Drugs, Health Technologies, Health Systems
Reimbursement Review
Pegunigalsidase Alfa (Elfabrio)
Sponsor: Chiesi Canada Corp.
Therapeutic area: Fabry disease
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
ACEi
angiotensin-converting enzyme inhibitor
ADA
antidrug antibody
ARB
angiotensin receptor blocker
BPI
Brief Pain Inventory
CDA-AMC
Canada’s Drug Agency
CFDI
Canadian Fabry Disease Initiative
CI
confidence interval
CKD
chronic kidney disease
CKD-EPI
Chronic Kidney Disease Epidemiology Collaboration
CrI
credible interval
eGFR
estimated glomerular filtration rate
ERT
enzyme replacement therapy
FD
Fabry disease
Gb3
globotriaosylceramide
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
Ig
immunoglobulin
IRR
infusion-related reaction
ITC
indirect treatment comparison
ITT
intention to treat
LVH
left ventricular hypertrophy
LVM
left ventricular mass
LVMI
Left Ventricular Mass Index
lyso-Gb3
globotriaosylsphingosine
MID
minimal important difference
MSSI
Mainz Severity Score Index
NMA
network meta-analysis
OLE
open-label extension
PAIC
population-adjusted indirect comparison
RCT
randomized controlled trial
RWE
real-world evidence
SAE
serious adverse event
SD
standard deviation
SE
standard error
SLR
systematic literature review
STC
simulated treatment comparison
TEAE
treatment-emergent adverse event
TEM
treatment effect modifier
TIA
transient ischemic attack
UPCR
urine protein-to-creatinine ratio
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Pegunigalsidase alfa (Elfabrio), 2 mg/mL, concentrate for solution (20 mg/10 mL and 5 mg/2.5 mL vial) for IV infusion |
Sponsor | Chiesi Canada Corp. |
Indication | Proposed: long-term enzyme replacement therapy in adult patients with a confirmed diagnosis of Fabry disease (deficiency of alpha-galactosidase) |
Reimbursement request | Per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | December 9, 2025 |
Recommended dosage | 1 mg/kg administered by IV infusion every 2 weeks, based on actual body weight |
NOC = Notice of Compliance.
Introduction
Fabry disease (FD) is a rare, progressive, X-linked, lysosomal storage disorder caused by a deficiency of the lysosomal enzyme alpha-galactosidase A due to a GLA variant. This deficiency leads to the progressive accumulation of glycolipids (mainly globotriaosylceramide [Gb3] and globotriaosylsphingosine [lyso-Gb3]), resulting in metabolic dysfunction, cellular death, and, eventually, progressive vital organ disease.1-4 FD is characterized by the development of hypertrophic cardiomyopathy, nephropathy with chronic renal failure, and stroke, and these conditions contribute to reduced health-related quality of life (HRQoL).5 Classic FD is the more severe form of the disease, characterized by severely reduced (< 5% of mean normal) alpha-galactosidase A activity in males; people with the nonclassic form of FD have residual alpha-galactosidase A activity that varies between 5% and 30% of normal levels.1,6 Hemizygous people tend to have a more severe phenotype, while heterozygotic people tend to have a milder phenotype. There is marked heterogeneity in clinical features in both sexes and among affected relatives. Patients with classic FD experience more severe disease, with early signs and symptoms manifesting during childhood and adolescence and including neuropathic pain, autonomic dysfunction, bloating, diarrhea, abdominal pain, angiokeratomas, hypohidrosis, and corneal opacity (cornea verticillata). Patients with nonclassic FD typically have milder disease and delayed onset of clinical manifestations, with symptoms emerging between the ages of 30 and 70 years.1,6,7 These patients tend to have single-system involvement, such as chronic kidney disease (CKD) or, more frequently, cardiac disease, with only mild findings in other organs. The global prevalence of FD is estimated to be between 1 in 117,000 to 1 in 37,000 live male births for classic FD.8-13 Classic phenotype FD has an estimated incidence of 1 in 50,000 males.11 The frequency of FD has been estimated in 1 study to be 1 in 117,000 in females and 1 in 40,000 in males.14 Published estimates on the prevalence and incidence of FD in Canada were not available, but the clinical experts consulted for this review estimated the prevalence to be between 1 in 74,000 and 1 in 50,000 in Canada. The testing requirements to confirm a diagnosis of FD (alpha-galactosidase A activity assay and genetic testing) are well established in Canada, are outlined in the Canadian Fabry Disease Treatment Guidelines, and involve the synthesis of clinical, biochemical, molecular, and pathologic criteria.5
According to the clinical experts consulted for this review, the goals of treatment are to delay heart failure, slow decline in kidney function (to –1 mL/min/1.73 m2 per year, the same as that of the general population), prevent stroke and transient ischemic attack (TIA), control pain and gastrointestinal symptoms, and improve HRQoL. Per the 2018 Canadian Fabry Disease Treatment Guidelines, it is only once an indication for treatment is confirmed that the choice of therapy is considered.5 The 2 types of disease-specific treatments for FD are enzyme replacement therapy (ERT) and pharmacologic chaperone therapy, but neither of these is completely effective.15 Agalsidase alfa and agalsidase beta are recombinant human ERTs, and the deficient enzyme (alpha-galactosidase A) is provided by IV infusion. Patients who have an indication for disease-specific therapy can receive ERT, regardless of the genetic variant.5 Migalastat is an oral chaperone therapy that binds and stabilizes specific forms of the enzyme with downstream effects of increased activity; therefore, migalastat is indicated only for patients with an amenable genetic variant (found in an estimated 35% of the patients with FD in Canada, as per the clinician group that provided input for this review).16 Identifying amenability to migalastat has been the main factor influencing the decision between treatment with an ERT and chaperone therapy. Other considerations include the method of administration, disease severity, and the potential for developing antidrug antibodies (ADAs) with ERT.5 Disease-specific therapies are aimed at treating early stages of disease progression (when there is evidence of end-organ involvement), rather than preventing disease before signs occur.5 Patients with FD may also be treated with nonspecific therapies to address pain and renal, cardiac, neurologic, or gastrointestinal disease.15
Pegunigalsidase alfa is a pegylated recombinant form of the human alpha-galactosidase A enzyme and is used to supplement or replace alpha-galactosidase A in patients with FD.17 The recommended dose is based on actual body weight at 1 mg/kg, administered by IV every 2 weeks. Pegunigalsidase alfa is currently undergoing a Health Canada review, and the proposed indication is for long-term ERT in adult patients with a confirmed diagnosis of FD (deficiency of alpha-galactosidase).
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of pegunigalsidase alfa, 2 mg/mL, for IV infusion in the treatment of FD in adults.
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of the input provided by the patient and clinician groups who responded to the call for input from Canada’s Drug Agency (CDA-AMC) and from the clinical experts consulted for the purpose of this review.
Patient Input
CDA-AMC received submissions from 1 patient group, the Canadian Fabry Association, and individual patients. Patient feedback was collected through 3 individual testimonials and semistructured interviews to learn from patients’ lived experience. During the interviews, patients reported positive impacts from ERTs and chaperone therapy. Patients described having more energy, fewer episodes of pain crisis, less gastrointestinal pain, and an ability to carry out everyday life activities. Overall, patients reported a reduction in their symptoms and stabilization of cardiac and renal disease.
Patients reported concerns about receiving ERT, including infusion-related reactions (IRRs), adverse events (e.g., nausea, fatigue, chills, fever), and the consequences of developing ADAs. According to the Canadian Fabry Association, important outcomes in the treatment of FD are less frequent infusions, improvement in IRRs, slowed disease progression, and prolonged and consistent symptom control.
Patients described the main unmet need in the treatment of FD as the need for additional treatments that slow disease progression, control symptoms, and are tolerable. Specifically for ERT, there is a need for drugs that are associated with fewer IRRs and have less risk of patients developing ADAs. Patients stated that while there is no cure for FD, they felt that pegunigalsidase alfa could improve patient HRQoL and health outcomes.
Clinician Input
Input From Clinical Experts Consulted for This Review
According to the clinical experts, although there are disease-specific treatments available for FD, not all patients experience a satisfactory response to ERT or chaperone therapy and there are no curative treatments. Males with classic disease tend to experience disease progression despite treatment, and the experts indicated that 1% to 5% of this subpopulation develop ADAs that make ERT futile. Other issues include the high cost of ERT and migalastat and the burden associated with ERT, which typically involves biweekly IV infusions that can last a few hours and have the potential for IRRs (estimated to occur in up to 30% of patients). Moreover, some symptoms (fatigue, neuropathic pain, gastrointestinal symptoms, hearing loss, and vertigo) have no treatment, can be difficult to treat, or are slow to respond to therapy. The experts stated that there is a need for treatment that is more effective, has less burdensome administration (e.g., oral drug, less frequent infusions), works for all forms of FD regardless of genetic variant, has few adverse reactions (including a low incidence of ADA development), and crosses the blood-brain barrier.
According to the clinical experts, pegunigalsidase alfa would be used as a first-line therapy, similar to other ERTs available, and is not expected to change the treatment paradigm for FD. The experts also indicated that it would not be necessary for patients to try other treatments before accessing pegunigalsidase alfa and that the drug would not be reserved for patients in whom other treatments are contraindicated or not tolerated. It was also noted that specific therapies for FD are not combined due to cost and other factors, despite the theoretical potential for better outcomes through using multiple mechanisms of action.
The clinical experts stated that adults with a confirmed FD diagnosis who have a clear indication for disease-specific therapy could receive pegunigalsidase alfa. Indications for treatment include evidence of kidney or heart disease, stroke, intractable neuropathic pain, or gastrointestinal symptoms. The experts noted that treatment eligibility for FD in Canada is largely, but not exclusively, determined by the Canadian Fabry Disease Treatment Guidelines and the Canadian Fabry Disease Initiative (CFDI) steering committee.
According to the experts, patients with severe disease are most in need of therapy. These patients tend to be males and may have limited or no residual enzyme activity, elevated plasma lyso-Gb3 levels, a severe GLA variant, older age (> 50 years), a low estimated glomerular filtration rate (eGFR) with advanced renal disease, proteinuria of 1.0 g/day or greater, advanced cardiac disease with myocardial fibrosis, dysrhythmias, stroke, and/or high-titre neutralizing ADAs.
Response to treatment may be assessed through stabilization of eGFR slope, CKD stage, or Left Ventricular Mass Index (LVMI) score; reduction in the rate of Fabry clinical events or Fabry Stabilization Index score and improvement in symptoms; increased exercise tolerance; and improved work or school attendance and survival. According to the experts, a clinically meaningful response includes a decrease in the Fabry clinical event rate; a decrease in pain by 1 to 2 points on a 7-point Likert scale; and stabilization of eGFR, CKD stage, left ventricular mass (LVM), and New York Heart Association class. Other indicators of treatment response are normalized sweating, return of normal gastrointestinal function, prevention of stroke, and improved HRQoL.
Because FD is a slowly progressing disease, the experts indicated that patients should be followed for at least 2 to 3 years before judging the degree of treatment response and whether the disease is stable or progressing. They also noted that treatment response to ERT varies depending on patient characteristics (e.g., age, sex, disease phenotype) and that the benefits of ERT may take a year or longer to be realized. In clinical practice, patients are typically seen every 6 to 12 months to monitor treatment and clinical changes, although children and young females who are asymptomatic with normal organ function may be assessed less frequently.
According to the Canadian Fabry Disease Treatment Guidelines, discontinuation of disease-specific treatment should be considered in a patient whose disease is not responding to treatment after at least 1 year, who has persistent and severe IRRs (despite prophylaxis) or IgE ADAs, or whose life expectancy is estimated to be less than 1 year. Other reasons for discontinuation include permanent and severe neurocognitive decline, severe reduction in HRQoL or functional status, lack of response to therapy for the organ involvement that mandated the initiation of treatment, poor adherence, and elevated plasma lyso-Gb3 levels that do not decline. The experts also discussed practical aspects that should be considered, such as the patient’s age and the burden of ERT compared to the benefit received from treatment. One expert stated that even though there are guidelines and suggested criteria for stopping treatment, these discussions are difficult once a patient has begun treatment and wants to continue. They also stated that there are older adults who are not eligible for (but would prefer) oral migalastat and decline ERT due to the burden of IV infusions.
Most patients in Canada receive ERT in an outpatient setting, either at home or at an infusion clinic. The experts stated that private nursing services that provide outpatient infusions are paid for by the ERT manufacturer. As with other ERTs, it is recommended that the first 6 to 10 infusions of pegunigalsidase alfa are administered in a hospital outpatient department or infusion clinic due to the risk of IRRs. Once deemed appropriate, most patients can receive at-home infusions.
Various specialists are involved in the diagnosis, treatment, and monitoring of FD, including medical geneticists, nephrologists, cardiologists, neurologists, general internists, pediatricians, and metabolic specialists. The experts stated that patients are best served in a centre of excellence where FD specialists are available. The experts also noted that the associated costs of infusions, ADA testing, and lyso-Gb3 monitoring should be covered by the sponsor or the drug plans because the current lack of coverage poses a significant barrier to effective FD care.
Clinician Group Input
Four clinicians on behalf of the CFDI submitted written input. The clinicians presented their assessment of peer-reviewed studies on FD, information from the CFDI registry, and input based on their professional expertise.
The group described the main unmet needs in the treatment of FD as the need for a new treatment with greater clinical efficacy than existing treatment and the need to reduce the frequency of ERT infusions. According to the clinician group, the treatment goals for FD are delaying dialysis and heart failure. The clinician group noted that there are other manifestations of the disease that are difficult to target with drugs, including abdominal pain, neuropathic pain, stroke, and mental health problems. In addition, cardiac disease continues to progress in people with FD, leading to high rates of atrial fibrillation and other arrhythmias, heart failure, and chest pain.
If pegunigalsidase alfa were available in Canada, the clinician group believed it would provide an alternative form of ERT at the 1 mg/kg dose, but the clinicians did not expect it to impact how treatment decisions are made. They noted that the drug may prove useful for some adult males with high-titre neutralizing ADAs who have reduced response to therapy.
The clinician group noted that reasons to stop treatment with pegunigalsidase alfa are outlined in the treatment guidelines; these reasons are consistent with those described by the clinical experts consulted for this review.
The clinician group recommended that FD be managed by a specialist with experience with the disease, which was consistent with the recommendation of the clinical experts consulted for this review.
Drug Program Input
The drug programs had comments to inform Canadian Drug Expert Committee deliberations relating to relevant comparators, considerations for the initiation and continuation of the drug under review, and system and economic issues. The programs had a question about whether patients younger than 18 years or older than 60 years (i.e., outside the eligible age range for the BALANCE trial) would be eligible for pegunigalsidase alfa. The clinical experts agreed that adult patients (no upper age limit) could receive treatment with pegunigalsidase alfa for FD. One expert indicated that patients younger than 18 years could also receive pegunigalsidase alfa for FD, while another expert noted that there would first need to be adequate evidence of safety and efficacy in a pediatric population before pediatric patients are treated with the drug. The proposed Health Canada indication is for adult patients with a confirmed diagnosis of FD.
Clinical Evidence
Systematic Review
Description of Studies
One phase III, double-blind, noninferiority, randomized controlled trial (RCT) (BALANCE trial; N = 78) evaluated the efficacy and safety of pegunigalsidase alfa (n = 53) compared to agalsidase beta (n = 25), both at a dosage of 1 mg/kg every 2 weeks, in adult patients with FD.18 The BALANCE trial was originally designed to test the noninferiority of pegunigalsidase alfa to agalsidase beta at the 12-month interim analysis and the superiority of the former at the 24-month final analysis. After agalsidase beta was given full market authorization in March 2021, and in consultation with the FDA (September 9, 2021), demonstration of superiority after 24 months was no longer required and noninferiority was assessed at the final analysis at a noninferiority margin for eGFR according to the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) formula of –3 mL/min/1.73 m2 per year. Efficacy was assessed through the primary end point of the median annualized change (slope) in CKD-EPI eGFR over the 2-year study period; other clinically relevant outcomes included incidence of Fabry clinical events, LVMI score, and measures of pain. Other outcomes relating to kidney health, exercise tolerance, pain medication use, plasma lyso-Gb3 concentration, and patient-reported outcomes were included as supportive evidence (Grading of Recommendations Assessment, Development and Evaluation [GRADE] was not applied).
The BALANCE trial only included adults with confirmed FD and reduced kidney function who had at least 1 year of treatment (stable dose for at least 6 months) with agalsidase beta, 1 mg/kg, every 2 weeks, before the study. The mean age of the patients was 43.9 years (standard deviation [SD] = 10.2 years) in the pegunigalsidase alfa group and 45.2 years (SD = 9.6 years) in the agalsidase beta group. There were more males than females in both treatment groups (by study design), and more than half the patients had classic disease. A larger proportion of patients in the pegunigalsidase alfa group (36.5%) than in the agalsidase beta group (24.0%) had an eGFR slope greater than –5 mL/min/1.73 m2 per year at baseline (i.e., had slower decline in kidney function), and proportionately fewer patients in the pegunigalsidase alfa group than in the agalsidase beta group were on angiotensin-converting enzyme inhibitors (ACEis) or angiotensin receptor blockers (ARBs) (50.0% versus 64.0%).
Efficacy Results
Incidence of Fabry Clinical Events
Fabry clinical events were classified into 4 categories: renal events (first occurrence of either initiation of dialysis or chronic dialysis [> 40 days] or renal transplant); cardiac events (cardiac-related death, myocardial infarction, first-time congestive heart failure, atrial fibrillation, ventricular tachycardia, evidence of progressive heart disease severe enough to require a pacemaker, implantation of pacemaker, bypass surgery, coronary artery dilatation, implantation of defibrillator); cerebrovascular events (hemorrhagic or ischemic stroke or TIA); and death due to non–cardiac-related causes.19 The 4 categories were combined into a composite end point: overall Fabry clinical events.
In the study, 9 patients (17.3%) in the pegunigalsidase alfa group and 2 patients (8.0%) in the agalsidase beta group experienced a Fabry clinical event. In the pegunigalsidase alfa group, 6 patients had 7 cardiac events (atrial fibrillation, angina pectoris, increased troponin, and second-degree atrioventricular block), 3 patients had 3 cerebrovascular events (TIA and cerebrovascular accident), and 1 patient had a renal event (end-stage renal disease necessitating a kidney transplant). In the agalsidase beta group, 2 patients had 2 cardiac events (ventricular tachycardia requiring a pacemaker and atrial fibrillation)
Annualized Change in CKD-EPI eGFR (Slope)
The median CKD-EPI eGFR slope was –2.51 mL/min/1.73 m2 per year (95% confidence interval [CI], –3.79 mL/min/1.73 m2 per year to –1.24 mL/min/1.73 m2 per year) in the pegunigalsidase alfa group and –2.16 mL/min/1.73 m2 per year (95% CI, –3.81 mL/min/1.73 m2 per year to –0.51 mL/min/1.73 m2 per year) in the agalsidase beta group. The treatment group difference was –0.36 mL/min/1.73 m2 per year (95% CI, –2.44 mL/min/1.73 m2 per year to 1.73 mL/min/1.73 m2 per year) for the intention-to-treat (ITT) population and –0.12 mL/min/1.73 m2 per year (95% CI, –2.45 mL/min/1.73 m2 per year to 2.21 mL/min/1.73 m2 per year) for the per-protocol population. Because the lower bound of the 95% CI for the treatment group difference was greater than the prespecified noninferiority margin of –3.0 mL/min/1.73 m2 per year, the sponsor concluded that pegunigalsidase alfa was noninferior to agalsidase beta.
LVMI Score
One of the cardiac complications of FD is thickening of the left ventricular wall. The LVMI score was calculated based on the LVM, as determined by MRI.
At baseline, the mean LVMI score was 75.97 g/m2 (standard error [SE] = 5.13 g/m2) in the pegunigalsidase alfa group and 82.22 g/m2 (SE = 6.34 g/m2) in the agalsidase beta group. At week 104, the mean LVMI score was 71.56 g/m2 (SE = 5.2 g/m2) in the pegunigalsidase alfa group and 82.43 g/m2 (SE = 8.39 g/m2) in the agalsidase beta group. The mean change from baseline was –0.64 g/m2 (SE = 2.69 g/m2) in the pegunigalsidase alfa group and 0.29 g/m2 (SE = 3.73 g/m2) in the agalsidase beta group. The difference between treatment groups was –0.92 g/m2 (95% CI, –10.26 g/m2 to 8.42 g/m2).
Short-Form Brief Pain Inventory Score
For the short-form Brief Pain Inventory (BPI) score, patients rate the severity of their pain from 0 to 10 points (from “no pain” to “worst imaginable pain”) and how the pain impacts their general activity, mood, walking, working, sleeping, relations with other people, and enjoyment of life from 0 to 10 (from “does not interfere” to “completely interferes”) for 9 pain-related questions.20 Scores are generated for pain at its worst in the last 24 hours, pain at its least in the last 24 hours, pain right now, and pain on average. Scores of 1 to 4 points indicate mild pain, of 5 or 6 points indicate moderate pain, and of 7 to 10 points indicate severe pain.
The mean change from baseline to week 104 for pain at its worst was –0.1 points (SE = 0.5 points) in the pegunigalsidase alfa group and 0.6 points (SE = 0.6 points) in the agalsidase beta group. The treatment group difference was –0.7 points (95% CI, –2.2 to 0.8 points).
The mean change from baseline to week 104 for pain at its least was 0.2 points (SE = 0.3 points) in the pegunigalsidase alfa group and 0.1 points (SE = 0.4 points) in the agalsidase beta group. The treatment group difference was 0.1 points (95% CI, –1.0 to 1.1 points).
The mean change from baseline to week 104 for current pain was 0.1 points (SE = 0.4 points) in the pegunigalsidase alfa group and 0.1 points (SE = 0.5 points) in the agalsidase beta group. The treatment group difference was –0.1 points (95% CI, –1.4 to 1.2 points).
The mean change from baseline to week 104 for average pain was 0.4 points (SE = 0.3 points) in the pegunigalsidase alfa group and 0.2 points (SE = 0.4 points) in the agalsidase beta group. The treatment group difference was 0.2 points (95% CI, –0.9 to 1.2 points).
Health-Related Quality of Life
There was no information on HRQoL in the BALANCE trial.
Other Efficacy Results
Plasma lyso-Gb3 concentration, urine protein-to-creatinine ratio (UPCR) category, Mainz Severity Score Index (MSSI) overall score, exercise tolerance (stress test) results, pain medication use, and EQ-5D-5L score were considered clinically relevant and were included as supporting evidence. The results are available in Table 32 of Appendix 1.
Harms Results
Treatment-emergent adverse events (TEAEs) were reported by 47 patients (90.4%) in the pegunigalsidase alfa group and 24 patients (96.0%) in the agalsidase beta group. The most common TEAEs were headache (21.2% of patients versus 20.0% of patients), nasopharyngitis (21.2% versus 16.0%), diarrhea (19.2% versus 24.0%), back pain (15.4% versus 20.0%), cough (11.5% versus 20.0%), and bronchitis (9.6% versus 20.0%). Eight patients (15.4%) in the pegunigalsidase alfa group reported 14 serious adverse events (SAEs), and 6 patients (24.0%) in the agalsidase beta group reported 11 SAEs. Two patients stopped treatment due to TEAEs (IRR and multiple events), and both were in the pegunigalsidase alfa group. There were no deaths in the study.
Six patients (11.5%) in the pegunigalsidase alfa group and 3 patients (12.0%) in the agalsidase beta group experienced an injection site reaction. Of the 11 patients (21.2%) reporting any IRR in the pegunigalsidase alfa group, 11 reported mild or moderate IRRs and 1 reported a severe or very severe IRR. Of the 6 patients (24.0%) reporting any IRR in the agalsidase beta group, all 6 reported mild or moderate IRRs. Six patients (11.5%) in the pegunigalsidase alfa group and 5 patients (20.0%) in the agalsidase beta group reported treatment-emergent ADAs. Of the 20 patients in the pegunigalsidase alfa group who had ADAs during the study, 15 (75.0%) had neutralizing ADAs. Of the 11 patients in the agalsidase beta group who had ADAs during the study, 9 (81.8%) had neutralizing ADAs.
Critical Appraisal
Randomization was stratified by baseline UPCR and no other characteristics, although there are many that impact FD outcomes. There were notable imbalances in the baseline characteristics between treatment groups, most of which point toward patients in the pegunigalsidase alfa group having less severe disease and better kidney function and toward biases in favour of the new drug. Imbalances may also be a result of the small study size (understandable given the rarity of the condition) and the heterogeneity of FD. The BALANCE trial was a noninferiority study aiming to determine if pegunigalsidase alfa was noninferior to agalsidase beta for the annualized change in CKD-EPI eGFR over 2 years at a noninferiority margin of –3.0 mL/min/1.73 m2 per year. The margin was based on natural history data for patients with FD and on European therapeutic goals.7,21 However, the FDA noted that the margin is unreliable for informing noninferiority due to the natural history data being from a clinically different population to the BALANCE trial and due to the methodology used in choosing the margin (i.e., not being based on preserving an established minimum treatment effect for agalsidase beta versus placebo in a population similar to that in the BALANCE trial).22 The FDA examined assay sensitivity using external data to support the expected treatment effect of agalsidase beta in the BALANCE trial population and to help contextualize the study results with an unreliable noninferiority margin; however, the noninferiority of pegunigalsidase alfa to agalsidase beta remains uncertain.22 Based on expert opinion and information from the literature, a smaller margin would be more appropriate; such a margin could be –1 mL/min/1.73 m2 per year, which is the eGFR slope of healthy populations aged 40 years and older (a treatment goal for patients with FD) and which has been suggested as a clinically meaningful difference in mean eGFR slopes for patients with CKD.23 There were differences between the 2 treatment groups in concomitant medication use, study withdrawals, and critical or major protocol deviations, all of which can bias toward a conclusion of noninferiority.24 However, the results for the ITT analyses (N = 77) and per-protocol analyses (N = 72) were similar, which supports the sponsor’s claim of noninferiority for the chosen margin. Although this was a noninferiority study, no noninferiority margins were provided for the secondary outcomes, and clinical meaningfulness was informed by clinical expert opinion or minimal important differences (MIDs) that have not been verified in FD. In the BALANCE trial, there were data missing for every outcome, there was no imputation for those data, and they were assumed to be missing at random, which is untestable and unlikely for all patients; this assumption would add further uncertainty into the results. The eGFR slope and the LVMI score, while objective outcomes, require years to produce a reliable estimate and vary based on patient and disease characteristics.
The BALANCE trial population does not represent the full spectrum of patients who could receive pegunigalsidase alfa in practice. There are pediatric and older adult patients who could be eligible for ERT but were not captured in the study. Additionally, not all patients with FD have kidney involvement (e.g., those with late-onset and cardiac variants), but they still need to be treated. All patients in the study had previously received treatment for FD and this study potentially informs treatment switching; however, this drug could be used in patients who have not previously received treatment for FD. There is also a risk of selection bias for patients who tolerate the clinical and nonclinical aspects of ERTs (premedication, travel to an infusion centre, IRRs, duration of infusion, and biweekly administration). The BALANCE trial eligibility criteria excluded patients with poorer health who might receive the drug in clinical practice in Canada, such as those receiving renal dialysis or transplant, those with proteinuria who are not receiving ACEi or ARB treatment, and those with cardiovascular events or heart failure. The proposed Health Canada indication is for adult patients with a confirmed diagnosis of FD and does not mention the need for patients to have symptoms of end-organ involvement, as noted in the Canadian Fabry Disease Treatment Guidelines.5 Input from patient and clinician groups highlighted the need for treatments that slow disease progression and improve clinical outcomes as well as FD symptoms and HRQoL. The BALANCE trial provides information on how pegunigalsidase alfa impacts Fabry clinical events, renal function, aspects of cardiac health, and pain, but there was a lack of evidence supporting the improvement of gastrointestinal issues and HRQoL. The BALANCE trial also does not provide information on the long-term use of pegunigalsidase alfa or its long-term comparability to other treatments.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for the outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined, as outlined by the GRADE Working Group.25,26
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The selection of outcomes for GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
incidence of Fabry clinical events
annualized change in CKD-EPI eGFR (slope)
LVMI score (g/m2), determined by MRI
short-form BPI score
IRRs and ADAs.
Table 2: Summary of Findings for Pegunigalsidase Alfa Versus Agalsidase Beta for Patients With FD
Outcome and follow-up | Patients (studies), N | Absolute effects | Certainty | What happens | ||
|---|---|---|---|---|---|---|
Agalsidase beta | Pegunigalsidase alfa | Difference | ||||
Fabry clinical events | ||||||
Incidence of overall Fabry clinical events, n Follow-up: 104 weeks | 77 (1 RCT) | 2 (8.0%) | 9 (17.3%) | 93 more per 1,000 (95% CI, 104 fewer per 1,000 to 243 more per 1,000) | Very lowa | The evidence is very uncertain about the effect of pegunigalsidase alfa on overall Fabry clinical events compared with agalsidase beta. |
Annualized CKD-EPI eGFR slope | ||||||
Annualized CKD-EPI eGFR slope (mL/min/1.73 m2 per year), median Follow-up: 104 weeks | 76 (1 RCT) | –2.16 (95% CI, –3.81 to –0.51) | –2.51 (95% CI, –3.79 to –1.24) | –0.36 (95% CI, –2.44 to 1.73) | Very lowb | The evidence is very uncertain about the effect of pegunigalsidase alfa on annualized CKD-EPI eGFR slope compared with agalsidase beta. |
LVMI score by MRI | ||||||
LVMI score (g/m2), mean Follow-up: 104 weeks | 47 (1 RCT) | 0.29 (SE = 3.73) | –0.64 (SE = 2.69) | –0.92 (95% CI, –10.26 to 8.42) | Very lowc | The evidence is very uncertain about the effect of pegunigalsidase alfa on LVMI score compared with agalsidase beta. |
Patient-reported outcomes | ||||||
Short-form BPI score for average pain (points): 0 (no pain) to 10 (worst imaginable pain) Follow-up: 104 weeks | 67 (1 RCT) | 0.2 (SE = 0.4) | 0.4 (SE = 0.3) | 0.2 (95% CI, –0.9 to 1.2) | Very lowd | The evidence is very uncertain about the effect of pegunigalsidase alfa on average pain based on the short-form BPI score compared with agalsidase beta. |
HRQoL | NA | NA | NA | NA | NA | There is no evidence for the effect of pegunigalsidase alfa on HRQoL. |
Harms | ||||||
Severe or very severe IRRs Follow-up: 104 weeks | 77 (1 RCT) | 0 per 1,000 patients | 19 per 1,000 patients | 19 more per 1,000 patients (95% CI, 122 fewer to 107 more per 1,000 patients) | Lowe | Pegunigalsidase alfa may result in an increase in the incidence of severe or very severe IRRs compared with agalsidase beta. The clinical importance of the increase is unclear. |
Neutralizing ADAs Follow-up: 104 weeks | 77 (1 RCT) | 818 per 1,000 patients | 750 per 1,000 patients | 68 fewer per 1,000 patients (95% CI, 360 fewer to 293 more per 1,000 patients) | Moderatef | Pegunigalsidase alfa likely results in a reduction in the incidence of neutralizing ADAs compared with agalsidase beta. The clinical importance of the reduction is unclear. |
ADA = antidrug antibody; BPI = Brief Pain Inventory; CI = confidence interval; CKD-EPI = Chronic Kidney Disease Epidemiology Collaboration; eGFR = estimated glomerular filtration rate; FD = Fabry disease; HRQoL = health-related quality of life; IRR = infusion-related reaction; LVMI = Left Ventricular Mass Index; NA = not applicable; RCT = randomized controlled trial; SE = standard error.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 1 level for study limitations (imbalances in baseline characteristics increase the potential for bias in the results, favouring pegunigalsidase alfa). Rated down 1 level for indirectness (all patients had experience with agalsidase beta before the BALANCE trial, and the Health Canada indication is not restricted to patients who have previously received treatment; no patients > 60 years were included; all patients had to have evidence of kidney function decline, which is not a characteristic of all patients with FD who require treatment; patients with poorer health who would be treated in practice were excluded). Rated down 1 level for imprecision (there was no known threshold for a clinically important effect, and the presence of an important effect was informed by expert opinion for this review; the results are difficult to interpret due to the small number of events, the wide CI, and the 2-year follow-up, which is relatively short for clinical outcomes that may take years to develop). The analysis of this outcome was not adjusted for multiplicity, and the results are considered supportive evidence.
bRated down 1 level for study limitations (imbalances in baseline characteristics increase the potential for bias in the results, favouring pegunigalsidase alfa). Rated down 1 level for indirectness (all patients had experience with agalsidase beta before the BALANCE trial, and the Health Canada indication is not restricted to patients who have previously received treatment; no patients > 60 years were included; all patients had to have evidence of kidney function decline, which is not a characteristic of all patients with FD who require treatment; patients with poorer health who would be treated in practice were excluded). Rated down 1 level for imprecision. (Noninferiority was reached based on the sponsor-suggested noninferiority margin. However, regulatory agencies have rejected the suggested noninferiority margin and indicated that a smaller margin would be more appropriate.22,27 Based on expert opinion and evidence from the literature,23 a noninferiority margin or meaningful difference of –1 mL/min/1.73 m2 per year would indicate that pegunigalsidase alfa is potentially meaningfully worse or better than agalsidase beta, based on the lower and upper bounds of the 95% CI.)
cRated down 1 level for study limitations (imbalances in baseline characteristics increase the potential for bias in the results, favouring pegunigalsidase alfa, and a notable amount of missing data biases toward a noninferiority conclusion). Rated down 1 level for indirectness (all patients had experience with agalsidase beta before the BALANCE trial, and the Health Canada indication is not restricted to patients who have previously received treatment; no patients > 60 years were included; patients with poorer health who would be treated in practice were excluded). Rated down 2 levels for imprecision. (There was no known threshold for a clinically important effect, and the presence of an important effect was informed by expert opinion for this review. Based on expert opinion for a clinically meaningful threshold, the bounds of the 95% CI suggest the possibility of both a meaningful improvement or worsening in LVMI score.) The analysis of this outcome was not adjusted for multiplicity, and the results are considered supportive evidence.
dRated down 1 level for study limitations (imbalances in baseline characteristics increase the potential for bias in the results, favouring pegunigalsidase alfa, and a notable amount of missing data biases toward a noninferiority conclusion). Rated down 1 level for indirectness (all patients had experience with agalsidase beta before the BALANCE trial, and the Health Canada indication is not restricted to patients who have previously received treatment; no patients > 60 years were included; patients with poorer health who would be treated in practice were excluded). Rated down 1 level for imprecision (the estimated threshold for a clinically important effect ranges from 1 to 2 points [but has not been verified in FD]; the bounds of the 95% CI approach or cross the estimated clinically meaningful threshold and suggest the possibility of both a meaningful improvement or worsening in average pain compared to agalsidase beta). The analysis of this outcome was not adjusted for multiplicity, and the results are considered supportive evidence.
eRated down 1 level for indirectness (all patients had experience with agalsidase beta before the BALANCE trial, and the Health Canada indication is not restricted to patients who have previously received treatment; no patients older than 60 years were included; all patients had to have evidence of kidney function decline, which is not a characteristic of all patients with FD who require treatment; patients with poorer health who would be treated in practice were excluded). Rated down 1 level for imprecision (there was no known threshold for a clinically important effect, and the presence of an important effect was informed by expert opinion for this review; few events were observed, and there were inadequate data to inform a higher certainty judgment). The analysis of this outcome was not adjusted for multiplicity, and the results are considered supportive evidence.
fRated down 1 level for indirectness (all patients had experience with agalsidase beta before the BALANCE trial, and the Health Canada indication is not restricted to patients who have previously received treatment; no patients > 60 years were included; patients with poorer health who would be treated in practice were excluded). The analysis of this outcome was not adjusted for multiplicity, and the results are considered supportive evidence.
Source: Clinical Study Report for BALANCE trial;18 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Long-Term Extension Studies
Description of Studies
Additional studies submitted by the sponsor provided evidence on patients who had not previously received ERT and included an additional open-label extension (OLE) study consisting of patients who had and had not previously received ERT.
The PB-102-F01 study (F01 study) and the PB-102-F02 study (F02 study) were open-label, multicentre, dose-ranging, single-arm studies of adults with symptomatic FD who had either never received ERT or not received it within the last 6 months. Enrolment in the F02 study required prior completion of the F01 study. The interventions were pegunigalsidase alfa, IV, at dosages of 0.2 mg/kg, 1.0 mg/kg, or 2.0 mg/kg every 2 weeks for 12 weeks (F01 study) or for 36 weeks (F02 study). The outcomes of interest were annualized change in CKD-EPI eGFR, pain as measured with the short-form BPI, and harms. The safety results revealed that pegunigalsidase alfa had an acceptable safety profile. Most TEAEs were mild or moderate in severity, and there was a low rate (18.8%) of treatment-induced antibody formation. The results from these trials were consistent with those in the BALANCE and BRIDGE studies.
Patients completing the F01 and F02 studies could enrol in the PB-102-F03 extension study (F03 study) (N = 15). The intervention was pegunigalsidase alfa 1.0 mg/kg, IV, every 2 weeks for up to 60 months. The outcomes measured in the F03 study were the same as in the F01 and F02 studies.
At the time of the submission, the PB-102-F60 study (F60 study) was an ongoing OLE study evaluating the safety, tolerability, and efficacy of pegunigalsidase alfa 1 mg/kg IV, every other week, in adult patients with FD (N = 87 patients).29 Since the submission, the F60 study has been completed. Patients eligible for the F60 study had to have completed the BALANCE or BRIDGE studies or at least 48 months of the F03 study. The total duration of the study treatment was up to 60 months, until pegunigalsidase alfa became commercially available, or at the sponsor’s discretion. The efficacy outcomes of interest included incidence of Fabry clinical events, annualized change in CKD-EPI eGFR (slope), and short-form BPI score. Safety outcomes — TEAEs, IRRs, and ADAs — were evaluated throughout the study.
Efficacy Results
Of the ██ patients enrolled in the F60 study at the interim data cut-off date (July 15, 2021), ██ patients were from the F03 study, ██ patients were from the BRIDGE study, and 59 patients were from the BALANCE trial. Overall, only ██████ patients had discontinued therapy at the time of the data cut-off (due to withdrawal of consent in ██████ patients and TEAEs in ██████ ████████). Overall, ██ ███████ patients completed the scheduled week 52 visit.
Incidence of Fabry Clinical Events
Overall, ██ ███████ patients reported ██ Fabry clinical events up to the data cut-off date. Cardiac events were the most common ███████, followed by cerebrovascular events ██████, and non–cardiac-related death ██████. There were ██ renal events. Overall, there was a ██████ █████████ of events in males than females.
Annualized Change in CKD-EPI eGFR (Slope)
Annualized change in CKD-EPI eGFR were estimated for each patient with at least 1 year of follow-up data. The mean annualized change in CKD-EPI eGFR in the ITT population was █████ ███████████████ ███ █ ████ ██████████████. The mean annualized change in CKD-EPI eGFR was ████ ████████ in males ██████ ██████████████ ███ █ ████ █████████████████ than females ██████ ███████████████ ███ █ ████ █████████████████.
LVMI Score
LVMI results were not available at the interim analysis for the F60 study.
Short-Form BPI Score
Pain severity domain results at week 52 in the overall ITT population had mean changes from baseline to week 52 of ████ ██████ ███ █ ████ ███████ for worst pain, ████ ██████ ███ █ ████ ███████ for least pain, ████ ██████ ███ █ ████ ███████ for pain right now, and ████ ██████ ███ █ ████ ███████ for average pain.
The short-form BPI scores were similar at baseline and week 52 in pain severity domains (████ ███████ █ ███ ██████) and pain interference domains (████ ███████ █ ███ ██████) based ██ ██ ██ ██ █████████ Most patients with data in the ITT population ███ ██ ██ ████████ ████████ had a numerical improvement or no change in average pain severity at week 52 compared to baseline.
Other Efficacy Results
Plasma lyso-Gb3 concentration, UPCR, MSSI score, and EQ-5D-5L score were considered clinically relevant as supporting evidence. The results are available in Table 33 of Appendix 1.
Harms Results
By week 52, █████ of patients reported at least 1 TEAE, █████ reported at least 1 SAE, ████ discontinued treatment due to a TEAE, and ████ of patients died.
A total of ██ IRRs within 2 hours postinfusion were reported in ██████ ████████ IRR took place between 2 and 24 hours postinfusion for a total of ██ ████ within 24 hours postinfusion in ██████ patients.
