 year, 2 years, and 3 years, respectively.38 The results shown in Table 17 and Appendix 1 (Table 27) demonstrate that patients experienced an increase in mFARS scores over time, with patients in the omaveloxolone-omaveloxolone group experiencing a greater mean increase from baseline than patients considered treatment-naive at the start of the OLE (i.e., the placebo-omaveloxolone group); this can indicate decaying efficacy of treatment on disease progression. It should be noted that the sample sizes of the subgroups of patients with and without pes cavus were small, and that results at week 144 are based on less than half of the patients enrolled in the OLE.
Table 17: Results and Mean Change From Baseline in mFARS (Safety Population)
Visit | Statistic | All patients | |
|---|---|---|---|
Placebo-omaveloxolone group (N = 106) | Omaveloxolone-omaveloxolone group (N = 43) | ||
Baseline | n | ███ | ██ |
Mean (SD) | █████ ████████ | █████ ████████ | |
Median | █████ | █████ | |
Minimum to maximum | █████ ████ | ████ ████ | |
Week 48 CFB | n | ██ | ██ |
Mean (SD) | ████ ███████ | ████ ███████ | |
Median | ████ | ████ | |
Minimum to maximum | █████ ████ | █████ ███ | |
Week 48 percentage CFB | Mean (SD) | ████ ████████ | ████ ████████ |
Median | ████ | ████ | |
Minimum to maximum | ██████ ████ | ██████ ████ | |
Week 144 CFB | n | ██ | ██ |
Mean (SD) | ████ ███████ | ████ ███████ | |
Median | ████ | ████ | |
Minimum to maximum | █████ ████ | ██████ ████ | |
Week 144 percentage CFB | Mean (SD) | ████ ████████ | ████ ████████ |
Median | ████ | ████ | |
Minimum to maximum | ██████ ████ | ██████ ████ | |
CFB = change from baseline; mFARS = modified Friedreich’s Ataxia Rating Scale; OLE = open-label extension; SD = standard deviation.
Note: The placebo-omaveloxolone group consisted of patients initially randomized to placebo in Part 2 or participating in Part 1 of the study. The omaveloxolone-omaveloxolone group consisted of patients initially randomized to omaveloxolone in Part 2.
Source: MOXIe OLE Clinical Study Report.37
Activities of Daily Living
Results for the ADL survey are summarized in Table 18. Lower scores suggest improved function. Baseline values for the mean total ADL scores were ██████ in the placebo-omaveloxolone group and ██████ in the omaveloxolone-omaveloxolone group, indicating that patients in the placebo-omaveloxolone group may have had slightly worse function at baseline.
Changes from baseline in total ADL score for the placebo-omaveloxolone group at week 48, week 96, and week 144 were ██████ █████, and █████, respectively. Changes from baseline in total ADL score for the omaveloxolone-omaveloxolone group at week 48, week 96, and week 144 were ██████ ██████ ███ █████, respectively. Based on natural history data, the average changes at 1 year, 2 years, and 3 years are █████ █████ ███ ████, respectively.38 Among all patients, the changes from baseline in total ADL score at week 48, week 96, and week 144 were ██████ ██████ ███ █████. Compared to natural history data at 3 years, patients treated with omaveloxolone had reduced long-term declines in total ADL scores; however, the sample sizes decreased over time.
9-Hole Peg Test
A longer time on the 9-HPT reflects more impairment on one’s upper-extremity function as a result of the disease; thus, a negative change from baseline suggests an improvement. Based on natural history data, an average change at 1 year is expected to be −0.001.38 Patients in the both the omaveloxolone-omaveloxolone and placebo-omaveloxolone groups observed improvements from baseline Appendix 1 (Table 28).
25-Foot Timed Walk Test
Results for the timed 25-foot walk test are summarized in Appendix 1 (Table 29). Longer test times reflect more impairment of the patient’s ability to walk; thus, a decrease from baseline suggests improvement. At baseline, the mean value for the average reciprocal time (units in 1 per second) needed to complete the 25-foot walk test was ██████/second in the placebo-omaveloxolone group and ██████/second in the omaveloxolone-omaveloxolone group. At week 48, the mean changes from baseline were –██████/second in the placebo-omaveloxolone group and ███████/second in the omaveloxolone-omaveloxolone group. At week 96, the mean changes from baseline were –██████/second in the placebo-omaveloxolone group and | ██████/second in the omaveloxolone-omaveloxolone group. At week 144, the mean changes from baseline were –0.0294/second in the placebo-omaveloxolone group and ███████/second in the omaveloxolone-omaveloxolone group. The average change in natural history data at 1 year, 2 years, and 3 years ██ ██████ ██████ ███ █████, respectively.38 Patients in both groups experienced improvements from baseline in their 25-foot timed walk test results over time.
Table 18: ADL Test by Study Visit (Safety Population)
Visit | Placebo-omaveloxolone group (N = 106) | Omaveloxolone-omaveloxolone group (N = 43) | Overall omaveloxolone group (N = 149) |
|---|---|---|---|
Baseline | |||
n | ███ | ██ | ███ |
Mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
████ ██ | |||
n | ██ | ██ | ███ |
Mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
CFB, mean (SD) | █████ ████████ | ██████ ████████ | █████ ████████ |
████ ██ | |||
n | ██ | ██ | ███ |
Mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
CFB, mean (SD) | █████ ████████ | █████ ████████ | █████ ████████ |
████ ██ | |||
n | ██ | ██ | ███ |
Mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
CFB, mean (SD) | █████ ████████ | █████ ████████ | █████ ████████ |
████ ██ | |||
n | ██ | ██ | ███ |
Mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
CFB, mean (SD) | █████ ████████ | █████ ████████ | █████ ████████ |
████ ███ | |||
n | ██ | ██ | ███ |
Mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
CFB, mean (SD) | █████ ████████ | █████ ████████ | █████ ████████ |
████ ███ | |||
n | ██ | ██ | ██ |
Mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
CFB, mean (SD) | █████ ████████ | █████ ████████ | █████ ████████ |
CFB = change from baseline; omav = omaveloxolone; SD = standard deviation.
Note: The placebo-omaveloxolone group consisted of patients initially randomized to placebo in Part 2 or participating in Part 1 of the study. The omaveloxolone-omaveloxolone group consisted of patients initially randomized to omaveloxolone in Part 2.
Source: MOXIe OLE Clinical Study Report.37
PGIC and CGIC
The PGIC and CGIC are 7-point scales that require the patient and clinician, respectively, to assess how much the patient’s illness has improved or worsened relative to baseline. PGIC and CGIC scores of less than 4 represent some measure of improvement; scores greater than 4 represent some measure of worsening; and a score of 4 represents no change.
Among all patients, the placebo-omaveloxolone group’s mean PGIC scores were ███ at week 48, ███ at week 96, and ███ at week 144. For the omaveloxolone-omaveloxolone group, mean PGIC scores were ███ at week 24, ███ at week 48 ███ at week 96, and ███ at week 144. Among all patients, mean CGIC scores at week 48, week 96, and week 144 for the placebo-omaveloxolone group were ████ ████ ███ ███, respectively. For the omaveloxolone-omaveloxolone group, mean CGIC scores at week 48, week 96, and week 144 were ████ ████ ███ ████ respectively.
Delayed-Start Analysis
An additional delayed-start analysis was conducted to evaluate the persistence of effect with omaveloxolone treatment by comparing the difference in mFARS scores at the end of the MOXIe Part 2 trial to the difference in mFARS scores after 72 weeks in the OLE. This analysis was based on data from 82 participants in the OLE FAS (placebo-omaveloxolone group: n = 42; omaveloxolone-omaveloxolone group: n = 40). The mean baseline mFARS score was higher in the omaveloxolone-omaveloxolone group (mean = 40.9; SD = 10.4) than in the placebo-omaveloxolone group (mean = 38.8; SD = 11.0). A noninferiority analysis using a single MMRM model was conducted using data from the 48-week MOXIe Part 2 trial and the OLE through extension week 144. Nearly parallel trajectories were observed between the 2 groups except from the data at week 48, which had many missing mFARS assessments due to interruptions caused by the COVID-19 pandemic (Figure 1). The change in mFARS scores of individuals in the omaveloxolone-omaveloxolone group were maintained relative to their extension baseline through week 144. There was a least squares mean difference of −2.91 at the end of the delayed-start period. There was an observed sustained treatment effect with patients who were initially treated with omaveloxolone during the MOXIe Part 2 trial and continued with omaveloxolone during the OLE (i.e., the omaveloxolone-omaveloxolone group) compared to patients who were initially randomized to placebo during Part 2, then started omaveloxolone during the OLE (i.e., the placebo-omaveloxolone group). However, sample sizes declined in both groups over time, indicating a risk of attrition bias. It is unclear whether those who were not benefiting from the treatment dropped out, skewing the results.
Figure 1: Change From Baseline in mFARS (FAS)39
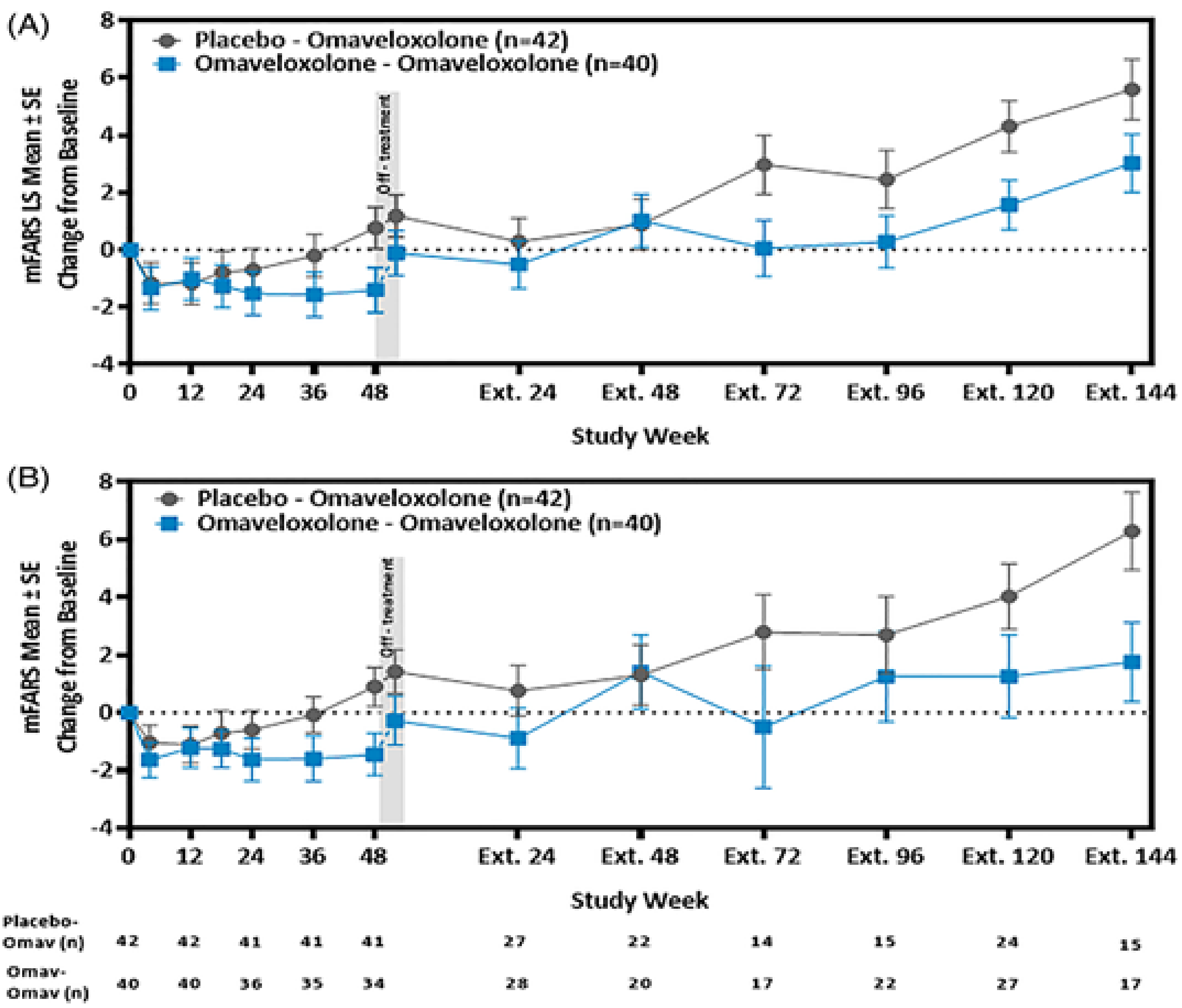
Ext. = extension; FAS = full analysis set; LS = least squares; mFARS = modified Friedreich’s Ataxia Rating Scale; MMRM = mixed model for repeated measures; omav = omaveloxolone; SE = standard error.
Notes: Part A of the figure depicts mean changes from baseline in mFARS score over time in the FAS for patients in the omaveloxolone-omaveloxolone (n = 40) or placebo-omaveloxolone (n = 42) groups, estimated using MMRM analysis.
Part B of the figure depicts observed mean changes from baseline in mFARS score over time in FAS for patients in the omaveloxolone-omaveloxolone (n = 40) or placebo-omaveloxolone (n = 42) groups.
Source: Lynch et al. Efficacy of Omaveloxolone in Friedreich's Ataxia: Delayed-Start Analysis of the MOXIe Extension. Mov Disord. 2023;38(2):313 to 320. Available from: https://movementdisorders.onlinelibrary.wiley.com/doi/10.1002/mds.29286. Reprinted in accordance with Creative Commons Attribution 4.0 International License (CC BY 4.0): https://creativecommons.org/licenses/by/4.0/deed.en.39
Harms
A summary of harms data from the OLE is provided in Table 19. As in the pivotal trial, most patients (96.0%) experienced at least 1 AE. The most frequently occurring AEs were coronavirus infection (18.8%), ALT increase (18.8%), headache (18.1%), upper respiratory tract infection (16.8%), nausea (16.1%), diarrhea (12.8%), fatigue (13.4%), arthralgia (12.1%), contusion (12.1%), excoriation (12.1%), and vaccination complication (10.1%). As in the pivotal trial, SAEs and AEs leading to permanent treatment discontinuation occurred infrequently; these were reported in 8.7% and 6.7% of patients, respectively. No deaths were observed during the OLE. Overall, long-term treatment with omaveloxolone was considered to be safe and tolerable for patients with FA.
Refer to Table 19 for harms data.
