Drugs, Health Technologies, Health Systems
Reimbursement Review
Odevixibat (Bylvay)
Sponsor: Medison Pharma Canada Inc.
Therapeutic area: Alagille syndrome
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
ADHD
attention-deficit/hyperactivity disorder
AE
adverse event
ALGS
Alagille syndrome
ALGSA
Alagille Syndrome Alliance
BMI
body mass index
CASL
Canadian Association for the Study of the Liver
CDA-AMC
Canada’s Drug Agency
CDEC
Canadian Drug Expert Committee
CI
confidence interval
CPHRG
Canadian Pediatric Hepatology Research Group
FAS
full analysis set
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
IBAT
ileal bile acid transporter
LS
least squares
LTE
long-term extension
MID
minimal important difference
MMRM
mixed model for repeated measures
ObsRO
observer-reported outcome
pCPA
pan-Canadian Pharmaceutical Alliance
PedsQL
Pediatric Quality of Life Inventory
PRO
patient-reported outcome
RCT
randomized controlled trial
SAE
serious adverse event
sBA
serum bile acid
SD
standard deviation
SE
standard error
SOC
standard of care
TEAE
treatment-emergent adverse event
UDCA
ursodeoxycholic acid
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Odevixibat (Bylvay), 200 mcg, 400 mcg, 600 mcg, and 1,200 mcg oral capsules |
Sponsor | Medison Pharma Canada Inc. |
Indication | For the treatment of cholestatic pruritus in patients aged 12 months or older with ALGS |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | July 22, 2025 |
Recommended dose | 120 mcg per kg administered orally once daily in the morning with a meal. Dose reduction to 40 mcg per kg per day may be considered if tolerability issues occur in the absence of other causes. Once tolerability issues stabilize, the dose may be increased to 120 mcg per kg per day again. |
ALGS = Alagille syndrome; NOC = Notice of Compliance.
Source: Sponsor submission for the review of odevixibat by Canada’s Drug Agency.1
Introduction
Alagille syndrome (ALGS) is a rare, life-threatening, autosomal dominant genetic disorder that presents with a range of features, including cholestatic liver disease, failure to thrive, cardiovascular disease, skeletal abnormalities, ocular abnormalities, renal and vascular abnormalities, and distinct facial features. In most cases, the liver dysfunction associated with ALGS is an early and serious feature of this genetic condition and typically presents within the first 3 months of life.2-4 Elevated levels of serum bile acid (sBA) and jaundice (elevated bilirubin) are hallmarks of ALGS and indicate the presence of impaired bile flow, also known as cholestasis. Clinically, the hepatic manifestations may range from mild cholestasis to progressive liver failure and usually include severe pruritus and, later, progression to cirrhosis. Cholestatic pruritus may be debilitating and intractable and have a considerable impact on sleep, growth, and school performance in children.5,6
ALGS prevalence and incidence are estimated to be from 1 in 30,000 to 1 in 100,000 individuals or births.7-11 Long-term follow-up studies report premature mortality rates of 7.5% to 35%, with a median age of death between 2.3 and 4 years.2,3,12-18 Diagnosis typically requires that at least 3 of 5 criteria be fulfilled: cholestatic liver disease, congenital heart disease or skeletal, ocular, or facial anomalies.5 While genetic testing (JAG1 or NOTCH2 mutations) supports diagnosis and is publicly funded in Canada, it is not required because a small subset of patients lacks detectable mutations despite meeting clinical criteria.5
In Canada, several drugs are prescribed off label to manage symptoms of ALGS, although many of these treatments have limited or transient efficacy and may cause undesirable adverse effects.19 The clinical experts consulted by Canada’s Drug Agency (CDA-AMC) noted that ursodeoxycholic acid (UDCA) is typically used early in cholestasis management to promote bile excretion, which may indirectly improve pruritus. Other antipruritic treatments prescribed include rifampin, cholestyramine, antihistamines, sertraline, and naltrexone. The clinical experts noted these drugs are often ineffective and have drug interactions and poor tolerability in pediatric patients. Surgical interventions include biliary diversion procedures and liver transplants, which are associated with significant morbidity and mortality. Maralixibat, an ileal bile acid transporter (IBAT) inhibitor like odevixibat, was approved in 2023 for the treatment of cholestatic pruritus in pediatric patients with ALGS. This novel class of drugs inhibits the absorption of bile acids in the small intestine.20 Maralixibat received a recommendation for reimbursement with conditions from the Canadian Drug Expert Committee (CDEC) in April 2024.21 At the start of this review, maralixibat was undergoing negotiations with the pan-Canadian Pharmaceutical Alliance (pCPA) and had not been reimbursed by public drug plans; on June 10, 2025, these negotiations concluded with a Letter of Intent.
The objective of this Clinical Review is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of odevixibat (Bylvay) oral capsules for the treatment of cholestatic pruritus in patients aged 12 months or older with ALGS. The focus will be placed on comparing odevixibat with relevant comparators and identifying gaps in the current evidence.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups that responded to the CDA-AMC call for input and the clinical experts consulted for the purpose of this review.