A total of ██ ███████ patients in the overall safety population were ADA positive at any postbaseline visit in the F60 study up to the interim analysis cut-off date. Treatment-emergent ADAs occurred in ██ ███████ patients, including ██████ patients with titre-boosted ADAs and ██████ patients with treatment-induced ADAs.
Critical Appraisal
Patient characteristics in the F60 study aligned with those in the F01, F02, F03, BALANCE, and BRIDGE studies. Overall, the F60 study had a small sample size (N = 87), which is a function of rare disease studies, and required patients to have completed the previous studies for enrolment. Because they had completed the requisite studies, the patients who enrolled in the F60 study were a highly enriched and selected sample. Given that only 11.5% of the F60 study patients had not previously received pegunigalsidase alfa, a majority of patients were likely to have had positive responses and tolerated pegunigalsidase alfa well. The largest percentage of patients came from the BALANCE trial (39 [44.8%] randomized to pegunigalsidase alfa and 20 [23.0%] randomized to agalsidase beta). The open-label design of the F60 study meant that patients were not blinded to treatment and may have reported more favourable patient-reported outcomes in HRQoL and pain and may have been less likely to report TEAEs in the follow-up period due to their knowledge of the treatment. The baseline characteristics appeared to be imbalanced between females and males, and the sponsor did not note the percentage of participants of each sex who had classic FD, which is a key effect modifier. In patients who switched from agalsidase beta to pegunigalsidase alfa, there is the potential for a 6-month carryover effect after discontinuation of the previous treatment, according to the clinical experts consulted for this review. It is unclear if a patient’s response to pegunigalsidase alfa was related to their prior drug doses (0.2 mg/kg versus 2.0 mg/kg) used in the F01 and F02 studies before starting at a dose of 1.0 mg/kg in the F60 study. Moreover, the lack of a comparator is a major limitation in this study. The outcomes in the F60 study were the same as in the BALANCE trial, but without LVMI score was missing. There were notable sex-based differences in annualized change in CKD-EPI eGFR. Females had a ███████ ██████ in pain scores measured using the short-form BPI from baseline to week 52, albeit with greater variability, as shown by the SE. Notable harms in the F60 study (████ ██████ █ █████ █████████████ ███ ██████████████████ ████) were infrequent. A possible explanation for the █████ TEAEs in the F60 study could be selection bias: individuals who previously experienced IRRs and ADAs in the F01, F02, and F03 studies may have dropped out before enrolling in the F60 study.30
The Health Canada indication under review is for long-term ERT in adult patients with a confirmed diagnosis of FD, which the F60 study aims to address. The patients enrolled in the F60 study included those previously receiving ERT (from the BALANCE and BRIDGE trials) and 10 patients (11.5%) from the F03 study who had not previously received ERT. Given that only 10 patients had not previously received ERT, subgroup analyses were not feasible and the results of the F60 study do not allow for meaningful conclusions to be drawn for this subgroup. Patients who started in the BALANCE trial and transitioned to the F60 study could have switched from agalsidase beta 1 mg/kg (n = 20; 23.0%) to pegunigalsidase alfa 1 mg/kg. According to the clinical experts consulted for this review, the practice of switching between ERTs varies among physicians; therefore, the results for patients who switched ERTs are limited in external generalizability, particularly in clinical contexts in which patients do not typically switch ERTs. According to clinical expert opinion, given the X-linked nature of FD, males have earlier disease onset that is often more serious, and they have a worse prognosis. Therefore, females who enrol in clinical studies tend to do better than males. Given that the F60 study had 55 males and 32 females, the study includes a larger proportion of participants with an expected worse prognosis than the estimated relative frequency of FD in the general population of 1 in 40,000 in males and 1 in 117,000 in females.14
Indirect Comparisons
Description of Studies
There is an absence of direct comparative efficacy and safety evidence between pegunigalsidase alfa and disease-specific treatments for FD other than agalsidase beta. To address this, the sponsor submitted a network meta-analysis (NMA) using a Bayesian framework comparing pegunigalsidase alfa to agalsidase beta, agalsidase alfa, and migalastat. The outcomes of interest included change in eGFR, LVMI score, BPI score, and ADAs.
In addition, the sponsor submitted unanchored population-adjusted indirect comparisons (PAICs) using simulated treatment comparison (STC) methods to adjust for population differences between patients receiving pegunigalsidase alfa in the BALANCE, F01, F02, F03, and BRIDGE studies and patients receiving comparator treatments. Regression models suitable for outcome type were fitted to the individual patient data of patients receiving pegunigalsidase alfa, adjusting for covariates based on treatment effect modifiers (TEMs) and prognostic variables at baseline in comparator trials. Adjustments could only be made for differences in treatment exposure status, age, sex, phenotype, migalastat-amenable variant status, and baseline eGFR, depending on what data were available for each outcome. Due to the limitations of unanchored PAICs, the results have not been reported or discussed in this review, and only the results of the Bayesian framework are presented.
Efficacy Results
The systematic literature review (SLR) found 120 records from 56 original studies. After study eligibility was applied, 7 studies were included in the NMA. For all outcomes of interest, there were only single studies comparing any 2 treatments, and a random-effects model could not be estimated. Therefore, the results are based on the fixed-effects model.
Change From Baseline in eGFR
The mean difference between pegunigalsidase alfa and the comparators was ████ ██████████████ ████████ ████████ ██████ █████ ██ ████ ██████████ for agalsidase beta after 104 weeks of treatment, ████ ██████████████ ████ ██████ ██ █████ ██████████ for agalsidase alfa after 52 weeks of treatment, and ██████ █████████████ ████ ██████ ██ █████ ██████████ for migalastat after 26 weeks of treatment. The sensitivity analyses that removed the real-world evidence (RWE) study and combined the 2 doses of agalsidase beta supported the base case.
LVMI Score
The mean difference between pegunigalsidase alfa and the comparators was ████ █████ ████ █████ ██ ████ ███ or agalsidase beta after 104 weeks of treatment, █████ ██████ ████ ██████ ██ █████ ███ for agalsidase alfa after 52 weeks of treatment, and █████ ██████ ████ ██████ ██ █████ ███ for migalastat after 26 weeks of treatment. The sensitivity analyses that removed the RWE study and combined the 2 doses of agalsidase beta supported the base case.
Short-Form BPI Score
The mean difference between pegunigalsidase alfa and the comparators was ████ ██████ ████ ████ █████ ██ ████ ███████ for agalsidase beta after 104 weeks of treatment and ████ ██████ ████ ████ █████ ██ ████ ███████ for agalsidase alfa after 24 weeks of treatment.
Harms Results
Antidrug Antibodies
The odds ratios of a patient developing ADAs were 1.26 (95% credible interval [CrI], 0.47 to 3.35), nominally favouring pegunigalsidase alfa over agalsidase beta, and 0.46 (95% CrI, 0.06 to 3.02), nominally favouring agalsidase alfa over pegunigalsidase alfa.
Critical Appraisal
Where the network was unconnected or there were important differences in TEMs between studies, PAICs were considered. Although the sponsor attempted to adjust for important TEMs and prognostic variables to control the risk of bias, data were unreported for many characteristics, and it was not possible to adjust for ADA status, ACEi or ARB treatment, UPCR category, baseline left ventricular hypertrophy (LVH) status, migalastat-amenable variant status, genotype, or lyso-Gb3 concentration. Most of the studies in the PAICs enrolled a small number of patients (< 30), and patient-level data were very limited for those who had not previously received treatment for FD (n = 6) or who had a migalastat-amenable variant (n = 9). Therefore, the unanchored PAICs using STC methods have limitations, putting their results at high risk of bias that increase the uncertainty of the comparisons, and the results have not been reported in this review. The uncertainties in the validity of the unanchored comparisons have also been acknowledged in the sponsor-submitted indirect treatment comparison (ITC) report.
The overall network was sparse: it included few drugs and few comparative trials. All studies had a small number of patients, which is understandable given that FD is rare, and most had fewer patients than the BALANCE trial (as few as 15 patients). For the outcomes of interest for this review, a random-effects model was not estimable; therefore, the comparisons used a fixed-effects model. Use of only a fixed-effects model, along with the wide CrIs, leads to a high level of uncertainty in the estimates.
To connect pegunigalsidase alfa and agalsidase alfa in some outcome networks, the normal and low doses of agalsidase beta had to be combined, which requires an assumption of similar efficacy and safety to be made that is unlikely to be true and introduces bias that cannot be adjusted for. Moreover, the lower doses of agalsidase beta are not used in clinical practice according to the clinical experts consulted for this review. The non-RCT or RWE studies that report on the agalsidase beta doses would need to be included in the network, but none of these studies were able to provide an unbiased estimate and therefore were not used.
For the results of an NMA to be valid, the studies and patients included in the network must be sufficiently similar; however, in this network there were clear differences in study dates, study design, geographic location, treatment duration, outcome assessment time points, and permission to switch treatments. There was also variability in the study eligibility criteria, which produced notably different study populations. This was confirmed when looking at the baseline characteristics across studies. There were differences in mean age, sex, mean baseline eGFR, and prior treatment exposure. Many characteristics that were identified as TEMs — such as disease phenotype, migalastat-amenable variant, LVH, mean UPCR, and presence of ADAs — were not reported across all studies. Moreover, some doses for migalastat and agalsidase beta in some studies were inconsistent with the Health Canada product monographs. There was also imputation of missing study values using the BALANCE trial data, potential variability in outcome assays or measurements (even for objective outcomes such as eGFR and LVMI score), and inconsistency in outcome definitions and time points. Overall, there were important differences across studies that cannot be adjusted for using NMA methods.
Due to the differences in study designs, study populations, comparator doses, and outcomes, the transitivity assumption is likely not valid. The homogeneity of the treatment effect could not be assessed because there were only single studies informing relevant direct comparisons (i.e., between active treatments for FD). The consistency of the treatment effect could not be assessed because there were no closed loops that contained pegunigalsidase alfa.
Outcomes of interest to patients and clinicians that were missing included eGFR slope, HRQoL, and IRRs. For eGFR slope and IRRs, an attempt was made to create networks, but it was not possible to connect pegunigalsidase alfa with agalsidase alfa or migalastat. Due to the lack of evidence from the ITC, it is unknown how these drugs compare for these outcomes. The study durations varied, and the long-term treatment effects are uncertain according to the results of the NMA.
Studies Addressing Gaps in the Evidence From the Systematic Review
Description of Studies
The BRIDGE study (N = 22) was a phase III, open-label, switch-over study evaluating the safety and efficacy of pegunigalsidase alfa, 1 mg/kg every 2 weeks, in adults with FD who had been previously treated for at least 2 years with agalsidase alfa and at a stable dose for at least 6 months. This study was included to address a gap in the evidence for patients treated with pegunigalsidase alfa who had not been previously treated with agalsidase beta (as in the BALANCE trial).
The eligibility criteria were generally similar to those used in the BALANCE trial, aside from all patients having prior treatment with agalsidase alfa in the BRIDGE study. Pegunigalsidase alfa was administered at a dosage consistent with that in the BALANCE trial for 12 months, and study outcomes were also consistent with the BALANCE trial.
Of the 22 patients enrolled in the BRIDGE study, 20 (90.9%) completed 12 months of treatment, and there were 2 discontinuations due to TEAEs. The mean age of the patients was 44.0 years (SD = 11.0 years). There were more males (68.2%) than females (31.8%), as per the study design, and more than half of patients had classic FD (63.6%). The mean eGFR was 82.5 mL/min/1.73 m2 (SD = 23.4 mL/min/1.73 m2), and the mean eGFR slope was –5.3 mL/min/1.73 m2 per year (SD = 6.3 mL/min/1.73 m2 per year). The mean time exposed to pegunigalsidase alfa in the study was 11.0 months (SD = 3.5 months).
Efficacy Results
Incidence of Fabry Clinical Events
One patient experienced 2 Fabry clinical events (cardiac and cerebrovascular events).
Annualized Change in CKD-EPI eGFR (Slope)
The preswitch mean eGFR slope was –5.90 mL/min/1.73 m2 per year (SE = 1.34 mL/min/1.73 m2 per year), based on historical data. The postswitch mean eGFR slope was –1.19 mL/min/1.73 m2 per year (SE = 1.77 mL/min/1.73 m2 per year).
LVMI Score
Of the 19 patients who contributed to the analysis, the mean LVMI score was 86.9 g/m2 (SE = 6.9 g/m2) at baseline and 89.4 g/m2 (SE = 6.1 g/m2) at month 12. The change from baseline to month 12 was 4.1 g/m2 (SE = 2.8 g/m2).
Short-Form BPI Score
The mean score for pain at its worst in the past 24 hours was 2.0 points (SE = 0.5 points) at baseline and 2.3 points (SE = 0.6 points) at month 12. The mean change from baseline was 0.4 points (SE = 0.4 points). The mean score for pain at its least in the past 24 hours was 0.9 points (SE = 0.3 points) at baseline and 1.2 points (SE = 0.4 points) at month 12. The mean change from baseline was 0.3 points (SE = 0.3 points). The mean score for current pain was 1.1 points (SE = 0.4 points) at baseline and 1.3 points (SE = 0.5 points) at month 12. The mean change from baseline was 0.2 points (SE = 0.2 points). The mean score for average pain was 1.9 points (SE = 0.4 points) at baseline and 1.9 points (SE = 0.5 points) at month 12. The mean change from baseline was 0.1 points (SE = 0.2 points).
Other Efficacy Results
Plasma lyso-Gb3 concentration, UPCR, MSSI score, exercise tolerance (stress test) results, pain medication use, and EQ‑5D‑5L score were considered clinically relevant as supporting evidence. The results are available in Table 35 of Appendix 1.
Harms Results
Twenty-one patients (95.5%) reported at least 1 TEAE during the study, with the 2 most common being headache (31.8% of patients) and nasopharyngitis (22.7% of patients). Four patients (18.2%) reported at least 1 SAE, which included 2 hypersensitivity events and 1 event each for infectious mononucleosis and urinary tract infection. Two patients (9.1%) withdrew due to TEAEs, of which both were IRRs (hypersensitivity reactions). There were no deaths in the study.
For the notable harms of interest for this review, 3 patients (13.6%) reported injection site reactions, 5 patients (22.7%) reported IRRs, and 7 patients (31.8%) had treatment-emergent ADAs; 2 (28.6%) of those 7 patients had neutralizing ADAs.
Critical Appraisal
The main limitations of the BRIDGE study were the open-label design and lack of comparator group. It is possible that knowledge of the treatment impacted subjective outcomes, such as the short-form BPI score and harms. Additionally, uncontrolled confounding puts the results at a high risk of bias. Historical values (up to 24 months before screening) were used to estimate the baseline eGFR and were likely unreliable. All patients in the BRIDGE study were previously receiving a stable dose of agalsidase alfa before the study, and there is the possibility of carryover effects during the first few months of the study. This is additionally concerning as the BRIDGE study was only 12 months long and carryover effects can persist for 6 months, according to the clinical experts.
The generalizability concerns discussed for the BALANCE trial are largely applicable to the BRIDGE study because the eligibility criteria were very similar. The age limits (18 to 60 years), the requirement for an eGFR of at least 40 mL/min/1.73 m2, the requirement for all patients to have had prior ERT for at least 2 years, and the exclusion of patients with poorer health may prevent the broad application of the results to those who do not meet these characteristics but could receive pegunigalsidase alfa in clinical practice. All patients switched from agalsidase alfa to pegunigalsidase alfa, and while the choice to switch varies in practice based on clinician and patient preference, the study does not provide information on patients who have not received prior ERT and does not provide an adequate (short-term or long-term) comparison between pegunigalsidase alfa and agalsidase alfa. The outcomes were generally the same as in the BALANCE trial, and the same limitations apply to the BRIDGE study. The study follow-up was 12 months and did not provide information on the long-term use of pegunigalsidase alfa, though patients were able to enrol in the OLE study (F60 study), which is discussed in this report.
Conclusions
FD is a rare, progressive, X-linked disease associated with kidney, cardiac, and cerebrovascular disease and reduced HRQoL. There is a need for safe and effective treatments that prevent progression to end-organ disease, reduce FD symptoms, improve HRQoL, and are less burdensome. Based on the evidence from 1 phase III, randomized, active comparator–controlled, noninferiority study of adults aged 18 to 60 years with symptomatic FD, kidney function decline, and previous experience with agalsidase beta (the BALANCE trial), 2 years of treatment with pegunigalsidase alfa 1.0 mg/kg, IV, every other week may result in a similar decline in eGFR slope as treatment with agalsidase beta 1.0 mg/kg. Compared to the sponsor-suggested noninferiority margin of –3.0 mL/min/1.73 m2 per year, pegunigalsidase alfa was considered to be noninferior to agalsidase beta; however, information from regulatory agencies, published literature, and clinical expert opinion indicated that a smaller margin would be more appropriate. At the suggested MID of 1.0 mL/min/1.73 m2 per year, pegunigalsidase alfa could have resulted in a clinically meaningful decline or increase in eGFR slope in the BALANCE trial. According to clinical expert opinion, the results from the study suggested that the 2 drugs could be similar for incidence in Fabry clinical events, cardiac complications (based on LVMI score), and pain (based on the short-form BPI score). Still, the evidence was highly uncertain due to the potential risk of bias in the study, limitations with generalizability, and imprecision in the effect estimates. Evidence from a 1-year, open-label study of patients who switched from agalsidase alfa to pegunigalsidase alfa (the BRIDGE study) showed a reduced rate of eGFR decline, an increase in LVMI score, and stable pain scores, but this evidence was based on few patients and a short duration. Longer-term evidence for an additional year of treatment with pegunigalsidase alfa (the F60 study) showed that patients continued to experience Fabry clinical events (as expected with a progressive disease and patients with pre-existing organ involvement), the eGFR slope continued to decline at a similar magnitude as in the BALANCE trial, and the pain scores generally remained stable. Low-quality evidence from the sponsor-submitted ITC showed that no FD-specific treatment was clearly favoured over another for change in eGFR, LVMI score, or BPI score, though the comparisons were hindered by the significant study and population heterogeneity, unverifiable NMA assumptions, and a high level of uncertainty in the estimates based on very wide CrIs. It is unclear what impact pegunigalsidase alfa has on HRQoL, and the long-term outcomes of what is expected to be a lifelong treatment remain unknown.
In the BALANCE trial, harms were comparable between pegunigalsidase alfa and agalsidase beta. Although there were numerically fewer IRRs and neutralizing ADAs in the pegunigalsidase alfa group, it was not clear if the differences were clinically meaningful between the 2 drugs, according to clinical expert opinion. There were no unexpected safety signals for patients who switched from agalsidase alfa in the BRIDGE study or for patients who continued treatment in the F60 study. The ITC for the development of ADAs between pegunigalsidase alfa and agalsidase alfa indicated that neither treatment was clearly favoured over the other.
Pegunigalsidase alfa offers patients and clinicians another treatment option for FD, but it is not clear if the drug provides any reduction in administrative or nonclinical burden over other ERTs.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of pegunigalsidase alfa, 2 mg/mL, for IV infusion in the treatment of FD in adults.
Disease Background
The contents within this section have been informed by the materials submitted by the sponsor and by clinical expert input. The following has been summarized and validated by the review team.
FD is a rare, progressive, X-linked lysosomal storage disorder caused by a deficiency of the lysosomal enzyme alpha-galactosidase A due to a GLA variant. This deficiency leads to the progressive accumulation of glycolipids (mainly Gb3 and lyso-Gb3) in plasma and lysosomes of a wide range of cells, resulting in metabolic dysfunction, cell death, and, eventually, progressive vital organ disease.1-4
FD is characterized by the development of hypertrophic cardiomyopathy, nephropathy with chronic renal failure, and stroke.5 Other symptoms include neuropathic pain, cerebrovascular disease, gastrointestinal symptoms, angiokeratomas, and hypohidrosis.28 In a survey of 280 patients with FD in the US and Canada, the most common symptoms were fatigue (72% of patients), tingling (62%), ringing in the ears or hearing loss (54%), general body pains or pain crises (51%), and abdominal pain (50%).31 These symptoms significantly affect individuals’ HRQoL and physical and emotional well-being and severely affect their ability to perform daily activities.32
The 2 types of FD, classic and nonclassic, are defined by GLA variant. Classic FD is the more severe form, characterized by severely reduced (< 5% of mean normal) enzyme activity in males; people with the nonclassic form of FD have residual enzyme activity that varies between 5% and 30% of normal levels.1,6 Due to the X-linked nature of the disease, hemizygous people tend to have a more severe phenotype, while heterozygotic people tend to have a milder phenotype.1,3,6 Patients with classic FD experience more severe disease, with early signs and symptoms manifesting during childhood and adolescence and including neuropathic pain, autonomic dysfunction, bloating, diarrhea, abdominal pain, angiokeratomas, hypohidrosis, and corneal opacity (cornea verticillata). Renal dysfunction also occurs early in life in a significant portion of children with classic FD, manifesting as proteinuria, leading to CKD in adulthood and progression to end-stage kidney disease.1,7 Other common organ complications emerge in adult patients and include cardiac manifestations (LVH, myocardial fibrosis, arrhythmias), auditory loss, TIA, strokes, and premature death.1 Patients with nonclassic FD typically have milder disease and delayed onset of clinical manifestations, with symptoms emerging between 30 and 70 years of age. These patients tend to have single-organ involvement, such as CKD or, more frequently, cardiac disease, with only mild findings in other organs.1,6,7
The global prevalence of FD is estimated to be between 1 in 117,000 and 1 in 37,000 live male births for classic FD.8-13 Classic FD has an estimated incidence of 1 in 50,000 males.11 Between the sexes, the frequency of FD has been estimated in 1 study to be 1 in 117,000 in females and 1 in 40,000 in males.14 Published prevalence and incidence rates of FD in Canada were not available, but the clinical experts consulted for this review estimated prevalence to be between 1 in 74,000 and 1 in 50,000.
The accumulation of Gb3 and related glycosphingolipids in lysosomes begins before birth and progressively affects organ function, leading to serious and life-threatening events with early mortality.33 According to the CFDI registry, there are 470 adults currently enrolled on the registry and, of these, 242 patients are currently receiving ERT or chaperone therapy.34
The testing requirements to confirm a diagnosis of FD (alpha-galactosidase A activity assay and genetic testing) are well established in Canada and are outlined in the Canadian Fabry Disease Treatment Guidelines.5 The diagnosis of FD requires the synthesis of clinical, biochemical, molecular, and pathologic criteria. Given the challenges with each of these criteria, it is recommended that an individual has at least 3 of the 4 criteria before an FD diagnosis is made.35 Current treatment criteria require a confirmed diagnosis of FD based on an unequivocal combination of DNA, enzyme, phenotypic, and biomarker evidence. Then, there must be clear evidence of stroke, renal disease, or cardiac disease consistent with FD or the presence of uncontrolled pain or gastrointestinal symptoms that interfere with the individual’s HRQoL.
Standards of Therapy
The contents within this section have been informed by the materials submitted by the sponsor and by clinical expert input. The following has been summarized and validated by the review team.
According to the clinical experts consulted for this review, the goals of treatment are to delay heart failure, slow decline in kidney function (to –1 mL/min/1.73 m2 per year, that of the general population), and prevent stroke and TIA. The experts also noted the importance of improving HRQoL; controlling neuropathic pain, gastrointestinal symptoms, dysrhythmias, and obstructive pulmonary disease; treating associated mental health disorders; reducing fatigue; and normalizing life expectancy.
According to the 2018 Canadian Fabry Disease Treatment Guidelines, once an FD diagnosis is confirmed and there is evidence of disease, a thorough evaluation takes place to determine whether disease-specific therapy is likely to provide clinical benefit.5 It is only once an indication for treatment is confirmed that the choice of therapy is considered.5 There are 2 types of disease-specific treatments for FD — ERT and pharmacologic chaperone therapy — but neither of these is completely effective.15 Agalsidase alfa and agalsidase beta are recombinant human ERTs, and the deficient enzyme is provided by IV infusion. Patients who have an indication for disease-specific therapy can receive ERT, regardless of the genetic variant.5 Migalastat is an oral chaperone therapy that binds and stabilizes specific forms of the enzyme with downstream effects of increased activity.16 Unstable enzymes result from various pathogenic GLA variants; therefore, migalastat is indicated only for patients with an amenable genetic variant (found in an estimated 35% of patients with FD in Canada, according to the clinician group that provided input for this review). A patient considering migalastat must undergo testing to confirm the disease would respond to the drug; however, it has been noted in the literature that in vitro testing does not perfectly predict in vivo outcomes and that clinical response must be assessed within a few months of initiating migalastat to determine continuation of therapy.15 Identifying amenability to migalastat has been the main factor influencing the decision between treatment with an ERT and chaperone therapy. Other considerations include the method of administration, the severity of the disease, and the potential for developing ADAs with ERT.5
As noted in the 2018 Canadian guidelines and by the clinical experts, indications for disease-specific therapies are aimed at treating early stages of disease progression (when there is evidence of end-organ involvement), rather than preventing disease before signs occur.5 Once a patient is identified, relatives who are at risk can also be tested. Those who do not meet the criteria for treatment should be followed regularly (every 2 years for females and annually for males), and disease-specific treatment can be introduced once the criteria for an indication are met.
Patients with FD may also be treated with nonspecific therapies to address pain and renal, cardiac, neurologic, or gastrointestinal disease.15 The clinical experts noted that antihypertensive drugs, antiplatelet drugs, cholesterol-lowering drugs, smoking cessation, vitamin D, sodium-glucose cotransporter-2 inhibitors, and mineralocorticoid receptor antagonists are used to lower proteinuria, stabilize eGFR, and treat heart failure. In patients with severe renal disease, dialysis and kidney transplant may be options.15 Treatments can also be used to address neuropathic pain, ischemic stroke, and other clinical manifestations of the skin, eyes, bones, or lungs.
Drug Under Review
The key characteristics of pegunigalsidase alfa and its proposed indication for FD are summarized in Table 3.
Pegunigalsidase alfa is a pegylated recombinant form of the human alpha-galactosidase A enzyme and is used to supplement or replace alpha-galactosidase A in patients with FD.17 The recommended dose is based on actual body weight at 1 mg/kg, administered by IV every 2 weeks. For the initial 4 to 6 infusions, pegunigalsidase alfa is recommended at the infusion rates described in the Health Canada product monograph for patients who have and have not previously received ERT.17 If the initial 4 to 6 infusions are well tolerated, the duration of every third infusion may be decreased in decrements of 30 minutes. The minimum recommended infusion duration is 1.5 hours. Home administration under the supervision of a health care provider may be considered for patients who have reached an infusion duration that is well tolerated. However, in the event of hypersensitivity reaction or IRR, the infusion rate may be slowed. For patients who have experienced hypersensitivity reactions to ERT, pretreatment with antihistamines and/or corticosteroids may be advisable. After the first 6 infusions of pegunigalsidase alfa, a stepwise discontinuation of the pretreatment regimen may be considered.
Pegunigalsidase alfa is currently undergoing a Health Canada review, and the proposed indication is for long-term ERT in adult patients with a confirmed diagnosis of FD (deficiency of alpha-galactosidase). A decision on the Notice of Compliance is anticipated on December 3, 2025. The sponsor is seeking reimbursement that aligns with the proposed Health Canada indication. The drug has not been previously reviewed by CDA-AMC.
Table 3: Key Characteristics of Pegunigalsidase Alfa and Other Treatments Available for FD
Characteristic | Pegunigalsidase alfa | Agalsidase beta | Agalsidase alfa | Migalastat |
|---|---|---|---|---|
Mechanism of action | A pegylated recombinant form of the human alpha-galactosidase A enzyme, which supplements or replaces alpha‑galactosidase A. Pegunigalsidase alfa catalyzes the hydrolysis of the terminal alpha‑galactosyl moieties of oligosaccharides and polysaccharides in the lysosome, reducing the amount of accumulation of Gb3 and lyso-Gb3. | A recombinant human alpha-galactosidase that catalyzes the hydrolysis of glycosphingolipids, including Gb3, in the lysosomes of multiple cell types and tissues. Agalsidase beta reduces Gb3 levels in the vascular endothelium and slows the rate of clinical progression in FD as manifested by renal, cardiac, and cerebrovascular outcomes. | Catalyzes the hydrolysis of Gb3, cleaving a terminal galactose residue from the molecule. Treatment with the enzyme has been shown to reduce the accumulation of Gb3 in many cell types, including endothelial and parenchymal cells. Agalsidase alfa has been produced in a human cell line to provide for a human glycosylation profile that influences biodistribution to allow preferential uptake by target cells. | An analogue of the terminal galactose of Gb3 that is a specific, potent, reversible, competitive inhibitor of human alpha‑galactosidase A. It is a specific structural stabilizer for the wild type and many mutant forms of alpha‑galactosidase A. |
Indicationa | Proposed: for long-term enzyme replacement therapy in adult patients with a confirmed diagnosis of Fabry disease (deficiency of alpha-galactosidase) | For long-term enzyme replacement therapy in patients with a confirmed diagnosis of Fabry disease | For long-term enzyme replacement therapy in patients with a confirmed diagnosis of Fabry disease | For long-term treatment of adults and adolescents aged 12 years and older with a confirmed diagnosis of Fabry disease and who have an alpha-galactosidase mutation determined to be amenable by an in vitro assay |
Route of administration | IV | IV | IV | Oral |
Recommended dosage | Based on actual body weight, 1 mg/kg. | Based on actual body weight, 1 mg/kg. | Based on actual body weight, 0.2 mg/kg. | One capsule taken once every other day at the same time of day. |
Serious adverse effects or safety issues | Hypersensitivity reactions, IRRs, ADAs, membranoproliferative glomerulonephritis. | Hypersensitivity reactions (anaphylaxis or anaphylaxis-like reactions), IRRs, ADAs. | Hypersensitivity reactions, IRRs, ADAs. | Contraindicated in patients who do not have an amenable variant. Not to be used concomitantly with ERT or in patients with severe renal insufficiency (eGFR < 30 mL/min/1.73 m2). |
ADA = antidrug antibody; eGFR = estimated glomerular filtration rate; ERT = enzyme replacement therapy; FD = Fabry disease; Gb3 = globotriaosylceramide; IRR = infusion-related reaction; lyso-Gb3 = globotriaosylsphingosine.
aHealth Canada–approved indication.
Source: Health Canada product monographs for pegunigalsidase alfa,17 agalsidase beta,36 agalsidase alfa,37 and migalastat.16
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
CDA-AMC received submissions from 1 patient group, the Canadian Fabry Association, and from individual patients.
Patient feedback was collected through 3 individual testimonials and semistructured interviews to learn from patients’ lived experience. The Canadian Fabry Association described the impact of FD, and its impacts on a patient’s life, as “a multi-systemic condition…[that] causes chronic pain, fatigue, heat intolerance, gastrointestinal issues, kidney disease, heart problems, and an increased risk of stroke, often starting in childhood. Many patients face misdiagnosis and emotional struggles.”
During the interviews, patients reported positive impacts of treatments, such as having more energy, fewer episodes of pain crisis, less gastrointestinal pain, and an ability to carry out everyday life activities. Overall, patients reported a reduction in their symptoms and a stabilization of cardiac and renal disease. These positive impacts were felt to be correlated to improvements in a person’s overall mental health.
Patients reported concerns about receiving ERT, including IRRs, adverse events (e.g., nausea, fatigue, chills, fever), and the consequences of developing ADAs. According to the Canadian Fabry Association, important outcomes in the treatment of FD are less frequent infusions, improvement in IRRs, slowed disease progression, and prolonged and consistent symptom control. Conversations with patients who have received ERT revealed that they experienced improved wellness, energy, and sleep and less pain.
Input from 3 patients documented their experiences with pegunigalsidase alfa. One patient who took pegunigalsidase alfa for a few years, first in the US then in Canada, reported that it reduced the frequency and duration of their gastrointestinal issues and that their cardiac and renal symptoms were well maintained. This patient described their transition from receiving infusions in hospital that lasted 8 hours every 2 weeks to at-home infusions that lasted 60 to 90 minutes, which the patient described as being much easier. A second patient described feeling noticeably better, with less severe symptoms, including their gastrointestinal symptoms being almost resolved. These 2 patients did not experience any associated adverse effects from the drug under review. A third patient indicated that pegunigalsidase alfa improved their HRQoL, their initial eye vortex disappeared, and they had fewer emergency department visits. This patient noted that less frequent infusions also meant reducing their travel to clinics and fewer days of missed work.
Patients described the main unmet need in the treatment of FD as the need for additional treatments that slow disease progression, control symptoms, and are tolerable. Specifically for ERT, there is a need for drugs that are associated with fewer IRRs and have less risk of patients developing ADAs. Patients stated that while there is no cure for FD, the drug under review could potentially improve patients’ HRQoL and health outcomes.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of FD.
Unmet Needs
Although there are disease-specific treatments available for FD, not all patients experience a satisfactory response to ERT or chaperone therapy and there are no curative treatments. Males with classic FD tend to experience disease progression despite treatment, and the experts indicated that 1% to 5% of this subpopulation develop ADAs, making ERT futile. Other issues with current treatments include the high cost of ERT and migalastat and the burden associated with ERT, which typically involves biweekly IV infusions that can last a few hours. Patients receiving ERT may also experience IRRs (estimated to occur in up to 30% of patients), which are more common in patients who are male, have large GLA deletions, lack residual enzyme activity, and receive higher dose ERT (1 mg/kg). Moreover, some symptoms (fatigue, neuropathic pain, gastrointestinal symptoms, hearing loss, and vertigo) have no treatment, can be difficult to treat, or are slow to respond. Clinicians and patients need treatments that are more effective, have less burdensome administration (e.g., oral drug, less frequent infusions), work for all forms of FD regardless of genetic variant, have few adverse reactions (including a low incidence of ADA development), and cross the blood‑brain barrier.
Place in Therapy
According to the clinical experts, pegunigalsidase alfa would be used as a first-line therapy, similar to other ERTs available, and is not expected to change the treatment paradigm for FD. The experts also indicated that it would not be necessary for patients to try other treatments before accessing pegunigalsidase alfa and that the drug would not be reserved for patients in whom other treatments are contraindicated or not tolerated. It was also noted that specific therapies for FD are not combined due to cost and other factors, despite the theoretical potential for better outcomes through the use of multiple mechanisms of action.
Patient Population
The clinical experts stated that adults diagnosed with FD who have a clear indication for disease-specific therapy could receive pegunigalsidase alfa. Indications for treatment include evidence of kidney or heart disease, stroke, intractable neuropathic pain, or gastrointestinal symptoms. The experts noted that treatment eligibility for FD in Canada is largely determined by, but not exclusive to, the Canadian Fabry Disease Treatment Guidelines and the CFDI steering committee.
The experts explained that individuals diagnosed with FD who show no evidence of disease are monitored for early signs of disease before starting treatment. The experts noted that early therapy, before irreversible organ tissue scarring and fibrosis develop, is associated with better outcomes.
According to the experts, patients with severe disease are most in need of therapy. These patients tend to be males and may have limited or no residual enzyme activity, elevated plasma lyso-Gb3 levels, a severe GLA variant, older age (> 50 years), a low eGFR with advanced renal disease, proteinuria of 1.0 g/day or greater, advanced cardiac disease with myocardial fibrosis, dysrhythmias, stroke, and/or high-titre IgG‑neutralizing ADAs.
Assessing the Response to Treatment
Response to treatment may be assessed through stabilization of the eGFR slope, CKD stage, or LVMI score; reduction in the rate of Fabry clinical events or Fabry Stabilization Index score and improvement in symptoms (neuropathic pain, fatigue, gastrointestinal issues, anhidrosis, vertigo, and mood); increased exercise tolerance; and improved work or school attendance and survival.
According to the experts, a clinically meaningful treatment response includes a decrease in the Fabry clinical event rate; a decrease in Fabry pain by 1 to 2 points on a 7-point Likert scale; and a stabilization of eGFR, CKD stage, LVM, and New York Heart Association class. Other indicators of treatment response are normalized sweating, return of normal gastrointestinal function, prevention of stroke, and improved HRQoL.
The experts noted that there are no recognized biomarkers that are accurate surrogates for clinical outcomes. Elevated plasma lyso-Gb3 levels correlate with greater risk of Fabry clinical events. However, it is unclear if reducing plasma lyso-Gb3 also lowers the risk of clinical events, and testing for this marker is not readily accessible in Canada. Although eGFR and LVMI score are outcomes in the BALANCE trial, these measures tend to be too variable to provide reliable data over short time frames and are more informative after 3 to 5 years of treatment.
Because FD is a slowly progressing disease, the experts indicated that patients should be followed for at least 2 to 3 years before judging the degree of treatment response and whether the disease is stable or progressing. They also noted that treatment response to ERT varies depending on patient characteristics (e.g., age, sex, disease phenotype) and that the benefits of ERT may take a year or longer to be realized. In clinical practice, patients are typically seen every 6 to 12 months, but children and young females who are asymptomatic with normal organ function may be seen less frequently.
Discontinuing Treatment
According to the Canadian Fabry Disease Treatment Guidelines, discontinuation of disease-specific treatment should be considered in a patient whose disease is not responding to treatment after at least 1 year, who has persistent and severe IRRs (despite prophylaxis) or persistent IgE ADAs, or whose life expectancy is estimated to be less than 1 year. Other reasons for discontinuation include permanent and severe neurocognitive decline, severe reduction in HRQoL and functional status, lack of response to therapy for the organ involvement that initially mandated the initiation of treatment, poor adherence, and elevated plasma lyso-Gb3 levels that do not decline.
The experts also discussed practical aspects that should be considered, such as the patient’s age and the burden of ERT compared to the benefit received from treatment. One expert stated that even though there are guidelines and suggested criteria for stopping treatment, these discussions are difficult once a patient has begun treatment and wants to continue. They also stated that there are older adults who are not eligible for (but would prefer) oral migalastat and decline ERT due to the burden of IV infusions.
Prescribing Considerations
Most patients in Canada receive ERT in an outpatient setting, either at home or in an infusion clinic. The experts stated that private nursing services that provide outpatient infusions are paid for by the ERT manufacturer. As with other ERTs, it is recommended that the first 6 to 10 infusions of pegunigalsidase alfa are administered in a medical setting (e.g., hospital outpatient department, infusion clinic) due to the risk of IRRs. Once deemed appropriate, most patients can receive at-home infusions.
Various specialists are involved in the diagnosis, treatment, and monitoring of FD, including medical geneticists, nephrologists, cardiologists, neurologists, general internists, pediatricians, and metabolic specialists. The experts also stated that patients are best served in a centre of excellence where physicians with expertise in FD are available.
The experts also noted that the associated costs of infusions, ADA testing, and lyso-Gb3 monitoring should be covered by the sponsor or the drug plans because the current lack of coverage poses a significant barrier to effective FD care.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
Four clinicians on behalf of the CFDI provided input. The clinicians presented their assessment of peer-reviewed studies on FD, information from the CFDI registry, and input based on their professional expertise.
The group described the main unmet needs in the treatment of FD as being the need for a new treatment with greater clinical efficacy than existing treatment and the need to reduce the frequency of ERT infusions. According to the clinician group, the treatment goals for FD are delaying dialysis and heart failure. The clinician group noted that there are other manifestations of the disease that are difficult to target with drugs, including abdominal pain, neuropathic pain, stroke, and mental health problems. In addition, cardiac disease continues to progress in people with FD, leading to high rates of atrial fibrillation and other arrhythmias, heart failure, and chest pain.
The clinician group was aligned with the perspectives of the clinical experts consulted by CDA-AMC on the drug’s place in therapy.
If pegunigalsidase alfa was available in Canada, the clinician group felt it would provide an alternative form of ERT at the 1 mg/kg dose, but the clinicians did not expect it to impact how treatment decisions are made. They noted that the drug may prove useful for some adult males with high-titre neutralizing ADAs who have reduced response to therapy. The group noted that, although less in vitro binding of ADAs to pegunigalsidase alfa compared to other ERTs has been shown, there are no in vivo data yet to support this hypothesis.
The clinician group stated that the best measure of treatment response is plasma lyso-Gb3, but this test is not accessible in most jurisdictions. Moreover, this test may be less informative in female patients and in individuals with late-onset disease variants. If clear disease progression is observed, clinicians may check for neutralizing ADAs. A decision is rarely made to stop treatment because of disease progression. Specialists in FD monitor for disease-related outcomes, as well as for information to inform the decision to initiate and stop therapy. The group noted that guideline-directed reasons to stop therapy are currently patient preference, severe drug intolerance, treatment futility, or very short life expectancy.
