Drugs, Health Technologies, Health Systems
Reimbursement Review
Dupilumab (Dupixent)
Sponsor: Sanofi-Aventis Canada Inc.
Therapeutic area: Chronic obstructive pulmonary disease
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
AESI
adverse event of special interest
CAT
COPD Assessment Test
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
COPD
chronic obstructive pulmonary disease
CTS
Canadian Thoracic Society
E-RS:COPD
Evaluating Respiratory Symptoms in COPD
FeNO
fractional exhaled nitric oxide
FEV1
forced expiratory volume in the first second
FVC
forced vital capacity
GOLD
Global Initiative for Chronic Obstructive Lung Disease
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
IA
interim analysis
ICS
inhaled corticosteroid
Ig
immunoglobulin
IL
interleukin
ITT
intention to treat
LABA
long-acting beta-agonist
LAMA
long-acting muscarinic antagonist
LS
least square
MCID
minimally clinically important difference
MID
minimal important difference
mMRC
modified Medical Research Council
MRC
Medical Research Council
PMM-MI
pattern mixture model–multiple imputation
RCT
randomized controlled trial
RR
relative risk
SABA
short-acting beta-agonist
SAE
serious adverse event
SC
subcutaneous
SCS
systemic corticosteroid
SD
standard deviation
SGRQ
St. George's Respiratory Questionnaire
TEAE
treatment-emergent adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Dupilumab (Dupixent), 300 mg single-use syringe or pen (300 mg/2 mL), solution for subcutaneous injection |
Sponsor | Sanofi-Aventis Canada Inc. |
Indication | Add-on maintenance treatment in adult patients with chronic obstructive pulmonary disease (COPD) characterized by raised blood eosinophils inadequately controlled by the combination of an inhaled corticosteroid (ICS), a long-acting beta2 agonist (LABA), and a long-acting muscarinic antagonist (LAMA), or on a combination of a LABA and a LAMA if ICS is not appropriate. |
Reimbursement request | As per the Health Canada indication, add-on maintenance treatment in adult patients with COPD characterized by raised blood eosinophils inadequately controlled by the combination of an ICS, a LABA, and a LAMA, or on a combination of a LABA and a LAMA if ICS is not appropriate, if the following conditions are met:
|
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | October 21, 2025 |
Recommended dose | 300 mg by subcutaneous injection once every other week |
COPD = chronic obstructive pulmonary disease; FEV1 = forced expiratory volume in the first second; FVC = forced vital capacity; ICS = inhaled corticosteroid; LABA = long-acting beta-agonist; LAMA = long-acting muscarinic antagonist; MRC = Medical Research Council; mMRC = modified Medical Research Council; NOC = Notice of Compliance.
Note: mMRC refers to the mMRC Dyspnea Scale grade. mMRC ≥ grade 1 is equivalent to MRC ≥ grade 2.
aModerate exacerbations defined as acute exacerbation of COPD that require either systemic corticosteroids (intramuscular, IV, or oral) and/or antibiotics. Severe exacerbations are defined as acute exacerbation of COPD requiring hospitalization or observation > 24 hours in an emergency department or urgent care facility.
Introduction
Chronic obstructive pulmonary disease (COPD) is a progressive inflammatory lung condition characterized by airflow obstruction, hyperinflation, and systemic effects, often associated with chronic bronchitis and emphysema.1,2 It manifests with dyspnea, wheezing, cough, sputum production, fatigue, and activity limitation, with exacerbations leading to worsened lung function, increased morbidity, and substantial health care costs.3 COPD affects approximately 8.7% of people living in Canada aged 35 years and older, with prevalence increasing over the past 2 decades.4 COPD remains a leading global cause of death, accounting for 3.23 million deaths in 2019.5,6 The disease is associated with comorbidities such as cardiovascular disease, osteoporosis, malnutrition, and metabolic syndrome,1 and up to 40% of patients exhibit type 2 inflammation, which correlates with worse outcomes.7-13 Standard therapy aims to reduce symptoms, prevent exacerbations, improve quality of life, and extend survival.3,14 Management strategies include smoking cessation, risk factor reduction, pulmonary rehabilitation, vaccinations, and patient education on symptom management.14,15 Pharmacological treatment follows guideline-based recommendations, with mild cases managed using a single inhaled long-acting muscarinic antagonist (LAMA) or long-acting beta-agonist (LABA).3,14,15 Patients with moderate to severe disease receive dual bronchodilator therapy (LABA-LAMA) if at low risk for exacerbations, while those at high risk require triple therapy with LABA, LAMA, and an inhaled corticosteroid (ICS).3,14,15 Additional therapies such as macrolides and roflumilast are reserved for specific populations, though concerns regarding antibiotic resistance and adverse effects limit their widespread use.16,17 The clinical experts noted that despite available treatments, COPD exacerbations remain common, and no currently approved therapies have demonstrated a significant reduction in exacerbations for patients already receiving triple therapy. Moreover, nondrug interventions such as smoking cessation, pulmonary rehabilitation, and noninvasive ventilation play critical roles in improving outcomes but remain underused due to accessibility challenges. Dupilumab has recently emerged as a potential add-on therapy for patients with elevated blood eosinophil levels (≥ 300 cells/µL) and symptoms of chronic bronchitis, as outlined in the Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2025 guidelines.15 Substantial gaps persist in achieving optimal symptom control, preventing disease progression, and addressing quality of life limitations in patients with COPD.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of dupilumab 300 mg/2 mL for subcutaneous (SC) injection as an add-on maintenance treatment in adult patients with COPD characterized by raised blood eosinophils inadequately controlled by the combination of an ICS, a LABA, and a LAMA, or on a combination of a LABA and a LAMA if ICS is not appropriate.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to our call for input and from clinical experts consulted by for the purpose of this review.
Patient Input
Two patient groups, the Chronic Obstructive Pulmonary Disease Association (COPD Canada) and the Lung Health Foundation, provided input for this review. COPD Canada gathered responses from an email survey conducted in November 2024, with 61 participants, while the Lung Health Foundation collected data through an online survey of 27 patients across Canada from December 2022 to December 2024, along with additional feedback from 7 individuals in Ontario. None of the patients had experience with the drug under review. Both groups highlighted the substantial burden of COPD on daily life, affecting fundamental activities such as breathing, working, and socializing. Shortness of breath, fatigue, and coughing were among the most debilitating symptoms, leading to difficulties in performing routine tasks and contributing to feelings of isolation. Patients emphasized the need for treatments that alleviate physical symptoms while improving overall quality of life. While current therapies offer some benefits, such as reduced shortness of breath and improved exercise capacity, residual symptoms, and side effects remain problematic. The input underscored the need for additional COPD treatments that minimize side effects, enhance treatment outcomes, and improve quality of life. Access to medications through public drug plans was also identified as a crucial concern for many older patients.
Clinician Input
Input From Clinical Experts Consulted for This Review
The clinical experts consulted for this review highlighted that despite the availability of triple therapy with LABA, LAMA, and ICS, many patients with COPD continue to experience exacerbations, and no current treatment fully eliminates them. According to the clinical experts, COPD remains a progressive disease with irreversible lung damage, and while smoking cessation and pulmonary rehabilitation offer some benefits, they do not halt disease progression. Poor inhaler technique, disease severity, and comorbidities often limit the real-world effectiveness of inhaled medications, leading to suboptimal adherence and symptom control. The clinical experts noted that given these limitations, there is an unmet need for additional therapies that improve exacerbation prevention, symptom relief, and long-term adherence. Dupilumab is considered an add-on therapy for patients who continue to experience exacerbations despite optimal inhaler therapy. It targets type 2 inflammation through systemic effects, complementing rather than replacing current treatments. The clinical experts emphasized that dupilumab should be reserved for patients with type 2 inflammation who remain at high risk despite triple therapy, aligning with clinical trial criteria.
The clinical experts suggested that the ideal patient population for dupilumab includes those with COPD and type 2 inflammation, particularly those with eosinophil levels of at least 300 cells/µL and a history of moderate to severe exacerbations. The clinical experts emphasized the importance of spirometry to confirm COPD diagnosis, as misdiagnosis rates are high. According to the clinical experts, treatment response is primarily assessed by reductions in exacerbations, improvement in quality of life, symptom relief, and increased functional capacity. The clinical experts noted that a lack of reduction in exacerbations within a year, unless attributed to another comorbidity, may indicate a lack of benefit, though improvements in validated COPD measures may still justify continued treatment. According to the clinical experts, discontinuation should also be considered in cases of substantial adverse effects. Dupilumab should be prescribed by respiratory specialists to ensure appropriate patient selection and monitoring.
Clinician Group Input
One clinician group, the Canadian Thoracic Society (CTS), provided input for this review, with a total of 5 clinicians contributing. In alignment with the clinical experts consulted for this review, the clinician group emphasized preventing exacerbations, improving symptoms, and optimizing treatment for patients with COPD. Also, the clinician group aligned with the clinical experts in identifying unmet needs, highlighting the importance of phenotyping patients with COPD to personalize treatment, and recognizing the need for additional therapies for those experiencing exacerbations despite optimized inhaled therapy. Both groups agreed that dupilumab should be reserved for patients with type 2 inflammation, specifically those with elevated blood eosinophil levels (≥ 300 cells/µL), and prescribed by respirologists managing COPD.
The clinician group stated that dupilumab would be the first biologic therapy available for COPD and could represent a paradigm shift by providing a targeted approach for patients with persistent exacerbations. They recommended that patients receive at least 6 months of treatment before considering discontinuation unless side effects necessitate earlier cessation. Additionally, they advised that the first doses should be administered in a clinical setting to ensure proper technique and patient tolerance, with subsequent doses potentially given at home.
Drug Program Input
Input was obtained from the drug programs that participate in the reimbursement review process of Canada’s Drug Agency (CDA-AMC). The following were identified as key factors that could potentially impact the implementation of a CDA-AMC recommendation for dupilumab:
relevant comparators
considerations for initiation of therapy
considerations for continuation or renewal of therapy
considerations for discontinuation of therapy
considerations for prescribing of therapy
generalizability
system and economic issues.
The clinical experts consulted for this review provided advice on the potential implementation issues raised by the drug programs. Refer to Table 3 for more details.
Clinical Evidence
Systematic Review
Description of Studies
The sponsor-conducted systematic literature review identified 2 multicentre, multinational, double-blind, identically designed, placebo-controlled, randomized controlled trials (RCTs) (BOREAS [N = 939] and NOTUS, a confirmatory study of the BOREAS study [N = 935]) that assessed the efficacy and safety of dupilumab relative to placebo in adults with uncontrolled COPD (i.e., at least 2 moderate or 1 severe exacerbations in the previous year and Medical Research Council [MRC] Dyspnea Scale grade ≥ 2, which is equivalent to modified MRC [mMRC] ≥ grade 1); moderate to severe airflow obstruction (i.e., postbronchodilator forced expiratory volume in the first second [FEV1] > 30% to ≤ 70% of predicted) despite receiving maximum standard of care background therapy, including LABA, LAMA, and ICS (unless ICS was contraindicated); and evidence of type 2 inflammation (with blood eosinophil count ≥ 300 cells/µL). The study population consisted of patients aged 40 to 80 years in the BOREAS study and aged 40 to 85 years in the NOTUS study who currently smoke or formerly smoked with a smoking history of at least 10 pack-years and who did not have either a history of or current diagnosis of asthma. In both studies, patients were randomized in a 1:1 ratio to receive SC dupilumab 300 mg every 2 weeks plus best supportive care or placebo plus best supportive care for a 52-week randomized study intervention period, followed by a 12-week safety postintervention follow-up period.
Outcomes of interest to this review included the annualized rate of moderate (requiring either systemic corticosteroid [SCS] and/or antibiotics) or severe (requiring hospitalization or observation for more than 24 hours in an emergency department or urgent care facility) COPD exacerbation, prebronchodilator FEV1 (at week 12 and week 52), St. George’s Respiratory Questionnaire (SGRQ) total score, and treatment-emergent adverse events of special interest (AESIs). At baseline, all patients in both studies had a history of smoking, with 30% of patients who currently smoke. The proportion of patients with moderate airflow limitation (i.e., GOLD grade 2 or predicted postbronchodilator FEV1 between ≥ 50% and < 80%) was █████ ██ █████ across the treatment arms in the BOREAS and NOTUS studies. The proportion of patients with severe airflow limitation (i.e., GOLD grade 3 or predicted postbronchodilator FEV1 of ≥ 30% and < 50%) was █████ ██ █████. A total of ██ ██████ patients in the BOREAS study and ██ ██████ patients in the NOTUS study had airflow limitation categorized as very severe (i.e., GOLD grade 4 or predicted postbronchodilator FEV1 < 30%).
Efficacy Results
Annualized Rate of Moderate or Severe COPD Exacerbations
As the primary end point in both trials, the adjusted annualized rate of moderate or severe COPD exacerbations over the 52-week intervention period in the intention-to-treat (ITT) population was lower in the dupilumab group compared with the placebo group: 0.78 (95% confidence interval [CI], 0.64 to 0.93) versus 1.10 (95% CI, 0.93 to 1.30) with a relative risk (RR) of 0.70 (95% CI, 0.58 to 0.86; P < 0.001) in the BOREAS study, and 0.86 (95% CI, 0.70 to 1.06) versus 1.30 (95% CI, 1.05 to 1.60) with an RR of 0.66 (95% CI, 0.54 to 0.82; P < 0.001) in the NOTUS study. The absolute difference between groups was ██████ ████ ███ ██████ ██ ███████ in the BOREAS study, and ██████ ████ ███ ██████ ██ ███████ in the NOTUS study.
Change From Baseline in Prebronchodilator FEV1 at Week 12
The least squares (LS) mean change from baseline to week 12 in prebronchodilator FEV1 in the ITT population was an increase of 0.160 L in the dupilumab group versus 0.077 L in the placebo group (between-group LS mean difference = 0.083 L; 95% CI, 0.042 L to 0.125 L; P < 0.001) in the BOREAS study, and an increase of 0.139 L in the dupilumab group versus 0.057 L in the placebo group (between-group LS mean difference = 0.082 L; 95% CI, 0.040 L to 0.124 L; P < 0.001) in the NOTUS study.
Change From Baseline in Prebronchodilator FEV1 at Week 52
The LS mean change from baseline to week 52 in prebronchodilator FEV1 in the ITT population was an increase of 0.153 L in the dupilumab group versus 0.070 L in the placebo group (between-group LS mean difference = 0.083 L; 95% CI, 0.038 L to 0.128 L; P < 0.001) in the BOREAS study, and an increase of 0.115 L in the dupilumab group versus 0.054 L in the placebo group (between-group LS mean difference = 0.062 L; 95% CI, 0.011 L to 0.113 L; P = 0.02) in the NOTUS study.
Change From Baseline in SGRQ Total Score at Week 52
The LS mean change from baseline to week 52 in SGRQ total score was −9.7 in the dupilumab group versus −6.4 in the placebo group (between-group LS mean difference = −3.4; 95% CI, −5.5 to −1.3; P = 0.002) in the BOREAS study, and −9.8 in the dupilumab group versus −6.4 in the placebo group (between-group LS mean difference = −3.4; 95% CI, −5.8 to −0.9; nominal █ ██████) in the NOTUS study.
Harms Results
Treatment-Emergent Adverse Events, Serious Adverse Events, and Withdrawal Due to Adverse Events
Treatment-emergent adverse events (TEAEs) were reported in 66% to 77% of patients across the treatment groups in the BOREAS and NOTUS studies and occurred in similar proportions of patients between the dupilumab and placebo groups. The most common TEAEs for dupilumab in either study were nasopharyngitis, upper respiratory tract infections, COVID-19, headache, diarrhea, and back pain. Treatment-emergent serious adverse events (SAEs) were reported in 13% to 16% of patients across the treatment groups in the 2 studies. The most common treatment-emergent SAE was █████ ████████████ ██ ████ ███ ██ ███ ██ █████ ███ █████████ █████████ ██ ████ █████████ ███ ███ ██████████ ██ ██ ██████ ██ ███ ███ ███████████ ██████████ █████████ █████ ██ ██████ ████████ █████ ██ ██████ ███ ████████ █████████ █████ ██ █████ ████ ███ ████ frequently reported serious infections. Withdrawal due to TEAEs (3% to 4%) were uncommon in the studies.
Mortality
In the BOREAS study, overall, 16 patients died during the on-study period, 8 (1.7%) in each intervention group. In the NOTUS study, overall, 21 patients died during the on-study period, 13 (2.8%) in the dupilumab group and 8 (1.7%) in the placebo group.
Treatment-Emergent AESIs
In the BOREAS study, the proportion of patients who experienced any treatment-emergent AESI was generally comparable between the groups: ████ ██████ ████ with dupilumab and placebo, respectively. Injection site reactions were more frequently reported in the dupilumab group compared to the placebo group (████ ██████ ████).
In the NOTUS study, the proportion of patients with any treatment-emergent AESI was numerically higher in the dupilumab group compared to the placebo group: ████ ██████ █████ respectively. The between-group difference was mostly driven by AESIs of ███████ █████████ ███████████ █████ ███████ █████ ███ ███ ███ ██ █████████ ████ ████████ ███ ██████████████ ███████████ █████ ████████ ██████.
Critical Appraisal
The BOREAS and NOTUS trials ensured allocation concealment through an interactive voice response or web response system. Randomization was stratified based on COPD prognosis factors, although the BOREAS study did not account for smoking status; however, its distribution remained balanced between the 2 treatment groups (those who currently smoke = 29% in the dupilumab group and 31% in the placebo group). Baseline characteristics were generally comparable between groups, except for notably higher serum immunoglobulin (Ig) E levels in the dupilumab arms compared with the placebo arms in both studies, a difference the clinical experts deemed unlikely to affect COPD-related outcomes. Blinding was maintained, yet patients might have inferred their treatment from symptom changes, potentially biasing subjective efficacy outcomes in favour of dupilumab and safety outcomes in favour of placebo. Objective measures, such as COPD exacerbation rates and prebronchodilator FEV1, had a low risk of bias. Sample size calculations were appropriate, although the NOTUS study reduced its week 52 end point population due to an interim analysis (IA). The primary reason for discontinuation was patient withdrawal (5% to 6% in the BOREAS study, and 4% to 5% in the NOTUS study), with small between-group differences unlikely to impact results. Missing data in key end points were handled through the restricted maximum likelihood method, and sensitivity analyses confirmed robust findings. Multiplicity control appeared adequate, minimizing risk of type I error. A negative binomial regression model was prespecified for COPD exacerbation counts, showing similar results using the Poisson regression in both trials. While subgroup analyses based on eosinophil counts were prespecified, the lack of sample size consideration and multiplicity control limits the reliability of these findings. The trials’ year-long duration minimized seasonal confounding, and the SGRQ total score’s minimal important difference (MID) estimate (4 points) was considered clinically meaningful, despite originating from within-group changes rather than direct between-group comparisons.
The BOREAS and NOTUS studies were multinational RCTs, with ██ ███ ██ ████████ patients, respectively. The clinical experts noted that trial eligibility criteria and the baseline characteristics of the patients enrolled broadly reflected the population of people living in Canada eligible for dupilumab treatment, although in practice, individuals outside the specified age ranges or with asthma may still be considered for treatment. The dupilumab dosing regimen in both studies aligned to the draft product monograph, and concomitant medications used (LABA, LAMA, ICS, SCS, and rescue therapies) mirrored standard COPD management in Canada, according to the clinical experts. The primary efficacy outcomes — annualized exacerbation rate, prebronchodilator FEV1, and health-related quality of life (HRQoL) — were clinically meaningful, with the clinical experts emphasizing that all exacerbations, regardless of severity, matter to patient health and health care utilization. The clinical experts noted that the 52-week randomized treatment period and the 12-week postrandomization safety follow-up were adequate to assess dupilumab’s efficacy. However, the clinical experts felt that a longer duration of follow-up would be required to capture the long-term safety of dupilumab, particularly for potential rare adverse events (AEs), such as helminth infections. Of note, the sponsor stated that dupilumab has been available in Canada since 2017, and for patients as young as 6 months since 2023. The BOREAS and NOTUS studies were the sole phase III RCTs submitted for review, with no head to head or indirect comparisons against other active therapies (azithromycin and roflumilast). Nonetheless, the absence of comparative evidence is unlikely to be a concern given that based on clinical expert input, there are currently no appropriate comparators of dupilumab in the treatment of uncontrolled COPD associated with type 2 inflammation in Canada.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, Grading of Recommendations Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group. Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
The selection of outcomes for GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
annualized rate of moderate or severe COPD exacerbation
mean change from baseline in prebronchodilator FEV1 to week 12 and week 52
HRQoL assessed with SGRQ total score
treatment-emergent AESIs.
Findings from the BOREAS and NOTUS studies were considered together and summarized narratively per outcome because these studies were similar in population, interventions, design, and outcome measures. The GRADE summary of findings for dupilumab versus placebo in adult patients with uncontrolled COPD associated with type 2 inflammation is presented in Table 2.
Table 2: Summary of Findings for Dupilumab vs. Placebo as an Add-on Maintenance Treatment in Adult Patients With Uncontrolled COPD Associated With Type 2 Inflammation
Outcome and follow-up | Patients (studies), N | Effect | Certainty | What happens |
|---|---|---|---|---|
Primary efficacy outcome | ||||
Annualized rate of moderate or severe COPD exacerbation, event rate (95% CI) Follow-up: 52 weeks | 1,874 (2 RCTs) | BOREAS trial
NOTUS trial
| High | Dupilumab results in a clinically important decrease in the annualized rate of moderate or severe COPD exacerbation when compared with placebo. |
Lung function | ||||
Prebronchodilator FEV1, change from baseline (L), LS mean (SE) Follow-up: 12 weeks | 1,860 (2 RCTs) | BOREAS trial
NOTUS trial
| Moderatea | Dupilumab likely results in little to no clinically meaningful difference in the prebronchodilator FEV1 when compared with placebo at 12 weeks. |
Prebronchodilator FEV1, change from baseline (L), LS mean (SE) Follow-up: 52 weeks | 1,650 (2 RCTs) | BOREAS trial
NOTUS trial
| Moderatea | Dupilumab likely results in little to no clinically meaningful difference in the prebronchodilator FEV1 when compared with placebo at 52 weeks. |
HRQoL (SGRQ) | ||||
SGRQ total score (0 [best HRQoL] to 100 [worst HRQoL]), change from baseline (points), LS mean (SE) Follow-up: 52 weeks | 1,604 (2 RCTs) | BOREAS trial
NOTUS trial
| Moderateb | Dupilumab likely results in little to no clinically meaningful difference in SGRQ improvement when compared with placebo. |
Harms | ||||
Proportion of patients with treatment-emergent AEs of special interestc (95% CI) Follow-up: 52 weeks | 1,872 (2 RCTs) | BOREAS trial ██████████ ██ ███ █████ ███ ██ ███ ███ ██████ ████████ ██ ███ █████ ███ ██ ███ ███ ██████ ███████████ █ █████ ███ █████ █████ ██ █████ ██ ██ ████ ███ ██████ NOTUS trial ██████████ ██ ███ █████ ███ ██ ███ ███ ██████ ████████ ██ ███ █████ ███ ██ ██ ███ ██████ ███████████ ██ ████ ███ █████ █████ ██ █████ ██ ██ ████ ███ ██████ | Lowd | Dupilumab may result in little to no difference in proportion of patients with treatment-emergent AEs of special interest when compared with placebo. |
AE = adverse event; CI = confidence interval; COPD = chronic obstructive pulmonary disease; FEV1 = forced expiratory volume in the first second; HRQoL = health-related quality of life; IMP = investigational medicinal product; LS = least square; MID = minimal important difference; NR = not reported; RCT = randomized controlled trial; SAE = serious adverse event; SE = standard error; SGRQ = St. George’s Respiratory Questionnaire; vs. = versus.
Notes: Study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes. In this table, the data presented for the BOREAS study are from the data cut-off date of February 8, 2023 (last patient end of treatment visit). The data presented for the NOTUS study are the primary efficacy analysis results based on the interim analysis data cut-off date of September 29, 2023.
aThe level of certainty was rated down 1 level for serious imprecision. A difference of 0.1 L between the groups was identified by the clinical expert consulted for this review as a threshold of clinical importance for this outcome. The point estimates suggested little to no difference, and the 95% CIs for the between-group difference crossed the MID threshold in the BOREAS and NOTUS studies.
bThe level of certainty was rated down 1 level for serious imprecision. Based on the MID identified in the literature (4 points based on within-group data),18,19 the point estimates suggested little to no difference, and the 95% CIs for the between-group difference crossed the MID threshold in the BOREAS and NOTUS studies.
cAn AE of special interest is an AE (serious or nonserious) of scientific and medical concern specific to the sponsor’s product or program, for which ongoing monitoring and immediate notification by the investigator to the sponsor is required. Specifically, AEs of special interest in the BOREAS and NOTUS studies included the following: clinically symptomatic eosinophilia (or eosinophilia associated with clinical symptoms), anaphylactic reactions or systemic allergic reactions that are related to IMP and require treatment, severe injection site reactions that last longer than 24 hours, and any infection meeting at least 1 of the criteria (any serious infection [SAE] that requires parenteral [IV, intramuscular, or subcutaneous] antimicrobial therapy; oral antimicrobial therapy for longer than 2 weeks; is a parasitic infection; or is an opportunistic infection).
dThe level of evidence was rated down 2 levels for very serious imprecision. The review team was unable to identify the MID to assess a between-group difference from the literature or the clinical experts consulted for this review; therefore, the null was used to assess certainty. The 95% CIs of the absolute effects included the null threshold of 0 in the BOREAS and NOTUS studies.
Sources: Clinical Study Reports for the BOREAS and NOTUS studies20,21 and sponsor’s submissions.22,23 Details included in the table are from the sponsor’s summary of clinical evidence.24
Long-Term Extension Studies
No long-term extension studies were submitted for this review.
Indirect Comparisons
No studies with indirect evidence were submitted for this review.
Studies Addressing Gaps in the Evidence From the Systematic Review
No studies addressing gaps in the pivotal and RCT evidence were submitted for this review.
Conclusions
Direct comparative evidence from 2 double-blind RCTs (BOREAS and NOTUS) demonstrated that in adult patients with uncontrolled COPD who had type 2 inflammation and moderate to severe airflow obstruction despite receiving triple inhaler therapy (LABA, LAMA, and ICS), dupilumab treatment resulted in a clinically meaningful reduction in moderate or severe exacerbations compared to placebo, when used in addition to best supportive care. Dupilumab likely results in little to no clinically meaningful benefits in lung function measured with prebronchodilator FEV1 compared to placebo. Patients’ quality of life as measured by SGRQ may be improved with dupilumab treatment; however, the magnitude of benefit is considered not clinically significant based on a recognized MID estimate. No notable safety concerns were identified with dupilumab treatment from the trials through week 52. The absence of longer-term efficacy and safety data beyond 1 year represents a gap in evidence given that COPD is a chronic disease. No comparative evidence for the efficacy and safety of dupilumab versus other therapies available in clinical practice was submitted.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of dupilumab 300 mg/2 mL for SC injection as an add-on maintenance treatment in adult patients with COPD characterized by elevated blood eosinophil levels inadequately controlled by the combination of an ICS, a LABA, and a LAMA, or patients receiving a combination of a LABA and a LAMA if ICS is not appropriate.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
COPD is a chronic inflammatory lung disease, often associated with chronic bronchitis and emphysema, that causes obstructed airflow from the lungs, lung hyperinflation, systemic manifestations, and increasing frequency and severity of exacerbations.1,2 Cigarette smoking is the leading cause of COPD.25,26 A number of factors may cause COPD and contribute to its complexity, including long-term cumulative exposure to occupational dusts and chemicals; second-hand smoke or wood smoke and other biomass fuels used for cooking; frequent lung infections as a child; or genetic reasons (alpha-1 antitrypsin deficiency).2 Patients with COPD present with dyspnea, wheezing, chest tightness, fatigue, activity limitation, and/or cough with or without sputum production. Exacerbations are acute events of increased respiratory symptoms and are key events in the course of COPD as they have substantial impact on health status, increase the rate of lung function decline, worsen the prognosis of the patient, and are associated with high health care costs.3 This disease is associated with several comorbidities, including ischemic heart disease, osteopenia and osteoporosis, glaucoma and cataracts, cachexia and malnutrition, anemia, peripheral muscle dysfunction, cancer, and metabolic syndrome.1 Patients who experience severe and/or frequent exacerbations are at even greater risk for rapid disease progression and poor outcomes including mortality and morbidity.27-29 A subset of up to 40% of patients with COPD have evidence of type 2 inflammation which is associated with higher mortality, increased risk of exacerbations, lower FEV1, higher impairment in health status, higher health care utilization, and increased risk of mortality.7-13
COPD is observed only in adults and most patients are at least 40 years old when symptoms of COPD first appear.3 Initially considered to be a disease occurring mainly in older adult males who smoked, COPD has become increasingly prevalent among women; however, there is still a higher prevalence in men; 11.8% in men and 8.5% in women.3 According to the Canadian Chronic Disease Surveillance System, the prevalence of COPD among people living in Canada aged 35 years and older in 2022 to 2023 was 8.74%, representing a 24% increase from 2000 to 2001.4 Globally, COPD was the cause of 3.23 million deaths in 2019 which represents the third leading cause of death worldwide.5 COPD accounts for 81.7% of deaths from chronic respiratory diseases.6
Patients with COPD have a high degree of disease-related morbidity (including work and activity limitation)30 and impaired health status or HRQoL, with effects seen independent of other comorbid diseases.31 Of note, COPD accounts for the majority of disability-adjusted life-years among patients with chronic respiratory diseases,32 and in 2019, a total of 2.9% of global disability-adjusted life-years were attributed to COPD.33 The high impact of the disease is also demonstrated in the rate of requested medical assistance in dying among patients with COPD. Respiratory conditions, including COPD, accounted for 13.2% of deaths through medical assistance in dying in Canada in 2022.34
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
According to the input from patient and clinician groups, and the clinical experts consulted for this review, the primary goals of treatment for COPD are to reduce symptoms such as dyspnea, wheeze, cough, and phlegm; reduce the frequency and severity of exacerbations; improve overall health status and quality of life (e.g., improved exercise tolerance and work attendance); and improve survival.3,14 The clinical experts noted that the most recent CTS guidelines have now explicitly added mortality reduction as a treatment goal, aligning with previous research indicating that patients with severe chronic diseases value extension of life expectancy. Exacerbation prevention remains a crucial goal, given its association with increased mortality and disease progression.
Initial COPD management includes reducing exposure to risk factors such as smoking and air pollutants; implementing smoking cessation strategies; administering vaccinations to reduce respiratory infections; and providing self-management education focused on breathlessness, stress management, pulmonary rehabilitation, and maintaining an active lifestyle.14,15
The 2023 CTS guidelines14 and the GOLD guidelines3,15 outline the pharmacological treatment of COPD as follows. Patients with mild COPD (defined as COPD Assessment Test [CAT] < 10, mMRC Dyspnea Scale grade = 1, FEV1 ≥ 80%, and low symptom burden in the 2023 CTS guidelines; aligned with the GOLD group A) should be treated with single inhaled LAMA or LABA.3,14,15 Patients with moderate and severe disease (defined as CAT ≥ 10, mMRC ≥ grade 1, and FEV1 < 80%) and a low risk of COPD exacerbation (≤ 1 moderate exacerbation in the past year in the 2023 CTS guidelines; aligned with GOLD group B) should be treated with inhaled double therapy of LABA plus LAMA.3,14,15 Patients with moderate and severe disease and a high risk of COPD exacerbation (≥ 2 moderate exacerbations or ≥ 1 severe exacerbation in the last year in the 2023 CTS guidelines; aligned with GOLD group E) should be treated with a combination of LABA, LAMA, and ICS administered as a single inhaled triple therapy (blood eosinophils levels ≥ 300 cells/µL as an additional indication for triple therapy in the 2025 GOLD guidelines compared to the 2023 CTS guidelines).3,14,15 While oral corticosteroids are commonly used to treat exacerbations in patients with COPD, high usage is associated with increased risks of both chronic (e.g., cardiovascular disease, diabetes, kidney disease, bone and eye disorders, sleep disorders, infections) and acute (e.g., heart attack, stroke, fractures, gastrointestinal issues, sepsis, depression) adverse effects.35
If patients continue to experience exacerbations despite receiving triple combination therapy of LABA, LAMA, and ICS, the addition of macrolide is recommended. Maintenance therapy with a macrolide, such as azithromycin, is only appropriate for patients who have normal QT interval on electrocardiogram, have no substantial drug interactions with concomitant medications, and no evidence of either indolent or active infection with atypical mycobacteria (“who are not current smokers” is an additional indication for macrolide treatment in the 2025 GOLD guidelines compared to the 2023 CTS guidelines).3,14,15 However, azithromycin lacks specific clinical evidence in the recommended population and is associated with a substantial incidence of AEs (e.g., hearing loss, gastrointestinal AEs, and QT interval prolongation). Additionally, prolonged antibiotic usage is linked to the rising rates for antimicrobial resistance seen in patients with COPD.16,17
The addition of roflumilast, a PDE4 inhibitor, to triple therapy is recommended in patients with COPD with a chronic bronchitis phenotype at high risk of exacerbations (FEV1 < 50% as an additional indication of roflumilast in the 2025 GOLD guidelines compared to the 2023 CTS guidelines),15 with a moderate to high symptom burden and/or health status impairment who continue to experience exacerbations despite being on the triple combination therapy of LABA, LAMA, and ICS.3,14,15 Of note, roflumilast received a Canadian Expert Drug Advisory Committee recommendation to not be listed for reimbursement in 2011.36
Of note, recommendations regarding the use of dupilumab were newly added in the recent 2025 update of the GOLD guidelines, indicating that if patients treated with triple therapy of LABA, LAMA, and ICS continue to experience exacerbations, dupilumab may be added for those with blood eosinophil levels of 300 cells/µL or higher and symptoms of chronic bronchitis.15
Drug Under Review
Dupilumab has been approved by Health Canada for the treatment of atopic dermatitis, asthma, chronic rhinosinusitis with nasal polyposis, eosinophilic esophagitis, prurigo nodularis, and chronic spontaneous urticaria. Dupilumab received a Notice of Compliance from Health Canada in October 2025 and is indicated as an add-on maintenance treatment for adult patients with uncontrolled COPD associated with type 2 inflammation.37 The recommended dose of dupilumab for adult patients is 300 mg given every other week by SC injection.37 The reimbursement criteria requested for dupilumab is as an add-on maintenance treatment in adult patients with uncontrolled COPD associated with type 2 inflammation, if the following conditions are met:
postbronchodilator FEV1 to forced vital capacity (FVC) ratio less than 0.70 and postbronchodilator FEV1 predicted to be greater than 30% and 70% or less
background therapy of the combination therapy of LABA, LAMA, and ICS, or the combination therapy of LABA plus LAMA if ICS is contraindicated
blood eosinophil levels of 300 cells/µL or higher
2 or more moderate or 1 or more severe exacerbations within the past year
mMRC of grade 1 or greater.