Table 19: Summary of Harms Results From the Long-Term Extension Study
Adverse events | Placebo-omaveloxolone group N = 106 n (%) | Omaveloxolone-omaveloxolone group N = 43 n (%) | Overall omaveloxolone group N = 149 n (%) |
|---|---|---|---|
Most common AEs, n (%)a | |||
Patients with at least 1 event | 103 (97.2) | 40 (93.0) | 143 (96.0) |
Gastrointestinal disorders | |||
Nausea | 17 (16.0) | 7 (16.3) | 24 (16.1) |
Diarrhea | 15 (14.2) | 4 (9.3) | 19 (12.8) |
Abdominal pain | 9 (8.5) | 7 (16.3) | 16 (10.7) |
Vomiting | 7 (6.6) | 5 (11.6) | 12 (8.1) |
Constipation | 3 (2.8) | 5 (11.6) | 8 (5.4) |
Gastroesophageal reflux disease | | █████ | | █████ | | █████ |
Dyspepsia | | █████ | | █████ | | █████ |
General disorders and administration-site conditions | |||
Fatigue | 14 (13.2) | 6 (14.0) | 20 (13.4) |
Pyrexia | | █████ | | █████ | | █████ |
Asthenia | | █████ | | █████ | | █████ |
Infections and infestations | |||
Coronavirus infection | 20 (18.9) | 8 (18.6) | 28 (18.8) |
Upper respiratory tract infection | 15 (14.2) | 10 (23.3) | 25 (16.8) |
Nasopharyngitis | 6 (5.7) | 7 (16.3) | 13 (8.7) |
Influenza | | █████ | | █████ | | █████ |
Urinary tract infection | | █████ | | █████ | | █████ |
Sinusitis | | █████ | | █████ | | █████ |
Injuries, poisonings, and procedural complications | |||
Excoriation | 16 (15.1) | 2 (4.7) | 18 (12.1) |
Contusion | 14 (13.2) | 4 (9.3) | 18 (12.1) |
Ligament sprain | 14 (13.2) | 3 (7.0) | 17 (11.4) |
Vaccination complication | 9 (8.5) | 6 (14.0) | 15 (10.1) |
Laceration | | █████ | | █████ | ██ █████ |
Joint injury | | █████ | | █████ | | █████ |
Joint dislocation | | █████ | | █████ | | █████ |
Foot fracture | | ████ | | █████ | | █████ |
Investigations | |||
ALT increase | 24 (22.6) | 4 (9.3) | 28 (18.8) |
AST increase | | █████ | | █████ | ██ █████ |
Weight decrease | | █████ | | █████ | | █████ |
Brain natriuretic peptide increased | | █████ | | █████ | | █████ |
Musculoskeletal and connective tissue disorders | |||
Arthralgia | 13 (12.3) | 5 (11.6) | 18 (12.1) |
Muscle spasms | 12 (11.3) | 2 (4.7) | 14 (9.4) |
Back pain | 6 (5.7) | 5 (11.6) | 11 (7.4) |
Pain in extremity | | █████ | | █████ | | █████ |
Myalgia | | █████ | | █████ | | █████ |
Musculoskeletal pain | | █████ | | █████ | | █████ |
Nervous system disorder | |||
Headache | 20 (18.9) | 7 (16.3) | 27 (18.1) |
Dizziness | | █████ | | █████ | | █████ |
Psychiatric disorders | |||
Depression | 4 (3.8) | 5 (11.6) | 9 (6.0) |
Insomnia | | █████ | | █████ | | █████ |
Renal and urinary disorders | |||
Micturition urgency | | █████ | | █████ | | █████ |
Respiratory, thoracic, and mediastinal disorders | |||
Cough | | █████ | | █████ | ██ █████ |
Epistaxis | | █████ | || | | █████ |
Skin and subcutaneous tissue disorders | |||
Eczema | | █████ | | █████ | | █████ |
Patients with ≥ 1 SAE, n(%) | |||
Patients with ≥ 1 SAE, n (%) | 8 (7.5) | 5 (11.6) | 13 (8.7) |
Cardiac disorders | |||
Cardiac failure, congestive | 0 | 1 (2.3) | 1 (0.7) |
Myocarditis | 1 (0.9) | 0 | 1 (0.7) |
Sinus tachycardia | 0 | 1 (2.3) | 1 (0.7) |
Gastrointestinal disorders | |||
Abdominal pain, lower | || | | █████ | | █████ |
Dental discomfort | | █████ | || | | █████ |
Nausea | | █████ | || | | █████ |
General disorders and administration-site conditions | |||
Asthenia | || | | █████ | | █████ |
Infections and infestations | |||
Coronavirus infection | | █████ | || | | █████ |
Gastroenteritis | 1 (0.9) | 0 | 1 (0.7) |
Influenza | | █████ | || | | █████ |
Patients who stopped treatment due to AEs, n(%) | |||
Patients who stopped treatment due to AEs, n (%) | | █████ | | █████ | ██ █████ |
ALT increase | | █████ | || | | █████ |
Fatigue | | █████ | || | | █████ |
Menstruation, irregular | | █████ | || | | █████ |
Alopecia | | █████ | || | | █████ |
AST increase | | █████ | || | | █████ |
Bipolar disorder | | █████ | || | | █████ |
Abdominal pain | || | | █████ | | █████ |
Constipation | || | | █████ | | █████ |
Frequent bowel movements | || | | █████ | | █████ |
AE = adverse event; ALT = alanine aminotransferase; AST = aspartate aminotransferase; OLE = open-label extension; SAE = serious adverse event.
Note: The placebo-omaveloxolone group consisted of patients initially randomized to placebo in Part 2 or participating in Part 1 of the study. The omaveloxolone-omaveloxolone group consisted of patients initially randomized to omaveloxolone in Part 2.
aAEs reported in greater than or equal to 3% of patients overall.
Source: MOXIe OLE Clinical Study Report.37 Details are from the sponsor’s Summary of Clinical Evidence.11
Critical Appraisal
Internal Validity
The long-term extension study was an open-label study without a comparator or placebo group. The lack of a control group precludes the ability to make causal statements about benefit and harm; increases in AE could not be interpreted relative to other interventions. The open-label nature of the study may increase the risk of bias in determining the magnitude of the subjective outcomes because the lack of blinding may influence patients’ expectations of the treatment, particularly for measures, including AEs, SAEs, and treatment-emergent AEs. Because completion of a pivotal trial was an eligibility criterion for enrolment in the extension study, patients who discontinued those trials due to AEs or lack of response were excluded. There is a risk of attrition bias, given that the number of patients contributing to the analyses declined steadily over time; final measures of the outcome are based on fewer than half of the enrolled patients. The COVID-19 pandemic also affected study visits and treatment continuity, with 14.8% of patients experiencing treatment interruptions.
External Validity
The clinical experts consulted for this review suggested that, aside from the exclusion of pediatric patients, the eligibility criteria of the OLE resulted in a study population comparable to patients in Canada. The trial’s strict inclusion and exclusion criteria, including constraints around cardiac involvement, may have led to a cohort that was healthier than what is typically encountered in routine Canadian practice. Furthermore, the study did not include any sites in Canada, which reduces the generalizability and applicability of the results to clinical practice in Canada.
Indirect Evidence
No indirect treatment comparisons were submitted for this review.
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
The limitations related to the lack of long-term comparative efficacy (given that the MOXIe Part 2 trial was 48 weeks in length) are outlined in Table 20. An RWE study by Lynch et al. (2023)40 used propensity score matching of patients from the FACOMS to provide an external control for the MOXIe trial and better understand the effects of omaveloxolone in FA. This study is summarized here.
Description of Studies
One propensity score–matched study was included to provide additional information about the longer-term efficacy of omaveloxolone compared to no treatment.
Table 20: Summary of Gaps in the Systematic Review Evidence
Gap in pivotal and RCT evidence | Study description | Summary of key results |
|---|---|---|
Because the MOXIe Part 2 trial lasted only 48 weeks, an OLE was conducted to evaluate the longer-term efficacy of omaveloxolone. In the OLE, all patients received active therapy; therefore, investigators conducted a comparative analysis using OLE data alongside data from historical control patients with FA. | The FACOMS is a large, ongoing, natural history study that began in 2003 to better understand FA in North America. It includes a large database of patients that can be used as an external control for comparison with clinical trial data; the FACOMS data were used in this manner to serve as an external control for patients in the MOXIe trial. A propensity score–matched analysis was conducted between the MOXIe extension data and the FACOMS natural history data to better understand the effects of omaveloxolone on FA at year 3.41 | Changes in mFARS scores were assessed and compared after 1 year, 2 years, and 3 years for patients in the MOXIe OLE study and matched patients in the FACOMS. At each time point, the mFARS scores were lower in patients treated with omaveloxolone compared to matched FACOMS controls. The results of this study suggest that omaveloxolone slows progression of disease by 55% at year 3 relative to patients in the natural history study.41 |
FA = Friedreich’s ataxia; FACOMS = Friedreich’s Ataxia Clinical Outcome Measures Study; mFARS = modified Friedreich’s Ataxia Rating Scale; OLE = open-label extension; RCT = randomized controlled trial.
Source: Sponsor’s Summary of Clinical Evidence.11
Table 21: Details of Studies Addressing Gaps in the Systematic Review Evidence
Detail | Propensity score–matched comparison of omaveloxolone treatment vs. FA natural history data (NCT03090789) |
|---|---|
Designs and populations | |
Study design | FACOMS (NCT03090789) is a large, ongoing natural history study following more than 1,250 patients with FA at 14 sites across North America, including 2 in Canada. Enrolled patients are evaluated annually using the FARS or mFARS tool, other neurologic outcomes, and quality of life measures. The time period of FACOMS overlaps with that of the MOXIe study. |
Enrolled, N | 272 (136 in the MOXIe extension and 136 in the FACOMS) |
Key inclusion criteria | To be included in this analysis, patients from the MOXIe OLE study and FACOMS must have had a baseline mFARS value available, at least 1 postbaseline mFARS value within 3 years after baseline, and values for the following characteristics: sex, baseline mFARS score, age at baseline, age at FA onset, and baseline gait score. All patients, regardless of presence of pes cavus, were included. |
Key exclusion criteria | Aligned with those of the MOXIe OLE study |
Drugs | |
Intervention | Omaveloxolone was administered to patients enrolled in the MOXIe OLE study. |
Comparator | None (absence of treatment) |
Outcomes | |
Primary end point | The primary end point of the FACOMS was change from baseline in mFARS score at year 3. |
Secondary end points | Secondary end points were changes from baseline in mFARS score at year 1 and year 2. |
Exploratory end points | None |
Notes | |
Publications | |
FA = Friedreich’s ataxia; FACOMS = Friedreich’s Ataxia Clinical Outcome Measures Study; FARS = Friedreich’s Ataxia Rating Scale; mFARS = modified Friedreich’s Ataxia Rating Scale; OLE = open-label extension; vs. = versus.
Source: Sponsor’s Summary of Clinical Evidence.11
Statistical Analysis
The data for this analysis are based on the interim analysis of the MOXIe extension trial (database lock: March 24, 2022). The data from the FACOMS were current as of March 24, 2021, and obtained from the Critical Path Institute.43
Two analysis populations were assessed:
the MOXIe extension population, which included patients from the MOXIe OLE study
the natural history population, which included patients from the FACOMS (as described in Table 21).
In addition, because the MOXIe OLE study included patients who were considered omaveloxolone-naive as well as previously treated patients, a different propensity score–matching approach was used for each analysis population. The primary analysis of this propensity score matching consisted of the natural history population and patients divided as follows:
The primary pooled group included all omaveloxolone-naive and -experienced patients.
The secondary placebo-omaveloxolone group included patients who had been enrolled in Part 1; these patients had had long off-treatment periods (of at least last 21 months), which meant they were considered treatment-naive. This group also included patients who had been randomized to placebo in the MOXIe Part 2 trial, then treated with omaveloxolone during the OLE study.
The secondary omaveloxolone-omaveloxolone group included patients who had been randomized to omaveloxolone in the MOXIe Part 2 trial and continued with omaveloxolone during the OLE.
Propensity Score Matching
The propensity score matching of patients from the FACOMS to those from the MOXIe OLE study was estimated using a logistic regression that was conditional on a given a set of covariates. The covariates used in the propensity score models included age, age of FA onset, sex, baseline mFARS score, and baseline gait score. These variables were used for matching because there was information available from both studies. It was noted that GAA1 repeat length may be a prognostic variable; however, it was not available for all patients. The FACOMS patients were matched to equivalent MOXIe OLE patients using optimal 1-to-1 matching without replacement. Diagnostics were then used to assess the similarity of the 2 groups and whether the propensity score model was adequately specified.
Primary Efficacy Analyses
The change from baseline in mFARS at year 3 was analyzed using an MMRM that included the following covariates: treatment group, baseline mFARS, visit, and interaction terms for visit-by-baseline and treatment group-by-visit. The model was fitted using restricted maximum likelihood, with a Toeplitz covariance structure (assuming that measurements taken closer together in time are more highly correlated than those taken farther apart). Mean changes from baseline, between-group differences in estimated changes from baseline, and associated P values estimated from the MMRM model are reported. Annual visits for the MMRM analysis were defined to align with the FACOMS assessment schedule. The mFARS assessment data collected closest to 1 year, 2 years, and 3 years after baseline were used for each of the annual assessments.
Secondary Efficacy Analyses
The same MMRM was used for both the primary and secondary analyses.
Results
Patient Disposition
A total of 136 patients had available data for key variables and were included in the propensity score–matched analysis. A total of 810 patients consented to have their data shared outside the core FACOMS; of these, 598 patients met the criteria for inclusion in the natural history study population and were considered potential matches for MOXIe OLE patients. After matching, 136 patients from the FACOMS natural history study were included. In total, the primary pooled population included 272 patients, with 136 patients from each of the MOXIe OLE and FACOMS.
Baseline Characteristics
A summary of baseline characteristics and covariates used for the propensity scores are reported in Table 22. Characteristics were balanced across the matched FACOMS (n = 136) and MOXIe extension (n = 136) groups. The trends observed in the primary pooled population (N = 272) were similar to those in the placebo-omaveloxolone (N = 190) and omaveloxolone-omaveloxolone (N = 182) groups (summarized in Supplementary Table 3 and Supplementary Table 4 of Lynch et al. [2023]).40 The mean baseline mFARS scores were similar across the matched FACOMS patients (41.0; SD = 16.1) and MOXIe OLE patients (42.2; SD = 12.6).
Table 22: Demographics and Baseline Characteristics Used as Covariates for Propensity Score Calculation (Primary Pooled Population)
Characteristic | Primary pooled population | |
|---|---|---|
Matched FACOMS N = 136 | MOXIe trial extension N = 136 | |
Age (years), mean (SD) | 26.2 (13.7) | 26.6 (7.3) |
Age at FA onset, mean (SD) | 15.2 (10.5) | 15.5 (5.3) |
Sex, n (%) | ||
Female | 70 (51.5%) | 70 (51.5%) |
Male | 66 (48.5%) | 66 (48.5%) |
mFARS, mean (SD) | 41.0 (16.1) | 42.2 (12.6) |
Gait (assessment 7 in FARS section E [upright stability]), mean (SD) | 2.7 (1.69) | 2.8 (1.36) |
Ethnicity, n (%) | ||
Hispanic or Latino | 6 (4.4) | 6 (4.4) |
Not Hispanic or Latino | 129 (94.9%) | 130 (95.6%) |
Not reported | 1 (0.7%) | 0 |
Race, n | 130 | 136 |
White, n (%) | 125 (96.2) | 133 (97.8) |
Nonwhite [wording from original source], n (%) | 5 (3.8) | 3 (2.2) |
Height (cm), n | 89 | 136 |
Mean (SD) | 165.1 (14.7) | 169.3 (10.4) |
Weight (kg), n | 95 | 136 |
Mean (SD) | 61.0 (20.7) | 69.1 (16.7) |
BMI (kg/m2), n | 89 | 136 |
Mean (SD) | 22.0 (5.7) | 24.0 (5.2) |
Systolic blood pressure (mm Hg), n | 82 | 136 |
Mean (SD) | 121.4 (15.0) | 121.1 (13.5) |
Diastolic blood pressure (mm Hg), n | 82 | 136 |
Mean (SD) | 73.2 (10.5) | 75.3 (8.7) |
Heart rate (beats/min), n | 82 | 136 |
Mean (SD) | 85.2 (15.4) | 79.8 (12.6) |
ADL total score, n | 124 | 136 |
Mean (SD) | 11.8 (5.9) | 12.5 (4.9) |
GAA1 repeat length, n | 129 | 119 |
Mean (SD) | 590 (246) | 721 (270) |
≥ 675, n (%) | 54 (41.9) | 66 (55.5) |
GAA2 repeat length, n | 121 | 116 |
Mean (SD) | 863 (232) | 728 (297) |
ADL = activities of daily living; BMI = body mass index; FA = Friedreich's ataxia; FACOMS = Friedreich’s Ataxia Clinical Outcome Measures Study; FARS = Friedreich’s Ataxia Rating Scale; GAA1 = guanosine-adenine-adenine 1; GAA2 = guanosine-adenine-adenine 2; mFARS = modified Friedreich’s Ataxia Rating Scale; SD = standard deviation.