The clinician group recommended that FD be managed by experienced specialists or by a physician under the supervision of a specialist (e.g., with a background in genetics, inherited metabolic diseases, nephrology, or cardiology) and that most patients’ treatment is managed through specialized treatment centres in their jurisdiction or the neighbouring jurisdiction. The group also recommended that the application for disease-modifying drug approval be reviewed by an expert committee, such as the CFDI or equivalent, before patients are started on therapy.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Both agalsidase beta and agalsidase alfa received negative recommendations from CDA-AMC but are reimbursed in some jurisdictions through exceptional access programs and funding programs for drugs for rare diseases. Eligibility may be based on criteria outlined in the Canadian Fabry Disease Treatment Guidelines, with patient assessment via the CFDI. | Comment from the drug program to inform CDEC deliberations. |
Considerations for initiation of therapy | |
The BALANCE and BRIDGE trials included patients aged 18 to 60 years. Should patients younger than 18 years or older than 60 years be eligible? | The clinical experts stated that patients aged 18 years or older could receive treatment with pegunigalsidase alfa for FD. One expert indicated that pediatric patients aged 17 years or younger could also receive the drug for FD, while another expert noted that there would first need to be adequate evidence of safety and efficacy in a pediatric population before pediatric patients are treated with the drug. The proposed Health Canada indication is for adult patients with a confirmed diagnosis of FD. |
Consider alignment with the initiation criteria used by plans for other ERT or migalastat, as appropriate. | Comment from the drug program to inform CDEC deliberations. |
Considerations for continuation or renewal of therapy | |
Consider alignment with the renewal criteria used by plans for other ERT or migalastat, as appropriate. | Comment from the drug program to inform CDEC deliberations. |
System and economic issues | |
Per the pan-Canadian BIA report:
Administration costs for the currently available ERTs are covered by the manufacturers, according to clinicians in Canada. Per the pan-Canadian BIA report:
| Comment from the drug program to inform CDEC deliberations. |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; CDEC = Canadian Drug Expert Committee; CFDI = Canadian Fabry Disease Initiative; ERT = enzyme replacement therapy; FD = Fabry disease.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of pegunigalsidase alfa, 2 mg/mL, for IV infusion in the treatment of FD in adults. The focus will be placed on comparing pegunigalsidase alfa to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of pegunigalsidase alfa is presented in 4 sections, with a critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes the pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes sponsor-submitted long-term extension studies. The third section includes indirect evidence from the sponsor. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
1 pivotal study identified in systematic review
1 OLE study
1 ITC
1 study addressing gaps in the evidence.
Systematic Review
The contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
One pivotal study was included in the systematic review of pegunigalsidase alfa and is summarized in Table 5. The BALANCE trial (N = 78) was a phase III, double-blind, noninferiority RCT evaluating the efficacy and safety of pegunigalsidase alfa (n = 53) compared to agalsidase beta (n = 25) in adult patients with FD who were previously treated with agalsidase beta for at least 1 year.18 The study consisted of a 1-month screening period and a 24-month treatment period, after which patients could enrol directly into an OLE study (the F60 study).29 The BALANCE trial was originally designed to test the noninferiority of pegunigalsidase alfa to agalsidase beta at the 12-month interim analysis and the superiority of the former at the 24-month final analysis. After agalsidase beta was given full market authorization in March 2021, and in consultation with the FDA (September 9, 2021), the demonstration of superiority after 24 months was no longer required and noninferiority was assessed at the final analysis. For the interim analysis, the last patient visit date was October 21, 2020 and database lock occurred in April 2021. Approximately 61% of patients had attended the 24-month visit (or discontinued early), and more than 90% of eGFR values had been collected by the time the interim analysis was conducted at 12 months. Furthermore, patients, investigators, and site staff remained blinded to treatment assignment and only were aware that the study had reached noninferiority.
Table 5: Details of Studies Included in the Systematic Review
Detail | BALANCE trial |
|---|---|
Designs and populations | |
Study design | Phase III, double-blind, active-controlled RCT |
Locations | 29 sites in 12 countries: Czechia, Finland, France, Hungary, Italy, Netherlands, Norway, Slovenia, Spain, Switzerland, UK, and US |
Patient enrolment dates | Start date: August 22, 2016 Last patient last visit: October 21, 2021 |
Randomized (N) | N = 78
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Pegunigalsidase alfa, 1 mg/kg, intravenously over 3 hours, every 2 weeks |
Comparator | Agalsidase beta, 1 mg/kg, intravenously over 3 hours, every 2 weeks |
Study duration | |
Screening phase | 1 month |
Treatment phase | 24 months |
Follow-up phase | Following completion of the trial, patients were eligible to continue treatment with pegunigalsidase alfa in the OLE study (F60 study) |
Outcomes | |
Primary end point | Annualized change (slope) in CKD-EPI eGFR during 30 planned visits over 2 years |
Secondary end points | Secondary:
|
Publication status | |
Publication | |
ACEi = angiotensin-converting enzyme inhibitor; AKI = acute kidney injury; ARB = angiotensin receptor blocker; BPI = Brief Pain Inventory; CKD‑EPI = Chronic Kidney Disease Epidemiology Collaboration; eGFR = estimated glomerular filtration rate; FD = Fabry disease; Gb3 = globotriaosylceramide; LVM = left ventricular mass; LVMI = Left Ventricular Mass Index; lyso-Gb3 = globotriaosylsphingosine; MSSI = Mainz Severity Score Index; NYHA = New York Heart Association; OLE = open-label extension; RCT = randomized controlled trial; TIA = transient ischemic attack; UPCR = urine protein-to-creatinine ratio.
Source: Clinical Study Report for BALANCE trial;18 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Populations
Inclusion and Exclusion Criteria
Eligible patients include adults with FD and reduced kidney function. Females must have had either genetic confirmation of a pathogenic FD variant or, in the case of a novel variant, a first-degree male relative with FD, and males must have had reduced plasma and/or leukocyte alpha-galactosidase activity (< 30% of the mean normal levels). Because the disease presents differently in females and males, the studies required that no more than 50% of enrolled patients be female. All patients must have displayed at least 1 of the characteristic features of FD (i.e., neuropathic pain, cornea verticillate, or clustered angiokeratoma). All patients had experience with ERT.
Individuals were not eligible if they had a history of renal dialysis or transplant, acute kidney injury in the year before enrolment, New York Heart Association Class IV congestive heart failure, or a cardiovascular or cerebrovascular event in the 6 months before enrolment. In the BALANCE trial, patients could not have an eGFR value between 91 mL/min/1.73 m2 and 120 mL/min/1.73 m2 at screening or a historical eGFR value of more than 120 mL/min/1.73 m2.
Interventions
Patients were randomized 2:1 to either switch to pegunigalsidase alfa or continue receiving agalsidase beta; both drugs had a dosage of 1 mg/kg every 2 weeks, IV, for 104 weeks. Randomization was stratified by UPCR (< 1 g/g versus ≥ 1 g/g). Patients receiving premedication with agalsidase beta to prevent IRRs had the same regimen used at the start of the study treatment, which could be tapered off during the initial 2 to 3 months at the investigator’s discretion and under careful observation. For the first 3 months, the study drug was infused over 3 hours; after 3 months, the infusion time could be gradually reduced to 1.5 hours if treatment was well tolerated. Initially, patients received the infusions at the study site, after which home infusions were possible if the investigator’s and sponsor’s medical monitor permitted it. Patients were clinically observed for up to 2 hours after dosing.
Patients permanently discontinued the study in the event of grade 3 or 4 toxicities considered to be associated with the study drug, in the event of progressive or severe hypersensitivity that was not mitigated by pretreatment, if adherence was poor, at the patient’s request, or at the investigator’s discretion.
Prohibited treatments included drugs for FD (i.e., agalsidase alfa). Patients were permitted to continue stable doses of ACEis or ARBs and could only initiate or discontinue these medications if permitted by the medical monitor.
Outcomes
A list of the efficacy end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures in Table 7. The summarized end points are based on the outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review according to the clinical experts consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, the end points selected were those considered to be most relevant to inform expert committee deliberations, and this list of end points was finalized in consultation with members of the expert committee. The summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE.
The following considerations went into the selection of the outcomes summarized in the report and assessed using GRADE:
outcomes important to patients that assess symptoms (short-form BPI) and outcomes identified by clinical experts as being important for decision-making (Fabry clinical events, annualized change in CKD-EPI eGFR, LVMI)
harms identified as being important in the input provided by patient groups, clinician groups, and clinical experts (IRRs, neutralizing ADAs).
Table 6: Outcomes Summarized From the BALANCE Trial
Outcome measure | Time point | Type of end pointa |
|---|---|---|
Incidence of Fabry clinical events | Through 104 weeks | Secondary |
Annualized change in CKD-EPI eGFR (slope) | From baseline to week 104 | Primary |
Change from baseline in LVMI (g/m2) by MRI | From baseline to week 104 | Secondary |
Change from baseline in short-form BPI score | From baseline to week 104 | Secondary |
Change from baseline in concentration of plasma lyso-Gb3 | From baseline to week 104 | Secondary |
Change from baseline in UPCR category by spot urine test | Through 104 weeks | Secondary |
Change from baseline in MSSI score | From baseline to week 104 | Secondary |
Change from baseline in exercise tolerance (stress test) | Through 104 weeks | Secondary |
Change from baseline in frequency of pain medication use | Through 104 weeks | Secondary |
Change from baseline in EQ-5D-5L | From baseline to week 104 | Secondary |
Safety (TEAEs, IRRs, ADAs) | From baseline to week 104 | Safety |
ADA = antidrug antibody; BPI = Brief Pain Inventory; CKD-EPI = Chronic Kidney Disease Epidemiology Collaboration; eGFR = estimated glomerular filtration rate; IRR = infusion-related reaction; LVMI = Left Ventricular Mass Index; lyso-Gb3 = globotriaosylsphingosine; MSSI = Mainz Severity Score Index; TEAE = treatment-emergent adverse event; UPCR = urine protein-to-creatinine ratio.
aNo end points were adjusted for multiplicity.
Source: Clinical Study Report for BALANCE trial;18 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Efficacy
Incidence of Fabry Clinical Events
Fabry clinical events were classified into 4 categories: renal events (first occurrence of either initiation of dialysis or chronic dialysis [> 40 days] or renal transplant), cardiac events (cardiac-related death, myocardial infarction, first-time congestive heart failure, atrial fibrillation, ventricular tachycardia, evidence of progressive heart disease severe enough to require a pacemaker, implantation of pacemaker, bypass surgery, coronary artery dilatation, implantation of defibrillator), cerebrovascular events (hemorrhagic or ischemic stroke or TIA), and death due to non–cardiac-related causes.19 Events were adjudicated by the sponsor’s medical monitor, who was blinded to treatment assignment. The 4 categories were combined into a composite end point: overall Fabry clinical events.
Annualized Change in CKD-EPI eGFR (Slope)
The eGFR was measured to estimate kidney function. The primary end point was the annualized change in eGFR over the 24-month follow-up period and was derived using the CKD-EPI formula, which uses serum creatinine levels and patient characteristics (sex, age, and race) and is standardized for body surface area. Serum creatinine measurements for determining eGFR were taken at screening, at baseline, and then either every 2 or every 4 weeks for a planned total of 30 assessments. Measurements were assessed at the central laboratory.
LVMI Score
One of the cardiac complications of FD is thickening of the left ventricular wall. The LVMI score was calculated based on the LVM determined by MRI. Hypertrophy was defined as an LVMI score greater than 91 g/m2 for males or greater than 77 g/m2 for females.40 MRIs were performed locally at baseline and then every 12 months. Image reading and quality control were centralized, and the analyses were conducted in a blinded manner by an independent contracted research organization.
Short-Form BPI Score
For the short-form BPI score, patients rate the severity of their pain from 0 to 10 points (from “no pain” to “worst imaginable pain”) and how the pain impacts their general activity, mood, walking, working, sleeping, relations with other people, and enjoyment of life from 0 to 10 (from “does not interfere” to “completely interferes”) for 9 pain-related questions.20 Scores are generated for pain at its worst in the last 24 hours, pain at its least in the last 24 hours, pain right now, and pain on average. Scores of 1 to 4 points indicate mild pain, of 5 or 6 points indicate moderate pain, and of 7 to 10 points indicate severe pain. The short-form BPI was completed at screening, at baseline, and every 6 months thereafter.
Other Efficacy Outcomes (GRADE Was Not Applied)
Plasma lyso-Gb3 concentration: In patients with FD, lyso-Gb3 accumulates in cells, tissues, and plasma. Measurements were taken preinfusion at baseline, at 1.5 months, every 3 months up to 1 year, and every 6 months up to 2 years. Plasma lyso-Gb3 concentration was quantified centrally.
UPCR category: The UPCR is used to estimate the extent of CKD. Creatinine is normally released into urine at a constant rate; therefore, the ratio of protein to creatinine is used to estimate how much protein is excreted. UPCR was assessed by spot urine test, and values were summarized into 3 categories (UPCR ≤ 0.5 g/g, 0.5 g/g < UPCR < 1 g/g, and 1 g/g ≤ UPCR) based on the Kidney Disease Improving Global Outcomes guidelines.41 Measurements were taken at screening, at baseline, and every 3 months, and analyses were performed centrally.
MSSI score: The MSSI is an investigator-administered, disease-specific instrument used to measure the severity of FD symptoms. Its 4 sections cover general, neurologic, cardiovascular, and renal signs and symptoms, and each section is weighted based on its contribution to morbidity. The overall score for the instrument ranges from 0 to 76; higher scores indicate greater impact of disease. A total MSSI score of less than 20 points indicates mild disease, of 20 to 40 points indicates moderate disease, and of more than 40 points indicates severe disease.42 The MSSI was completed at baseline and every 6 months thereafter.
Exercise tolerance (stress test): Patients underwent an exercise tolerance (stress test) to assess cardiovascular function, and tests were conducted locally according to the Bruce protocol.43,44 The stress test was conducted at baseline and every 12 months thereafter.
Frequency of pain medication use: Pain medication was recorded as part of all concomitant medications used at each study visit. The focus was on neuropathic and chronic pain, particularly the classes of anticonvulsant and antidepressant drugs.
EQ-5D-5L: This questionnaire addresses 5 dimensions: mobility, self-care, usual activities, pain or discomfort, and anxiety or depression. Patients rate their health state for each dimension based on 5 levels (no problems, slight problems, moderate problems, severe problems, and extreme problems or inability). The responses for each of the 5 dimensions are scored to describe a patient’s overall health state on a 0 to 1 scale (0 = dead; 1 = perfect health). A patient’s overall health was also rated on a visual analogue scale from 0 to 100. According to the sponsor, the EQ-5D-5L was used because there are no disease‑specific HRQoL instruments for FD. Patients completed the EQ-5D-5L at baseline and every 6 months thereafter.
Safety
The safety outcomes included the proportion of patients who reported TEAEs (coded using the Medical Dictionary for Regulatory Activities, version 19.0), SAEs, withdrawals from treatment due to TEAEs, and deaths. TEAEs included any pre-existing medical condition or adverse event that worsened in severity from the start of the study treatment to the final dose. SAEs were any untoward medical occurrence or effect that resulted in death, was life-threatening, required inpatient hospitalization or prolongation of existing hospitalization, resulted in persistent or significant disability or incapacity, was associated with a congenital anomaly, or was an important medical event.
Notable Harms
Based on discussion with the clinical experts and information from the Health Canada product monograph, IRRs and ADAs were deemed to be notable harms relevant to the current review of pegunigalsidase alfa.
IRRs were TEAEs that occurred during drug infusion or within a defined time after completion (either 2 or 24 hours) and were not considered injection site reactions.
Testing for treatment-induced ADAs or seroconversion (change from ADA-negative status at baseline to ADA-positive status postbaseline) and ADA boosting (a 4-fold or greater increase in titre compared to the baseline value in patients who were ADA positive at baseline) was conducted for each patient’s assigned drug at screening, at baseline, 2 weeks after baseline, each month from month 1 to 5, and every 3 months to the end of the study. Patients who were ADA positive were subsequently tested for neutralizing antibodies.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
Short-form BPI score | An instrument that captures the sensory and reactive dimensions of pain. | Validity and reliability: The short‑form BPI has established reliability and validity in patients with osteoarthritis, rotator cuff injury, and a range of pain conditions.45 Responsiveness: In FD, the short‑form BPI severity and interference scores were significantly different between alpha-galactosidase A and placebo groups from baseline to week 24 (P < 0.05) in an RCT.20 | A change of 1 point on the interference scale of the short-form BPI, about one‑half of its standard deviation, is a reasonable benchmark for future studies to identify an MID.46 The sponsor suggested an MID of 2 points. Neither of these has been validated in FD. |
MSSI score | A FD-specific instrument that measures the response to ERT. | Validity: There is evidence of known-group construct validity. Patients with FD scored higher on the MSSI than patients without FD (P < 0.0001). The MSSI score of patients with FD was positively correlated with age (r = 0.55). The MSSI differentiated between female and male patients with FD. Male patients with FD had significantly higher scores on the neurologic subscale than female patients both before (P = 0.035) and after (P = 0.006) 1 year of treatment with agalsidase alfa. Most male patients were rated as severely or moderately affected on the MSSI, while most female patients were rated as moderately affected. Reliability: No evidence. Responsiveness: The MSSI score demonstrated responsiveness to ERT. One year of treatment with agalsidase alfa was associated with a reduction in MSSI score (median = 9 points).47 | Not established. |
EQ-5D-5L | A generic instrument used to assess an individual’s health state in 5 dimensions (mobility, self‑care, usual activities, pain or discomfort, and anxiety or depression). | Validity, reliability, and responsiveness: No evidence in patients with FD. | Not established in patients with FD. |
BPI = Brief Pain Inventory; ERT = enzyme replacement therapy; FD = Fabry disease; MID = minimal important difference; MSSI = Mainz Severity Score Index; RCT = randomized controlled trial.
Statistical Analysis
The statistical analyses are summarized in Table 8.
Sample Size and Power Calculation
A sample size of 66 patients randomized 2:1 would provide at least 90% power to demonstrate the noninferiority of pegunigalsidase alfa versus agalsidase beta for the annualized change (slope) in eGFR. The power calculation assumed a 1‑sided, 2-sample t test with a 1-sided alpha level of 0.025 and a noninferiority margin of –3.0 mL/min/1.73 m2 per year. The true difference in slopes was assumed to be 1.1 mL/min/1.73 m2 per year favouring pegunigalsidase alfa, with an assumed SD of 1.5 mL/min/1.73 m2 per year in each arm. A 1.1 mL/min/1.73 m2 per year reduction in the rate of decline (i.e., improvement) in renal function was expected to be equal to a mean annualized change (slope) in eGFR of –1.9 mL/min/1.73 m2 per year with pegunigalsidase alfa, which represents an approximate 30% improvement — considered a clinically relevant improvement. To account for a 15% dropout rate, 78 patients needed to be randomized.
Noninferiority Margin
The sponsor did not identify an established, clinically relevant noninferiority margin for the annualized change in eGFR. Therefore, the noninferiority margin used in the BALANCE trial was based on the natural history of the disease and the published data on the effect of available treatments on renal function deterioration in patients with FD. Noninferiority was indicated if the lower bound of the 95% CI for the treatment difference (pegunigalsidase alfa minus agalsidase beta) in the annualized change in CKD-EPI eGFR was greater than or equal to –3.0 mL/min/1.73 m2 per year; this margin was based on the following:
Evidence on the natural history of FD indicated that individuals who were not treated presented with progressive kidney deterioration and an eGFR slope worse (more negative) than –3 mL/min/1.73 m2 per year (between approximately –4 mL/min/1.73 m2 per year and –12 mL/min/1.73 m2 per year). Therefore, –3 mL/min/1.73 m2 per year was deemed a relevant threshold.7,21
The European therapeutic goals used the same threshold to define patients whose kidney function was considered clinically stable (a main goal for the long-life treatment of progressive disease).7
Statistical Testing
In the BALANCE trial, the primary end point — the mean eGFR slope over the 24-month follow-up as estimated from a longitudinal mixed model — was derived from the level of serum creatinine using the CKD-EPI formula. The primary efficacy analysis compared the eGFR slope between the treatment arms using a 2-stage approach. All data points for a patient were used to estimate the slope, excluding eGFR values measured during an acute kidney injury episode, which were considered missing. At the first stage, the individual eGFR slope was estimated using a linear regression model for all patients with at least 4 eGFR measurements (after the exclusion of eGFR during acute kidney injury). For patients with fewer than 4 measurements, the slope was considered missing. At the second stage, the eGFR slopes between the 2 treatment arms were compared using quantile regression to estimate the median slopes. The dependent variable was the slope of each individual patient, and the model included the treatment arm and the intercept as covariates.
The study was originally designed to assess noninferiority at the 12-month interim analysis and the superiority of pegunigalsidase alfa to agalsidase beta at the 24-month final analysis. There was no description of adjustment for P values for multiple looks at the data.
Multiple Testing Procedure
In the BALANCE trial, the primary efficacy end points for both the interim and final analyses was the annualized change (slope) in eGFR. Both the interim and final analyses evaluated noninferiority, and the assessment used a 1-sided alpha level of 0.025. No multiplicity adjustment was made to other efficacy end points, time points, sensitivity analyses, or subgroup analyses.
Data Imputation Methods
There was no imputation for missing data for the primary efficacy analyses in the study, and missing data were assumed to be missing at random.
Sensitivity Analyses
In the BALANCE trial, sensitivity analyses were conducted on the ITT and per-protocol analysis sets. A sensitivity analysis using 2-stage model with quantile regression was repeated using the same estimand as the primary analysis and UPCR (UPCR < 1 g/g and UPCR ≥ 1 g/g) as an additional covariate. A second sensitivity analysis was conducted in which eGFR values associated with elevated serum creatinine events were excluded from the analysis; an event was defined as a 1.5‑fold or greater increase compared to the immediate previous serum creatinine value if taken within 34 days. A third sensitivity analysis used multiple imputation with the assumption that data were missing at random to assess the impact of missing data (for patients who terminated early), and missing data were imputed within each treatment group.
Subgroup Analyses
Prespecified subgroup analyses were conducted for the following:
sex (female or male)
baseline status of ADAs to the assigned study drug (negative or positive)
FD classification (classic or nonclassic)
baseline eGFR (≤ 60 mL/min/1.73 m2, between 60 mL/min/1.73 m2 and 90 mL/min/1.73 m2, or > 90 mL/min/1.73 m2)
baseline annualized change in CKD-EPI eGFR (≤ –5 mL/min/1.73 m2 per year or > –5 mL/min/1.73 m2 per year)
baseline ACEi or ARB treatment (yes or no)
region (US or not US)
UPCR categories (≤ 0.5 g/g, between 0.5 g/g and 1 g/g, or ≥ 1 g/g).
The clinical experts confirmed that all the subgroups, except region, are known TEMs in FD. One expert noted that analysis based on variant severity would be relevant, but this was not explored in the study. Based on clinical expert opinion, the subgroups were deemed unlikely to impact treatment and reimbursement decisions.
Secondary Outcomes of the Studies
For secondary end points of interest (e.g., LVMI score, short-form BPI score), the 2 treatment groups were compared and a 95% CI was calculated based on 2 independent sample t-distributions for continuous end points and on Clopper-Pearson methods for binary end points. None of the secondary end points were adjusted for multiplicity. Missing data were not imputed. Descriptive statistics (number of patients, mean, SE, SD, median, and range) were reported for continuous variables; counts and percentages were reported for categorical variables.
Table 8: Statistical Analysis of Efficacy End Points From the BALANCE Trial
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Incidence of Fabry clinical events | Descriptive statistics | None | No imputation | None |
Annualized change in CKD-EPI eGFR (slope) | Longitudinal mixed model | Covariates included the treatment arm and slope intercept | No imputation |
|
Change from baseline to 104 weeks in LVMI score (g/m2) by MRI | Descriptive statistics | None | No imputation | None |
Change from baseline to 104 weeks in short-form BPI score | Descriptive statistics | None | No imputation | None |
Change from baseline to 104 weeks in plasma lyso-Gb3 concentration | MMRM | None | No imputation | None |
Change from baseline to 104 weeks in UPCR categories | Descriptive statistics | None | No imputation | None |
Change from baseline to 104 weeks in MSSI score | Descriptive statistics | None | No imputation | None |
Change from baseline in exercise tolerance (stress test) | Qualitative evaluation | None | No imputation | None |
Change from baseline in frequency of pain medication use | Descriptive statistics | None | No imputation | None |
Change from baseline in EQ-5D-5L | Descriptive statistics | None | No imputation | None |
BPI = Brief Pain Inventory; CI = confidence interval; CKD-EPI = Chronic Kidney Disease Epidemiology Collaboration; eGFR = estimated glomerular filtration rate; LVMI = Left Ventricular Mass Index; lyso-Gb3 = globotriaosylsphingosine; MAR = missing at random; MI = multiple imputation; MMRM = mixed model for repeated measures; MSSI = Mainz Severity Score Index; UPCR = urine protein-to-creatinine ratio.
Source: Clinical Study Report for BALANCE trial;18 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Analysis Populations
The analysis populations are summarized in Table 9.
Table 9: Analysis Populations From the BALANCE Trial
Study | Population | Definition | Application |
|---|---|---|---|
BALANCE trial | ITT | All randomized patients who received ≥ 1 dose of study medication, based on the assigned treatment arm in the randomization. | Efficacy analyses |
Per protocol | All ITT patients who completed ≥ 24 months of treatment with study drug adherence of at least 80% and with no major protocol deviations that could have impacted the primary end point or were prespecified in the statistical analysis plan. | Sensitivity analyses for the primary end point | |
Safety | All patients who were randomized and who received ≥ 1 partial dose of study medication. Assignment was by treatment received. | Safety analyses |
ITT = intention to treat.
Source: Clinical Study Report for BALANCE trial;18 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Results
Patient Disposition
Patient disposition is summarized in Table 10. In the BALANCE trial, 48 of 53 patients (90.6%) randomized to the pegunigalsidase alfa group completed 24 months of treatment, and 24 of 25 patients (96.0%) randomized to the agalsidase beta group completed 24 months of treatment. Less than 10% of patients discontinued from either group, and these discontinuations were due to TEAEs or withdrawn consent.
Table 10: Summary of Patient Disposition From the BALANCE Trial (ITT Population)
Patient disposition | Pegunigalsidase alfa (N = 52) | Agalsidase beta (N = 25) |
|---|---|---|
Screened, N | 127 | |
Reason for screening out, n (%) | 49 (38.6) | |
Patient did not meet eligibility criteria | 39 (30.7) | |
Patient withdrew consent | 3 (2.4) | |
Other | 7 (5.5) | |
Randomized, N | 53 | 25 |
Exposed to treatment, n (%) | 52 (98.1) | 25 (100.0) |
Completed 12 months, n (%) | 49 (92.5) | 25 (100.0) |
Completed 24 months, n (%) | 48 (90.6) | 24 (96.0) |
Discontinued from study, n (%) | 5 (9.4) | 1 (4.0) |
Reason for discontinuation, n (%) | ||
Adverse events | 2 (3.8) | 0 (0.0) |
Withdrawal of consent | 3 (5.7) | 1 (4.0) |
ITT analysis set, N | 52 | 25 |
Per-protocol analysis set, N | 48 | 24 |
Safety analysis set, N | 52 | 25 |
ITT = intention to treat.
Source: Clinical Study Report for BALANCE trial;18 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Baseline Characteristics
The baseline characteristics summarized in Table 11 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results. The mean age of the patients was 43.9 years (SD = 10.2 years) in the pegunigalsidase alfa group and 45.2 years (SD = 9.6 years) in the agalsidase beta group. There were more males than females in both treatment groups (as part of the study design), and more than half the patients had classic disease. The mean duration of the last continuous agalsidase beta treatment was shorter in the pegunigalsidase alfa group (65.0 months; SD = 48.0 months) than in the agalsidase beta group (77.3 months; SD = 41.3 months). A larger proportion of patients in the pegunigalsidase alfa group (36.5%) than in the agalsidase beta group (24.0%) had an eGFR slope less negative than –5 mL/min/1.73 m2 per year (i.e., had slower decline in kidney function). Proportionately fewer patients in the pegunigalsidase alfa group than in the agalsidase beta group were on ACEis or ARBs (50.0% versus 64.0%) or were using premedication for infusions (38.5% versus 60.0%). Elevated UPCR, a risk factor for kidney disease progression, was also imbalanced (a larger proportion of patients in the pegunigalsidase alfa group were in higher UPCR categories).
Table 11: Summary of Baseline Characteristics From the BALANCE Trial (ITT Population)
Characteristic | Pegunigalsidase alfa (N = 52) | Agalsidase beta (N = 25) |
|---|---|---|
Demographic characteristics | ||
Age (years) | ||
Mean (SD) | 43.9 (10.2) | 45.2 (9.6) |
Median (range) | 44.0 (20 to 60) | 48.0 (18 to 58) |
Sex, n (%) | ||
Female | 23 (44.2) | 7 (28.0) |
Male | 29 (55.8) | 18 (72.0) |
Race, n (%) | ||
Asian | 2 (3.8) | 0 |
Black | 1 (1.9) | 2 (8.0) |
White | 49 (94.2) | 23 (92.0) |
Weight (kg) | ||
Mean (SD) | 77.8 (17.1) | 81.2 (18.5) |
Median (range) | 73.4 (52.0 to 129.0) | 79.3 (47.9 to 135.0) |
Region, n (%) | ||
US | 33 (63.5) | 18 (72.0) |
Not US | 19 (36.5) | 7 (28.0) |
Clinical characteristics | ||
Duration of the last continuous agalsidase beta treatment (months) | ||
Mean (SD) | 65.0 (48.0) | 77.3 (41.3) |
Median (range) | 51.5 (12.6 to 236.9) | 67.8 (27.6 to 168.3) |
FD phenotype, n (%) | ||
Classic | 27 (51.9) | 14 (56.0) |
Nonclassic | 25 (48.1) | 11 (44.0) |
eGFR (mL/min/1.73 m2) | ||
Mean (SD) | 73.5 (20.2) | 74.2 (21.0) |
Median (range) | 73.5 (30.2 to 125.9) | 74.9 (34.1 to 107.6) |
eGFR slope at baseline (mL/min/1.73 m2 per year)a | ||
Mean (SD) | –8.0 (6.6) | –8.3 (4.3) |
Median (range) | –6.7 (–30.5 to 6.3) | –7.8 (–20.3 to –2.8) |
Baseline eGFR slope category (mL/min/1.73 m2 per year), n (%) | ||
≤ –5 | 33 (63.5) | 19 (76.0) |
> –5 | 19 (36.5) | 6 (24.0) |
UPCR stratification at screening, n (%) | ||
< 1 g/g | 41 (78.8) | 21 (84.0) |
≥ 1 g/g | 11 (21.2) | 4 (16.0) |
UPCR categories at baseline, n (%) | ||
Mild proteinuria: UPCR ≤ 0.5 g/g | 36 (69.2) | 20 (80.0) |
Moderate proteinuria: UPCR between 0.5 g/g and 1 g/g | 9 (17.3) | 2 (8.0) |
Severe proteinuria: UPCR ≥ 1 g/g | 7 (13.5) | 3 (12.0) |
Treatment with ACEi or ARB, n (%) | ||
Yes | 26 (50.0) | 16 (64.0) |
No | 26 (50.0) | 9 (36.0) |
Plasma lyso-Gb3 (nM) | ||
Mean (SD) | 26.2 (27.3) | 32.1 (35.4) |
Median (range) | 15.2 (0.8 to 143.9) | 17.6 (2.1 to 142.0) |
Premedication use for infusion before enrolment, n (%) | ||
Yes | 20 (38.5) | 15 (60.0) |
No | 32 (61.5) | 10 (40.0) |
Organ system involvement, n (%) | ||
Nervous system | 50 (96.2) | 25 (100) |
Skin | 48 (92.3) | 24 (96.0) |
Cardiovascular | 41 (78.8) | 23 (92.0) |
Gastrointestinal | 41 (78.8) | 22 (88.0) |
Eyes | 41 (78.8) | 18 (72.0) |
Pulmonary | 22 (42.3) | 8 (32.0) |
ACEi = angiotensin-converting enzyme inhibitor; ARB = angiotensin II receptor blocker; eGFR = estimated glomerular filtration rate; FD = Fabry disease; ITT = intention to treat; lyso-Gb3 = globotriaosylsphingosine; SD = standard deviation; UPCR = urine protein-to-creatinine ratio.
aThe eGFR slope at baseline was based on historical, screening, and baseline serum creatinine levels.
Source: Clinical Study Report for BALANCE trial;18 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Exposure and Adherence to Study Treatments
Exposure and adherence to the study drug are summarized in Table 12. The median duration of exposure to the study treatment was similar between treatment groups, as was adherence to the study drug.
Table 12: Summary of Patient Exposure and Adherence From the BALANCE Trial (Safety Population)
Exposure | Pegunigalsidase alfa (N = 52) | Agalsidase beta (N = 25) |
|---|---|---|
Cumulative exposure (months) | 1,176.2 | 596.4 |
Exposure (months) | ||
Mean (SD) | 22.6 (5.2) | 23.9 (1.4) |
Median (range) | 24.0 (0.9 to 27.4) | 24.0 (17.7 to 26.0) |
Adherence (%), mean (SD) | 99.2 (2.0) | 99.0 (2.3) |
SD = standard deviation.
Source: Clinical Study Report for BALANCE trial;18 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Concomitant Treatments
Concomitant treatments are summarized in Table 13. Nearly all patients (96.2% and 100% in the pegunigalsidase alfa and agalsidase beta groups, respectively) received at least 1 concomitant medication. The use of concomitant treatments was generally proportionately lower in the pegunigalsidase alfa group than in the agalsidase beta group. The most common concomitant treatments were paracetamol, losartan, acetylsalicylic acid, and diphenhydramine.
Table 13: Summary of Concomitant Treatments From the BALANCE Trial (Safety Population)
Concomitant treatments | Pegunigalsidase alfa (N = 52) | Agalsidase beta (N = 25) |
|---|---|---|
Patients with ≥ 1 concomitant treatment, n (%) | 50 (96.2) | 25 (100.0) |
Concomitant treatments,a n (%) | ||
Paracetamol | 33 (63.5) | 19 (76.0) |
Losartan | 15 (28.8) | 10 (40.0) |
Acetylsalicylic acid | 14 (26.9) | 12 (48.0) |
Diphenhydramine | 14 (26.9) | 8 (32.0) |
Salbutamol | 9 (17.3) | 9 (36.0) |
Atorvastatin | 8 (15.4) | 7 (28.0) |
Cholecalciferol | 5 (9.6) | 9 (36.0) |
aUsed in at least 25% of patients in either group.
Source: Clinical Study Report for BALANCE trial;18 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Protocol Deviations
In the BALANCE trial, 73.1% of patients in the pegunigalsidase alfa group and 68.0% of patients in the agalsidase beta group had at least 1 critical or major protocol deviation. The most common critical or major protocol deviations in the pegunigalsidase alfa and agalsidase beta groups related to study procedures (46.2% versus 32.0%), laboratory assessments (42.3% versus 36.0%), visit schedule criteria (30.8% versus 24.0%), and investigational product adherence (9.6% versus 16.0%). Other critical or major protocol deviations were generally balanced between treatment groups and reported in 5 or fewer patients per group.
Efficacy
The main efficacy outcomes are summarized in Table 14.
Incidence of Fabry Clinical Events
During the 104-week study, 9 patients (17.3%) in the pegunigalsidase alfa group and 2 patients (8.0%) in the agalsidase beta group experienced a Fabry clinical event. In the pegunigalsidase alfa group, 6 patients had 7 cardiac events (atrial fibrillation, angina pectoris, increased troponin, and second-degree atrioventricular block), 3 patients had 3 cerebrovascular events (TIA and cerebrovascular accident), and 1 patient had a renal event (end-stage renal disease necessitating a kidney transplant). In the agalsidase beta group, 2 patients had 2 cardiac events (ventricular tachycardia requiring a pacemaker and atrial fibrillation).
Annualized Change in CKD-EPI eGFR (Slope)
The median annual CKD-EPI eGFR slope was –2.51 mL/min/1.73 m2 per year (95% CI, –3.79 mL/min/1.73 m2 per year to –1.24 mL/min/1.73 m2 per year) in the pegunigalsidase alfa group and –2.16 mL/min/1.73 m2 per year (95% CI, –3.81 mL/min/1.73 m2 per year to –0.51 mL/min/1.73 m2 per year) in the agalsidase beta group. The treatment group difference was –0.36 mL/min/1.73 m2 per year (95% CI, –2.44 mL/min/1.73 m2 per year to 1.73 mL/min/1.73 m2 per year) for the ITT population and –0.12 mL/min/1.73 m2 per year (95% CI, –2.45 mL/min/1.73 m2 per year to 2.21 mL/min/1.73 m2 per year) for the per-protocol population. Because the lower bound of the 95% CI for the treatment group difference was greater than the prespecified noninferiority margin of –3.0 mL/min/1.73 m2 per year, the sponsor concluded that pegunigalsidase alfa was noninferior to agalsidase beta.
The results of the sensitivity analyses using the same model with stratification by UPCR (treatment group difference = 0.28 mL/min/1.73 m2 per year; 95% CI, –1.79 mL/min/1.73 m2 per year to 2.35 mL/min/1.73 m2 per year), removing eGFR values associated with elevated serum creatinine events (treatment group difference = –0.36 mL/min/1.73 m2 per year; 95% CI, –2.52 mL/min/1.73 m2 per year to 1.80 mL/min/1.73 m2 per year), and using multiple imputation methods (treatment group difference = 0.02 mL/min/1.73 m2 per year; 95% CI, –2.04 mL/min/1.73 m2 per year to 2.08 mL/min/1.73 m2 per year) supported those of the primary analysis.
A forest plot of the subgroup analyses for the primary outcome is shown in Figure 3 of Appendix 1. Overall, there was no clear trend suggesting that 1 treatment was favoured over the other because of the wide CIs, which crossed the null.
LVMI Score
At baseline, the mean LVMI score was 75.97 g/m2 (SE = 5.13 g/m2) in the pegunigalsidase alfa group and 82.22 g/m2 (SE = 6.34 g/m2) in the agalsidase beta group. At week 104, the mean LVMI score was 71.56 g/m2 (SE = 5.2 g/m2) in the pegunigalsidase alfa group and 82.43 g/m2 (SE = 8.39 g/m2) in the agalsidase beta group. The mean change from baseline was –0.64 g/m2 (SE = 2.69 g/m2) in the pegunigalsidase alfa group and 0.29 g/m2 (SE = 3.73 g/m2) in the agalsidase beta group. The treatment group difference was –0.92 g/m2 (95% CI, –10.26 g/m2 to 8.42 g/m2).
Short-Form BPI Score
For pain at its worst in the last 24 hours, the baseline and week 104 scores were 3.5 points (SE = 0.4 points) and 3.3 points (SE = 0.5 points), respectively, for the pegunigalsidase alfa group and 2.6 points (SE = 0.6 points) and 3.0 points (SE = 0.7 points), respectively, for the agalsidase beta group. The mean change from baseline to week 104 score was –0.1 points (SE = 0.5 points) in the pegunigalsidase alfa group and 0.6 points (SE = 0.6 points) in the agalsidase beta group. The treatment group difference was –0.7 points (95% CI, –2.2 to 0.8 points).
For pain at its least in the last 24 hours, the baseline and week 104 scores were 1.1 points (SE = 0.2 points) and 1.4 points (SE = 0.3 points), respectively, for the pegunigalsidase alfa group and 1.2 points (SE = 0.4 points) and 1.3 points (SE = 0.5 points), respectively, for the agalsidase beta group. The mean change from baseline to week 104 score was 0.2 points (SE = 0.3 points) in the pegunigalsidase alfa group and 0.1 points (SE = 0.4 points) in the agalsidase beta group. The treatment group difference was 0.1 points (95% CI, –1.0 to 1.1 points).
For current pain, the baseline and week 104 scores were 1.8 points (SE = 0.3 points) and 1.9 points (SE = 0.4 points), respectively, for the pegunigalsidase alfa group and 1.5 points (SE = 0.5 points) and 1.6 points (SE = 0.6 points), respectively, for the agalsidase beta group. The mean change from baseline to week 104 score was 0.1 points (SE = 0.4 points) in the pegunigalsidase alfa group and 0.1 points (SE = 0.5 points) in the agalsidase beta group. The treatment group difference was –0.1 points (95% CI, –1.4 to 1.2 points).