Note that moderate exacerbations are defined as acute exacerbation of COPD that require either SCS (intramuscular, IV, or oral) and/or antibiotics. Severe exacerbations are defined as acute exacerbation of COPD requiring hospitalization or observation for longer than 24 hours in an emergency department or urgent care facility.
Dupilumab is a recombinant human monoclonal IgG4 monoclonal antibody that inhibits interleukin (IL) 4 and IL-13 signalling by specifically binding to the IL-4R-alpha subunit shared by the IL-4 and IL-13 receptor complexes. Dupilumab inhibits IL-4 signalling via the type I receptor (IL-4R-alpha/gamma c), and both IL-4 and IL-13 signalling through the type II receptor (IL-4R-alpha/IL-13R-alpha).37
Dupilumab should not be used to treat acute symptoms or acute exacerbations of asthma or COPD, and not to be used to treat acute bronchospasm or status asthmaticus. Serious adverse effects or safety issues include hypersensitivity reactions, including anaphylaxis, serum sickness or serum sickness-like reactions, and angioedema; elevation of blood eosinophil levels leading to eosinophilic conditions, such as eosinophilic pneumonia or vasculitis consistent with eosinophilic granulomatosis with polyangiitis; conjunctivitis and keratitis related events; and arthralgia.
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
Two patient groups, the COPD Canada and the Lung Health Foundation, provided input for this review. COPD Canada gathered information through an email survey conducted in November 2024. A total of 61 people participated in the survey. The Lung Health Foundation collected data through an online survey completed by 27 patients across Canada between December 2022 and December 2024, as well as feedback from 7 individuals living with COPD in Ontario through monthly support group sessions, 4 patients through one-on-one virtual appointments with certified respiratory educators between March and November 2024, and 2 patients through virtual interviews. No patients from these inputs had any experience with the drug under review.
The 2 groups highlighted that COPD is associated with a considerable burden of disease, affecting many things that are fundamental to everyday life, such as the ability to breathe, talk, sleep, work, climb stairs, play sports, socialize, and so forth. Referring to their online survey data, the Lung Health Foundation highlighted that the top symptoms experienced by patients included shortness of breath (88.9%), fatigue (74.1%), and coughing (44.4%). Similar insights were observed in the data collected from the support group and one-on-one appointments with the certified respiratory educators. Those who responded to the online survey conducted by the Lung Health Foundation also expressed their most common challenges to be their inability to perform daily activities due to shortness of breath (77.8%), fatigue (66.7%), and feelings of isolation (48.1%). The Lung Health Foundation group emphasized that patients need treatments that not only address physical symptoms but also enhance their ability to engage in meaningful activities and improve their overall quality of life.
Those who responded to the Lung Health Foundation identified some benefits of current COPD treatments, with the most common ones being reduced shortness of breath (70.4%), increased ability to exercise (40.7%), improved participation in daily activities (25.9%), and reduced coughing (25.9%). When asked about side effects from current treatments, 44% of people who responded to the online survey reported no side effects. Among those who did, voice hoarseness (33%), difficulty sleeping (26%), and low energy (15%) were the most common. Those who responded from both inputs expressed concerns for prednisone’s side effects. While the Lung Health Foundation input noted that the current COPD treatments provide meaningful benefits such as reduced shortness of breath and increased ability to exercise, they also indicated that patients continue to face limitations in their daily lives due to residual symptoms and side effects. The input reiterated the urgent need for additional COPD medications that could address unmet needs, minimize side effects, enhance overall treatment outcomes, and improve quality of life.
When asked about the key treatment outcomes that are important, those who responded to the online survey conducted by the Lung Health Foundation prioritized improved quality of life (77.8%), followed by reduction in symptoms (59.3%), increased energy (44.4%), and better symptom management (40.7%). The support group feedback by this input underscored reduction in symptoms (86%), enhanced quality of life (57%), and improved energy (57%) as critical needs for patients with COPD. Moreover, COPD Canada input noted that most of their members were aged older than 65 years, and for many, having COPD medication coverage by public drug plans is financially crucial.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of COPD.
Unmet Needs
According to the clinical experts consulted for this review, despite the currently available combination therapy of LABA, LAMA, and ICS, many patients with COPD continue to experience exacerbations, with annual rates of 1.08 for patients at high risk and 0.46 for patients at low risk,38 and no current treatment fully eliminates exacerbations. The clinical experts noted that while smoking cessation and some medications can slow disease progression, no treatment reverses COPD, and triple therapy provides only partial and incomplete improvement in lung function (FEV1). Many patients continue to experience substantial symptoms such as dyspnea, cough, and sputum production, which impair daily activities and well-being, and although pulmonary rehabilitation offers some benefit, its effects diminish over time. The clinical experts indicated that the effectiveness of inhaled medications in real-world settings is often compromised by poor inhaler technique, disease severity (e.g., inadequate inspiratory flow), and comorbidities (e.g., osteoarthritis affecting device use), contributing to declining compliance with therapy over time. The clinical experts noted that overall, current treatments remain suboptimal for exacerbation prevention, disease modification, symptom relief, and long-term adherence, underscoring the need for new therapeutic options.
Place in Therapy
The clinical experts noted that dupilumab would serve as an add-on therapy for patients with COPD who continue to experience exacerbations despite optimal inhaled triple or double combination therapy. According to the clinical experts, dupilumab does not replace existing treatments but complements them by targeting IL-4 and IL-13 pathways, which mediate type 2 inflammation. While ICSs also act against type 2 inflammation, they do so primarily at the airway level, whereas dupilumab has systemic effects that contribute to its therapeutic benefits. The clinical experts further indicated that type 2 inflammation represents only a portion of the complex inflammatory mechanisms driving COPD, and dupilumab does not fundamentally alter disease progression beyond its impact on exacerbations. Given this, dupilumab should be considered for patients with type 2 inflammation who remain at high risk for exacerbations despite first-line triple therapy. The clinical experts pointed out that dupilumab should not be a first-line or last-resort option, nor should it be used outside the parameters established by the BOREAS and NOTUS studies. According to the clinical experts, while it expands treatment options, its introduction does not represent a paradigm shift but rather an extension of the current treatment approach. Patients should undergo a reasonable trial of triple therapy before initiating dupilumab, ensuring that its use aligns with the evidence from clinical trials.
Patient Population
The clinical experts indicated that patients best suited for treatment with dupilumab are those with COPD who exhibit type 2 inflammation, particularly those with elevated eosinophil levels (≥ 300 cells/µL) and a history of moderate to severe exacerbations despite the triple or double combination therapy. The clinical experts noted that patients with combined asthma and COPD, as well as those with chronic rhinosinusitis with nasal polyps, are also likely to benefit, although specific clinical trial data are lacking. In the opinion of the clinical experts, while the BOREAS and NOTUS trials set criteria such as age, FEV1 impairment, and dyspnea symptoms, these factors do not directly influence dupilumab’s mechanism and should not be used to determine eligibility. The clinical experts also noted that patients with higher FEV1 may experience substantial benefits by preventing exacerbations and reducing lung function decline.
The clinical experts pointed out that spirometry is essential to confirm COPD diagnosis due to the high false-positive rate based on clinical diagnosis alone. Other diagnostic markers, such as fractional exhaled nitric oxide (FeNO), have not been extensively studied in COPD and are not reliable for patient selection.
Assessing the Response to Treatment
The clinical experts noted that in clinical practice, treatment response to dupilumab in patients with COPD is primarily assessed by a reduction in exacerbation frequency, improvement in quality of life, decrease in COPD symptoms, or an increase in functional capacity. According to the clinical experts, there would be no fixed period to determine its effectiveness because dupilumab is mainly used to prevent exacerbations. According to the clinical experts, patients who do not show a reduction in exacerbations may still be considered to have responded to treatment if they exhibit improvements in validated COPD outcomes. These include a decrease in SGRQ score by at least 4 points (quality of life), a decrease in CAT score by at least 2 points (symptoms), or an increase in 6-minute walk test by at least 30 m (functional capacity).
Discontinuing Treatment
The clinical experts pointed out that discontinuing dupilumab should be based on the absence of a meaningful clinical response, which is primarily assessed through a reduction in exacerbations, improvement in quality of life, decrease in COPD symptoms, or an increase in functional capacity. According to the clinical experts, a failure to achieve a reduction in exacerbations within the first year compared to the previous year, unless explained by another medical comorbidity, would indicate a lack of treatment benefit. However, patients may still be considered to have responded to treatment if they demonstrate clinically substantial improvements in validated COPD measures. Additionally, the clinical experts pointed out that treatment discontinuation should be considered in cases of substantial adverse effects, such as uncontrolled uveitis or severe infusion reactions, although no standardized clinical algorithm exists. The clinical experts noted that the criteria for determining loss of response or absence of benefit would align with those used for dupilumab and other biologics in patients with asthma under public drug program formularies in Canada.
Prescribing Considerations
The clinical experts noted that dupilumab treatment should be prescribed by respiratory medicine specialists.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
One input from the CTS was provided for this review. A total of 5 clinicians provided input for this review.
According to the clinician group, the key goals for COPD treatment are improving symptoms and health status and reducing exacerbations and mortality. The group further divided these goals into 2 broad categories — reducing symptoms and reducing future risk. Reducing symptoms included relieving dyspnea, wheeze, cough, and phlegm; improving overall health status and quality of life; and improving exercise tolerance. Reducing future risk included preventing disease progression, preventing and treating exacerbations, and reducing mortality. The group noted that a common reason for patients living with COPD to be admitted to hospital is due to COPD exacerbations (also known as acute exacerbation of COPD), which can be triggered by many factors, such as bacteria and viruses, environmental exposures, comorbid conditions, and/or progression of their disease. The group highlighted that the consequences of exacerbations include accelerated loss of lung function, loss of physical function and frailty, worse quality of life, social isolation, increased risk of cardiovascular events, and death.
Regarding the unmet needs, the group emphasized that preventing exacerbations is one of the essential treatment goals for both clinicians and patients living with COPD, which aligns with the input from the clinical experts consulted for this review. The group further noted that because COPD is a heterogeneous disease, different factors interact within individuals with COPD and impact their prognosis. The clinician group emphasized that being able to phenotype a patient with COPD would allow a clinician to potentially personalize the treatment approach for individual patients to optimize their outcomes. The clinician group added that there has been a shift in the treatment paradigm to target the suppression of eosinophils with inhaled or oral steroids; however, in many cases, persistent exacerbations had been observed among patients despite optimal therapy.
The clinician group specified that dupilumab would be the first biologic therapy available for patients with COPD and would represent a paradigm shift by providing a more targeted therapy, if approved. The group further added that dupilumab would be a valuable addition to the current treatment framework as an add-on therapy for patients who remain at high risk of exacerbations despite optimized inhaled triple therapy and have evidence of type 2 inflammation as measured by a blood eosinophil count of 300 cells/µL or higher. The group further noted that these patients would be the best suited for treatments with dupilumab. The group recommended that the use of this drug should be restricted to respirologists who manage patients with COPD.
The clinician group indicated that the discontinuation of dupilumab treatment depends on several factors, which include the absence of clinically meaningful improvements within the expected time frame (such as annual exacerbation rates remaining consistent with pretreatment levels), substantial worsening in prebronchodilator FEV1, and patient symptom scores. In addition, the group recommended that patients should receive at least 6 months of biologic treatment before deciding to discontinue treatment (exceptions would be important side effects). The clinician group also recommended that the first 1 or more doses of dupilumab should be administered by a health care professional in a clinical setting to ensure correct technique for administration and tolerance by the patient; subsequent doses could be administered in a community setting or at home.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by for this review are summarized in Table 3.
Table 3: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The efficacy and safety of dupilumab was assessed in 2 multinational, randomized, double-blind, placebo-controlled, phase III trials (BOREAS and NOTUS). Both trials were designed and conducted to investigate the efficacy and safety of dupilumab 300 mg SC q.2.w. over 52 weeks in patients with uncontrolled COPD (i.e., ≥ 2 moderate or ≥ 1 severe exacerbations in the previous year and MRC ≥ grade 2) and moderate to severe airflow obstruction (i.e., postbronchodilator FEV1 > 30% to ≤ 70% of predicted) despite receiving maximum best supportive care background therapy, including LABA, LAMA, and ICS (unless ICS was contraindicated). The primary end point of both trials was annualized rate of acute exacerbation of COPD over the 52-week treatment period compared to placebo. In both studies, dupilumab 300 mg q.2.w. led to a significant and clinically relevant reduction in the annualized rate of acute exacerbation of COPD during the 52-week intervention period (30% reduction in the BOREAS study and 34% reduction in the NOTUS study compared with placebo). The key secondary end points were prebronchodilator FEV1 over 12 weeks compared to placebo; HRQoL, assessed by the change from baseline to week 52 in the SGRQ and E-RS:COPD scale; and prebronchodilator FEV1 over 52 weeks compared to placebo. Both trials were placebo-controlled. The trials did not compare dupilumab add-on therapy to add-on therapy with azithromycin, N-acetylcysteine, or roflumilast.a | This is a comment from the drug plans to inform CDEC deliberations. According to the clinical experts, azithromycin, although used to reduce exacerbations, is an off-label treatment, poses risks such as QTc prolongation and antibiotic resistance, and does not target underlying inflammation. Roflumilast, a PDE4 inhibitor, is limited by modest efficacy, GI intolerance, and lack of reimbursement by jurisdictions in Canada. While other biologics like mepolizumab, benralizumab, and tezepelumab have been studied in COPD, none have demonstrated significant clinical benefits. The clinical experts noted that there is no evidence to show that N-acetylcysteine confers benefit when added on to contemporary first-line COPD therapy and it is rarely used in this context in clinical practice in Canada. The clinical experts commented that there are no appropriate comparators to dupilumab for COPD treatment. |
Roflumilast is not covered by any jurisdictions in Canada; however, as noted previously, roflumilast is rarely used and may not be considered a relevant comparator. | This is a comment from the drug plans to inform CDEC deliberations. The clinical experts noted that roflumilast is not a relevant comparator of dupilumab for COPD treatment. |
Considerations for initiation of therapy | |
Inclusion criteria for the BOREAS and NOTUS trials:
Questions for clinical experts: 1. Should all inclusion criteria used in the trials be required to be eligible for treatment with dupilumab? 2. Are there any other parameters that should be considered to determine eligibility for treatment with dupilumab? | The clinical experts suggested that not all inclusion criteria from the trials should be required for dupilumab eligibility in clinical practice, and there are no other parameters to consider beyond these. According to the clinical experts, key considerations include:
|
Questions for clinical experts: Would dupilumab be considered in patients who fall outside the target populations of the clinical trials (e.g., those who have a history of or a current diagnosis of asthma, outside the age range used in trials, and so forth)? | The clinical experts suggest that dupilumab could be considered for patients outside the strict inclusion criteria of the BOREAS and NOTUS studies. The clinical experts noted the following conditions should not be an exclusion of COPD treatment with dupilumab: a history or current diagnosis of asthma should not be an exclusion, as type 2 inflammation remains the key factor for treatment response; age should not be a limiting factor; and low FEV1 should not be a reason for exclusion. While FEV1 > 70% may raise concerns, exceptions could be made for patients with emphysema-dominant COPD, such as those with alpha-1 antitrypsin deficiency. According to the clinical experts, given that dupilumab is already approved in Canada for asthma, patients with COPD with asthma-COPD overlap may already qualify for treatment. Other trial criteria that are not directly tied to dupilumab’s mechanism of action or the clinical phenotype most likely to benefit (i.e., age and FEV1 thresholds), should not necessarily restrict treatment eligibility. |
Considerations for continuation or renewal of therapy | |
It was noted in the sponsor’s submission that onset of the dupilumab treatment effects as add-on treatment to current best supportive care was apparent within 2 weeks of initiation of dupilumab treatment and was maintained at week 52. A return to baseline COPD, including increased exacerbations, decreased prebronchodilator FEV1, and increased SGRQ, was noted after dupilumab treatment discontinuation, supporting the need for long-term dupilumab therapy. In addition to the primary benefit in reducing the frequency of moderate or severe exacerbations, the sponsor noted that dupilumab also led to a statistically significant and clinically relevant improvement in patients’ lung function, as demonstrated by statistically significant improvements in prebronchodilator FEV1 at week 12 and week 52 as compared to placebo. It also improved health status or HRQoL. Questions for clinical experts:
| The clinical experts noted that in clinical practice, treatment response to dupilumab in COPD is determined by a reduction in exacerbation frequency, improved quality of life, decreased COPD symptoms, or increased functional capacity. The clinical experts further indicated that patients who do not show a reduction in exacerbations may still be considered to have responded to treatment if they demonstrate meaningful improvements in other validated COPD outcomes, such as a decrease in SGRQ score of ≥ 4 points, a decrease in CAT score of ≥ 2 points, or an increase in 6-minute walk distance of ≥ 30 m. The clinical experts suggested that follow-up should occur at approximately 12 weeks to assess early response, with ongoing monitoring for the first year to evaluate long-term benefit. |
Considerations for discontinuation of therapy | |
The sponsor noted that in the BOREAS and NOTUS trials, consistent benefit was seen across subgroups of baseline biomarkers (including biomarkers of type 2 inflammation), with a trend toward numerically greater improvements with increased type 2 biomarker levels. The benefits of dupilumab were consistently observed in the overall population and in patients with potentially higher morbidity and mortality outcomes and in populations who formerly smoked or currently smoke, according to the sponsor. Question for clinical experts: What would be considered an absence of clinical benefit or loss of response? | The clinical experts pointed out that an absence of clinical benefit or loss of response to dupilumab would be determined by the treating physician based on adverse side effects, such as uncontrolled uveitis or substantial infusion reactions, although no standardized clinical algorithm exists. Additionally, failure to achieve key treatment goals including reducing exacerbation frequency, improving quality of life, decreasing COPD symptoms, or increasing functional capacity, would indicate a lack of response, unless worsening is attributable to another medical comorbidity. The clinical experts noted that these criteria align with those already used by public drug program formularies in Canada to assess treatment response for dupilumab and other biologics in patients with asthma. |
As per the indication, the patient should be currently using triple therapy (LABA, LAMA, and ICS) or dual therapy (LABA plus LAMA) if ICS is contraindicated. Question for clinical experts: What should be the duration of triple (or dual) therapy use that would be required before add-on therapy with dupilumab? | The clinical experts noted that the required duration of triple (or dual) therapy use before considering add-on therapy with dupilumab should be at least 3 months, as per the inclusion criteria of the BOREAS and NOTUS trials. In the opinion of the clinical experts, prior studies have shown that the benefit or lack of benefit of triple therapy on exacerbation risk can typically be assessed within 12 weeks. Therefore, because the exacerbation rate of an individual patient with COPD remains stable over time in the absence of treatment changes, a severe exacerbation occurring within the first 3 months suggests a continued high risk of exacerbations despite ongoing triple therapy, warranting consideration of add-on therapy like dupilumab. |
Considerations for prescribing of therapy | |
For information — dosing of dupilumab is 300 mg SC q.2.w. | This is a comment from the drug plans to inform CDEC deliberations. |
For information — per sponsor, both spirometry and blood eosinophil tests are widely available across Canada. However, spirometry is only performed in specialty respiratory clinics or hospitals and therefore there may be limitations in accessing this test for some patients. | This is a comment from the drug plans to inform CDEC deliberations. |
Generalizability | |
Question for clinical experts: Could dupilumab be considered in patients with uncontrolled COPD who do not have type 2 inflammation? | The clinical experts do not recommend dupilumab for patients who do not have type 2 inflammation, due to the lack of evidence regarding its efficacy and safety in these patients. |
System and economic issues | |
The drug programs stated results of the sponsor’s base-case analysis of the estimated incremental cost-effectiveness ratio and the budget impact analysis, for CDEC’s consideration. | This is a comment from the drug plans to inform CDEC deliberations. |
CAT = COPD Assessment Test; CDEC = Canadian Drug Expert Committee; COPD = chronic obstructive pulmonary disease; E-RS:COPD = Evaluating Respiratory Symptoms in chronic obstructive pulmonary disease; FeNO = fractional exhaled nitric oxide; FEV1 = forced expiratory volume in the first second; FVC = forced vital capacity; GI = gastrointestinal; HRQoL = health-related quality of life; ICS = inhaled corticosteroid; LABA = long-acting beta-agonist; LAMA = long-acting muscarinic antagonist; mMRC = modified Medical Research Council; MRC = Medical Research Council; q.2.w. = every 2 weeks; SC = subcutaneous; SGRQ = St. George's Respiratory Questionnaire.
Note: mMRC ≥ grade 1 is equivalent to MRC ≥ grade 2.39
aClinical experts in Canada indicated that roflumilast and azithromycin are rarely used in the maintenance treatment of COPD (secondary to adverse events, safety concerns, microbial resistance).
bModerate exacerbations were recorded by the investigator and defined as acute exacerbation of COPD that require either systemic corticosteroids (intramuscular, IV, or oral) and/or antibiotics. One of the 2 required moderate exacerbations had to require the use of systemic corticosteroids.
cSevere exacerbations were recorded by the investigator and defined as acute exacerbation of COPD requiring hospitalization or observation > 24 hours in an emergency department or urgent care facility.
Clinical Evidence
The objective of this Clinical Review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of dupilumab 300 mg single-use syringe or pen (300 mg/2mL solution) for SC injection, in the add-on maintenance treatment in adult patients with uncontrolled COPD associated with type 2 inflammation. The focus will be placed on comparing dupilumab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of dupilumab is presented in 1 section with the CDA-AMC critical appraisal of the evidence included at the end of it. This section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The assessment by CDA-AMC of the certainty of the evidence in this section using the GRADE approach follows the critical appraisal of the evidence. No long-term extension studies, indirect evidence, or additional studies were submitted to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
2 RCTs identified in the sponsor-conducted systematic review (BOREAS, the pivotal study; NOTUS, a confirmatory study of the BOREAS study).40,41
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
Description of Studies
Characteristics of the included studies are summarized in Table 4.
Two multicentre, multinational, double-blind, identically designed RCTs (BOREAS, N = 939; NOTUS, N = 935)40,41 aiming to assess the efficacy and safety of dupilumab relative to placebo in adult patients with uncontrolled COPD associated with type 2 inflammation, were identified by the systematic review conducted by the sponsor.24
The BOREAS study was a randomized, placebo-controlled phase III clinical study that aimed to investigate the efficacy and safety profile of dupilumab over a 52-week intervention period in patients with uncontrolled COPD (i.e., at least 2 moderate or 1 severe exacerbations in the previous year and MRC Dyspnea Scale grade ≥ 2, which is equivalent to mMRC ≥ grade 1); moderate to severe airflow obstruction (i.e., postbronchodilator FEV1:FVC ratio < 0.70 and postbronchodilator FEV1 > 30% to ≤ 70% of predicted) despite receiving maximum standard of care background therapy, including LABA, LAMA, and ICS (unless ICS was contraindicated); and evidence of type 2 inflammation (with blood eosinophils count ≥ 300 cells/µL). The NOTUS study was conducted as a confirmatory trial, using the same study design (Figure 1), because it was a requirement by the FDA to provide substantial evidence in replicate in 2 trials.24 The study population consisted of patients aged 40 to 80 years in the BOREAS study and 40 to 85 years in the NOTUS study who currently smoked or formerly smoked with a smoking history of at least 10 pack-years and who did not have either a history of or current diagnosis of asthma.
The primary objective of the BOREAS and NOTUS studies was to evaluate the efficacy of dupilumab 300 mg every 2 weeks in patients with moderate or severe COPD as measured by annualized rate of acute exacerbation of COPD. The key secondary objectives of the studies were to evaluate the effect of dupilumab 300 mg every 2 weeks on prebronchodilator FEV1 over 12 weeks compared to placebo; HRQoL, assessed by the change from baseline to week 52 in the SGRQ scores; and prebronchodilator FEV1 over 52 weeks compared to placebo.20,21
In both the BOREAS and NOTUS studies, all patients were centrally assigned to randomized study intervention using an interactive response technology. Patients were randomized to dupilumab 300 mg every 2 weeks plus best supportive care or placebo every 2 weeks plus best supportive care. In both studies, patients were randomized (1:1) to receive either dupilumab or matching placebo. Randomization was stratified by country and ICS dose (high-dose ICS [yes or no]) at baseline. The total number of patients who currently smoke (as defined by smoking status at screening visit) was capped at 30%.20,21
The BOREAS and NOTUS trials consisted of a 4-week (± 1 week) screening period, and a 52-week (± 3 days) randomized study intervention period. Upon completion of the 52-week study intervention period, patients continued their triple background of combination therapy of LABA, LAMA, and ICS (unless ICS was contraindicated) and entered the 12-week (± 5 days) safety postintervention follow-up period. Adjustment of background medication was allowed at the discretion of the investigator as clinically indicated during the postintervention follow-up period.20,21
In this report, the data presented for the BOREAS study in the following are from the data cut-off date of February 8, 2023 (last patient end of treatment visit). At the time of this cut-off date, some patients in the BOREAS study were still ongoing in the postintervention follow-up period. The data presented for the NOTUS study are the primary efficacy analyses results based on the IA data because the study met its multiplicity-controlled primary efficacy end point. The IA data cut-off date of the NOTUS study was September 29, 2023 (i.e., when the information fraction for the primary efficacy end point was 0.92 based on follow-up time for moderate or severe COPD exacerbations over the 52-week treatment period). At that time, the first 721 patients randomized (n = 362 [77.0%] patients in the dupilumab group and n = 359 [77.2%] patients in the placebo group) in the NOTUS study had the opportunity to complete 52 weeks of study intervention.24
Figure 1: BOREAS and NOTUS Trials Study Design
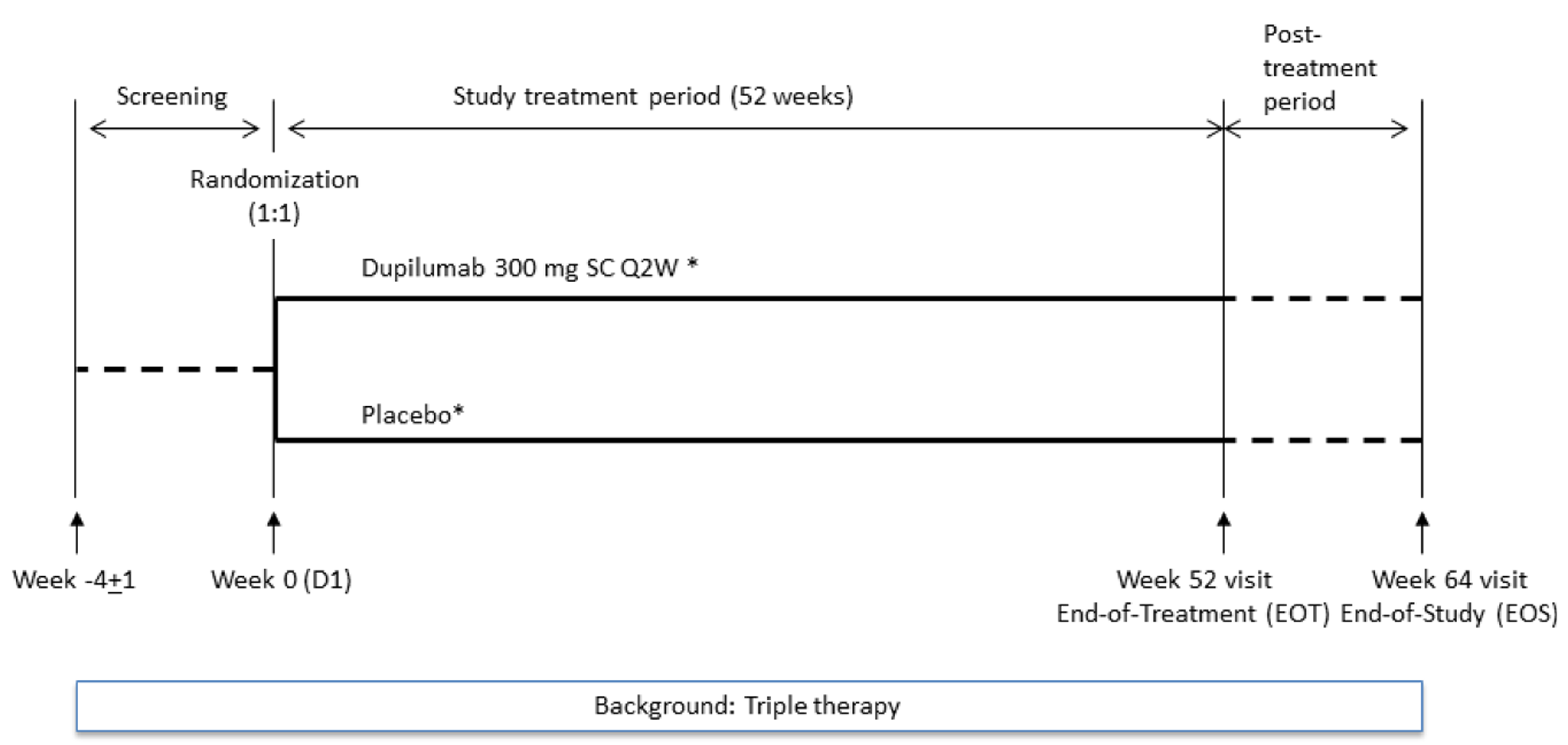
D1 = day 1; EOS = end of study; EOT = end of treatment; Q2W = every 2 weeks; SC = subcutaneous.
Notes: Dupilumab(*) 300 mg Q2W, administered as 1 SC injection of dupilumab 300 mg (2 mL). Placebo(*), administered as 1 SC injection; placebo matched dupilumab 300 mg (2 mL).
Sources: Clinical Study Reports for the BOREAS and NOTUS studies.20,21
Table 4: Details of Studies Included in the Systematic Review
Detail | BOREAS trial | NOTUS trial |
|---|---|---|
Designs and populations | ||
Study design | Phase III, multicentre, double-blind, placebo-controlled trial | Phase III, multicentre, double-blind, placebo-controlled trial |
Locations | 275 centres in 24 countries from continents of Asia, Europe, North America (Canada, 21 sites with 16 patients), and South America | 329 centres in 29 countries from continents of Africa, Europe, North America (Canada, 11 sites with 12 patients), Oceania, and South America |
Patient enrolment dates | Start date: May 19, 2019 End date: February 8, 2023 | Start date: July 6, 2020 End date: February 28, 2024 |
Data cut-off date for this review | February 8, 2023 | September 29, 2023 (IA data cut-off date) |
Randomized | Randomized: N = 939
| Randomized: N = 935
|
Key inclusion criteria | Aged ≥ 40 to ≤ 80 years in the BOREAS study, and aged ≥ 40 to ≤ 85 years in the NOTUS study, at the time of signing the informed consent. In both the BOREAS and NOTUS studies:
| |
Key exclusion criteria | In both the BOREAS and NOTUS studies:
| |
Drugs | ||
Intervention | Dupilumab 300 mg by subcutaneous injection every 2 weeks for 52 weeks | Dupilumab 300 mg by subcutaneous injection every 2 weeks for 52 weeks |
Comparator | Placebo by subcutaneous injection every 2 weeks for 52 weeks | Placebo by subcutaneous injection every 2 weeks for 52 weeks |
Study duration | ||
Screening phase | 4 weeks ± 1 week | 4 weeks ± 1 week |
Treatment phase | 52 weeks ± 3 days | 52 weeks ± 3 days |
Follow-up phase | 12 weeks ± 5 days | 12 weeks ± 5 days |
Outcomes | ||
Primary end point | Annualized rate of moderate or severe COPD exacerbations over the 52-week treatment period compared to placebo | |
Secondary and exploratory end points | Controlled secondary:
Other secondary:
Exploratory:
| |
Publication status | ||
Publications | Bhatt et al. (2023)41 Clinicaltrials.gov identifier: NCT03930732 | Bhatt et al., 202440 Clinicaltrials.gov identifier: NCT04456673 |
BiPAP = bilevel positive airway pressure; COPD = chronic obstructive pulmonary disease; E-RS:COPD = Evaluating Respiratory Symptoms in chronic obstructive pulmonary disease; EXACT = Exacerbations of Chronic Pulmonary Disease tool; FEF = forced expiratory flow; FeNO = fractional exhaled nitric oxide; FEV1 = forced expiratory volume in the first second; FVC = forced vital capacity; IA = interim analysis; ICS = inhaled corticosteroid; LABA = long-acting beta-agonist; LAMA = long-acting muscarinic antagonist; mMRC = modified Medical Research Council; MRC = Medical Research Council; SGRQ = St. George's Respiratory Questionnaire.
aModerate exacerbations were recorded by the investigator and defined as acute exacerbation of COPD that require either systemic corticosteroids (intramuscular, IV, or oral) and/or antibiotics. One of the 2 required moderate exacerbations had to require the use of systemic corticosteroids.
bSevere exacerbations were recorded by the investigator and defined as acute exacerbation of COPD requiring hospitalization or observation > 24 hours in an emergency department or urgent care facility.