Source: Sponsor’s Summary of Clinical Evidence.11
Exposure to Study Treatments
The mean follow-up durations were similar among patients in both studies. Among the matched FACOMS patients, the mean follow-up was 2.54 years (SD = 0.786), while among patients in the MOXIe OLE, it was 2.60 years (SD = 0.524).
Efficacy
A summary of mean change in mFARS scores after 1 year, 2 years, and 3 years is provided in Table 23. There was an observed improvement among patients treated with omaveloxolone in the MOXIe OLE trial compared to matched FACOMS patients at each time point. Patients from the MOXIe OLE trial experienced slower increases in mFARS scores than matched FACOMS patients (i.e., indicative of slowed disease progression).
Change From Baseline in mFARS Score at Year 3
After 3 years, the estimated changes from baseline in mFARS scores were 6.61 (SE = 0.65) in matched FACOMS patients and 3.00 (SE = 0.66) in OLE patients, representing a statistically significant difference of −3.61 (95% CI, −1.79 to −5.43) in favour of omaveloxolone (Figure 2). These results were consistent among the placebo-omaveloxolone and omaveloxolone-omaveloxolone groups (Table 23).
Figure 2: Change in mFARS From Baseline Over Time (Primary Pooled Population)
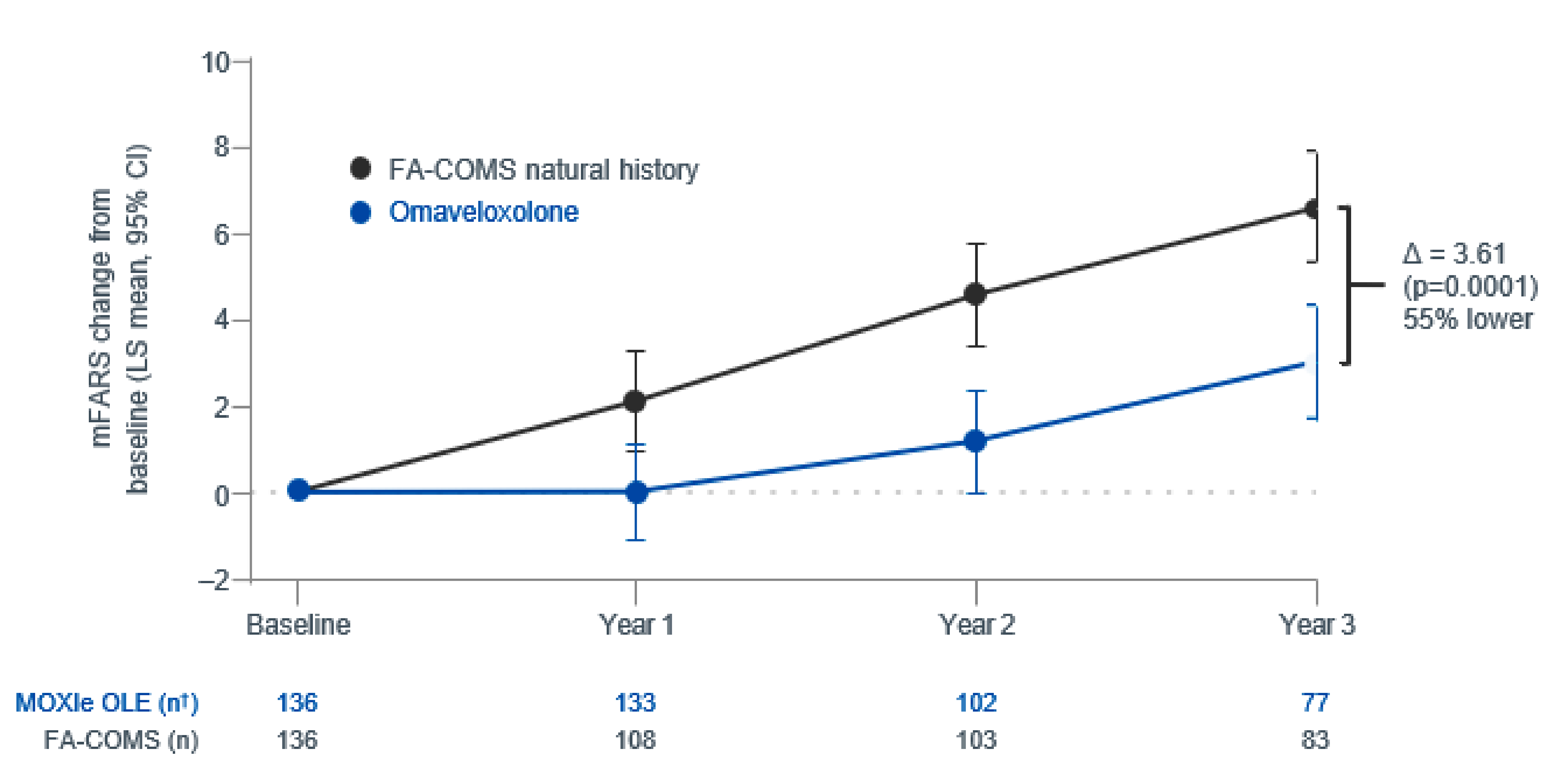
CI = confidence interval; FA-COMS = Friedreich’s Ataxia Clinical Outcome Measures; LS = least squares; mFARS = modified Friedreich’s Ataxia Rating Scale; MMRM = mixed model for repeated measures; OLE = open-label extension.
Note: LS refers to MMRM estimates.
Source: Sponsor’s Summary of Clinical Evidence.11
Table 23: Change in mFARS Over 3 Years
Population | Baseline | mFARS change from baseline | ||||||
|---|---|---|---|---|---|---|---|---|
Year 1 | Year 2 | Year 3 | ||||||
N | Mean (SD) | N | Meana(SE) | N | Meana (SE) | N | Meana (SE) | |
Primary pooled population | ||||||||
MOXIe extension study | 136 | 42.2 (12.6) | 133 | 0.015 (0.56) | 102 | 1.18 (0.59) | 77 | 3.00 (0.66) |
Matched FACOMS | 136 | 41.0 (16.1) | 108 | 2.11 (0.59) | 103 | 4.58 (0.59) | 83 | 6.61 (0.65) |
Difference | NA | NR | NA | −2.10 (0.18) P = 0.010 | NA | −3.41 (0.84) P < 0.0001 | NA | −3.61 (0.93) P = 0.0001 |
Placebo-omaveloxolone population | ||||||||
MOXIe extension | 95 | 42.8 (12.8) | 95 | −0.43 (0.63) | 69 | 1.18 (0.69) | 56 | 3.21 (0.76) |
Matched FACOMS | 95 | 44.5 (18.0) | 72 | 2.23 (0.69) | 71 | 4.23 (0.68) | 64 | 7.29 (0.72) |
Difference | NA | NR | NA | −2.75 (0.94) P = 0.0035 | NA | −3.06 (0.97) P = 0.0017 | NA | −4.09 (1.05) P = 0.0001 |
Omaveloxolone-omaveloxolone population | ||||||||
MOXIe extension | 41 | 40.9 (12.2) | 38 | 1.05 (1.09) | 33 | 1.10 (1.13) | 21 | 2.38 (1.33) |
Matched FACOMS | 41 | 39.6 (16.8) | 34 | 2.48 (1.12) | 33 | 3.57 (1.13) | 25 | 6.14 (1.24) |
Difference | NA | NR | NA | −1.43 (1.56) P = 0.36 | NA | −2.47 (1.60) P = 0.13 | NA | −3.76 (1.82) P = 0.0400 |
FACOMS = Friedreich’s Ataxia Clinical Outcome Measures Study; mFARS = modified Friedreich’s Ataxia Rating Scale; MMRM = mixed model for repeated measures; NA = not applicable; NR = not reported; SD = standard deviation; SE = standard error.
aThe mean difference in change from baseline was estimated using an MMRM.
Source: Details are from the sponsor’s Summary of Clinical Evidence.11
Harms
Harms data were not assessed in this study.
Critical Appraisal
The International Society for Pharmacoepidemiology (ISPE) RWE appraisal tool — modified for use by CDA-AMC according to the guidance documentation — was used to assess the quality of the propensity scored–matched analysis.44 The Guidance for Real-World Evidence reporting was not completed before submission by the CDA-AMC; thus, it was not reviewable. Key excluded sections are noted as follows.
Setting and Context
No information was provided about differences in health systems, access to care, available health care resources, or other factors that may affect the care of patients with FA and, in turn, the applicability of findings to the Canadian context.
Data Specifications
No information was provided about data access, cleaning, or linkage.
Data Sources, Data Dictionary, and Variables
Detailed descriptions of data sources, data dictionary and variables were not provided. This means important variables were not captured, potentially affecting study results.
An assessment of how the RWE study follows the CDA-AMC Guidance for Reporting Real-World Evidence checklist was conducted.
Study Design
The study design was considered appropriate because it used comparative evidence and described the efficacy of treatment compared to no treatment in a treatment-naive population. However, the choice of the first visit as baseline in the FACOMS group can introduce indication bias because it is unclear whether there are unmeasured confounding factors that would cause patients to enroll in the FACOMS. While no issues were identified with the timing of treatment initiation, the primary analysis cohort includes patients who were randomized to omaveloxolone as well as patients who were randomized to placebo and completed 48 weeks of follow-up before initiating treatment. The patients who completed 48 weeks of follow-up may be systematically different from those included in FACOMS because the matching strategy did not adjust for these unmeasured criteria; bias may result.
Methods and Results
There is limited potential for bias due to exposure or for outcome misclassification due to study design. However, there is the possibility of measurement error in the results, particularly at year 3; the mean study day of collection of mFARS results was 2 months to 3 months later in matched FACOMS patients than in the MOXIe cohort, which may bias the results slightly in favour of omaveloxolone. The variables included in the propensity score matching were deemed sufficient by CDA-AMC clinical experts. The diagnostic results indicated that the FACOMS cohort constructed using matching was sufficiently comparable to the MOXIe cohort, based on the included covariates. Covariate values were nearly completely known; this reduces the risk of bias being introduced due to the exclusion of patients based on important but unknown information.
The estimation of progression at 3 years is reliant on a MAR assumption, while the numbers of observed scores at the 3-year mark are 77 (57%) in the omaveloxolone group and 83 (61%) in the matched FACOMS natural history study cohort. The sensitivity of the conclusion to the MAR assumption was not reported, which could be important. No analyses describing the causes of missing outcomes or dropouts are provided; this raises the question of whether the benefit observed is due to a depletion of the patients remaining in the study or AEs inducing dropout.
Discussion
Summary of Available Evidence
The primary evidence for omaveloxolone as a treatment for FA comes from 1 pivotal, phase II, randomized, double-blinded, placebo-controlled trial (the MOXIe Part 2 trial, N = 103) comparing omaveloxolone 150 mg (3 capsules of 50 mg each) to placebo over 48 weeks. Additional long-term information is provided from an OLE study (N = 77), with evidence from a real-world external control analysis comparing patients in the MOXIe OLE study (N = 136) to matched natural history controls from the FACOMS database (N = 136) over 3 years. The trial analyses evaluated differences in disease progression using mFARS scores at 48 weeks from baseline, while the comparison of the MOXIe OLE study to FACOMS evaluated changes in mFARS at year 1, year 2, and year 3.
The MOXIe Part 2 trial and MOXIe OLE study populations consisted primarily of young adults with a mean age of around 26 years and a mean of 14 years since diagnosis. Most, but not all, of the key baseline characteristics were generally well-balanced between treatment groups in the MOXIe Part 2 trial, with mean baseline mFARS scores of 40.1 and 37.9 in the groups receiving omaveloxolone and placebo, respectively, in the all-randomized population. In the external control analysis, propensity score matching was used to identify FACOMS controls with similar baseline characteristics to the MOXIe cohort across factors, including age, sex, GAA repeat length, disease duration, baseline mFARS score, and ambulatory status.
Interpretation of Results
Efficacy
The primary treatment goal was to identify a treatment benefit of omaveloxolone through change in mFARS score. The clinical experts consulted indicated that a change of 2 points in this score is considered clinically meaningful, but also that stabilization of disease over a long period would be considered beneficial, given the progressive nature of FA. The MOXIe Part 2 trial demonstrated a statistically significant improvement in mFARS scores after 48 weeks of treatment with omaveloxolone compared to placebo (i.e., a difference of −2.40 points; P = 0.014). In addition, according to the GRADE framework, the certainty of evidence is moderate, indicating that compared to placebo, omaveloxolone likely results in slower progression in mFARS scores. In the OLE study, a comparison of treatment-naive patients and patients maintaining treatment supports this finding but illustrates that the treatment effects may wain or decrease, given that the newly treated patients had better mFARS scores after 48 weeks. The RWE comparison, in which patients maintained an observed improvement in mFARS scores for up to 3 years compared to an external cohort, illustrated that treated and nontreated patients progressed at similar rates after the first year. While this provides supportive evidence of effectiveness in clinical practice, there are important limitations — including potential selection bias in the FACOMS cohort and missing data at later time points — that warrant cautious interpretation.
Key outcomes identified as important by patient groups included maintaining independence in ADLs, slowing disease progression, improving quality of life, managing fatigue and energy levels, and maintaining the ability to work and/or attend school. While the mFARS score captures aspects of physical function and disease progression (encapsulated in the mFARS end point), direct measures of quality of life, fatigue, and ADLs were either not assessed in the clinical trial or did not show significant treatment effects. This includes the key secondary outcomes of PGIC and CGIC. Beyond the primary outcomes, all outcomes tested in the MOXIe trial either included the null or fell outside the statistical testing hierarchy, suggesting that the only evidence of efficacy for omaveloxolone was seen in the mFARS end point. This represents a gap in the evidence for the outcomes most meaningful to patients. The clinical experts noted that improvements in mFARS would be expected to translate to functional benefits; however, this relationship has not been formally established.
Other notable evidence gaps include effectiveness in patients with more advanced disease, impact on non-neurologic manifestations, effects on disease progression in very early disease, and comparative long-term efficacy (i.e., beyond 3 years).
Harms
Common AEs included headache, nausea, fatigue, and abdominal pain. SAEs were numerically higher among patients receiving omaveloxolone than among those receiving placebo (9.8% versus 5.8%), with no deaths reported. Discontinuations due to AEs were also numerically higher among patients receiving omaveloxolone than among those receiving placebo (7.8% versus 3.8%).
The product monograph recommends monitoring liver function regularly and exercising caution when omaveloxolone is coadministered with CYP3A4 inhibitors or inducers. It also advises caution in patients with hepatic impairment. The clinical experts agreed that these risks can be managed effectively through routine monitoring and dose adjustments, as needed, in typical clinical practice. Patient groups have expressed that the observed side effects are acceptable, given the progressive nature of the disease and the limited treatment options available. Commonly experienced symptoms, such as headache and nausea, were considered manageable with supportive care measures.