For average pain, the baseline and week 104 scores were 2.2 points (SE = 0.3 points) and 2.6 points (SE = 0.4 points), respectively, for the pegunigalsidase alfa group and 2.2 points (SE = 0.4 points) and 2.5 points (SE = 0.5 points), respectively, for the agalsidase beta group. The mean change from baseline to week 104 score was 0.4 points (SE = 0.3 points) in the pegunigalsidase alfa group and 0.2 points (SE = 0.4 points) in the agalsidase beta group. The treatment group difference was 0.2 points (95% CI, –0.9 to 1.2 points).
Other Efficacy Outcomes
Other outcomes of interest, which were not assessed using GRADE (plasma lyso-Gb3 concentration, UPCR category, overall MSSI score, exercise tolerance, frequency of pain medication use, and EQ-5D-5L score), are summarized in Table 32 of Appendix 1.
Table 14: Summary of Key Efficacy Results From the BALANCE Trial (ITT Population)
Variable | Pegunigalsidase alfa (N = 52) | Agalsidase beta (N = 25) |
|---|---|---|
Incidence of Fabry clinical events | ||
Overall, n (%) | 9 (17.3) | 2 (8.0) |
Cardiac events, n (%) | 6 (11.5) | 2 (8.0) |
Cerebrovascular events, n (%) | 3 (5.8) | 0 |
Renal events, n (%) | 1 (1.9) | 0 |
Non–cardiac-related death, n (%) | 0 | 0 |
Annualized CKD-EPI eGFR slopea | ||
Patients contributing to the analysis, n (%) | 51 (98.1) | 25 (100) |
Week 104 estimated annual CKD-EPI eGFR slope (mL/min/1.73 m2 per year), mean (SD) | –2.38 (8.90) | –2.31 (3.56) |
Week 104 estimated annual CKD-EPI eGFR slope (mL/min/1.73 m2 per year), median (95% CI) | –2.51 (–3.79 to –1.24) | –2.16 (–3.81 to –0.51) |
Treatment group vs. control CKD-EPI eGFR slope (mL/min/1.73 m2 per year), median difference (95% CI)b | –0.36 (–2.44 to 1.73) | |
LVMI score by MRIc | ||
Patients contributing to the analysis, n (%) | 28 (53.8) | 19 (76.0) |
Baseline LVMI score (g/m2), mean (SE) | 75.97 (5.13) | 82.22 (6.34) |
Week 104 LVMI score (g/m2), mean (SE) | 71.56 (5.2) | 82.43 (8.39) |
Change from baseline LVMI score to week 104 (g/m2), mean (SE) | –0.64 (2.69) | 0.29 (3.73) |
Treatment group vs. control LVMI score (g/m2), treatment group difference (95% CI)d | –0.92 (–10.26 to 8.42) | |
Short-form BPI score | ||
Pain at its worst in last 24 hours | ||
Patients contributing to the analysis, n (%) | 45 (86.5) | 22 (88.0) |
Baseline BPI score (points), mean (SE) | 3.5 (0.4) | 2.6 (0.6) |
Week 104 BPI score (points), mean (SE) | 3.3 (0.5) | 3.0 (0.7) |
Change from baseline BPI score to week 104 (points), mean (SE) (95% CI)e | –0.1 (0.5) (–1.1 to 0.8) | 0.6 (0.6) (–0.7 to 1.8) |
Treatment group vs. control BPI score (points), treatment group difference (95% CI)f | –0.7 (–2.2 to 0.8) | |
Pain at its least in last 24 hours | ||
Patients contributing to the analysis, n (%) | 45 (86.5) | 22 (88.0) |
Baseline BPI score (points), mean (SE) | 1.1 (0.2) | 1.2 (0.4) |
Week 104 BPI score (points), mean (SE) | 1.4 (0.3) | 1.3 (0.5) |
Change from baseline BPI score to week 104 (points), mean (SE) (95% CI)e | 0.2 (0.3) (–0.4 to 0.8) | 0.1 (0.4) (–0.7 to 1.0) |
Treatment group vs. control BPI score (points), treatment group difference (95% CI)f | 0.1 (–1.0 to 1.1) | |
Pain right now | ||
Patients contributing to the analysis, n (%) | 45 (86.5) | 22 (88.0) |
Baseline BPI score (points), mean (SE) | 1.8 (0.3) | 1.5 (0.5) |
Week 104 BPI score (points), mean (SE) | 1.9 (0.4) | 1.6 (0.6) |
Change from baseline BPI score to week 104 (points), mean (SE) (95% CI)e | 0.1 (0.4) (–0.7 to 0.8) | 0.1 (0.5) (–1.0 to 1.2) |
Treatment group vs. control BPI score (points), treatment group difference (95% CI)f | –0.1 (–1.4 to 1.2) | |
Average pain | ||
Patients contributing to the analysis, n (%) | 45 (86.5) | 22 (88.0) |
Baseline BPI score (points), mean (SE) | 2.2 (0.3) | 2.2 (0.4) |
Week 104 BPI score (points), mean (SE) | 2.6 (0.4) | 2.5 (0.5) |
Change from baseline BPI score to week 104 (points), mean (SE) (95% CI)e | 0.4 (0.3) (–0.3 to 1.0) | 0.2 (0.4) (–0.6 to 1.0) |
Treatment group vs. control BPI score (points), treatment group difference (95% CI)f | 0.2 (–0.9 to 1.2) | |
BPI = Brief Pain Inventory; CI = confidence interval; CKD-EPI = Chronic Kidney Disease Epidemiology Collaboration; eGFR = estimated glomerular filtration rate; ITT = intention to treat; LVMI = Left Ventricular Mass Index; SD = standard deviation; SE = standard error; vs. = versus.
aThe noninferiority margin for the primary end point was –3.0 mL/min/1.73 m2 per year.
bThe analysis is based on a quantile regression for the median, with the eGFR slope of each individual patient as a dependent variable and the treatment arm as a covariate of the model. All observations are used, including unscheduled visits.
cHypertrophy was defined as an LVMI score > 77 g/m2 for females and > 91 g/m2 for males. For patients with a missing evaluation at baseline, only the outcomes at visits 27 and 53 are presented and no change from baseline is available.
dThe 95% CIs for the difference between the 2 groups are presented using the t-distribution for 2 samples.
eThe 95% CI for the change from baseline is based on a t-distribution for a paired sample.
fThe 95% CIs for the difference between the 2 arms are presented using a t-distribution for 2 samples only for visit 27 and visit 53 (week 104).
Source: Clinical Study Report for BALANCE trial;18 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Harms
Harms are summarized in Table 15.
Treatment-Emergent Adverse Events
TEAEs were reported by 47 patients (90.4%) in the pegunigalsidase alfa group and 24 patients (96.0%) in the agalsidase beta group. The most common TEAEs were headache (21.2% of patients in the pegunigalsidase alfa group versus 20.0% of patients in the agalsidase beta group), nasopharyngitis (21.2% versus 16.0%), diarrhea (19.2% versus 24.0%), back pain (15.4% versus 20.0%), cough (11.5% versus 20.0%), and bronchitis (9.6% versus 20.0%).
Serious Adverse Events
Eight patients (15.4%) in the pegunigalsidase alfa group and 6 patients (24.0%) in the agalsidase beta group reported an SAE. SAEs in the pegunigalsidase alfa group included atrioventricular block, protein-losing gastroenteropathy, hypothermia, hypersensitivity, bronchitis, contusion, femur fracture, increased hepatic enzyme, dehydration, acute kidney injury, medical device battery replacement, nephrectomy, aortic stenosis, and deep vein thrombosis. SAEs in the agalsidase beta group included atrial fibrillation, tachycardia, ventricular tachycardia, chest pain (reported by 2 patients), pneumonia, sepsis, altered state of consciousness, suicidal ideation, acute respiratory failure, and chronic obstructive pulmonary disease.
Withdrawals Due to Adverse Events
Two patients stopped treatment due to a TEAE, and both were in the pegunigalsidase alfa group. The reasons included IRR (hypersensitivity reaction) and multiple events (hypertension, renal impairment, anemia, kidney fibrosis, and decreased platelet count).
Mortality
There were no deaths in the study.
Notable Harms
Six patients (11.5%) in the pegunigalsidase alfa group and 3 patients (12.0%) in the agalsidase beta group experienced an injection site reaction. Of the 11 patients (21.2%) reporting any IRRs in the pegunigalsidase alfa group, 11 reported mild or moderate IRRs and 1 reported severe or very severe IRRs. Of the 6 patients (24.0%) reporting IRRs in the agalsidase beta group, all 6 reported mild or moderate IRRs. Six patients (11.5%) in the pegunigalsidase alfa group and 5 patients (20.0%) in the agalsidase beta group reported treatment-emergent ADAs. Of the 20 patients in the pegunigalsidase alfa group who had ADAs during the study, 15 (75.0%) had neutralizing ADAs. Of the 11 patients in the agalsidase beta group who had ADAs during the study, 9 (81.8%) had neutralizing ADAs.
Table 15: Summary of Harms Results From the BALANCE Trial (Safety Population)
Harms | Pegunigalsidase alfa (N = 52) | Agalsidase beta (N = 25) |
|---|---|---|
Most common TEAEs,a n (%) | ||
Patients with ≥ 1 TEAE | 47 (90.4) | 24 (96.0) |
Headache | 11 (21.2) | 5 (20.0) |
Nasopharyngitis | 11 (21.2) | 4 (16.0) |
Diarrhea | 10 (19.2) | 6 (24.0) |
Back pain | 8 (15.4) | 5 (20.0) |
Cough | 6 (11.5) | 5 (20.0) |
Bronchitis | 5 (9.6) | 5 (20.0) |
SAEs, n (%) | ||
Patients with ≥ 1 SAE | 8 (15.4) | 6 (24.0) |
Patients who stopped treatment due to a TEAE, n (%) | ||
Patients who stopped treatment | 2 (3.8) | 0 |
Deaths, n (%) | ||
Patients who died | 0 | 0 |
TEAEs of special interest, n (%) | ||
Injection site reaction | 6 (11.5) | 3 (12.0) |
Any IRRs | 11 (21.2) | 6 (24.0) |
Mild or moderate IRRs | 11 (21.2) | 6 (24.0) |
Severe or very severe IRRs | 1 (1.9) | 0 |
Treatment-emergent ADAs | 6 (11.5) | 5 (20.0) |
Titre boostedb,c | 3 (50.0) | 2 (40.0) |
Treatment inducedb,d | 3 (50.0) | 3 (60.0) |
ADAs at any postbaseline visit | 20 (38.5) | 11 (44.0) |
Neutralizing ADAse | 15 (75.0) | 9 (81.8) |
ADA = antidrug antibody; IRR = infusion-related reaction; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aOccurring in ≥ 20% of patients.
bPercentage is based on patients who had treatment-emergent ADAs.
cA 4-fold or greater increase in titre compared to the baseline value in patients who were ADA positive at baseline.
dA change from ADA-negative status at baseline to ADA-positive status postbaseline.
eOnly assessed in patients who were immunoglobulin G–positive (i.e., the number of patients with ADAs at any postbaseline visit).
Source: Clinical Study Report for BALANCE trial;18 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Critical Appraisal
Internal Validity
Overall, the process for generating and concealing the randomization sequence seemed appropriate. Randomization was stratified by baseline UPCR and no other characteristics, although there are many that impact FD outcomes. Patients and staff administering the drugs were blinded to the assigned treatment, and according to the clinical experts consulted for this review, unblinding was not a concern. When looking at the baseline characteristics, there were notable imbalances between treatment groups. In the pegunigalsidase alfa group, there was, compared with the agalsidase beta group, a smaller proportion of males, a shorter duration of last continuous agalsidase beta treatment, a larger proportion of patients with an eGFR slope less negative than –5 mL/min/1.73 m2 per year, a smaller proportion of patients receiving ACEi or ARB treatment, and a lower plasma lyso-Gb3 concentration among patients. Most of these differences point toward the patients in the pegunigalsidase alfa group having less severe disease and better kidney function than those in the agalsidase beta group. In contrast, a larger proportion of the patients in the pegunigalsidase alfa group were in higher UPCR categories, which is a risk factor for kidney disease progression. The clinical experts explained that patients with more advanced disease are known to experience faster disease progression and less response to treatment; thus, most of the imbalances tend to bias in favour of the pegunigalsidase alfa group. Imbalances may also be a result of the small study size (understandable given the rarity of the condition) and the heterogeneity of FD.
The BALANCE trial was a noninferiority study aiming to determine if pegunigalsidase alfa was noninferior to agalsidase beta for the primary outcome: annualized change in CKD-EPI eGFR over 2 years. For the sample size and power calculation, the difference in slopes was assumed to be 1.1 mL/min/1.73 m2 per year favouring pegunigalsidase alfa, and the noninferiority margin was –3.0 mL/min/1.73 m2 per year. According to the sponsor, the noninferiority margin was based on natural history data21 for patients with FD and on European therapeutic goals, which suggest that a patient experiencing an eGFR slope decline of 1 mL/min/1.73 m2 per year to 3 mL/min/1.73 m2 per year indicates stable function.7 According to the FDA, the natural history data were based on untreated males, and the data cannot be adjusted for differences in the BALANCE trial population.22 Additionally, the margin was not based on preserving an established minimum treatment effect for agalsidase beta versus placebo in a population similar to that in the BALANCE trial, and the assumption is that agalsidase beta had its expected effect in the noninferiority study.22 Because this assumption is unverified, the noninferiority of pegunigalsidase alfa to agalsidase beta is uncertain. The FDA examined assay sensitivity using external data to support the expected treatment effect of agalsidase beta in the BALANCE trial population.22 The FDA identified observational evidence showing that patients receiving agalsidase beta at a dose less than half of the normal dose experienced worsening eGFR and that patients receiving agalsidase beta had a slower eGFR decline than individuals who were untreated or patients who received placebo.48-50 However, limitations to this assessment include the naive comparison across studies, the nonrandomized study populations, and the varying baseline characteristics of the study populations (particularly for prior treatment status and kidney function) compared to the BALANCE trial. Moreover, it is likely that the constancy assumption has been violated in that the BALANCE trial population is different from the populations of previous agalsidase beta studies.22 The agalsidase beta studies enrolled patients who had not previously received treatment for FD and had worse kidney function than in the BALANCE study; nearly all patients in these studies had classic disease. The BALANCE trial only enrolled patients who had previously received treatment for FD and who had less severe kidney impairment than in the agalsidase beta studies; 53.2% of the patients had classic disease. Although the analysis for assay sensitivity does not validate the noninferiority margin in the BALANCE trial, the FDA indicated that this information helps contextualize the results when the noninferiority margin may be unreliable. The FDA and the European Medicines Agency also noted that, based on the evidence for using agalsidase beta to treat FD over no treatment, an acceptable and robust noninferiority margin would be so small that it would require a prohibitively large sample size that would not be feasible for a rare disease like FD.22,27
Based on expert opinion obtained for this review, 1 of the goals of FD treatment is for the patient’s eGFR slope to be the same as that of a healthy population (i.e., approximately –1 mL/min/1.73 m2 per year for patients aged 40 years and older) — a slope more negative than that would be considered abnormal. Furthermore, a change of 0.5 mL/min/1.73 m2 per year to 1 mL/min/1.73 m2 per year over 2 to 3 years has been suggested as a clinically meaningful difference in mean eGFR slopes.23 It was also suggested that this clinically meaningful difference applies to populations at high risk of progressive kidney disease and in studies that include at least 2 years of follow-up, such as the BALANCE trial. The sponsor has noted that, based on the European expert consensus statement, for patients with existing deteriorating kidney function, a response to treatment would be indicated by a reduction in annual eGFR slope to less than 3 mL/min/1.73 m2 per year.7
For a noninferiority study, differences between the 2 treatment groups (e.g., in background care, use of concomitant medications, treatment adherence, patient withdrawals, and protocol deviations) increase the uncertainty of the results and can bias toward a conclusion of noninferiority.24 There were imbalances in the proportions of patients receiving the most common concomitant medications (Table 13), in study withdrawals (Table 10), and in reports of critical or major protocol deviations (73.1% of patients in the pegunigalsidase alfa group versus 68.0% in the agalsidase beta group). Noninferiority studies often compare the results of the ITT population (patients as they were originally randomized) and the per-protocol population (patients who adhered to their randomization assignment). The results of the treatment group differences in annualized change in CKD-EPI eGFR for the ITT population (N = 77) and the per-protocol population (N = 72) were similar: –0.36 mL/min/1.73 m2 per year (95% CI, –2.44 mL/min/1.73 m2 per year to 1.73 mL/min/1.73 m2 per year) and –0.12 mL/min/1.73 m2 per year (95% CI, –2.45 mL/min/1.73 m2 per year to 2.21 mL/min/1.73 m2 per year), respectively, which supports the sponsor’s claim of noninferiority for the chosen noninferiority margin.
In the BALANCE trial, there were data missing for every outcome, there was no imputation for those data, and those data were assumed to be missing at random. This assumption is untestable and unlikely to be true for all patients who discontinued or did not have data. In the study, 5 patients (9.4%) discontinued from the pegunigalsidase alfa group: 2 discontinued due to TEAEs and 3 due to withdrawal of consent. The FDA suggested that it was reasonable to assume that data were missing at random for the latter 3 patients, but not for the 2 patients who withdrew due to TEAEs.22 When the reasons for data being missing were compared between groups for secondary outcomes of interest, early discontinuations was 1 of the main causes for imbalances.
One patient was randomized to the pegunigalsidase alfa group but did not receive the study drug and therefore was not included in the ITT analysis. For the primary outcome, data were missing for 1 patient (who was in the pegunigalsidase alfa group) due to not having at least 4 values from which to calculate the eGFR slope. Based on the available information, it was not possible to determine if the reason for not having sufficient values was random or would bias the results, but because it was a single patient, it likely does not make a meaningful difference in the interpretation of the results. The results of 3 sensitivity analyses (using the same model with stratification by UPCR, removing eGFR values associated with elevated serum creatinine events, and using multiple imputation methods) supported those of the primary analysis. For the LVMI score, the amount of missing data was large and imbalanced: 46.2% in the pegunigalsidase alfa group versus 24.0% in the agalsidase beta group. Many of the reasons were likely unrelated to the treatment (e.g., related to COVID-19, a missing baseline value), but not all. In the pegunigalsidase alfa group, 4 patients discontinued early (2 due to TEAEs, as discussed earlier) and 4 had implanted pacemakers or a cardioverter-defibrillator (likely these patients had worse cardiovascular health than the agalsidase beta group, potentially leading to bias in favour of pegunigalsidase alfa. There were missing data for the short-form BPI score (13.5% in the pegunigalsidase alfa group versus 12.0% in the agalsidase beta group), and most of the reasons indicated that the missingness was unrelated to the treatment, aside from the early discontinuations.
The results of the secondary outcomes were considered supportive. Interpretation of the secondary outcome results is complicated by the fact that there were no noninferiority margins provided by the sponsor; clinical meaningfulness was informed by clinical expert opinion or MIDs that have not been verified in FD.
The eGFR slope and the LVMI are objective measures and were centrally assessed in the study; however, 1 clinical expert noted that these outcomes can have low precision over short follow-up times. The clinical experts indicated that 2 years was adequate to produce a reliable eGFR slope but noted that it may take longer than 2 years to observe a change in patients with slowly progressing disease. The mean baseline eGFR slopes were –8.03 mL/min/1.73 m2 per year for pegunigalsidase alfa and –8.25 mL/min/1.73 m2 per year for agalsidase beta. It was noted that these values are likely based on unreliable measurements taken before baseline (relying on historical data, irregular measurements, and assessments performed at different labs) compared to those taken in the study (regular measurements, central assessment, and possibly improved adherence to concomitant medications).51 Moreover, all patients were receiving agalsidase beta for at least 1 year (with a stable dose of at least 6 months) before the study, and patients who continued with agalsidase beta showed a decline in eGFR during the study despite not changing treatment. Randomization was not stratified by baseline LVMI score, and the results are at risk of bias due to the likelihood of treatment groups being dissimilar, the small number of patients in the study, disease heterogeneity, and many patients having missing evaluations. The experts also noted that there are sex-related differences for this biomarker: females have a lower risk of LVH, and the higher LVMI scores in the study could be attributed to males and there being a higher proportion of them in the agalsidase beta group. One expert indicated that a difference of 5 g/m2 in LVMI score is clinically meaningful, though interpretation of the results is uncertain due to the wide 95% CIs and previously discussed issues that increase uncertainty.
Fabry clinical events, short-form BPI score, and harms have an element of subjectivity in their reporting, and the differences in the use of concomitant medication, study withdrawals, and critical and major protocol deviations would also be expected to affect these end points, although the direction or magnitude of the potential bias is unknown. The sponsor did not identify MIDs from the literature for any outcomes that were specific to a population with FD. Fabry clinical events were based on harms reported in the study and adjudicated by the sponsor’s medical monitor, who was blinded to treatment. The clinical experts had mixed opinions on whether the higher proportion of Fabry clinical events in the pegunigalsidase alfa group was meaningful. The short-form BPI score is a patient-reported questionnaire that produces 4 scores. There is evidence of its validity and reliability in other diseases, and the score was shown to be responsive to ERT in patients with FD. Although there was no established MID for this outcome, the sponsor suggested that a 2-point difference could be clinically meaningful. This difference being clinically meaningful has not been validated in FD, but the clinical experts were of the opinion that a 2-point change could be reasonably applied to patients with FD. They also noted that it can take years for pain to decline, whereas pain can return within months if a treatment is not effective at controlling this outcome, and this likely would be observed if pegunigalsidase alfa was ineffective.
Although most of the subgroups explored in the study were for TEMs, the analyses were small and underpowered and randomization was not preserved, putting the analyses at high risk of bias. Moreover, treatment and reimbursement decisions were unlikely to be restricted based on any particular subgroup.
Patients in the study went directly from receiving agalsidase beta before the study to their assigned study treatment, understandably without a washout period. According to the clinical experts consulted for this review, the carryover effects of the original treatment could last as long as 6 months, which could limit early changes (within the first 6 months) observed in the outcomes of patients who switched from agalsidase beta to pegunigalsidase alfa. However, the experts indicated that the 2-year study duration should be long enough to observe a treatment effect for pegunigalsidase alfa after switching.
External Validity
The BALANCE trial enrolled patients aged 18 to 60 years with declining kidney function but at least some kidney function (i.e., an eGFR slope more negative than –2 mL/min/1.73 m2 per year and an eGFR ≥ 40 mL/min/1.73 m2). All enrolled patients had received ERT for at least 1 year and had been receiving a stable dose of agalsidase beta 1 mg/kg for at least 6 months. It is unclear if any patients had previously received agalsidase alfa or migalastat. In reality, not all patients who need treatment for FD have these characteristics. There are pediatric and older adult patients who could be eligible for ERT, but these patients were not captured in the study. According to the clinical experts, it would be reasonable for patients aged 60 years and older to receive pegunigalsidase alfa, but there would need to be adequate evidence of safety and efficacy in patients aged 17 years or younger before they are treated with the drug as per the Health Canada indication. Additionally, not all individuals with FD have kidney involvement that would qualify them for entry into the BALANCE trial, and there are forms of FD (e.g., late-onset and cardiac variants) that still need to be treated. All patients in the study had previously received ERT, and there is a risk of selection bias toward patients who tolerate the clinical and nonclinical aspects of ERTs (premedication, travel to an infusion centre, duration of infusion, and biweekly administration). Patients with an indication for treatment but who do not have experience with agalsidase beta were not enrolled in the BALANCE trial; however, treatment options should be available to them. Moreover, patients and clinicians may or may not decide to switch between treatments, but this study only provides evidence for patients switching from agalsidase beta to pegunigalsidase alfa. The BALANCE trial eligibility criteria excluded patients with poorer health who, based on expert opinion, could receive the drug in clinical practice in Canada, such as those treated through renal dialysis or transplant, those with proteinuria without ACEi or ARB treatment, and those with cardiovascular events or heart failure. Overall, the generalizability of the results is less certain in populations outside the BALANCE trial eligibility criteria due to the lack of data.
Based on the baseline characteristics, the clinical experts indicated that the patients in the study are generally representative of those treated in clinical practice in Canada. The proposed Health Canada indication for pegunigalsidase alfa is for adult patients with a confirmed diagnosis of FD and does not mention the need for patients to have symptoms of end-organ involvement. Although CFDI does not decide whether individuals receive treatment or which treatment they receive, the organization’s guidelines indicate that only individuals with a confirmed diagnosis of FD and evidence of clinical disease would be treated, considering the potential burden and invasiveness of ERT and the costs associated with any disease-specific treatments.5
Input from patient and clinician groups highlighted the need for treatments that slow disease progression and improve clinical outcomes as well as FD symptoms and HRQoL. As such, the outcomes included in this report were chosen based on their ability to provide information on the most important outcomes to patients and clinicians. The BALANCE trial provides information on how pegunigalsidase alfa impacts Fabry clinical events, renal function, aspects of cardiac health, and pain, but there was limited evidence supporting the improvement of gastrointestinal issues and HRQoL.
The clinical experts indicated that the 2-year study duration is adequate for providing information on the short-term duration of switching from agalsidase beta to pegunigalsidase alfa and on the comparison of the 2 drugs but that it does not provide information on the long-term use of pegunigalsidase alfa or its long-term comparability to other treatments.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for the outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:25,26
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — the true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — the true effect is likely to be substantially different from the estimate of the effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for pegunigalsidase alfa versus agalsidase beta.
Long-Term Extension Studies
The contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
Three open-label studies provided by the sponsor (F01, F02, and F03 studies) are summarized briefly to provide evidence on patients who have not previously received ERT. An additional OLE study consisting of patients who had and had not previously received ERT (the F60 study) is also described.
F01. and F02 Studies
The F01 and F02 studies were open-label, multicentre, dose-ranging, single-arm studies of adult patients with symptomatic FD who had never received ERT or had not received ERT in the 6 months before screening. Enrolment in the F02 study required prior completion of the F01 study. Each dose category included 6 to 9 patients; the F01 study had 19 patients, and the F02 study had 16 patients. Dosages were 0.2 mg/kg, 1.0 mg/kg, or 2.0 mg/kg, IV, every 2 weeks for 12 weeks (F01 study) and for 36 weeks (F02 study). The outcomes investigated were annualized change in CKD-EPI eGFR, pain severity measured with the short-form BPI, and patient safety markers.
The F01 and F02 studies showed the efficacy and safety of pegunigalsidase alfa in patients with FD who had not previously received ERT. All patients exhibited stable renal and cardiac function with favourable trends after 12 months of treatment with pegunigalsidase alfa. Pain severity and interference scores, as assessed by the short-form BPI, indicated improvements. The safety results of this 12-month study revealed that pegunigalsidase alfa is tolerated with an acceptable safety profile. The majority of TEAEs were mild or moderate in severity, and there was a low rate of treatment-induced ADA formation (18.8% of patients). The results from these trials were consistent with those in the BALANCE and BRIDGE trials.
F03. Study: Enrolment of Patients From the F01 and F02 Studies
Patients who completed the F01 and F02 studies could enrol in an OLE study: the F03 study (N = 15). The intervention was pegunigalsidase alfa, 1 mg/kg, IV, every 2 weeks for up to 60 months. The outcomes measured in the F03 study were the same as in the F01 and F02 studies.
The F03 study reported efficacy results consistent with those previously observed in the F01 and F02 studies and showed that pegunigalsidase alfa continued to be an effective treatment in patients who have never received ERT or have not received ERT in the past 6 months. The safety and immunogenicity profile of pegunigalsidase alfa was aligned with the F01 and F02 studies.
F60. Study: OLE Study
The F60 study was an OLE study (N = 87 patients; n = 32 females; n = 55 males).29 Patients eligible for the F60 study had FD and had completed the F03, BALANCE, or BRIDGE studies. Details of study participants, interventions, outcomes, statistical analyses, and results are found in the following sections.
Populations
At the time of the submission, the F60 study was an ongoing OLE study evaluating the safety, tolerability, and efficacy of pegunigalsidase alfa at 1 mg/kg every other week, administered by IV infusion, in adult patients with FD. The total duration of the study treatment was up to 60 months, until pegunigalsidase alfa became commercially available, or at the sponsor’s discretion. Patients eligible to enrol in the F60 study had completed the BALANCE or BRIDGE studies or at least 48 months of the F03 study. Patients were excluded if a medical, emotional, behavioural, or psychological condition would, in the judgment of the investigator and medical monitor, interfere with adherence to the study requirements.
The results of the interim analyses are presented in this report, with a clinical cut-off date of July 15, 2021 (up to 52 weeks of data). The interim database lock was on June 21, 2022. At the data cut-off date, ██ ████████ were participating and enrolment was ongoing. The screening visit is the last visit of the previous study (i.e., the last study visit of the F03, BALANCE, or BRIDGE studies). Figure 1 shows the flow of patients into the F60 study from the parent studies.
Figure 1: Flow of Study Patients to the F60 Study (PB-102-F60)
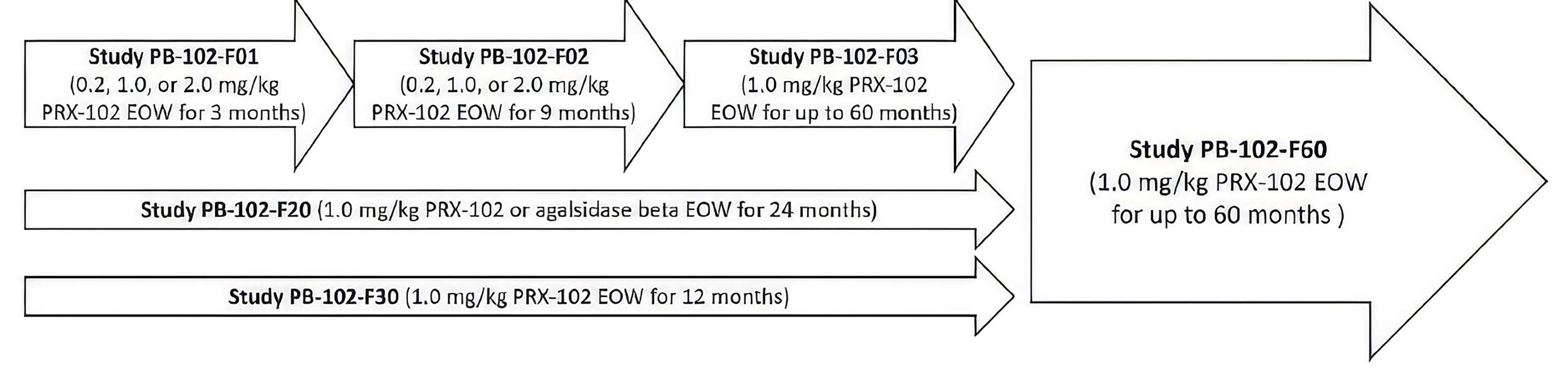
EOW = every other week; PRX-102 = pegunigalsidase alfa.
Interventions
All patients received pegunigalsidase alfa at a dosage of 1 mg/kg every other week, administered by IV infusion.
Patients who were enrolled from the F03 or BRIDGE studies could continue the same premedication regimen and infusion duration as used in the previous study; the infusion duration could not be less than 60 minutes, with a postdosing observation time of an additional 60 minutes.
All patients who completed the BALANCE trial received pegunigalsidase alfa at 1 mg/kg every other week. To not compromise the blinding of the BALANCE trial, the first infusion of pegunigalsidase alfa was administered via IV infusion over 3 hours, with 2 hours of postdosing clinical observation. From the second infusion onward, a reduction of the infusion time up to 60 minutes could be conducted in a stepwise manner depending on the patient’s tolerance, at the investigator’s discretion and after the medical monitor’s approval (if required by the protocol). The infusion was managed according to the guidance in the BALANCE study protocol for the transition of infusion time and premedication, the investigator’s evaluation, and the medical monitor’s approval. If premedication was used with the infusions in the BALANCE trial, it was continued with the first infusions in this study and then tapered down at the investigator’s discretion depending on the patient’s tolerance and according to the guidance in the study protocol. Patients returned to the previous setting for drug administration, either at home or in a predefined clinical centre, once the investigator and the medical monitor agreed that it was safe to do so.
Outcomes
The key outcomes used in the F60 study were consistent with those in the pivotal trial. The outcomes assessed at the week 52 interim analysis are listed in Table 16. Although changes in LVM and LVMI were assessed as part of the F60 study, data on these end points were not reported in this interim analysis. The definitions of the efficacy outcomes were previously outlined in the Systematic Review section.
Table 16: Summary of Outcomes From the F60 Study
Outcome measure | Time point | Type of end point |
|---|---|---|
Change in eGFR | Baseline to week 52 | Secondary |
eGFR slope | Baseline to week 52 | Secondary |
Change in short-form BPI score | Baseline to week 52 | Secondary |
TEAEs | Throughout study | Secondary |
IRRs | Throughout study | Secondary |
ADAs | Throughout study | Secondary |
ADA = antidrug antibody; BPI = Brief Pain Inventory; eGFR = estimated glomerular filtration rate; IRR = infusion-related reaction; TEAE = treatment-emergent adverse event.
Source: Clinical Study Report for F60 study;29 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Statistical Analysis
Summarized definitions of the study populations are presented in Table 17.
Table 17: Analysis Populations From the F60 Study
Study | Population | Definition |
|---|---|---|
F60 | Enrolled | All patients who provided consent |
Intention to treat | All patients who provided informed consent and received any dose (including a partial dose) of pegunigalsidase alfa | |
Per protocol | Patients who completed the study with no major protocol violations | |
Safety | All patients who provided informed consent and received any dose (including a partial dose) of pegunigalsidase alfa |
Source: Clinical Study Report for F60 study;29 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
All efficacy and safety analyses were summarized using descriptive statistics for continuous variables and using counts and percentages for categorical variables. For descriptive analyses of most continuous efficacy variables, quartiles and interquartile ranges were presented.
The baseline values for the OLE study were generally defined as the last assessment in the parent study (i.e., F03, BALANCE, or BRIDGE studies). If the last assessment was recorded more than 3 months before the last infusion in the parent study or the last assessment was not applicable, the assessment from the F60 study before the first infusion of the F60 study was used.
Results
Patient Disposition
Baseline demographics and disease characteristics for the ITT population of the F60 study are presented in Table 18. There were more males ███████ than females ███████. Overall, the mean age was ████ ██████ ███████ ████ ██ ██ ██ █████. At the baseline of the current study, ██ ███████ patients were ADA positive. The mean age at FD diagnosis in the overall ITT population was ████ █████ ███ █ ████ ██████. The last ERT before the first pegunigalsidase alfa treatment was agalsidase beta in the majority of patients (██ ███████ █████████ ███ ████████ ████ ███████), while ██ ███████ patients had received agalsidase alfa (all patients from the BRIDGE study) and ██ ███████ patients had not received prior treatment for FD (all patients from the F03 study). The mean eGFR at baseline was █████ █████████████ █ █████ ██████████. Around half the patients ███████ had an eGFR between ██ ███ ██ ███████████, indicating stage ██ CKD, according to the clinical practice guidelines for CKD.41 Furthermore, █████ of patients had an eGFR ██ ██ █████████████ █████ (including CKD stages █ or with moderate to severe reductions in eGFR), mostly comprising patients with eGFR between ██ ███ ██ ██████.
Table 18: Summary of Baseline Characteristics From the F60 Study (ITT Population)
Characteristic | Females (N = 32) | Males (N = 55) | Overall (N = 87) |
|---|---|---|---|
Demographic characteristics | |||
Age (years) | |||
Mean (SD) | ████ ██████ | ████ ██████ | ████ ██████ |
Median (range) | ████ ██████ | ████ ██████ | ████ ██████ |
Sex, n (%) | |||
Female | ████ ██████ | ████ ██████ | ████ ██████ |
Male | ████ ██████ | ████ ██████ | ████ ██████ |
Race, n (%) | |||
Asian | ████ ██████ | ████ ██████ | ████ ██████ |
Black | ████ ██████ | ████ ██████ | ████ ██████ |
White | ████ ██████ | ████ ██████ | ████ ██████ |
Height (cm) | |||
Mean (SD) | ████ ██████ | ████ ██████ | ████ ██████ |
Median (range) | ████ ██████ | ████ ██████ | ████ ██████ |
Weight (kg) | |||
Mean (SD) | ████ ██████ | ████ ██████ | ████ ██████ |
Median (range) | ████ ██████ | ████ ██████ | ████ ██████ |
Region, n (%) | |||
US | ████ ██████ | ████ ██████ | ████ ██████ |
Not US | ████ ██████ | ████ ██████ | ████ ██████ |
Clinical characteristics | |||
Age at FD diagnosis (years) | |||
Mean (SD) | ████ ██████ | ████ ██████ | ████ ██████ |
Median (range) | ████ ██████ | ████ ██████ | ████ ██████ |
Type of last ERT before first ever pegunigalsidase alfa treatment, n (%) | |||
Agalsidase alfa | ████ ██████ | ████ ██████ | ████ ██████ |
Agalsidase beta | ████ ██████ | ████ ██████ | ████ ██████ |
None | ████ ██████ | ████ ██████ | ████ ██████ |
Duration of last continuous treatment with ERT (years), n | ████ ██████ | ████ ██████ | ████ ██████ |
Mean (SD) | ████ ██████ | ████ ██████ | ████ ██████ |
Median (range) | ████ ██████ | ████ ██████ | ████ ██████ |
Pegunigalsidase alfa treatment before F60 study, n (%) | |||
Yes | ████ ██████ | ████ ██████ | ████ ██████ |
No | ████ ██████ | ████ ██████ | ████ ██████ |
Duration of the pegunigalsidase alfa treatment before F60 study (years), n | ████ ██████ | ████ ██████ | ████ ██████ |
Mean (SD) | ████ ██████ | ████ ██████ | ████ ██████ |
Median (range) | ████ ██████ | ████ ██████ | ████ ██████ |
Baseline eGFR (mL/min/1.73 m2) | |||
Mean (SD) | ████ ██████ | ████ ██████ | ████ ██████ |
Median (range) | ████ ██████ | ████ ██████ | ████ ██████ |
Baseline eGFR (mL/min/1.73 m2), n (%) | |||
≤ 60 | ████ ██████ | ████ ██████ | ████ ██████ |
≤ 30 | ████ ██████ | ████ ██████ | ████ ██████ |
> 90 | ████ ██████ | ████ ██████ | ████ ██████ |
> 120 | ████ ██████ | ████ ██████ | ████ ██████ |
Between 30 and 45 | ████ ██████ | ████ ██████ | ████ ██████ |
Between 45 and 60 | ████ ██████ | ████ ██████ | ████ ██████ |
Between 60 and 90 | ████ ██████ | ████ ██████ | ████ ██████ |
Between 90 and 120 | ████ ██████ | ████ ██████ | ████ ██████ |
Premedication at last infusion in parent study, n (%) | |||
Yes | ████ ██████ | ████ ██████ | ████ ██████ |
No | ████ ██████ | ████ ██████ | ████ ██████ |
Missing | ████ ██████ | ████ ██████ | ████ ██████ |
ADA status for pegunigalsidase alfa, n (%) | |||
Positive | ████ ██████ | ████ ██████ | ████ ██████ |
Negative | ████ ██████ | ████ ██████ | ████ ██████ |
ADA = antidrug antibody; eGFR = estimated glomerular filtration rate; ERT = enzyme replacement therapy; FD = Fabry disease; ITT = intention to treat; SD = standard deviation.
Source: Clinical Study Report for F60 study;29 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Patient Disposition
Eighty-seven patients were enrolled at the interim data cut-off: 10 patients from the F03 study, 18 patients from the BRIDGE study, and 59 patients from the BALANCE trial (Table 19). Overall, only ██████ patients had discontinued therapy at the time of the data cut-off (due to withdrawal of consent in ██████ patients and TEAEs in ██████ patients). Overall, ██ ███████ patients completed the week 52 visit.
Table 19: Patient Disposition From the F60 Study (Enrolled Set)
Patient disposition | F03 (pegunigalsidase alfa; N = 10) | BRIDGE (pegunigalsidase alfa; N = 18) | BALANCE (pegunigalsidase alfa; N = 39) | BALANCE (agalsidase beta; N = 20) | Overall (N = 87) |
|---|---|---|---|---|---|
Completed the study, n (%) | ████ | ████ | ████ | ████ | ████ |
Discontinued, n (%) | ████ | ████ | ████ | ████ | ████ |
Adverse event | ████ | ████ | ████ | ████ | ████ |
Protocol violation | ████ | ████ | ████ | ████ | ████ |
Withdrawal of consent | ████ | ████ | ████ | ████ | ████ |
Lost to follow-up | ████ | ████ | ████ | ████ | ████ |
Ongoing at the cut-off date, n (%) | ████ | ████ | ████ | ████ | ████ |
Enrolled set, n (%) | ████ | ████ | ████ | ████ | ████ |
ITT or safety population, n (%) | ████ | ████ | ████ | ████ | ████ |
ITT = intention to treat.