Sources: Clinical Study Reports for the BOREAS and NOTUS studies,20,21 and sponsor’s submission.42 Details included in the table are from the sponsor’s summary of clinical evidence.24
Populations
Inclusion and Exclusion Criteria
The BOREAS and NOTUS studies enrolled patients with a diagnosis of moderate to severe COPD and with evidence of type 2 inflammation. The BOREAS study included patients aged 40 to 80 years and the NOTUS study included patients aged 40 to 85 years. Patients had moderate to severe COPD defined as postbronchodilator FEV1:FVC ratio less than 0.70 and postbronchodilator FEV1 predicted to be greater than 30% and less than or equal to 70%, and MRC Dyspnea Scale grade of 2 or greater (equivalent to mMRC ≥ grade 1).43 Additionally, the patients had been at a high risk of exacerbations, defined as exacerbation history of 2 or more moderate or 1 or more severe exacerbations within the year before inclusion; at least 1 exacerbation should have occurred while the patient was receiving the triple combination therapy of LABA, LAMA, and ICS (or LABA and LAMA if ICS was contraindicated). A moderate exacerbation was defined as requiring either SCS (intramuscular, IV, or oral) and/or antibiotics and a severe exacerbation was defined as requiring hospitalization or observation for more than 24 hours in an emergency department or urgent care facility. Patients who currently smoke or formerly smoked with a smoking history of greater than 10 pack-years. Patients were to be receiving a stable dose of background triple therapy (LABA, LAMA, and ICS) for 3 months before randomization. Background double therapy (LABA and LAMA) was permitted if ICS use was contraindicated. Evidence of type 2 inflammation was also required, defined as a blood eosinophil count of 300 cells/µL or greater at screening. In both trials, patients must have had a COPD diagnosis for more than 12 months and no current or historical diagnosis of asthma.
Interventions
In both the BOREAS and NOTUS studies, patients in the dupilumab group received dupilumab 300 mg for SC administration supplied as a 150 mg/mL solution in 2.25 mL prefilled glass syringes, delivering 300 mg in 2.0 mL. Patients in the placebo group received placebo provided in identically matched glass prefilled syringes to deliver 2 mL matching dupilumab 300 mg. Study drugs were given every 14 days (± 3 days).20,21
Permitted concomitant medications included:20,21
maintenance treatment of COPD with ICS, LABA, and LAMA, at a stable dosage
SCS in case of acute exacerbation up to a maximum of 6 weeks
rescue medication with short-acting beta-agonists (SABAs) or short-acting antimuscarinics (e.g., inhaled ipratropium).
Use of PDE4 inhibitors and methylxanthines was allowed during the studies if the dose was stable for more than 6 months before the screening visit.20,21
Outcomes
A list of efficacy end points assessed in this Clinical Review report is provided in Table 5, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review according to the clinical experts consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, end points were selected that were considered to be the most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE.
Considerations that informed the selection of outcomes to be summarized and assessed using GRADE include the following:
The annualized rate of moderate or severe COPD exacerbation in the overall ITT population was identified by the patient and clinician group input, and the clinical experts consulted for this review.
The mean change from baseline in prebronchodilator FEV1 to week 12 and week 52 in the overall ITT population was identified by the patient and clinician group input, and the clinical experts consulted for this review.
HRQoL outcomes were identified by the patient and clinician group input and specified by the clinical experts to include change from baseline in the SGRQ total score in the overall ITT population.
The outcome of any treatment-emergent AESIs was identified by the patient group input, and the clinical experts consulted for this review.
Table 5: Outcomes Summarized From the BOREAS and NOTUS Trials
Outcome measure | Time point | BOREAS trial | NOTUS trial |
|---|---|---|---|
Annualized rate of moderate and severe COPD exacerbations | Over 52 weeks | Primarya | Primarya |
Change in prebronchodilator FEV1 from baseline | To week 12 | Secondarya | Secondarya |
Change in prebronchodilator FEV1 from baseline | To week 52 | Secondarya | Secondarya |
Change from baseline in SGRQ total score | To week 52 | Secondarya | Secondarya |
Proportion of patients with SGRQ improvement ≥ 4 points | To week 52 | Secondarya | Secondarya |
Change in E-RS:COPD total score | To week 52 | Secondarya | Secondarya |
COPD = chronic obstructive pulmonary disease; E-RS:COPD = Evaluating Respiratory Symptoms in chronic obstructive pulmonary disease; FEV1 = forced expiratory volume in the first second; SGRQ = St. George's Respiratory Questionnaire.
aStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
Sources: Clinical Study Reports for the BOREAS and NOTUS studies.20,21 Details included in the table are from the sponsor’s summary of clinical evidence.24
Efficacy Outcomes
The BOREAS and NOTUS studies investigated the same outcomes. A description of the efficacy outcome measures and the measurement properties that were used in both studies are presented in Table 6.
Exacerbation Events
In both the BOREAS and NOTUS studies, the primary efficacy end point was the annualized rate of moderate or severe COPD exacerbations (also referred to as acute exacerbation of COPD) over the 52-week treatment period compared to placebo. The definitions of moderate and severe exacerbations are in alignment with clinical guidelines.14,15
A moderate exacerbation was defined as an acute exacerbation of COPD event that required either SCS (such as intramuscular, IV, or oral) and/or antibiotics.
A severe exacerbation was defined as acute exacerbation of COPD requiring hospitalization, observation for more than 24 hours in an emergency department or urgent care facility, or resulting in death.
Two acute exacerbation of COPD events were considered as distinct events if they were separated by at least 14 days. Any acute exacerbation of COPD event recorded in the electronic case report form was reviewed by an independent adjudication committee. Only acute exacerbation of COPD events confirmed by the adjudication committee were included in the analysis.
The reduction of the annualized rate of moderate or severe exacerbations is a commonly used end point in pivotal COPD studies that demonstrates a meaningful benefit to patients.44 Acute exacerbation of COPDs (i.e., moderate or severe exacerbations) are medically important events that are associated with an inflammatory response leading to tissue destruction and disease progression. Exacerbations can require treatment with medications such as SCS and/or antibiotics which carry risks of systemic side effects, especially when multiple courses are required per year. Exacerbations can be life-threatening and may result in emergency department visits and hospitalization.3 Moderate and severe exacerbations can accelerate loss of lung function, and frequent exacerbations (> 2 per year) are associated with a greater risk of worsening respiratory symptoms, impairment in health status or HRQoL, and mortality.27,45
There is no validated minimal clinically important difference (MCID) for exacerbations because reduction in rates between 9% and 53.3% have indicated meaningful clinical benefit and this remains a challenge due to a lack of uniform definition for exacerbations and severity grading, seasonal variation, and substantial interpatient and intrapatient variability in frequency.19 The clinical experts consulted for this review noted that a between-group difference of 11% could be considered a reasonable MCID in this review.46
Lung Function
FEV1 is a measure of the extent of airflow obstruction and is a predictor of mortality in COPD.47,48 FEV1 is a widely accepted end point of efficacy in COPD studies.24 FEV1 is a well-accepted assessment to determine the effect of a drug on lung function.14,15 In patients with chronic severe lung disease, such as COPD, rapid relief of bronchial obstruction may offer a meaningful clinical benefit, and therefore week 12 was selected as the time point for this analysis.49,50 An additional analysis at week 52 was performed to show maintenance of dupilumab treatment effect on lung function.49,50
In the BOREAS and NOTUS studies, key secondary end points included changes in prebronchodilator FEV1 from baseline to week 12 and week 52 compared to placebo.
Although there are uncertainties around the evidence for MCID of FEV1 in patients with COPD,24,51,52 an MCID of 100 mL for FEV1 for within-group change based on clinical anchoring to exacerbations, dyspnea, and decline in lung function in asthma was proposed.19,53 Also, the clinical experts consulted for this review noted that a between-group difference of 100 mL in FEV1 is reasonable and applicable in clinical practice.
HRQoL as Assessed by the SGRQ
In the BOREAS and NOTUS studies, key secondary end points also included HRQoL or health status assessments using the SGRQ. The SGRQ (version 14, 2003) is a 50-item self-administered questionnaire designed to measure and quantify health status and HRQoL in adult patients with chronic airflow limitation.54,55 The SGRQ has been recognized as a clinically important patient-reported outcome assessment tool providing supportive evidence of efficacy in a clinical trial56 and has been used in a range of disease groups including asthma, COPD, and bronchiectasis.24 The SGRQ is a validated tool with demonstrated validity, reliability, and sensitivity. Scores range from 0 to 100, where higher scores represent worse health status. Scores by dimension are calculated for 3 domains: symptoms, activity, and impacts (psychosocial), as well as a total or global score. The symptoms domain evaluates symptomatology, including frequency and severity of cough, sputum production, wheeze, breathlessness, and the duration and frequency of episodes of breathlessness or wheeze. The activity domain assesses disturbances to patients’ daily physical activities. The impacts domain covers a range of effects that chest troubles may have on patients’ daily life and psychosocial functions (e.g., daily life activities and functioning, employment, physical functioning, emotional impact, stigmatization, and patients’ perceptions when treated). In both the BOREAS and NOTUS studies, patients completed the SGRQ at baseline and at weeks 4, 12, 24, 36, and 52.20,21
In the BOREAS and NOTUS studies, the following health status and HRQoL measurements were defined as key secondary end points:49,50
change from baseline to week 52 in SGRQ total score
proportion of patients with improvement in SGRQ score by 4 or more points at week 52.
The widely used MCID for the SGRQ is a reduction of 4 units in the total score, within group.18,19
Harms Outcomes
AEs were predefined and reported in both the BOREAS and NOTUS studies, including TEAEs, serious TEAEs, TEAEs leading to discontinuation, death, and treatment-emergent AESIs (including systemic hypersensitivity reactions, severe injection site reactions, serious infections, significant alanine aminotransferase elevation, conjunctivitis, keratitis, injection site reaction, malignancy, and helminthic infections).20,21
Table 6: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MCID |
|---|---|---|---|
SGRQ | The SGRQ is a 76-item self-administered questionnaire designed to measure and quantify health status in adult patients with chronic airflow limitation. Scores range from 0 to 100, where higher scores represent worse health status. Scores by dimension are calculated for 3 domains: symptoms, activity, and impacts (psychosocial), as well as a total or global score. The symptoms domain evaluates symptomatology, including frequency and severity of cough, sputum production, wheeze, breathlessness, and the duration and frequency of episodes of breathlessness or wheeze. The activity domain assesses disturbances to patients’ daily physical activities. The impacts domain covers a range of effects that respiratory symptoms may have on patients’ daily life and psychosocial functions.54,55 | Validity: The cross-sectional validity of the SGRQ total scores in patients with COPD has been established in different studies. In a review assessing evidence for the validity of the measurement, the correlations between changes in health status score and changes in other measures of disease activity for the SGRQ were found to be weaker than the corresponding cross-sectional comparisons due to the smaller range of scores (r value ranged between 0.14 to 0.7). The pattern of between-patient and within-patient correlations were similar for the SGRQ questionnaire.54,55,57 Reliability: In a study testing the repeatability of the SGRQ questionnaire, the intraclass correlation coefficient for patients with COPD was 0.92. The coefficient values for the component domains were between 0.87 to 0.91.54,55 Responsiveness: The SGRQ questionnaire was found to be responsive in COPD.57 | Based on anchor-based (MRC Dyspnea grade, CRQ dyspnea domain, mortality rate) estimates as well as expert and patient preference estimates, reduction of 4 units in the SGRQ total score were established as a validated MCID for patients with COPD.18,19 |
COPD = chronic obstructive pulmonary disease; CRQ = Chronic Respiratory Questionnaire; MCID = minimal clinically important difference; MRC = Medical Research Council; SGRQ = St. George's Respiratory Questionnaire.
Statistical Analysis
Table 7 summarizes the statistical analysis methods of the efficacy end points in the BOREAS and NOTUS trials.
Sample Size and Power Calculation
The sample size calculation was the same for both studies and was determined based on power calculations for the primary end point of annualized rate of moderate or severe COPD exacerbations over the 52-week intervention period. Assuming the number of exacerbations follow a negative binomial distribution with a dispersion parameter of 1; a placebo annualized rate of exacerbations of 1.5; an average treatment duration of 0.95 per year (to account for an average of 5% of the planned study intervention period with missing data); and a randomization ratio of 1:1 to the 2 study intervention groups, with 924 patients randomized (462 for each study intervention group), the study had 90% power to detect a 25% RR reduction (i.e., annualized rate of 1.125 for the dupilumab group) in the annualized rate of moderate or severe COPD exacerbations at the 2-sided significance level with an alpha of 0.05.20,21
Statistical Testing
In the BOREAS and NOTUS studies, the annualized rate of moderate or severe COPD exacerbation events (primary end point) was analyzed using a negative binomial regression model. The model included the total number of events that occurred during the 52-week study intervention period as the response variable, with the following covariates: treatment group, region (pooled country), ICS dose at baseline (high-dose ICS [yes or no]), smoking status at screening (those who currently smoke or do not smoke), baseline disease severity (as predicted postbronchodilator FEV1), and number of moderate or severe COPD exacerbation events within 1 year before the study (≤ 2, 3, or ≥ 4). Log-transformed observation duration was used as an offset variable. The estimated annualized event rate for each treatment group and its 2-sided 95% CIs was derived. The event rate ratio of the dupilumab group against the placebo group, its 2-sided 95% CI, and P value were provided.20,21
In the BOREAS and NOTUS studies, the change from baseline in prebronchodilator FEV1 and change from baseline in SGRQ total score were analyzed using a mixed-effect model with repeated measures. The proportion of patients with an improvement in SGRQ score of 4 or more points was analyzed using a logistic regression model.20,21
In the BOREAS study, a hierarchical testing procedure was used to control the overall type I error rate for testing the primary, key secondary, and selected other end points at a 2-sided significance level of 0.049. As a prespecified nonbinding futility IA was conducted during the study, an administrative penalty of 0.001 was taken from the significance level used at final analysis; the overall alpha was 0.05.20 The prespecified hierarchy of 9 items of the testing procedure was as follows: annualized rate of moderate or severe COPD exacerbation over the 52-week treatment period (primary end point), followed by secondary end points of change in prebronchodilator FEV1 from baseline to week 12 and then to week 52 in the ITT population, change in prebronchodilator FEV1 from baseline to week 12 and then to week 52 in the subgroup of patients with baseline FeNO 20 or more parts per billion, change from baseline to week 52 in SGRQ total score, proportion of patients with improvement in SGRQ score of 4 or more points at week 52, change in the Evaluating Respiratory Symptoms in COPD (E-RS:COPD) total score from baseline to week 52, and annualized rate of moderate or severe COPD exacerbation over the 52-week treatment period in the subgroup of patients with baseline FeNO of 20 parts per billion or more.20,24
For the NOTUS study, multiplicity was considered for performing an IA and for testing multiple end points. The overall type I error rate was controlled at the 2-sided 0.05 level. Based on an information fraction of 0.92, the primary end point was tested at the level of an alpha of 0.018 based on the Kim-DeMets spending function with a power parameter of a rho of 12. A hierarchical testing procedure that was identical to that in the BOREAS study was used to control the overall type I error rate for testing the primary and key secondary end points, and an additional selected other end point. As the primary end point demonstrated efficacy at this IA based on the alpha allocated, the rest of the multiplicity-controlled end points were subsequently tested with an alpha of 0.05 because this was the final analysis and no further testing were performed.21
Subgroup Analyses
In the BOREAS and NOTUS studies, prespecified and additional subgroup analyses (by baseline demographic and disease characteristics, and by biomarker levels) were performed, including the assessment of interactions between the subgroups and treatment groups for the primary end point and key secondary efficacy end point of change from baseline in prebronchodilator FEV1 at week 12. The specific, prespecified subgroup analyses in the BOREAS and NOTUS studies included the following: age group (aged < 65 years or ≥ 65 years; or 40 to 64 years, 65 to 74 years, or 75 to 85 years, respectively); sex (female or male); region; territory; race (white or other racial groups); ethnicity (Hispanic or Latino, or not); baseline weight (< 70 kg, ≥ 70 kg to < 90 kg, or ≥ 90 kg; or < 60 kg or ≥ 60 kg, respectively); baseline body mass index (< 25 mg/m2, ≥ 25 mg/m2 to < 30 mg/m2, or ≥ 30 mg/m2); ICS high-dose level at baseline (high-dose ICS, non–high-dose ICS, or no ICS); ICS dose level at baseline (< median or ≥ median); smoking status at screening (currently smoke or formerly smoked); number of moderate or severe COPD exacerbation events within 1 year before visit 1 (≤ 2, 3, or ≥ 4); number of severe COPD exacerbation events within 1 year before visit 1 (0, 1, or ≥ 2 or 0 or ≥ 1, respectively); baseline predicted postbronchodilator FEV1 (< median or ≥ median); baseline FEV1 reversibility (< 12% or ≥ 12%; or < median or ≥ median, respectively); baseline FeNO (< 20 parts per billion or ≥ 20 parts per billion); baseline eotaxin-3 (< median or ≥ median); baseline serum IgE level (< 100 IU/mL or ≥ 100 IU/mL); baseline pulmonary and activation-regulated chemokine (< median or ≥ median); baseline fibrinogen (< 350 mg/dL or ≥ 350 mg/dL); maximum eosinophil count during screening (< 0.5 giga/L or ≥ 0.5 giga/L [i.e., < 500 cells/µL or ≥ 500 cells/µL]); emphysema ongoing at baseline (yes or no); and baseline body mass index, airflow obstruction, dyspnea, and exercise capacity (≤ 4 or > 4).49,50 Among these, the clinical experts consulted for this review noted maximum eosinophil count during screening (< 500 cells/µL or ≥ 500 cells/µL) as an important subgroup analysis.
Sensitivity Analyses
The BOREAS and NOTUS studies applied the same sensitivity analyses (Table 7). For the primary end point, if patients withdrew from the study before week 52, moderate or severe exacerbation events that might occur after study discontinuation would not be observed. These patients were considered as patients with missing data on moderate or severe exacerbation. In addition, the sensitivity analyses were reconducted to assess the robustness of the conclusion based on the primary analysis.49,50
Pattern Mixture Model–Multiple Imputation
For each patient with missing data of moderate or severe exacerbation events, individual biweekly event probability was estimated using observed data with adjustment of the planned treatment group, region (pooled country), ICS dose at baseline (high-dose ICS [yes or no]), smoking status at screening (currently smoke or do not smoke), baseline disease severity (as predicted postbronchodilator FEV1), and number of moderate or severe COPD exacerbation events within 1 year before the study (≤ 2, 3, or ≥ 4). The total number of acute exacerbation of COPD moderate or severe events (on a biweekly basis) were calculated based on data imputed using multiple imputation.
Control-Based Pattern Mixture Model–Multiple Imputation
For each patient with missing data, individual biweekly event probability was estimated using observation in the placebo arms only, with adjustment of the same variables as in pattern mixture model–multiple imputation (PMM-MI) except for 1 variable (“the planned treatment group” was not included as an adjustment variable in the control-based PMM-MI). The total number of acute exacerbation of COPD moderate or severe events (on a biweekly basis) were calculated based on data imputed using multiple imputation.
Tipping Point Analysis
For each patient with missing data, the biweekly event was imputed in a similar fashion as PMM-MI based on various odds values. If the patient was receiving dupilumab, the predicted odds were increased; if the patient was on placebo, the predicted odds value was decreased. The adjusted rate was then used to impute the number of events that occurred during the missing observation period. A sequence of increasing or decreasing ratio was used to generate different imputed datasets.
For each of the methods previously listed, a negative binomial model was fitted using each of the complete datasets composed of observed and imputed data, including the total number of observed and imputed events during the 52 weeks as the response variable, with the same adjustment variables described in PMM-MI as covariates. Log-transformed observation duration was used as the offset variable for patients who completed the 52-week treatment or study period, and log-transformed 52 weeks was used as the offset variable for patients who discontinued the study before week 52. The SAS MIANALYZE procedure was used to generate statistical inferences by combining results from the analysis with each dataset using Rubin’s formula. Details of sensitivity analyses for the key end points are not presented in this report but are available in the statistical analysis plan documents of the 2 studies.49,50
Table 7: Statistical Analysis of Efficacy End Points in the BOREAS and NOTUS Trials
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
BOREAS and NOTUS trials | ||||
Annualized rate of moderate or severe COPD exacerbation over 52-week treatment period | Negative binomial regression model | Treatment group, region (pooled country), ICS dose at baseline (high-dose ICS [yes/no]), smoking status at screening (currently smoke or do not smoke), baseline disease severity (as predicted postbronchodilator FEV1), and number of moderate or severe COPD exacerbation events within 1 year before the study (≤ 2, 3, or ≥ 4) | No imputation was performed for the unobserved events that might happen after study discontinuation and up to week 52 |
|
Change in prebronchodilator FEV1 from baseline to week 12 compared to placebo | MMRM | Treatment group, age, sex, height, region (pooled country), ICS dose at baseline (high-dose ICS [yes/no]), smoking status at screening (currently smoke or do not smoke), visit, treatment by visit interaction, baseline prebronchodilator FEV1, and FEV1 baseline by visit interaction | Restricted maximum likelihood method using the Newton-Raphson algorithm |
|
Change in prebronchodilator FEV1 from baseline to week 52 compared to placebo | MMRM | Treatment group, age, sex, height, region (pooled country), ICS dose at baseline (high-dose ICS [yes/no]), smoking status at screening (currently smoke or do not smoke), visit, treatment by visit interaction, baseline prebronchodilator FEV1, and FEV1 baseline by visit interaction | Restricted maximum likelihood method using the Newton-Raphson algorithm |
|
Change from baseline to week 52 in SGRQ total score compared to placebo | MMRM | Treatment group, region (pooled country), ICS dose at baseline (high-dose ICS [yes/no]), smoking status at screening (currently smoke or do not smoke), visit, treatment by visit interaction, baseline SGRQ total score, and SGRQ baseline by visit interaction | Restricted maximum likelihood method using the Newton-Raphson algorithm |
|
Proportion of patients with SGRQ improvement ≥ 4 points at week 52 compared to placebo | Logistic regression model | Treatment group, region (pooled country), ICS dose at baseline (high-dose ICS [yes/no]), smoking status at screening (currently smoke or do not smoke), and baseline SGRQ total score | Patients with missing SGRQ total score at week 52 were considered to not have responded to treatment |
|
Change in E-RS:COPD total score from baseline to week 52 compared to placebo | MMRM | Treatment group, region (pooled country), ICS dose at baseline (high-dose ICS [yes/no]), smoking status at screening (currently smoke or do not smoke), visit, treatment by visit interaction, baseline SGRQ total score, and SGRQ baseline by visit interaction | Restricted maximum likelihood method using the Newton-Raphson algorithm |
|
COPD = chronic obstructive pulmonary disease; E-RS:COPD = Evaluating Respiratory Symptoms in chronic obstructive pulmonary disease; FEV1 = forced expiratory volume in the first second; ICS = inhaled corticosteroid; MMRM = mixed-effect model with repeated measures; PMM-MI = pattern mixture model–multiple imputation; SGRQ = St. George's Respiratory Questionnaire.
Sources: Clinical Study Reports for the BOREAS and NOTUS studies.20,21 Details included in the table are from the sponsor’s summary of clinical evidence.24
Analysis Populations
The primary analysis population for the efficacy assessment in the BOREAS and NOTUS studies was the ITT population, which included all patients who were randomized. The analysis was based on the treatment group allocated at randomization regardless of whether the treatment kit was used or not. The week 52 efficacy end points (continuous and proportion type) were analyzed using the ITT population for the BOREAS study and the ITT population who had an opportunity to complete 52 weeks of study intervention for the NOTUS study. All safety analyses were descriptive and performed on the safety population, defined as all patients who received at least 1 dose of study intervention analyzed according to the intervention actually received (Table 8).49,50
Table 8: Analysis Populations of the BOREAS and NOTUS Trials
Population | Definition | Application |
|---|---|---|
ITT | Randomized population analyzed according to the treatment group allocated by randomization, analyzed in the treatment group to which they are randomized. | Efficacy analyses |
Safety analysis set | All patients who actually received at least 1 dose or a partial dose of the study drug, analyzed according to the treatment patients actually received. | Safety analyses |
ITT = intention to treat.
Sources: Clinical Study Reports for the BOREAS and NOTUS studies.20,21 Details included in the table are from the sponsor’s summary of clinical evidence.24
Results
Results of the 52-week double-blind, placebo-controlled treatment phase of the BOREAS and NOTUS studies are summarized in this section.
Patient Disposition
Patient disposition in the BOREAS and NOTUS trials in the double-blind treatment period are summarized in Table 9. In the BOREAS and NOTUS studies, 63.9% and 66.2% of patients did not meet the inclusion criteria, respectively, mostly due to not having evidence of type 2 inflammation (i.e., did not have a blood eosinophil count ≥ 300 cells/µL at screening).
In the BOREAS study, a total of 939 patients were randomized to placebo (n = 471) and dupilumab (n = 468). All patients who were randomized were exposed to study intervention. There were no patients who were exposed but not randomized. Study intervention discontinuation in the placebo group (11.5%) was higher numerically compared with the dupilumab group (9.0%), with withdrawal by patient being the most common reason in both groups (5.9% in the placebo group; 4.7% in the dupilumab group).
In the NOTUS study, a total of 935 patients were randomized to placebo (n = 465) and dupilumab (n = 470). All patients were exposed to study intervention, with the exception of 2 patients in the dupilumab group who were randomized by error but not exposed to the study intervention. Study intervention discontinuation in the dupilumab group (9.6%) was higher numerically compared with the placebo group (8.4%), with withdrawal by patient being the most common reason in both groups (4.5% in the placebo group; 4.3% in the dupilumab group).
In both trials, the ITT population consisted of all patients who were randomized in the treatment groups. The safety population consisted of all patients who received at least 1 full or partial dose of dupilumab or placebo; the analysis was performed according to the treatment each patient received. In the BOREAS study, 1 patient assigned to the placebo group inadvertently received a single dose of dupilumab on day 40, which was reported as a major protocol deviation. This patient was included in the dupilumab group for the safety analyses, so the safety population in the BOREAS study included 470 patients in the placebo group and 469 patients in the dupilumab group. In the NOTUS study, 1 patient allocated to the placebo group inadvertently received a single dose of dupilumab on day 113, which was reported as a major protocol deviation; this patient was included in the dupilumab group for the safety analyses. Overall, the safety population in the NOTUS study consisted of close to all patients who were randomized (99.8%) in the treatment groups.
Table 9: Summary of Patient Disposition From the BOREAS and NOTUS Trials
Patient disposition | BOREAS trial | NOTUS trial | ||
|---|---|---|---|---|
Placebo n = 471 | Dupilumab n = 468 | Placebo n = 465 | Dupilumab n = 470 | |
Screened, N | 2,599 | 2,599 | 2,769 | 2,769 |
Unsuccessful screening, n (%) | 1,660 (63.9) | 1,660 (63.9) | 1,834 (66.2) | 1,834 (66.2) |
Reason for unsuccessful screening, n (%) | ||||
Not having evidence of type 2 inflammation, ≥ 300 cells/µL | 877 (52.8) | 877 (52.8) | 929 (50.7) | 929 (50.7) |
Patients not meeting specific criteria at screening related to smoking history, lung function, symptoms, exacerbations, and background therapy | 399 (24.0) | 399 (24.0) | 558 (30.4) | 558 (30.4) |
Other | 384 (23.1) | 384 (23.1) | 347 (18.9) | 347 (18.9) |
Randomized and exposed, N (%) | 471 (100) | 468 (100) | 465 (100) | 468 (99.6)a |
Completed the study intervention period, n (%) | 417 (88.5) | 426 (91.0) | 333 (71.6) | 330 (70.2) |
Discontinued from study intervention period, n (%) | 54 (11.5) | 42 (9.0) | 39 (8.4) | 45 (9.6) |
Still on-study intervention at data cut-off date, n (%) | 0 | 0 | 93 (20.0) | 93 (19.8) |
Reason for intervention discontinuation, n (%) | ||||
Adverse events | 15 (3.2) | 14 (3.0) | 14 (3.0) | 17 (3.6) |
Lack of efficacy | 1 (0.2) | 0 | 1 (0.2) | 1 (0.2) |
Progressive disease | 1 (0.2) | 1 (0.2) | 0 | 1 (0.2) |
Poor compliance to protocol | 2 (0.4) | 2 (0.4) | 2 (0.4) | 0 |
Withdrawal by patient | 28 (5.9) | 22 (4.7) | 21 (4.5) | 20 (4.3) |
Other reasonsb | 7 (1.5) | 3 (0.6) | 1 (0.2) | 6 (1.3) |
Completed 52-week study period | 440 (93.4) | 445 (95.1) | 337 (72.5) | 344 (73.2) |
Ongoing in the 52-week study period | 0 | 0 | 102 (21.9) | 97 (20.6) |
Discontinued from the study before week 52 | 31 (6.6) | 23 (4.9) | 26 (5.6) | 29 (6.2) |
Reason for discontinuation before week 52, n (%) | ||||
Adverse event | 8 (1.7) | 5 (1.1) | 6 (1.3) | 12 (2.6) |
Poor compliance to protocol | 0 | 1 (0.2) | 1 (0.2) | 0 |
Withdrawal by patient | 18 (3.8) | 11 (2.4) | 18 (3.9) | 14 (3.0) |
Other reason | 5 (1.1) | 5 (1.1) | 1 (0.2) | 3 (0.6) |
Completed the study period | 402 (85.4) | 410 (87.6) | 292 (62.8) | 304 (64.7) |
Ongoing in study | 33 (7.0) | 30 (6.4) | 138 (29.7) | 131 (27.9) |
Discontinued from the study period | 36 (7.6) | 28 (6.0) | 35 (7.5) | 35 (7.4) |
Reason for study discontinuation, n (%) | ||||
Adverse event | 9 (1.9) | 7 (1.5) | 7 (1.5) | 13 (2.8) |
Poor compliance to protocol | 0 | 1 (0.2) | 1 (0.2) | 0 |
Withdrawal by patient | 22 (4.7) | 11 (2.4) | 25 (5.4) | 17 (3.6) |
Other reason | 5 (1.1) | 9 (1.9) | 2 (0.4) | 5 (1.1) |
ITT, N | 471 | 468 | 465 | 470 |
Safety, N | 470c | 469c | 464 | 469 |
ITT = intention to treat.
Note: Data cut-off date was February 8, 2023, for the BOREAS study, and September 29, 2023, (interim analysis) for the NOTUS study, respectively.
aTwo patients (0.4%) in the dupilumab group were randomized but not exposed to the study drug.
bNone of the “other” reasons for permanent study intervention discontinuation were related to safety.
cOne patient who was allocated to the placebo group inadvertently received a single dose of dupilumab on day 40 and was included in the dupilumab group for the safety analyses.