However, long-term safety data (i.e., beyond 3 years) remain limited. Clinical experts have highlighted several areas for ongoing monitoring, including cardiovascular effects, liver function, and growth and development in pediatric populations. There is also interest in better understanding the potential effects of omaveloxolone on non-neurologic manifestations of the disease as more long-term data become available.
Other Considerations
There are currently no approved disease-modifying treatments for FA. A significant unmet need for such a treatment is consistently highlighted by patient groups and clinical experts. The oral administration of omaveloxolone was viewed favourably by patients compared to potential injectable therapies in development, although some noted challenges with the current requirement for 150 mg (3 capsules of 50 mg each) taken orally once daily.
Several ongoing studies are evaluating omaveloxolone, including:
OLE studies following patients for up to 5 years
studies in patients with more advanced disease
pediatric studies
studies examining cardiac effects.
Results from these studies will help address current evidence gaps.
The clinical experts noted that, while questions remain about long-term benefits, the available evidence supports offering this treatment option to eligible patients, given the serious nature of the condition and the lack of alternatives.
Conclusion
Evidence from 1 phase II RCT suggests that, compared to placebo, omaveloxolone likely results in slower decline in neurologic function as measured by mFARS scores over 48 weeks in patients with FA aged 16 years and older. It likely results in improvements in ADL measures, which include activities involving upper limb function, mobility, and frequency of falls. While this is suggestive of functional relevance, with the clinical experts suggesting 2-point and 1-point potentially meaningful thresholds for mFARS and ADL, respectively, the actual clinical significance of these observations is uncertain due to the lack of an established minimal clinically important difference in mFARS and ADL scores. Other functional outcomes, including upper limb function, mobility, and frequency of falls, did not show significant improvements with treatment.
Comparison with natural history controls suggests that the treatment benefit of omaveloxolone may be maintained for up to 3 years. However, these studies do not allow clear conclusions to be drawn about whether disease progression will continue to be slower among patients treated with omaveloxolone than among matched controls. A long-term extension study suggests that benefit may be observed within the first year, given that treatment-naive patients were observed to have better responses. However, these findings are limited by potential selection bias and the study’s open-label design; in addition, the magnitude of these group differences is uncertain. The safety profile of omaveloxolone appears to be manageable, with liver enzyme elevations being the primary concern requiring monitoring. Common adverse effects, including gastrointestinal symptoms and fatigue, were generally mild to moderate in severity.
Important evidence gaps remain, particularly regarding efficacy in pediatric populations (which represent a significant proportion of newly diagnosed patients), patients with more advanced disease or significant cardiac involvement, and long-term comparative efficacy beyond 3 years. Additionally, while mFARS captures aspects of neurologic function, there is limited evidence on outcomes identified as important by patients, such as HRQoL, fatigue, and ability to maintain independence. Given that FA is a progressive, debilitating condition with no approved disease-modifying treatment, even a modest slowing of progression could be meaningful to patients. However, uncertainty remains about the magnitude and durability of benefit.
References
1.Durr A, Cossee M, Agid Y, et al. Clinical and genetic abnormalities in patients with Friedreich's ataxia. N Engl J Med. 1996;335(16):1169-75. doi:10.1056/NEJM199610173351601 PubMed
2.Opal P, Zoghbi HY. Post T, ed. Friedreich Ataxia. UpToDate; 2023. Accessed October 29, 2024. http://www.uptodate.com
3.Buesch K, Zhang R. A systematic review of disease prevalence, health-related quality of life, and economic outcomes associated with Friedreich's Ataxia. Curr Med Res Opin. 2022;38(10):1739-1749. doi:10.1080/03007995.2022.2112870 PubMed
4.Polek B, Roach MJ, Andrews WT, Ehling M, Salek S. Burden of Friedreich's Ataxia to the Patients and Healthcare Systems in the United States and Canada. Front Pharmacol. 2013;4:66. doi:10.3389/fphar.2013.00066 PubMed
5.Ataxia Canada. Biogen Data on file [confidential information; sponsor supplied reference]. Biogen Canada, Inc.; 2024.
6.Indelicato E, Nachbauer W, Eigentler A, et al. Onset features and time to diagnosis in Friedreich's Ataxia. Orphanet J Rare Dis. 2020;15(1):198. doi:10.1186/s13023-020-01475-9 PubMed
7.Parkinson MH, Boesch S, Nachbauer W, Mariotti C, Giunti P. Clinical features of Friedreich's ataxia: classical and atypical phenotypes. J Neurochem. 2013;126 Suppl 1:103-17. doi:10.1111/jnc.12317 PubMed
8.Lynch DR, Schadt K, Kichula E, McCormack S, Lin KY. Friedreich Ataxia: Multidisciplinary Clinical Care. J Multidiscip Healthc. 2021;14:1645-1658. doi:10.2147/jmdh.S292945 PubMed
9.National Institute of Neurological Disorders and Stroke (NINDS). Friedreich Ataxia [sponsor supplied reference]. 2023. Accessed April 28, 2023. https://www.ninds.nih.gov/health-information/disorders/friedreich-ataxia
10.Tsou AY, Paulsen EK, Lagedrost SJ, et al. Mortality in Friedreich ataxia. J Neurol Sci. 2011;307(1-2):46-9. doi:10.1016/j.jns.2011.05.023 PubMed
11.Biogen Canada Inc. Sponsor Summary of Clinical Evidence for Omaveloxolone (Brand name TBD) for Friedreich’s Ataxia [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Skyclarys (omaveloxolone), 50 mg oral capsule. Biogen Canada; 2023.
12.Silva AM, Brown JM, Buckle VJ, Wade-Martins R, Lufino MM. Expanded GAA repeats impair FXN gene expression and reposition the FXN locus to the nuclear lamina in single cells. Hum Mol Genet. 2015;24(12):3457-71. doi:10.1093/hmg/ddv096 PubMed
13.Bidichandani SI, Delatycki MB. Friedreich ataxia [sponsor supplied reference]. GeneReviews®; 2017.
14.Rummey C, Corben LA, Delatycki M, et al. Natural History of Friedreich Ataxia: Heterogeneity of Neurologic Progression and Consequences for Clinical Trial Design. Neurology. 2022;99(14):e1499-e1510. doi:10.1212/wnl.0000000000200913 PubMed
15.Reetz K, Dogan I, Hilgers RD, et al. Progression characteristics of the European Friedreich's Ataxia Consortium for Translational Studies (EFACTS): a 4-year cohort study. Lancet Neurol. 2021;20(5):362-372. doi:10.1016/S1474-4422(21)00027-2 PubMed
16.Burk K. Friedreich Ataxia: current status and future prospects. Cerebellum Ataxias. 2017;4:4. doi:10.1186/s40673-017-0062-x PubMed
17.Boghdeh NA, McGraw B, Barrera MD, et al. Inhibitors of the Ubiquitin-Mediated Signaling Pathway Exhibit Broad-Spectrum Antiviral Activities against New World Alphaviruses. Viruses. 2023;15(3):655. doi:10.3390/v15030655 PubMed
18.Corben LA, Collins V, Milne S, et al. Clinical management guidelines for Friedreich ataxia: best practice in rare diseases. Orphanet J Rare Dis. 2022;17(1):415. doi:10.1186/s13023-022-02568-3 PubMed
19.National Organization for Rare Disorders (NORD). Friedreich’s Ataxia. 2023. Accessed April 28, 2023. https://rarediseases.org/rare-diseases/friedreichs-ataxia/
20.Biogen Canada Inc. Skyclarys (omaveloxolone): capsule, 50 mg, oral [draft product monograph; sponsor supplied reference]. 2024. Accessed 2024.
21.Lynch DR, Chin MP, Delatycki MB, et al. Safety and Efficacy of Omaveloxolone in Friedreich Ataxia (MOXIe Study). Ann Neurol. 2021;89(2):212-225. doi:10.1002/ana.25934 PubMed
22.Lynch D. Correction to Safety and Efficacy of Omaveloxolone in Friedreich Ataxia (MOXIe Study). Ann Neurol. 2023;94(6):1190. doi:10.1002/ana.26808 PubMed
23.Hendrix S, Goldsberry A, Meyer C, Lynch D. Efficacy of Omaveloxolone in Friedreich' s Ataxia: Post-Hoc Analysis Using Global Statistics Test to Strengthen Secondary Endpoint Analyses. Neurology. 2023;100(17_supplement_2):1. doi:10.1212/WNL.0000000000203544
24.Reata Pharmaceuticals Inc. Clinical Study Report. MOXIe Part 2 CSR: RTA 408 (omaveloxolone) 408-C-1402 Part 2: A phase 2 study of the safety, efficacy, and pharmacodynamics of RTA 408 in the treatment of Friedreich’s ataxia [internal sponsor's report]. November 5, 2020.
25.Azevado M. Part One of Reata’s MOXIe Trial for Friedreich’s Ataxia Shows Promising Results [sponsor supplied reference]. Friedreich's Ataxia News: A Bionews Brand. 2017. https://friedreichsataxianews.com/news/part-one-of-moxie-trial-of-omaveloxolone-for-friedreichs-ataxia-shows-promising-results/
26.Reata Pharmaceuticals Inc. Clinical Study Report. RTA 408 (OMAVELOXOLONE) 408-C-1402 Part 1: A Phase 2 Study of the Safety, Efficacy, and Pharmacodynamics of RTA 408 in the Treatment of Friedreich’s Ataxia [internal sponsor's report]. September 29, 2020.
27.Reata Pharmaceuticals Inc. Protocol. RTA 408 (408-C-1402) A phase 2 study of the safety, efficacy, and pharmacodynamics of RTA 408 in the treatment of Friedreich's ataxia: Study Procedure Manual Version 13.0 [internal sponsor's report]. April 11, 2024.
28.Rummey C, Farmer JM, Lynch DR. Predictors of loss of ambulation in Friedreich's ataxia. EClinicalMedicine. 2020;18:100213. doi:10.1016/j.eclinm.2019.11.006 PubMed
29.Rummey C, Corben LA, Delatycki MB, et al. Psychometric properties of the Friedreich Ataxia Rating Scale. Neurol Genet. 2019;5(6):371. doi:10.1212/nxg.0000000000000371 PubMed
30.Rummey C, Zesiewicz TA, Perez-Lloret S, Farmer JM, Pandolfo M, Lynch DR. Test-retest reliability of the Friedreich's ataxia rating scale. Ann Clin Transl Neurol. 2020;7(9):1708-1712. doi:10.1002/acn3.51118 PubMed
31.Tai G, Corben LA, Woodcock IR, Yiu EM, Delatycki MB. Determining the Validity of Conducting Rating Scales in Friedreich Ataxia through Video. Mov Disord Clin Pract. 2021;8(5):688-693. doi:10.1002/mdc3.13204 PubMed
32.Fahey MC, Corben LA, Collins V, Churchyard AJ, Delatycki MB. The 25-foot walk velocity accurately measures real world ambulation in Friedreich ataxia. Neurology. 2007;68(9):705-6. doi:10.1212/01.wnl.0000256037.63832.6f PubMed
33.Traschutz A, Fleszar Z, Hengel H, et al. FARS-ADL across Ataxias: Construct Validity, Sensitivity to Change, and Minimal Important Change. Mov Disord. 2024;39(6):965-974. doi:10.1002/mds.29788 PubMed
34.Reata Pharmaceuticals Inc. Clinical Study Report Addendum. 408-C-1402 Part 2 Clinical Study Report. A phase 2 study of the safety, efficacy, and pharmacodynamics of RTA 408 in the treatment of Friedreich’s ataxia [internal sponsor's report]. November 5, 2020.
35.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi:10.1016/j.jclinepi.2010.07.015 PubMed
36.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi:10.1016/j.jclinepi.2019.10.014 PubMed
37.Reata Pharmaceuticals Inc. MOXIe OLE Interim CSR: RTA 408 (Omaveloxolone) 408-C-1402 Extension. A Phase 2 Study of the Safety, Efficacy, and Pharmacodynamics of RTA 408 in the Treatment of Friedreich’s Ataxia. Interim Clinical Study Report [internal sponsor's report]. 2021.
38.Patel M, Isaacs CJ, Seyer L, et al. Progression of Friedreich ataxia: quantitative characterization over 5 years. Ann Clin Transl Neurol. 2016;3(9):684-94. doi:10.1002/acn3.332 PubMed
39.Lynch DR, Chin MP, Boesch S, et al. Efficacy of Omaveloxolone in Friedreich's Ataxia: Delayed-Start Analysis of the MOXIe Extension. Mov Disord. 2023;38(2):313-320. doi:10.1002/mds.29286 PubMed
40.Lynch DR, Goldsberry A, Rummey C, et al. Propensity matched comparison of omaveloxolone treatment to Friedreich ataxia natural history data. Ann Clin Transl Neurol. 2024;11(1):4-16. doi:10.1002/acn3.51897 PubMed
41.De Michele G, Perrone F, Filla A, et al. Age of onset, sex, and cardiomyopathy as predictors of disability and survival in Friedreich's disease: a retrospective study on 119 patients. Neurology. 1996;47(5):1260-4. doi:10.1212/wnl.47.5.1260 PubMed
42.Lynch D, Goldsberry A, Rummey C, et al. Direct utility of natural history data in analysis of clinical trials: Propensity match-based analysis of Omaveloxolone in Friedreich ataxia using the FA-COMS dataset. medRxiv. 2022:2022.08.12.22278684. doi:10.1101/2022.08.12.22278684
43.Critical Path Institute (C-Path), Friedreich’s Ataxia Research Alliance (FARA). C-Path and FARA Launch Friedreich's Ataxia Integrated Clinical Database to Advance the Development of Treatments for FA. 2019. Accessed October 24, 2024. https://c-path.org/c-path-and-fara-launch-friedreichs-ataxia-integrated-clinical-database-to-advance-the-development-of-treatments-for-fa/
44.CADTH. Guidance for Reporting Real-World Evidence. 2023. Accessed December 27, 2024. https://www.cda-amc.ca/sites/default/files/RWE/MG0020/MG0020-RWE-Guidance-Report-Secured.pdf
45.Reata Pharmaceuticals Inc. Clinical Study Report: MOXIe Extension 408-C-1402_CSR_Tables [internal sponsor's report]. 2022.
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Patient Global Impression of Change
The PGIC is a 7-point scale that requires patients to assess how much their illness has improved or worsened relative to a baseline state at the beginning of an intervention. The PGIC is assessed by completing the following statement “since I began trial treatment, my overall status is: very much improved (1), much improved (2), minimally improved (3), no change (4), minimally worse (5), much worse (6), and very much worse (7).” Patients completed this assessment following completion of the FARS neurologic assessment or the CGIC.
The PGIC measures the patient’s subjective perception of overall improvement or decline in their condition from baseline. It is scored from 1 (“very much improved”) to 7 (“very much worse”), with lower scores indicating better perceived improvement and higher scores indicating greater perceived worsening.