Source: Clinical Study Report for F60 study;29 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Exposure to Study Treatments
The cumulative total exposure to pegunigalsidase alfa (i.e., including previous studies) for patients in the safety population was ███████ months overall (Table 20). The median exposure was ████ ██████ ███████ █ ██ ██ ███████. The cumulative exposure to pegunigalsidase alfa in the safety population was ███████ months overall. The median exposure was ████ ██████ ███████ █ ██ ██ ███████.
Table 20: Summary of Patient Exposure to Pegunigalsidase Alfa From the F60 Study (Safety Population)
Exposure | F03 (pegunigalsidase alfa; N = 10) | BRIDGE (pegunigalsidase alfa; N = 18) | BALANCE (pegunigalsidase alfa; N = 39) | BALANCE (agalsidase beta; N = 20) | Overall (N = 87) |
|---|---|---|---|---|---|
Cumulative exposure across all studies (months) | ████ | ████ | ████ | ████ | ████ |
Cumulative exposure in F60 study (months) | ████ | ████ | ████ | ████ | ████ |
Exposure across all studies (months) | ████ | ████ | ████ | ████ | ████ |
Mean (SD) | ████ | ████ | ████ | ████ | ████ |
Median (range) | ████ | ████ | ████ | ████ | ████ |
Exposure in F60 study (months) | ████ | ████ | ████ | ████ | ████ |
Mean (SD) | ████ | ████ | ████ | ████ | ████ |
Median (range) | ████ | ████ | ████ | ████ | ████ |
SD = standard deviation.
Source: Clinical Study Report for F60 study;29 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Concomitant Medications and Co-Interventions
Nearly all patients in the safety population (85 patients; 97.7%) had at least 1 concomitant medication. The most common concomitant medications overall were paracetamol (50.6% of patients), acetylsalicylic acid (34.5%), and clopidogrel (26.4%).
Efficacy
The main efficacy outcomes are summarized in Table 21.
Table 21: Summary of Key Efficacy Results From the F60 Study (ITT Population)
Variable | Females (N = 32) | Males (N = 55) | Overall (N = 87) |
|---|---|---|---|
Fabry clinical events | |||
Overall, n (%) | ████ ███ | ████ ███ | ████ ███ |
Cardiac events, n (%) | ████ ███ | ████ ███ | ████ ███ |
Cerebrovascular events, n (%) | ████ ███ | ████ ███ | ████ ███ |
Renal events, n (%) | ████ ███ | ████ ███ | ████ ███ |
Non–cardiac-related death, n (%) | ████ ███ | ████ ███ | ████ ███ |
eGFR absolute values and changes from baseline to week 52 (mL/min/1.73 m2) | |||
Patients at baseline, n | ████ ███ | ████ ███ | ████ ███ |
Mean (SE) | ████ ███ | ████ ███ | ████ ███ |
Median (range) | ████ ███ | ████ ███ | ████ ███ |
Patients at week 52, n | ████ ███ | ████ ███ | ████ ███ |
Mean (SE) | ████ ███ | ████ ███ | ████ ███ |
Median (range) | ████ ███ | ████ ███ | ████ ███ |
Change from baseline to week 52 | |||
Mean (SE) | ████ ███ | ████ ███ | ████ ███ |
Median (range) | ████ ███ | ████ ███ | ████ ███ |
Percent change from baseline to week 52 | |||
Mean (SE) | ████ ███ | ████ ███ | ████ ███ |
Median (range) | ████ ███ | ████ ███ | ████ ███ |
Annualized change in CKD-EPI eGFR (mL/min/1.73 m2 per year)a | |||
Patients, n | ████ ███ | ████ ███ | ████ ███ |
Mean (SE) [95% CI] | ████ ███ | ████ ███ | ████ ███ |
Median (range) [95% CI] | ████ ███ | ████ ███ | ████ ███ |
Short-form BPI score (points) | |||
Pain at its worst in last 24 hours | |||
Baseline, n | NR | NR | 86 |
Mean (SE) | NR | NR | ████ ███ |
Week 52, n | NR | NR | ████ ███ |
Mean (SE) | NR | NR | ████ ███ |
Change from baseline to week 52, mean (SE) | NR | NR | ████ ███ |
95% CI for the change from baseline | NR | NR | ████ ███ |
Pain at its least in last 24 hours | |||
Baseline, n | NR | NR | ████ ███ |
Mean (SE) | NR | NR | ████ ███ |
Week 52, n | NR | NR | ████ ███ |
Mean (SE) | NR | NR | ████ ███ |
Change from baseline to week 52, mean (SE) | NR | NR | ████ ███ |
95% CI for the change from baseline | NR | NR | ████ ███ |
Pain right now | |||
Baseline, n | NR | NR | ████ ███ |
Mean (SE) | NR | NR | ████ ███ |
Week 52, n | NR | NR | ████ ███ |
Mean (SE) | NR | NR | ████ ███ |
Change from baseline to week 52, mean (SE) | NR | NR | ████ ███ |
95% CI for the change from baseline | NR | NR | ████ ███ |
Pain on average | |||
Baseline, n | 54 | ████ ███ | ████ ███ |
Mean (SE) | ████ ███ | ████ ███ | ████ ███ |
Week 52, n | ████ ███ | ████ ███ | ████ ███ |
Mean (SE) | ████ ███ | ████ ███ | ████ ███ |
Change from baseline to week 52, mean (SE) | ████ ███ | ████ ███ | ████ ███ |
95% CI for the change from baseline | ████ ███ | ████ ███ | ████ ███ |
BPI = Brief Pain Inventory; CI = confidence interval; eGFR = estimated glomerular filtration rate; ITT = intention to treat; NR = not reported; SE = standard error.
aThe individual annualized mean change in eGFR (slope) was estimated for each patient with ≥ 1 year of follow-up data using a linear regression model and excluding any eGFR values measured during an acute kidney injury episode.
Source: Clinical Study Report for F60 study;29 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Fabry Clinical Events
In the F60 study, ██ ███████ patients reported ██ Fabry clinical events up to the data cut-off date. Cardiac events were the most common ███████, followed by cerebrovascular events ██████, non–cardiac-related death ██████; there were no renal events. Overall, there was a higher incidence of events in males than in females.
Change in eGFR and eGFR Slope From Baseline to Week 52
The mean eGFR was █████ █████████████ █ ████ ██████████ at baseline and decreased ██ █████ █████████████ █ ████ ███████████ at week 52. The mean change from baseline at week 52 of █████ █████████████ █ ████ ███████████. The absolute mean eGFR values at baseline and week 52 tended to be lower in males (█████ █████████████ █ ████ ███████████ and █████ ██████████████ █ ████ ███████████, respectively) than in females (█████ ██████████████ █ ████ ███████████ and █████ ██████████████ █ ████ ███████████, respectively). The mean change from baseline to week 52 ███ █████ █████████████ █ ████ ██████████ in males and █████ █████████████ █ ████ ███████████) in females.
Annualized changes in CKD-EPI eGFR were estimated for each patient with at least 1 year of follow-up data. The mean annualized change in CKD-EPI eGFR in the overall ITT population was █████ ███████████████ ███ █ ████ ████████████████). The annualized change in CKD-EPI eGFR declined in male and female patients, with a slightly lower slope loss in male patients (mean = █████ ███████████████ ███ █ ████ ███████████████ than in female patients (mean = █████ ███████████████ ███ █ ████ █████████████████).
Short-Form BPI Score
Pain severity domain results at week 52 in the overall ITT population had mean changes from baseline to week 52 of ████ ██████ ███ █ ████ ███████ for worst pain, ████ ██████ ███ █ ████ ███████ for least pain, ████ ██████ ███ █ ████ ███████ for pain right now, and ████ ██████ ███ █ ████ ███████ for average pain.
The short-form BPI score showed no major changes from baseline to week 52 in pain severity domains █████ ███████ █ ████ and pain interference domains █████ ███████ █ ████ for the 53 of 87 patients with data. The majority of patients with available data in the ITT population ███ ██ ██ ████████ ████████ had improvement or no change in average pain severity compared to baseline at week 52.
Other Efficacy Outcomes
Other outcomes of interest (e.g., UPCR category, overall MSSI score, EQ-5D-5L score) are summarized in Table 32 of Appendix 1.
Harms
Harms data are summarized in Table 22.
Most patients ███████ reported at least 1 TEAE by week 52 in the F60 study. The most common TEAEs (occurring in █████ of patients) were arthralgia, cough, and coronavirus infection.
SAEs occurred in █████ of patients by week 52. The SAEs reported more than once included acute myocardial infarction ██████, cerebrovascular accident ██████, and sepsis ██████.
TEAEs leading to withdrawal from treatment (cardiac failure, sudden death, and cerebrovascular accident) occurred in 3.4% of patients, and all events were fatal.
IRRs were analyzed according to 2 time frames: IRRs occurring during infusion or within 2 hours postinfusion and IRRs occurring within 24 hours postinfusion. A total of ███████ IRRs within 2 hours postinfusion were reported in ██████ patients. ███████ IRR took place between 2 and 24 hours postinfusion for a total of ██████ ██ IRRs within 24 hours postinfusion reported in ██████ patients.
A total of ██ ███████ patients in the overall safety population were ADA positive at any postbaseline visit in the F60 study up to the interim analysis cut-off date. Treatment-emergent ADAs were observed in ██ ███████ patients, including ██████ patients with titre-boosted ADAs and ██████ patients with treatment-induced ADAs.
Table 22: Summary of Harms Results From the F60 Study Up to Week 52 (Safety Population)
Harms | Safety population (N = 87) | |
|---|---|---|
Number of patients (%) | Number of events (rate)a | |
Most common TEAEsb | ||
Patients with ≥ 1 TEAE | ██ ██████ | ██ ██████ |
Arthralgia | ██ ██████ | ██ ██████ |
Cough | ██ ██████ | ██ ██████ |
Coronavirus infection | ██ ██████ | ██ ██████ |
Headache | ██ ██████ | ██ ██████ |
Back pain | ██ ██████ | ██ ██████ |
Diarrhea | ██ ██████ | ██ ██████ |
Fatigue | ██ ██████ | ██ ██████ |
Pain in extremity | ██ ██████ | ██ ██████ |
Vomiting | ██ ██████ | ██ ██████ |
Pyrexia | ██ ██████ | ██ ██████ |
Nasopharyngitis | ██ ██████ | ██ ██████ |
Abdominal pain | ██ ██████ | ██ ██████ |
Bradycardia | ██ ██████ | ██ ██████ |
Infusion site extravasation | ██ ██████ | ██ ██████ |
SAEs | ||
Patients with ≥ 1 SAE | ██ ██████ | ██ ██████ |
TEAEs leading to treatment discontinuation | ||
Patients who stopped treatment | ██ ██████ | ██ ██████ |
Death | ||
Patients who died | ██ ██████ | ██ ██████ |
Adverse events of special interest, n (%) | ||
Injection site reactions | ██ ██████ | ██ ██████ |
IRR 2 hours postinfusion | ██ ██████ | ██ ██████ |
ADA positive at any postbaseline visit | ██ ██████ | ██ ██████ |
Treatment-emergent ADAs | ██ ██████ | ██ ██████ |
Titre boosted | ██ ██████ | ██ ██████ |
Treatment induced | ██ ██████ | ██ ██████ |
ADA = antidrug antibody; IRR = infusion-related reaction; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aThe rate was calculated as the adjusted number of events to 100 person-years exposed.
bTEAEs reported for ≥ 5% of patients in either treatment group.
Source: Clinical Study Report for F60 study;29 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Critical Appraisal
Internal Validity
The description of patients in the F60 study aligned with patients in the F01, F02, F03, BALANCE, and BRIDGE trials. Overall, the F60 study had a small sample size (N = 87), which is common for rare disease studies, and required patients to have completed the previous studies for enrolment. Because they had completed the requisite studies (100% of patients from the F03 study, 81.9% from the BALANCE trial, and 90% from the BRIDGE study), the patients who elected to continue receiving pegunigalsidase alfa and enrolled in the F60 study were a highly enriched and selected sample. Given that only 11.5% of the F60 study patients had not previously received treatment for FD, the majority of patients in the study were likely to have experienced positive responses to and good tolerance of pegunigalsidase alfa. The largest percentage of patients came from the BALANCE trial (39 [44.8%] randomized to pegunigalsidase alfa and 20 [23.0%] randomized to agalsidase beta); the next largest percentage was from the BRIDGE study (18 [20.7%] received pegunigalsidase alfa), and the smallest percentage was from the F03 study (10 [11.5%] received pegunigalsidase alfa). The open-label design of the F60 study meant that patients were not blinded to treatment and may have reported more favourable patient-reported outcomes in HRQoL and pain and may have been less likely to report TEAEs in the follow-up period due to their knowledge of the treatment. There was a potential imbalance of baseline characteristics between females and males, which would be expected to contribute to some of the observed differences in outcomes. The sponsor did not note the percentage of patients who had classic FD among the sexes, which is a key effect modifier. It is likely that a larger percentage of classic disease in males than in females is the reason that males were diagnosed younger and have more compromised kidney function, as well as why a significantly higher percentage have positive ADA status.
There is a potential 6-month treatment carryover effect in patients who switched treatments from agalsidase beta to pegunigalsidase alfa, according to the clinical experts consulted for this review. It is unclear if a patient’s response to pegunigalsidase alfa was related to their prior drug doses used in the F01 and F02 studies (whether they were receiving the 0.2 mg/kg or 2.0 mg/kg doses) compared with the F60 study dose of 1.0 mg/kg. The lack of a comparator is a major limitation in this study. An ideal comparator is a similar active comparator (e.g., agalsidase beta) used for the same indication and place in therapy. In the absence of an active comparator, this study is susceptible to residual confounding.30
The outcomes in the F60 study were the same as those in the BALANCE trial, except that LVMI score was not reported. Because the F60 study was a single-arm extension study without a comparator, the change in scores from baseline to week 52 are difficult to interpret. There were notable sex-based differences in absolute change in eGFR from baseline to week 52 and in annualized changes in CKD-EPI eGFR. Females had a larger absolute change in eGFR from baseline to week 52 ██████ ██████████████ █ ████ █████████████ than males ██████ ██████████████ █ ████ █████████████.
Compared with males, females had a ████ ████████ mean eGFR slope and lower 95% CI, suggesting that females had a █████ ████ ██ ██████ ████████ ███████: females █████ ███████████████ ███ █ ████ ████████████████ ███ ███ ████ ██ ████ ████████████████ compared to males ███████████████████ ███ █ ████ ████████████████ ███ ███ ████ ██ ████ ████████████████. Those with FD who are untreated are expected to present with progressive deterioration and an eGFR slope more negative than ████ ███████████████; the mean eGFR slope and the lower bound 95% CIs of both sexes is lower than that, suggesting that the mean eGFR slope and the lower bound of effectiveness may be similar to natural deterioration without treatment.
Females had a smaller change in pain measured using the short-form BPI from baseline to week 52, albeit with greater uncertainty, where mean values were ████ ██████ ███ █ ████ ███████ ███ ███ ████ ██ ███ ███████ for females and ████ ██████ ███ █ ████ ███████ ███ ███ ███ ██ ███ ███████ for males. Given that the MID for the short-form BPI score is estimated to be 1 to 2 points,46 the mean change in pain scores for males and females is ████ than the MID. In females, the upper bound of the 95% CI of the short-form BPI ███████ the MID, suggesting there may be █████████ ██ ████ from baseline to week 52 that is clinically meaningful. This worsening was not observed in males.
At baseline, the distribution of sexes was ██ females and ██ males. There was a smaller proportion of females than males with eGFR measures at 52 weeks (██ females and ██ males for change from baseline in eGFR; ██ females and ██ males for annualized change in CKD-EPI eGFR). Similarly, a smaller proportion of females than males had short-form BPI measurements at week 52: ██ females and ██ males. The smaller sample sizes of females at 52 weeks is 1 reason that SE measurements are higher in females, reflecting greater uncertainty. The substantial number of patients without eGFR absolute values and changes from baseline to week 52, annualized changes in CKD-EPI eGFR, and short-form BPI measurements might explain the observed changes in measurement. Participants with worse outcomes may have elected to discontinue treatment or discontinue being actively followed for assessments.
IRRs within 2 hours postinfusion and treatment-emergent ADAs were relatively infrequent. IRRs within 2 hours postinfusion were reported in ██████ patients, while treatment-emergent ADAs were reported in ██ ███████ patients, both lower than in the BALANCE trial. A possible explanation for the lower number of events in the F60 study could be selection bias: individuals who previously experienced IRRs and ADAs in the F01, F02, and F03 studies may have dropped out before enrolling in the F60 study.30
External Validity
The Health Canada indication under review is for long-term ERT in adult patients with a confirmed diagnosis of FD, which the F60 study aims to address. Patients enrolled in the F60 study included those previously treated with ERT (from the BALANCE and BRIDGE trials) and 10 patients (11.5%) who had not previously received ERT (from the F03 study). Given that only 10 patients had not previously received ERT, subgroup analyses were not feasible. Therefore, the results of the F60 study do not allow for meaningful conclusions to be drawn for this subgroup.
Eighteen patients from the BRIDGE study switched from agalsidase alfa 1 mg/kg to pegunigalsidase alfa 1 mg/kg in the F60 study. According to expert opinion, the practice of switching between ERTs varies across physicians; therefore, the results from patients who switched ERTs are limited in external generalizability, particularly in clinical contexts where patients do not typically change treatments.
The F60 study enrolled 32 females and 55 males; therefore, the study includes a larger proportion of participants with an expected worse prognosis than the estimated relative frequency of FD in the general population of 1 in 117,000 in females and 1 in 40,000 in males.14 Given the X-linked nature of FD, males have earlier disease onset, which is often more serious, and they have a worse prognosis. Therefore, females who enrol in clinical studies tend to do better than males. Yet there was a sex-based difference in mean annualized changes in CKD-EPI eGFR; the slope for females was ████ ████████ than males: females █████ ███████████████ ███ █ ████ ████████████████ ███ ███ ████ ██ ████ ████████████████ and males █████ ██████████████ ███ █ ████ ████████████████ ███ ███ ████ ██ ████ ████████████████. There are 2 hypothesized explanations for this outcome according to the clinical experts. First, the ████ ████████ █████ observed in females could be due to a larger proportion of females with an eGFR greater than 90 mL/min/1.73 m2 at baseline; a decline in eGFR to less than 90 mL/min/1.73 m2 could lead to a relatively large decline in eGFR. Second, the smaller slope observed in males suggests that there could be males with nonclassic disease, and these males would be expected to have better outcomes than those with classic disease. The absence of reporting classic versus nonclassic disease in the female and male subgroups is a limitation to adequately interpreting these results.
Indirect Evidence
The contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Objectives for the Summary of Indirect Evidence
The BALANCE trial provided a direct comparison between pegunigalsidase alfa and agalsidase beta among adults aged 18 to 60 years with symptomatic FD. Still, there is an absence of direct comparative efficacy and safety evidence between pegunigalsidase alfa and other treatments for FD. The sponsor submitted an NMA and unanchored PAICs (using STC methods) between pegunigalsidase alfa and agalsidase beta, agalsidase alfa, or migalastat to address this gap in the evidence.
Description of ITC
One NMA was submitted that estimated the relative effectiveness of pegunigalsidase alfa versus other treatments for FD. The study selection criteria and methods used in the sponsor-submitted ITC are summarized in Table 23.
Table 23: Study Selection Criteria and Methods Used in the ITC Submitted by the Sponsor
Characteristics | Clinical trials SLR | RWE SLR |
|---|---|---|
Population | Patients with FD, irrespective of age and previous treatment status | Patients diagnosed with FD (Anderson FD, Anderson disease, Fabry syndrome, alpha-galactosidase deficiency, Fabry dyslipidosis) |
Interventions |
|
|
Comparators |
|
|
Outcomes |
|
|
Study designs |
|
|
Publication characteristics |
|
|
Exclusion criteria |
|
|
Databases searched |
| |
Selection process | The selection process was performed in 2 phases:
| |
Data extraction process | Data were extracted by a single reviewer in the preagreed data extraction template and independently checked against the original study report by a second reviewer. Multiple publications identified for the same study, setting, and reporting data, for the same intervention, were linked together and extracted as a single reference. Full-text articles, irrespective of the publication year, were extracted along with the linked publications. | |
Quality assessment | Two independent reviewers performed a descriptive quality assessment of the included RCTs using comprehensive assessment criteria based on the recommendations in the NICE manufacturer’s submission template. The assessment of the included non-RCTs was performed using a checklist by Downs and Black. | |
ACEi = angiotensin-converting enzyme inhibitor; ACP = American College of Physicians; ADA = antidrug antibody; AMI = acute myocardial infarction; CDSR = Cochrane Database of Systematic Reviews; CENTRAL = Cochrane Central Register of Controlled Trials; CNS = central nervous system; DARE = Database of Abstracts of Reviews of Effects; eGFR = estimated glomerular filtration rate; ERT = enzyme replacement therapy; ESRD = end-stage renal disease; FD = Fabry disease; Gb3 = globotriaosylceramide; GL-3 = globotriaosylceramide; GI = gastrointestinal; HRQoL = health-related quality of life; HTA = health technology assessment; IRR = infusion-related reaction; ITC = indirect treatment comparison; LVEDD = left ventricular end diastolic distance; LVMI = Left Ventricular Mass Index; lyso-Gb3 = globotriaosylsphingosine; MSSI = Mainz Severity Score Index; NICE = National Institute for Health and Care Excellence; PK = pharmacokinetic; RCT = randomized controlled trial; RWE = real-world evidence; SLR = systematic literature review; TIA = transient ischemic attack.
Source: Sponsor’s ITC technical report;52 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
ITC Design
Objectives
The sponsor-submitted ITC aimed to estimate the efficacy of pegunigalsidase alfa relative to other treatments for FD.
Study Selection Methods
The sponsor conducted 2 SLRs: 1 for clinical trials (conducted in May 2022 and updated in April 2023) and 1 for RWE studies (conducted in August 2022 and updated in April 2023), in accordance with the methodological guidelines and requirements of the National Institute for Health and Care Excellence and the Cochrane Handbook.53,54 Details of the study selection methods are summarized in Table 23. According to the sponsor, the relevant comparators for the ITC included agalsidase alfa, agalsidase beta, and migalastat because these drugs are approved by Health Canada and funded by the public drug plans for the treatment of FD, though the sponsor noted that other comparators were included in the search.
Analysis Methods
████████ ███████ ███ ██████████ ██ █████ ███ █ ███████████ ██████████ ███ █████████ ██ ████████ ██ ███████████ ██ ███████ ███ █████████ ██ ███ ████ ███ ████ ████ █████████ █████ ███ ██████████ ███ █████ ███ ███████ ███ ███████████ ██ █████ ████ █████████ ███████████ ██ ████ ██████ █████ █ ████ ███ ███████████████ ████ ████ █████████ █████ █ ████████ █████████ ████ ██████ █████ █████ █████ ██████████ ████████ █████ ███████ ████ ███ █████ █ ██████ ████ █ ███████ ██████ ██ ██████ ███████████ ███████████ ███ ████████ █████ ███ ███████████████████ ██████████ ███ ███████ ███████████ ██████████ ██████ ██ ███████ ██████████ ███ ████ █████ ███ ████ ██ ██████ ███ ████████ ██ ██████ ████ ██████ ██ ████████ ████ ███ ██ █████ █ ███████ █████████ ███ █████████████████████ ████████ █████ ██████ ██████████ ████ ███ ███ ██████ ██ ██ ████████ ███ ██████ ████ █████ █ █████████ █████ ██ ██████████ ████ ████ █████ ███ ███ ██████ ███ ██ ██ ████████ ██ █ ██████ ████ ██ █████ ██ ██████ █ ██████████ ██ ███ ████████ █████████ ███ █████ ███████ ████ █████ ███ ███████ ████████ ███████ ███ █████ ███ ███ ████████ ██████ ████ █████████ █████████ ██████ █ ███ █████ █████ ██ ████████ ██ ███ ███████ ██ ██ ████████ ███ █████ ███ ████████ ██ ███ ███████ ██ ████████████ ████ ███ ██████████ ██ ███ ███ ████████ ███████████ ████████ ████ █████████ ██ ████████ ███ ████████ ███████ ████ ████ ███ ████ ███ ███ █████████ ████ ████ ███ ████ ████ ███ █████████ ███ █ ██████████ ████ █████ ████ █ ██████ ███████████████████████ ████████ ███████████ ██████████████ ███ █████ ████████ ███ █████████ ████████ ██████ ███ ███████████████████ ███████ ███████ ███ █████████ ██████ █████ ████ ██████ ███ █████ ██ ██████ ███████████████████ █████████ ███████████ ██ ██████████ ██████ ██████████ ██ ███ ████ ██ ██████████ ████ ████████ ████ ████████ █████████ ███ ██████ ███ ████ ██████████ ████████ ███████ ████ ███ ███ ███████ ████ ███ ██████ ██ ███ ██████████ ███████ ████ ██ ███ ███████████████ ████ █████████ ███ ██████████████████ ██ ███ ██████████ █████ ████████ ██ ████████████ ██ ████████ ████ ███ ███ ███████ ██ ████ ██████████ ██████ ███ ████████ ███████ ██ ███ ████████ █████████████████████ ████ ████████ ██ ███ ██████████ ███████ ███████████ ███ ██████████ █████████ ███ █████████ ██ █████ ███ ████ █████████ ██████ ████████ ██ ███ ██████████ █████████████ ████ ███ ██████████ ████████████ ██ █ █████ ███ ███ █████ ████ █ ███ ███████ ███████ ███ ███████ █████ ███ █████████ ██ ████ ██ ███ █████ ██████ █████████ ███ ██████████████████ ██ ███ ██████████ █████ ████████ ██ ████████████ ██ ████████ ████ ███ ███ ███████ ██ ████ ██████████ ██████ ███ ████████ ███████ ██ ███ ████████ █████████████████████ ████ ████████ ██ ███ ██████████ ███████ ███████████ ███ ██████████ █████████ ███ █████████ ██ █████ ███ ████ █████████ ██████ ████████ ██ ███ ██████████ █████████████ ████ ███ ██████████ ████████████ ██ █ █████ ███ ███ █████ ███████ ████ ████ ██████████ ████████ ████ ███████ █████ █████ ███ ██ ██████████ ██████████ ████████ ██ █████ ████████ ████████████ ███ ███████████████████ ████████.
Table 24: ITC Analysis Methods
Methods | Description |
|---|---|
Analysis methods | ███ ███ ███ █████████ █████ █ ████████ █████████ ████ ████ ██████████ ███████ ███████████ ██ ████ █████ ███ ███ ██████ ███████ ██ ██ |
Priors | ███████████████ ██████ ███ ██████ ████ ████████ ██ ███ ██████████████ ████████ ███████ ███ █████ ████████ ██████ ███████████ █ ███████ ███████ ██ █████ ███ ████████ ██ ███ █████████████ ████████ █████████ █ |
Assessment of model fit | ███ ████ ████ ██ ███ ██ ██████ ████ █████ |
Assessment of consistency | ██████ ██ ███████ |
Assessment of convergence | ███████████ ██ ███ ███ ███ ████████ █████ ███ ███████████████████ ██████████ ███ ███████ ██████ |
Outcomes | ██████████ █████████ ██████ ████ ████████ ██ █████ █████ ███ ███ ████████ █████ █ ██████ ██████████ ███ ████████ ███████████ █████████ ████ ████████ █████ █ |
Follow-up time points | ████ ███████ █ ███ ██ █████████████████ ████ ██████ ████ ████ █████ ███████ ██ ███ ██ ████████████ ███████ █ ███ ██ ███████████ ███████ █ ███ ██ ██████ |
Construction of nodes | ████ ████ ███████████ █ █████████ ████ █ ██████ ██████ ██████ ███ ███ █████████ ██████████ ████ ███ █████ ██████ ███ ██████████ ████ ███ █████ ███████ ███ ██████████ ████ ███ ██████████ █ ███ ███████ |
Sensitivity analyses | ███ ███ ████ ███████ ███████ ████ ███ █████ ███ █████████ ██████████ ████ █████ ████ ████████ ████ █ █████████ ███ ████ ███████ █████████ ████ ███ █████ ███ █████████ |
Subgroup analysis | ███ ██████████ |
Simulated treatment comparison | ██████████ ████ ████ █████████ ██ ██████ ███ ██████████ ████████████ ██████████ ██████ ████████ ███ █████ |
ADA = antidrug antibody; BPI = Brief Pain Inventory; DIC = deviance information criteria; eGFR = estimated glomerular filtration rate; FE = fixed effects; Gb3 = globotriaosylceramide; IPD = individual patient data; ITC = indirect treatment comparison; LVMI = Left Ventricular Mass Index; MCMC = Markov chain Monte Carlo; NMA = network meta-analysis; PV = prognostic variable; q.2.w = every other week; RE = random-effects; RWE = real-world evidence; STC = simulated treatment comparison; TEM = treatment effect modifier.
Source: Sponsor’s ITC technical report;52 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Results of the ITC
Summary of Included Studies
████ ███ ████████ ████ █████ ████ ███ ███████ ████ ██ ████████ ████████ █████████ ██ ████ ███ ██ █████████ ██ ███ ██ █████ █ ████ ████████ ████ ███ ███████████ ███████████ █████ ██ ███ █████████ ████ ███ ███ ██████ ██ ██ ███████ ██ ████████ ████ ███ ████ ████████ ██ ███████ ███ ████ █████████ ███ ████████ ██ ████████ ████████ ██████ ████ ████████ ██ █████ ██████████ ████ ██████ ██████ ████ ████████ ██ █████ ██████ ████ ████████ ██ ████ █████ ███ █████ ███ ███████ ███████ ██ ████ ████████ ███ ███ ███ ██ ██ ██████ ██████ ███ ███ ████ █████ ████ ███ ███████ ███████████ ███ ███ ████ ██████████ ██████████ ███ █████████ ██ ███ ███ ████ ███ ████ ███████ █████ ███ ██ ███████████████ █████ ███████ ███ ███████████ ████████ ███████ ███ ██████ ██ █████████ ███████ ███ ██ ████████ ████ ██████████ ██ ███ ████ ██ ███ ██ ███████ ███████████ ██ ████ ███████████ ██ ███ ██ ██████████ █ ████████ ███████████████ ███████████ ███████████████ ██ ███ ████ ████████ ███ ███ ███ ███ ██████████ ██ █████ ███ ████ ██ ███ █ ███████ ██████████ █████ ███ ███ █████████ █ ████ ███████████ ███ ██████ █████ ███ ████ ████ ███ ███ ████ ██████ ███ ███████ ████ █████ █████████ ████████ ██████ ████ █ ██████ ██ ██ ███████ █████ ███████ ███████ █████████ █████████ ██████ ██████ ███ █████ ██ ██ █████████████ ███████████████ ██ ████████ ██ ███ ████ ███ ██████████ ██ █████ ███ █████ ██ ███ █ ███████ ████████ ████████ ███ ███ █████ █████████ █████████ ████ ███ ██ ████████ ██████ ████ ██ ██ ██ █████ ██████ ███████ ███ ███ ███████████ ██ ████ ███ ████ ████████ ████ ████ █████████ ████ ████ ███ █████ ██ ██ ████ ██████ ████ ████ ██████ ████ ██ ██ ███ ████████████████ ████████ ███ ██████████ ██ ████████ ███ ███ █████████ ████████ ████ ███ ████ ████ █████ ███ █████████ ██ ██████████ ██ ███████████ ███ ███ ███████ ████████ ██ ███ ███ ██ ██████████ ██ █████ ██.
Figure 2: Global Network Diagram of RCTs Eligible for the ITC
EOW = every other week; ERT = enzyme replacement therapy; OD = once daily; Peg. = pegunigalsidase.
Source: Sponsor’s ITC technical report;52 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Table 25: Assessment of Homogeneity for the ITC
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Disease severity | The mean time since disease diagnosis ranged from 5 to 15 years and was not reported in 3 studies. The proportion of males in treatment groups ranged from 32% to 100%. The proportion of patients with classic phenotype ranged from 36% to 64% but was not reported in 4 studies. The mean eGFR ranged from 52.4 mL/min/1.73 m2 (SD = 17.7 mL/min/1.73 m2) to 106.3 mL/min/1.73 m2 (SD = 36.8 mL/min/1.73 m2) across the studies. |
Treatment history | Two studies required all patients to have been previously treated with an ERT before enrolment (BALANCE trial and Hughes 2017),18,60 1 study only allowed patients to have been previously treated if they stopped treatment ≥ 6 months before enrolment (Germain [2016]),61 and 1 study explicitly did not allow patients to be previously treated with an ERT (Banikazemi [2007]).62 The sponsor assumed that the remaining studies did not allow prior treatment with an ERT (Hughes [2008], Schiffmann [2001], Vedder [2007]).20,63,64 |
Trial eligibility criteria | Diagnosis of FD based on enzyme activity varied across studies. The 2 studies for migalastat required all patients to have migalastat-amenable variants (Germain [2016], Hughes [2017]).60,61 One study recruited patients with a linear eGFR slope more negative than –2 mL/min/1.73 m2 per year based on ≥ 3 creatinine values over 9 to 18 months (BALANCE trial).18 The eGFR slope was not reported in other studies. In 6 of 7 studies, patients were excluded if they had a history of renal dialysis or transplant; this criterion was unreported in 1 study (Hughes [2008]).63 Cardiac and cerebrovascular events were not permitted within 3 to 6 months of study enrolment for 3 studies (BALANCE trial, Banikazemi [2007], Hughes [2017])18,60,62 and were not reported in the others. Congestive heart failure NYHA Class IV was not permitted in 4 studies (BALANCE trial, Banikazemi [2007], Hughes [2008], Hughes [2017])18,60,62,63 and was not reported in the others. |
Dosing of comparators | Two dosages for agalsidase beta were included in the NMA (1.0 mg/kg q.2.w. and 0.2 mg/kg q.2.w.). To connect the networks for eGFR and LVMI outcomes, the dosages had to be combined into a single node. This implies that the dosages have similar efficacy and safety, which is not likely valid.65 |
Placebo response | Not reported. |
Definitions of end points | The eGFR was measured using different methods (e.g., CKD-EPI equation, iothalamate and hippuran or iohexol infusions, and creatinine clearance). The Vedder (2007)64 study only reported LVM, and LVMI had to be imputed using the mean body surface area of all patients in the BALANCE trial. The Hughes (2008)63 study did not report a measure of variance for mean change in LVMI; therefore, imputation was performed using the mean of the variances in the BALANCE trial. Different BPI scores were used (i.e., worst pain in last 24 hours and worst pain in past week) to connect the treatments. |
Timing of end point evaluation | Time points varied between 3 and 24 months. The longest treatment duration ranged from 6 to 35 months. |
Withdrawal frequency | Not reported. |
Clinical trial setting | The oldest study was Schiffmann (2001),20 and the most recent study was the BALANCE trial (2024).39 Studies took place in Argentina, Australia, Canada, Egypt, Europe, the Netherlands, Norway, and the US. One study was a single-centre study, 5 were multicentre, and 1 was unreported. |
Study design | The NMA included 2 open-label studies and 5 double-blind studies. Three studies allowed treatment switching: 1 during the study and 2 for the OLE portion. Bias due to treatment switching cannot be mitigated through ITC methods. |
BPI = Brief Pain Inventory; CKD-EPI = Chronic Kidney Disease Epidemiology Collaboration; eGFR = estimated glomerular filtration rate; ERT = enzyme replacement therapy; FD = Fabry disease; ITC = indirect treatment comparison; LVM = left ventricular mass; LVMI = Left Ventricular Mass Index; NMA = network meta-analysis; NYHA = New York Heart Association; OLE = open-label extension; q.2.w. = every 2 weeks; SD = standard deviation.
Source: Sponsor’s ITC technical report;52 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Results
The results of the NMA for the outcomes of interest are included in Table 27. For all outcomes of interest discussed in this section, there were only single studies comparing any 2 treatments, and a random-effects model could not be estimated. Therefore, the results are based on the fixed-effects model. In the sponsor-submitted ITC, unanchored STCs could only adjust for differences in treatment exposure status, age, sex, phenotype, migalastat-amenable variant status, and baseline eGFR, depending on what data were available for each outcome. Due to the various limitations with the unanchored STCs, the results have not been reported.
Annualized Change in eGFR (Slope)
Pegunigalsidase alfa could not be connected to agalsidase alfa or migalastat using RCTs. To compare pegunigalsidase alfa to migalastat, an unanchored PAIC was necessary.
Change From Baseline in eGFR
The network included 5 studies, 1 of which was an RWE study (Lenders [2016])66 used to link the standard and lower doses of agalsidase beta. Three different methods were used to calculate eGFR across the various studies: the CKD-EPI equation, iothalamate and hippuran or iohexol infusions, and creatinine clearance. Time points included 24, 26, 52, or 104 weeks. Week 52 (or month 12) was chosen to compare pegunigalsidase alfa and agalsidase alfa. The comparison between pegunigalsidase alfa and migalastat compared week 52 data with week 26 data, respectively.
The mean difference between pegunigalsidase alfa and the comparators was ████ ██████████████ ████ ████████ ███████████ for agalsidase beta after 104 weeks of treatment, ████ ██████████████ ████ ██████ ██ █████ ███████████ for agalsidase alfa after 52 weeks of treatment, and ██████ ██████████████ ████ ██████ ██ █████ ███████████ for migalastat after 26 weeks of treatment. The sensitivity analyses that removed the RWE study and combined the 2 doses of agalsidase beta supported the base case.
LVMI Score
The network included 5 studies, 1 of which was an RWE study (Weidemann [2014])48 used to link the standard and lower doses of agalsidase beta. Vedder (2007)64 only reported LVM, and LVMI score had to be imputed using the mean body surface area of all patients in the BALANCE trial. Hughes (2008)63 did not report a measure of variance for mean change in LVMI score; therefore, imputation was performed using the mean of the variances in the BALANCE trial. Time points included 26, 52, 78, or 104 weeks. Week 52 (or month 12) was chosen to compare pegunigalsidase alfa and agalsidase alfa. The comparison between pegunigalsidase alfa and migalastat compared week 52 data with week 26 data, respectively.
The mean difference between pegunigalsidase alfa and the comparators was 3.82 g/m2 (95% CrI, –1.26 g/m2 to 8.91 g/m2) for agalsidase beta after 104 weeks of treatment, 21.00 g/m2 (95% CrI, –21.68 g/m2 to 63.73 g/m2) for agalsidase alfa after 52 weeks of treatment, and 28.69 g/m2 (95% CrI, –34.01 g/m2 to 91.38 g/m2) for migalastat after 26 weeks of treatment. The sensitivity analyses that removed the RWE study and combined the 2 doses of agalsidase beta supported the base case.
BPI Score
The network included 3 studies and did not include migalastat. To connect treatments, different scores were used (i.e., worst pain in last 24 hours and worst pain in past week). Additionally, for 1 study, values had to be transcribed from a figure, which introduces potential human error.20 Time points included 26, 52, 78, or 104 weeks. The comparison between pegunigalsidase alfa and agalsidase alfa compared week 104 (or month 24) data with week 24 data, respectively.
The mean difference between pegunigalsidase alfa and the comparators was ████ ██████ ████ ████ █████ ██ ████ ███████ for agalsidase beta after 104 weeks of treatment and ████ ██████ ████ ████ █████ ██ ████ ███████ for agalsidase alfa after 24 weeks of treatment.
Detail | CFB in eGFR | LVMI | BPI | ADAs |
|---|---|---|---|---|
Studies, N | 5 | 5 | 3 | 2 |
Model | FE | FE | FE | FE |
Deviance | NA | NA | NA | NA |
DIC | NA | NA | NA | NA |
ADA = antidrug antibody; BPI = Brief Pain Inventory; CFB = change from baseline; DIC = deviance information criterion; eGFR = estimated glomerular filtration rate; FE = fixed effects; ITC = indirect treatment comparison; LVMI = Left Ventricular Mass Index; NA = not applicable.