Sources: Clinical Study Reports for the BOREAS and NOTUS studies.20,21 Details included in the table are from the sponsor’s summary of clinical evidence.24
Baseline Characteristics
The baseline characteristics outlined in Table 10 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
The baseline characteristics of the patients enrolled in general were balanced between treatment arms in the BOREAS and NOTUS trials, except for several variables like the serum IgE levels. The mean age of patients was 65.1 years (standard deviation [SD] = 8.1 years) and 65.0 years (SD = 8.3 years) in the BOREAS and NOTUS studies, respectively. The proportion of male patients (66.0% in the BOREAS study; 67.6% in the NOTUS study) were higher than female patients (34.0% in the BOREAS study; 32.4% in the NOTUS study) in both studies. Most patients were white (84.1% in the BOREAS study; 89.6% in the NOTUS study), followed by Asian (14.3% in the BOREAS study; 1.1% in the NOTUS study), American Indian or Alaska Native [wording from original source] (0.7% in the BOREAS study; 5.1% in the NOTUS study), Black or of African descent [wording from original source] (0.5% in the BOREAS study; 1.3% in the NOTUS study), Native Hawaiian or Other Pacific Islander [wording from original source] (0.1% in the BOREAS study; 0.1% in the NOTUS study), multiple races (0.2% in the BOREAS study; 2.1% in the NOTUS study), and not reported (0% in the BOREAS study; 0.6% in the NOTUS study). All patients in both studies had a history of smoking, with 30.0% of patients in the BOREAS study and 29.5% of patients in the NOTUS study who currently smoke. The mean predicted postbronchodilator FEV1 was 50.6% in the BOREAS study and 50.1% in the NOTUS study. The proportion of patients with moderate airflow limitation (i.e., GOLD grade 2; predicted postbronchodilator FEV1 between ≥ 50% and < 80%) was █████ ██ █████ across the treatment arms in the BOREAS and NOTUS studies. The proportion of patients with severe airflow limitation (i.e., GOLD grade 3; predicted postbronchodilator FEV1 between ≥ 30% and < 50%) was █████ ██ ██████ A total of ██ ██████ patients in the BOREAS study and ██ ██████ patients in the NOTUS study had airflow limitation categorized as very severe (i.e., GOLD grade 4; predicted postbronchodilator FEV1 < 30%). The mean prebronchodilator FEV1 was 1.3 L and 1.4 L in the BOREAS and NOTUS studies, respectively; the mean predicted prebronchodilator FEV1 was 47.1% and 47.2% in the BOREAS and NOTUS studies, respectively.20,21 Most patients had MRC Dyspnea Scale grade 3 (equivalent to mMRC = grade 2; 55.6% to 66.0% across the groups in the BOREAS and NOTUS studies) or MRC Dyspnea Scale grade 4 (equivalent to mMRC = grade 3; 31.0% to 36.2% across the groups in the BOREAS and NOTUS studies).
In both the BOREAS and NOTUS studies, randomization was stratified by ICS dose at baseline. Overall, 27.4% of patients in the BOREAS study and 27.9% of patients in the NOTUS study were receiving high-dose ICS while 72.6% and 72.1% of patients were not receiving high-dose ICS at baseline in the 2 studies, respectively. This latter group also included patients who were receiving double therapy (LABA and LAMA; 2.1% and 1.1% of all patients in the BOREAS and NOTUS studies, respectively).20,21
In the BOREAS study, approximately 22% of patients had a history of at least 1 other type 2 inflammatory disease, with allergic rhinitis being the most frequent (13.0%). In the NOTUS study, 12.4% of patients had a history of at least 1 other type 2 inflammatory disease, with allergic rhinitis being the most frequent (6.5%). In the NOTUS study, 2 patients in the dupilumab group had an ongoing medical history of asthma, which was reported as a major protocol deviation.20,21
Several between-group imbalances in baseline characteristics were identified in the trials. In the BOREAS study, compared with the placebo group, the dupilumab group had a higher proportion of patients who were female (31.6% in the placebo group; 36.3% in the dupilumab group), had at least 1 severe COPD exacerbation experienced within 1 year before visit 1 (24.2% in the placebo group; 27.6% in the dupilumab group), had allergic rhinitis (10.8% in the placebo group; 14.5% in the dupilumab group), and mean serum IgE level (417.4 IU/mL [SD = 1,063.0 IU/mL ] in the placebo group; 486.6 IU/mL [SD = 1,868.1 IU/mL] in the dupilumab group). In the NOTUS study, compared with the placebo group, the dupilumab group had a lower proportion of patients with emphysema (32.3% in the placebo group; 28.5% in the dupilumab group), a lower proportion of patients with moderate airflow limitation (GOLD grade 2; █████ in the placebo group; █████ in the dupilumab group), a higher proportion of patients with severe airflow limitation (GOLD grade 3; 46.6% in the placebo group; 51.2% in the dupilumab group), and higher mean serum IgE level (429.1 IU/mL [SD = 1,273.1 IU/mL ] in the placebo group; 532.3 IU/mL [SD = 1,299.5 IU/mL] in the dupilumab group). No other notable between-group imbalances were observed for other baseline characteristics in the trials.
Table 10: Summary of Baseline Characteristics From the BOREAS and NOTUS Trials
Characteristic | BOREAS trial | NOTUS trial | ||
|---|---|---|---|---|
Placebo n = 471 | Dupilumab n = 468 | Placebo n = 465 | Dupilumab n = 470 | |
Demographic characteristics | ||||
Age (years) | ||||
Mean (SD) | 65.2 (8.1) | 65.0 (8.0) | 64.9 (8.5) | 65.2 (8.1) |
40 to 64 years, n (%) | 201 (42.7) | 192 (41.0) | 213 (45.8) | 196 (41.7) |
65 to 74 years, n (%) | 204 (43.3) | 216 (46.2) | 190 (40.9) | 218 (46.4) |
75 to 80 years, n (%) | 66 (14.0) | 60 (12.8) | 57 (12.3) | 52 (11.1) |
81 to 85 years, n (%) | 0 | 0 | 5 (1.1) | 4 (0.9) |
Sex, n (%) | ||||
Female | 149 (31.6) | 170 (36.3) | 153 (32.9) | 150 (31.9) |
Male | 322 (68.4) | 298 (63.7) | 312 (67.1) | 320 (68.1) |
Race, n (%) | ||||
American Indian or Alaska Native | 4 (0.8) | 3 (0.6) | 26 (5.6) | 22 (4.7) |
Asian | 67 (14.2) | 67 (14.3) | 3 (0.6) | 7 (1.5) |
Black or of African descent | 2 (0.4) | 3 (0.6) | 8 (1.7) | 4 (0.9) |
Native Hawaiian or other Pacific Islander | 1 (0.2) | 0 | 0 | 1 (0.2) |
White | 397 (84.3) | 393 (84.0) | 416 (89.5) | 422 (89.8) |
Multiple | 0 | 2 (0.4) | 8 (1.7) | 12 (2.6) |
Not reported | 0 | 0 | 4 (0.9) | 2 (0.4) |
Weight and BMI | ||||
Mean weight, kg (SD) | 77.51 (17.60) | 76.51 (17.25) | 78.74 (18.29) | 79.74 (17.11) |
Mean BMI, kg/m2 (SD) | 27.65 (5.73) | 27.51 (5.44) | 27.78 (5.56) | 28.09 (5.29) |
Smoking status | ||||
Formerly smoked, n (%) | 323 (68.6) | 334 (71.4) | 331 (71.2) | 328 (69.8) |
Currently smoke, n (%) | 148 (31.4) | 134 (28.6) | 134 (28.8) | 142 (30.2) |
Mean smoking pack-years (SD) | 41.38 (24.36) | 39.60 (22.32) | 42.12 (30.18) | 38.56 (23.67) |
Disease characteristics | ||||
Baseline high-dose ICS from medication history, yes, n (%) | 126 (26.8) | 131 (28.0) | 134 (28.8) | 127 (27.0) |
Baseline prebronchodilator FEV1 (L) | n = 471 | n = 467 | n = 465 | n = 469 |
Mean (SD) | 1.32 (0.46) | 1.28 (0.45) | 1.38 (0.50) | 1.35 (0.49) |
Baseline postbronchodilator FEV1 (L) | n = 471 | n = 468 | n = 464 | n = 467 |
Mean (SD) | 1.41 (0.47) | 1.39 (0.47) | 1.46 (0.50) | 1.43 (0.49) |
Baseline prebronchodilator FEV1 predicted | n = 471 | n = 467 | n = 465 | n = 469 |
Mean (SD) | 47.27 (13.17) | 46.91 (13.56) | 47.88 (13.01) | 46.59 (13.02) |
Baseline postbronchodilator FEV1 predicted | n = 471 | n = 468 | n = 465 | n = 469 |
Mean (SD) | 50.63 (12.97) | 50.57 (13.26) | 50.68 (12.58) | 49.49 (12.58) |
Baseline prebronchodilator FVC (L) | n = 471 | n = 467 | n = 465 | n = 469 |
Mean (SD) | 2.83 (0.90) | 2.71 (0.74) | 2.86 (0.85) | 2.82 (0.87) |
Baseline prebronchodilator FEV1:FVC ratio | n = 471 | n = 467 | n = 465 | n = 469 |
Mean (SD) | 0.48 (0.11) | 0.48 (0.12) | 0.49 (0.12) | 0.49 (0.12) |
Baseline postbronchodilator FEV1:FVC ratio | n = 471 | n = 468 | n = 464 | n = 467 |
Mean (SD) | 0.49 (0.11) | 0.49 (0.12) | 0.50 (0.12) | 0.50 (0.12) |
Baseline SGRQ total score | n = 461 | n = 461 | n = 449 | n = 456 |
Mean (SD) | 48.41 (17.80) | 48.42 (17.04) | 51.12 (16.51) | 51.95 (17.51) |
Mean time since first diagnosis of COPD, years (SD) | 8.96 (6.10) | 8.59 (5.96) | 8.96 (6.66) | 9.57 (6.01) |
Mean age (years) at onset of COPD (SD) | 56.8 (9.2) | 57.0 (8.8) | 56.5 (9.1) | 56.1 (9.1) |
Number of moderate or severe COPD exacerbationsa experienced within 1 year before visit 1 | n = 471 | n = 468 | n = 465 | n = 470 |
Mean (SD) | 2.3 (1.0) | 2.2 (1.1) | 2.1 (0.7) | 2.2 (1.0) |
1, n (%) | 46 (9.8) | 59 (12.6) | 59 (12.7) | 58 (12.3) |
2, n (%) | 303 (64.3) | 312 (66.7) | 343 (73.8) | 322 (68.5) |
3, n (%) | 90 (19.1) | 57 (12.2) | 50 (10.8) | 57 (12.1) |
≥ 4, n (%) | 32 (6.8) | 40 (8.5) | 13 (2.8) | 32 (6.8) |
Number of moderate COPD exacerbationsa experienced within 1 year before visit 1 | n = 471 | n = 468 | n = 464 | n = 470 |
Mean (SD) | 2.0 (1.1) | 1.9 (1.3) | 1.8 (1.0) | 1.9 (1.1) |
1, n (%) | 30 (6.4) | 30 (6.4) | 21 (4.5) | 27 (5.7) |
2, n (%) | 280 (59.4) | 280 (59.8) | 320 (69.0) | 303 (64.5) |
3, n (%) | 77 (16.3) | 51 (10.9) | 44 (9.5) | 50 (10.6) |
≥ 4, n (%) | 25 (5.3) | 29 (6.2) | 9 (1.9) | 21 (4.5) |
Number of severe COPD exacerbationsa experienced within 1 year before visit 1 | n = 471 | n = 468 | n = 465 | n = 470 |
Mean (SD) | 0.3 (0.6) | 0.4 (0.7) | 0.3 (0.6) | 0.3 (0.6) |
≥ 1, n (%) | 114 (24.2) | 129 (27.6) | 100 (21.5) | 113 (24.1) |
1, n (%) | 95 (20.2) | 107 (22.9) | 86 (18.5) | 91 (19.4) |
2, n (%) | 13 (2.8) | 15 (3.2) | 9 (1.9) | 17 (3.6) |
3, n (%) | 4 (0.8) | 3 (0.6) | 2 (0.4) | 3 (0.6) |
≥ 4, n (%) | 2 (0.4) | 4 (0.9) | 3 (0.6) | 2 (0.4) |
Baseline E-RS:COPD total score | n = 467 | n = 461 | n = 446 | n = 455 |
Mean (SD) | 13.0 (6.9) | 12.9 (7.2) | 13.3 (7.2) | 13.4 (6.7) |
GOLD severityb at baseline | ||||
GOLD grade 1, n (%) | 12 (2.5) | 12 (2.6) | 7 (1.5) | 4 (0.9) |
GOLD grade 2, n (%) | 219 (46.5) | 229 (48.9) | 229 (████) | 213 (████) |
GOLD grade 3, n (%) | 229 (████) | 213 (████) | 216 (46.6) | 239 (51.2) |
GOLD grade 4, n (%) | 11 (2.3) | 14 (3.0) | 12 (2.6) | 11 (2.4) |
Ongoing COPD-related comorbid and type 2 conditions | ||||
Chronic bronchitis, n (%) | 438 (93.0) | 437 (93.4) | 458 (98.5) | 462 (98.3) |
Emphysema, n (%) | 151 (32.1) | 155 (33.1) | 150 (32.3) | 134 (28.5) |
Bronchiectasis, n (%) | 0 | 6 (1.3) | 4 (0.9) | 3 (0.6) |
Allergic rhinitis, n (%) | 51 (10.8) | 68 (14.5) | 27 (5.8) | 32 (6.8) |
Chronic sinusitis, n (%) | 16 (3.4) | 19 (4.1) | 14 (3.0) | 17 (3.6) |
Nasal polyps, n (%) | 10 (2.1) | 12 (2.6) | 0 | 8 (1.7) |
Food allergy, n (%) | 12 (2.5) | 11 (2.4) | 3 (0.6) | 1 (0.2) |
Atopic dermatitis, n (%) | 5 (1.1) | 10 (2.1) | 2 (0.4) | 7 (1.5) |
Hives, n (%) | 6 (1.3) | 2 (0.4) | 1 (0.2) | 2 (0.4) |
Hypersensitivity to NSAID, n (%) | 3 (0.6) | 7 (1.5) | 6 (1.3) | 2 (0.4) |
Allergic conjunctivitis, n (%) | 4 (0.8) | 5 (1.1) | 1 (0.2) | 0 |
Hypersensitivity to Aspirin, n (%) | 3 (0.6) | 3 (0.6) | 1 (0.2) | 3 (0.6) |
Eosinophilic esophagitis, n (%) | 1 (0.2) | 0 | 0 | 1 (0.2) |
Biomarkers at baseline | ||||
Baseline blood eosinophil levels | n = 471 | n = 468 | n = 465 | n = 469 |
Mean (SD), giga/L | 0.41 (0.33) | 0.39 (0.26) | 0.40 (0.31) | 0.41 (0.36) |
< 300 cells/µL, n (%) | 186 (39.5) | 180 (38.5) | 188 (40.4) | 184 (39.2) |
≥ 300 cells/µL, n (%) | 285 (60.5) | 288 (61.5) | 277 (59.6) | 285 (60.8) |
FeNO (parts per billion) | n = 442 | n = 433 | n = 423 | n = 429 |
Mean (SD) | 23.51 (22.00) | 25.18 (22.79) | 24.35 (23.38) | 24.76 (28.31) |
< 20 parts per billion, n (%) | 254 (57.5) | 238 (55.0) | 240 (56.7) | 257 (59.9) |
≥ 20 parts per billion, n (%) | 188 (42.5) | 195 (45.0) | 183 (43.3) | 172 (40.1) |
Serum IgE (IU/mL) | n = 444 | n = 442 | n = 441 | n = 455 |
Mean (SD) | 417.43 (1,063.00) | 486.58 (1,868.08) | 429.05 (1,273.08) | 532.25 (1,299.50) |
Mean fibrinogen (SD), mg/gL | n = 469 | n = 464 | n = 459 | n = 461 |
Mean (SD) | 471.97 (144.67) | 460.61 (143.60) | 483.46 (136.55) | 491.32 (133.84) |
MRC or mMRC Dyspnea Scale grade at baseline | ||||
N | 470 | 464 | 462 | 466 |
MRC grade 2 (equivalent to mMRC grade 1), n (%) | 0 | 2 (0.4) | 6 (1.3) | 7 (1.5) |
MRC grade 3 (equivalent to mMRC grade 2), n (%) | 269 (57.2) | 258 (55.6) | 305 (66.0) | 291 (62.4) |
MRC grade 4 (equivalent to mMRC grade 3), n (%) | 164 (34.9) | 168 (36.2) | 143 (31.0) | 164 (35.2) |
MRC grade 5 (equivalent to mMRC grade 4), n (%) | 37 (7.9) | 36 (7.8) | 8 (1.7) | 4 (0.9) |
BMI = body mass index; COPD = chronic obstructive pulmonary disease; E-RS:COPD = Evaluating Respiratory Symptoms in chronic obstructive pulmonary disease; FeNO = fractional exhaled nitric oxide; FEV1 = forced expiratory volume in the first second; FVC = forced vital capacity; GOLD = Global Initiative for Chronic Obstructive Lung Disease; ICS = inhaled corticosteroid; IgE = immunoglobulin E; mMRC = modified Medical Research Council; MRC = Medical Research Council; NSAID = nonsteroidal anti-inflammatory drug; SD = standard deviation; SGRQ = St. George's Respiratory Questionnaire.
Notes: 1 giga/L is equivalent to 1,000 cells/µL.
Racial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
aModerate exacerbations are recorded by the investigator and defined as acute exacerbation of COPD that require either systemic corticosteroids (such as intramuscular, IV, or oral) and/or antibiotics. Severe exacerbations are recorded by the investigator and defined as acute exacerbation of COPD requiring hospitalization, treatment for > 24 hours in an emergency department or urgent care facility, or result in death. For both moderate and severe events to be counted as separate events, they must be separated by at least 14 days.
bAirflow limitation severity is based on the postbronchodilator value of FEV1 (% reference) and assigned into GOLD grade 1 to 4. GOLD grade 1 refers to mild airflow limitation (predicted postbronchodilator FEV1 ≥ 80%), GOLD 2 refers to moderate (predicted postbronchodilator FEV1 between ≥ 50% and < 80%), GOLD 3 refers to severe (predicted postbronchodilator FEV1 between ≥ 30% and < 50%), and GOLD 4 refers to very severe (predicted postbronchodilator FEV1 < 30%).15
Sources: Clinical Study Reports for the BOREAS and NOTUS studies.20,21 Details included in the table are from the sponsor’s summary of clinical evidence.24
Exposure to Interventions
Study Treatments
Patient exposure to study treatment in the double-blind treatment period of the BOREAS and NOTUS studies are summarized in Table 11. No notable between-group difference was observed in the cumulative duration and mean duration of patient exposure to treatment in both trials. A very small proportion of patients (0 to 1.1% across the treatment arms) had a treatment compliance rate of less than 80%. A total of 427 patients (91.0%) in the dupilumab group and 418 (88.9%) in the placebo group in the BOREAS study, and 338 (72.1%) in the dupilumab group and 341 (73.5%) in the placebo group in the NOTUS study had at least 48 weeks of study treatment exposure.20,21
Table 11: Summary of Patient Exposure From the BOREAS and NOTUS Trials
Exposure | BOREAS trial | NOTUS trial | ||
|---|---|---|---|---|
Placebo n = 470 | Dupilumab n = 469 | Placebo n = 464 | Dupilumab n = 469 | |
Cumulative duration to treatment exposure, patient-years | 439 | 444 | 409 | 410 |
Duration (days), mean (SD) | 340.78 (76.25) | 346.07 (66.55) | 321.88 (84.54) | 319.58 (85.68) |
Mean % injections with compliance to IMP administration (SD) | 98.45 (3.55) | 98.40 (5.07) | 98.53 (4.04) | 98.82 (2.72) |
Patients with < 80% compliance | 2 (0.4) | 5 (1.1) | 3 (0.6) | 0 |
Patients with ≥ 80% compliance | 468 (99.6) | 464 (98.9) | 461 (99.4) | 469 (100) |
IMP = investigational medicinal product; SD = standard deviation.
Sources: Clinical Study Reports for the BOREAS and NOTUS studies.20,21 Details included in the table are from the sponsor’s summary of clinical evidence.24
Concomitant Medications
The use of concomitant medications to control COPD in the double-blind treatment period of the BOREAS and NOTUS studies is summarized in Table 12. In both studies, most patients (97.4% to 99.1% across the treatment arms) were receiving ICS controller medications, and the triple therapy of LABA, LAMA, and ICS (97.2% to 99.1%). A small proportion of patients (0.9% to 2.6%) were receiving LABA and LAMA (with no ICS). No notable imbalance was observed between treatment groups.
The proportion of patients who received SCS over the 52-week intervention period regardless of cause was slightly lower in the dupilumab group compared with the placebo group in the BOREAS study (36.8% in the dupilumab group; 42.3% in the placebo group)20 and the NOTUS study (30.4% in the dupilumab group; 35.9% in the placebo group).21 Data of rescue medication with SABAs and short-acting antimuscarinics were not reported in the BOREAS and NOTUS studies.20,21
Table 12: Summary of Controller Medication Use From the BOREAS and NOTUS Trials
Controller medication | BOREAS trial | NOTUS trial | ||
|---|---|---|---|---|
Placebo n = 471 | Dupilumab n = 468 | Placebo n = 465 | Dupilumab n = 470 | |
Patients with ICS controller medication, n (%) | 463 (98.3) | 456 (97.4) | 459 (98.7) | 466 (99.1) |
High-dose ICS, n (%) | 126 (26.8) | 131 (28.0) | 134 (28.8) | 127 (27.0) |
Non–high-dose ICS, n (%) | 337 (71.5) | 325 (69.4) | 325 (69.9) | 339 (72.1) |
ICS dose in fluticasone propionate equivalent dose (mcg) | ||||
n | 463 | 456 | 459 | 466 |
Mean (SD) | 547.49 (303.91) | 553.83 (293.65) | 815.61 (5,816.59)a | 1,398.65 (18,507.00)a |
Patients with controller medication other than ICS | ||||
LABA, n (%) | 471 (100) | 468 (100) | 464 (99.8) | 470 (100) |
LAMA, n (%) | 469 (99.6) | 467 (99.8) | 464 (99.8) | 470 (100) |
Antileukotrienes, n (%) | 7 (1.5) | 11 (2.4) | 5 (1.1) | 1 (0.2) |
Methylxanthines, n (%) | 16 (3.4) | 12 (2.6) | 14 (3.0) | 22 (4.7) |
PDE4 inhibitor, n (%) | 9 (1.9) | 2 (0.4) | 5 (1.1) | 2 (0.4) |
Other, n (%)b | 1 (0.2) | 1 (0.2) | 0 | 1 (0.2) |
Patients with combinations of controller medications | ||||
ICS, LABA, and LAMA, n (%) | 461 (97.9) | 455 (97.2) | 458 (98.5) | 466 (99.1) |
LABA and LAMA (no ICS), n (%) | 8 (1.7) | 12 (2.6) | 6 (1.3) | 4 (0.9) |
Percentage of days with compliance to overall controller medication(s) (ICS, LABA, and LAMA or LABA and LAMA if ICS contraindicated) | ||||
n | 470 | 469 | 464 | 468 |
Mean (SD) | 91.31 (10.93) | 91.82 (10.83) | 90.67 (11.87) | 90.33 (11.44) |
ICS = inhaled corticosteroid; LABA = long-acting beta-agonist; LAMA = long-acting muscarinic antagonist; SD = standard deviation.
aIn the NOTUS study, 2 patients (1 in the placebo group and 1 in dupilumab group) had an incorrect unit reported, which led to incorrect high fluticasone propionate equivalent doses.
b“Other” included ipratropium and salbutamol sulphate in the BOREAS study and included salbutamol in the NOTUS study.
Sources: Clinical Study Reports for the BOREAS and NOTUS studies.20,21 Details included in the table are from the sponsor’s summary of clinical evidence.24
Efficacy
Key efficacy results from the double-blind treatment period of the BOREAS and NOTUS studies are summarized in Table 13. Additional efficacy results of the 2 studies are summarized in Table 15. According to the clinical experts consulted for this review, none of the prespecified subgroup analysis factors have been shown as a treatment effect modifier. The clinical experts noted that the eosinophil count is a biomarker of type 2 inflammation. In this report, subgroup results are narratively summarized for the subgroup analyses of maximum eosinophil counts during screening (< 500 cells/µL or ≥ 500 cells/µL) only.
Annualized Rate of Moderate or Severe COPD Exacerbations During the 52-Week Treatment Period
As the primary end point in both trials, the adjusted annualized rate of moderate or severe COPD exacerbations over the 52-week intervention period in the ITT population was lower in the dupilumab group compared with the placebo group: 0.78 (95% CI, 0.64 to 0.93) versus 1.10 (95% CI, 0.93 to 1.30) with an RR of 0.70 (95% CI, 0.58 to 0.86; P < 0.001) in the BOREAS study, and 0.86 (95% CI, 0.70 to 1.06) versus 1.30 (95% CI, 1.05 to 1.60) with an RR of 0.66 (95% CI, 0.54 to 0.82; P < 0.001) in the NOTUS study. The absolute difference between groups was ██████ ████ ███ ██████ ██ ███████ in the BOREAS study, and ██████ ████ ███ ██████ ██ ███████ in the NOTUS study. The total number of moderate or severe COPD exacerbation events reported during the 52-week intervention period was lower in the dupilumab group as compared to the placebo group (296 versus 422 in the BOREAS study, and 263 versus 352 in the NOTUS study) (Table 15). Of these, most were moderate COPD exacerbation events (268 in the dupilumab group versus 385 in the placebo group in the BOREAS study, and 237 in the dupilumab group versus 313 in the placebo group in the NOTUS study). The total number of severe COPD exacerbation events was low in the dupilumab and placebo groups (28 and 37 events, respectively, the BOREAS study; 26 and 39 events, respectively, in the NOTUS study). In both groups of the BOREAS and NOTUS studies, all severe COPD exacerbation events required hospitalization or an emergency medical care visit, and none led to death (Table 15). In both trials, during the 52-week intervention period, a lower mean cumulative number of moderate or severe COPD exacerbation events was observed in the dupilumab group compared to the placebo group. In both trials, the difference between the 2 groups was apparent by week 4 and progressively increased up to week 52 (Figure 2 and Figure 3).
In both trials, the results from the prespecified sensitivity analyses of the annualized rate of moderate or severe COPD exacerbation events in the ITT population were consistent with the primary analysis results.20,21
In the BOREAS study, the point estimate of the RR was lower in patients with the maximum eosinophils counts of 500 cells/µL or greater during screening (RR = 0.505; 95% CI, 0.345 to 0.737) compared with the patients with eosinophil counts of less than 500 cells/µL (RR = 0.797; 95% CI, 0.637 to 0.997) with a nominally significant treatment by subgroup quantitative interaction (nominal P value = 0.0141).20 In the NOTUS study, the point estimates of the RR were similar between the 2 subgroups: patients with the maximum eosinophil counts of 500 cells/µL or greater during screening (RR = 0.699; 95% CI, 0.490 to 0.998) and patients with eosinophil counts of less than 500 cells/µL (RR = 0.646; 95% CI, 0.492 to 0.848).21
Change From Baseline in Prebronchodilator FEV1 at Week 12 in the ITT Population
The mean prebronchodilator FEV1 values at baseline were similar between study groups in both the BOREAS study (1.28 L in the dupilumab group; 1.32 L in the placebo group) and the NOTUS study (1.35 L in the dupilumab group and 1.38 L in the placebo group). In the BOREAS study, the LS mean change from baseline to week 12 in prebronchodilator FEV1 was an increase of 0.160 L in the dupilumab group versus 0.077 L in the placebo group (between-group LS mean difference = 0.083 L; 95% CI, 0.042 L to 0.125 L; P < 0.001), and an increase of 0.139 L in the dupilumab group versus 0.057 L in the placebo group (between-group LS mean difference = 0.082 L; 95% CI, 0.040 L to 0.124 L; P < 0.001) in the NOTUS study.
In both trials, the results from the prespecified sensitivity analyses of the change from baseline in prebronchodilator FEV1 at week 12 in the ITT population were overall consistent with the primary analysis results.20,21
In the BOREAS study, the point estimate of the LS mean difference in the change from baseline in prebronchodilator FEV1 at week 12 was numerically greater in patients with maximum eosinophil counts of 500 cells/µL or higher during screening (0.142; 95% CI, 0.060 to 0.223) compared with the patients with the eosinophil counts of less than 500 cells/µL (0.053; 95% CI, 0.006 to 0.100) with a nominally significant treatment by subgroup quantitative interaction (nominal P value = 0.0382).20 Similarly, in the NOTUS study, the point estimates of the LS mean difference were numerically greater in patients with the maximum eosinophil counts of 500 cells/µL or higher during screening (0.122; 95% CI, 0.050 to 0.195) compared with the patients with the eosinophil counts of less than 500 cells/µL (0.059; 95% CI, 0.007 to 0.111) with no treatment by subgroup interaction observed.21
Change From Baseline in Prebronchodilator FEV1 at Week 52 in the ITT Population
The LS mean change from baseline to week 52 in prebronchodilator FEV1 was an increase of 0.153 L in the dupilumab group versus 0.070 L in the placebo group (between-group LS mean difference = 0.083 L; 95% CI, 0.038 L to 0.128 L; P < 0.001) in the BOREAS study, and an increase of 0.115 L in the dupilumab group versus 0.054 L in the placebo group (between-group LS mean difference = 0.062 L; 95% CI, 0.011 L to 0.113 L; P = 0.02) in the NOTUS study.
The results of sensitivity analyses and the subgroup analyses by maximum eosinophil counts during screening (< 500 cells/µL or ≥ 500 cells/µL) were not reported for the change from baseline in prebronchodilator FEV1 at week 52.20,21
Health-Related Quality of Life
Change From Baseline in SGRQ Total Score at Week 52
The mean SGRQ total score was similar between the dupilumab and placebo groups in both the BOREAS study (48.42 and 48.41 points, respectively) and the NOTUS study (52.66 and 51.01, respectively). The LS mean change from baseline to week 52 in SGRQ total score was −9.7 in the dupilumab group versus −6.4 in the placebo group (between-group LS mean difference = −3.4; 95% CI, −5.5 to −1.3; P = 0.002) in the BOREAS study, and −9.8 in the dupilumab group versus −6.4 in the placebo group (between-group LS mean difference = −3.4; 95% CI, −5.8 to −0.9; nominal P = ██████) in the NOTUS study.
The results of sensitivity analyses and the subgroup analyses by maximum eosinophils counts during screening were not reported for the change from baseline in the SGRQ total score at week 52.20,21
Table 13: Summary of Key Efficacy Results From the BOREAS and NOTUS Trials
Outcomes | BOREAS trial | NOTUS trial | ||
|---|---|---|---|---|
Placebo n = 471 | Dupilumab n = 468 | Placebo n = 465 | Dupilumab n = 470 | |
Annualized rate of moderate or severe COPD exacerbations during the 52-week treatment period | ||||
Adjusted annualized moderate or severe exacerbation event rate (95% CI) | 1.10 (0.93 to 1.30) | 0.78 (0.64 to 0.93) | 1.30 (1.05 to 1.60) | 0.86 (0.70 to 1.06) |
RR vs. placebo (95% CI) | 0.705 (0.581 to 0.857) | 0.705 (0.581 to 0.857) | 0.664 (0.535 to 0.823) | 0.664 (0.535 to 0.823) |
P value | 0.0005a | 0.0005a | 0.0002a | 0.0002a |
Risk difference vs. placebo (95% CI) | ██████ ████████ ███████ | ██████ ████████ ███████ | ██████ ████████ ███████ | ██████ ████████ ███████ |
Change from baseline in prebronchodilator FEV1 at week 12 | ||||
Baseline number of patients | 471 | 467 | 465 | 469 |
Baseline prebronchodilator FEV1 (L), mean (SD) | 1.32 (0.46) | 1.28 (0.45) | 1.38 (0.50) | 1.35 (0.49) |
Week 12 number of patients | 439 | 449 | 444 | 449 |
Mean at week 12, L (SD) | 1.38 (0.51) | 1.44 (0.55) | 1.44 (0.56) | 1.49 (0.59) |
Change from baseline (L), mean (SD) | 0.06 (0.30) | 0.15 (0.37) | 0.05 (0.31) | 0.14 (0.35) |
Number of patients in the model | 469 | 466 | 461 | 464 |
Change from baseline (L), LS mean (SE) | 0.077 (0.018) | 0.160 (0.018) | 0.057 (0.017) | 0.139 (0.017) |
LS mean difference (L), mean (95% CI) | 0.083 (0.042 to 0.125) | 0.083 (0.042 to 0.125) | 0.082 (0.040 to 0.124) | 0.082 (0.040 to 0.124) |
P value vs. placebo | < 0.0001a | < 0.0001a | 0.0001a | 0.0001a |
Change from baseline in prebronchodilator FEV1 at week 52 | ||||
Baseline number of patients | 471 | 467 | 359 | 361 |
Baseline prebronchodilator FEV1 (L), mean (SD) | 1.32 (0.46) | 1.28 (0.45) | 1.40 (0.49) | 1.36 (0.49) |
Week 52 number of patients | 420 | 426 | 324 | 333 |
Mean at week 52, L (SD) | 1.39 (0.53) | 1.44 (0.57) | 1.46 (0.58) | 1.47 (0.61) |
Change from baseline (L), mean (SD) | 0.05 (0.32) | 0.14 (0.39) | 0.05 (0.34) | 0.11 (0.35) |
Number of patients in the model | 469 | 466 | 356 | 359 |
Change from baseline (L), LS mean (SE) | 0.070 (0.019) | 0.153 (0.019) | 0.054 (0.020) | 0.115 (0.021) |
LS mean difference (L), mean (95% CI) | 0.083 (0.038 to 0.128) | 0.083 (0.038 to 0.128) | 0.062 (0.011 to 0.113) | 0.062 (0.011 to 0.113) |
P value vs. placebo | 0.0003a | 0.0003a | 0.0182a | 0.0182a |
Change from baseline in the SGRQ total score at week 52b | ||||
Baseline number of patients | 461 | 461 | 347 | 352 |
Mean baseline SGRQ total score (SD) | 48.41 (17.80) | 48.42 (17.04) | 51.01 (16.14) | 52.66 (17.03) |
Week 52 number of patients | 406 | 420 | 318 | 325 |
Mean at week 52 (SD) | 42.08 (18.51) | 38.52 (19.01) | 44.25 (17.71) | 42.35 (19.88) |
Change from baseline number of patients | 400 | 415 | 310 | 317 |
Mean change from baseline (SD) | −6.16 (17.59) | −9.44 (18.30) | −5.86 (15.76) | −9.87 (18.04) |
Number of patients in the model | 456 | 456 | 342 | 350 |
Change from baseline, LS mean (SE) | −6.4 ███████ | −9.7 ███████ | −6.4 ███████ | −9.8 ███████ |
LS mean difference, mean (95% CI) | −3.4 (−5.5 to −1.3) | −3.4 (−5.5 to −1.3) | −3.4 (−5.8 to −0.9) | −3.4 (−5.8 to −0.9) |
P value vs. placebo | 0.002a | 0.002a | ██████c | ██████c |
CI = confidence interval; COPD = chronic obstructive pulmonary disease; FEV1 = forced expiratory volume in the first second; ITT = intention to treat; LS = least squares; RR = relative risk; SD = standard deviation; SE = standard error; SGRQ = St. George's Respiratory Questionnaire; vs. = versus.