Table 24: Key Secondary Analysis — CGIC at Week 48
CGIC outcome at week 48 | Full analysis set | All-randomized population | ||
|---|---|---|---|---|
Omaveloxolone (n = 40) | Placebo (n = 42) | Omaveloxolone (n = 51) | Placebo (n = 52) | |
LS mean (95% CI) | 3.92 (█████ ████) | 4.06 (3████ ████) | ████ ██████ █████ | ████ ██████ █████ |
LS mean difference between treatment groups (95% CI), P valuea | −0.13 (█████ ████) P = 0.5259 | █████ ███████ ███████ █ ██████ | ||
CGIC = Clinical Global Impression of Change; CI = confidence interval; LS = least squares; PGIC = Patient Global Impression of Change.
aComparison of PGIC and CGIC for patients treated with omaveloxolone 150 mg and placebo was estimated using analyses of covariance, with the following fixed factors: site, pes cavus status, and treatment. Missing data were imputed using multiple imputation.
Source: MOXIe trial Part 2 Clinical Study Report.24
Table 25: Key Secondary Analysis — PGIC at Week 48
PGIC outcome at week 48 | Full analysis set | All-randomized population | ||
|---|---|---|---|---|
Omaveloxolone (n = 40) | Placebo (n = 42) | Omaveloxolone (n = 51) | Placebo (n = 52) | |
LS mean (95% CI) | 3.89 (█████ ████) | 4.32 (█████ ████) | ████ ██████ █████ | ████ ██████ █████ |
LS mean difference between treatment groups (95% CI), p-valuea | −0.43 (█████ ████) P = 0.1300 | █████ ███████ ████████ █ ██████ | ||
CGIC = Clinician Global Impression of Change; CI = confidence interval; LS = least squares; PGIC = Patient Global Impression of Change.
aComparison of PGIC and CGIC for patients treated with omaveloxolone 150 mg and placebo was estimated using analyses of covariance, with the following fixed factors: site, pes cavus status, and treatment. Missing data were imputed using multiple imputation.
Source: MOXIe trial Part 2 Clinical Study Report,24 MOXIe trial Part 2 Clinical Study Report Erratum 1.34
Clinical Global Impression of Change
The CGIC is a 7-point scale that requires the clinician to assess how much the patient's illness has improved or worsened relative to a baseline state at the beginning of an intervention (Guy, 1976). The CGIC is assessed by completing the following statement “Compared to the patient’s condition at the start of the trial, this patient’s overall status is: very much improved (1), much improved (2), minimally improved (3), no change (4), minimally worse (5), much worse (6), and very much worse (7).” Clinicians could take into account prior results of study assessments to influence their clinical judgment of the CGIC. Clinicians completed this assessment following the patient’s completion of the FARS neurologic assessment or the PGIC.
The CGIC reflects the clinician’s overall impression of how the patient’s condition has changed from baseline. Similar to the PGIC, it is scored on a 7-point scale (1 = “very much improved,” 7 = “very much worse”), where lower scores represent greater clinical improvement and higher scores represent greater clinical worsening.
Table 26: Duration of Study Treatment and Exposure to Omaveloxolone (Safety Population)
Exposure | Placebo – omaveloxolone N = 106 n (%) | Omaveloxolone – omaveloxolone N = 43 n (%) | Overall omaveloxolone N = 149 n (%) |
|---|---|---|---|
Cumulative dose dispensed (mg), mean (SD) | █████████ ███████████ | █████████ ███████████ | █████████ ███████████ |
Number of doses received, mean (SD) | ████ ███████ | ██████ ██████ | ████████████ |
Duration of treatment (days), mean (SD) | █████ ███████ | █████ ███████ | █████ ██████ |
Study drug compliance (%), mean (SD) | █████ ███████ | █████ ████████ | █████ ███████ |
Total number of doses dispensed, mean (SD) | ██████ ██████ | ██████ ███████ | ██████ ██████ |
Total number of doses returned, mean (SD) | █████ ███████ | █████ ███████ | █████ ██████ |
SD = standard deviation.
Note: The placebo-omaveloxolone group consisted of patients initially randomized to placebo in Part 2 or participating in Part 1 of the study. The omaveloxolone-omaveloxolone group consisted of patients initially randomized to omaveloxolone in Part 2.
Source: MOXIe OLE Clinical Study Report.37
Table 27: mFARS Results and Mean Change From Baseline (Safety Population)
Visit | Statistic | All patients | |
|---|---|---|---|
Placebo – omaveloxolone (N = 106) | Omaveloxolone – omaveloxolone (N = 43) | ||
Baseline | n | ███ | ██ |
Mean (SD) | █████ ████████ | █████ ████████ | |
Median | █████ | █████ | |
Min, Max | █████ ████ | ████ ████ | |
Week 24 CFB | n | ██ | ██ |
Mean (SD) | █████ ███████ | █████ ███████ | |
Median | █████ | ████ | |
Min, Max | ██████ ████ | █████ ███ | |
Week 24 percentage CFB | Mean (SD) | █████ ████████ | ████ ████████ |
Median | █████ | ████ | |
Min, Max | ██████ ████ | ██████ ████ | |
Week 48 CFB | n | ██ | ██ |
Mean (SD) | ████ ███████ | ████ ███████ | |
Median | ████ | ████ | |
Min, Max | █████ ████ | █████ ███ | |
Week 48 percentage CFB | Mean (SD) | ████ ████████ | ████ ████████ |
Median | ████ | ████ | |
Min, Max | ██████ ████ | ██████ ████ | |
Week 72 CFB | n | ██ | ██ |
Mean (SD) | ████ ███████ | ████ ███████ | |
Median | ████ | ████ | |
Min, Max | █████ ████ | ██████ ███ | |
Week 72 percentage CFB | Mean (SD) | ████ ████████ | ████ ████████ |
Median | ████ | ████ | |
Min, Max | ██████ ████ | ██████ ████ | |
Week 96 CFB | n | ██ | ██ |
Mean (SD) | ████ ███████ | ████ ███████ | |
Median | ████ | ████ | |
Min, Max | █████ ████ | ██████ ███ | |
Week 96 percentage CFB | Mean (SD) | ████ ████████ | ████ ████████ |
Median | ████ | ████ | |
Min, Max | ██████ ████ | ██████ ████ | |
Week 120 CFB | n | ██ | ██ |
Mean (SD) | ████ ███████ | ████ ███████ | |
Median | ████ | ████ | |
Min, Max | ██████ ████ | ██████ ████ | |
Week 120 percentage CFB | Mean (SD) | ████ ████████ | ████ ████████ |
Median | ████ | ████ | |
Min, Max | ██████ ████ | ██████ ████ | |
Week 144 CFB | n | ██ | ██ |
Mean (SD) | ████ ███████ | ████ ███████ | |
Median | ████ | ████ | |
Min, Max | █████ ████ | ██████ ████ | |
Week 144 percentage CFB | Mean (SD) | ████ ████████ | ████ ████████ |
Median | ████ | ████ | |
Min, Max | ██████ ████ | ██████ ████ | |
CFB = change from baseline; Max = maximum; Min = minimum; Omav = omaveloxolone; SD = standard deviation.
Note: The placebo-omaveloxolone group consisted of patients initially randomized to placebo in Part 2 or participating in Part 1 of the study. The omaveloxolone-omaveloxolone group consisted of patients initially randomized to omaveloxolone in Part 2.
Source: MOXIe OLE Clinical Study Report.37
Table 28: 9-HPT Results and Change From Baseline by Study Visit (Safety Population)
Visit | Placebo-omaveloxolone (N = 106) | Omaveloxolone-omaveloxolone (N = 43) | Overall omaveloxolone (N = 149) |
|---|---|---|---|
Reciprocal of average time (1/second): Nondominant hand | |||
Baseline | |||
n | ███ | ██ | ███ |
Mean (SD) | ██████ ████████ | ██████ █████████ | ██████ █████████ |
Week 24 | |||
n | ██ | ██ | ███ |
Mean (SD) | ██████ ████████ | ██████ █████████ | ██████ █████████ |
CFB, mean (SD) | ██████ ████████ | ██████ ████████ | █████ █████████ |
Week 48 | |||
n | ██ | ██ | ██ |
Mean (SD) | █████ ████████ | █████ ████████ | █████ ████████ |
CFB, mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
Week 72 | |||
n | ██ | ██ | ██ |
Mean (SD) | █████ ████████ | ██████ █████████ | ██████ █████████ |
CFB, mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
Week 96 | |||
n | ██ | ██ | ██ |
Mean (SD) | █████ ████████ | ██████ █████████ | ██████ █████████ |
CFB, mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
Week 120 | |||
n | ██ | ██ | ██ |
Mean (SD) | █████ ████████ | ██████ █████████ | ██████ █████████ |
CFB, mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
Week 144 | |||
n | ██ | ██ | ██ |
Mean (SD) | ██████████████ | ██████ █████████ | ██████ █████████ |
CFB, mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
Reciprocal of average time (1/second): Dominant hand | |||
Baseline | |||
n | ███ | ██ | ███ |
Mean (SD) | █████ ████████ | ██████ █████████ | ██████ █████████ |
Week 24 | |||
n | ██ | ██ | ███ |
Mean (SD) | █████ ████████ | ██████ █████████ | ██████ █████████ |
CFB, mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
Week 48 | |||
n | ██ | ██ | ██ |
Mean (SD) | █████ ████████ | ██████ █████████ | ██████ █████████ |
CFB, mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
Week 72 | |||
n | ██ | ██ | ██ |
Mean (SD) | █████ ████████ | ██████ █████████ | ██████ █████████ |
CFB, mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
Week 96 | |||
n | ██ | ██ | ██ |
Mean (SD) | █████ ████████ | ██████ █████████ | ██████ █████████ |
CFB, mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
Week 120 | |||
n | ██ | ██ | ██ |
Mean (SD) | █████ ████████ | ██████ █████████ | ██████ █████████ |
CFB, mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
Week 144 | |||
n | ██ | ██ | ██ |
Mean (SD) | █████ ████████ | ██████ █████████ | ██████ █████████ |
CFB, mean (SD) | ██████ ████████ | ███████ █████████ | ███████ █████████ |
CFB = change from baseline; OLE = open-label extension; SD = standard deviation.
Note: The placebo-omaveloxolone group consisted of patients initially randomized to placebo in Part 2 or participating in Part 1 of the study. The omaveloxolone-omaveloxolone group consisted of patients initially randomized to omaveloxolone in Part 2.
Source: MOXIe OLE Clinical Study Report37 and OLE extension tables.45
Table 29: Timed 25-Foot Walk Test Results and Change From Baseline by Study Visit (Safety Population)
Visit | Placebo-omaveloxolone (N = 106) | Omaveloxolone-omaveloxolone (N = 43) | Overall omaveloxolone (N = 149) |
|---|---|---|---|
Baseline | |||
n | ██ | ██ | ███ |
Mean (SD) | ██████ ████████ | ██████ █████████ | ██████ █████████ |
Week 24 | |||
n | ██ | ██ | ██ |
Mean (SD) | ██████ ████████ | ██████ █████████ | ██████ █████████ |
n | ██ | ██ | ██ |
CFB, mean (SD) | ███████ ███████ | ███████ ████████ | ███████ ████████ |
Week 48 | |||
n | ██ | ██ | ██ |
Mean (SD) | ██████ ████████ | ██████ █████████ | ██████ █████████ |
n | ██ | ██ | ██ |
CFB, mean (SD) | ███████ ███████ | ███████ ████████ | ███████ ████████ |
Week 72 | |||
n | ██ | ██ | ██ |
Mean (SD) | ██████ ███████ | ██████ █████████ | ██████ █████████ |
n | ██ | ██ | ██ |
CFB, mean (SD) | ███████ ███████ | ███████ ███████ | ██████ ████████ |
Week 96 | |||
n | ██ | ██ | ██ |
Mean (SD) | ██████ ████████ | ██████ █████████ | ██████ █████████ |
n | ██ | ██ | ██ |
CFB, mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
Week 120 | |||
n | ██ | ██ | ██ |
Mean (SD) | █████ ████████ | ██████ █████████ | ██████ █████████ |
n | ██ | ██ | ██ |
CFB, mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
Week 144 | |||
n | ██ | ██ | ██ |
Mean (SD) | █████ ████████ | ██████ █████████ | ██████ █████████ |
n | ██ | ██ | ██ |
CFB, mean (SD) | ██████ ████████ | ██████ ████████ | ██████ ████████ |
CFB = change from baseline; OLE = open-label extension; SD = standard deviation.
Note: The placebo-omaveloxolone group consisted of patients initially randomized to placebo in Part 2 or participating in Part 1 of the study. The omaveloxolone-omaveloxolone group consisted of patients initially randomized to omaveloxolone in Part 2.
Source: MOXIe OLE Clinical Study Report.37
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
EFACTS
European Friedreich's Ataxia Consortium for Translational Studies
FA
Friedreich’s ataxia
FACOMS
Friedreich’s Ataxia Clinical Outcome Measures Study
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
LY
life-year
MDC
Muscular Dystrophy Canada
mFARS
modified Friedreich’s Ataxia Rating Scale
QALY
quality-adjusted life-year
SARA
Scale for the Assessment and Rating of Ataxia
SOC
standard of care
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Omaveloxolone (Skyclarys), 50 mg oral capsule |
Indication | Proposed: For the treatment of Friedreich’s ataxia in adults and adolescents aged 16 years and older |
Health Canada approval status | Under review (pre-NOC) |
Health Canada review pathway | Priority review |
NOC date | March 13, 2025 |
Reimbursement request | As per indication |
Sponsor | Biogen Canada Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Regression-based model |
Target population | Adults and adolescents aged 16 years and older with FA |
Treatment | Omaveloxolone plus SOC, assumed by the sponsor to comprise treatments for symptom and comorbidity management |
Dose regimen | Omaveloxolone: 150 mg once daily |
Submitted price | Omaveloxolone: $364.30 per capsule |
Submitted treatment cost | Omaveloxolone: $346,933 per yeara |
Comparator | SOC |
Perspective | Canadian publicly funded health care payer Societal perspectiveb |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (84 years) |
Key data sources |
|
Submitted results |
|
Key limitations |
|
CDA-AMC reanalysis results |
|
CDA-AMC = Canada’s Drug Agency; EFACTS = European Friedreich's Ataxia Consortium for Translational Studies; FA = Friedreich’s ataxia; FACOMS = Friedreich’s Ataxia Clinical Outcome Measures Study; HRQoL = health-related quality of life; ICER = incremental cost-effectiveness ratio; LY = life-year; mFARS = modified Friedreich's Ataxia Rating Scale; QALY = quality-adjusted life-year; SARA = Scale for the Assessment and Rating of Ataxia; SF-36 = Short Form (36) Health Survey; SOC = standard of care; vs. = versus; WTP = willingness to pay.
aAssuming 87% relative dose intensity. Annual cost without adjustment: $399,180.
bOmaveloxolone is being reviewed by CDA-AMC through the complex review pathway. As such, CDA-AMC has appraised 2 cost-effectiveness analyses submitted by the sponsor: 1 adopting a publicly funded health care payer perspective and 1 adopting a societal perspective.
Conclusions
The clinical review by Canada’s Drug Agency (CDA-AMC) concluded that omaveloxolone likely results in slower neurologic decline compared to placebo, as measured by modified Friedreich’s Ataxia Rating Scale (mFARS) scores compared to placebo over 48 weeks in patients with Friedreich's ataxia (FA) aged 16 years and older. However, the clinical importance of this finding is uncertain due to the lack of an established minimum clinically important difference. Other functional outcomes, including upper limb function, mobility, and frequency of falls, did not show significant improvements with omaveloxolone treatment in the MOXIe trial.