Source: Sponsor’s ITC technical report;52 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Comparator | CFB in eGFR MD (95% CrI)a | LVMI MD (95% CrI)b | BPI MD (95% CrI)b | ADAs OR (95% CrI)c |
|---|---|---|---|---|
NMA results: pegunigalsidase alfa vs. comparator | ||||
Agalsidase beta | ████ ███ | 3.82 (–1.26 to 8.91) | ████ ███ | ████ ███ |
Agalsidase alfa | ████ ███ | 21.00 (–21.68 to 63.73) | ████ ███ | ████ ███ |
Migalastat | ████ ███ | 28.69 (–34.01 to 91.38) | ████ ███ | ████ ███ |
Sensitivity analysis results: pegunigalsidase alfa vs. comparator | ||||
Agalsidase alfa | ████ ███ | 18.06 (–21.13 to 57.09) | ████ ███ | ████ ███ |
Migalastat | ████ ███ | 26.81 (–34.52 to 88.41) | ████ ███ | ████ ███ |
ADA = antidrug antibody; BPI = Brief Pain Inventory; CFB = change from baseline; CrI = credible interval; eGFR = estimated glomerular filtration rate; LVMI = Left Ventricular Mass Index; MD = mean difference; NA = not applicable; NMA = network meta-analysis; OR = odds ratio; vs. = versus.
aMDs < 0 indicate that there is a nominal trend favouring pegunigalsidase alfa (i.e., on average, patients receiving pegunigalsidase alfa may experience less deterioration or more improvement in the outcome than the comparator). A 95% CrI of the MD that includes the null effect (0) indicates that there is not a difference in the efficacy outcome of the treatments.
bMDs > 0 indicate that there is a nominal trend favouring pegunigalsidase alfa (i.e., on average, patients receiving pegunigalsidase alfa may experience less deterioration or more improvement in the outcome than the comparator). A 95% CrI of the MD that includes the null effect (0) indicates that there is not a difference in the efficacy outcome of the treatments.
cORs > 1 indicate that there is a nominal trend favouring pegunigalsidase alfa (i.e., on average, fewer patients develop ADAs). A 95% CrI of the OR that includes the null effect (1) indicates that there is not a difference in the development of ADAs for the treatments.
Source: Sponsor’s ITC technical report;52 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Antidrug Antibodies
ADAs were only reported in a subset of patients (29 of 34 patients) in the Vedder (2007) study.64 To compare pegunigalsidase alfa to agalsidase alfa, the 2 doses of agalsidase beta needed to be combined in 1 node.
The odds ratios of a patient developing ADAs were 1.26 (95% CrI, 0.47 to 3.35), nominally favouring pegunigalsidase alfa over agalsidase beta, and 0.46 (95% CrI, 0.06 to 3.02), nominally favouring agalsidase alfa over pegunigalsidase alfa.
Critical Appraisal of ITC
Due to the lack of direct evidence comparing pegunigalsidase alfa to other relevant comparators (besides agalsidase beta in the BALANCE trial), the sponsor conducted an NMA to inform indirect comparisons. When the network was unconnected or there were important differences in TEMs between studies, PAICs were considered. To better control the risk of bias, unanchored PAICs should control for all TEMs and prognostic variables. When the information was available, the sponsor attempted to adjust for the following characteristics: treatment exposure status, age, sex, phenotype, migalastat-amenable variant status, and baseline eGFR. However, this is not a comprehensive list, according to clinical expert opinion, and other important characteristics include ADA status, ACEi or ARB treatment, UPCR category, baseline LVH status, migalastat-amenable variant status, genotype, and lyso-Gb3 concentration. Understandably, not every study reported every effect modifier, and it was not possible to make the necessary adjustments. Most of the studies in the PAICs enrolled a small number of patients (< 30 patients) and are therefore more likely to produce unreliable comparisons. Moreover, the patient-level data for patients who had not previously received treatment for FD or who had a migalastat-amenable variant were only available from single-arm studies and were limited to a small number of patients (n = 6 and n = 9, respectively), further reducing confidence in the results. Therefore, the unanchored PAICs that used STC methods have limitations, putting their results at high risk of bias, which increases the uncertainty of the comparisons; therefore, the results have not been reported in this review. The uncertainties in the validity of the unanchored comparisons have also been acknowledged in the sponsor-submitted ITC report.
For the SLR and the NMA, the search strategy, screening, and data extraction methods appeared to be adequate. The study eligibility criteria for the SLR and the feasibility assessment (including the reasons for excluding studies) were reasonable and relevant to the Reimbursement Review. All relevant comparators that have a Health Canada–approved indication for FD were included. The sponsor-submitted technical report indicated that there was an appraisal of the risk of bias of the included studies, but it was unclear if there was a plan investigating the impact of studies with a high risk of bias. The inclusion of studies that vary in quality can violate the exchangeability assumption.
The overall network was sparse: it included few drugs and few comparative trials. All studies had a small number of patients, which is understandable given that FD is rare, and most had fewer patients than the BALANCE trial (some as few as 15 patients). For the outcomes of interest for the Reimbursement Review report, a random-effects model was not estimable; therefore, the comparisons used a fixed-effects model. The use of only a fixed-effects model, along with the wide CrIs, leads to a high level of uncertainty in the estimates.
The comparison between pegunigalsidase alfa and agalsidase alfa required the inclusion of the Banikazemi (2007) or Vedder (2007) studies.62,64 For outcome networks in which the Banikazemi (2007) study did not report on the outcome, the Vedder (2007) study had to be used (i.e., for networks on change from baseline in eGFR, LVMI score, and ADAs). This meant that the normal and low doses of agalsidase beta had to be combined, which requires assumptions to be made that are unlikely to be true and introduces bias that cannot be adjusted for. Combining the doses implies that they have similar efficacy and safety, which is not valid,65 as has been acknowledged in the sponsor’s report, and the lower doses of agalsidase beta are not used in clinical practice according to the clinical experts consulted for this review. To evaluate the normal and low doses of agalsidase beta separately, non-RCT or RWE studies that report on the agalsidase beta doses would need to be included in the network, but none of these studies identified from the searches would provide an unbiased estimate, and therefore they were not used.
For the results of an NMA to be valid, the studies and patients included in the network must be sufficiently similar. Table 34 and Table 35 compare the RCTs included in the NMA and the baseline characteristics of the patients from those RCTs, and there are clear differences among the studies and patients. The oldest study was from 2001, and it is likely that clinical practice and patient characteristics have changed since then. Two of the 7 studies were open label, which increases the risk of bias for subjective outcomes. Most studies were multicentre studies in Europe and North America, but they also included centres in Argentina, Australia, and Egypt, and there was a single-centre study in the US. Only 1 study was specified as taking place in Canada. There could be variability in standard of care across different geographic regions. The maximum treatment duration ranged from 6 to 35 months. Follow-up times varied across studies, and not all comparisons used the same time point. In addition, treatment effects may not be noticeable for some outcomes over a short duration (e.g., changes in LVMI and pain scores). Treatment switching was permitted in 3 studies, which introduces bias because it cannot be adjusted for using ITC methods.
There was also variability in study eligibility criteria for prior FD treatment, confirmation of a migalastat-amenable variant, and eGFR, thus producing different study populations. This was confirmed when looking at the baseline characteristics across studies. There were differences in mean ages (ranging from 34 to 50 years), sex (ranging from 32% to 100% males), mean baseline eGFR (ranging from 52 mL/min/1.73 m2 to 106 mL/min/1.73 m2), and prior treatment exposure (ranging from 0% to 100%). Many characteristics that were identified as TEMs were not reported across all studies, such as disease phenotype, migalastat-amenable variant, LVH, mean UPCR, and presence of ADAs. Overall, there were important differences in patient characteristics.
The doses used in some studies were not consistent with the Health Canada product monographs. The Germain (2016) study looked at migalastat 150 mg once daily, but the product monograph recommends that 1 capsule (150 mg migalastat hydrochloride) be taken once every other day at the same time of day for patients aged 12 years or older weighing 45 kg or more.61,67 As discussed previously, the Vedder (2007) study looked at a low dosage of agalsidase beta (0.2 mg/kg every other week), but the product monograph recommends a dosage of 1.0 mg/kg body weight every 2 weeks.36,64 The network included a mixture of RCTs and RWE studies for some outcomes, and the latter are expected to be at increased risk of bias as they cannot control for all known and unknown prognostic variables important in FD. Although the sponsor conducted sensitivity analyses to remove the RWE studies, the CrIs in the base case were very wide, and it would be difficult to detect changes in the models within the sensitivity analyses.
Although eGFR and LVMI score are objective outcomes, there is the potential for variability when different assays or methods of measurement are used. The Vedder (2007) study only reported LVM, and the Hughes (2008) study did not report LVMI variance.63,64 For both, missing data were imputed using values from the BALANCE trial, which is unlikely to be a valid approach given the large differences in baseline characteristics observed among the studies. For BPI score, there was the potential for human error when transcribing data from a figure from the Schiffmann (2001) study.20 Additionally, the long-form and short-form versions of the questionnaire were used and were treated as the same outcome. The questionnaires produce various scores, and the scores reported were not consistent among all studies. The studies looked at the worst pain, but over different durations: the last 24 hours or last week. For ADAs, a subset of patients (29 of 34) from the Vedder (2007) study contributed data for the comparison.64 The time points used to compare pegunigalsidase alfa and migalastat were different for eGFR, LVMI, and BPI outcomes. The differences in outcomes and how they were measured cannot be adjusted for using NMA methods.
Due to the differences in study designs, study populations, comparator dosages, and outcomes, the transitivity assumption is likely not valid.
Homogeneity of treatment effect could not be assessed because there were only single studies informing relevant direct comparisons (i.e., between active treatments).
Consistency of treatment effect could not be assessed because there were no closed loops that contained pegunigalsidase alfa.
Outcomes of interest to patients and clinicians that were missing include eGFR slope, HRQoL, and IRRs. For eGFR slope and IRRs, an attempt was made to create networks, but it was not possible to connect pegunigalsidase alfa with agalsidase alfa or migalastat. Due to the lack of evidence from the ITC, it is unknown how these drugs compare for these outcomes. Study durations varied, and the long-term persistence of the treatment effect is uncertain based on the results of the NMA.
The networks were sparse, and most of the mean differences had very wide CrIs (ranging from substantial benefit to substantial lack of benefit), adding to the high degree of uncertainty in the NMA results. Thus, interpretation of the data is highly limited. This, along with the various other limitations (differences in study and patient characteristics, comparator dosages, and outcomes, as well as NMA assumptions that could not be validated), show that the quality of evidence is low on the comparability of pegunigalsidase alfa to other FD-specific drugs.
Studies Addressing Gaps in the Systematic Review Evidence
The contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
One study was included that addresses a gap in the evidence for patients treated with pegunigalsidase alfa who were not previously treated with agalsidase beta (as in the pivotal study, the BALANCE trial). The BRIDGE study (N = 22) was a phase III, open-label, switch-over study evaluating the safety and efficacy of pegunigalsidase alfa, 1 mg/kg, every 2 weeks in adults with FD who had previously been treated for at least 2 years with agalsidase alfa at a stable dose for at least 6 months (Table 28).68 There was a 3‑month screening period, followed by a 12‑month treatment period; upon completion, patients could continue treatment in the F60 study.
Table 28: Details of Studies Addressing Gaps in the Systematic Review Evidence
Detail | BRIDGE study |
|---|---|
Designs and populations | |
Study design | Phase III, open-label, switch-over study |
Enrolled, N | 22 |
Key inclusion criteria |
|
Key exclusion criteria |
|
Drugs | |
Intervention | Pegunigalsidase alfa, 1 mg/kg, intravenously over 3 hours, every 2 weeks |
Comparator(s) | NA |
Outcomes | |
Primary end point | Safety of pegunigalsidase alfa:
|
Secondary end points | Secondary:
|
Notes | |
Publication | |
ACEi = angiotensin-converting enzyme inhibitor; AKI = acute kidney injury; ARB = angiotensin receptor blocker; BPI = Brief Pain Inventory; CKD-EPI = Chronic Kidney Disease Epidemiology Collaboration; ECG = electrocardiogram; eGFR = estimated glomerular filtration rate; FD = Fabry disease; Gb3 = globotriaosylceramide; IRR = infusion-related reaction; ISR = injection site reaction; LVMI = Left Ventricular Mass Index; lyso-Gb3 = globotriaosylsphingosine; MSSI = Mainz Severity Score Index; NA = not applicable; NYHA = New York Heart Association; TEAE = treatment-emergent adverse event; TIA = transient ischemic attack; UPCR = urine protein-to-creatinine ratio.
Source: Clinical Study Report for BRIDGE study;68 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Populations
Patients eligible for the BRIDGE study were adults aged 18 to 60 years with FD and reduced kidney function. Males must have had reduced plasma and/or leukocyte alpha-galactosidase activity (less than the lower limit or normal); females must have had either genetic confirmation of a pathogenic FD variant or, in the case of a novel variant, a first-degree male relative with FD. All patients must have displayed at least 1 of the characteristic features of FD (i.e., neuropathic pain, cornea verticillate, or clustered angiokeratoma). The eGFR slope was not an eligibility criterion, and patients must have been treated with agalsidase alfa for at least 2 years.
Individuals were not eligible if they had a history of renal dialysis or transplant, acute kidney injury in the year before enrolment, congestive heart failure of New York Heart Association Class IV, or a cardiovascular or cerebrovascular event in the 6 months before enrolment.
Interventions
All patients switched to pegunigalsidase alfa, 1 mg/kg, every 2 weeks for 12 months. The infusion protocol and reasons for permanent discontinuation were similar to those in the BALANCE trial. The use of agalsidase alfa and agalsidase beta was prohibited.
Outcomes
CKD-EPI eGFR (to calculate slope) was measured monthly during the 3-month screening period and at visits 1, 3, 5, 7, 9, 11, 14, 16, 18, 20, 22, 24, and 27. The baseline annualized CKD-EPI eGFR slope while receiving agalsidase alfa (i.e., before the switch to pegunigalsidase alfa) was compared to the annualized CKD-EPI eGFR slope after the switch to pegunigalsidase alfa.
Other efficacy and safety outcomes were similar to those described in the BALANCE trial and were assessed from baseline to 12 months.
Statistical Analysis
No formal sample size calculation was performed. The sample size of 22 patients was considered by the sponsor to be adequate to evaluate the safety of switching from agalsidase alfa to pegunigalsidase alfa. Challenges with patient recruitment for a rare disease were considered.
The eGFR slope per patient was estimated using linear regression. Comparison of the change in annualized changes in CKD‑EPI eGFR between the preswitch period (while receiving agalsidase alfa) and the postswitch period (while receiving pegunigalsidase alfa) was performed using a paired t test; patients were their own controls. All other end points were presented using descriptive statistics.
There were no adjustment factors or sensitivity analyses for any end points. There was no imputation for missing data for the primary efficacy analyses in the studies; missing data were assumed to be missing at random.
Prespecified subgroup analyses were conducted for sex (female or male), treatment-emergent immunogenicity status (ADA positive or ADA negative), and disease manifestation at baseline (classic or nonclassic). Based on clinical expert opinion, the subgroups were deemed unlikely to impact treatment and reimbursement decisions.
Analysis Populations
The analysis populations were as follows:
Safety population (N = 22): all patients who received any dose of pegunigalsidase alfa in the study. This population was used for safety analyses.
Efficacy population (N = 20): all patients who had at least 1 visit with an efficacy evaluation after the first pegunigalsidase alfa infusion. This population was used for efficacy analyses.
Per-protocol efficacy population (N = 20): all patients who completed the 12-month treatment period with efficacy data available and with no major protocol violations. This population was also used for efficacy analyses.
Results
Patient Disposition
In the BRIDGE study, 27 individuals were screened, of whom 22 were enrolled in the study. Of the 22 enrolled patients, 20 (90.9%) completed 12 months of treatment; there were 2 discontinuations due to TEAEs.
Baseline Characteristics
The patients’ baseline characteristics are summarized in Table 29. The mean age of the patients was 44.0 years (SD = 11.0 years). There were more males (68.2%) than females (31.8%), as per the study design, and more than half of the patients had classic FD (63.6%). The mean eGFR was 82.5 mL/min/1.73 m2 (SD = 23.4 mL/min/1.73 m2), and the mean eGFR slope was –5.3 mL/min/1.73 m2 per year (SD = 6.3 mL/min/1.73 m2 per year) based on all eGFR values obtained up to 24 months before screening to preinfusion of the study drug at visit 1.
Table 29: Summary of Baseline Characteristics From the BRIDGE Study (Safety Population)
Characteristic | Pegunigalsidase alfa (N = 22) |
|---|---|
Demographic characteristics | |
Age (years) | |
Mean (SD) | 44.0 (11.0) |
Median (range) | 44.5 (24 to 60) |
Sex, n (%) | |
Female | 7 (31.8) |
Male | 15 (68.2) |
Race, n (%) | |
White | 22 (100.0) |
Weight (kg) | |
Mean (SD) | 74.8 (15.0) |
Median (range) | 75.0 (41.9 to 105.0) |
Region, n (%) | |
Not US | 22 (100.0) |
Clinical characteristics | |
FD phenotype, n (%) | |
Classic | 14 (63.6) |
Nonclassic | 8 (36.4) |
eGFR (mL/min/1.73 m2) | |
Mean (SD) | 82.5 (23.4) |
Median (range) | 87.0 (49.4 to 124.2) |
eGFR slope at baseline (mL/min/1.73 m2 per year)a | |
Mean (SD) | –5.3 (6.3) |
Median (range) | –4.3 (–20.5 to 6.3) |
Plasma lyso-Gb3 (nmol/L) | |
Mean (SD) | 38.3 (41.2) |
Median (range) | 27.6 (1.2 to 189.4) |
UPCR categories at baseline, n (%) | |
No protein detectable: UPCR < 4 mg/dL | 12 (54.5) |
Normal to mildly increased: UPCR < 0.15 g/g | 3 (13.6) |
Moderately increased: UPCR between 0.15 g/g and 0.5 g/g | 3 (13.6) |
Severely increased: UPCR > 0.5 g/g | 4 (18.2) |
Treatment with ACEi or ARB, n (%) | |
Yes | 12 (54.5) |
No | 10 (45.5) |
Premedication use for infusion before enrolment, n (%) | |
Yes | 1 (4.5) |
No | 21 (95.5) |
Organ system involvement, n (%) | |
Nervous system | 19 (86.4) |
Skin | 18 (81.8) |
Cardiovascular | 17 (77.3) |
Gastrointestinal | 13 (59.1) |
Eyes | 18 (81.8) |
Pulmonary | 10 (45.5) |
ACEi = angiotensin-converting enzyme inhibitor; ARB = angiotensin II receptor blocker; eGFR = estimated glomerular filtration rate; FD = Fabry disease; lyso-Gb3 = globotriaosylsphingosine; SD = standard deviation; UPCR = urine protein-to-creatinine ratio.
aThe eGFR slope at baseline was based on historical (up to 24 months before screening), screening, and baseline serum creatinine levels.
Source: Clinical Study Report for BRIDGE study;68 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Exposure to Study Treatments
The cumulative exposure time to pegunigalsidase alfa for all patients was 241.7 months. The mean exposure time was 11.0 months (SD = 3.5 months), and the median exposure time was 12.0 months (range, 0.0 to 12.5 months). Drug adherence was 97.8% (SD = 3.6%).
Concomitant Treatments
All patients received at least 1 concomitant medication during the study. The most common concomitant treatments were paracetamol (54.5%), ibuprofen (36.4%), omeprazole (27.3%), and cholecalciferol (27.3%).
Protocol Deviations
All 22 patients had major protocol deviations. The major protocol deviations were related to study procedures (40.9% of patients), visit schedule criteria (36.4%), study drug adherence (31.8%), laboratory assessments (31.8%), and informed consent (27.3%). There were 2 critical protocol deviations related to study drug adherence (n = 1) and eligibility and entry criteria (4.5%).
Efficacy
The main efficacy outcomes are summarized in Table 29.
Incidence of Fabry Clinical Events
One patient experienced 2 Fabry clinical events (cardiac and cerebrovascular events).
Annualized Change in CKD-EPI eGFR (Slope)
The preswitch mean eGFR slope was –5.90 mL/min/1.73 m2 per year (SE = 1.34 mL/min/1.73 m2 per year), and the postswitch mean eGFR slope was –1.19 mL/min/1.73 m2 per year (SE = 1.77 mL/min/1.73 m2 per year). The change from preswitch to postswitch mean eGFR slope was 4.70 mL/min/1.73 m2 per year (SE = 2.26 mL/min/1.73 m2 per year).
LVMI Score
Of the 19 patients who contributed to the analysis, the LVMI score was 86.9 g/m2 (SE = 6.9 g/m2) at baseline and 89.4 g/m2 (SE = 6.1 g/m2) at month 12. The change from baseline to month 12 was 4.1 g/m2 (SE = 2.8 g/m2).
Short-Form BPI Score
The mean score for pain at its worst in the past 24 hours was 2.0 points (SE = 0.5 points) at baseline and 2.3 points (SE = 0.6 points) at month 12. The mean change from baseline was 0.4 points (SE = 0.4 points). The mean score for pain at its least in the past 24 hours was 0.9 points (SE = 0.3 points) at baseline and 1.2 points (SE = 0.4 points) at month 12. The mean change from baseline was 0.3 points (SE = 0.3 points). The mean score for current pain was 1.1 points (SE = 0.4 points) at baseline and 1.3 points (SE = 0.5 points) at month 12. The mean change from baseline was 0.2 points (SE = 0.2 points). The mean score for average pain was 1.9 points (SE = 0.4 points) at baseline and 1.9 points (SE = 0.5 points) at month 12. The mean change from baseline was 0.1 points (SE = 0.2 points).
Other Efficacy Outcomes
Other outcomes are summarized in Table 36 of Appendix 1.
Table 30: Summary of Key Efficacy Results From the BRIDGE Study (Efficacy Population)
Variable | Pegunigalsidase alfa (N = 20) |
|---|---|
Fabry clinical eventsa | |
Overall, n (%) | 1 (5.0) |
Cardiac events, n (%) | 1 (5.0) |
Cerebrovascular events, n (%) | 1 (5.0) |
Renal events, n (%) | 0 |
Non–cardiac-related death, n (%) | 0 |
Annualized CKD-EPI eGFR slope | |
Patients contributing to the analysis, n (%) | 20 (100) |
Preswitch (mL/min/1.73 m2 per year), mean (SE) | –5.90 (1.34) |
Postswitch (mL/min/1.73 m2 per year), mean (SE) | –1.19 (1.77) |
Change from pre- to postswitch (mL/min/1.73 m2 per year), mean (SE) (95% CI) | 4.70 (2.26) (–0.03 to 9.43) |
LVMI (g/m2) by MRI | |
Patients contributing to the analysis, n (%) | 19 (95.0) |
Baseline (g/m2), mean (SE) | 86.9 (6.9) |
Month 12 (g/m2), mean (SE) | 89.4 (6.1) |
Change from baseline to month 12 (g/m2), mean (SE) | 4.1 (2.8) |
Short-form BPI score | |
Pain at its worst in last 24 hours | |
Patients contributing to the analysis, n (%) | 20 (100) |
Baseline (points), mean (SE) | 2.0 (0.5) |
Month 12 (points), mean (SE) | 2.3 (0.6) |
Change from baseline (points), mean (SE) | 0.4 (0.4) |
Pain at its least in last 24 hours | |
Patients contributing to the analysis, n (%) | 20 (100) |
Baseline (points), mean (SE) | 0.9 (0.3) |
Month 12 (points), mean (SE) | 1.2 (0.4) |
Change from baseline (points), mean (SE) | 0.3 (0.3) |
Pain right now | |
Patients contributing to the analysis, n (%) | 20 (100) |
Baseline (points), mean (SE) | 1.1 (0.4) |
Month 12 (points), mean (SE) | 1.3 (0.5) |
Change from baseline (points), mean (SE) | 0.2 (0.2) |
Average pain | |
Patients contributing to the analysis, n (%) | 20 (100) |
Baseline (points), mean (SE) | 1.9 (0.4) |
Month 12 (points), mean (SE) | 1.9 (0.5) |
Change from baseline (points), mean (SE) | 0.1 (0.2) |
BPI = Brief Pain Inventory; CI = confidence interval; CKD-EPI = Chronic Kidney Disease Epidemiology Collaboration; eGFR = estimated glomerular filtration rate; LVMI = Left Ventricular Mass Index; SE = standard error.
aA patient with > 1 Fabry clinical event is counted only once in the overall summary.
Source: Clinical Study Report for BRIDGE study;68 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Harms
The harms results are summarized in Table 31. Twenty-one (95.5%) patients reported at least 1 TEAE during the study, with the 2 most common being headache (31.8% of patients in the study) and nasopharyngitis (22.7% of patients in the study). Four patients (18.2%) reported at least 1 SAE, which included 2 events of hypersensitivity and 1 event each for infectious mononucleosis and urinary tract infection. Two patients (9.1%) withdrew due to TEAEs, both of which were IRRs (hypersensitivity reactions). There were no deaths in the study.
For notable harms of interest for this review, 3 patients (13.6%) reported injection site reactions, 5 patients (22.7%) reported IRRs, 7 patients (31.8%) had treatment-emergent ADAs, and 2 of those 7 patients (28.6%) had neutralizing ADAs.
Table 31: Summary of Harms Results From the BRIDGE Study (Safety Population)
Harms | Pegunigalsidase alfa (N = 22) |
|---|---|
Most common TEAEs,a n (%) | |
Patients with ≥ 1 TEAE | 21 (95.5) |
Nasopharyngitis | 7 (31.8) |
Headache | 5 (22.7) |
SAEs, n (%) | |
Patients with ≥ 1 SAE | 4 (18.2) |
Patients who stopped treatment due to a TEAE, n (%) | |
Patients who stopped treatment | 2 (9.1) |
Deaths, n (%) | |
Patients who died | 0 |
TEAEs of special interest, n (%) | |
Injection site reaction | 3 (13.6) |
Any IRRs | 5 (22.7) |
Mild or moderate IRRs | 3 (13.6) |
Severe or very severe IRRs | 2 (9.1) |
Treatment-emergent ADAs | 7 (31.8) |
ADAs at any postbaseline visit | 7 (31.8) |
Neutralizing ADAsb | 2 (28.6) |
ADA = antidrug antibody; IRR = infusion-related reaction; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aOccurring in ≥ 15% of patients.
bOnly assessed in patients who were immunoglobulin G–positive (i.e., the number of patients with ADAs at any postbaseline visit).
Source: Clinical Study Report for BRIDGE study;68 sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Critical Appraisal
Internal Validity
The main limitations of the BRIDGE study were the open-label design and the lack of comparator group. It is possible that knowledge of the treatment impacted subjective outcomes, such as short-form BPI score and harms, although the degree of bias is uncertain. Additionally, uncontrolled confounding puts the results at a high risk of bias. All 22 patients had major protocol deviations, which increases the uncertainty of the results. As discussed in the critical appraisal of the BALANCE trial, historical values (up to 24 months before screening) were used to estimate the baseline eGFR slope and were likely unreliable. Similar to the BALANCE trial, all patients in the BRIDGE study were previously receiving a stable dose of agalsidase alfa before the study, and there is the possibility of carryover effects during the first few months of the study. This is additionally concerning as the BRIDGE study was only 12 months long and carryover effects can be as long as 6 months, according to the clinical experts.
External Validity
The generalizability concerns discussed for the BALANCE trial are largely applicable to the BRIDGE study because the eligibility criteria were very similar. The age limit (18 to 60 years), the requirement of an eGFR of at least 40 mL/min/1.73 m2, all patients having prior ERT experience for at least 2 years, and the exclusion of patients with poorer health may prevent broad application of the results to those who do not meet these characteristics but could otherwise receive pegunigalsidase alfa in clinical practice. All patients switched from agalsidase alfa to pegunigalsidase alfa, and while the choice to switch varies in practice based on clinician and patient preference, the study does not provide information on patients who have not previously received treatment for FD and does not provide an adequate (short-term or long-term) comparison between pegunigalsidase alfa and agalsidase alfa. The outcomes were generally the same as those in the BALANCE trial, and the same limitations apply to the BRIDGE study. The study follow-up was 12 months, and therefore does not provide information on the long-term use of pegunigalsidase alfa, though patients were able to enrol in the OLE study (the F60 study), which is discussed in the Long‑Term Extension Studies section of this report.
Discussion
Summary of Available Evidence
The evidence included in this review consisted of 1 pivotal study identified in the sponsor’s systematic review (the BALANCE trial), 1 OLE study (the F60 study), 1 ITC, and 1 study addressing gaps in the evidence (the BRIDGE study).
The BALANCE trial (N = 78) was a phase III, double-blind, active control, noninferiority study of pegunigalsidase alfa in patients aged 18 to 60 years with FD. Patients were randomized 2:1 to 104 weeks of treatment with pegunigalsidase alfa 1 mg/kg every 2 weeks (n = 53) or agalsidase beta 1 mg/kg every 2 weeks (n = 25). To be eligible, patients must have had a confirmed diagnosis of FD that was symptomatic, an CKD-EPI eGFR between 40 mL/min/1.73 m2 and 120 mL/min/1.73 m2, and a linear CKD-EPI eGFR slope more negative than –2 mL/min/1.73 m2 per year, and they must have been receiving agalsidase beta for at least 1 year with a stable dose for at least the last 6 months before the study. The primary end point was the annualized change (slope) in CKD-EPI eGFR over 2 years of study treatment. Noninferiority was indicated if the lower bound of the 95% CI for the treatment difference (pegunigalsidase alfa minus agalsidase beta) was greater than or equal to –3.0 mL/min/1.73 m2 per year. Secondary end points relevant to the Reimbursement Review included the incidence of Fabry clinical events, LVMI score, short-form BPI score, and notable harms (IRRs and ADAs). The mean age of patients across the study was 44.3 years (SD = 10.0 years), there were more males (61.0%) than females (39.0%), and more than half the patients had classic FD (53.2%).
At the time of the sponsor’s submission, the F60 study (N = 87) was an ongoing OLE study assessing the longer-term safety and efficacy of pegunigalsidase alfa for up to 60 months, until pegunigalsidase alfa became commercially available, or at the sponsor’s discretion. Eligible patients must have completed the BALANCE trial, the BRIDGE study, or at least 48 months of the F03 study (which included patients with symptomatic FD who had either never received ERT or had not received it in the past 6 months). The mean age of the patients was ████ █████ ███ █ ████ ██████, there were more males ███████ than females ███████ in the study, and █████ of patients had not previously received ERT. The mean total exposure (including parent studies) to pegunigalsidase alfa was ████ ██████ ███ █ ████ ███████ and the mean exposure in the F60 study was ████ ██████ ███ █ ███ ███████. The outcomes of interest aligned with those in the parent studies.
Due to the lack of evidence directly comparing pegunigalsidase alfa with relevant therapies for the treatment of FD, a sponsor-submitted ITC was summarized and appraised. An NMA was used to assess the relative efficacy and safety of pegunigalsidase alfa versus disease-specific treatments (agalsidase beta, agalsidase alfa, and migalastat) for change from baseline in eGFR, LVMI score, short-form BPI score, and ADAs. Unanchored PAICs (using STC methods) were used when the network was unconnected for an outcome or when there were important differences in TEMs across RCTs; however, due to the high risk of bias of unanchored PAICs, the results were not included in this report.
The BRIDGE study was a phase III, open-label, single-arm, switch-over study evaluating the safety and efficacy of pegunigalsidase alfa, 1 mg/kg, every 2 weeks in adults with FD who were previously treated for at least 2 years with agalsidase alfa. Patient eligibility was similar to that of the BALANCE trial, aside from all patients previously receiving agalsidase alfa. All patients received the standard dosage of pegunigalsidase alfa, 1 mg/kg every 2 weeks, for 12 months. The primary outcome was safety, and the efficacy outcomes of interest for the review aligned with those in the BALANCE trial. The mean age of the patients was 44.0 years (SD = 11.0 years). There were more males (68.2%) than females (31.8%) and more than half the patients had classic FD (63.6%).
Interpretation of Results
Efficacy
FD is rare, and despite the availability of disease-specific treatments, input from patient and clinician groups stated a need for therapies that reduce symptoms, prevent progression to end-organ failure, improve HRQoL, and reduce IRRs and ADAs. Input from the patient and clinician groups also noted the importance of having multiple treatment options to choose from.
The key efficacy outcomes from the pivotal trial assessed using GRADE in this report were considered very low certainty evidence, and the reasons for this were similar across the outcomes. Baseline imbalances between treatment groups in the BALANCE trial increased the risk of bias in results, which potentially favoured pegunigalsidase alfa. There was also notable missing data for LVMI and short-form BPI outcomes. The clinical experts indicated that the patients in the BALANCE trial were generally representative of those treated in clinical practice in Canada; however, generalizability to a broader population who could receive the new drug was limited by the study’s eligibility criteria. All patients had experience with agalsidase beta, there were no data for patients older than 60 years, all patients had evidence of kidney function decline, and patients with poor health were excluded. However, the clinical experts noted that patients who do not have these characteristics still require treatment. Imprecision in the treatment effect (wide 95% CIs and/or lack of established MIDs for FD) also contributed to the uncertainty of the evidence.
Preventing Fabry clinical events, which included renal, cardiac, and cerebrovascular events and noncardiac deaths, was 1 of the most important outcomes to patients and clinicians who provided input for this review. In the BALANCE trial, the proportion of patients experiencing overall Fabry clinical events was larger in the pegunigalsidase alfa group (17.3%) than in the agalsidase beta group (8.0%) for the 2-year follow-up. However, the clinical experts consulted for this review did not feel that the difference was clinically meaningful for the context of the study. They noted that there was a small number of patients (all of whom were receiving ERT before and during the study), that there were few events, and that the duration of follow-up was too short to show a meaningful difference between treatment groups because it can take years for these events to develop. Thus, it is challenging to attribute the imbalance in Fabry clinical events to pegunigalsidase alfa or to other possible reasons. In the BRIDGE study, only 1 patient reported a Fabry clinical event during the 1-year follow-up. In the F60 study, 15 patients reported Fabry clinical events by the 1-year data cut-off. The short follow-up time and lack of comparator groups in these studies make it difficult to draw meaningful conclusions about how pegunigalsidase alfa impacts the incidence of clinically meaningful events. This was not an outcome included in the sponsor-submitted ITC, and it is unknown how the drug compares to other specific treatments for FD.
Kidney function decline is a common characteristic of FD, and kidney outcomes were noted as being important to patients and clinicians. The primary outcome for assessing the noninferiority of pegunigalsidase alfa to agalsidase beta in the BALANCE trial was the median annualized change in CKD-EPI eGFR. Based on the lower bound of the 95% CI of the between-group change difference (–2.44 mL/min/1.73 m2 per year) being greater than the sponsor-suggested noninferiority margin of –3.0 mL/min/1.73 m2 per year, noninferiority was indicated. The margin was based on natural history data21 for male patients with FD who were untreated (which is different from the BALANCE trial population) and European therapeutic goals.7 Additionally, the margin was not based on preserving an established minimum treatment effect for agalsidase beta versus placebo in a population similar to that in the BALANCE trial, and the assumption is that agalsidase beta had its expected effect in the noninferiority study, which is unverifiable.22 Regulatory agencies have rejected the noninferiority margin, stating that a smaller margin would be more acceptable and robust, though they also acknowledge that having a smaller margin would require a large sample size, which would be unfeasible for a rare disease.22,27 Based on clinical expert opinion and evidence from the literature for patients with progressive kidney disease,23 an estimated clinically meaningful threshold for median annualized change in CKD-EPI eGFR of –0.5 mL/min/1.73 m2 per year to –1 mL/min/1.73 m2 per year over 2 to 3 years has been suggested. The 95% CI for the between-group change difference was –2.44 mL/min/1.73 m2 per year to 1.73 mL/min/1.73 m2 per year, suggesting the possibility of both a clinically meaningful worsening or improvement in eGFR slope. Using the MID suggested from the literature and the clinical experts of –0.5 mL/min/1.73 m2 per year to –1 mL/min/1.73 m2 per year as a noninferiority margin, it is not possible to conclude that pegunigalsidase alfa is not worse than agalsidase beta. Both the BRIDGE and F60 study results showed that patients continued to experience kidney function decline (a negative eGFR slope) while receiving pegunigalsidase alfa, but with different magnitudes that were greater than the estimated MID (means of –1.19 mL/min/1.73 m2 per year [SE = 1.77 mL/min/1.73 m2 per year] and █████ ███████████████ ███ █ ████ ████████████████, respectively). Once again, there are limitations with the study designs of the BRIDGE and F60 studies that increase the uncertainty of these results for patients who switch ERT and for longer-term use of pegunigalsidase alfa. It was not possible to connect treatments of interest in the ITC for the eGFR slope outcome network, and it is unknown how pegunigalsidase alfa compares to other disease-specific treatments for FD. Additionally, generalizability for this outcome may be limited because these studies selected patients with declining kidney function and not all patients with FD experience this.
Advanced cardiac disease is another marker of FD that patients and clinicians would like new treatments to address. In the BALANCE trial, there was no noninferiority margin to inform the comparison between pegunigalsidase alfa and agalsidase beta for LVMI score, but clinical expert opinion suggested a clinically meaningful threshold of 5 g/m2. The 95% CI for the between-group difference (–10.26 g/m2 to 8.42 g/m2) indicated the possibility of either a meaningful improvement or decline for this outcome. Randomization was not stratified by hypertrophy, there was a considerable amount of missing data, and imbalances in baseline characteristics increase the risk of bias for these results. Considering that all patients in the BALANCE trial were receiving active treatment before and during the BALANCE trial and that enrolled patients did not have significant end-stage cardiovascular disease (patients in poorer health were excluded), based on the 2-year study, the clinical experts indicated that there was likely not a meaningful difference between the treatment groups. In the BRIDGE study, LVMI scores increased from baseline to month 12 (mean = 4.1 g/m2; SE = 2.8 g/m2), but this was based on data from 19 patients and there was no comparator group. There were no results for LVMI score from the F60 study, and the long-term effect of pegunigalsidase alfa on LVMI score remains unknown. Results from the ITC comparing pegunigalsidase alfa to agalsidase alfa and migalastat showed a nominal trend favouring the new ERT (with wide 95% CrIs that crossed the null), but limitations with the NMA make these results highly uncertain. Based on the results, it is unclear what effect pegunigalsidase alfa has on cardiac outcomes, such as LVMI score.
FD-associated pain is a symptom patients would like treatments to control yet is hard to manage with existing therapies. In the BALANCE trial, both bounds of the 95% CIs for the between-group differences for the 4 BPI scores approached or crossed the estimated MID of 1 to 2 points. Thus, compared to agalsidase beta, pegunigalsidase alfa could produce either a clinically meaningful benefit or harm in terms of pain after 2 years of treatment. In the BRIDGE study, the mean change from baseline for all 4 BPI scores was modest, ranging from 0.1 points (SE = 0.2 points) to 0.4 points (SE = 0.4 point), indicating that pain may not have changed when patients switched from agalsidase alfa to pegunigalsidase alfa for 1 year. Longer-term results from the F60 study indicated that pain scores ████████ ██████ ██████ with another year of treatment with pegunigalsidase alfa; however, there were missing data for this outcome, which would introduce bias into the estimates. The sponsor-submitted ITC could only compare pegunigalsidase alfa with agalsidase alfa, and while the point estimate suggests a meaningful benefit (relative to the estimated MID of 1 to 2 points) favouring the new drug, the wide 95% CrI indicated the possibility of either benefit or lack of benefit versus agalsidase alfa. All patients in the BALANCE, BRIDGE, and F60 studies had been receiving some form of ERT for at least 2 years, and according to the clinical experts, pain with FD is slow to decline with treatment, if at all. Conversely, when patients stop treatment (or if a treatment does not work), the clinical experts indicated that FD-associated pain would return within a few months. The experts also noted that pain is subjective and that scores on pain questionnaires can vary considerably.
Improvement in HRQoL was noted as being important to both patients and clinicians, but no comprehensive measure of HRQoL was used in any of the studies or the ITC. Although the EQ-5D is not considered a comprehensive measure of HRQoL, the results from the BALANCE trial suggest that pegunigalsidase alfa and agalsidase beta may result in similar overall health scores at week 104 (mean between-group difference = 0.8 points; 95% CI, –7.2 to 8.8 points), though the wide 95% CI and the lack of MID reduce the certainty in this estimate. After 52 additional weeks of treatment with pegunigalsidase alfa in the F60 study, there was minimal change in patients’ overall health score (mean = –0.39 points; SE = 1.95 points). In the BRIDGE study, there was a numerical increase (improvement) in overall health score (mean = 5.1 points; SE = 3.3 points) 12 months after switching from agalsidase alfa to pegunigalsidase alfa.