Note: For the SGRQ outcome, in both the BOREAS and NOTUS studies, a participant whose condition responded to treatment is defined as a participant with improvement from baseline in SGRQ total score at week 52 by ≥ 4 points. Participants with improvement < 4 points or with missing values are considered as patients who did not respond to treatment.
aP value has been adjusted for multiple testing.
bData for change from baseline in the SGRQ total score were reported for the population who were randomized in the BOREAS study, and the ITT population with an opportunity to reach week 52 in the NOTUS study.
cNominal P value.
Sources: Clinical Study Reports for the BOREAS and NOTUS studies.20,21 Details included in the table are from the sponsor’s summary of clinical evidence.24
Harms
At data cut-off for the BOREAS study (February 8, 2023), all patients had completed the 52-week intervention period (or prematurely discontinued the study intervention), and 63 patients (6.71%) were ongoing in the 12-week postintervention follow-up period. At the IA cut-off for the NOTUS study (September 29, 2023), of the 935 patients who were randomized, 186 patients (19.89%) were still on-study intervention, and 70 (7.49%) were ongoing in the 12-week postintervention follow-up period. The harms data of the BOREAS and NOTUS studies are summarized in Table 14.
Treatment-Emergent Adverse Events
In the BOREAS study, there was a similar proportion of patients with any TEAE between the dupilumab (77.4%) and placebo (76.0%) groups. The following TEAEs were more frequently reported in the dupilumab group compared to the placebo group (≥ 2% in the dupilumab group and a difference of ≥ 1% versus the placebo group): urinary tract infection (4.9% of patients in the dupilumab group versus 2.1% of patients in the placebo group), headache (8.1% in the dupilumab group versus 6.8% in the placebo group), diarrhea (5.3% in the dupilumab group versus 3.6% in the placebo group), toothache (2.6% in the dupilumab group versus 1.1% in the placebo group), gastritis (2.3% in the dupilumab group versus 0.4% in the placebo group), and back pain (5.1% in the dupilumab group versus 3.4% in the placebo group). By contrast, the following TEAEs were reported with a lower frequency in the dupilumab group compared to the placebo group (≥ 2% in the placebo group and a difference of ≥ 1% versus the dupilumab group): upper respiratory tract infection (7.9% versus 9.8% of patients in the dupilumab and placebo groups, respectively), COVID-19 (4.1% versus 5.7% in the dupilumab and placebo groups, respectively), pneumonia (2.8% versus 4.0% in the dupilumab and placebo groups, respectively), lower respiratory tract infection (1.3% versus 2.3% in the dupilumab and placebo groups, respectively), gastroenteritis (1.1% versus 2.3% in the dupilumab and placebo groups, respectively), and hypertension (3.6% versus 6.0% in the dupilumab and placebo groups, respectively).
In the NOTUS study, there was a similar proportion of patients with any TEAE between the dupilumab (66.7%) and placebo (65.9%) groups. The following TEAEs were more frequently reported in the dupilumab group as compared to the placebo group (≥ 2% in the dupilumab group and a difference of ≥ 1% versus the placebo group): COVID-19 (9.4% in the dupilumab group versus 8.2% patients in the placebo group), headache (7.5% in the dupilumab group versus 6.5% in the placebo group), nasopharyngitis (6.2% in the dupilumab group versus 5.2% in the placebo group), back pain (3.8% in the dupilumab group versus 2.8% in the placebo group), rhinitis (3.2% in the dupilumab group versus 1.1% in the placebo group), viral upper respiratory tract infections (3.0% in the dupilumab group versus 1.1% in the placebo group), and COVID-19 pneumonia (2.1% in the dupilumab group versus 0.4% in the placebo group). By contrast, the following TEAEs were reported with a lower frequency in the dupilumab group compared to the placebo group (≥ 2% in the placebo group and a difference of ≥ 1% versus the dupilumab group): COPD (4.9% in the dupilumab group versus 7.8% in the placebo group), lower respiratory tract infection bacterial (0.9% in the dupilumab group versus 2.4% in the placebo group), and osteoarthritis (0.9% in the dupilumab group versus 2.2% in the placebo group).
Treatment-Emergent SAEs
In the BOREAS study, treatment-emergent SAEs were reported for 13.6% patients in the dupilumab group and 15.5% patients in the placebo group. The most frequently reported treatment-emergent SAE was COPD (i.e., █████ ████████████ ██ █████ ████ ██ ███ █████████ █████ ███ ████ ███████ █████). ██ █████ ███ █████████ █████████ ██ ████ █████████ ███ ███ ██████████ ██ ██ ██████ ██ ███ ███ ███████████ █████████ █████████ ███████████ █████ ████████ ██████ ████████ █████ ███ ██████ ███ ████████ █████████ █████ ███ █████ ████ ███ ████ ██████████ ████████ ███████ ██████████.
In the NOTUS study, treatment-emergent SAEs were less frequently reported in the dupilumab group compared to the placebo group (13.0% versus 15.9%, respectively). The most frequently reported treatment-emergent SAE was COPD (4.9% of patients in the dupilumab group; 7.8% of patients in the placebo group). Of note, per protocol, worsening of COPD was not considered an AE unless it met the seriousness criteria. Pneumonia (1.5% of patients in the dupilumab group; 0.9% of patients in the placebo group), COVID-19 (0.6% in the dupilumab group and 0.9% in the placebo group), and COVID-19 pneumonia (1.3% in the dupilumab group and 0.4% in the placebo group) were the most frequently reported serious infections.
Withdrawals Due to AEs
In the BOREAS study, the proportion of patients with any TEAEs reported by the investigator as the reason for permanent study intervention discontinuation was similar in the dupilumab and placebo groups (14 [3.0%] and 16 [3.4%], respectively). All events were reported in 1 or 2 patients in each study group, and no particular pattern of TEAEs leading to discontinuation of the study drugs was observed. Nine patients in the dupilumab group and 13 patients in the placebo group experienced an SAE that led to permanent study intervention discontinuation. The most frequently reported treatment-emergent SAE that led to permanent study intervention discontinuation was COVID-19 pneumonia (0 in the dupilumab group; 0.4% in the placebo group).
In the NOTUS study, the proportion of patients with any TEAEs reported by the investigator as the reason for permanent study intervention discontinuation was higher in the dupilumab group compared to the placebo group (18 [3.8%] versus 12 [2.6%], respectively). A total of 13 patients in the dupilumab group and 11 patients in the placebo group experienced a treatment-emergent SAE that led to permanent study intervention discontinuation. The most frequent treatment-emergent SAE that led to permanent study intervention discontinuation was COVID-19 pneumonia (0.4% in the dupilumab group; 0.2% in the placebo group).
Mortality
In the BOREAS study, overall, 16 patients died during the on-study period, 8 (1.7%) in each intervention group. Of the 16 deaths reported during the on-study period, 11 occurred during the TEAE period (from the start of treatment up to last investigational medicinal product date plus 98 days) and 5 during the posttreatment period (including 4 that were attributed to TEAEs and 1 that was attributed to a posttreatment AE). In the BOREAS study, 7 patients (1.5%) in the dupilumab group and 8 (1.7%) in the placebo group experienced TEAEs leading to death. Of note, 1 patient in the dupilumab group experienced an SAE leading to death that started during the posttreatment period (cardiogenic shock). Four patients (1 in the dupilumab group and 3 in the placebo group) died due to infections during the study. Five patients (4 in the dupilumab group and 1 in the placebo group) died due to cancer; the majority (4) were lung cancer.
In the NOTUS study, overall, 21 patients died during the on-study period, 13 (2.8%) in the dupilumab group and 8 (1.7%) in the placebo group. Of the 21 deaths reported during the on-study period, 18 occurred during the TEAE period and 3 during the posttreatment period (including 1 that was attributed to TEAEs and 2 that were attributed to posttreatment AEs). In the NOTUS study, there was a higher proportion of patients in the dupilumab group (2.6%) than that in the placebo group (1.5%) who experienced TEAEs leading to death.
Adverse Events of Special Interest
In the BOREAS study, the proportion of patients who experienced any treatment-emergent AESI was generally comparable between the groups: ████ ██████ ████ with dupilumab and placebo, respectively. Injection site reactions were more frequently reported in the dupilumab group compared to the placebo group (████ ██████ ████).
In the NOTUS study, the proportion of patients with any treatment-emergent AESI was numerically higher in the dupilumab group compared to the placebo group: ████ ██████ ████, respectively. The between-group difference was mostly driven by AESIs of ███████ █████████ ███████████ █████ ████████ █████ ███ ███ ███ ██ █████████ ████ ████████ ███ ██████████████ ███████ ███████████ █████ ████████ ██████.
Table 14: Summary of Harms Results in the BOREAS and NOTUS Trials (Safety Population)
AEs | BOREAS trial | NOTUS trial | ||
|---|---|---|---|---|
Placebo n = 470 | Dupilumab n = 469 | Placebo n = 464 | Dupilumab n = 469 | |
Most common TEAEs, n (%) | ||||
Patients with ≥ 1 TEAE | 357 (76.0) | 363 (77.4) | 306 (65.9) | 313 (66.7) |
Infections and infestations | 227 (48.3) | 205 (43.7) | 179 (38.6) | 197 (42.0) |
Nasopharyngitis | 45 (9.6) | 44 (9.4) | 24 (5.2) | 29 (6.2) |
Upper respiratory tract infection | 46 (9.8) | 37 (7.9) | NR | NR |
Urinary tract infection | 10 (2.1) | 23 (4.9) | NR | NR |
Bronchitis | 22 (4.7) | 19 (4.1) | 20 (4.3) | 18 (3.8) |
COVID-19 | 27 (5.7) | 19 (4.1) | 38 (8.2) | 44 (9.4) |
Pneumonia | 19 (4.0) | 13 (2.8) | NR | NR |
Rhinitis | 12 (2.6) | 9 (1.9) | 5 (1.1) | 15 (3.2) |
Lower respiratory tract infection | 11 (2.3) | 6 (1.3) | 10 (2.2) | 7 (1.5) |
Gastroenteritis | 11 (2.3) | 5 (1.1) | NR | NR |
Viral upper respiratory tract infection | NR | NR | 5 (1.1) | 14 (3.0) |
Influenza | NR | NR | 9 (1.9) | 13 (2.8) |
COVID-19 pneumonia | NR | NR | 2 (0.4) | 10 (2.1) |
Lower respiratory tract infection bacterial | NR | NR | 11 (2.4) | 4 (0.9) |
Nervous system disorders | 60 (12.8) | 61 (13.0) | 48 (10.3) | 47 (10.0) |
Headache | 32 (6.8) | 38 (8.1) | 30 (6.5) | 35 (7.5) |
Vascular disorders | 45 (9.6) | 33 (7.0) | 28 (6.0) | 21 (4.5) |
Hypertension | 28 (6.0) | 17 (3.6) | 15 (3.2) | 15 (3.2) |
Respiratory, thoracic, and mediastinal disorders | 80 (17.0) | 75 (16.0) | 53 (11.4) | 48 (10.2) |
COPD | 28 (6.0) | 27 (5.8) | 36 (7.8) | 23 (4.9) |
Gastrointestinal disorders | 70 (14.9) | 81 (17.3) | 60 (12.9) | 54 (11.5) |
Diarrhea | 17 (3.6) | 25 (5.3) | 11 (2.4) | 10 (2.1) |
Toothache | 5 (1.1) | 12 (2.6) | NR | NR |
Gastritis | 2 (0.4) | 11 (2.3) | NR | NR |
Musculoskeletal and connective tissue disorders | 62 (13.2) | 69 (14.7) | 59 (12.7) | 57 (12.2) |
Back pain | 16 (3.4) | 24 (5.1) | 13 (2.8) | 18 (3.8) |
Arthralgia | 12 (2.6) | 12 (2.6) | 13 (2.8) | 17 (3.6) |
Osteoarthritis | NR | NR | 10 (2.2) | 4 (0.9) |
Injury, poisoning, and procedural complications | 77 (16.4) | 64 (13.6) | 61 (13.1) | 55 (11.7) |
Accidental overdose | 30 (6.4) | 26 (5.5) | 32 (6.9) | 31 (6.6) |
Fall | 10 (2.1) | 6 (1.3) | NR | NR |
Treatment-emergent SAE, n (%) | ||||
Patients with ≥ 1 treatment-emergent SAE | 73 (15.5) | 64 (13.6) | 74 (15.9) | 61 (13.0) |
Infections and infestations | 26 (5.5) | 19 (4.1) | 19 (4.1) | 27 (5.8) |
Pneumonia | 12 (2.6) | 5 (1.1) | 4 (0.9) | 7 (1.5) |
COVID-19 | 4 (0.9) | 3 (0.6) | 4 (0.9) | 3 (0.6) |
COVID-19 pneumonia | 5 (1.1) | 2 (0.4) | 2 (0.4) | 6 (1.3) |
Nervous system disorders | 6 (1.3) | 5 (1.1) | 7 (1.5) | 2 (0.4) |
Ischemic stroke | 2 (0.4) | 1 (0.2) | 0 | 0 |
Cardiac disorders | 12 (2.6) | 9 (1.9) | 12 (2.6) | 9 (1.9) |
Cardiac failure | 2 (0.4) | 2 (0.4) | 1 (0.2) | 1 (0.2) |
Coronary artery disease | 1 (0.2) | 2 (0.4) | 2 (0.4) | 0 |
Respiratory, thoracic, and mediastinal disorders | 32 (6.8) | 30 (6.4) | 38 (8.2) | 23 (4.9) |
Chronic obstructive pulmonary disease | 26 (5.5) | 27 (5.8) | 36 (7.8) | 23 (4.9) |
Acute respiratory failure | 2 (0.4) | 1 (0.2) | NR | NR |
Patients who stopped treatment due to AEs, n (%) | ||||
Patients who stopped treatment | 16 (3.4) | 14 (3.0) | 12 (2.6) | 18 (3.8) |
Infections and infestations | 5 (1.1) | 2 (0.4) | 2 (0.4) | 7 (1.5) |
COVID-19 pneumonia | 2 (0.4) | 0 | 1 (0.2) | 2 (0.4) |
COVID-19 | NR | NR | 1 (0.2) | 1 (0.2) |
Neoplasms benign, malignant, and unspecified (including cysts and polyps) | 4 (0.9) | 6 (1.3) | 4 (0.9) | 4 (0.9) |
Lung neoplasm malignancy | 1 (0.2) | 1 (0.2) | NR | NR |
Adenocarcinoma of the colon | NR | NR | 1 (0.2) | 1 (0.2) |
Prostate cancer | NR | NR | 1 (0.2) | 1 (0.2) |
Deaths, n (%) | ||||
Death on studya | 8 (1.7) | 8 (1.7) | 8 (1.7) | 13 (2.8) |
Death occurred during the TEAE periodb | 7 (1.5) | 4 (0.9) | 7 (1.5) | 11 (2.3) |
Death occurred during the posttreatment periodc | 1 (0.2) | 4 (0.9) | 1 (0.2) | 2 (0.4) |
TEAE leading to death in the posttreatment period | 1 (0.2) | 3 (0.6) | 0 | 1 (0.2) |
Posttreatment AE leading to death in the posttreatment period | 0 | 1 (0.2) | 1 (0.2) | 1 (0.2) |
Patients with any TEAE leading to death | 8 (1.7) | 7 (1.7) | 7 (1.5) | 12 (2.6) |
AESIs, n (%) | ||||
Patients with ≥ 1 AESI | ██ █████ | ██ █████ | 33 (7.1) | 39 (8.3) |
95% CI of proportion of patients with ≥ 1 AESI (%) | 6.5 to 11.8 | 6.0 to 11.1 | 4.8 to 9.5 | 5.8 to 10.8 |
Risk difference vs. placebo, % (95% CI) | −0.62 (−4.26 to 3.02) | −0.62 (−4.26 to 3.02) | 1.20 (−2.22 to 4.63) | 1.20 (−2.22 to 4.63) |
P value | 0.7379 | 0.7379 | 0.4909 | 0.4909 |
Systemic hypersensitivity reactions (medically reviewed) | 2 (0.4) | 2 (0.4) | 2 (0.4) | 1 (0.2) |
Severe injection site reactions that last longer than 24 hours | 0 | 1 (0.2) | 0 | 0 |
AESI infections | 39 (8.3) | 37 (7.9) | 31 (6.7) | 38 (8.1) |
Serious infections | 26 (5.5) | 19 (4.1) | 19 (4.1) | 27 (5.8) |
Parasitic infections | 0 | 0 | 0 | 1 (0.2) |
Infection requires parenteral (IV, intramuscular, subcutaneous) antimicrobial therapy | 30 (6.4) | 29 (6.2) | 24 (5.2) | 24 (5.1) |
Infection requires oral antimicrobial therapy for longer than 2 weeks | 11 (2.3) | 6 (1.3) | 8 (1.7) | 12 (2.6) |
Opportunistic infection | 2 (0.4) | 4 (0.9) | 0 | 2 (0.4) |
Significant ALT elevation | 2 (0.4) | 0 | 0 | 0 |
Any severe type of conjunctivitis | 0 | 0 | 0 | 1 (0.2) |
Keratitis | 0 | 0 | 0 | 1 (0.2) |
Other selected AEs | 20 (4.3) | 26 (5.5) | 11 (2.4) | 23 (4.9) |
Injection site reaction | ██████ | ██ █████ | 2 (0.4) | 9 (1.9) |
Malignancy | 9 (1.9) | 8 (1.7) | 5 (1.1) | 5 (1.1) |
Conjunctivitis (narrow) | 7 (1.5) | 4 (0.9) | 2 (0.4) | 9 (1.9) |
Conjunctivitis (broad) | 9 (1.9) | 5 (1.1) | 4 (0.9) | 10 (2.1) |
Conjunctivitis (FDA)d | 7 (1.5) | 4 (0.9) | 2 (0.4) | 9 (1.9) |
Helminthic infections | 0 | 0 | 0 | 0 |
AE = adverse event; AESI = adverse event of special interest; CI = confidence interval; COPD = chronic obstructive pulmonary disease; IMP = investigational medicinal product; NR = not reported; SAE = serious adverse event; TEAE = treatment-emergent adverse event; USPI = US Prescribing Information; vs. = versus.
Notes: An AESI is an AE (serious or nonserious) of scientific and medical concern specific to the sponsor’s product or program, for which ongoing monitoring and immediate notification by the investigator to the sponsor is required. Specifically, AESIs in the BOREAS and NOTUS studies included the following: clinically symptomatic eosinophilia (or eosinophilia associated with clinical symptoms); anaphylactic reactions or systemic allergic reactions that are related to IMP and require treatment; severe injection site reactions that last longer than 24 hours; and any infection meeting at least 1 of the following criteria: any serious infection (SAE), requires parenteral (IV, intramuscular, subcutaneous) antimicrobial therapy, requires oral antimicrobial therapy for longer than 2 weeks, is a parasitic infection, or is an opportunistic infection.
aIncludes all deaths that occurred after the start of treatment up to the end of study (defined as last protocol planned visit or the resolution or stabilization of all treatment-emergent SAEs and AEs of prespecified monitoring).
bIncludes all deaths that occurred after the start of treatment up to last IMP date plus 98 days.
cIncludes all deaths that occurred after the last IMP date plus 98 days.
dLabelling subgroup of preferred terms included in the USPI for dupilumab.
Sources: Clinical Study Reports for the BOREAS and NOTUS studies,20,21 and sponsor’s submission.22 Details included in the table are from the sponsor’s summary of clinical evidence.24
Critical Appraisal
Internal Validity
The BOREAS and NOTUS studies were randomized, double-blind, placebo-controlled trials. The method used for the centralized randomization consisted of an interactive voice response or web response system, which enabled concealment of the allocation sequence. Factors relevant to COPD prognosis were used in randomization stratification. Of note, in the BOREAS study, randomization was not stratified by smoking status; however, the distribution was generally balanced between the 2 treatment groups (29% in the dupilumab group and 31% in the placebo group currently smoked). The NOTUS study included smoking status at screening as a stratification factor. The baseline patient demographics and disease characteristics were generally balanced between the treatment groups, except for serum IgE levels, which were notably higher in the dupilumab group compared to the placebo group in both the BOREAS study (mean in the dupilumab group = 486.6 IU/mL [SD = 1,868.1 IU/mL]; mean in the placebo group = 417.4 IU/mL [SD = 1,063.0 IU/mL]) and the NOTUS study (mean in the dupilumab group = 532.3 IU/mL [SD = 1,299.5 IU/mL ]; mean in the placebo group = 429.1 IU/mL [SD = 1,273.1 IU/mL ]). The clinical experts consulted for this review noted that this imbalance is unlikely to substantially impact study results, as IgE is a biomarker associated with allergy rather than COPD. Additionally, other baseline parameters including eosinophil count, FeNO levels, and fibrinogen, were generally balanced between the treatment groups.
In both studies, blinding of patients and study personnel was appropriately maintained. However, because a placebo was used as the control, patients may have inferred their treatment assignment based on changes in self-perceived symptoms over time. This could have introduced bias in favour of dupilumab for efficacy outcomes reliant on subjective self-reporting, such as HRQoL, and in favour of placebo for harms outcomes such as headache, back pain, and arthralgia. The risk of bias in outcome measurement remains low for objective measures, such as the annualized rate of moderate or severe COPD exacerbations, prebronchodilator FEV1, and safety outcomes like infections, which were assessed by investigators.
An adequate sample size was achieved in the BOREAS study based on a priori sample size calculations for the primary end point. In the NOTUS study, the sample size for the week 52 end points was reduced owing to the IA, with approximately 30% of patients still ongoing in the study.
In both studies, the most common reason for discontinuation from the 52-week treatment period was patient withdrawal (5% to 6% in the BOREAS study and 4% to 5% in the NOTUS study). No imputation was performed for unobserved events that might have occurred after study discontinuation and up to week 52 for the annualized rate of moderate or severe COPD exacerbations. The restricted maximum likelihood method, using the Newton-Raphson algorithm, was applied to handle missing data in the assessment of change from baseline in prebronchodilator FEV1 and the SGRQ total score. Sensitivity analyses (PMM-MI and tipping point analysis) were conducted to evaluate the impact of missing outcome data on primary and secondary end points, with results consistent with the primary analyses. Additionally, the clinical experts noted that between-group differences in discontinuation rates were small and unlikely to substantially impact the efficacy and safety results of the 2 studies. No notable between-group differences were observed in patient exposure to treatment, or concomitant medications in both trials.
Generally, multiplicity control appeared to be adequate in the BOREAS and NOTUS studies. Given the significant differences between treatment arms, including findings based on IAs on primary and secondary end points in the NOTUS study, the potential for inflated type I error is small.
In both trials, a negative binomial regression was used to model the count of moderate or severe COPD exacerbations, as prespecified in the protocol and statistical analysis plan. A post hoc sensitivity analysis with Poisson regression model generated similar results for the annualized rate of moderate or severe exacerbation and the analyses using negative binomial regression in the BOREAS study (RR = 0.700; 95% CI, 0.603 to 0.813) and the NOTUS study (RR = 0.662; 95% CI, 0.562 to 0.779). Subgroup analyses of interest (by baseline eosinophil counts) were specified a priori; however, there is a lack of sample size consideration and control for multiplicity for these subgroup analyses, which preclude definitive conclusions on subgroup effects.
For the primary end point in both the BOREAS and NOTUS studies — the annualized rate of moderate or severe COPD exacerbations — the 52-week intervention period helps minimize potential confounding effects of seasonality by capturing data across all seasons of 1 entire year.18,19 Evidence supporting the validity and MID estimate of the SGRQ total score in patients with COPD was available. Although the literature-based MID estimate (4 points)18 was derived from within-group changes rather than data from direct comparisons between dupilumab and placebo, the clinical experts considered it a reasonable threshold for assessing clinical importance to both patients and clinicians.
External Validity
Both studies were multinational RCTs. A total of 16 patients in the BOREAS study and 12 patients in the NOTUS study were from Canada. The clinical experts consulted for this review noted that the inclusion and exclusion criteria of the studies generally reflected the patient population eligible for dupilumab treatment in Canada. According to the clinical experts, as clinical trials often have stricter eligibility criteria, the study populations in the BOREAS study (aged 40 to 80 years) and the NOTUS study (aged 40 to 85 years) consisted of people who currently smoke or formerly smoked with a smoking history of at least 10 pack-years and no history or current diagnosis of asthma; however, in clinical practice, adults outside of these age ranges, those with different smoking histories, and patients with asthma would not necessarily be excluded from dupilumab treatment. The clinical experts noted that generalizability of the study results is unlikely to be substantially influenced by these eligibility restrictions.
According to the clinical experts, the baseline disease characteristics of the trial populations were representative of patients with uncontrolled COPD and evidence of type 2 inflammation. The clinical experts noted that overall, these characteristics aligned with the patient population expected to receive dupilumab treatment. In the clinical experts’ opinion, the conditions outlined in the sponsor’s reimbursement requests — reflecting COPD severity and markers of type 2 inflammations — aligned with the eligibility criteria in both trials and are appropriate and applicable to clinical practice in Canada.
The dosing regimen of dupilumab used in both studies aligns with the draft product monograph. Additionally, the clinical experts commented that the concomitant medications used — such as LABA, LAMA, and ICS as maintenance treatment for COPD, SCS for acute exacerbation, and rescue medication (SABA or short-acting antimuscarinics) — are consistent with the standard clinical practice in Canada.
The efficacy outcomes assessed in both studies were of clinical importance to patients and clinicians, including the annualized rate of moderate to severe COPD exacerbations, prebronchodilator FEV1 levels, and HRQoL. The clinical experts emphasized that all exacerbations, regardless of severity, are important. On the other hand, the assessment of moderate to severe exacerbations in these studies was appropriate given their substantial impact on patient health and health care resource utilization. The SGRQ is a validated tool commonly used in the patients with COPD. The clinical experts noted that the 52-week randomized treatment period and the 12-week postrandomization safety follow-up were adequate to assess dupilumab’s efficacy. However, the clinical experts felt that a longer duration of follow-up would be required to capture the long-term safety of dupilumab, particularly for potential rare AEs such as helminth infections. Of note, the sponsor stated that dupilumab has been available in Canada since 2017, and for patients aged as young as 6 months since 2023.
It should be noted that the BOREAS and NOTUS trials are the only phase III RCT evidence submitted for review. Both studies were placebo-controlled, and no head to head or indirect comparisons with other active therapies (azithromycin and roflumilast) were submitted. Nonetheless, the absence of comparative evidence is unlikely to be a concern given that based on clinical expert input, there are currently no appropriate comparators for dupilumab in the treatment of uncontrolled COPD associated with type 2 inflammation in Canada.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.58,59
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — the true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — the true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. For this review, the target of the certainty of evidence assessment was the presence or absence of an important effect based on thresholds (MID) identified in the literature for the SGRQ total score. The target of the certainty of evidence assessment was based on presence or absence of an important effect as informed by thresholds identified based on clinical expert input (annualized rate of moderate or severe COPD exacerbations, and prebronchodilator FEV1). For the proportion of patients with at least 1 AESI, the target of the certainty of evidence assessment was based on the null because there was uncertainty in the thresholds suggested by the clinical experts. Findings from the BOREAS and NOTUS studies were considered together and summarized narratively per outcome because these studies were similar in population, interventions, design, and outcome measures.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for dupilumab versus placebo in adult patients with uncontrolled COPD associated with type 2 inflammation.
Long-Term Extension Studies
No long-term extension studies were submitted for this review.
Indirect Evidence
No studies with indirect evidence were submitted for this review.
Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in the pivotal and RCT evidence were submitted for this review.
Discussion
This report summarizes the evidence for dupilumab as an add-on maintenance treatment in adult patients with uncontrolled COPD associated with type 2 inflammation based on 2 phase III RCTs.
Summary of Available Evidence
Two studies, BOREAS (N = 939) and NOTUS (N = 935), met the inclusion criteria for the systematic review conducted by the sponsor. The BOREAS and NOTUS studies were double-blind, identically designed RCTs that assessed the efficacy and safety of 52 weeks of treatment with SC dupilumab 300 mg every 2 weeks relative to placebo in adult patients who had uncontrolled COPD associated with type 2 inflammation. Patients eligible for both studies must meet the following criteria: a postbronchodilator FEV1:FVC ratio less than 0.70 and a postbronchodilator FEV1 predicted to be greater than 30% and 70% or less; background therapy consisting of either a triple therapy with an ICS, LABA, and LAMA or dual therapy with a LABA and LAMA if ICS was contraindicated; blood eosinophil levels of at least 300 cells/µL; a history of at least 2 moderate (requiring treatment with either SCS and/or antibiotics) or 1 severe (requiring hospitalization or observation for more than 24 hours in an emergency department or urgent care facility) exacerbations within the past year; and an MRC Dyspnea Scale grade of at least 2 (equivalent to mMRC of at least grade 1).
The annualized rates of moderate or severe COPD exacerbation, prebronchodilator FEV1 (at week 12 and week 52), SGRQ total score, and the treatment-emergent AESIs were outcomes of interest to this review. At baseline, all patients in both studies had a history of smoking, with 30% of patients who currently smoke. The proportion of patients with moderate airflow limitation (i.e., GOLD grade 2; predicted postbronchodilator FEV1 between ≥ 50% and < 80%) was █████ ██ █████ across the treatment arms in the BOREAS and NOTUS studies. The proportion of patients with severe airflow limitation (i.e., GOLD grade 3; predicted postbronchodilator FEV1 between ≥ 30% and < 50%) was █████ ██ █████. A total of ██ ██████ patients in the BOREAS study and ██ ██████ patients in the NOTUS study had airflow limitation categorized as very severe (i.e., GOLD grade 4; predicted postbronchodilator FEV1 < 30%).
No long-term extension studies, indirect comparative evidence, and other studies addressing gaps in the systematic review evidence were submitted for this review.
Interpretation of Results
Efficacy
The patient and clinician groups emphasized the urgent need for novel therapeutic strategies for COPD due to its high prevalence, chronic progression, and life-threatening nature. Current treatments have limited efficacy, particularly for patients with uncontrolled COPD (GOLD grade E), where few effective options exist to prevent exacerbations. Frequent COPD exacerbations contribute to accelerated lung function decline, worsened health status, increased mortality, and higher health care costs. Preventing or reducing the severity of exacerbations is a critical objective in COPD management. For patients already receiving maximal inhaled therapy (LABA, LAMA, and ICS), additional treatment options are scarce, with no approved therapies addressing the immunologic underpinnings of COPD. Among patients with COPD and a history of exacerbations, approximately 50% continue to experience moderate or severe exacerbations despite triple therapy.60 This need is even more pronounced in the subset of patients with type 2 inflammation, who face higher exacerbation rates, increased mortality, and impaired lung function.8,12
The BOREAS and NOTUS trials, conducted in patients without other substantial pulmonary diseases (notably excluding asthma), evaluated clinically relevant outcomes through a 52-week randomized treatment period. The clinical experts commented that the exclusion of asthma was likely to ensure results were not confounded by patients already eligible for dupilumab for asthma treatment. The reimbursement request aligns with the inclusion criteria in the RCTs, incorporating features of type 2 inflammation. Of note, the BOREAS and NOTUS studies included patients with an MRC grade of at least 2, while the reimbursement request specified an mMRC grade of at least 1. This discrepancy is not an issue, as both scales assess breathlessness severity similarly. The original MRC scale ranges from 1 to 5, while the mMRC scale ranges from 0 to 4, with higher grades indicating more severe breathlessness. The wording for each grade is comparable between the 2 scales, and an MRC grade of at least 2 corresponds to an mMRC grade of at least 1. Specifically, in the mMRC scale, grade 1 is defined as experiencing breathlessness when hurrying on level ground or walking up a slight hill.39
The definition of moderate and severe exacerbations in the reimbursement request conditions aligns with CTS and GOLD standards, distinguishing them based on the level of medical intervention required. Both trials used placebo as a comparator, and the sponsor stated that there is an absence of head to head, direct evidence comparing dupilumab with other COPD pharmacotherapies relevant in practice in Canada. The clinical experts consulted for this review agree that there are no appropriate comparators to dupilumab for COPD treatment. Azithromycin, although used to reduce exacerbations, lacks a Health Canada indication, poses risks such as QTc prolongation and antibiotic resistance, and does not target underlying inflammation.61-63 Roflumilast, a PDE4 inhibitor,64 is limited by modest efficacy, gastrointestinal intolerance, and lack of reimbursement by public drug programs.65 While other biologics like mepolizumab, benralizumab, and tezepelumab have been studied in COPD, none have demonstrated substantial benefits. Dupilumab remains the only approved biologic in Canada that inhibits IL-4 and IL-13 signalling, distinguishing it from existing treatments.