The sponsor submitted an economic analysis comparing the cost-effectiveness of omaveloxolone plus standard of care (SOC) to SOC alone, with effectiveness (based on change in mFARs score) informed by data from the submitted propensity score–matched analysis. Results of the sponsor’s base case suggest that omaveloxolone plus SOC will be more effective over a patient’s lifetime than SOC alone (i.e., incremental life-years [LYs] = 1.15; incremental quality-adjusted life-years [QALYs] = 2.59); however, owing to the identified uncertainty in the clinical evidence — including uncertainty as to whether omaveloxolone improves health-related quality of life (HRQoL) — and the magnitude and durability of change in mFARS scores, the incremental QALYs predicted by the sponsor’s economic modelling are uncertain.
Based on the sponsor’s results, omaveloxolone plus SOC is not cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per QALY gained when either the public health care payer or a societal perspective is adopted. Price reductions of 95% to 97% would be required for omaveloxolone plus SOC to be cost-effective compared to SOC from the societal and public payer perspectives, respectively, at a WTP threshold of $50,000 per QALY gained. Given the uncertainty in the clinical evidence base, it is highly uncertain whether and to what extent the predicted gains in LYs and QALYs will be realized in clinical practice, especially considering that approximately 96% of the incremental QALYs predicted by the sponsor’s model were gained in the extrapolated period after the MOXIe trial; that the incremental cost-effectiveness ratio (ICER) is highly sensitive to the choice of utility values; and that the sponsor’s model did not include the impact of treatment on non-neurologic outcomes. Therefore, further price reductions may be required.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient group input was received from Ataxia Canada – Claude-St-Jean Foundation, the Friedreich’s Ataxia Research Alliance, Muscular Dystrophy Canada (MDC), and the National Ataxia Foundation through interviews and surveys of patients with FA and caregivers. Respondents indicated that FA symptoms severely affect daily life by decreasing quality of life, reducing mobility, impairing muscle coordination, causing fatigue and slurred speech, curving the spine, worsening mental health, and diminishing the ability to perform daily activities. Respondents noted that there are no approved or effective therapies for FA. They described using treatments (e.g., anticoagulation medications, baclofen, tizanidine, benzodiazepines, and intramuscular botulinum toxin injection) to manage symptoms of the disease, such as paroxysmal atrial fibrillation or muscle cramps and spasms. Patients additionally reported requiring occupational therapy, physiotherapy, speech therapy, and exercise or balance training. Patients and caregivers indicated that the most important outcomes for new treatment options include symptom reduction and prevention of disease progression to maintain a certain quality of life, specifically as it relates to maintaining mobility. Respondents with experience with omaveloxolone described slower progression of disease, increased ability to cope with fatigue, improved quality of life, and general stabilization of their condition, with minor adverse events (AEs), including elevated cholesterol, headache, diarrhea, and nausea.
Clinician input was received from the Neuromuscular Disease Network for Canada. Clinicians indicated that current treatment is limited to the management of complications of FA and rehabilitation related to associated challenges, such as motor impairment, vision and hearing loss, psychological and cognitive issues, cardiomyopathy, diabetes, and skeletal abnormalities. Clinicians noted that omaveloxolone will be the first approved treatment for FA and that it would ideally delay FA progression.
CDA-AMC–participating drug plans noted the lack of an active comparator in the MOXIe trial and expressed concerns about the availability of genetic testing for FA. Plans questioned whether the initiation criteria for omaveloxolone should reflect the inclusion criteria for the MOXIe trial (i.e., patients aged 16 years to 40 years with mFARS scores from 20 to 80, without significant cardiac issues or severe pes cavus). Plans also questioned how treatment response would be defined in clinical practice and whether treatment interruptions, dose reductions, and re-treatment after discontinuation (e.g., because of AEs) would be considered. Lastly, plans expressed concern about the potential budget impact of reimbursing omaveloxolone.
Several of these concerns were addressed in the sponsor’s model:
The sponsor’s model incorporated change in mFARS score from baseline, which considers change in neurologic function.
HRQoL and health care resource use were included in the model.
Costs related to occupational therapy, physiotherapy, and speech therapy were included in the societal perspective.
CDA-AMC was unable to address the following concerns:
There was a lack of head-to-head data informing the clinical effectiveness of omaveloxolone plus SOC versus SOC alone.
Long-term efficacy data were lacking.
Costs related to genetic testing were not included in the model.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of omaveloxolone plus SOC compared to SOC alone in patients aged 16 years and older with FA.1 The modelled population is aligned with the inclusion criteria of the MOXIe studies, which enrolled patients aged 16 years to 40 years with FA.2 In the pharmacoeconomic model, SOC was assumed by the sponsor to comprise treatments for symptom and comorbidity management, and was assumed not to differ between patients receiving omaveloxolone plus SOC and those receiving SOC alone.
Omaveloxolone is available as 50 mg oral capsules, with a recommended dose of 150 mg once daily.3 At the submitted price of $364.30 per 50 mg capsule, the sponsor estimated the annual cost of omaveloxolone to be $346,933, based on a relative dose intensity of 87%.1 No drug acquisition costs were included for SOC.
The clinical outcomes were QALYs and LYs estimated over a lifetime horizon (i.e., 84 years; 1-year cycle length). Discounting (1.5% per annum) was applied to both costs and outcomes. The sponsor provided base-case analyses from the perspective of public health care payers in Canada and from a societal perspective.
Model Structure
The sponsor submitted a regression-based model that consisted of 2 treatment-related states (i.e., treatment with omaveloxolone plus SOC or SOC alone) and dead.1 Patients who started on omaveloxolone plus SOC could discontinue omaveloxolone, based on data from the MOXIe trial, after which time they were assumed to receive SOC alone until death. Patients who entered the model on SOC remained on SOC until death. The sponsor used a regression-based approach to account for differences in disease progression and risk of death based on age of onset (i.e., ≤ 7 years, 8 years to 14 years, 15 years to 24 years, or > 24 years). Patients in each age group were tracked separately in the model to estimate LYs, QALYs, and costs based on mFARS score and whether they were receiving omaveloxolone plus SOC or SOC alone.
Model Inputs
Baseline characteristics and distribution of patients differed across subgroups based on age of FA onset (≤ 7 years: 34%; 8 years to 14 years: 40%; 15 years to 24 years: 18%; > 24 years: 7%), based on data from the Friedreich’s Ataxia Clinical Outcome Measures Study (FACOMS) database,4 an ongoing natural history study conducted in North America.
The sponsor assumed that patients with onset of FA at less than 7 years of age and those with onset between 8 years and 14 years of age would start treatment at 16 years of age (i.e., the starting age in the model was 16 years).1 The sponsor used cohort characteristics (age of FA onset, baseline age, percentage of males, baseline modified mFARS score, and baseline gait score) to inform a regression equation to predict the average mFARS trajectory for patients receiving SOC alone. The mFARS trajectory for patients receiving omaveloxolone was calculated by the sponsor by applying the treatment effect (rate ratios) to the mFARS trajectory for SOC patients. The data to inform this were derived from a sponsor-conducted, propensity score–matched analysis that compared disease progression (based on change in mFARS) in patients in the FACOMS cohort (assumed to represent SOC) to those who received omaveloxolone in the MOXIe trials. In the base case, the sponsor used the rate ratio based on the observed cumulative change in mFARS over 3 years of treatment and assumed that the rate of change would remain constant for the remainder of the model horizon (i.e., no waning of treatment effectiveness). Discontinuation rates were based on data from the MOXIe Part 2 trial and the sponsor’s open-label extension study.2,5 Mortality risk was predicted based on mFARS using published literature and patient-level data from the European Friedreich's Ataxia Consortium for Translational Studies (EFACTS) and FACOMS.4,6,7
The sponsor used Scale for the Assessment and Rating of Ataxia (SARA) scores from EFACTS to derive utility values for the model.7 The sponsor mapped the SARA scores for patients with FA to mFARS scores using a published algorithm8 and then estimated EQ-5D values by linear regression of EQ-5D data from EFACTS and the mapped SARA scores. AEs in the model were nausea, diarrhea, oropharyngeal pain, and influenza; however, disutility was included only for influenza.9 In the societal perspective, the sponsor applied caregiver disutilities for nonambulatory patients. Disutilities were obtained from the literature.10
Costs included in the model were those associated with omaveloxolone acquisition, AEs, background medication use, and health care resource use. Drug acquisition costs for omaveloxolone were based on the sponsor’s submitted price, with the total estimated cost based on the monograph-recommended dose adjusted by relative dose intensity (87%) from the MOXIe trial.1,2 Other costs were derived from Alberta Health Costing, the Ontario Schedule of Benefits, the Canadian Institute for Health Information patient cost estimator, and the published literature.11-15 The societal perspective additionally included costs related to patient productivity and caregiver costs (estimated using the proportion of patients who required caregiving, hours estimated, amount of time off required, and hourly wages), obtained from the published literature and clinician input.16,17
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (5,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented here. Disaggregated results are available in Appendix 3.
Base-Case Results
From the publicly funded health care payer perspective, omaveloxolone plus SOC was associated with an incremental gain of 2.59 QALYs and incremental costs of $3,975,265 compared to SOC alone, resulting in an ICER of $1,534,503 per QALY gained. Results were driven by the acquisition cost of omaveloxolone. More than 96% of the total incremental QALYs predicted for omaveloxolone plus SOC were accrued based on extrapolation (i.e., after 48 weeks, the duration of the MOXIe trial).
From the societal perspective, omaveloxolone plus SOC was associated with an incremental gain of 3.00 QALYs and incremental costs of $3,899,888 compared to SOC alone, resulting in an ICER of $1,297,851 per QALY gained (Table 3).
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. SOC ($/QALY) |
|---|---|---|---|---|---|
Publicly funded health care payer perspective | |||||
SOC | 1,037,040 | Reference | 13.65 | Reference | Reference |
Omaveloxolone plus SOC | 5,012,305 | 3,975,265 | 16.24 | 2.59 | 1,534,503 |
Societal perspective | |||||
SOC | 2,532,040 | Reference | 12.15 | Reference | Reference |
Omaveloxolone plus SOC | 6,431,928 | 3,899,888 | 15.15 | 3.00 | 1,297,851 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several scenario analyses, including varying the age of FA onset and adopting alternate inputs for mortality risk, extrapolation of mFARS score, treatment effect, and utility values. Adopting alternative utility values derived from the FACOMS registry had a meaningful impact on the ICER (i.e., health care payer perspective ICER = $2,812,158 per QALY gained; societal perspective ICER = $3,020,214 per QALY gained).
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The comparative clinical efficacy of omaveloxolone plus SOC to SOC alone is uncertain: As noted in the CDA-AMC Clinical Review, evidence from the MOXIe trial suggests that omaveloxolone likely results in slower neurologic decline (based on mFARS score) compared to placebo over 48 weeks. Comparison by the sponsor of patients from the MOXIe trial to natural history controls suggests that patients who receive omaveloxolone may have slower disease progression over 3 years compared to matched controls. However, these findings are limited by potential selection bias and the open-label design; in addition, the magnitude of these group differences is uncertain. Further, the clinical importance of these observations is uncertain due to the lack of an established minimum clinically important difference. Other functional outcomes, including upper limb function, mobility, and frequency of falls, did not show significant improvements with omaveloxolone treatment.
CDA-AMC could not address uncertainty in the clinical evidence. The sponsor’s model was based on change in mFARS score and did not include other functional outcomes noted to be important to patients.
Uncertainty in the long-term effectiveness of omaveloxolone. The submitted comparison of patients who received omaveloxolone versus natural history controls suggests that the treatment benefit of omaveloxolone may be maintained for up to 3 years. However, no data are available to support the continued effectiveness of omaveloxolone beyond 3 years, and this propensity score–matched analysis is subject to the limitations noted in the previous limitation (i.e., potential selection bias and open-label design). In the pharmacoeconomic model, the sponsor assumed that the estimated effectiveness of omaveloxolone plus SOC based on up to 3 years of propensity score–matched data would be maintained indefinitely, with no effectiveness waning. In the sponsor’s model, approximately 96% of the incremental QALYs predicted to be gained with omaveloxolone plus SOC relative to SOC alone were accrued after the duration of the MOXIe trial (i.e., beyond 48 weeks), and 89% were accrued beyond the 3 years of propensity score–matched data.
CDA-AMC was unable to address this limitation owing to a lack of comparative data beyond 3 years and uncertainties in the submitted clinical evidence. Whether the treatment effect is maintained indefinitely is highly uncertain, owing to a lack of long-term data.
The impact of omaveloxolone on HRQoL is uncertain: The sponsor’s base case predicts an incremental gain of 2.59 QALYs with omaveloxolone plus SOC compared to SOC alone. As noted in the CDA-AMC Clinical Review, there is limited evidence on outcomes identified as important by patients, such as quality of life. In the MOXIe trial, the mean change in Short Form (36) Health Survey scores from baseline to week 48 were small and similar between omaveloxolone and placebo. Thus, whether the use of omaveloxolone in clinical practice will lead to improved HRQoL for patients is uncertain. Further, the health state utility values used in the sponsor’s base case were derived by mapping, which is not recommended practice because the predicted utilities can vary dramatically depending on the instruments being mapped, algorithms used, and the severity of the health states included.18 In the sponsor-conducted scenario analyses, the use of alternate health state utility values resulted in ICERs that were approximately $1 million higher than in the sponsor’s base case (i.e., payer perspective ICER = $2,812,158 per QALY gained; societal perspective ICER = $3,020,214 per QALY gained). This highlights the impact of uncertainty in the utility values on the estimated cost-effectiveness.
The magnitude of incremental QALYs predicted in the sponsor’s base case is uncertain owing to uncertainties about the impact of omaveloxolone on HRQoL and the utility values. If incremental QALYs were lower than predicted, the ICER would be correspondingly higher.
The impact of omaveloxolone on costs and outcomes relevant to the societal perspective analysis is highly uncertain: The sponsor’s submitted analysis from a societal perspective, which included indirect costs and the impact of treatment on caregivers, required assumptions about the impact of FA on caregivers, hours worked by patients and caregivers, the proportion of patients and caregivers employed, and the costs paid by patients versus the health care system. There are several sources of uncertainty related to the values adopted for each of these inputs, including the length of the friction period (i.e., the time it takes to replace a worker in the economy); the proportion of patients and caregivers employed (assumed by the sponsor to be 50% of patients and 86% of caregivers); the number of hours worked per week (assumed to be 40 hours for patients and 26 hours for caregivers); the cost of home modifications, aids, and medical devices; the costs paid by the patient versus the health care system for psychiatrists, occupational therapists, dieticians, physiotherapists, and speech therapists; and caregiver disutilities. Indirect costs and caregiver outcomes were not assessed in the MOXIe trial, and no evidence was submitted by the sponsor to support any impact of omaveloxolone on these costs or outcomes.
The impact of omaveloxolone on costs and outcomes related to the societal perspective is highly uncertain due to a lack evidence of the impact of omaveloxolone and uncertainty associated with the chosen model inputs.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
No reanalyses were performed by CDA-AMC owing to uncertainty in the clinical evidence that could not be resolved through reanalysis. Based on the sponsor’s submission, the ICERs for omaveloxolone plus SOC are $1,534,503 per QALY gained when a health care payer perspective is adopted and $1,297,851 per QALY gained when a societal perspective is adopted (Table 3). In both analyses, results were driven by the acquisition cost of omaveloxolone.
Scenario Analysis Results
Based on the sponsor’s submitted health care payer base case, a 97% price reduction would be required to achieve cost-effectiveness of omaveloxolone plus SOC relative to SOC alone at a WTP threshold of $50,000 per QALY. From the societal perspective, a 95% price reduction would be required to achieve cost-effectiveness of omaveloxolone plus SOC relative to SOC alone at this threshold. This would translate to a price of $10 to $19 per 50 mg tablet.