Other outcomes of interest included plasma lyso-Gb3, UPCR category, overall MSSI score, exercise tolerance (stress test), frequency of pain medication used, and the EQ-5D-5L. The results from the BALANCE trial were mixed: while plasma lyso-Gb3 concentration appeared to favour agalsidase beta, overall MSSI score seemed to favour pegunigalsidase alfa. Other outcomes did not show a clear difference between the 2 treatments after 2 years. The results from the BRIDGE study were less informative due to the lack of comparator arm and the small number of patients (N = 20). Longer-term results from the F60 study seemed to suggest that outcomes were stable with continued use of the drug, but missing data was an issue.
Although noninferiority was indicated by the study results, it is uncertain if pegunigalsidase alfa is truly not worse than agalsidase beta. However, based on the results from the BALANCE trial, it is reasonable to conclude that pegunigalsidase alfa has some treatment effect similar to agalsidase beta because the outcomes for eGFR slope, pain, and plasma lyso-Gb3 concentration would not occur spontaneously without treatment. The sponsor-submitted ITC did not demonstrate a difference in favour of 1 treatment over another, and definitive conclusions related to the treatment effect of pegunigalsidase alfa compared to agalsidase alfa or migalastat could not be drawn due to the various limitations with the ITC (small number of studies contributing to the network; heterogeneity in study designs, patient characteristics, and outcomes).
Clinical expert input noted that pegunigalsidase alfa offers another treatment option for patients with FD, which is important because some patients’ disease continues to progress while receiving treatment and patients desire less administrative burden (infusion times, intolerable IRRs, travel for infusions). As pegunigalsidase alfa is another infused ERT, it may not meet all these needs.
Harms
The types and incidences of TEAEs were comparable between pegunigalsidase alfa and agalsidase beta in the BALANCE trial, and the clinical experts consulted for this review indicated that they were manageable and did not raise new concerns with the drug. There were proportionately fewer SAEs but more withdrawals due to TEAEs in the pegunigalsidase alfa group than in the comparator group. The results from the BRIDGE study for patients who switched from agalsidase alfa were similar to those in the BALANCE trial, and the F60 study did not show new safety signals with an additional year of pegunigalsidase alfa treatment.
IRRs and the development of ADAs (particularly neutralizing ADAs, which reduce treatment efficacy) were of particular concern to patients and clinicians. The evidence relating to severe or very severe IRRs in the BALANCE trial was assessed using GRADE as being of low certainty due to the few events (1 event in the pegunigalsidase alfa group) and the generalizability limitations with the overall study, as previously discussed. Proportionately fewer patients with neutralizing ADAs were receiving pegunigalsidase alfa compared to agalsidase beta; however, the experts consulted for this review did not feel that the difference between groups was clinically meaningful. The results of the ITC trended toward patients being less likely to develop ADAs with pegunigalsidase alfa than with agalsidase beta but more likely to develop ADAs than with agalsidase alfa, but the 95% CrIs included the null and no treatment was clearly favoured over the others for this outcome.
There is the possibility of selection bias for the BALANCE, BRIDGE, and F60 studies because all patients were previously treated with an ERT for at least 1 year before enrolment. The study patients likely had a tolerance to ERT (both clinical and nonclinical aspects) as well as to ERT-associated TEAEs, allowing them to continue receiving infusions, and these results may not represent what is expected for patients who have not previously received ERT.
Conclusion
FD is a rare, progressive, X-linked disease associated with kidney, cardiac, and cerebrovascular disease and reduced HRQoL. There is a need for safe and effective treatments that prevent progression to end-organ disease, reduce FD symptoms, improve HRQoL, and are less burdensome. Based on the evidence from 1 phase III, randomized, active comparator–controlled, noninferiority study of adults aged 18 to 60 years with symptomatic FD, kidney function decline, and previous experience with agalsidase beta (the BALANCE trial), 2 years of treatment with pegunigalsidase alfa 1.0 mg/kg, IV, every other week may result in a similar decline in eGFR slope as treatment with agalsidase beta 1.0 mg/kg. Compared to the sponsor-suggested noninferiority margin of –3.0 mL/min/1.73 m2 per year, pegunigalsidase alfa was considered to be noninferior to agalsidase beta; however, information from regulatory agencies, published literature, and clinical expert opinion indicated that a smaller margin would be more appropriate. At the suggested MID of 1.0 mL/min/1.73 m2 per year, pegunigalsidase alfa could have resulted in a clinically meaningful decline or increase in eGFR slope in the BALANCE trial. According to clinical expert opinion, the results from the study suggested that the 2 drugs could be similar for incidence of Fabry clinical events, cardiac complications (based on LVMI score), and pain (as per the short-form BPI). Still, the evidence was highly uncertain due to the potential risk of bias in the study, limitations with generalizability, and imprecision in the effect estimates. Evidence from a 1-year, open-label study of patients who switched from agalsidase alfa to pegunigalsidase alfa (the BRIDGE study) showed a reduced rate of eGFR decline, an increase in LVMI score, and stable pain scores, but this was based on only a few patients and a short duration. Longer-term evidence (the F60 study) for an additional year of treatment with pegunigalsidase alfa showed that patients continued to experience Fabry clinical events (as expected with a progressive disease and patients with preexisting organ involvement), eGFR slope continued to decline at a similar magnitude as in the BALANCE trial, and pain scores generally remained stable. Low-quality evidence from the sponsor-submitted ITC showed that no FD-specific treatment was clearly favoured over another for change in eGFR, LVMI score, or BPI score, though the comparisons were hindered by the significant study and population heterogeneity, unverifiable NMA assumptions, and a high level of uncertainty in the estimates based on very wide CrIs. It is unclear what impact pegunigalsidase alfa has on HRQoL, and the long-term outcomes of what is expected to be a lifelong treatment remain unknown.
In the BALANCE trial, harms were comparable between pegunigalsidase alfa and agalsidase beta. Although there were numerically fewer IRRs and neutralizing ADAs in the pegunigalsidase alfa group, it was not clear if the differences were clinically meaningful between the 2 drugs, according to clinical expert opinion. There were no unexpected safety signals for patients who switched from agalsidase alfa to pegunigalsidase alfa in the BRIDGE study or for patients who continued treatment with pegunigalsidase alfa in the F60 study. The ITC for the development of ADAs between pegunigalsidase alfa and agalsidase alfa indicated that neither treatment was clearly favoured over the other.
Pegunigalsidase alfa offers patients and clinicians another treatment option for FD, but as an ERT, it is not clear if the drug provides any reduction in administrative or nonclinical burden over other ERTs.
References
1.Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab. 2018;123(4):416-427. doi:10.1016/j.ymgme.2018.02.014 PubMed
2.Biegstraaten M, Arngrimsson R, Barbey F, et al. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: the European Fabry Working Group consensus document. Orphanet J Rare Dis. 2015;10:36. doi:10.1186/s13023-015-0253-6 PubMed
3.Laney DA, Bennett RL, Clarke V, et al. Fabry disease practice guidelines: recommendations of the National Society of Genetic Counselors. J Genet Couns. 2013;22(5):555-64. doi:10.1007/s10897-013-9613-3 PubMed
4.Wanner C, Germain DP, Hilz MJ, Spada M, Falissard B, Elliott PM. Therapeutic goals in Fabry disease: Recommendations of a European expert panel, based on current clinical evidence with enzyme replacement therapy. Mol Genet Metab. 2019;126(3):210-211. doi:10.1016/j.ymgme.2018.04.004 PubMed
5.Sirrs S, Bichet DG, Iwanochko RM, et al. Canadian Fabry Disease Treatment Guidelines 2018. 2019.
6.El-Abassi R, Singhal D, England JD. Fabry's disease. J Neurol Sci. 2014;344(1-2):5-19. doi:10.1016/j.jns.2014.06.029 PubMed
7.Wanner C, Arad M, Baron R, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab. 2018;124(3):189-203. doi:10.1016/j.ymgme.2018.06.004 PubMed
8.Vardarli I, Rischpler C, Herrmann K, Weidemann F. Diagnosis and Screening of Patients with Fabry Disease. Ther Clin Risk Manag. 2020;16:551-558. doi:10.2147/TCRM.S247814 PubMed
9.Beck M, Ramaswami U, Hernberg-Stahl E, et al. Twenty years of the Fabry Outcome Survey (FOS): insights, achievements, and lessons learned from a global patient registry. Orphanet J Rare Dis. 2022;17(1):238. doi:10.1186/s13023-022-02392-9 PubMed
10.Giugliani R, Niu D-M, Ramaswami U, et al. A 15-Year Perspective of the Fabry Outcome Survey. Journal of Inborn Errors of Metabolism and Screening. 2016;4:1-12. doi:https://doi.org/10.1177/2326409816666298
11.Spada M, Pagliardini S, Yasuda M, et al. High incidence of later-onset fabry disease revealed by newborn screening. Am J Hum Genet. 2006;79(1):31-40. doi:10.1086/504601 PubMed
12.Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281(3):249-54. doi:10.1001/jama.281.3.249 PubMed
13.Lin HY, Chong KW, Hsu JH, et al. High incidence of the cardiac variant of Fabry disease revealed by newborn screening in the Taiwan Chinese population. Circ Cardiovasc Genet. 2009;2(5):450-6. doi:10.1161/CIRCGENETICS.109.862920 PubMed
14.Snit M, Przyludzka M, Grzeszczak W. Fabry disease - a genetically conditioned extremely rare disease with a very unusual course. Intractable Rare Dis Res. 2022;11(1):34-36. doi:10.5582/irdr.2021.01132 PubMed
15.Mauer M, Wallace E, Schiffmann R. UpToDate, ed. Fabry disease: Treatment and prognosis. 2025. Accessed April 7, 2025. https://www.uptodate.com/contents/fabry-disease-treatment-and-prognosis
16.Amicus Therapeutics Canada Inc. GALAFOLD (migalastat): capsules, 123 mg migalastat (as migalastat hydrochloride), oral [product monograph]. November 25, 2024. Accessed May 20, 2025. https://pdf.hres.ca/dpd_pm/00077857.PDF
17.Chiesi Canada Corp. TBC (pegunigalsidase alfa): 2 mg/mL concentrate for solution for intravenous infusion [product monograph] DRAFT. TBD. Updated January 1, 1800.
18.Chiesi Canada Corp. Clinical Study Report: PB-102-F20. A Randomized, Double-blind, Active Control Study of the Safety and Efficacy of PRX-102 Compared to Agalsidase Beta on Renal Function in Patients with Fabry Disease Previously Treated with Agalsidase Beta (The BALANCE Study) [internal sponsor's report]. July 22, 2022.
19.Hopkin RJ, Cabrera G, Charrow J, et al. Risk factors for severe clinical events in male and female patients with Fabry disease treated with agalsidase beta enzyme replacement therapy: Data from the Fabry Registry. Mol Genet Metab. 2016;119(1-2):151-9. doi:10.1016/j.ymgme.2016.06.007 PubMed
20.Schiffmann R, Kopp JB, Austin HA, 3rd, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285(21):2743-9. doi:10.1001/jama.285.21.2743 PubMed
21.Schiffmann R, Hughes DA, Linthorst GE, et al. Screening, diagnosis, and management of patients with Fabry disease: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2017;91(2):284-293. doi:10.1016/j.kint.2016.10.004 PubMed
22.Center for Drug Evaluation Research. M-drs. (FDA) USFaDA, ed. TBC (pegunigalsidase alfa) 2 mg/mL, for IV infusion. Company: Chiesi Canada Corp. Application No.:761161Orig1s000. Approval date: 05/09/2023 (FDA approval package). . 2023. Accessed June 02, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2023/761161Orig1s000MultidisciplineR.pdf
23.Levey AS, Gansevoort RT, Coresh J, et al. Change in Albuminuria and GFR as End Points for Clinical Trials in Early Stages of CKD: A Scientific Workshop Sponsored by the National Kidney Foundation in Collaboration With the US Food and Drug Administration and European Medicines Agency. Am J Kidney Dis. 2020;75(1):84-104. doi:10.1053/j.ajkd.2019.06.009 PubMed
24.Mo Y, Lim C, Watson JA, White NJ, Cooper BS. Non-adherence in non-inferiority trials: pitfalls and recommendations. BMJ. 2020;370:m2215. doi:10.1136/bmj.m2215 PubMed
25.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi:10.1016/j.jclinepi.2019.10.014 PubMed
26.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi:10.1016/j.jclinepi.2010.07.015 PubMed
27.alfa). CfMPfHUArTp. Agency EM, ed. (European public assessment report). 2023. Accessed June 2, 2025. https://www.ema.europa.eu/en/documents/assessment-report/elfabrio-epar-public-assessment-report_en.pdf
28.Chiesi Canada Corp. Sponsor's Summary of Clinical Evidence Template TBC (pegunigalsidase alfa) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: TBC (Pegunigalsidase alfa): 2 mg/mL concentrate for solution for intravenous infusion. 2025.
29.Chiesi Canada Corp. Interim Clinical Study Report: PB-102-F60. Open-Label Extension Study to Evaluate the Long-Term Safety and Efficacy of Pegunigalsidase Alfa (PRX-102) in Patients with Fabry Disease [internal sponsor's report]. December 1, 2022.
30.Bykov K, Jaksa A, Lund J, et al. APPRAISE: a tool for appraising potential for bias in real-world evidence studies on medication effectiveness or safety. 2024. https://osf.io/a4nhd/
31.Berry L, Walter J, Johnson J, et al. Patient-reported experience with Fabry disease and its management in the real-world setting: results from a double-blind, cross-sectional survey of 280 respondents. Orphanet J Rare Dis. 2024;19(1):153. doi:10.1186/s13023-024-03090-4 PubMed
32.CADTH. Patient Group Input Submission: Migalastat (GALAFOLD). 2017. Accessed September 2024. https://www.cda-amc.ca/sites/default/files/cdr/relatedinfo/SR0522_Galafold_Patient_Input.pdf
33.Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30. doi:10.1186/1750-1172-5-30 PubMed
34.Canadian Fabry Disease I. Patient Registry Data up to July 2022.
35.van der Tol L, Smid BE, Poorthuis BJ, et al. A systematic review on screening for Fabry disease: prevalence of individuals with genetic variants of unknown significance. J Med Genet. 2014;51(1):1-9. doi:10.1136/jmedgenet-2013-101857 PubMed
36.Inc. S-AC. FABRAZYME (agalsidase beta) (Recombinant human α-galactosidase A), lyophilized powder, 5 mg and 35 mg, intravenous [product monograph]. 2024. Accessed June 3, 2025. https://pdf.hres.ca/dpd_pm/00068925.PDF
37.Takeda Canada I. Product Monograph of REPLAGAL (agalsidase alfa). 2021.
38.Chiesi Canada Corp. NCT02795676: Study of the Safety and Efficacy of PRX-102 Compared to Agalsidase Beta on Renal Function (BALANCE). ClinicalTrials.gov; 2023. Accessed May 20, 2025. https://clinicaltrials.gov/study/NCT02795676
39.Wallace EL, Goker-Alpan O, Wilcox WR, et al. Head-to-head trial of pegunigalsidase alfa versus agalsidase beta in patients with Fabry disease and deteriorating renal function: results from the 2-year randomised phase III BALANCE study. J Med Genet. 2024;61(6):520-530. doi:10.1136/jmg-2023-109445 PubMed
40.Kawel-Boehm N, Maceira A, Valsangiacomo-Buechel ER, et al. Normal values for cardiovascular magnetic resonance in adults and children. J Cardiovasc Magn Reson. 2015;17(1):29. doi:10.1186/s12968-015-0111-7 PubMed
41.Kidney Disease Improving Global Outcomes CKAWG. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. 2013. https://kdigo.org/wp-content/uploads/2017/02/KDIGO_2012_CKD_GL.pdf
42.Beck M. The Mainz Severity Score Index (MSSI): development and validation of a system for scoring the signs and symptoms of Fabry disease. Acta Paediatr Suppl. 2006;95(451):43-6. doi:10.1080/08035320600618825 PubMed
43.Garner KK, Pomeroy W, Arnold JJ. Exercise Stress Testing: Indications and Common Questions. Am Fam Physician. 2017;96(5):293-299. PubMed
44.Wolk MJ, Bailey SR, Doherty JU, et al. ACCF/AHA/ASE/ASNC/HFSA/HRS/SCAI/SCCT/SCMR/STS 2013 multimodality appropriate use criteria for the detection and risk assessment of stable ischemic heart disease: a report of the American College of Cardiology Foundation Appropriate Use Criteria Task Force, American Heart Association, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, Society of Cardiovascular Computed Tomography, Society for Cardiovascular Magnetic Resonance, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2014;63(4):380-406. doi:10.1016/j.jacc.2013.11.009 PubMed
45.Law C, Wang S, Mani R, Chapple CM, Zeng J, Ribeiro DC. Reliability and validity of the Brief Pain Inventory-Short Form in individuals with rotator cuff-related shoulder pain. Disabil Rehabil. 2025;47(7):1854-1860. doi:10.1080/09638288.2024.2387688 PubMed
46.Dworkin RH, Turk DC, Wyrwich KW, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. J Pain. 2008;9(2):105-21. doi:10.1016/j.jpain.2007.09.005 PubMed
47.Whybra C, Kampmann C, Krummenauer F, et al. The Mainz Severity Score Index: a new instrument for quantifying the Anderson-Fabry disease phenotype, and the response of patients to enzyme replacement therapy. Clin Genet. 2004;65(4):299-307. doi:10.1111/j.1399-0004.2004.00219.x PubMed
48.Weidemann F, Krämer J, Duning T, et al. Patients with Fabry disease after enzyme replacement therapy dose reduction versus treatment switch. J Am Soc Nephrol. 2014;25(4):837-49. doi:10.1681/asn.2013060585 PubMed
49.Ortiz A, Kanters S, Hamed A, et al. Agalsidase beta treatment slows estimated glomerular filtration rate loss in classic Fabry disease patients: results from an individual patient data meta-analysis. Clin Kidney J. 2021;14(4):1136-1146. doi:10.1093/ckj/sfaa065 PubMed
50.Administration USFaD. Prescribing information: TBC (pegunigalsidase alfa) 2 mg/mL, for IV infusion. Company: Chiesi Canada Corp. . Accessed June 2, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/103979s5309lbl.pdf
51.Center for Drug Evaluation Research. Ors. (FDA) USFaDA, ed. TBC (pegunigalsidase alfa) 2 mg/mL, for IV infusion. Company: Chiesi Canada Corp. Application No.:761161Orig1s000. Approval date: 05/09/2023 (FDA approval package). . 2023. Accessed June 02, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2023/761161Orig1s000OtherR.pdf
52.Chiesi Canada Corp. Sponsor's indirect treatment comparisons involving TBC (pegunigalsidase alfa) in Fabry disease [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: TBC (pegunigalsidase alfa): 2 mg/mL concentrate for solution for intravenous infusion December 21, 2023.
53.Excellence NIfHaC, ed. NICE health technology evaluations: the manual. 2025. Accessed June 3, 2025. https://www.nice.org.uk/process/pmg36/resources/nice-health-technology-evaluations-the-manual-pdf-72286779244741
54.Collaboration TC, ed. Cochrane Handbook for Systematic Reviews of Interventions. 2025. Accessed June 3, 2025. https://training.cochrane.org/handbook
55.Brooks SP, and Gelman A. General Methods for Monitoring Convergence of Iterative Simulations. Journal of Computational and Graphical Statistics. 1998;7(4):434-455. doi:10.1080/10618600.1998.10474787
56.Gelman A, Rubin DB. Inference from Iterative Simulation Using Multiple Sequences. Statist Sci 1992;7(4):457-472. doi:10.1214/ss/1177011136
57.Phillippo D, Ades T, Dias S, Palmer S, Abrams KR, Welton N. NICE DSU Technical Support Document 18: Methods for population-adjusted indirect comparisons in submissions to NICE. 2016;
58.Ishak KJ, Proskorovsky I, Benedict A. Simulation and matching-based approaches for indirect comparison of treatments. Pharmacoeconomics. 2015;33(6):537-49. doi:10.1007/s40273-015-0271-1 PubMed
59.Azimpour K, Tordoff-Gibson C, Dorling P, Koulinska I, Laliman-Khara V, Forsythe A. Impact of Baseline Characteristics on Indirect Treatment Comparisons (ITCS) in Fabry Disease: A Systematic Literature Review (SLR). 2023. Accessed June 3, 2025. https://www.ispor.org/heor-resources/presentations-database/presentation/intl2023-3665/125979
60.Hughes DA, Nicholls K, Shankar SP, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet. 2017;54(4):288-296. doi:10.1136/jmedgenet-2016-104178 PubMed
61.Germain DP, Hughes DA, Nicholls K, et al. Treatment of Fabry's Disease with the Pharmacologic Chaperone Migalastat. N Engl J Med. 2016;375(6):545-55. doi:10.1056/NEJMoa1510198 PubMed
62.Banikazemi M, Bultas J, Waldek S, et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007;146(2):77-86. doi:10.7326/0003-4819-146-2-200701160-00148 PubMed
63.Hughes DA, Elliott PM, Shah J, et al. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson-Fabry disease: a randomised, double-blind, placebo-controlled clinical trial of agalsidase alfa. Heart. 2008;94(2):153-8. doi:10.1136/hrt.2006.104026 PubMed
64.Vedder AC, Linthorst GE, Houge G, et al. Treatment of Fabry disease: outcome of a comparative trial with agalsidase alfa or beta at a dose of 0.2 mg/kg. PLoS One. 2007;2(7):e598. doi:10.1371/journal.pone.0000598 PubMed
65.Warnock DG, Mauer M. Fabry disease: dose matters. J Am Soc Nephrol. 2014;25(4):653-5. doi:10.1681/asn.2013121322 PubMed
66.Lenders M, Canaan-Kühl S, Krämer J, et al. Patients with Fabry Disease after Enzyme Replacement Therapy Dose Reduction and Switch-2-Year Follow-Up. J Am Soc Nephrol. 2016;27(3):952-62. doi:10.1681/asn.2015030337 PubMed
67.Amicus Therapeutics Canada Inc. GALAFOLD (migalastat), 123 mg migalastat (as migalastat hydrochloride), oral, alpha-galactosidase A (alpha-Gal A) pharmacological chaperone [product monograph]. 2023. Accessed June 3, 2025. https://pdf.hres.ca/dpd_pm/00071852.PDF
68.Chiesi Canada Corp. Clinical Study Report: PB-102-F30. An Open Label Study to Assess the Safety and Efficacy of PRX-102 in Patients with Fabry Disease Currently Treated With REPLAGAL® (Agalsidase Alfa) [internal sponsor's report]. November 4, 2020.
69.Chiesi Canada Corp. NCT03018730: Safety and Efficacy of PRX-102 in Patients With Fabry Disease Currently Treated With REPLAGAL® (Agalsidase Alfa). ClinicalTrials.gov; 2023. Accessed May 20, 2025. https://clinicaltrials.gov/study/NCT03018730
70.Linhart A, Dostalova G, Nicholls K, et al. Safety and efficacy of pegunigalsidase alfa in patients with Fabry disease who were previously treated with agalsidase alfa: results from BRIDGE, a phase 3 open-label study. Orphanet J Rare Dis. 2023;18(1):332. doi:10.1186/s13023-023-02937-6 PubMed
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Table 32: Summary of Other Efficacy Results From the BALANCE Trial (ITT Population)
Variable | Pegunigalsidase alfa (N = 52) | Agalsidase beta (N = 25) |
|---|---|---|
Plasma lyso-Gb3 concentration | ||
Number of patients contributing to the analysis, n (%) | 51 (98.1) | 25 (100) |
Baseline (nM), mean (SE) | 26.22 (3.78) | 32.14 (7.08) |
Week 104 (nM), mean (SE) | 29.22 (4.48) | 19.65 (3.60) |
Change from baseline to week 104 (nM), mean (SE) | 3.30 (1.38) | –8.74 (4.85) |
Adjusted treatment group change from baseline to week 104 of log-transformed values, adjusted mean (95% CI)a | 0.113 (–0.003 to 0.229) | –0.148 (–0.314 to 0.017) |
Adjusted treatment group change difference vs. control of log-transformed values (95% CI)a | 0.261 (0.058 to 0.463) | |
UPCRb | ||
Baseline, n (%) | 52 (100) | 25 (100) |
≤ 0.5 g/g | 36 (69.2) | 20 (80.0) |
0.5 to < 1 g/g | 9 (17.3) | 2 (8.0) |
≥ 1 g/g | 7 (13.5) | 3 (12.0) |
Week 104, n (%) | 45 (86.5) | 24 (96.0) |
≤ 0.5 g/g | 34 (75.6) | 18 (75.0) |
0.5 to < 1 g/g | 5 (11.1) | 2 (8.3) |
≥ 1 g/g | 6 (13.3) | 4 (16.7) |
Overall MSSI score | ||
Number of patients contributing to the analysis, n (%) | 44 (84.6) | 23 (92.0) |
Baseline (points), mean (SE) | 23.18 (1.42) | 25.16 (2.14) |
Week 104 (points), mean (SE) | 22.11 (1.80) | 27.09 (2.30) |
Change from baseline to week 104 (points), mean (SE) (95% CI)c | –2.07 (0.77) (–3.62 to –0.52) | 2.04 (1.10) (–0.24 to 4.33) |
Treatment group change difference vs. control (points) (95% CI)d | –4.11 (–6.8 to –1.4) | |
Exercise tolerance (stress test) | ||
Baseline, n (%) | 29 (55.8) | 12 (48.0) |
Normal | 18 (34.6) | 6 (24.0) |
Not normal | 11 (21.2) | 6 (24.0) |
Missing | 23 (44.2) | 13 (52.0) |
Week 104, n (%) | 38 (73.1) | 17 (68.0) |
Normal | 22 (42.3) | 11 (44.0) |
Not normal | 16 (30.8) | 6 (24.0) |
Missing | 14 (26.9) | 8 (32.0) |
Pain medication used during the study, n (%) | ||
Number of pain medications used | ||
0 | 14 (26.9) | 3 (12.0) |
1 | 11 (21.2) | 7 (28.0) |
2 | 11 (21.2) | 5 (20.0) |
3 | 4 (7.7) | 5 (20.0) |
4 | 7 (13.5) | 0 |
5 | 3 (5.8) | 2 (8.0) |
6 | 1 (1.9) | 2 (8.0) |
7 | 1 (1.9) | 0 |
8 | 0 | 1 (4.0) |
Pain medication use — shift from baseline to last visit | ||
Pain medication used at baseline, n | ||
0 | 23 | 8 |
1 | 17 | 9 |
≥ 2 | 12 | 8 |
Overall | 52 | 25 |
Pain medication used postbaseline, n (%) | ||
Used 0 medications at baseline and 0 medications at last visit | 20 (87.0) | 8 (100) |
Used 0 medications at baseline and 1 medication at last visit | 2 (8.7) | 0 |
Used 0 medications at baseline and ≥ 2 medications at last visit | 1 (4.3) | 0 |
Used 1 medication at baseline and 0 medications at last visit | 2 (11.8) | 2 (22.2) |
Used 1 medication at baseline and 1 medication at last visit | 15 (88.2) | 7 (77.8) |
Used 1 medication at baseline and ≥ 2 medications at last visit | 0 | 0 |
Used ≥ 2 medications at baseline and 0 medications at last visit | 1 (8.3) | 0 |
Used ≥ 2 medications at baseline and 1 medication at last visit | 0 | 0 |
Used ≥ 2 medications at baseline and ≥ 2 medications at last visit | 11 (91.7) | 8 (100) |
EQ-5D-5L overall health scoree | ||
Baseline, n (%) | 52 (100) | 25 (100) |
Score (points), mean (SE) | 74.6 (3.1) | 75.9 (2.9) |
Week 104, n (%) | 46 (88.5) | 22 (88.0) |
Score (points), mean (SE) | 75.8 (2.5) | 78.0 (3.8) |
Change from baseline at week 104 (points), mean (SE) | 2.0 (1.9) | 1.2 (3.5) |
Treatment group change difference vs. control (points), mean (95% CI)c | 0.8 (–7.2 to 8.8) | |
CI = confidence interval; ITT = intention to treat; lyso-Gb3 = globotriaosylsphingosine; MSSI = Mainz Severity Score Index; nM = nanomolar; SE = standard error; UPCR = urine protein-to-creatinine ratio.
aAnalysis is based on a Mixed Model Repeated Measures with change of log of plasma lyso-Gb3 at each visit as dependent variable, log of baseline lyso-Gb3 value, visit, treatment arm, treatment arm by visit interaction as fixed part of the model. An unstructured covariance matrix is considered to model the within patient correlations and the Kenward-Roger adjustment is used for the degrees of freedom.
bThe laboratory limit of detection for urine protein was 4 mg/dL, and for a large number of the measurements, the protein was undetectable (i.e., < 4 mg/dL), resulting in an uninformative UPCR value (< x g/g). These observations were classified in 1 of the predefined categories, ignoring the ‘ < ’ sign in the observation, conservatively.
cThe 95% CI for the change from baseline is based on the t-distribution for a paired sample.
dThe 95% CIs for the difference between the 2 arms is presented using t-distribution for 2 samples only for visit 27 and visit 53 (week 104).
eOverall health score ranges between 0 (the worst health you can imagine) to 100 (the best health you can imagine).
Source: Clinical Study Report for BALANCE trial18 and sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Table 33: Summary of Other Efficacy Outcomes From the F60 Study (ITT Population)
Variable | Females (N = 32) | Males (N = 55) | Overall (N = 87) |
|---|---|---|---|
Plasma lyso-Gb3 concentration (nM)a | |||
Baseline, n (%) | ██ ██████ | ██ ██████ | ██ ██████ |
Mean (SE) | ██ ██████ | ██ ██████ | ██ ██████ |
Week 52, n (%) | ██ ██████ | ██ ██████ | ██ ██████ |
Mean (SE) | ██ ██████ | ██ ██████ | ██ ██████ |
Mean change from baseline at week 52 (SE) [95% CI] | ██ ██████ | ██ ██████ | ██ ██████ |
UPCR | |||
Baseline, n (%) | ██ ██████ | ██ ██████ | ██ ██████ |
UPCR < 0.15 g/g | ██ ██████ | ██ ██████ | ██ ██████ |
UPCR ≥ 0.15 g/g to ≤ 0.5 g/g | ██ ██████ | ██ ██████ | ██ ██████ |
UPCR > 0.5 g/g | ██ ██████ | ██ ██████ | ██ ██████ |
UPCR ≤ 0.5 g/g | ██ ██████ | ██ ██████ | ██ ██████ |
UPCR > 0.5 g/g to ˂ 1 g/g | ██ ██████ | ██ ██████ | ██ ██████ |
UPCR ≥ 1 g/g | ██ ██████ | ██ ██████ | ██ ██████ |
Week 52, n (%) | ██ ██████ | ██ ██████ | ██ ██████ |
UPCR < 0.15 g/g | ██ ██████ | ██ ██████ | ██ ██████ |
UPCR ≥ 0.15 g/g to ≤ 0.5 g/g | ██ ██████ | ██ ██████ | ██ ██████ |
UPCR > 0.5 g/g | ██ ██████ | ██ ██████ | ██ ██████ |
UPCR ≤ 0.5 g/g | ██ ██████ | ██ ██████ | ██ ██████ |
UPCR > 0.5 g/g to ˂ 1 g/g | ██ ██████ | ██ ██████ | ██ ██████ |
UPCR ≥ 1 g/g | ██ ██████ | ██ ██████ | ██ ██████ |
Overall MSSI score | |||
Baseline, n (%) | ██ ██████ | ██ ██████ | ██ ██████ |
Mean (SE) | ██ ██████ | ██ ██████ | ██ ██████ |
Median (range) | ██ ██████ | ██ ██████ | ██ ██████ |
Week 52, n (%) | ██ ██████ | ██ ██████ | ██ ██████ |
Mean (SE) | ██ ██████ | ██ ██████ | ██ ██████ |
Median (range) | ██ ██████ | ██ ██████ | ██ ██████ |
Mean change from baseline at week 52 (SE) | ██ ██████ | ██ ██████ | ██ ██████ |
95% CI | ██ ██████ | ██ ██████ | ██ ██████ |
Changes from baseline to 12 months in of EQ-5D-5L overall health score | |||
Baseline, n (%) | ██ ██████ | ██ ██████ | ██ ██████ |
Mean (SE) | ██ ██████ | ██ ██████ | ██ ██████ |
Change from baseline to week 52, n (%) | ██ ██████ | ██ ██████ | ██ ██████ |
Mean (SE) | ██ ██████ | ██ ██████ | ██ ██████ |
Median (range) | ██ ██████ | ██ ██████ | ██ ██████ |
CI = confidence interval; ITT = intention to treat; lyso-Gb3 = globotriaosylsphingosine; MSSI = Mainz Severity Score Index; nM = nanomolar; SE = standard error; UPCR = urine protein-to-creatinine ratio.
aPlasma lyso-Gb3 in the F03 study was assessed by a different laboratory compared to the values assessed within the F60 study. Consequently, for study cohort the F03 study (and thus overall), baseline and postbaseline values are not readily comparable and change from baseline needs to be interpreted with that in mind.
Source: Clinical Study Report for PB-102-F6029 and sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Figure 3: Forest Plot of Difference in eGFR Slopes — Subgroup Analyses for ITT Population
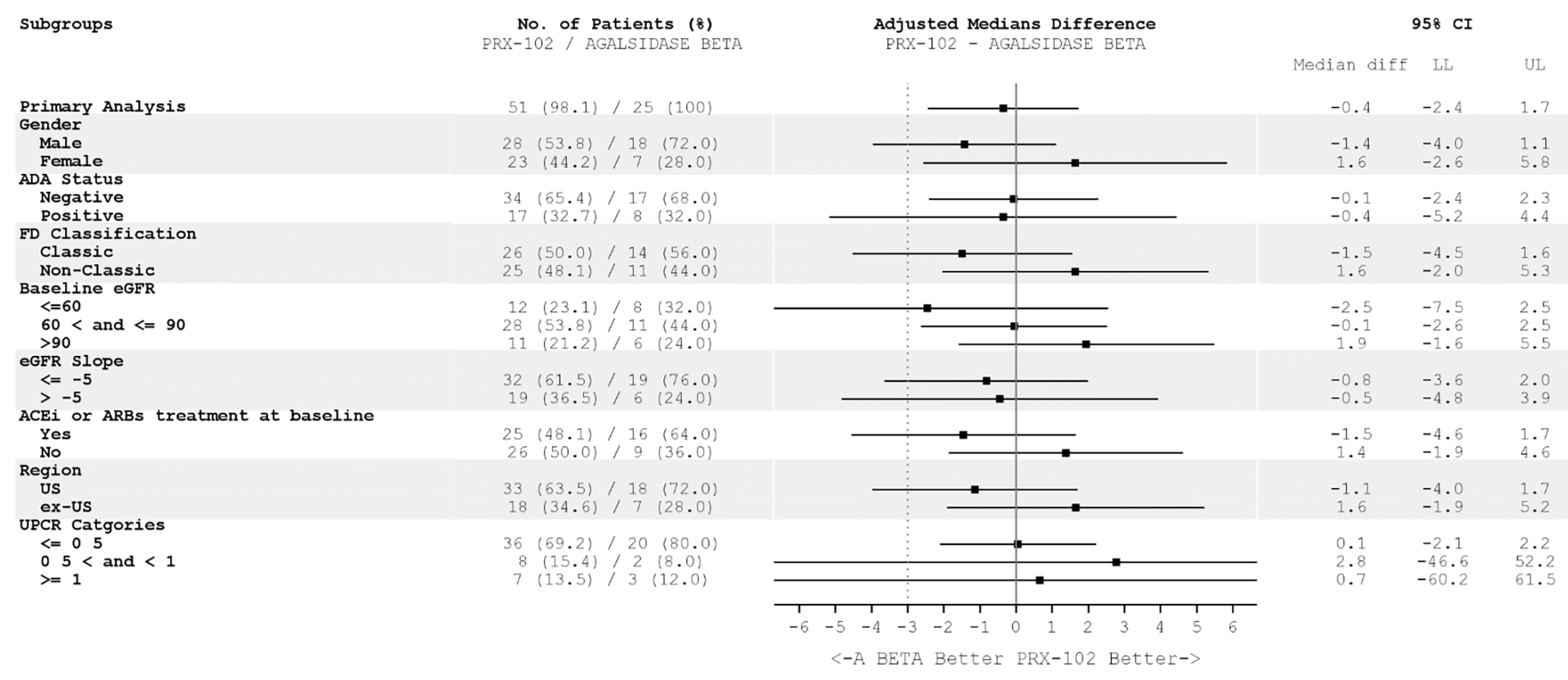
A BETA = agalsidase beta; ACEi = angiotensin-converting enzyme inhibitor; ADA = antidrug antibody; ARB = angiotensin receptor blocker; CI = confidence interval; eGFR = estimated glomerular filtration rate; FD = Fabry disease; LL = lower limit; PRX-102 = pegunigalsidase alfa; UL = upper limit; UPCR = urine protein-to-creatinine ratio.
Note: Vertical dotted line represents the prespecified noninferiority margin of –3.0 mL/min/1.73 m2 per year.
Source: Sponsor’s ITC technical report.52
Table 34: Study Characteristics of RCTs Eligible for ITC
Study reference | Intervention (n) | Comparator (n) | Blinding (phase) | Region | Number of centres | Maximum treatment duration (months) | Switch permitted | Switch time point | Switch comment |
|---|---|---|---|---|---|---|---|---|---|
CSR [PB-102_F20] (BALANCE trial) | Pegunigalsidase alfa 1.0 mg/kg e.o.w. (n = 52) | Agalsidase beta 1.0 mg/kg e.o.w. (n = 25) | Double (III) | Europe, US | Multicentre | 24 | No | NA | NA |
Banikazemi (2007) | Agalsidase beta, 1.0 mg/kg e.o.w. (n = 51) | Placebo (n = 31) | Double (IV) | Europe, North America | Multicentre | 35 | Yes | Trial completion | OLE where placebo patients were allowed to switch to Fabrazyme |
Germain (2016) | Migalastat, 150 mg OD (n = 34) | Placebo (n = 33) | Double (III) | Argentina, Australia, Canada, Egypt, Europe, US | Multicentre | 6 | Yes | 6 months | OLE where placebo patients were allowed to switch to Galafold |
Hughes (2008) | Agalsidase alfa 0.2 mg/kg e.o.w. (n = 7) | Placebo (n = 8) | Double (NR) | NR | NR | 6 | No | NA | NA |
Hughes (2017) | Migalastat, 150 mg q.o.d. (n = 36) | ERT (agalsidase alfa 0.2 mg/kg e.o.w. or agalsidase beta 1.0 mg/kg e.o.w.) (n = 24) | Open (III) | Australia, Europe, US | Multicentre | 18 | No | NA | NA |
Schiffmann (2001) | Agalsidase alfa 0.2 mg/kg e.o.w. (n = 14) | Placebo (n = 12) | Double (NR) | US | Single centre | 6 | No | NA | NA |
Vedder (2007) | Agalsidase alfa, 0.2 mg/kg e.o.w. (n = 18) | Agalsidase beta, 0.2 mg/kg e.o.w. (n = 16) | Open (NR) | Netherlands, Norway | Multicentre | 24 | Yes | Treatment failure | Upon treatment failure (efficacy or safety), patients were allowed to switch to Fabrazyme 1.0 mg/kg e.o.w. |
e.o.w. = every other week, ERT = enzyme replacement therapy; FD = Fabry disease; ITC = indirect treatment comparison; NA = not applicable; NR = not reported; OD = once daily; OLE = open-label extension; q.o.d. = every other day.