At baseline, patients in the BOREAS and NOTUS studies were at high risk of acute exacerbation of COPD (GOLD group E), with a mean number of 2.3 (SD = 1.0) and 2.1 (SD = 0.9) moderate or severe acute exacerbation of COPD events, respectively, in the year before screening. Dupilumab demonstrated a statistically significant and clinically meaningful reduction in the annualized rate of moderate or severe exacerbations sustained over 52 weeks. Based on an MID noted by the clinical experts and literature, 11% (or 0.11), the point estimates and both bounds of 95% CIs of the risk differences between the dupilumab and placebo groups were lower than the MID estimate in both the BOREAS study (−0.32; 95% CI, −0.51 to −0.14) and the NOTUS study (−0.44; 95% CI, −0.68 to −0.19) at week 52, suggesting the treatment effect is larger than a threshold that is important to clinicians and patients. Consistent with the mechanism of action of dupilumab, treatment benefit of dupilumab was observed in a broad population with type 2 inflammation, and a greater treatment benefit of dupilumab versus placebo was observed in a subgroup of patients with eosinophil levels of 500 cells/µL or higher during screening (higher levels of biomarker indicating type 2 inflammation) compared to those with eosinophil levels of less than 500 cells/µL (RR = 0.505; 95% CI, 0.345 to 0.737 and RR = 0.797; 95% CI, 0.637 to 0.997, respectively; nominal treatment by subgroup interaction P = 0.0141) in the BOREAS study. The point estimates of the RR were similar between these 2 subgroups in the NOTUS study for annualized rate of moderate or severe COPD exacerbations. However, due to limited sample sizes and lack of control for multiplicity, definitive conclusions on subgroup effects cannot be drawn.
FEV1 is a measure widely accepted for assessing lung function response to treatment. Prebronchodilator FEV1 assessed in both trials was identified as important by patient and clinical groups, and clinical experts. The clinical experts suggested an MID of 100 mL (or 0.1 L), as smaller differences are not typically perceptible to patients. The between-group differences in increase from baseline at week 12 and week 52 in the prebronchodilator FEV1 were not regarded as clinically meaningful, as 95% CIs included the MID estimate, with LS mean difference of 0.08 L (95% CI, 0.04 L to 0.13 L) at both week 12 and week 52 in the BOREAS study, and 0.08 L (95% CI, 0.04 L to 0.12 L) at week 12 and 0.06 L (95% CI, 0.01 L to 0.11 L) at week 52 in the NOTUS study. Subgroup analyses indicated a more pronounced response in patients with eosinophil levels of 500 cells/µL or greater at screening. While numerically greater effects were seen in this subgroup, no statistically significant interaction was observed in the NOTUS study. Nonetheless, the lack of sample size considerations and multiplicity control limits the interpretability of these findings.
The patient and clinician groups, and clinical experts, regarded HRQoL an outcome of importance. Based on the SGRQ, a validated measurement comprising domains of symptoms, activity, and impacts, patients in the dupilumab group reported greater improvement from baseline than those in the placebo group at week 52 in both studies. However, the between-group differences in the SGRQ total score change from baseline were not regarded as clinically meaningful, as the 95% CIs included the MID estimate of 4 points, a threshold deemed reasonable by the clinical experts for assessing the effect of dupilumab relative to placebo.
Harms
The BOREAS and NOTUS studies demonstrated that the occurrence of TEAEs at week 52 was likely similar between the dupilumab and placebo groups, aligning with the established profile of dupilumab. The most commonly reported TEAEs of dupilumab included nasopharyngitis, upper respiratory tract infections, headache, diarrhea, and back pain (all of which were reported in less than 10% of patients across treatment groups).
The clinical experts consulted for this review noted that treatment-emergent SAEs were consistent with the established profile of dupilumab, with no specific pattern among them and no new safety concerns identified. The clinical experts noted that the reported TEAEs leading to death — such as infections, cancer, and cardiogenic shock — were generally consistent with what is anticipated in an older patient population with uncontrolled COPD.
The clinical experts also noted that the AESIs reported in both studies were consistent with those anticipated for dupilumab, including hypersensitivity, serious and opportunistic infections, injection site reactions, conjunctivitis, and keratitis. However, a longer duration of follow-up is needed to adequately capture the long-term safety of dupilumab, particularly for rare AEs, such as helminth infections, which are highlighted as a safety warning in the draft product monograph.
No safety outcomes comparing dupilumab and placebo beyond 52 weeks of treatment plus 12 weeks of safety follow-up were submitted. Additionally, no direct or indirect comparative evidence on harms was submitted for dupilumab versus active therapies such as azithromycin or roflumilast.
Conclusion
Direct comparative evidence from 2 double-blind RCTs (BOREAS and NOTUS) demonstrated that in adult patients with uncontrolled COPD who had type 2 inflammation and moderate to severe airflow obstruction despite receiving triple inhaler therapy (LABA, LAMA, and ICS), dupilumab treatment resulted in a clinically meaningful reduction in moderate or severe exacerbations compared to placebo, when used in addition to best supportive care. Dupilumab likely results in little to no clinically meaningful benefits in lung function measured with a prebronchodilator FEV1 compared to placebo. Patients’ quality of life as measured by SGRQ may be improved with dupilumab treatment; however, the magnitude of benefit is considered not clinically significant based on a recognized MID estimate. No notable safety concerns were identified with dupilumab treatment from the trials through week 52. The absence of longer-term efficacy and safety data beyond 1 year represents a gap in evidence given that COPD is a chronic disease. No comparative evidence for the efficacy and safety of dupilumab versus other therapies available in clinical practice was submitted.
References
1.O'Donnell DE, Hernandez P, Kaplan A, et al. Canadian Thoracic Society recommendations for management of chronic obstructive pulmonary disease - 2008 update - highlights for primary care. Can Respir J. 2008;15 Suppl A(Suppl A):1a-8a.
2.Canadian Lung Association. Lung disease A-Z. 2025; https://www.lung.ca/lung-health/lung-diseases/chronic-obstructive-pulmonary-disease-copd/causes-and-symptoms-copd. Accessed January 16, 2025.
3.Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease (2024 Report). Global initiative for chonic obstructive lung disease, Inc.; 2024: https://goldcopd.org/2024-gold-report/. Accessed 11 Jan, 2024.
4.Government of Canada. Canadian Chronic Disease Surveillance System (CCDSS): COPD age-standardized prevalence. https://health-infobase.canada.ca/ccdss/data-tool/. Accessed 28 Oct 2024.
5.World Health Organization. The top 10 causes of death. 2020; https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death. Accessed 05 Jun 2024.
6.Levine SM, Marciniuk DD. Global Impact of Respiratory Disease: What Can We Do, Together, to Make a Difference? Chest. 2022;161(5):1153-1154. PubMed
7.Brightling CE. Biomarkers that predict and guide therapy for exacerbations of chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2013;10 Suppl:S214-219. PubMed
8.Singh D, Kolsum U, Brightling CE, et al. Eosinophilic inflammation in COPD: prevalence and clinical characteristics. Eur Respir J. 2014;44(6):1697-1700. PubMed
9.Vedel-Krogh S, Nielsen SF, Lange P, Vestbo J, Nordestgaard BG. Blood Eosinophils and Exacerbations in Chronic Obstructive Pulmonary Disease. The Copenhagen General Population Study. Am J Respir Crit Care Med. 2016;193(9):965-974. PubMed
10.Hastie AT, Martinez FJ, Curtis JL, et al. Association of sputum and blood eosinophil concentrations with clinical measures of COPD severity: an analysis of the SPIROMICS cohort. Lancet Respir Med. 2017;5(12):956-967. PubMed
11.Yun JH, Lamb A, Chase R, et al. Blood eosinophil count thresholds and exacerbations in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2018;141(6):2037-2047 e2010.
12.Belanger M, Couillard S, Courteau J, et al. Eosinophil counts in first COPD hospitalizations: a comparison of health service utilization. Int J Chron Obstruct Pulmon Dis. 2018;13:3045-3054. PubMed
13.Brightling CE, Monteiro W, Ward R, et al. Sputum eosinophilia and short-term response to prednisolone in chronic obstructive pulmonary disease: a randomised controlled trial. Lancet. 2000;356(9240):1480-1485. PubMed
14.Bourbeau J, Bhutani M, Hernandez P, et al. 2023 Canadian Thoracic Society Guideline on Pharmacotherapy in Patients With Stable COPD. Chest. 2023;164(5):1159-1183. PubMed
15.Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2025 Report). Global initiative for chronic obstructive lung disease, Inc.; 2025.
16.Pailhories H, Herrmann JL, Velo-Suarez L, et al. Antibiotic resistance in chronic respiratory diseases: from susceptibility testing to the resistome. Eur Respir Rev. 2022;31(164). PubMed
17.Smith D, Gill A, Hall L, Turner AM. Prevalence, Pattern, Risks Factors and Consequences of Antibiotic Resistance in COPD: A Systematic Review. Copd. 2021;18(6):672-682. PubMed
18.Jones PW. St. George's Respiratory Questionnaire: MCID. COPD. 2005;2(1):75-79. PubMed
19.Jones PW, Beeh KM, Chapman KR, Decramer M, Mahler DA, Wedzicha JA. Minimal clinically important differences in pharmacological trials. Am J Respir Crit Care Med. 2014;189(3):250-255. PubMed
20.Clinical Study Report: EFC15804. A randomized, double-blind, placebo-controlled, parallel-group, 52-week pivotal study to assess the efficacy, safety, and tolerability of dupilumab in patients with moderate-to-severe chronic obstructive pulmonary disease (COPD) with type 2 inflammation (BOREAS). Chilly-Mazarin (FR): Sanofi Group; August 8, 2023.
21.Clinical Study Report: EFC15805. A randomized, double-blind, placebo-controlled, parallel-group, 52-week pivotal study to assess the efficacy, safety, and tolerability of dupilumab in patients with moderate-to-severe chronic obstructive pulmonary disease (COPD) with type 2 inflammation (NOTUS). Chilly-Mazarin (FR): Sanofi Group; December 14, 2023.
22.Sanofi-aventis Canada Inc. response to February 4, 2025 CDA-AMC request for additional information regarding dupilumab for COPD CDA-AMC review [internal additional sponsor's information]. Toronto (ON): Sanofi-aventis Canada Inc.; February 7, 2025.
23.Sanofi-aventis Canada Inc. response to February 11, 2025 CDA-AMC request for additional information regarding dupilumab for COPD CDA-AMC review [internal additional sponsor's information]. Toronto (ON): Sanofi-aventis Canada Inc.; February 21, 2025.
24.Sponsor's clinical summary report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Dupilumab (Dupixent), 300 mg single-use syringe/pen (300 mg/2 mL), solution for subcutaneous injection. Toronto (ON): Sanofi-aventis Canada Inc.; 2024.
25.Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet. 2007;370(9589):765-773. PubMed
26.Raherison C, Girodet PO. Epidemiology of COPD. Eur Respir Rev. 2009;18(114):213-221. PubMed
27.Celli B, Locantore N, Yates JC, et al. Markers of disease activity in COPD: an 8-year mortality study in the ECLIPSE cohort. Eur Respir J. 2021;57(3). PubMed
28.Suissa S, Dell'Aniello S, Ernst P. Long-term natural history of chronic obstructive pulmonary disease: severe exacerbations and mortality. Thorax. 2012;67(11):957-963. PubMed
29.Halpin DMG, Decramer M, Celli BR, Mueller A, Metzdorf N, Tashkin DP. Effect of a single exacerbation on decline in lung function in COPD. Respir Med. 2017;128:85-91. PubMed
30.Wheaton AG, Cunningham TJ, Ford ES, Croft JB. Employment and activity limitations among adults with chronic obstructive pulmonary disease--United States, 2013. MMWR Morb Mortal Wkly Rep. 2015;64(11):289-295. PubMed
31.van Manen JG, Bindels PJ, Dekker FW, et al. The influence of COPD on health-related quality of life independent of the influence of comorbidity. J Clin Epidemiol. 2003;56(12):1177-1184. PubMed
32.Prevalence and attributable health burden of chronic respiratory diseases, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Respir Med. 2020;8(6):585-596. PubMed
33.GBD 2019 Diseases and Injuries Collaborators. Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2020;396(10258):1204-1222. PubMed
34.Health Canada. Fourth annual report on Medical Assistance in Dying in Canada 2022. 2023: https://www.canada.ca/en/health-canada/services/publications/health-system-services/annual-report-medical-assistance-dying-2022.html. Accessed 11 Jan 2024.
35.Bazell C, Pollack M, Comellas AP, et al. A 4-Year Retrospective Claims Analysis of Oral Corticosteroid Use and Health Conditions in Newly Diagnosed Medicare FFS Patients with COPD. Int J Chron Obstruct Pulmon Dis. 2022;17:2635-2652. PubMed
36.Drug Reimbursement Review clinical guidance report: CEDAC FINAL RECOMMENDATION, ROFLUMILAST (Daxas — Nycomed Canada Inc.) Indication: Chronic Obstructive Pulmonary Disease. Ottawa (ON): CADTH; 2011: https://www.cadth.ca/sites/default/files/cdr/complete/cdr_complete_Daxas_August-3-2011_e.pdf. Accessed December 23, 2024.
37.DRAFT: DUPIXENT (dupilumab) solution for subcutaneous injection Product Monograph. Toronto (ON): Sanofi-aventis Canada Inc.; April 30, 2024.
38.Heo YA. Budesonide/Glycopyrronium/Formoterol: A Review in COPD. Drugs. 2021;81(12):1411-1422. PubMed
39.Williams N. The MRC breathlessness scale. Occup Med (Lond). 2017;67(6):496-497. PubMed
40.Bhatt SP, Rabe KF, Hanania NA, et al. Dupilumab for COPD with Blood Eosinophil Evidence of Type 2 Inflammation. N Engl J Med. 2024;390(24):2274-2283. PubMed
41.Bhatt SP, Rabe KF, Hanania NA, et al. Dupilumab for COPD with Type 2 Inflammation Indicated by Eosinophil Counts. N Engl J Med. 2023;389(3):205-214. PubMed
42.Sanofi-aventis Canada Inc. response to January 27, 2025 CDA-AMC request for additional information regarding dupilumab for COPD CDA-AMC review [internal additional sponsor's information]. Toronto (ON): Sanofi-aventis Canada Inc.; February 3, 2025.
43.U. K. Research Innovation. MRC Dyspnoea Scale. https://www.ukri.org/councils/mrc/facilities-and-resources/find-an-mrc-facility-or-resource/mrc-dyspnoea-scale/. Accessed 04 Dec 2024.
44.European Medicines Agency. Guideline on clinical investigation of medicinal products in the treatment of chronic obstructive pulmonary disease (COPD). 2012: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-investigation-medicinal-products-treatment-chronic-obstructive-pulmonary-disease_en.pdf. Accessed 11 Jan 2024.
45.Anzueto A, Leimer I, Kesten S. Impact of frequency of COPD exacerbations on pulmonary function, health status and clinical outcomes. Int J Chron Obstruct Pulmon Dis. 2009;4:245-251. PubMed
46.Chapman KR, Bergeron C, Bhutani M, et al. Do we know the minimal clinically important difference (MCID) for COPD exacerbations? Copd. 2013;10(2):243-249. PubMed
47.Celli BR. Predictors of mortality in COPD. Respir Med. 2010;104(6):773-779. PubMed
48.Fletcher C, Peto R. The natural history of chronic airflow obstruction. Br Med J. 1977;1(6077):1645-1648. PubMed
49.Sanofi G. Statistical Analysis Plan: EFC15804 A Randomized, Double-blind, Placebo-controlled, Parallel-group, 52-week Pivotal Study to Assess the Efficacy, Safety, and Tolerability of Dupilumab in Patients With Moderate-to-severe Chronic Obstructive Pulmonary Disease (COPD) with Type 2 inflammation. 2022.
50.Sanofi G. Statistical Analysis Plan: EFC15805 A Randomized, Double-blind, Placebo-controlled, Parallel-group, 52-week Pivotal Study to Assess the Efficacy, Safety, and Tolerability of Dupilumab in Patients With Moderate-to-severe Chronic Obstructive Pulmonary Disease (COPD) with Type 2 inflammation. 2023.
51.Crim C, Frith LJ, Midwinter D, Donohue JF. FEV(1) Minimum Important Difference versus Minimal Detectable Difference? In Search of the Unicorn. Am J Respir Crit Care Med. 2021;203(12):1573-1576. PubMed
52.Kostikas K, Greulich T, Mackay AJ, et al. Treatment response in COPD: does FEV(1) say it all? A post hoc analysis of the CRYSTAL study. ERJ Open Res. 2019;5(1). PubMed
53.Donohue JF. Minimal clinically important differences in COPD lung function. Copd. 2005;2(1):111-124. PubMed
54.Jones PW, Quirk FH, Baveystock CM. The St George's Respiratory Questionnaire. Respir Med. 1991;85 Suppl B:25-31; discussion 33-27.
55.Jones PW, Quirk FH, Baveystock CM, Littlejohns P. A self-complete measure of health status for chronic airflow limitation. The St. George's Respiratory Questionnaire. Am Rev Respir Dis. 1992;145(6):1321-1327. PubMed
56.Food and Drug Administration. Chronic Obstructive Pulmonary Disease: Use of the St. George’s Respiratory Questionnaire as a PRO Assessment Tool Guidance for Industry. Silver Spring (MD)2018: https://www.fda.gov/media/71121/download. Accessed 11 Jan 2024.
57.Jones PW. Health status measurement in chronic obstructive pulmonary disease. Thorax. 2001;56(11):880-887. PubMed
58.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
59.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
60.Halpin DMG, Dransfield MT, Han MK, et al. The effect of exacerbation history on outcomes in the IMPACT trial. Eur Respir J. 2020;55(5). PubMed
61.Ahmadian S, Sin DD, Lynd L, Harrison M, Sadatsafavi M. Benefit-harm analysis of azithromycin for the prevention of acute exacerbations of chronic obstructive pulmonary disease. Thorax. 2022;77(11):1079-1087. PubMed
62.Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med. 2011;365(8):689-698. PubMed
63.Rodríguez-Arce I, Morales X, Ariz M, et al. Development and multimodal characterization of an elastase-induced emphysema mouse disease model for the COPD frequent bacterial exacerbator phenotype. Virulence. 2021;12(1):1672-1688. PubMed
64.Singh D, Martinez FJ, Watz H, Bengtsson T, Maurer BT. A dose-ranging study of the inhaled dual phosphodiesterase 3 and 4 inhibitor ensifentrine in COPD. Respir Res. 2020;21(1):47. PubMed
65.Martinez FJ, Rabe KF, Calverley PMA, et al. Determinants of Response to Roflumilast in Severe Chronic Obstructive Pulmonary Disease. Pooled Analysis of Two Randomized Trials. Am J Respir Crit Care Med. 2018;198(10):1268-1278. PubMed
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Figure 2: Cumulative Mean Number of Moderate or Severe COPD Exacerbation Events During the 52-Week Treatment Period in the BOREAS Trial (ITT Population)
COPD = chronic obstructive pulmonary disease; q2w = every 2 weeks; ITT = intention to treat.
Source: Clinical Study Report for BOREAS.20
Figure 3: Cumulative Mean Number of Moderate or Severe COPD Exacerbation Events During the 52-Week Treatment Period in the NOTUS Trial (ITT Population)
COPD = chronic obstructive pulmonary disease; q2w = every 2 weeks; ITT = intention to treat.
Source: Clinical Study Report for NOTUS.21
Table 15: Summary of Other Efficacy Results From the BOREAS and NOTUS Trials
Outcomes | BOREAS trial | NOTUS trial | ||
|---|---|---|---|---|
Placebo (N = 471) | Dupilumab (N = 468) | Placebo (N = 465) | Dupilumab (N = 470) | |
Annualized rate of moderate or severe COPD exacerbations during the 52-week treatment period | ||||
Number of patients with ≥ 1 moderate or severe exacerbation event, n (%) | 212 (45.0) | 182 (38.9) | 182 (39.1) | 156 (33.2) |
Mean number of moderate or severe exacerbation events, mean (SD) | 0.9 (1.4) | 0.6 (1.0) | 0.8 (1.3) | 0.6 (1.0) |
Median number of moderate or severe exacerbation events | 0.0 | 0.0 | 0.0 | 0.0 |
Minimum:maximum | 0:8 | 0:8 | 0:7 | 0:6 |
Patients with 0 events, n (%) | 259 (55.0) | 286 (61.1) | 283 (60.9) | 314 (66.8) |
Patients with 1 event, n (%) | 106 (22.5) | 113 (24.1) | 98 (21.1) | 93 (19.8) |
Patients with 2 events, n (%) | 55 (11.7) | 47 (10.0) | 41 (8.8) | 36 (7.7) |
Patients with 3 events, n (%) | 25 (5.3) | 9 (1.9) | 18 (3.9) | 15 (3.2) |
Patients with ≥ 4 events, n (%) | 26 (5.5) | 13 (2.8) | 25 (5.4) | 12 (2.6) |
Total number of moderate or severe exacerbation events | 422 | 296 | 352 | 263 |
Adjusted annualized moderate or severe exacerbation event rate (95% CI) | 1.10 (0.93 to 1.30) | 0.78 (0.64 to 0.93) | 1.30 (1.05 to 1.60) | 0.86 (0.70 to 1.06) |
RR vs. placebo (95% CI) | 0.705 (0.581 to 0.857) | 0.705 (0.581 to 0.857) | 0.664 (0.535 to 0.823) | 0.664 (0.535 to 0.823) |
P value | 0.0005a | 0.0005a | 0.0002 a | 0.0002 a |
Risk difference vs. placebo (95% CI) | ██████ ████████ ███████ | ██████ ████████ ███████ | ██████ ████████ ███████ | ██████ ████████ ███████ |
Number of severe events | 37 | 28 | 39 | 26 |
Requiring hospitalization, n (%) | 36 (97.3) | 27 (96.4) | 39 (100) | 26 (100) |
Requiring an emergency medical care visit, n (%) | 1 (2.7) | 1 (3.6) | 0 | 0 |
Resulting in death, n (%) | 0 | 0 | 0 | 0 |
Number of moderate events | 385 | 268 | 313 | 237 |
Only requiring use of systemic corticosteroid medications, n (%) | 130 (33.8) | 84 (31.3) | 107 (34.2) | 70 (29.5) |
Only requiring use of systemic antibiotics, n (%) | 68 (17.7) | 52 (19.4) | 56 (17.9) | 40 (16.9) |
Requiring use of both systemic corticosteroid medications and systemic antibiotics, n (%) | 187 (48.6) | 132 (49.3) | 150 (47.9) | 127 (53.6) |
Time to first moderate or severe COPD exacerbation | ||||
Number of participants with moderate or severe exacerbation, n (%) | 212 (45.01) | 182 (38.89) | 182 (39.14) | 156 (33.19) |
Number of participants censored, n (%) | 259 (54.99) | 286 (61.11) | 283 (60.86) | 314 (66.81) |
Kaplan-Meier estimates for probability of a participant with ≥ 1 event (95% CI) up to: | ||||
12 weeks | 0.205 (0.170 to 0.243) | 0.142 (0.112 to 0.175) | 0.172 (0.139 to 0.207) | 0.108 (0.081 to 0.138) |
24 weeks | 0.307 (0.266 to 0.349) | 0.254 (0.215 to 0.294) | 0.265 (0.225 to 0.306) | 0.206 (0.170 to 0.244) |
36 weeks | 0.386 (0.341 to 0.430) | 0.326 (0.284 to 0.369) | 0.342 (0.298 to 0.387) | 0.292 (0.250 to 0.335) |
52 weeks | 0.454 (0.408 to 0.499) | 0.394 (0.350 to 0.439) | 0.424 (0.375 to 0.471) | 0.361 (0.315 to 0.407) |
Hazard ratio (95% CI) | 0.803 (0.658 to 0.980) | 0.803 (0.658 to 0.980) | 0.714 (0.574 to 0.887) | 0.714 (0.574 to 0.887) |
P value vs. placebo | 0.0309b | 0.0309b | 0.0024b | 0.0024b |
Annualized rate of severe COPD exacerbations over the 52-week treatment period | ||||
Number of patients with ≥ 1 severe exacerbation event, n (%) | 28 (5.9) | 20 (4.3) | 32 (6.9) | 21 (4.5) |
≥ 1 severe exacerbation event, no (%) | 443 (94.1) | 448 (95.7) | 433 (93.1) | 449 (95.5) |
Mean number of severe exacerbations events (SD) | 0.1 (0.4) | 0.1 (0.3) | 0.1 (0.3) | 0.1 (0.3) |
Patients with 0 events, n (%) | 443 (94.1) | 448 (95.7) | 433 (93.1) | 449 (95.5) |
Patients with 1 event, n (%) | 23 (4.9) | 15 (3.2) | 26 (5.6) | 17 (3.6) |
Patients with 2 events, n (%) | 3 (0.6) | 3 (0.6) | 5 (1.1) | 3 (0.6) |
Patients with 3 events, n (%) | 1 (0.2) | 1 (0.2) | 1 (0.2) | 1 (0.2) |
Patients with ≥ 4 events, n (%) | 1 (0.2) | 1 (0.2) | 0 | 0 |
Total number of severe exacerbations | 37 | 28 | 39 | 26 |
Adjusted annualized severe exacerbation event rate estimate (95% CI) | 0.086 (0.050 to 0.147) | 0.072 (0.040 to 0.132) | 0.124 (0.072 to 0.215) | 0.070 (0.039 to 0.123) |
RR vs. placebo (95% CI) | 0.847 (0.447 to 1.602) | 0.847 (0.447 to 1.602) | 0.559 (0.308 to 1.018) | 0.559 (0.308 to 1.018) |
P value | 0.6085b | 0.6085b | 0.0571b | 0.0571b |
Risk difference vs. placebo (95% CI) | −0.013 (−0.063 to 0.037) | −0.013 (−0.063 to 0.037) | −0.055 (−0.117 to 0.008) | −0.055 (−0.117 to 0.008) |
Proportion of patients with SGRQ improvement by ≥ 4 points at week 52 — ITT population | ||||
Patients whose condition responded to treatment at week 52 , n (%) | 203 (43.1) | 241 (51.5) | 167 (46.5) | 186 (51.4) |
Patients who did not respond to treatment at week 52 , n (%) | 268 (56.9) | 227 (48.5) | 192 (53.5) | 176 (48.6) |
Imputed patients who did not respond to treatment at week 52 , n (%) | 71 (15.1) | 53 (11.3) | 49 (13.6) | 45 (12.4) |
Odds ratio vs. placebo (95% CI) | 1.439 (1.096 to 1.890) | 1.439 (1.096 to 1.890) | 1.164 (0.856 to 1.581) | 1.164 (0.856 to 1.581) |
P value vs. placebo | 0.0089a | 0.0089a | 0.3329b | 0.3329b |
Change in E-RS:COPD scores from baseline to week 52 — ITT population | ||||
Baseline number of patients | 467 | 461 | 355 | 356 |
Baseline mean (SD) | 12.95 (6.94) | 12.94 (7.19) | 13.11 (7.14) | 13.71 (6.74) |
Number of patients at week 52 | 381 | 386 | 287 | 288 |
Week 52 mean (SD) | 10.95 (7.15) | 9.70 (7.05) | 10.96 (7.34) | 11.50 (7.14) |
Change from baseline number of patients | 379 | 380 | 285 | 284 |
Change from baseline mean (SD) | −1.61 (4.99) | −3.00 (5.75) | −1.63 (5.23) | −2.18 (5.90) |
Number of patients in the model | 467 | 461 | 355 | 356 |
LS mean (SE) | −1.558 (0.256) | −2.694 (0.257) | −1.770 (0.300) | −2.388 (0.301) |
LS mean difference (95% CI) | −1.137 (−1.823 to −0.450) | −0.618 (−1.430 to 0.193) | ||
P value vs. placebo | 0.0012a | 0.1351b | ||
CI = confidence interval; COPD = chronic obstructive pulmonary disease; E-RS = Evaluating Respiratory Symptoms; ITT = intention to treat; LS = least squares; RR = relative risk; SD = standard deviation; SE = standard error; SGRQ = St. George's Respiratory Questionnaire; vs. = versus.
Notes: For the SGRQ outcome, in both BOREAS and NOTUS, a participant whose condition responded to treatment is defined as a participant with improvement from baseline in SGRQ total score at week 52 by ≥ 4 points. Participants with improvement < 4 points or with missing values are considered as patients who did not respond to treatment.
aP value has been adjusted for multiple testing.
bNominal P value.
Sources: Clinical Study Reports for BOREAS and NOTUS.20,21 Details included in the table are from the sponsor’s summary of clinical evidence.24
Annualized Rate of Severe COPD Exacerbations Over the 52-Week Treatment Period
In the BOREAS trial, few patients experienced a severe acute exacerbation of COPD event during the 52-week treatment period in both the dupilumab and placebo groups (4.3% and 5.9%, respectively). The adjusted annualized rate of severe acute exacerbation of COPD events over the 52-week treatment period in the ITT population was 0.072 (95% CI, 0.040 to 0.132) in the dupilumab group and 0.086 (95% CI, 0.050 to 0.147) in the placebo group (nominal P = 0.6085). There was no difference between the dupilumab and placebo groups in the time to first severe COPD exacerbation event (hazard ratio = 0.744; 95% CI, 0.416 to 1.331; nominal P = 0.3185).
In the NOTUS trial, few patients experienced a severe acute exacerbation of COPD event during the 52-week treatment period in both the dupilumab and placebo groups (4.5% and 6.9%, respectively). The adjusted annualized rate of severe acute exacerbation of COPD events over the 52-week treatment period in the ITT population was numerically lower in the dupilumab group compared with the placebo group (0.070; 95% CI, 0.039 to 0.123 versus 0.124; 95% CI, 0.072 to 0.215). Despite the small number of severe COPD exacerbation events observed, dupilumab 300 mg every 2 weeks led to a large reduction (i.e., 44%) in the annualized rate of severe acute exacerbation of COPD events as compared to placebo (nominal P = 0.0571).
Proportion of Patients With SGRQ Improvement by 4 Points or More at Week 52
In the BOREAS trial, the proportion of patients who achieved a clinically meaningful response in SGRQ total score (i.e., reduction [improvement] by ≥ 4 points) was higher in the dupilumab group as compared to the placebo group (51.5% versus 43.1%); the difference was statistically significant (odds ratio = 1.439; 95% CI, 1.096 to 1.890; P = 0.0089).
In the NOTUS trial, the proportion of patients who achieved a clinically meaningful response in SGRQ total score (i.e., reduction [improvement] by ≥ 4 points) was numerically higher in the dupilumab group as compared to the placebo group (51.4% versus 46.5%); the treatment difference was not significant (odds ratio = 1.164; 95% CI, 0.856 to 1.581; nominal P = 0.3329). Note that a high rate of response was observed in the placebo group and that although not significant, an improvement over placebo was observed in the dupilumab group.
At week 24, the proportion of patients who achieved a clinically meaningful reduction (improvement) by 4 points or more was 55.2% in the dupilumab group versus 46.0% in the placebo group, corresponding to a nominally significant treatment difference (odds ratio = 1.429; 95% CI, 1.046 to 1.952; nominal P = 0.0249).
Change in E-RS:COPD Scores From Baseline to Week 52
In the BOREAS trial at baseline, the mean E-RS:COPD total score was 12.94 in the dupilumab group and 12.95 in the placebo group. A significantly greater reduction in the E-RS:COPD total score, indicating an improvement in the severity of COPD-related respiratory symptoms, was observed in the dupilumab group as compared to the placebo group, with LS mean changes from baseline to week 52 of −2.694 and −1.558, respectively, and an LS mean difference versus placebo.
The reduction (improvement) in E-RS:COPD total score was rapid, with a difference in favour of dupilumab versus placebo observed as early as week 1 (LS mean difference versus placebo: −0.373; nominal P = 0.0366) and sustained and progressive over the 52-week intervention period of −1.137 (95% CI, −1.823 to −0.450; P = 0.0012).
Similar to the beneficial effect on the E-RS:COPD total score, reductions (improvements) were also observed at week 52 in all 3 E-RS:COPD domain scores in the dupilumab group as compared to the placebo group (E-RS:COPD RS-Breathlessness [nominal P = 0.0005], E-RS:COPD RS-Cough and Sputum [nominal P = 0.0170], and E-RS:COPD RS-Chest Symptoms [nominal P = 0.0171]). These differences in favour of dupilumab versus placebo were observed as early as week 2.
In the NOTUS trial, at baseline, the mean E-RS:COPD total score was 13.71 in the dupilumab group and 13.11 in the placebo group. A numerically greater reduction in the E-RS:COPD total score was observed in the dupilumab group as compared to the placebo group, with LS mean changes from baseline to week 52 of −2.388 and −1.770, respectively; the LS mean difference versus placebo did not reach nominal significance (−0.618; 95% CI, −1.430 to 0.193; nominal P = 0.1351). The numerical reduction (improvement) in E-RS:COPD total score was nominally significant from week 8 to week 36 (LS mean difference versus placebo: −0.854; nominal P = 0.0318).