Table 4: CDA-AMC Price Reduction Analyses
Analysis | Unit drug cost ($) | Sponsor’s ICERs for omaveloxolone plus SOC | |
|---|---|---|---|
Price reduction | $ | Health care payer perspective | Societal perspective |
No price reduction | 364 | 1,534,503 | 1,297,851 |
10% | 328 | 1,381,751 | 1,166,160 |
20% | 291 | 1,229,000 | 1,034,469 |
30% | 255 | 1,076,248 | 902,777 |
40% | 219 | 923,497 | 771,086 |
50% | 182 | 770,745 | 639,395 |
60% | 146 | 617,993 | 507,704 |
70% | 109 | 465,242 | 376,013 |
80% | 73 | 312,490 | 244,321 |
90% | 36 | 159,739 | 112,630 |
100% | 19 | 6,987 | Dominant |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care; vs. = versus.
Issues for Consideration
Clinical expert input received by CDA-AMC indicated that the mFARS is not commonly used in clinical practice in Canada. Experts noted that if assessment using the mFARS is required for reimbursement of omaveloxolone, training will be required in the use of this scale, and the amount of time spent with each patient will likely increase. The use of mFARS to determine reimbursement may increase costs to the health care system.
The Health Canada indication for omaveloxolone is for use in patients aged 16 years and older. The clinical experts noted that FA is typically diagnosed around 11 years of age, and indicated a preference for intervention earlier than 16 years of age. The cost-effectiveness of omaveloxolone if used off-label in patients younger than 16 years of age is unknown, owing to a lack of clinical data; however, cost-effectiveness in this group of patients is outside the scope of the current review. There is an ongoing trial for omaveloxolone in the pediatric population.19
Patients with severe pes cavus were excluded by the sponsor from the full analysis set of the MOXIe trial (and, thus, the pharmacoeconomic evaluation). As noted in the CDA-AMC Clinical Review, the analysis of a dataset from the MOXIe trial that included patients with severe pes cavus showed a smaller magnitude of benefit of omaveloxolone compared to placebo in this population. If the clinical effectiveness of omaveloxolone in the full Health Canada–indicated population (i.e., not excluding patients with severe pes cavus) is lower than incorporated in the economic analysis, then the ICER would be higher than predicted by the sponsor.
Costs of genetic testing were not included in the submitted economic model. Genetic testing for confirmation of FA is broadly available across Canada; however, the clinician group input noted that non-neurologic presentations of FA may delay recognition and that genetic testing is performed only if FA is suspected based on clinical presentation. Additional genetic testing would increase costs to the health care system.
Overall Conclusions
The CDA-AMC Clinical Review concluded that, based on observations from the MOXIe trial, omaveloxolone likely results in slower neurologic decline compared to placebo, as measured by mFARS scores over 48 weeks in patients aged 16 years and older with FA; however, the clinical significance of this finding is uncertain. Other functional outcomes, including upper limb function, mobility, and frequency of falls, did not show significant improvements with omaveloxolone compared to placebo. A comparison submitted by the sponsor of patients who received omaveloxolone versus natural history controls suggests that the treatment benefit of omaveloxolone may be maintained for up to 3 years, with treated patients possibly showing slower disease progression compared to matched controls. A long-term extension study suggests that the benefit may be observed within the first year, given that treatment-naive patients were observed to have a better response. However, these findings are limited by potential selection bias and the trial’s open-label design; in addition, the magnitude of differences between omaveloxolone and placebo is uncertain. Thus, the magnitude and durability of omaveloxolone benefit are highly uncertain.
The sponsor submitted an economic analysis comparing the cost-effectiveness of omaveloxolone plus SOC to SOC alone, with effectiveness (as measured by change in mFARs score) informed by data from its submitted propensity score–matched analysis. The results of the sponsor’s base case suggest that omaveloxolone plus SOC will be more effective over a patient’s lifetime than SOC alone (incremental QALYs = 2.59); however, owing to the identified uncertainty in the clinical evidence — including uncertainty as to whether omaveloxolone improves HRQoL and uncertainty regarding the magnitude and durability of changes in mFARS scores — the magnitude of benefit predicted by the sponsor’s economic modelling is uncertain.
Based on the sponsor’s results, omaveloxolone plus SOC is not cost-effective at a WTP threshold of $50,000 per QALY gained. A 95% to 97% price reduction for omaveloxolone would be required for omaveloxolone plus SOC to be cost-effective compared to SOC alone at this WTP threshold. Given the uncertainty in the clinical evidence base, it is highly uncertain whether and to what extent the gains in QALYs will be realized in clinical practice, especially considering that approximately 96% of the incremental QALYs predicted by the sponsor’s model were gained during the extrapolated period and that the sponsor’s model did not include the impact of treatment on non-neurologic outcomes. Therefore, further price reductions may be required.
References
1.Biogen Canada Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: omaveloxolone, capsule, 50 mg oral [internal sponsor's package]. October 2, 2024.
2.Reata Pharmaceuticals Inc. Clinical Study Report. MOXIe Part 2 CSR: RTA 408 (omaveloxolone) 408-C-1402 Part 2: A phase 2 study of the safety, efficacy, and pharmacodynamics of RTA 408 in the treatment of Friedreich’s ataxia [internal sponsor's report]. November 5, 2020.
3.Biogen Canada Inc. Skyclarys (omaveloxolone): capsule, 50 mg oral [product monograph]. March 13, 2025.
4.Friedreich’s Ataxia Research Alliance. Friedreich's Ataxia Integrated Clinical Database (FA-ICD) – Clinical Outcome Measures in Friedreich’s Ataxia (FACOMS) study [sponsor supplied reference]. https://www.curefa.org/research/research-resources/
5.Lynch DR, Chin MP, Boesch S, et al. Efficacy of Omaveloxolone in Friedreich's Ataxia: Delayed-Start Analysis of the MOXIe Extension. Mov Disord. 2023;38(2):313-320. doi:10.1002/mds.29286 PubMed
6.Indelicato E, Reetz K, Maier S, et al. Predictors of Survival in Friedreich's Ataxia: A Prospective Cohort Study. Mov Disord. 2024;39(3):510-518. doi:10.1002/mds.29687 PubMed
7.Reetz K, Dogan I, Hilgers RD, et al. Progression characteristics of the European Friedreich's Ataxia Consortium for Translational Studies (EFACTS): a 4-year cohort study. Lancet Neurol. 2021;20(5):362-372. doi:10.1016/S1474-4422(21)00027-2 PubMed
8.Rummey C, Harding IH, Delatycki MB, Tai G, Rezende T, Corben LA. Harmonizing results of ataxia rating scales: mFARS, SARA, and ICARS. Ann Clin Transl Neurol. 2022;9(12):2041-2046. doi:10.1002/acn3.51686 PubMed
9.National Institute for Health and Care Excellence. Single technology appraisal: Ocrelizumab for treating relapsing multiple sclerosis (ID937). Committee Papers [sponsor supplied reference]. 2018. https://www.nice.org.uk/guidance/ta533/documents/committee-papers
10.Pennington BM. Inclusion of carer health-related quality of life in National Institute for Health and Care Excellence appraisals. Value Health. 2020;23(10):1349-1357. doi:10.1016/j.jval.2020.05.017 PubMed
11.Government of Alberta. Interactive drug benefit list [sponsor supplied reference]. 2023. https://idbl.ab.bluecross.ca/idbl/load.do
12.Alberta Health Interactive Health Data Team. Hospital Inpatient Care Case Costs - CMG/Plex [sponsor supplied reference]. 2015. https://open.alberta.ca/opendata/hospital-inpatient-care-case-costs-cmgplex
13.Ontario Ministry of Health. Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 24, 2023)) [sponsor supplied reference]. 2023. https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf
14.CIHI. Patient Cost Estimator [sponsor supplied reference]. 2020. https://www.cihi.ca/en/patient-cost-estimator
15.ClosingtheGap Healthcare. Home Care Costs in Ontario—A Complete Breakdown [sponsor supplied reference]. 2020. https://www.closingthegap.ca/home-care-costs-in-ontario-a-complete-breakdown/
16.Giunti P, Greenfield J, Stevenson AJ, et al. Impact of Friedreich's Ataxia on health-care resource utilization in the United Kingdom and Germany. Orphanet J Rare Dis. 2013;8(1):38. doi:10.1186/1750-1172-8-38 PubMed
17.Statistics Canada. Average weekly earnings, average hourly wage rate and average usual weekly hours by union status, annual [sponsor supplied reference]. 2024. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1410013401
18.CADTH. Guidelines for the economic evaluation of health technologies: Canada. 2017. Accessed December 15, 2024. https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition
19.Biogen. NCT06054893: A Study to Find Out How BIIB141 (Omaveloxolone) is Processed in the Body and to Learn More About Its Safety in Participants With Friedreich's Ataxia Aged 2 to 15 Years Old (BOLD). ClinicalTrials.gov. Accessed December 15, 2024. https://clinicaltrials.gov/study/NCT06054893
20.Biogen Canada Inc. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: omaveloxolone, capsule, 50 mg oral [internal sponsor's package]. October 2, 2024.
21.Tsou AY, Paulsen EK, Lagedrost SJ, et al. Mortality in Friedreich ataxia. Journal of the neurological sciences. 2011;307(1-2):46-9. doi:10.1016/j.jns.2011.05.023 PubMed
22.Statistics Canada. Table 17-10-0005-01 Population estimates on July 1, by age and gender [sponsor supplied reference]. 2024. https://doi.org/10.25318/1710000501-eng
23.Williams CT, De Jesus O. Friedreich Ataxia. StatPearls. 2024.
24.Delatycki MB, Corben LA. Clinical features of Friedreich ataxia. J Child Neurol. 2012;27(9):1133-7. doi:10.1177/0883073812448230 PubMed
25.Cossée M, Schmitt M, Campuzano V, et al. Evolution of the Friedreich's ataxia trinucleotide repeat expansion: founder effect and premutations. Proc Natl Acad Sci U S A. 1997;94(14):7452-7. doi:10.1073/pnas.94.14.7452 PubMed
26.Statistics Canada. Canada's population estimates: Strong population growth in 2023 [sponsor supplied reference]. 2023. https://www150.statcan.gc.ca/n1/daily-quotidien/240327/dq240327c-eng.htm
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 5: CDA-AMC Cost Comparison Table for Friedreich’s Ataxia
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Omaveloxolone | 50 mg | Oral capsule | 364.2987 | 150 mg (3 capsules of 50 mg each) taken orally once daily | 1,092.90 | 399,180 |
CDA-AMC = Canada’s Drug Agency.
Note: Price of omaveloxolone is from the sponsor’s pharmacoeconomic submission1 and does not include dispensing fees. Recommended dose is from the product monograph.3 Annual cost calculation uses 365.25 days per year.
Appendix 2: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of the Sponsor’s Base Case
Table 6: Disaggregated Summary of the Sponsor’s Economic Evaluation Results — Health Care Payer Perspective
Parameter | Omaveloxolone plus SOC | SOC |
|---|---|---|
Discounted LYs | ||
Total | 28.14 | 26.99 |
Discounted QALYs | ||
Total | 16.24 | 13.65 |
Patient utilities | 16.24 | 13.65 |
Adverse events | 0.00 | 0.00 |
Discounted costs ($) | ||
Total | 5,012,305 | 1,037,040 |
Acquisition costs | 3,968,287 | 0 |
Administration costs | 0 | 0 |
Adverse event costs | 70 | 10 |
Background medication costs | 0 | 0 |
Medical resource use costs | 428,776 | 442,169 |
Comorbidity costs | 615,173 | 594,861 |
LY = life-year; QALY = quality-adjusted life-year; SOC = standard of care.
Source: Sponsor’s pharmacoeconomic submission.1
Table 7: Disaggregated Summary of the Sponsor’s Economic Evaluation Results — Societal Perspective
Parameter | Omaveloxolone plus SOC | SOC |
|---|---|---|
Discounted LYs | ||
Total | 28.17 | 27.03 |
Discounted QALYs | ||
Total | 15.15 | 12.15 |
Patient utilities | 16.24 | 13.65 |
Carer disutilitiesa | −1.09 | −1.50 |
Adverse events | 0.00 | 0.00 |
Discounted costs ($) | ||
Total | 6,431,928 | 2,532,040 |
Acquisition costs | 3,968,287 | 0 |
Administration costs | 0 | 0 |
Adverse event costs | 70 | 10 |
Background medication costs | 0 | 0 |
Medical resource use costs | 428,776 | 442,169 |
Comorbidity costs | 615,173 | 594,861 |
Nonmedical resource use costsa | 0 | 0 |
Patient productivity lossesa | 1,166,053 | 1,225,097 |
Informal caregiver costsa | 184,111 | 176,578 |
LY = life-year; QALY = quality-adjusted life-year; SOC = standard of care.
aIncluded in the societal perspective analysis only.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 3: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 8: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a budget impact analysis (BIA) assessing the expected budgetary impact of reimbursing omaveloxolone for the treatment of FA in adults and adolescents aged 16 years and older.20 The BIA was undertaken using an epidemiologic approach from the perspective of a public payer in Canada over a 3-year time horizon (2025 to 2027). Data to inform the model were obtained from various sources, including the published literature, the sponsor’s internal data, and input from clinical experts consulted by the sponsor.20 To derive the eligible population, the sponsor applied the incidence of FA to the number of people aged 16 years and older. The resulting number of patients with FA was adjusted by the sponsor using a “survival adjustment factor” of 45%, which was intended to adjust for early mortality among FA patients. The sponsor estimated this adjustment factor by dividing the average age of death for FA patients (37 years) by the overall average age of death in Canada (82 years).21 The sponsor additionally used a “provincial prevalence adjustment factor,” derived based on the number of patients with FA registered with MDC,20 to estimate the number of eligible patients in each CDA-AMC–participating jurisdiction. Additional key inputs to the BIA are documented in Table 8.
The sponsor compared a reference scenario in which patients received SOC alone to a new drug scenario in which omaveloxolone was reimbursed as an add-on therapy to SOC. SOC was assumed by the sponsor to comprise treatments for symptom and comorbidity management and to not differ with the addition of omaveloxolone. The uptake of omaveloxolone in the new drug scenario was assumed to be 75% by year 3, based on the sponsor’s internal estimates. The analysis included drug acquisition costs for omaveloxolone based on the sponsor’s submitted price and excluded dispensing fees and markups. No costs for SOC were included in the model.
Table 9: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
People aged 16 and older (CDA-AMC–participating jurisdictions) | 26,093,25622 |
Incidence of FA | |
Survival adjustment factor | 45%a,21 |
Proportion diagnosed with FA | 100%b |
Eligibility for treatment | 100%b |
Public coverage | 80% for all CDA-AMC–participating jurisdictions with exception of 100% NIHBb |
Population growth factor (annual) | 2.40%26 |
Number of patients eligible for drug under review | 219 / 224 / 230 |
Market uptake (3 years) | |
Uptake (reference scenario) Omaveloxolone plus SOC SOC | 0% / 0% / 0% 100% / 100% / 100% |
Uptake (new drug scenario) Omaveloxolone plus SOC SOC | 45% / 65% / 75% 55% / 35% / 25% |
Cost of treatment (per patient, per year) | |
Omaveloxolone SOC | $399,180 $0c |
CDA-AMC = Canada’s Drug Agency; FA = Friedreich’s Ataxia; SOC = standard of care.
aThe sponsor calculated a survival adjustment factor by dividing the average age of death of FA patients by the average age of death in the general population of patients in Canada (i.e., 37 years divided by 82 years).21
bBased on sponsor assumption, clinical expert opinion, or internal estimates.
cSOC was assumed by the sponsor to include treatment of FA symptoms but no disease-specific pharmacological agents.