Source: Sponsor’s ITC technical report.52
Table 35: Baseline Characteristics of Patients in the RCTs Eligible for ITC
Study reference | Intervention | Treatment-exposed (%) | Time since FD diagnosis (years), mean (SD) | Age (years), mean (SD) | Males (%) | Classic phenotype (%) | Migalastat-amenable variant (%) | LVH (%) | eGFR (mL/min/ | UPCR (g/g), mean (SD) | Presence of ADAs (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
CSR [PB-102_F20] (BALANCE trial) | Pegunigalsidase alfa | 100 | 15.7 (13.1) | 43.9 (10.2) | 55.8 | 51.9 | 5.8 | 40.4 | 73.5 (20.2) | 0.72 (0.69) | 34.6 |
Agalsidase beta | 100 | 13.2 (8.5) | 45.2 (9.6) | 72 | 56.0 | NR | 56.0 | 74.2 (21.0) | 0.54 (0.62) | 32.0 | |
Banikazemi (2007) | Agalsidase beta | 0 | NR | 46.9 (9.8) | 88 | NR | NR | NR | 53.0 (17.7) | 1.5 (1.5) | NR |
Placebo | 0 | NR | 44.3 (9.2) | 87 | NR | NR | NR | 52.4 (17.7) | 1.1 (1.4) | NR | |
Germain (2016) | Migalastat | 14.3 | 5.6 (6.9) | 41.5 (13.0) | 32.1 | 64.3 | 100 | NR | 94.4 (27.0) | NR | NR |
Placebo | 31.8 | 7.3 (8.8) | 45.1 (8.0) | 40.9 | 54.5 | 100 | NR | 90.6 (17.1) | NR | NR | |
Hughes (2008) | Agalsidase alfa | 0 | NR | 37.1 (10.0) | 100 | NR | NR | NR | 106.3 (36.8) | NR | NR |
Placebo | 0 | NR | 37.3 (8.2) | 100 | NR | NR | NR | 100.6 (45.0) | NR | NR | |
Hughes (2017) | Migalastat | 100 | 10.2 (12.0) | 50.5 (13.8) | 44.4 | 36 | 100 | NR | 89.6 (22.2) | NR | NR |
ERT | 100 | 13.4 (11.9) | 46.3 (15.1) | 42.9 | 36 | 100 | NR | 95.8 (18.8) | NR | NR | |
Schiffmann (2001) | Agalsidase alfa | 0 | 12.8 (8.9) | 34.0 (8.5) | 100 | NR | NR | NR | 92.7 (22.4) | NR | NR |
Placebo | 0 | 12.1 (9.4) | 34.4 (7.7) | 100 | NR | NR | NR | 100.6 (40.5) | NR | NR | |
Vedder (2007) | Agalsidase beta | 0 | NR | 48.3 (13.8) | 56.3 | NR | NR | NR | 103.4 (31.3) | NR | NR |
Agalsidase alfa | 0 | NR | 42.1 (13.4) | 50 | NR | NR | NR | 91.9 (32.9) | NR | NR |
ADA = antidrug antibody; eGFR = estimated glomerular filtration rate; ERT = enzyme replacement therapy; FD = Fabry disease; ITC = indirect treatment comparison; LVH = left ventricular hypertrophy; NR = not reported; RCT = randomized controlled trial; SD = standard deviation; UPCR = urine protein-to-creatinine ratio.
Table 36: Summary of Other Efficacy Results From the BRIDGE Study (Efficacy Population)
Variable | Pegunigalsidase alfa (N = 20) |
|---|---|
Plasma lyso-Gb3 | |
Number of patients contributing to the analysis, n (%) | 20 (100) |
Baseline (nM), mean (SE) | 38.51 (9.68) |
Month 12 (nM), mean (SE) | 24.20 (5.10) |
Change from baseline (nM), mean (SE) | –14.31 (5.13) |
UPCR | |
Baseline, n (%) | 20 (100) |
Protein undetectable | 10 (50.0) |
< 0.15 g/g (normal to mildly increased) | 3 (15.0) |
0.15 g/g to ≤ 0.5 g/g (moderately increased) | 3 (15.0) |
> 0.5 g/g (severely increased) | 4 (20.0) |
Month 12, n (%) | 20 (100) |
Protein undetectable | 9 (45.0) |
< 0.15 g/g (normal to mildly increased) | 4 (20.0) |
0.15 g/g to ≤ 0.5 g/g (moderately increased) | 2 (10.0) |
> 0.5 g/g (severely increased) | 5 (25.0) |
Overall MSSI score | |
Number of patients contributing to the analysis, n (%) | 20 (100) |
Baseline (points), mean (SE) | 5.1 (0.8) |
Month 12 (points), mean (SE) | 5.0 (0.8) |
Change from baseline (points), mean (SE) (95% CI) | –1.0 (0.9) |
Exercise tolerance (stress test) | |
Baseline, n (%) | 20 (100) |
Normal | 13 (65.0) |
Not normal | 7 (35.0) |
Missing | 0 |
Month 12, n (%) | 18 (90.0) |
Normal | 10 (50.0) |
Not normal | 8 (40.0) |
Missing | 2 (10.0) |
Pain medication use, n (%) | |
0 | 4 (20.0) |
1 | 6 (30.0) |
2 | 5 (25.0) |
3 | 3 (15.0) |
4 | 1 (5.0) |
5 | 1 (5.0) |
Pain medication use — shift from baseline to last visit | |
Pain medication used at baseline, n | |
0 | 6 |
1 | 6 |
≥ 2 | 8 |
Pain medication used postbaseline, n (%) | |
Used 0 medications at baseline and 0 medications at last visit | 4 (66.7) |
Used 0 medications at baseline and 1 medication at last visit | 2 (33.3) |
Used 0 medications at baseline and ≥ 2 medications at last visit | 0 |
Used 1 medication at baseline and 0 medications at last visit | 1 (16.7) |
Used 1 medication at baseline and 1 medication at last visit | 5 (83.3) |
Used 1 medication at baseline and ≥ 2 medications at last visit | 0 |
Used ≥ 2 medications at baseline and 0 medications at last visit | 0 |
Used ≥ 2 medications at baseline and 1 medication at last visit | 0 |
Used ≥ 2 medications at baseline and ≥ 2 medications at last visit | 8 (100) |
EQ-5D-5L overall health scored | |
Baseline, n (%) | 20 (100) |
Score (points), mean (SE) | 71.8 (4.3) |
Month 12, n (%) | 20 (100) |
Score (points), mean (SE) | 76.9 (4.5) |
Change from baseline at month 12 (points), mean (SE) | 5.1 (3.3) |
CI = confidence interval; lyso-Gb3 = globotriaosylsphingosine; MSSI = Mainz Severity Score Index; nM = nanomolar; SE = standard error; UPCR = urine protein-to-creatinine ratio.
Source: Clinical Study Report for BRIDGE study68 and sponsor’s summary of clinical evidence.28 Details included in the table are from the sponsor’s summary of clinical evidence.
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
CFDI
Canadian Fabry Disease Initiative
CMA
cost-minimization analysis
NMA
network meta-analysis
Economic Review
The objective of the economic review is to review and critically appraise the pharmacoeconomic evidence submitted by the sponsor on the cost and budget impact of pegunigalsidase alfa compared to agalsidase beta, agalsidase alfa, and migalastat for adult patients with a confirmed diagnosis of Fabry disease. Additional information about the sponsor’s submission is summarized in Appendix 2.
Item | Description |
|---|---|
Drug product | Pegunigalsidase alfa (Elfabrio), 2 mg/mL, concentrate for solution (20 mg/10 mL and 5 mg/2.5 mL vial) for IV infusion |
Indication | Proposed: long-term enzyme replacement therapy in adult patients with a confirmed diagnosis of Fabry disease (deficiency of alpha-galactosidase) |
Submitted price | $706.48 per 2.5 mL single-dose vial $2,825.93 per 10 mL single-dose vial |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | December 9, 2025 |
Reimbursement request | Per indication |
Sponsor | Chiesi Canada Corp. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Key Messages
Pegunigalsidase alfa is available as a 2 mg/mL concentrate for solution for IV infusion.1 At the submitted price of $706.48 and $2,825.93 per 2.5 mL and 10 mL single-dose vial, respectively, the annual cost of pegunigalsidase alfa is expected to be $293,897 per patient, based on the Health Canada–recommended dosage.1,2
Based on the results of the Canada’s Drug Agency (CDA-AMC) base case, pegunigalsidase alfa is predicted to be associated with both higher total costs (compared with agalsidase alfa, incremental costs = $1,232) and lower total costs (compared with migalastat and agalsidase beta, incremental savings = $42,488 and $113,972, respectively), which is entirely driven by costs associated with drug acquisition.
CDA-AMC estimates that reimbursing pegunigalsidase alfa for the treatment of Fabry disease will result in a budgetary saving of approximately $2 million over the first 3 years of reimbursement compared to the amount currently spent on agalsidase beta, migalastat, and agalsidase alfa, with an estimated expenditure of $30 million on pegunigalsidase alfa over this period. The actual budget impact of reimbursing pegunigalsidase alfa will depend on the size of the eligible patient population, the distribution of patient weights, which comparators pegunigalsidase alfa captures market share from, and the prices paid by public drug plans for comparator treatments.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-minimization analysis (CMA) comparing pegunigalsidase alfa to agalsidase beta, agalsidase alfa, and migalastat from the perspective of a public drug plan payer in Canada over a 3-year time horizon.2 The modelled population comprised adult patients with a confirmed diagnosis of Fabry disease, which is aligned with the Health Canada indication, and was based on the participants in the BALANCE trial.3 The baseline patient weight was informed by the Canadian Fabry Disease Initiative (CFDI) national patient registry.4 The sponsor’s base-case analysis included costs related to drug acquisition only (submitted price for pegunigalsidase alfa and prices from past CADTH reviews for comparators).2,5
In the sponsor’s base case, pegunigalsidase alfa was associated with cost savings ranging from $5,336 to $107,813 relative to comparators.2 Additional information about the sponsor’s submission is summarized in Appendix 2.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 2; full details are provided in Appendix 3). A revised base case was therefore developed.
Table 2: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The clinical similarity of pegunigalsidase alfa to relevant comparators is uncertain. | Head-to-head evidence for pegunigalsidase alfa compared with agalsidase beta was available, and the CDA-AMC Clinical Review concluded that the drugs may be similar across some outcomes. However, the evidence was highly uncertain due to potential risk of bias in the study, limitations with generalizability, and imprecision in the effect estimates. There have been no head-to-head trials comparing pegunigalsidase alfa to other relevant comparators. The results of the sponsor’s submitted NMA are highly uncertain owing to various limitations in the analysis. As a result, CDA-AMC was unable to conclude whether pegunigalsidase alfa was clinically similar to agalsidase alfa and migalastat. | CDA-AMC could not address uncertainty in the comparative clinical evidence. | No scenario analysis was conducted because of a lack of more robust clinical data. |
The sponsor’s approach to incorporating weight into the analysis was inappropriate. | The sponsor used a median weight from the CFDI patient registry4 to estimate drug costs and used a normal distribution to incorporate this parameter into the probabilistic analysis. Use of median weight is inappropriate because it cannot be incorporated into probabilistic analyses. The use of the normal distribution is also inappropriate because it can permit numbers less than 0. | CDA-AMC incorporated the mean and standard deviation for patient weight from the BALANCE trial and used a lognormal distribution to incorporate weight into the probabilistic analysis. | No scenario analysis was conducted because uncertainty in the weight parameters was incorporated into the probabilistic analysis. |
The sponsor’s analyses rely on publicly accessible list prices and do not reflect existing confidential prices negotiated by public plans. | According to the drug plan input received for this review, comparators to pegunigalsidase alfa have successfully undergone price negotiations for the treatment of Fabry disease. Therefore, it is likely that the current unit costs paid by public drug plans for these treatments are lower than the prices used in the sponsor’s analysis. | CDA-AMC was unable to incorporate the presence of confidential negotiated prices in the reanalysis. | No scenario analyses were conducted. Whether there will be cost savings and the extent of any savings realized by the drug plans is highly uncertain. |
CDA-AMC = Canada’s Drug Agency; CFDI = Canadian Fabry Disease Initiative; NMA = network meta-analysis.
Note: Full details of the issues identified by CDA-AMC are provided in Appendix 3.
CDA-AMC Assessment of Cost
The CDA-AMC base case included drug acquisition costs only. The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to Table 6), in consultation with clinical experts. Detailed information about the CDA-AMC base case is provided in Appendix 3.
The acquisition cost of pegunigalsidase alfa is expected to be $293,897 per patient per year, with a total per-patient cost of $877,087 over a 3-year period, compared to a total cost of $875,855, $919,575, and $991,059 per patient for agalsidase alfa, migalastat, and agalsidase beta, respectively, over the same time period (Table 3). The use of pegunigalsidase alfa is expected to result in incremental costs of $1,232 (relative to agalsidase alfa) and incremental savings of $42,488 and $113,972 (relative to migalastat and agalsidase beta, respectively) per patient. The differences in health care spending result from differences in drug acquisition costs between comparators. Results are highly influenced by patient weight, which influences the number of vials used and the wastage amounts.
Table 3: Summary of CDA-AMC Results
Drug | Total costs ($)a,b |
|---|---|
Pegunigalsidase alfa | 877,087 |
Agalsidase alfa | 875,855 |
Migalastat | 919,575 |
Agalsidase beta | 991,059 |
CDA-AMC = Canada’s Drug Agency.
aIncludes drug acquisition costs only.
bTotal costs over a 3-year period.
Summary of the Budget Impact
The sponsor submitted a budget impact analysis (BIA) to estimate the 3-year (2026 to 2028) budget impact of reimbursing pegunigalsidase alfa for use in the Health Canada–indicated population.6 The sponsor assumed that the payer would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The price of pegunigalsidase alfa was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on the publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in Appendix 4.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (Appendix 4). CDA-AMC estimated that the following numbers of patients would be eligible for treatment with pegunigalsidase alfa over a 3-year period: 294 in year 1; 324 in year 2; 359 in year 3. Of these patients, the following are expected to receive pegunigalsidase alfa: 103 in year 1; 114 in year 2; 125 in year 3. The estimated incremental budgetary savings associated with reimbursing pegunigalsidase alfa are expected to be approximately $2 million over the first 3 years, with an expected expenditure of $30 million on pegunigalsidase alfa. The actual budget impact of reimbursing pegunigalsidase alfa will depend on the size of the eligible patient population, the distribution of patient weights, which comparators pegunigalsidase alfa captures market share from, and the prices paid by public drug plans for comparator treatments.
Conclusions
At the submitted price, pegunigalsidase alfa is expected to be more costly than agalsidase alfa and less costly than migalastat and agalsidase beta for the treatment of adult patients with a confirmed diagnosis of Fabry disease. However, confidential pricing agreements exist for comparators, and a price reduction for pegunigalsidase alfa may be required such that no additional costs are incurred by the health care system. The results are highly sensitive to patient weight, which influences the number of vials required and the amount of wastage associated with each comparator.
The estimated incremental budgetary savings associated with reimbursing pegunigalsidase alfa to the public drug plans in the first 3 years are estimated to be approximately $2 million. The 3-year expenditure on pegunigalsidase alfa (i.e., not accounting for current expenditure on comparators) is estimated to be $30 million. The estimated budget impact is uncertain due to uncertainty in the size of the eligible population, the uncertainty in the actual distribution of patient weights, and the confidential prices of comparators.
References
1.BioScript Logistics Inc. Elfabrio (pegunigalsidase alfa for injection): 2 mg/mL concentrate for solution for intravenous infusion [product monograph]. 2025.
2.Chiesi Canada Corp. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Elfabrio (pegunigalsidase alfa for injection), 2 mg/mL concentrate for solution for intravenous infusion. April 4, 2025.
3.Protalix Ltd. Clinical Study Report: PB-102-F20. A Randomized, Double-blind, Active Control Study of the Safety and Efficacy of PRX-102 Compared to Agalsidase Beta on Renal Function in Patients with Fabry Disease Previously Treated with Agalsidase Beta (The BALANCE Study) [internal sponsor's report]. July 22, 2022.
4.Canadian Fabry Disease Initiative. CFDI Registry Data [sponsor supplied reference]. 2024.
5.CADTH. Drug Reimbursement Review pharmacoeconomic report: migalastat (Galafold) for Fabry disease [sponsor supplied reference]. 2018. https://www.cda-amc.ca/sites/default/files/cdr/pharmacoeconomic/SR0522_Galafold_PE_Report.pdf
6.Chiesi Canada Corp. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Elfabrio (pegunigalsidase alfa for injection), 2 mg/mL concentrate for solution for intravenous infusion. April 4, 2025.
7.Institut national d'excellence en santé et en services sociaux. INESSS Submission Guide for Drugs, Blood System Products and Medical Devices Related to the Administration of Drugs. 2024. Accessed June 23, 2026. https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Fiches_inscription/en/Submission_guidance_document.pdf
8.IQVIA. DeltaPA. 2023. Accessed June 23, 2025. https://www.iqvia.com/
9.Ontario Ministry of Health. Exceptional Access Program product prices. Accessed June 23, 2025. https://www.ontario.ca/page/exceptional-access-program-product-prices
10.Protalix Ltd. Clinical Study Report: PB-102-F20. A Randomized, Double-blind, Active Control Study of the Safety and Efficacy of PRX-102 Compared to Agalsidase Beta on Renal Function in Patients with Fabry Disease Previously Treated with Agalsidase Beta (The BALANCE Study) [internal sponsor's report]. July 22, 2022.
11.Chiesi Canada Corp. Indirect treatment comparisons involving Elfabrio (Pegunigalsidase alfa) in Fabry disease, version 16.0 [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Elfabrio (pegunigalsidase alfa for injection), 2 mg/mL concentrate for solution for intravenous infusion. December 21, 2023.
12.Ontario Ministry of Health. Ontario Schedule of Benefits for Physician Services [sponsor supplied reference]. 2025. https://www.ontario.ca/files/2025-03/moh-schedule-benefit-2025-03-19.pdf
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and CDA-AMC–participating public drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans
Table 4: Cost Comparison for Fabry Disease
Treatment | Strength and/or concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Average annual cost ($) |
|---|---|---|---|---|---|---|
Pegunigalsidase alfa (Elfabrio) | 5 mg 20 mg | Concentrate for solution for IV infusion | 706.4835a 2,825.9340a | 1.0 mg/kg IV infusion every 2 weeks | 804.65 | 293,897 |
Enzyme replacement therapies | ||||||
Agalsidase alfa (Replagal) | 3.5 mg | Vial for IV infusion | 2,300b | 0.2 mg/kg IV infusion every other week | 819.18 | 299,000 |
Agalsidase beta (Fabrazyme) | 5 mg 35 mg | Vial for IV infusion | 798.2900b 5,588.0000b | 1.0 mg/kg IV infusion every 2 weeks | 852.96 | 311,332 |
Chaperone therapy | ||||||
Migalastat (Galafold) | 123 mg | Capsule | 1,700.0000c | 123 mg orally once every other day | 850.00 | 310,250 |
Note: All prices do not include dispensing fees. All weight based doses assumed a patient weight of 76 kg.7
aSponsor submitted price.2
bDeltaPA (accessed June 2025).8
cOntario Exceptional Access Program (accessed June 2025).9
Appendix 2: Additional Details of the Sponsor’s Submission
Please note that this appendix has not been copy-edited.
Table 5: Key Components of the Sponsor’s Economic Evaluation
Component | Description |
|---|---|
Treatment information | |
Drug under review | Pegunigalsidase alfa (Elfabrio), 2 mg/mL, concentrate for solution (20 mg/10 mL and 5 mg/2.5 mL vial) for IV infusion |
Submitted price of drug under review | $706.48 per 2.5 mL single-dose vial $2,825.93 per 10 mL single-dose vial |
Regimen | 1 mg/kg administered by IV infusion every 2 weeks |
Annual cost of drug under review | $311,332 per patient, assuming a weight of 74.2 kg, 26 administrations annually and accounting for wastage |
Model information | |
Type of economic evaluation | Cost-minimization analysis |
Treatment | Pegunigalsidase alfa |
Included comparators |
|
Perspective | Publicly funded health care payer |
Time horizon | 3 years |
Modelled population | Derived from the CFDI patient registry (median weight 74.2 kg)4 |
Model health states | NA |
Data sources | |
Comparative efficacy | The assumption of similar clinical efficacy was based on the BALANCE trial, which compared pegunigalsidase alfa compared with agalsidase beta in adult patients with Fabry disease.10 For all other comparators, the assumption of clinical efficacy was informed by the sponsor’s submitted NMA, which compared pegunigalsidase alfa with agalsidase alfa andmigalastat.11 |
Costs |
|
Summary of the submitted results | |
Base-case results |
|
Scenario analysis results |
|
CFDI = Canadian Fabry Disease Initiative; NA = not available; NMA = network meta-analysis.
Appendix 3: Additional Details of CDA-AMC Reanalyses
Please note that this appendix has not been copy-edited.
Clinical Data in the Economic Model
Assuming similar clinical efficacy and safety for pegunigalsidase alfa compared to agalsidase beta, agalsidase alfa, and migalastat, the sponsor submitted a CMA comparing drug acquisition costs for the Health Canada–indicated population (i.e., adult patients with a confirmed diagnosis of Fabry disease).2 Based on the CDA-AMC Clinical Review of the BALANCE trial, pegunigalsidase alfa may result in a similar decline in estimated glomerular filtration rate slope compared with agalsidase beta. However, the evidence was highly uncertain due to potential risk of bias in the study, limitations with generalizability, and imprecision in the effect estimates. In BALANCE, harms were comparable between pegunigalsidase alfa and agalsidase beta.
Based on the CDA-AMC Clinical Review of the sponsor-submitted network meta-analysis (NMA), the sponsor’s NMA did not demonstrate a difference in favour of 1 treatment over another. However, the assumption of comparable clinical efficacy and safety is highly uncertain because definitive conclusions related to the treatment effect of pegunigalsidase alfa compared to agalsidase alfa or migalastat could not be drawn due to the various limitations with the indirect treatment comparison (i.e., small number of studies contributing to the network; heterogeneity in study designs, patient characteristics, and outcomes; and wide credible intervals resulting in imprecise estimates). Overall, the CDA-AMC Clinical Review concluded that evidence from the submitted NMA is inadequate to inform whether pegunigalsidase alfa will result in similar or different effects compared to other currently funded treatment options for Fabry disease.
Key Issues of the Submitted Economic Evaluation
CDA-AMC identified the following key issues with the sponsor’s analysis:
The clinical similarity of pegunigalsidase alfa to relevant comparators is uncertain: The sponsor submitted a CMA assuming clinical similarity between pegunigalsidase alfa and comparators.2 For the comparison with agalsidase beta, this assumption was based on the BALANCE trial which examined the efficacy and safety of pegunigalsidase alfa versus agalsidase beta.10 Based on the CDA-AMC Clinical Review of the BALANCE trial, pegunigalsidase alfa may result in a similar decline in estimated glomerular filtration rate slope compared with agalsidase beta. However, the evidence was highly uncertain due to potential risk of bias in the study, limitations with generalizability, and imprecision in the effect estimates. In BALANCE, harms were comparable between pegunigalsidase alfa and agalsidase beta. The evidence was rated as being very low certainty for all outcomes, apart from for neutralizing antidrug antibodies, which was rated as having moderate certainty, and severe or very severe infusion-related reactions, which was rated as having low certainty, using the Grading of Recommendations Assessment, Development and Evaluation approach. As such, it is uncertain whether pegunigalsidase alfa is clinically similar to agalsidase beta.
For the comparison with other treatment for Fabry disease (i.e., agalsidase alfa and migalastat), the assumption of clinical equivalence was based on a sponsor-submitted NMA.11 According to the CDA-AMC Clinical Review, the sponsor-submitted indirect treatment comparison did not demonstrate a difference in favour of 1 treatment over another. Overall, definitive conclusions related to the treatment effect of pegunigalsidase alfa compared to agalsidase alfa or migalastat could not be drawn due to the various limitations with the indirect treatment comparison (small number of studies contributing to the network; heterogeneity in study designs, patient characteristics, and outcomes; and wide credible intervals resulting in imprecise estimates). As well, health-related quality of life, which was noted as being important to both patients and clinicians, was not included in the NMA; as such, comparative evidence for pegunigalsidase alfa and agalsidase alfa and migalastat for this outcome was not available. CDA-AMC notes that the sponsor also submitted unanchored population-adjusted indirect comparisons. The CDA-AMC Clinical Review notes that the population-adjusted indirect comparisons have methodological limitations that put their results at high risk of bias that increase the uncertainty of the comparisons; thus, the results were not reported in the clinical review report. Taken together, CDA-AMC was unable to conclude whether pegunigalsidase alfa was clinically equivalent to agalsidase alfa and migalastat.
If differences between pegunigalsidase alfa and comparators exist, a CMA would be insufficient to assess cost-effectiveness.
CDA-AMC was unable to address this limitation through reanalyses.
Method for incorporating patient weight into probabilistic analysis was inappropriate: The sponsor appropriately conducted a probabilistic analysis to account for uncertainty regarding patient weight. This is appropriate because CMA results are sensitive to differences in patient weights due to the number of vials required and accounting for wastage. For example, based on a deterministic analysis using the sponsor’s base-case weight (74.2 kg), pegunigalsidase alfa is $35,803, $23,471 and $35,571 less costly than agalsidase beta, agalsidase alfa, and migalastat, respectively, annually. However, at a weight of 70 kg, pegunigalsidase alfa is $33,416 and $53,940 less costly than agalsidase beta and migalastat, respectively, however, it is $17,960 more costly than agalsidase alfa at this patient weight. Therefore, patient weight is an important driver and it is appropriate for this parameter to be incorporated via probabilistic analyses.
However, the way weight was incorporated in the probabilistic analyses was not appropriate as the sponsor used the median weight from the CFDI patient registry and applied a normal distribution to sample parameters. One, CDA-AMC was not able to validate the median weight or range used with the sponsor’s supporting evidence.4 Two, this approach is inappropriate because the normal distribution should only be used in cases where samples are normally distributed about the mean. As the sponsor used the median rather than the mean for the normal distribution, and because the spread around the median (35.0 kg to 175.6 kg) indicated that the data are skewed, a normal distribution would be inappropriate to use with a median. Further, it is unclear how the standard deviation used to sample was calculated by the sponsor without a mean. Three, use of the normal distribution could permit sampling of values less than 0, which is not clinically plausible for patient weight.
The median weight in the BALANCE trial (74.4 kg) was similar to the median weight cited by the sponsor from the CFDI registry (74.2 kg) meaning that the BALANCE trial may be representative of patients in the CFDI registry (however, there was less spread).3,4
CDA-AMC conducted reanalyses that used the mean patient weight and standard deviation from the BALANCE trial. CDA-AMC also changed the distribution to a lognormal distribution.
Administration costs for Fabry disease are uncertain: Administration costs were not included in the sponsor’s base case; however, the sponsor included administration costs as a scenario analysis.2 As indicated in the sponsor’s submission and confirmed by clinical expert feedback and drug plan input received for this review, the majority of administration costs for current Fabry disease comparators are paid for through various manufacturer patient support programs; therefore, administration costs are not borne by the public health care payer. Whether the sponsor for pegunigalsidase alfa will also cover administration costs is uncertain.
CDA-AMC maintained the sponsor’s assumption that administration costs are excluded from the CMA. If administration costs for pegunigalsidase alfa are not covered by the sponsor, this is likely to result in incremental costs compared with currently reimbursed treatments for Fabry disease as these costs are not typically born by the public health care payer.
Confidential pricing agreements: The sponsor’s base case and CDA-AMC reanalysis is based on publicly available prices from DeltaPA and Ontario EAP.8,9 However, according to drug plan input received for this review, agalsidase beta, agalsidase alfa, and migalastat have successfully gone through price negotiations for Fabry disease. Therefore, the submitted price of pegunigalsidase alfa may require a further price reduction to avoid incurring additional costs relative to its comparators.
CDA-AMC was unable to address this limitation as the negotiated prices of the comparators are unknown.
CDA-AMC Cost Analysis
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. CDA-AMC undertook reanalyses that addressed key limitations within the submitted economic model (Table 6). CDA-AMC reanalyses are presented deterministically and probabilistically.
Results of the CDA-AMC cost comparison are presented in Table 7 and Table 8. At an average annual cost of $293,897 per patient, or $877,087 over a 3-year time horizon, treatment with pegunigalsidase alfa is expected to be $42,488 and $113,972 less costly than migalastat and agalsidase beta, respectively, and $1,232 more costly than agalsidase alfa over a 3-year time horizon. Results of the analysis are sensitive to patient weight, as patient weight influences the number of vials used per dose, influencing the magnitude of cost savings or incremental costs associated with pegunigalsidase alfa.
Table 6: Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Weight | Median = 74.2 kg Standard deviation = 19.85 | Mean = 78.24 kg Standard deviation = 18.14 |
2. Probabilistic distribution used | Normal | Lognormal |
CDA-AMC base case | ― | ― |
CDA-AMC = Canada’s Drug Agency.
Table 7: Summary of the Stepped Analysis
Stepped analysis | Drug | Total costs ($) |
|---|---|---|
Sponsor’s base case | Pegunigalsidase alfa | 814,430 |
Agalsidase alfa | 883,809 | |
Migalastat | 919,575 | |
Agalsidase beta | 920,260 | |
CDA-AMC reanalysis 1 | Pegunigalsidase alfa | 868,726 |
Agalsidase alfa | 883,809 | |
Migalastat | 919,575 | |
Agalsidase beta | 981,611 | |
CDA-AMC reanalysis 2 | Pegunigalsidase alfa | 814,430 |
Agalsidase alfa | 883,809 | |
Migalastat | 919,575 | |
Agalsidase beta | 920,260 | |
CDA-AMC base case (deterministic) (reanalysis 1 + 2) | Pegunigalsidase alfa | 814,430 |
Agalsidase alfa | 883,809 | |
Migalastat | 919,575 | |
Agalsidase beta | 920,260 | |
CDA-AMC base case (probabilistic) (reanalysis 1 + 2) | Agalsidase alfa | 875,855 |
Pegunigalsidase alfa | 877,087 | |
Migalastat | 919,575 | |
Agalsidase beta | 991,059 |
CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
Table 8: Summary of the CDA-AMC Cost Analysis
Drug | Unit drug cost ($) | 3-year drug cost ($) | 3-year total cost ($) | Incremental total costs vs. pegunigalsidase alfa ($) |
|---|---|---|---|---|
Pegunigalsidase alfa | 5 mg vial = 706.4835 20 mg vial = 2,825.9340 | 877,087 | 877,087 | Reference |
Agalsidase alfa | 3.5 mg vial = 2,300.0000 | 875,855 | 875,855 | 1,232 |
Migalastat | 123 mg capsule = 1,700.000 | 919,575 | 919,575 | −42,488 |
Agalsidase beta | 5 mg vial = 798.2900 35 mg vial = 5,588.0000 | 991,059 | 991,059 | −113,972 |
CDA-AMC = Canada’s Drug Agency; vs. = versus.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatment. Probabilistic results reported.
aSponsor’s submitted price for pegunigalsidase alfa.2
Issues for Consideration
Generalizability concerns: All patients enrolled in the BALANCE trial had experience with agalsidase beta and there was limited evidence regarding pegunigalsidase alfa in patients who are treatment naive.3 Clinical expert input received for this review indicated that treatment naive patients could be treated with pegunigalsidase alfa, which is aligned with the Health Canada indication. As well, in the BALANCE trial, patients between age 18 and 60 years were enrolled, and clinical experts indicated that they would consider using pegunigalsidase alfa in patients outside of this age range.
Appendix 4: Budget Impact Analysis
Please note that this appendix has not been copy-edited.
Summary of the Sponsor’s BIA
The sponsor submitted a BIA that estimated the expected incremental budgetary impact of reimbursing pegunigalsidase alfa for the treatment of adult patients with a confirmed diagnosis of Fabry disease.6
The BIA was conducted from the perspective of public drug plan payers over a 3-year time horizon (2026 to 2028), with 2025 as the base year. The sponsor’s estimate reflects the aggregated results from the jurisdictional provincial budgets (excluding Quebec) as well as the Non-Insured Health Benefits Program. The sponsor estimated the eligible population using an epidemiological approach using data from the CFDI national patient registry.4 The sponsor estimated the population size by taking the number of patients with active Fabry disease enrolled in the registry in 2024 and applying an annual growth rate, which was derived by dividing the number of active patients with Fabry disease enrolled over the past 3 years by the total number of active patients with Fabry disease (both adults and children) in each province. The sponsor then estimated the proportion of patients taking disease specific therapy by dividing the number of treated patients in each jurisdiction by the number of enrolled active adults in the registry.
The sponsor’s base case included drug acquisition costs. Market shares in the reference scenario were estimated based on the CFDI registry.4 The market uptake for pegunigalsidase alfa was estimated using sponsor commercial assumption and clinical expert validation. The key inputs to the BIA are documented in Table 9.
The sponsor estimated the 3-year incremental budgetary savings associated with reimbursing pegunigalsidase alfa for the treatment of adult patients with Fabry disease would be $3,033,626 (year 1 = $442,011; year 2 = $975,257; year 3 = $1,616,358).
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Number of patients with active Fabry disease in 2024 | █ █4 |
Fabry disease population growth | Jurisdiction specific, ranged from 0% to 16%4 |
Proportion of patients enrolled in registry with disease specific therapy | Jurisdiction specific, ranged from 11% to 67%4 |
Number of patients eligible for drug under review | █ █ / █ █ / █ █ |
Market shares (reference scenario)a | |
Pegunigalsidase alfa | 0% / 0% / 0% |
Agalsidase beta | █ █% / █ █% / █ █% |
Migalastat | █ █% / █ █% / █ █% |
Agalsidase alfa | █ █% / █ █% / █ █% |
Market shares (new drug scenario)a | |
Pegunigalsidase alfa | 5% / 10% / 15% |
Agalsidase beta | █ █% / █ █% / █ █% |
Migalastat | █ █% / █ █% / █ █% |
Agalsidase alfa | █ █% / █ █% / █ █% |
Cost of treatment (per patient per year) | |
Pegunigalsidase alfa | $275,529 |
Agalsidase beta | $311,332 |
Migalastat | $311,100 |
Agalsidase alfa | $299,000 |
aMarket shares were jurisdiction specific and based on actual treated numbers from CFDI registry.4 Presented numbers are for the Non-Insured Health Benefits Program, which are based on the average market share in Canada.
Key Issues of the Submitted BIA
CDA-AMC identified several key issues to the sponsor’s analysis that have notable implications on the results of the BIA:
Using registry data to estimate market size is uncertain: According to the sponsor, the CFDI registry is a voluntary national patient registry.6 Based on clinical expert feedback received by CDA-AMC for this review, not all patients who are treated for Fabry disease in Canada consent to enrol in the registry. Additionally, based on clinical expert feedback, there are some physicians in Canada who treat Fabry disease who are not part of the registry and therefore do not enrol patients in the registry. Therefore, the number of patients with active disease who are treated in the registry is lower than the total number of patients who are being treated in Canada.
The sponsor’s 2024 values, used to project future cases, also included pediatric patients, who are not included in the proposed Health Canada indication; therefore, inclusion of these cases may overestimate the total number of eligible patients.
Based on clinical expert feedback received for this review, prevalence estimates for Fabry disease vary, and may range between 1 in 74,000 to 1 in 50,000. Of these patients, approximately 55% will be treated, according to clinical expert feedback received for this review.
As well, CDA-AMC observed vastly different growth rates among jurisdictions, which were used to project the number of cases over the time horizon, ranging from 0% to 16%. Clinical expert feedback indicated that as Fabry disease is a rare condition, small additions to the registry, especially in small jurisdictions, are likely to result in large changes to growth rates based on the sponsor’s approach. Clinical expert feedback also indicated that growth in patient cases is likely to exceed population growth for the next 5 to 10 years as access to genetic testing continues to increase; this was also reflected in the sponsor’s estimates. However, expert feedback also indicated that not all newly identified cases will be treated, adding uncertainty to the sponsor’s approach. Taken together, clinical expert feedback indicated that the sponsor’s approach may have overestimated the number of active cases over the time horizon.
CDA-AMC could not address this limitation in reanalyses. The size of the population eligible for treatment may be an underestimate, as the registry does not capture all active or treated cases of Fabry disease, or an overestimate, because the sponsor did not exclude pediatric patients. The sponsor’s 2025 total population size was estimated to be 266. Using a prevalence of 1 in 74,000 and 1 in 50,000 and a treatment rate of 55%, this would result in 241 and 357 active, treated cases, respectively. As pegunigalsidase alfa is lower in cost compared with other comparators at public list prices, a smaller population size will result in less cost savings and a larger population size may increase cost savings anticipated with reimbursing pegunigalsidase alfa.
Patient weight is uncertain and highly influential: Similar to the CMA, the sponsor used the median patient weight from the CFDI registry to estimate dosage.4 CDA-AMC was unable to validate this weight with the information provided by the sponsor.
To align with the CMA, CDA-AMC conducted reanalyses using the mean weight in the BALANCE trial.3
Market capture assumptions are uncertain: The sponsor estimated uptake for pegunigalsidase alfa was deemed to be potentially reasonable based on clinical expert feedback obtained by CDA-AMC for this review. However, the sponsor assumed that pegunigalsidase alfa would take market share proportionally from each comparator.6 Based on clinical expert feedback received for this review, this was deemed inappropriate as market capture is expected to be proportionately taken from agalsidase alfa and agalsidase beta and pegunigalsidase alfa is less likely to capture market from migalastat.
CDA-AMC did not adjust market capture in reanalyses, in the absence of evidence regarding the proportion of uptake that will come from migalastat. CDA-AMC presented a scenario analysis conducted by the sponsor in which no capture is assumed to come from migalastat to demonstrate the impact of this assumption.
The price of drugs paid by public plans is uncertain: The sponsor’s analysis incorporated pricing available past CDA-AMC reviews.5 However, according to drug plan input received for this review, comparators to pegunigalsidase alfa have successfully undergone price negotiations for the treatment of Fabry disease. Therefore, it is likely that the current unit cost paid by public drug plans for these treatments are lower than the prices used in the sponsor’s analysis.
CDA-AMC was unable to address this limitation as the negotiated prices of the comparators are unknown. The budgetary impact of reimbursing pegunigalsidase alfa at its submitted price is likely underestimated.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analyses by making changes in model parameter values and assumptions, in consultation with clinical experts, as outlined in Table 10.
Table 10: Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Patient weight | Median weight, CFDI registry | Mean weight, BALANCE trial |
CDA-AMC base case | ― | Reanalysis 1 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; CFDI = Canadian Fabry Disease Initiative.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 11 and a more detailed breakdown is presented in Table 12. In the CDA-AMC base case, the 3-year incremental budgetary savings of reimbursing pegunigalsidase alfa for adult patients with a confirmed diagnosis of Fabry disease was $1,915,791 (year 1 = $279,654; year 2 = $616,233; year 3 = $1,019,903).
Table 11: Summary of the Stepped Analysis of the CDA-AMC Base Case
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | −3,033,626 |
CDA-AMC reanalysis 1 | −1,915,791 |
CDA-AMC base case: reanalysis 1 | −1,915,791 |
CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments.
CDA-AMC used the CDA-AMC base case to conduct scenario analyses to explore uncertainty in the estimated budget impact of reimbursing pegunigalsidase alfa. The results are provided in Table 12.
Assume none of the uptake of pegunigalsidase alfa is captured from migalastat.
Table 12: Disaggregated Summary of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference total | 81,425,986 | 89,762,997 | 99,102,660 | 109,579,547 | 298,445,204 |
Pegunigalsidase alfa | 0 | 0 | 0 | 0 | 0 | |
All other comparators | 81,425,986 | 89,762,997 | 99,102,660 | 109,579,547 | 298,445,204 | |
New drug total | 81,425,986 | 89,320,710 | 98,126,505 | 107,961,318 | 295,408,532 | |
Pegunigalsidase alfa | 0 | 4,045,863 | 8,934,110 | 14,818,703 | 27,798,677 | |
All other comparators | 81,425,986 | 85,274,847 | 89,192,394 | 93,142,615 | 267,609,856 | |
Budget impact | 0 | –442,287 | –976,156 | –1,618,229 | –3,036,671 | |
CDA-AMC base case | Reference total | 83,375,253 | 91,904,833 | 101,459,507 | 112,176,800 | 305,541,140 |
Pegunigalsidase alfa | 0 | 0 | 0 | 0 | 0 | |
All other comparators | 83,375,253 | 91,904,833 | 101,459,507 | 112,176,800 | 305,541,140 | |
New drug total | 83,375,253 | 91,625,178 | 100,843,274 | 111,156,897 | 303,625,349 | |
Pegunigalsidase alfa | 0 | 4,315,587 | 9,529,718 | 15,806,617 | 29,651,922 | |
All other comparators | 83,375,253 | 87,309,591 | 91,313,556 | 95,350,280 | 273,973,427 | |
Budget impact | 0 | –279,654 | –616,233 | –1,019,903 | –1,915,791 | |
CDA-AMC scenario analyses | ||||||
Scenario 1: no uptake from migalastat | Reference total | 83,375,253 | 91,904,833 | 101,459,507 | 112,176,800 | 305,541,140 |
New drug total | 83,375,253 | 91,617,020 | 100,825,606 | 111,128,187 | 303,570,813 | |
Budget impact | 0 | –287,812 | –633,901 | –1,048,613 | –1,970,327 | |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@cda-amc.ca.