A reduction (improvement) in the E-RS:COPD RS-Breathlessness domain score at week 52 was observed in the dupilumab group compared to the placebo group (nominal P = 0.0326); the treatment effect was observed as early as week 8. No significant differences between intervention groups were observed on the E-RS: COPD RS-Cough and Sputum and RS-Chest Symptoms domain scores.
Analysis of SCS Use
In the BOREAS trial, the adjusted annualized total number of SCS courses for COPD exacerbations was lower in the dupilumab group as compared to the placebo group (0.667; 95% CI, 0.536 to 0.829 versus 0.948; 95% CI, 0.780 to 1.151), corresponding to a reduction of 30% in the annualized total number of SCS courses for COPD exacerbations with dupilumab treatment as compared to placebo (nominal P = 0.0026).
In the NOTUS trial, the adjusted annualized total number of SCS courses for COPD exacerbations was lower in the dupilumab group as compared to the placebo group (0.682; 95% CI, 0.527 to 0.883 versus 1.114; 95% CI, 0.856 to 1.451), corresponding to a reduction of 39% in the annualized total number of SCS courses for COPD exacerbations with dupilumab treatment as compared to placebo (nominal P = 0.0003).
Analysis of Antibiotic Use
In the BOREAS trial, the adjusted annualized total number of antibiotic courses for COPD exacerbations was lower in the dupilumab group as compared to the placebo group (0.594; 95% CI, 0.476 to 0.742 versus 0.818; 95% CI, 0.671 to 0.996), corresponding to a reduction of 27% in the annualized total number of antibiotic courses for COPD exacerbations with dupilumab treatment as compared to placebo (nominal P = 0.0076).
In the NOTUS trial, the adjusted annualized total number of antibiotic courses for COPD exacerbations was lower in the dupilumab group as compared to the placebo group (0.470; 95% CI, 0.344 to 0.642 versus 0.723; 95% CI, 0.526 to 0.995), corresponding to a reduction of 35% in the annualized total number of antibiotic courses for COPD exacerbations with dupilumab treatment as compared to placebo (nominal P = 0.0092).
Pharmacoeconomic Review
Abbreviations
AE
adverse event
CDA-AMC
Canada’s Drug Agency
COPD
chronic obstructive pulmonary disease
CV
cardiovascular
FEV1
forced expiratory volume in the first second
GOLD
Global Initiative for Chronic Obstructive Lung Disease
ICER
incremental cost-effectiveness ratio
ICS
inhaled corticosteroid
LABA
long-acting beta-agonist
LAMA
long-acting muscarinic agonist
QALY
quality-adjusted life-year
SGRQ
St. George’s Respiratory Questionnaire
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Dupilumab (Dupixent), 300 mg/2 mL single-use syringe or pen for subcutaneous injection |
Indication | Add-on maintenance treatment in adult patients with chronic obstructive pulmonary disease (COPD) characterized by raised blood eosinophils inadequately controlled by the combination of an inhaled corticosteroid (ICS), a long-acting beta2 agonist (LABA), and a long-acting muscarinic antagonist (LAMA), or on a combination of a LABA and a LAMA if ICS is not appropriate. |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | October 21, 2025 |
Reimbursement request | As per indication |
Sponsor | Sanofi-Aventis Canada Inc. |
Submission history | Yesa Indication: As an add-on maintenance treatment in patients aged 12 years and older with severe asthma with a type 2/eosinophilic phenotype or oral corticosteroid-dependent asthma. Recommendation date: June 8, 2021 Recommendation: Reimburse with clinical criteria and/or condition Indication: As an add-on maintenance treatment in patients aged 6 years and older with severe asthma with a type 2/eosinophilic phenotype or oral corticosteroid-dependent asthma. Recommendation date: January 20, 2023 Recommendation: Reimburse with clinical criteria and/or condition Indication: As add-on maintenance treatment with intranasal corticosteroids in adult patients with severe chronic rhinosinusitis with nasal polyposis inadequately controlled by systemic corticosteroids and/or surgery. Recommendation date: TBD (currently an active review) Recommendation: TBD |
CDA-AMC = Canada’s Drug Agency; COPD = chronic obstructive pulmonary disease; NOC = Notice of Compliance; TBD = to be determined.
Notes: The sponsor’s application was filed on a pre-NOC basis and the pharmacoeconomic submission (economic evaluation and budget impact analysis) is reflective of the proposed indication and information in the draft product monograph. The wording of the indication was revised to specify inadequately controlled on specific treatments and removed reference to type-2 inflammation. The CDA-AMC appraisal was undertaken based on the submitted information, and the appraisal was not revised after the NOC and final product monograph was received.
aDupilumab had also been under review by CDA-AMC for atopic dermatitis and is currently under review for prurigo nodularis.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov |
Target population | Patients with uncontrolled COPD associated with type 2 inflammation |
Treatment | Dupilumab as an add-on to background therapy |
Dose regimen | 300 mg given every other week |
Submitted price | Dupilumab: $978.70 per single-use syringe or pen |
Submitted treatment cost | $27,078 per patient annually |
Comparator | Background therapy (LABA + LAMA + ICS) |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (35 years) |
Key data sources | BOREAS and NOTUS clinical trials |
Submitted results | ICER = $64,601 per QALY gained (incremental QALYs = 1.24; incremental costs = $79,757) |
Key limitations |
|
CDA-AMC reanalysis results |
|
CDA-AMC = Canada's Drug Agency; COPD = chronic obstructive pulmonary disease; FEV1 = forced expiratory volume in the first second; ICER = incremental cost-effectiveness ratio; ICS = inhaled corticosteroid; LABA = long-acting beta-agonist; LAMA = long-acting muscarinic antagonist; LY = life-year; QALY = quality-adjusted life-year.
Conclusions
Direct comparative evidence from 2 pivotal, double-blind, randomized controlled trials (BOREAS and NOTUS) demonstrated that in adult patients with uncontrolled chronic obstructive pulmonary disease (COPD) who had type 2 inflammation and moderate to severe airflow obstruction despite receiving triple inhaler therapy (long-acting beta-agonist [LABA], long-acting muscarinic agonist [LAMA], and inhaled corticosteroid [ICS]), dupilumab treatment used in addition to background therapy resulted in a clinically meaningful reduction in moderate or severe exacerbations compared to placebo. Dupilumab likely results in little to no clinically meaningful benefits in lung function measured with prebronchodilator forced expiratory volume in the first second (FEV1) compared to placebo. Patients’ quality of life as measured by St. George’s Respiratory Questionnaire (SGRQ) may be improved with the addition of dupilumab treatment; however, the magnitude of benefit is considered not clinically significant based on a recognized minimal important difference estimate. No notable safety concerns were identified with dupilumab treatment from the pivotal trials through week 52.
Results of the reanalysis by Canada’s Drug Agency (CDA-AMC) suggest the incremental cost-effectiveness ratio (ICER) for dupilumab as an add-on to background therapy was $720,337 per quality-adjusted life-year (QALY) gained compared to background therapy alone (incremental costs = $129,389; incremental QALYs = 0.180). Over a patient’s lifetime, the model estimates that on average patients will spend 5.24 years receiving dupilumab and this will prevent 1.92 exacerbations (1.52 moderate and 0.40 severe). Incremental QALYs were driven by the mortality benefit associated with preventing severe exacerbations (an additional 0.22 life-years for patients receiving dupilumab relative to background therapy alone). Incremental costs were driven largely by additional drug costs (an additional $135,644 in lifetime drug costs per patient relative to background therapy alone). The addition of dupilumab to background therapy does result in some cost savings due to the reduction in exacerbations, but these are small relative to the incremental drug costs ($6,151 in lifetime cost savings per patient from reducing exacerbations). Based on the CDA-AMC reanalysis, the annual cost of dupilumab as an add-on to background therapy would need to be $2,708 per patient (90% price reduction from list price) to be considered cost-effective at a $50,000 per QALY threshold versus background therapy alone.
Based on the clinical interpretation of the evidence, dupilumab provides no clinically meaningful benefit with regard to lung function. Therefore, in the CDA-AMC base case, results are largely driven by the reduction in exacerbations which are associated with a reduction in quality of life, a mortality risk, and costs to health care systems. Of these 3 impacts, the CDA-AMC analysis may overestimate the mortality impact as there is no direct evidence from the trial to support mortality reductions. The analysis may therefore overestimate the cost-effectiveness of dupilumab as an add-on to background therapy.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient group input was received from the Chronic Obstructive Pulmonary Disease Association (also known as COPD Canada) and the Lung Health Foundation. Input was gathered from a total of 88 participants (27 and 61, respectively) across Canada via online surveys or one-on-one discussions or interviews. Those who responded noted that COPD is associated with symptoms such as fatigue, shortness of breath, and coughing that may have a large impact on their daily life. While some participants noted that currently available therapies provide some symptom relief, others noted that there is still an urgent need for additional COPD medications that address unmet needs, minimize side effects, and improve overall treatment outcomes and quality of life. No participants reported experience with dupilumab as add-on maintenance treatment for uncontrolled COPD associated with type 2 inflammation.
Clinician input was received from the Canadian Thoracic Society. Clinician input noted that the key treatment goals for COPD are to improve symptoms, reduce exacerbations, and reduce mortality. Clinician input noted that despite the availability of treatments such as short-acting beta-agonists, short-acting muscarinic antagonists, LABAs, LAMAs, and ICSs, many patients continue to experience exacerbations that have an important impact on quality of life and resource use. Clinician input noted that dupilumab is a targeted biologic therapy, the first of its kind of patients with COPD, and may be beneficial to patients who remain at high risk of exacerbation despite optimized treatment.
Drug plan input received for this review raised concerns with the choice of comparator in the submitted trials, as both trials were placebo controlled and did not compare dupilumab add-on therapy to add-on therapy with azithromycin, roflumilast, or N-acetylcysteine. The drug plans questioned if patients in clinical practice should meet all the trial’s inclusion criteria before initiating dupilumab and questioned the use of dupilumab in patients with COPD who do not have type 2 inflammation. Finally, the drug plans further inquired what would be considered an absence of clinical benefit or a loss of response.
Several of these concerns were addressed in the sponsor’s model.
The impact of disease and treatment on patient’s quality of life was captured with utility values.
The impact of COPD and exacerbations on mortality was considered.
Although the sponsor provided an analysis that included a response assessment, this was not deemed appropriate by clinical experts consulted by CDA-AMC.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis to assess the cost-effectiveness of dupilumab as an add-on to background therapy for the treatment of adult patients with uncontrolled COPD associated with type 2 inflammation. The model population aligns with the Health Canada–approved indication and represents the sponsor’s reimbursement request.
Dupilumab is available as 300 mg/2 mL single-use syringes or pens.1 The recommended dose is 300 mg given every other week.1 The submitted price for dupilumab is $1,957.40 per syringe or pen or $27,078 per year. The comparator in this analysis is background therapy defined as LABA plus LAMA plus ICS and was estimated to cost approximately $1,545 per year.
Outcomes of the model included QALYs and life-years over a lifetime horizon of 35 years. Discounting (1.5% per annum) was applied for both costs and outcomes and a cycle length of 1 year was used with a half-cycle correction applied.
Model Structure
The sponsor submitted a decision tree plus Markov model to track a cohort of adult patients with uncontrolled COPD associated with type 2 inflammation. The decision tree represented the first 52 weeks (trial period). At model entry, patients were split across 4 COPD severity health states (i.e., mild, moderate, severe, and very severe) based on their COPD severity and exacerbation rate. After 52 weeks, patients were distributed into the Markov model based on their response to dupilumab plus background therapy. Those who responded to treatment continued to receive dupilumab plus background therapy in the Markov model; whereas, those who did not respond to treatment transitioned to background therapy alone.2 Within the Markov model, patients could transition between 12 health states defined by their COPD status and exacerbation status (Figure 2). As such, patients could transition due to a change in exacerbation status (e.g., from “mild COPD with no exacerbations” to “mild COPD with moderate exacerbations”) or across COPD status if they experienced a change in FEV1 (e.g., from “mild COPD with no exacerbations” to “moderate COPD with no exacerbations”). Patients were assumed to only experience a decline or deterioration of their COPD severity over time and therefore could not transition to a less severe COPD health state. At any point, patients could transition to the “death” health state. Likewise, upon experiencing an exacerbation, patients faced an additional mortality risk. Patients could discontinue dupilumab treatment at the end of the decision tree (i.e., week 52) and throughout the Markov model annually.2
Model Inputs
The baseline characteristics used to inform the model were based on the pooled patient population of the BOREAS study (interim cut-off date of February 8, 2023) and the NOTUS study (interim cut-off date of September 29, 2023), which enrolled adult patients with uncontrolled COPD with type 2 inflammation. The mean age of the pooled cohort was 65 years and 66.8% were male.
Clinical inputs for dupilumab as an add-on to background therapy were informed by data pooled from the BOREAS and NOTUS clinical trials, which included a total of 1,874 participants. Patients were distributed into the health states based on Global Initiative for Chronic Obstructive Lung Disease (GOLD) severity at week 2 for the decision tree. Upon entry into the Markov model, patients were additionally distributed based on their response rate, exacerbation rate, and the number of exacerbations patients experienced. Those who responded to treatment were defined as a patient with an annualized rate of on-treatment severe exacerbations lower than the number of severe exacerbations before the study and represented 94.2% of patients treated with dupilumab as an add-on to background therapy.2 Those who did not respond to treatment were assumed to have the same clinical outcomes of those receiving background therapy only. In the base-case analysis, the sponsor informed exacerbation rates from the year before randomization of the trials for background therapy and used a relative rate versus standard of care derived from the trials for dupilumab plus background therapy.2 Finally, the distribution of patients by number of exacerbations was also informed by pooled trial data based on the number of exacerbation events patients experienced within the first 52 weeks.
In the absence of data from the clinical trials, transitions between health states due to changes in exacerbation status (i.e., no exacerbation, ≥ 1 moderate exacerbation [without severe], or ≥ 1 severe exacerbation) were informed by the Whittaker et al. study (2022).3 The Whittaker et al. study (2022) reported adjusted incidence rate ratios for patients with 1, 2, and 3 or more exacerbations compared to patients with no exacerbations.3 The exacerbation rate for patients with no recent prior exacerbation was estimated from the Wallace et al. study, (2019).4 Health state transitions due to changes in COPD severity (i.e., mild, moderate, severe, very severe) were informed by annual probabilities reported by Fenwick et al. (2021),5 adjusted to account for the presence of type 2 inflammation and the characteristics of the patient population observed in the BOREAS and NOTUS trials.
Patients receiving dupilumab as an add-on to background therapy were able to discontinue treatment and transition to background therapy throughout the model. The annual discontinuation rate was 9.3% for the first year (informed by the pooled trial data) and was implemented at the end of the decision tree.2 The annual discontinuation rate was 15.0% for each year onward.2 Discontinuation was only applied to dupilumab.2
Background mortality was informed by Statistics Canada life tables.6 The model incorporated an increased mortality risk associated with COPD and exacerbation. The impact of COPD on mortality was incorporated using standardized mortality ratios obtained from the Whittaker et al. study (2024),3 where patients in the moderate, severe, and very severe COPD health states had a standardized mortality ratio of 1.5, 2.3, and 4.1 applied, respectively.7 Based on data from the Hoogendoorn et al. study, 15.6% of severe exacerbations were assumed to be fatal and this was an additional mortality risk applied every time a patient experienced a severe exacerbation.8
The model included headache, back pain, urinary tract infection, gastritis, and hypertension as adverse events (AEs). Rates were obtained from pooled data from the BOREAS and NOTUS clinical trials for dupilumab as an add-on to background therapy and background therapy alone. Cardiovascular (CV) events (i.e., myocardial infarction, stroke, unstable angina, and transient ischemic attack) were also considered. The risk of nonfatal CV events during the trial period was informed by pooled data from the BOREAS and NOTUS clinical trials. Posttrial nonfatal CV event risk was derived from the Kunisaki et al. study.9
Health state utilities were derived using SGRQ scores pooled from the BOREAS and NOTUS trials.2 SGRQ scores were converted to EQ-5D utilities based on a mapping algorithm using UK tariff values.10 Therefore, patients receiving dupilumab in addition to background therapy with mild, moderate, severe, and very severe COPD had utility values of 0.8327, 0.8106, 0.7887, and 0.7314, respectively. Patients receiving background therapy along with mild, moderate, severe, and very severe COPD had utility values of 0.8286, 0.7961, 0.7749, and 0.7192, respectively.2 The sponsor further incorporated disutilities associated with exacerbations, AEs, and CV events. Disutilities associated with acute moderate and acute severe exacerbations were assumed to be 0.0154 and 0.0506, respectively. Disutilities associated with CV events (i.e., myocardial infarction, stroke, unstable angina, and transient ischemic attack) were informed by published literature and assumptions.11
Costs included in the model consisted of treatment acquisition costs, administration costs, disease management and routine follow-up care costs, and the cost of unplanned events (i.e., exacerbations, AEs, and CV events). Acquisition costs were based on the sponsor’s submitted price for dupilumab, whereas other drug costs were obtained from the Ontario Drug Benefit Formulary/Comparative Drug Index.2,12 Inhaled treatments were assumed to be self-administered and thus not associated with any administration costs. The sponsor included a 1-time subcutaneous administration training cost for dupilumab, assumed equal to the cost of 1 hour of an Ontario nurse’s pay.2 Costs associated with exacerbations were calculated from the sponsor’s burden of illness and health care resource utilization study based on data accessed through the ICES.13 Disease management costs appear to be derived from a 2003 study and ranged from $147 to $1,287 for mild to very severe COPD health states.14 Patients who experienced nonfatal CV events (i.e., myocardial infarction, stroke, unstable angina, and transient ischemic attack) incurred management costs informed by the Canadian Institute for Health Information’s patient cost estimator whereas patients experiencing AEs were assumed to incur the cost of 1 visit by a family physician per event.12,15
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically with 1,000 iterations. The deterministic results were aligned with submitted probabilistic results. The probabilistic findings are presented in the following.
Base-Case Results
As part of an additional information request, the sponsor resubmitted an economic model that revised errors identified by CDA-AMC. In the sponsor’s updated deterministic base-case analysis, dupilumab in addition to background therapy was associated with an additional 1.22 QALYs at an additional cost of $78,545 compared to background therapy alone. Therefore, the ICER was $64,447 per QALY gained. Based on the deterministic results, the majority (90%) of the incremental QALYs associated with dupilumab in addition to background therapy were found to be accrued during the extrapolation period. The result was driven primarily by extending life (1.55 additional life-years). In the submitted model, the sponsor did not rerun the probabilistic analysis after fixing an identified error. The results presented in Table 3 are the sponsor’s results with the probabilistic analysis rerun.
At a threshold of $50,000 per QALY, dupilumab in addition to background therapy had a 3% probability of being cost-effective.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total LYs | Total QALYs | Incremental QALYs | ICER vs. reference ($/QALY) |
|---|---|---|---|---|---|---|
Background therapy | 159,509 | Reference | 7.758 | 5.729 | Reference | Reference |
Dupilumab plus background therapy | 239,377 | 79,868 | 9.305 | 6.967 | 1.24 | 64,499 |
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.2
Sensitivity and Scenario Analysis Results
Updated scenario analyses were not submitted with the updated model that resolved errors identified by CDA-AMC.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis.
Response assessment to inform treatment discontinuation is uncertain. In the sponsor’s base-case analysis, patients were categorized as those who responded to treatment or who did not respond to treatment at the end of week 52. This was based on the BOREAS and NOTUS clinical trials where those who responded to treatment were defined as patients with an annualized rate of on-treatment severe exacerbation strictly lower than the number of severe exacerbations before the study. Based on the pooled trials, in the intention-to-treat population, 94.2% of patients responded to treatment and continued to receive dupilumab whereas those who did not respond to treatment transitioned to background therapy. Clinical expert feedback received by CDA-AMC indicated that there is no ideal way to identify those who respond to treatment in clinical practice in Canada and it would therefore not be used to inform treatment discontinuation. For example, even if a patient experiences an exacerbation, this is not necessarily indicative of treatment failure as the treatment reduces the risk of exacerbations rather than eliminates their likelihood of occurring. As such, use of a response assessment in the model may bias results in favour of dupilumab plus background therapy as there is no practical way to identify those who are responding and not responding to treatment.
Finally, the way the sponsor implemented efficacy for those who did not respond to treatment in the model likely overestimates the benefit of dupilumab as an add-on to background therapy. That is because the sponsor assumes patients who do not respond to treatment experience a treatment effect equivalent to those receiving background therapy alone. This assumes that 100% of patients who receive background therapy alone do not respond to treatment. This is not accurate as some patients who receive background therapy alone responded to treatment in the trial period. If a patient therefore does not respond to dupilumab plus background therapy, they will likely experience the same treatment effect as those who did not respond to background therapy alone as opposed to the average patient on background therapy.
In the CDA-AMC base case, response assessment was excluded from the analysis.
Dupilumab’s impact on lung function is uncertain. In the sponsor’s base-case analysis, in the second year of the analysis patients who receive dupilumab cannot progress through COPD stages; patients who receive background therapy alone can progress. The sponsor notes that in the poststudy observation period extended to 64 weeks, FEV1 began to decline for the patients treated with background therapy alone. At study end (week 52), dupilumab was withdrawn from patients so these data are not available for the dupilumab arm. The sponsor therefore assumes that while patients who received background therapy alone will continue to progress, patients who received dupilumab will not. There are various concerns with this assumption.
First, conclusions from the clinical report note that dupilumab likely results in little to no clinically meaningful benefits in lung function measured with prebronchodilator FEV1 compared to placebo. Although the trial shows a statistically significant difference in FEV1, this was noted as not clinically meaningful and unlikely to be perceptible by patients. Whereas the model suggests that FEV1 improvements alone are associated with dupilumab resulting in improvements in utility and a reduction in mortality risk. Therefore, assuming patients who received dupilumab do not progress whereas patients receiving background therapy alone suggests a large treatment effect occurs in the second year of treatment, not evidenced in the first.
Second, this assumption goes against other assumptions used in the model. The sponsor assumes that after experiencing a moderate or severe exacerbation, the likelihood of COPD progressing increases. As patients who receive dupilumab experience moderate and severe exacerbations this would indicate they are at higher risk of progressing to more severe COPD stages. Likewise, evidence from the trial shows that in the trial period some patients moved to more severe COPD states indicating that lung function deteriorates for some patients while receiving dupilumab.
Finally, this is not an evidence-based assumption. As noted by the sponsor, there is no evidence of differences in lung function between background therapy alone and dupilumab after 52 weeks. The decline in lung function in year 2 of the model is also taken from an external source and is not the decline seen in the trial data. If lung function did not change for any patient receiving dupilumab in year 2, the decline in lung function for those on placebo should be based on the trial population.
Overall, if dupilumab improved lung function, relative to background therapy alone, then this should be modelled based on the available data. However, as noted in the clinical report, differences in lung function were not seen as clinically meaningful when comparing the placebo and dupilumab arms of both the NOTUS and BOREAS trials. By assuming lung function continues to decline for those receiving background therapy but ceases to decline at all for those receiving dupilumab, this creates a treatment effect not evidenced in the trial.
In the CDA-AMC base case, the assumption that no patients who received dupilumab would progress in the second year of the model was removed. The base case therefore only focuses on the benefits derived from preventing exacerbations.
Discontinuation of dupilumab for weeks 52 and after were uncertain. In the base-case analysis, the sponsor used an annual rate of 15% to inform dupilumab discontinuation occurring beyond week 52. The sponsor notes that this rate was informed based on real-world data for dupilumab as an add-on to background therapy for the treatment of adult patients with asthma due to the absence of long-term COPD data.2 While CDA-AMC acknowledges that the absence of long-term discontinuation data for dupilumab in patients with COPD is a challenge, the use of data from patients with asthma is inappropriate. This is primarily due to differences between the asthma and COPD disease spaces. Within the asthma disease space there are alternative treatments options available; therefore, patients with asthma may discontinue dupilumab in favour of transitioning to an alternative therapy. This is not an option for the treatment of COPD where dupilumab is the first biologic with a Health Canada–approved indication for COPD treatment. If treatment discontinuation rates increase over time, then this might be due to a loss in treatment efficacy or an increase in more severe AEs which has not been modelled.
In the CDA-AMC base case, CDA-AMC set the dupilumab discontinuation for beyond week 52 equal to the discontinuation rate applied for the trial period (i.e., 9.3%). This assumes the rate of discontinuation seen in the trial remains constant over time.
Baseline exacerbation rates informed by the year before randomization are inappropriate. Baseline exacerbation rates for background therapy in the sponsor’s base case were informed from a pooled analysis for the BOREAS and NOTUS studies for the year before randomization. The sponsor argued that the year before randomization was used to maintain the benefits of a trial population given that the exacerbations rates observed within the trials may not be reflective of real-world rates. However, patients were required to experience an exacerbation before entry into the trials. Relative to the year prior, exacerbation rates decreased for those receiving background therapy within the trial. This indicates that these patients may have been receiving suboptimal disease management before the trial. For example, a patient may have been receiving a suboptimal dose or was nonadherent to their medication in the year before the trial. This may have been resolved after entering the trial. The randomized period of the trial therefore represents the best data for exacerbation rates for those who received background therapy alone as this incorporates any potential change in medical management that would occur in the real world following an exacerbation. This same change in medical management would have also applied to the dupilumab plus background therapy arm. Ignoring this potential effect might either overestimate the number of exacerbations or misrepresent the treatment effect associated with dupilumab as an add-on to background therapy.
In the CDA-AMC base case, baseline exacerbation for background therapy was set to be informed by the selected trial.
CDA-AMC acknowledges that exacerbation rates in the real world may be higher; therefore, CDA-AMC additionally conducted a scenario where baseline exacerbation was set to data collected from real-world evidence.
The impact of exacerbation on mortality is overestimated. In the NOTUS trial, 21 patients died during the on-study period. Of those, 13 (2.8%) were in the dupilumab plus background therapy group and 8 (1.7%) were in the placebo group. In the BOREAS study, overall, 16 patients died during the on-study period, 8 (1.7%) in each intervention group. Therefore, no evidence was available from the trial to demonstrate a mortality benefit associated with dupilumab as an add-on to background therapy. However, the trial demonstrated a reduction in hospitalizations. Pooling across both the NOTUS and BOREAS studies, 53 patients experienced hospitalization in the dupilumab plus background therapy arm versus 75 in the placebo arm. Clinical expert feedback received by CDA-AMC noted that hospitalized patients with COPD have a higher risk of death and therefore reducing the number of hospitalizations may improve mortality. Mortality improvements underpin the large degree of benefit in the sponsor’s base case which estimates treatment with dupilumab will extend life on average by 1.5 years.
In the sponsor’s submitted analysis, mortality associated with COPD was incorporated in 2 ways. The first was as standardized mortality ratios applied to the general population mortality for the various COPD stages informed by the Whittaker et al. study (2024).7 This was to capture the increased mortality risk associated with COPD as the disease progresses. The second was applied as an instantaneous risk of death (15.6%) that occurs after each severe exacerbation. This was informed by the study by Hoogendoorn et al.8
The first limitation with the preceding approach is that the standardized mortality ratios applied to the GOLD stages already include any mortality risk associated with exacerbations. Therefore, additionally applying a mortality adjustment to account for excess mortality due to an exacerbation event results in double counting of mortality associated with COPD. Second, the 15.6% value is applied regardless of GOLD stage or age. This means a patient aged 60 years with mild COPD has the same risk of death postexacerbation as a patient aged 80 years with very severe COPD. This is inappropriate, as the study by Hoogendoorn et al.8 notes age as an important predictor of mortality after a hospitalization.
The sponsor additionally provided an option to model excess mortality due to exacerbations as an incident rate ratio based on the Whittaker et al. study (2022).3 Because this incident ratio is applied to general population mortality, the probability of death after hospitalization increases with age.
In the CDA-AMC base case, excess mortality due to exacerbations was informed by the Whittaker et al. study (2022), as per the sponsor’s provided option. Mortality is still double counted as the hazard ratio applied to the GOLD stage still likely captures the mortality risk associated with exacerbations. The results therefore remain biased in favour of dupilumab.
CDA-AMC additionally conducted 2 scenario analyses: 1 exploring the impact of removing the excess mortality associated with exacerbations and 1 assuming no mortality benefit associated with dupilumab as an add-on to background therapy.
Exacerbation management costs were overestimated. Exacerbation management costs in the sponsor’s base-case analysis were informed by a sponsor-conducted burden of illness health care resource utilization study.2 Using data from ICES, the study looked at all health care costs, regardless of whether they were related to COPD, in the year following a moderate or severe COPD exacerbation. Costs attributed to an exacerbation, as opposed to COPD management or non-COPD related costs, were then derived by subtracting an average annual background cost of COPD management (informed by Chapman et al.14) from the total annual health care costs derived from the ICES analysis.2,14 The assumption is that costs not related to the exacerbation are fully captured in the Chapman et al. study; therefore, by removing these costs, the remaining estimate must be fully attributed to the exacerbation. This is unlikely to be true for several reasons.
First, because the study by Chapman et al.14 derived health care costs using surveys, it is unlikely individuals would recall every instance of health care utilization; whereas, the sponsor’s study using ICES data is robust in capturing all health care utilization. Second, the study by Chapman et al. notes the aim was to establish the direct cost of COPD, excluding costs unrelated to the disease. Therefore, the study does not identify all health care costs unrelated to an exacerbation, which are included in the ICES study. Third, the study is based on data that are more than 20 years old and is unlikely to be reflective of health care costs today. Finally, the patient populations analyzed in both studies are different. The mean age of a patient in the ICES study was 75 years; whereas, in the Chapman et al. study, the mean age was 62 years. Health care costs are likely to be higher in a cohort aged 75 years than a cohort aged 62 years.
Overall, the approach taken by the sponsor likely overestimates the attributable costs related to exacerbations. A preferred approach to establishing attributable costs using observational data would be to establish a control cohort while controlling for confounding. As part of an additional information request, CDA-AMC requested data on costs associated with the baseline period of the study (i.e., 12 months before the initial index event [first exacerbation experienced by a patient]) to help better understand the health care resource use associated with an exacerbation. The sponsor noted these inputs would not be robust and therefore would not be provided. Without a robust control arm to isolate the costs attributed to the exacerbation, the data from the ICES study could not be used.
A microcosting approach to cost out exacerbation management costs was also provided by the sponsor. However, when attempting to validate the unit cost of an inpatient hospital stay, CDA-AMC found that the value reported from the Canadian Institute for Health Information for patients with COPD aged 60 to 79 years was $8,255 (before adjusting for inflation) versus the sponsor’s reported $18,255. It is unclear if this was an error as no source could be identified for the $18,255 value.
In the CDA-AMC base case, exacerbation costs were informed by the microcosting option provided by the sponsor. Additionally, the unit cost of an inpatient hospital stay was set to $8,255 (preinflated value) as informed by the Canadian Institute for Health Information’s patient cost estimator for patients with COPD.
Use of treatment-specific utilities is inappropriate. As part of the base case, the sponsor assumed that health state utilities differed by treatment. Utility values were informed by pooled data from the BOREAS and NOTUS studies. According to the CDA-AMC Guidelines for the Economic Evaluation of Health Technologies: Canada, health state utilities should reflect the health states in the model rather than the treatment comparators.16 The use of treatment-specific utilities implies that a patient’s quality of life will differ as a result of the treatment received independent of disease severity, AEs, and exacerbation status. Given all of these factors have been accounted for, there is limited justification for applying treatment-specific utilities. Additionally, clinical expert feedback received by CDA-AMC noted that there is likely no clinically meaningful difference in the quality of life between treatment arms and thus there is limited justification for the inclusion of this in the economic model.
In the CDA-AMC base case, CDA-AMC used nontreatment-specific health state utility values provided by the sponsor in a request for additional information.
Trial period distribution for patients receiving dupilumab as an add-on to background therapy is uncertain. In the base-case analysis, the sponsor pools the distribution of patients by GOLD stage across all treatment arms in both the NOTUS and BOREAS studies. At the start of the model, it is assumed that 2% have mild COPD, 47% have moderate, 48% have severe, and 3% have very severe disease. After 52 weeks, for those receiving background therapy alone, 6% have mild COPD, 47% have moderate, 42% have severe, and 5% have very severe disease. Of the patients who also receive dupilumab, 10% have mild COPD, 44% have moderate, 41% have severe, and 5% have very severe disease. There are several issues with the way this has been used in the model.
First, the sponsor assumes patients cannot regress through health states, meaning patients cannot return to a less severe disease stage. This was confirmed to be a valid assumption by the clinical experts consulted by CDA-AMC because loss of FEV1 is typically irreversible. However, using the sponsor’s approach of applying trial distributions, some patients return to a less severe disease stage within the first 52 weeks. This assumes that patients experience an improvement in their quality of life as well as a reduction in their mortality risk regardless of whether they have experienced an exacerbation or not. Second, the sponsor assumes no difference at baseline between treatment arms. Given the randomized nature of the trials, it is unlikely there are large differences between staging at baseline between arms, but these should be accounted for, especially as the differences in staging between the 2 arms are small. Third, health states in the model are derived using discrete intervals of FEV1. This means a very small change in FEV1 can cause a patient to move between 2 states which has large consequences in terms of mortality risk and quality of life; this occurs even if the change is small and not clinically meaningful. This may be driving movement between some of the health states. Finally, the Clinical Review concludes that dupilumab, in addition to background therapy, likely results in little to no clinically meaningful benefits in lung function or improvement in severe respiratory symptoms compared to placebo. Small differences in lung function may cause patients to move between health states; however, these small differences are unlikely to result in improved mortality and better quality of life.