Summary of the Sponsor’s BIA Results
The sponsor estimated that the 3-year budget impact of reimbursing omaveloxolone for the treatment of FA in patients aged 16 years and older would be $166,382,586 (year 1 = $39,364,303; year 2 = $58,224,178; year 3 = $68,794,105).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The number of patients eligible for omaveloxolone is uncertain. The sponsor used an epidemiologic approach to derive the eligible population, which resulted in an estimated 214 eligible patients in CDA-AMC–participating jurisdictions in the base year (2024). This estimate is lower than that based on registry data from MDC provided by the sponsor. Clinical expert input received by CDA-AMC for this review indicated that, although not all patients with FA may choose to become registered with MDC, these registry data represent the most reliable estimates for Canada. Based on MDC registry data provided by the sponsor, in 2024 there were 361 patients aged 16 years and older with FA in Canada (excluding Quebec), of whom 289 would be considered eligible for treatment (assuming 80% are eligible for public coverage). This suggests that the number of eligible patients in the sponsor’s base case is likely underestimated. CDA-AMC notes that this is likely due to the use of incidence data combined with a sponsor-derived “survival adjustment factor” to estimate the prevalence.
In the base case, CDA-AMC used the registry data provided by the sponsor from MDC to determine the number of patients aged 16 years and older with FA in CDA-AMC–participating jurisdictions. CDA-AMC notes that the true number of patients may still be underestimated based on clinician input.
The uptake of omaveloxolone is uncertain. The sponsor anticipates that 45%, 65%, and 75% of eligible patients would receive omaveloxolone in year 1, year 2, and year 3, respectively, based on internal estimates. Clinician input obtained by CDA-AMC indicated that the uptake of omaveloxolone is likely to be rapid, given the high unmet need (i.e., the absence of other disease modifying treatments) and that market share in year 1 (45%) may be an underestimate. As such, the number of patients who receive omaveloxolone in the sponsor’s analysis may be underestimated.
In scenario analysis, CDA-AMC adopted alternative uptake values, as supplied by the sponsor, to explore the impact of uncertainty in the uptake of omaveloxolone (49.5% in year 1, 71.5% in year 2, and 82.5% in year 3).
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analyses by using the number of patients aged 16 years and older with FA in CDA-AMC–participating jurisdictions to derive the number of eligible patients. The changes made to derive the CDA-AMC base case are described in Table 10.
Table 10: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Number of patients eligible for omaveloxolone | Year 1 = 219 Year 2 = 224 Year 3 = 230 | Year 1 = 296a Year 2 = 303a Year 3 = 310a |
CDA-AMC base case | Reanalysis 1 | |
CDA-AMC = Canada’s Drug Agency.
aIn the CDA-AMC reanalysis, the number of eligible patients in 2024 (the base year; 289 patients) was derived from sponsor-submitted registry data from Muscular Dystrophy Canada. In each subsequent year (years 1 to 3), the population growth rate of 2.40% submitted by the sponsor26 was used to derive the number of eligible patients.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 11 and a more detailed breakdown is presented in Table 12.
In the CDA-AMC base case, the 3-year budget impact of reimbursing omaveloxolone for the treatment of FA in patients aged 16 and older is expected to be $224,535,025 (year 1 = $53,122,535; year 2 = $78,574,132; year 3 = $92,838,358).
Table 11: Summary of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 166,382,586 |
CDA-AMC reanalysis 1 | 224,535,025 |
CDA-AMC base case | 224,535,025 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
CDA-AMC conducted the following scenario analyses to explore remaining uncertainty associated with the potential budgetary impact of reimbursing omaveloxolone, using the CDA-AMC base case. Results are provided in Table 12.
Assuming 100% public coverage.
Assuming uptake of omaveloxolone is 49.5% in year 1, 71.5% in year 2, and 82.5% in year 3 among eligible patients.
Table 12: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 39,364,303 | 58,224,178 | 68,794,105 | 166,382,586 | |
Budget impact | 0 | 39,364,303 | 58,224,178 | 68,794,105 | 166,382,586 | |
CDA-AMC base case | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 53,122,535 | 78,574,132 | 92,838,358 | 224,535,025 | |
Budget impact | 0 | 53,122,535 | 78,574,132 | 92,838,358 | 224,535,025 | |
CDA-AMC scenario analysis 1: 100% public coverage | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 66,403,169 | 98,217,664 | 116,047,948 | 280,668,781 | |
Budget impact | 0 | 66,403,169 | 98,217,664 | 116,047,948 | 280,668,781 | |
CDA-AMC scenario analysis 2: increased market share of omaveloxolone | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 58,434,788 | 86,431,545 | 102,122,194 | 246,988,527 | |
Budget impact | 0 | 58,434,788 | 86,431,545 | 102,122,194 | 246,988,527 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
Ethics Review
Abbreviations
FA
Friedreich’s ataxia
mFARS
modified Friedreich’s Ataxia Rating Scale
Ethical Considerations
Friedreich’s ataxia (FA) is a rare, genetic, life-shortening neuromuscular disorder caused by a trinucleotide repeat expansion in the frataxin gene, which reduces frataxin expression and leads to mitochondrial dysfunction. FA typically manifests in childhood with progressive neurologic impairment and may also involve cardiomyopathy, skeletal abnormalities, and metabolic disturbances, ultimately affecting patients’ mobility, independence, and life expectancy.
This brief report is informed by the sponsor’s submission as well as input received by Canada’s Drug Agency from patient groups, clinician groups, and drug plans. It is also informed by direct consultation with 4 clinical experts (3 pediatric neurologists and 1 neurologist specializing in adults) who have experience treating pediatric patients with FA.
This brief report highlights ethical considerations regarding the use of omaveloxolone for the treatment of FA in adults and adolescents aged 16 years or older. It outlines considerations that are relevant for decision-making regarding the public reimbursement and implementation of omaveloxolone in Canada. However, it does not present an exhaustive list of all ethical considerations associated with FA and its treatment.
Diagnosis, Treatment, and Experiences of People Living With FA
FA substantially reduces life expectancy, with most patients worldwide living an average of 37 years.1 Cardiac complications account for the majority of deaths, alongside risks such as pneumonia, stroke, and diabetic coma.1 Beyond its life-limiting nature, FA imposes significant physical and psychosocial burdens on patients and their caregivers. The progressive loss of coordination, balance, and muscle strength typically leads to reliance on mobility aids within 8 years to 10 years of symptom onset, with many patients requiring a wheelchair 11 years to 15 years after diagnosis.2,3 Comorbidities, such as cardiomyopathy, diabetes, and scoliosis, further contribute to fatigue, pain, and reduced mobility. The patient group input noted that as the condition progresses, people slowly lose their ability to function independently and often become unable to participate in social activities or the workplace due to impacts on mobility, coordination, and speech. This may result not only in social isolation and a lost sense of autonomy, but in mental health issues; the patient group input reports experiences of anxiety and depression among patients with FA. Caregivers also face challenges as they strive to balance the demands of care with their own mental health, work responsibilities, and financial stability.
Both the patient and clinician group input indicated that, while FA is typically diagnosed in childhood or adolescence, disparities in diagnosis and referral pathways can delay access to essential care. Although the requisite genetic testing for confirmation of FA is broadly available across Canada, the clinician group input noted that non-neurologic presentations may delay recognition and that testing occurs only when clinical suspicion arises.
There are currently no therapies indicated to slow or halt the progression of FA. Instead, patients and clinicians rely on treatment options oriented toward rehabilitation (e.g., physiotherapy and occupational therapy) and managing complications. Both the patient group input and clinical experts suggested that existing care is inadequate, with supportive therapies providing inconsistent benefits and access often limited by factors such as out-of-pocket costs, lack of insurance coverage, and variable geographic availability of specialized services. In advanced stages, logistical and financial barriers to accessing specialized care centres compound these challenges. Taken together, this underscores a high unmet need for an accessible treatment option that can address the progression of FA and improve quality of life for patients and their families.
The patient group input and clinical experts indicated that people with FA want an intervention that can slow or stop the progression of FA. Interventions that could improve symptom control (which may come with slowed progression), preserve mobility, improve energy levels, preserve quality of life, and prevent long-term complications (e.g., scoliosis, diabetes, and heart problems) would also be highly valued.
Clinical Evidence Used in the Evaluation of Omaveloxolone
The safety and efficacy of omaveloxolone in patients aged 16 years to 40 years with genetically confirmed FA were evaluated in the MOXIe trial (N = 103), a pivotal, phase II, randomized, double-blinded, placebo-controlled study. The trial’s primary objective was to assess whether a daily oral 150 mg dose of omaveloxolone improved modified Friedreich’s Ataxia Rating Scale (mFARS) scores by week 48 when compared to placebo. The scale is a composite measure that evaluates neurologic function in patients with FA, including speech, upper and lower limb coordination, and standing and walking ability. The results demonstrated a statistically significant improvement in mFARS scores for patients receiving omaveloxolone versus placebo. However, the Clinical Review notes that the clinical significance of this improvement remains uncertain due to the absence of an established minimal clinically important difference for mFARS scores. Additionally, other functional measures included as secondary outcomes (e.g., changes in performance on a 9-hole peg test and 25-foot timed walk test) did not demonstrate meaningful improvements with treatment. No end points examined changes in health-related quality of life or experiences with symptoms such as fatigue, which are important outcomes to patients. Further details on the MOXIe trial are provided in the Clinical Review.
The sponsor-submitted real-world evidence suggests that the benefits observed in mFARS scores during the initial 48 weeks of treatment may be maintained over 3 additional years of treatment, with treated patients showing slower disease progression compared to natural history. However, long-term data beyond 3 years are limited. Ongoing data collection and monitoring are essential for understanding whether omaveloxolone offers sustained clinical value.
The clinical experts considered the MOXIe trial results to be broadly generalizable to the population of patients with FA; the predominantly white trial population (98%) was seen as appropriate, given that the condition occurs primarily in people who are white. However, the exclusion of patients with clinically significant cardiac disease — a common FA comorbidity — represents a notable gap in the evidence. While this absence would not deter clinical experts from prescribing omaveloxolone to patients with comorbid cardiac disease, the experts emphasized the importance of careful monitoring and the collection of real-world data to inform future clinical decision-making in this population. Similarly, individuals aged younger than 16 years were excluded from the MOXIe trial, leaving a significant data gap in the understanding of the safety and efficacy of omaveloxolone for this population.
Clinical Use of Omaveloxolone
Omaveloxolone represents the first potential disease-modifying treatment for FA, addressing a critical unmet need in this population. The clinical experts indicated that they would prescribe it as a first-line treatment for patients aged 16 years and older, given the lack of alternatives. They expressed confidence in its safety profile and considered the observed slowing of disease progression, as measured by changes in mFARS scores over the first 48 weeks of treatment, to be promising. Real-world evidence suggests that the initial benefit in delayed progression may be sustained for up to 3 years. Both the clinical expert and patient group input underscored the significance of this potential benefit, with patients emphasizing their hope for treatments that could slow disease progression.
While omaveloxolone appears to be well-tolerated overall, the clinical experts highlighted uncertainties in managing specific adverse events and pre-existing comorbidities. Elevated liver enzymes were noted as a particular challenge, with the lack of clear guidance from the sponsor making decisions about discontinuing treatment difficult in cases of prolonged elevation. Similarly, the exclusion of individuals with significant cardiac disease limits the understanding of the safety of omaveloxolone in this group, leaving providers to navigate potential risks without robust evidence. These gaps underscore the importance of thorough informed consent processes and close monitoring to address patient-specific complexities.
The clinical experts anticipate that if omaveloxolone is recommended for public reimbursement, caregivers will be interested in having it prescribed off-label to patients younger than 16 years of age, despite the current absence of data in this population. This scenario poses an ethical dilemma for providers, who may experience moral distress when trying to balance an uncertain risk-benefit profile in this population against the potential harm of waiting to treat an early-onset, progressive disease. Clinical experts noted that this could lead to disparate prescribing practices, with some providers offering off-label treatment and others adhering strictly to the indicated population, potentially creating inequities in access and care. This variability could risk eroding trust in health care providers because families navigating these decisions may encounter conflicting recommendations. The clinical experts noted that if providers choose to prescribe to this population, they will need to engage in transparent conversations with families, clearly addressing the known risks, lack of evidence, and uncertainties surrounding potential outcomes.
The clinical experts noted that patients with FA are typically managed in specialized settings by multidisciplinary care teams with specialization in FA. With the introduction of omaveloxolone, the experts indicated that continued reliance on these specialized clinics will be essential to ensure appropriate monitoring and treatment. However, this requirement could create barriers for patients in rural or underserved regions who may already face challenges accessing care. Additionally, implementing intensive or standardized outcome measures, such as mFARS, could strain clinical capacity and limit accessibility. Ensuring equitable access to omaveloxolone will require careful consideration of how monitoring protocols can be adapted to balance feasibility and fairness across diverse care settings.
Health Systems Impact
Introducing omaveloxolone raises ethical considerations about resource allocation and health system sustainability, given the therapy’s high cost and limited long-term safety and efficacy evidence. The patient group input suggested potential downstream savings, such as fewer emergency visits and prolonged workforce participation by patients; however, there is currently no evidence to support this. The clinical experts emphasized the importance of collecting real-world data through registries to better understand omaveloxolone’s impact on function, symptoms, quality of life, mortality, and health care costs. However, they cautioned that data-collection efforts could place great financial and administrative burdens on clinicians and health systems, particularly in under-resourced settings.
The potential use of mFARS as a standardized tool to guide treatment initiation, monitoring, and renewal raises significant practical and resource challenges, given that it risks overburdening the health system while offering unknown benefit. While mFARS was the primary end point in the MOXIe trial, it is not routinely used in clinical practice and would require specialized training, extended appointment times, and greater administrative coordination to implement effectively. These requirements could strain under-resourced clinics, particularly those in underserved areas, and bias prescribing toward larger, well-equipped centres. Further, because the mFARS is reliant on clinician interpretation and is subject to variability, its use in clinical practice may create inconsistencies in treatment decisions. As such, the clinical experts questioned whether such intensive monitoring efforts justify the significant resources required, particularly given the subjective nature of the measures and their view that providers would be reluctant to discontinue therapy in the absence of serious harms. Instead, the clinical experts suggested that, instead of depending solely on trial scales, long-term monitoring should rely on clinical judgment, assessments of patient function (e.g., gait, upper limb coordination, and bulbar function), and possibly the use of adapted objective measures every 6 months to 12 months.
References
1.Tsou AY, Paulsen EK, Lagedrost SJ, et al. Mortality in Friedreich ataxia. J Neurol Sci. 2011;307(1-2):46-9. doi:10.1016/j.jns.2011.05.023 PubMed
2.Rummey C, Farmer JM, Lynch DR. Predictors of loss of ambulation in Friedreich's ataxia. EClinicalMedicine. 2020;18:100213. doi:10.1016/j.eclinm.2019.11.006 PubMed
3.Bürk K. Friedreich Ataxia: current status and future prospects. Cerebellum Ataxias. 2017;4:4. doi:10.1186/s40673-017-0062-x PubMed
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.