Based on the preceding information, it is unlikely that patients experience an improvement in disease severity (i.e., patients with moderate COPD regress to mild).
In the CDA-AMC base case, CDA-AMC set the distributions for both the end of the FEV1 amelioration phase and the end of the FEV1 maintenance phase for dupilumab as an add-on to background therapy as equal to background therapy. This does not resolve the fact that in the first 52 weeks some patients move to mild disease, but this is unlikely to substantially influence the results.
Additionally, Table 4 details key assumptions made by the sponsor, which have been appraised by CDA-AMC.
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
AE management costs were assumed to be equal to a visit to a family physician as AEs included in the model are not severe. | Reasonable. AEs included in the model include headache, back pain, urinary tract infection, gastritis, and hypertension which are likely to be addressed with a visit to a family physician. |
AE = adverse event; CDA-AMC = Canada's Drug Agency.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
The CDA-AMC base-case analysis was derived by making changes in the model parameter value and assumptions, in consultation with clinical experts. The changes, summarized in Table 5, include excluding response assessment, allowing patients receiving dupilumab to progress in year, changing the rate of treatment discontinuation after week 52, modifying the source used to inform baseline exacerbation rate for background therapy, setting the distribution of patients at the end of the FEV1 maintenance phase as equal between the 2 treatment arms, modifying mortality inputs and assumptions, using alternative exacerbation management costs, and using nontreatment-specific health state utility values.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Response assessment | Include | Exclude |
2. No progression for patients who received dupilumab in year 2 | Include | Exclude |
3. Treatment discontinuation for weeks after week 52 | 15% | 9.3% |
4. Baseline exacerbation rate for background therapy | Year before randomization of selected trial | Period during the selected trial |
5. Morality due to exacerbation | 15% excess mortality for severe | Increased risk of death applied using a hazard ratio from Whittaker et al. (2022)3 |
6. Exacerbation management costs | Aggregate | Microcosting approach Hospital stay unit cost = $8,679 ($8,255 uninflated) |
7. Health state utilities | Treatment-specific values | Nontreatment-specific values Mild = 0.8307 (SE = 0.005) Moderate = 0.8033 (SE = 0.003) Severe = 0.7818 (SE = 0.003) Very severe = 0.7253 (SE = 0.005) |
8. Distribution of patients at the end of FEV1 amelioration and maintenance phase | Informed by trial data by treatment arm | Dupilumab plus background therapy distribution set equal to distribution of background therapy only |
CDA-AMC base case | ― | Reanalysis 1 + 2 + 3 + 4 + 5 + 6 + 7 + 8 |
CDA-AMC = Canada's Drug Agency; FEV1 = forced expiratory volume in the first second; SE = standard error.
In the CDA-AMC reanalysis, dupilumab plus background therapy was associated with an ICER of $720,337 per QALY gained compared to background therapy only (incremental costs = $129,389; incremental QALYs = 0.18). Incremental costs were primarily due to the drug acquisition cost of dupilumab.
Table 6: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | Background therapy | 159,509 | 5.729 | Reference |
Dupilumab plus background therapy | 239,377 | 6.967 | 64,499 | |
CDA-AMC reanalysis 1 | Background therapy | 160,628 | 5.69 | Reference |
Dupilumab plus background therapy | 248,904 | 6.52 | 106,706 | |
CDA-AMC reanalysis 2 | Background therapy | 160,628 | 5.69 | Reference |
Dupilumab plus background therapy | 235,556 | 6.61 | 81,782 | |
CDA-AMC reanalysis 3 | Background therapy | 160,628 | 5.69 | Reference |
Dupilumab plus background therapy | 262,824 | 7.08 | 73,477 | |
CDA-AMC reanalysis 4 | Background therapy | 155,988 | 6.11 | Reference |
Dupilumab plus background therapy | 238,099 | 7.23 | 73,358 | |
CDA-AMC reanalysis 5 | Background therapy | 197,966 | 6.92 | Reference |
Dupilumab plus background therapy | 263,723 | 7.70 | 84,873 | |
CDA-AMC reanalysis 6 | Background therapy | 59,088 | 5.69 | Reference |
Dupilumab plus background therapy | 148,025 | 6.91 | 72,974 | |
CDA-AMC reanalysis 7 | Background therapy | 160,628 | 5.74 | Reference |
Dupilumab plus background therapy | 239,172 | 6.93 | 65,952 | |
CDA-AMC reanalysis 8 | Background therapy | 160,628 | 5.69 | Reference |
Dupilumab plus background therapy | 238,143 | 6.85 | 66,958 | |
CDA-AMC base case (reanalysis 1 + 2 + 3 + 4 + 5 + 6 + 7 + 8; deterministic) | Background therapy | 70,377 | 7.10 | Reference |
Dupilumab plus background therapy | 199,425 | 7.25 | 844,798 | |
CDA-AMC base case (reanalysis 1 + 2 + 3 + 4 + 5 + 6 + 7 + 8; probabilistic) | Background therapy | 70,748 | 7.13 | Reference |
Dupilumab plus background therapy | 200,137 | 7.31 | 720,337 |
CDA-AMC = Canada's Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. The results of all steps are presented deterministically unless otherwise indicated, while the cumulative CDA-AMC base case is always presented both deterministically and probabilistically.
Scenario Analysis Results
CDA-AMC undertook price reduction analyses based on the sponsor’s results and the CDA-AMC base case (Table 7). Results of the CDA-AMC reanalysis suggest a price reduction of approximately 90% would be required for dupilumab used in combination with background therapy to achieve cost-effectiveness versus background therapy alone at a $50,000 per QALY willingness-to-pay threshold. The unit cost of dupilumab would be $196 with a 90% price reduction.
CDA-AMC conducted additional scenario analyses to explore the impact of alternative baseline exacerbation rates, removing the excess mortality associated with exacerbations, and removing all mortality benefit associated with dupilumab plus background therapy.
When baseline exacerbation rates for background therapy are based on real-world evidence (via the sponsor’s provided option), the ICER for dupilumab as an add-on therapy to background therapy decreases to $694,431 per QALY gained compared to background therapy alone. Overall, a higher baseline rate of exacerbation does not substantially change the results with incremental QALYs increasing from 0.180 to 0.186.
When excess mortality associated with exacerbations was removed, the ICER for dupilumab as an add-on to background therapy increases to $1,783,873 per QALY gained compared to background therapy alone. After exacerbation, the sponsor assumes that a patient is more likely to progress to a more severe stage. Therefore, preventing exacerbations delays progression to more severe disease stages. This scenario analysis demonstrates that the mortality benefit associated with this assumption contributes to more than 50% of the QALY gain in the base-case analysis.
Finally, in the scenario that removed all mortality benefit associated with dupilumab, the ICER for dupilumab as an add-on therapy to background therapy increases to $5,373,844 per QALY gained compared to background therapy alone. This assumes all patients in moderate, severe, and very severe states experience an elevated standardized mortality ratio of 2, relative to the general public. This scenario analysis demonstrates that the mortality benefit associated with dupilumab constitutes more than 95% of the health benefit in the analysis.
Table 7: CDA-AMC Price Reduction Analyses
Analysis Price reduction | Unit drug cost ($) | ICERs for dupilumab in combination with background therapy vs. background therapy ($/QALY) | |
|---|---|---|---|
Sponsor base case | CDA-AMC reanalysis | ||
No price reduction | $1,957 | 64,601 | 720,337 |
10% | $1,762 | 56,747 | 645,815 |
20% | $1,566 | Dominant | 571,293 |
30% | $1,370 | Dominant | 496,771 |
40% | $1,174 | Dominant | 422,249 |
50% | $979 | Dominant | 347,726 |
60% | $783 | Dominant | 273,204 |
70% | $587 | Dominant | 198,682 |
80% | $391 | Dominant | 124,160 |
90% | $196 | Dominant | 49,638 |
CDA-AMC = Canada's Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Issues for Consideration
Dupilumab is currently being reviewed by CDA-AMC for 2 other distinct indications: the treatment of adult patients with moderate to severe prurigo nodularis whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable, and as add-on maintenance treatment with intranasal corticosteroids in adult patients with severe chronic rhinosinusitis with nasal polyposis inadequately controlled by systemic corticosteroids and/or surgery.17,18
Dupilumab has a letter of intent from the pan-Canadian Pharmaceutical Alliance.19
Overall Conclusions
Direct comparative evidence from 2 pivotal double-blind, randomized controlled trials (BOREAS and NOTUS) demonstrated that in adult patients with uncontrolled COPD who had type 2 inflammation and moderate to severe airflow obstruction despite receiving triple inhaler therapy (LABA, LAMA, and ICS), dupilumab treatment used in addition to background therapy resulted in a clinically meaningful reduction in moderate or severe exacerbations compared to placebo. Dupilumab likely results in little to no clinically meaningful benefits in lung function measured with prebronchodilator FEV1 compared to placebo. Patients’ quality of life as measured by SGRQ may be improved with the addition of dupilumab treatment; however, the magnitude of benefit is considered not clinically significant based on a recognized minimal important difference estimate. No notable safety concerns were identified with dupilumab treatment from the pivotal trials through week 52.
Results of the CDA-AMC reanalysis suggest the ICER for dupilumab as an add-on to background therapy was $720,337 per QALY gained compared to background therapy alone (incremental costs = $129,389; incremental QALYs = 0.180). Over a patient’s lifetime, the model estimates that, on average, patients will spend 5.24 years receiving dupilumab and this will prevent 1.92 exacerbations (1.52 moderate and 0.40 severe). Incremental QALYs were driven by the mortality benefit associated with preventing severe exacerbations (an additional 0.22 life-years for patients receiving dupilumab relative to background therapy alone). Incremental costs were driven largely by additional drug costs (an additional $135,644 in lifetime drug costs per patient relative to background therapy alone). The addition of dupilumab to background therapy does result in some cost savings due to the reduction in exacerbations, but these are small relative to the incremental drug costs ($6,151 in lifetime cost savings per patient from reducing exacerbations). Based on the CDA-AMC reanalysis, the annual cost of dupilumab as an add-on to background therapy would need to be $2,708 per patient (90% price reduction from list price) to be considered cost-effective at a threshold of $50,000 per QALY versus background therapy alone.
Based on the clinical interpretation of the evidence, dupilumab provides no clinically meaningful benefit with regard to lung function. Therefore, in the CDA-AMC base case, results are largely driven by the reduction in exacerbations which are associated with a reduction in quality of life, a mortality risk, and costs to the health care system. Of those 3 impacts, the analysis may overestimate the mortality impact as there is no direct evidence from the trial to support mortality reductions. The analysis may therefore overestimate the cost-effectiveness of dupilumab as an add-on to background therapy. In a scenario where excess mortality associated with exacerbations was removed, the ICER increased to $5,373,844 per QALY gained compared to background therapy alone. Therefore, if the mortality benefits associated with dupilumab as an add-on to background therapy are not realized or are lower than estimated by the model, higher price reductions may be required.
References
1.Dupixent (dupilumab): 300 mg/ 2mL single-use or pen for subcutaneous injection [product monograph]. Sanofi-aventis Canada Inc.; 2024.
2.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Dupixent COPD, 300 mg/ 2mL single-use syringe or pen for subcutaneous injection. North York (ON): sanofi-aventis Canada; 2024 Dec 11
3.Whittaker H, Rubino A, Müllerová H, et al. Frequency and severity of exacerbations of COPD associated with future risk of exacerbations and mortality: a UK routine health care data study. Int J Chron Obstruct Pulmon Dis. 2022;17:427-437. doi:10.2147/COPD.S346591 PubMed
4.Wallace AE, Kaila S, Bayer V, et al. Health care resource utilization and exacerbation rates in patients with COPD stratified by disease severity in a commercially insured population. J Manag Care Spec Pharm. 2019;25(2):205-217. doi:10.18553/jmcp.2019.25.2.205 PubMed
5.Fenwick E, Martin A, Schroeder M, Mealing SJ, Solanke O, Risebrough N, Ismaila AS. Cost-effectiveness analysis of a single-inhaler triple therapy for COPD in the UK. ERJ Open Research. 2021;7(1):00480-2020. doi:10.1183/23120541.00480-2020. PubMed
6.Statistics Canada. Table 13-10-0114-01 Life expectancy and other elements of the complete life table, three-year estimates, Canada, all provinces except Prince Edward Island. Updated 31 Oct 2024. Accessed 31 Oct 2024, https://doi.org/10.25318/1310011401-eng
7.Whittaker H, Rothnie KJ, Quint JK. Cause-specific mortality in COPD subpopulations: a cohort study of 339 647 people in England. Thorax. 2024;79(3):202-208. doi:10.1136/thorax-2022-219320 PubMed
8.Hoogendoorn M, Rutten-van Molken MP, Hoogenveen RT, Al MJ, Feenstra TL. Developing and applying a stochastic dynamic population model for chronic obstructive pulmonary disease. Value Health. 2011;14(8):1039-47. doi:10.1016/j.jval.2011.06.008 PubMed
9.Kunisaki KM, Dransfield MT, Anderson JA, et al. Exacerbations of chronic obstructive pulmonary disease and cardiac events. A post hoc cohort analysis from the SUMMIT randomized clinical trial. Am J Respir Crit Care Med. 2018;198(1):51-57. doi:10.1164/rccm.201711-2239OC PubMed
10.Hernández Alava M, Wailoo A, Pudney S, Gray L, Manca A. Mapping clinical outcomes to generic preference-based outcome measures: development and comparison of methods. Health Technol Assess. 2020;24(34):1-68. doi:10.3310/hta24340 PubMed
11.Sterne JA, Bodalia PN, Bryden PA, et al. Oral anticoagulants for primary prevention, treatment and secondary prevention of venous thromboembolic disease, and for prevention of stroke in atrial fibrillation: systematic review, network meta-analysis and cost-effectiveness analysis. Health Technol Assess. 2017;21(9):1-386. doi:10.3310/hta21090 PubMed
12.Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 24, 2023)). Ontario Ministry of Health; 2023. Accessed 2025 Jan 27. https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf
13.Sanofi. [Data on File] Burden of Illness and Health Resource Utilization for Chronic Obstructive Pulmonary Disease in Ontario. Final Results v1.0. 2024.
14.Chapman KR, Bourbeau J, Rance L. The burden of COPD in Canada: results from the confronting COPD survey. Respir Med. 2003;97:S23-S31. doi:10.1016/s0954-6111(03)80022-7 PubMed
15.Canadian Institute for Health Information. Patient Cost Estimator. Accessed 24 Oct 2024, https://www.cihi.ca/en/patient-cost-estimator
16.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. CADTH; 2017. Accessed Feb 3. 2025. https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition
17.CDA-AMC. Dupilumab Prurigo nodularis (PN). Accessed March 18. 2025, https://www.cda-amc.ca/dupilumab-8
18.CDA-AMC. Dupilumab chronic rhinosinusitis with nasal polyps. Accessed March 18. 2025, https://www.cda-amc.ca/dupilumab-7
19.pCPA. Dupixent (dupilumab). Accessed March 27, 2025, https://www.pcpacanada.ca/negotiation/22270
20.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. Accessed Feb 18, 2025, https://www.formulary.health.gov.on.ca/formulary/
21.Statistics Canada. Table 17-10-0005-01 Population estimates on July 1, by age and gender. 2024.
22.Canadian Chronic Disease Surveillance System. Data Tool. Chronic obstructive pulmonary disease, age-standardized prevalence, percent, age 35 years and older, Canada. 2024.
23.Haughney J, Gruffydd-Jones K, Roberts J, Lee AJ, Hardwell A, McGarvey L. The distribution of COPD in UK general practice using the new GOLD classification. The European respiratory journal. 2014;43(4):993-1002. doi:10.1183/09031936.00065013 PubMed
24.Adelphi. GOLD Classification (GOLD 2, or 3) (pts. with FEV1pp 50% - 79%, & 30% - 49%). 2018.
25.Sanofi. DATA ON FILE: Burden of Illness and Health Resource Utilization for Chronic Obstructive Pulmonary Disease in Ontario. Final Results v1.0. 2024.
26.Sutherland G, Dihn T. Understanding the gap: a pan-Canadian analysis of prescription drug insurance coverage. The Conference Board of Canada; 2017. Accessed Feb 28, 2025. https://www.conferenceboard.ca/e-library/abstract.aspx?did=9326
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison Table for COPD
Treatment | Strength/concentration | Form | Price | Recommended dosage | Daily cost ($) | Annual cost ($)a |
|---|---|---|---|---|---|---|
Dupilumab (Dupixent) | 300 mg/2mL | Single-use vial or pen for injection | 978.7000a | 300 mg every 2 weeks | 37.09 | 27,078.20 |
ICS-LAMA-LABA fixed-dose combinations | ||||||
Budesonide-glycopyrronium-formoterol fumarate (Breztri Aerosphere) | 160/7.2/5 mcg | MDI (120 doses) | 127.0000 | Two inhalations twice daily | 4.23 | 1,545 |
Fluticasone furoate-umeclidinium-vilanterol (Trelegy Ellipta) | 100/62.5/25 mcg | Inhalant powder (30 doses) | 137.6700 | 100/62.5/25 mcg once daily | 4.59 | 1,675 |
LAMA-LABA fixed-dose combinations | ||||||
Aclidinium-formoterol (Duaklir Genuair) | 400/12 mcg | Inhalant powder (60 doses) | 62.7224 | 400/12 mcg twice daily | 2.09 | 763 |
Indacaterol-glycopyrronium (Ultibro Breezhaler) | 110/50 mcg | Inhalant powder capsule | 2.5830 | 110/50 mcg daily | 2.58 | 943 |
Tiotropium-olodaterol (Inspiolto Respimat) | 2.5/2.5 mcg | Inhalation solution (60 doses) | 64.3740 | 5/5 mcg once daily | 1.07 | 392 |
Umeclidinium-vilanterol (Anoro Ellipta) | 62.5/25 mcg | Inhalant powder (30 doses) | 88.5100 | 62.5/25 mcg daily | 2.95 | 1,077 |
COPD = chronic obstructive pulmonary disease; ICS = inhaled corticosteroid; LABA = long-acting beta-agonist; LAMA = long-acting muscarinic antagonist; MDI = metered-dose inhaler.
Notes: All prices are from the Ontario Drug Benefit Formulary (accessed February 2025), unless otherwise indicated, and do not include dispensing fees.20 Recommended dosage are from respective product monographs, unless otherwise indicated. Calculations assume 365 days per year.
aSponsor submitted price.
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing. | Yes | No comment |
Model has been adequately programmed and has sufficient face validity. | No | CDA-AMC identified errors in the model that required the submission of an updated model. |
Model structure is adequate for decision problem. | Yes | No comment |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis). | Yes | No comment |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem. | Yes | No comment |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details). | No | Certain elements of the submitted Pharmacoeconomic Review report did not match what was in the model (i.e., utility values reported, wording about how the sponsor conducted a scenario analysis assuming “year prior to randomization” for baseline exacerbation rate, which was used in the base case, and so forth). |
CDA-AMC = Canada’s Drug Agency.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Figure 1: Model Structure — Decision Tree
COPD = chronic obstructive pulmonary disease.
Source: Sponsor’s pharmacoeconomic submission.2
Figure 2: Model Structure — Markov Model
COPD = chronic obstructive pulmonary disease.
Source: Sponsor’s pharmacoeconomic submission.2
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Dupilumab plus background therapy | Background therapy |
|---|---|---|
Discounted LYs | ||
Total LY | 9.305 | 7.758 |
LY – mild COPD | 0.541 | 0.062 |
No exacerbation | 0.136 | 0.153 |
Moderate exacerbation | 0.333 | 0.034 |
Severe exacerbation | 0.072 | 2.427 |
LY – moderate COPD | 3.315 | 0.403 |
No exacerbation | 1.193 | 1.622 |
Moderate exacerbation | 1.805 | 0.402 |
Severe exacerbation | 0.317 | 2.128 |
LY – severe COPD | 2.761 | 0.053 |
No exacerbation | 0.525 | 1.500 |
Moderate exacerbation | 1.789 | 0.575 |
Severe exacerbation | 0.447 | 2.954 |
LY – very severe COPD | 2.687 | 0.006 |
No exacerbation | 0.434 | 1.581 |
Moderate exacerbation | 1.317 | 1.366 |
Severe exacerbation | 0.937 | 0.249 |
Discounted QALYs | ||
Total QALY | 6.967 | 5.729 |
QALY – mild COPD | 0.439 | 0.202 |
No exacerbation | 0.111 | 0.051 |
Moderate exacerbation | 0.270 | 0.124 |
Severe exacerbation | 0.058 | 0.027 |
QALY – moderate COPD | 2.604 | 1.900 |
No exacerbation | 0.947 | 0.316 |
Moderate exacerbation | 1.411 | 1.270 |
Severe exacerbation | 0.246 | 0.314 |
QALY – severe COPD | 2.110 | 1.620 |
No exacerbation | 0.409 | 0.041 |
Moderate exacerbation | 1.365 | 1.143 |
Severe exacerbation | 0.336 | 0.436 |
QALY – very severe COPD | 1.846 | 2.043 |
No exacerbation | 0.309 | 0.004 |
Moderate exacerbation | 0.903 | 1.097 |
Severe exacerbation | 0.634 | 0.942 |
Adverse event utility decrement | 0.000 | 0.000 |
Caregiver utility decrement | 0.000 | 0.000 |
CV event utility decrement | −0.031 | −0.036 |
Discounted costs ($) | ||
Total costs | 239,377 | 159,509 |
Drug acquisition costs | 113,117 | 11,970 |
Drug administration costs | 66 | 0 |
Adverse events | 41 | 32 |
Exacerbation management | 116,768 | 138,722 |
COPD management | 7,560 | 6,972 |
Indirect costs | 0 | 0 |
CV event costs | 1,825 | 1,813 |
Respiratory failure cost | 0 | 0 |
COPD = chronic obstructive pulmonary disease; CV = cardiovascular; LY = life-year; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.2
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CDA-AMC Base Case
Table 11: Disaggregated Summary of the CDA-AMC Economic Evaluation Results
Parameter | Dupilumab plus background therapy | Background therapy |
|---|---|---|
Discounted LYs | ||
Total LY | 9.812 | 9.589 |
LY – mild COPD | 0.291 | 0.297 |
No exacerbation | 0.103 | 0.117 |
Moderate exacerbation | 0.156 | 0.148 |
Severe exacerbation | 0.032 | 0.032 |
LY – moderate COPD | 3.051 | 2.857 |
No exacerbation | 1.233 | 0.707 |
Moderate exacerbation | 1.517 | 1.772 |
Severe exacerbation | 0.301 | 0.378 |
LY – severe COPD | 2.621 | 2.516 |
No exacerbation | 0.464 | 0.181 |
Moderate exacerbation | 1.665 | 1.701 |
Severe exacerbation | 0.492 | 0.634 |
LY – very severe COPD | 3.849 | 3.919 |
No exacerbation | 0.428 | 0.019 |
Moderate exacerbation | 1.726 | 2.070 |
Severe exacerbation | 1.695 | 1.831 |
Discounted QALYs | ||
Total QALY | 7.313 | 7.133 |
QALY – mild COPD | 0.236 | 0.241 |
No exacerbation | 0.085 | 0.096 |
Moderate exacerbation | 0.126 | 0.119 |
Severe exacerbation | 0.025 | 0.025 |
QALY – moderate COPD | 2.403 | 2.252 |
No exacerbation | 0.979 | 0.561 |
Moderate exacerbation | 1.190 | 1.395 |
Severe exacerbation | 0.235 | 0.296 |
QALY – severe COPD | 2.008 | 1.929 |
No exacerbation | 0.361 | 0.141 |
Moderate exacerbation | 1.275 | 1.305 |
Severe exacerbation | 0.373 | 0.483 |
QALY – very severe COPD | 2.675 | 2.726 |
No exacerbation | 0.304 | 0.013 |
Moderate exacerbation | 1.200 | 1.444 |
Severe exacerbation | 1.172 | 1.269 |
Adverse event utility decrement | 0.000 | 0.000 |
Caregiver utility decrement | 0.000 | 0.000 |
CV event utility decrement | −0.011 | −0.015 |
Discounted costs ($) | ||
Total costs | 200,137 | 70,748 |
Drug acquisition costs | 150,500 | 14,855 |
Drug administration costs | 65 | 0 |
Adverse events | 44 | 39 |
Exacerbation management | 42,354 | 48,505 |
COPD management | 5,100 | 5,083 |
Indirect costs | 0 | 0 |
CV event costs | 2,074 | 2,266 |
Respiratory failure cost | 0 | 0 |
COPD = chronic obstructive pulmonary disease; CV = cardiovascular; LY = life-year; QALY = quality-adjusted life-year.
Scenario Analyses
Table 12: Scenario Analyses Conducted on the CDA-AMC Base Case
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | Background therapy | 160,701 | 5.74 | Reference |
Dupilumab plus background therapy | 240,458 | 6.98 | 64,601 | |
CDA-AMC base case (probabilistic) | Background therapy | 70,748 | 7.13 | Reference |
Dupilumab plus background therapy | 200,137 | 7.31 | 720,337 | |
CDA-AMC Scenario 1: baseline exacerbation informed by RWE | Background therapy | 71,669 | 7.07 | Reference |
Dupilumab plus background therapy | 200,933 | 7.25 | 694,431 | |
CDA-AMC Scenario 2: No excess mortality associated with exacerbations | Background therapy | 84,692 | 8.06 | Reference |
Dupilumab plus background therapy | 221,039 | 8.14 | 1,783,873 | |
CDA-AMC Scenario 3: No mortality | Background therapy | 107,465 | 9.29 | Reference |
Dupilumab plus background therapy | 252,260 | 9.32 | 5,373,844 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; RWE = real-world evidence.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 13: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
CDA-AMC = Canada’s Drug Agency; COPD = chronic obstructive pulmonary disease.
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a budget impact analysis to estimate the 3-year budget impact of reimbursing dupilumab as an add-on background therapy in adult patients with uncontrolled COPD associated with type 2 inflammation. The analysis was taken from the perspective of the public drug plan in Canada. A 3-year time horizon was used from 2026 to 2028 (with 2025 as the baseline year). The target population size was derived with an epidemiological approach based on published literature and a sponsor submitted data. Key inputs to the budget impact analysis are documented in Table 14.
Key assumptions:
Growth rate for the COPD population is equal to that of the general population of Canada.
Prevalence of COPD in patients aged 34 years and younger is 0%.
Background therapy was defined as ISC + LAMA + LABA.
Group E is defined as those with uncontrolled disease (i.e., a history of ≥ 2 moderate exacerbations or ≥ 1 exacerbation leading to hospitalization in the previous year).
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Pan-Canadian population | 18,403,05921 |
Prevalence of COPD | 8.7%22 |
Patients treated, % | 83.2%23 |
Patients with GOLD 2 or GOLD 3, % | 63.0%24 |
Patients with GOLD group E, % | 29.3%25 |
Patients receiving triple therapy or LAMA-LABA, % | 45.9%25 |
Patients with type 2 inflammation, % | 71.8%25 |
Patients with public coverage, % | 83.8% |
Number of patients eligible for drug under review | 59,304 / 60,205 / 61,120 |
Market uptake (3 years) | |
Uptake (reference scenario) Background therapy | 100% / 100% / 100% |
Uptake (new drug scenario) Dupilumab + background therapy Background therapy | 4.5% / 11.5% / 19.5% 95.5% / 88.5% / 80.5% |
Cost of treatment (per patient annually) | |
Dupilumab + background therapy Background therapy | $25,534 $0 |
COPD = chronic obstructive pulmonary disease; GOLD = Global Initiative for Chronic Obstructive Lung Disease; LABA = long-acting beta-agonist; LAMA = long-acting muscarinic antagonist.
Summary of the Sponsor’s Budget Impact Analysis Results
In the sponsor’s base-case analysis, the estimated incremental budget impact of funding dupilumab as an add-on to background therapy for the treatment of adult patients with uncontrolled COPD associated with type 2 inflammation was $68,140,419 in year 1, $176,783,503 in year 2, and $304,319,733 in year 3. Therefore, the 3-year budget impact was $549,243,655.
CDA-AMC Appraisal of the Sponsor’s Budget Impact Analysis
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the budget impact analysis:
Uncertainty in the sponsor patient population estimate. In the sponsor’s base-case analysis, the number of patients eligible for treatment was derived using an epidemiological approach based on published literature and sponsor-submitted data. CDA-AMC noted that the sponsor reported and used a value of 83.2% to inform the number of patients receiving COPD medication (informed by Haughney et al.23); however, when validating the estimate CDA-AMC found that Haughney et al.23 reported the proportion of patients receiving any COPD medication was 87.4%.
Finally, to inform the proportion of patients covered by public plans, the sponsor used estimates derived from data from IQVIA claims. While CDA-AMC notes that this approach is likely reasonable, the sponsor did not provide a technical document outlining the methodology and data used. Therefore, CDA-AMC conducted a scenario analysis using estimates from Sutherland and Dihn (2017) to explore the uncertainty in the sponsor’s patient population.26
Market share estimates for dupilumab as an add-on to background therapy is uncertain. In the sponsor’s base-case analysis, the market uptake of dupilumab as an add-on to background therapy was assumed to be 4.5%, 11.5%, and 19.5% in year 1, year 2, and year 3, respectively. These estimates were informed by the sponsor’s internal market share estimates and validated by clinical expert feedback received by the sponsor. Clinical expert feedback received by CDA-AMC noted that these estimates may be reasonable based on current clinical practice; however, it is anticipated that more patients will be referred to a specialist upon dupilumab reimbursement and therefore the uptake of dupilumab may be higher. To assess the impact of this on the budget impact, CDA-AMC conducted a scenario analysis assuming the uptake of dupilumab would be twice as high.
CDA-AMC conducted a scenario analysis assuming higher uptake of dupilumab based on the assumption that more patients may gain access to specialists upon dupilumab reimbursement.
CDA-AMC Reanalyses of the Budget Impact Analysis
CDA-AMC revised the sponsor’s submitted analysis by modifying the proportion of patients estimated to receive a COPD medication. The changes made to derive the CDA-AMC base case are described in Table 15.
Table 15: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Patients receiving COPD medication, % | 83.2% | 87.4% |
CDA-AMC base case | Reanalysis 1 | |
CDA-AMC = Canada’s Drug Agency; COPD = chronic obstructive pulmonary disease.
The results of the CDA-AMC step-wise reanalyses are presented in summary format in Table 16 and a more detailed breakdown is presented in Table 17. In the CDA-AMC base-case analysis, the estimated incremental budget impact of funding dupilumab as an add-on to background therapy for adult patients with uncontrolled COPD associated with type 2 inflammation was $71,498,300 in year 1, $185,495,190 in year 2, and $319,316,259 in year 3. Therefore, the 3-year budget impact is $576,309,748.
Table 16: Summary of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 549,243,655 |
CDA-AMC reanalysis 1 | 576,309,748 |
CDA-AMC base case | 576,309,748 |
CDA-AMC = Canada’s Drug Agency.
CDA-AMC anticipates that the budget impact of dupilumab as an add-on to background therapy will be sensitive to inputs and assumptions related to the proportion of patients eligible for treatment along with the market share of dupilumab. This was reflected in a scenario analysis conducted by CDA-AMC that explored the impact of alternative proportion of patients eligible for public coverage and increased market uptake of dupilumab if reimbursed.
Results of the CDA-AMC scenario using the proportion of patients eligible for public coverage from Sutherland and Dihn, (2017)25 found that compared to the sponsor’s estimates, the three-year budget impact would decrease to $473,859,238. Additionally, when exploring a higher uptake of dupilumab as an add-on to background therapy, the results suggest that if the market share was doubled compared to the sponsor’s estimates in year 1, 2, and 3, then the three-year budget impact would increase to $1,153,939,794. Results of the analysis can be found in Table 16.
Table 17: Detailed Breakdown of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 68,140,419 | 176,783,503 | 304,319,733 | 549,243,655 | |
Budget impact | 0 | 68,140,419 | 176,783,503 | 304,319,733 | 549,243,655 | |
CDA-AMC base case | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 71,498,300 | 185,495,190 | 319,316,259 | 576,309,748 | |
Budget impact | 0 | 71,498,300 | 185,495,190 | 319,316,259 | 576,309,748 | |
CDA-AMC scenario analysis 1: Proportion of patients eligible for public coverage | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 58,788,056 | 152,519,733 | 262,551,448 | 473,859,238 | |
Budget impact | 0 | 58,788,056 | 152,519,733 | 262,551,448 | 473,859,238 | |
CDA-AMC scenario analysis 2: Increased Market Uptake of Dupilumab | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 143,160,399 | 371,415,340 | 639,364,055 | 1,153,939,794 | |
Budget impact | 0 | 143,160,399 | 371,415,340 | 639,364,055 | 1,153,939,794 |
CDA-AMC = Canada’s Drug Agency.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.