Patient Input
Input for this review was submitted by 1 patient group, the Alagille Syndrome Alliance (ALGSA), a global nonprofit organization that supports the global ALGS community through resources, programs, and research and support initiatives. ALGSA gathered information for this submission through direct interviews with patients and caregivers and responses to an international September 2020 survey that included respondents living in Canada. Through interactions with patients and family members in international ALGSA support networks, some of whom were living in Canada, the patient group obtained feedback from those with first-hand experience with odevixibat.
The patient group highlighted ALGS as a complex, multisystem genetic disorder that profoundly impacts the day-to-day lives and emotional well-being of patients and caregivers. The most burdensome symptom of ALGS is chronic liver disease, resulting in symptoms such as jaundice, severe itch (pruritus), loss of sleep, discomfort, and malabsorption. The itch was described as “relentless, consuming, and life-altering” and results in open wounds from scratching, sleep disruption, difficulty concentrating, and mental health challenges. Patients often face lengthy diagnostic journeys involving invasive diagnostic procedures and misdiagnoses. Beyond liver complications, ALGS can affect the heart, kidney, bones, eyes, and thyroid, sometimes requiring invasive interventions such as open-heart surgery, dialysis, and catheterizations. Nutritional challenges, including malabsorption, poor growth, and feeding issues, introduce further emotional and time burdens on patients and caregivers. The patient group noted that families often feel trapped in “survival mode,” managing complex medical regimens, frequent appointments, and financial stress.
Before maralixibat and odevixibat were approved in the US, itch was typically managed through the systemic antibiotic rifampin and combined with medications like hydroxyzine, cholestyramine, naltrexone, or UDCA, alongside nonprescription remedies such as oatmeal baths, melatonin, and diphenhydramine. However, patients indicated that these approaches rarely provided significant relief from itch, and the risks of long-term use of antibiotics remain unclear. The patient group noted that the Health Canada approval of maralixibat improved treatment options for ALGS, and that the approval of odevixibat would offer patients greater flexibility in administration methods and provide an alternative treatment option if treatment efficacy is lost over time.
According to the patient group, an “ideal treatment” would provide meaningful and consistent relief from severe itch, reduce the need for multiple medications, avoid major adverse effects (particularly sedation, “liver strain,” and serious infection), minimize disruption to daily life, and offer a convenient administration method. Patients noted they would be willing to accept mild adverse effects if the treatment significantly improves itching and quality of life, while treatments with more substantial risks would be considered only as a last resort.
Overall, families who have had experience with odevixibat report that it substantially reduced or eliminated pruritus, improved appetite and weight gain, and reduced stress, anxiety, and depression. The reported adverse effects were related primarily to mild to moderate gastrointestinal discomfort, including cramping and diarrhea, which typically improved within 1 to 2 weeks of treatment. Families indicated that the benefit of itch relief provided by odevixibat outweighed the initial adverse effects. Families of patients who had discontinued odevixibat reported a rapid return of pruritus, which they felt emphasized its importance for maintaining quality of life.
Clinician Input
Input From Clinical Experts Consulted for This Review
According to the clinical experts consulted by CDA-AMC, the main treatment goals for a patient with cholestasis are to reduce the severity of symptoms (specifically pruritus), to support normal growth and development, and to delay disease progression and need for a liver transplant. The clinical experts emphasized the importance of pruritus to patients, which impacts sleep, school attendance and performance, and the overall quality of life of patients and their families. According to the clinical experts, the current standard treatment options for pruritus (sedatives, antihistamines, cholestyramine, rifampicin, naltrexone and, rarely, sertraline) are of limited effectiveness, have adverse effects, and do not address the mechanism of cholestatic pruritus. Given the lack of effective pruritus treatments, biliary diversion surgery or a liver transplant may be performed to manage symptoms in patients with severe symptoms. In such cases, a liver transplant may be performed before the onset of liver failure. Maralixibat has recently been approved for the management of cholestatic pruritus in patients with ALGS; however, the majority of patients lack access due to cost and reliance on inconsistent private insurance coverage. According to the clinical experts, there are currently no specific treatments that have been demonstrated to modify the natural history of ALGS, although they noted that longer-term data on IBAT inhibitors are still developing.
The clinical experts stated that odevixibat would be used in combination with standard treatment options to control cholestatic pruritus. The patients who are best suited to treatment with odevixibat are those with ALGS, significant or severe pruritus, and consequent impairment in patient and family quality of life, according to the clinical experts consulted by CDA-AMC. These patients would be identified based on the clinical history provided by the patient and/or their family. Other criteria may include high sBA levels. Patients with ALGS and any of the genetic mutations involved (JAG1 or NOTCH2) would be suitable for odevixibat therapy. One clinical expert suggested that odevixibat would be used as a second-line or third-line therapy after starting UDCA (which may improve cholestasis) and a trial of rifampin (to manage pruritus). The other clinical expert identified specific cases where a first-line IBAT inhibitor may be warranted.
In clinical practice, the experts identified improvement in pruritus, sleep, and quality of life and improved growth as the most important outcomes for assessing response to therapy. Improved liver function on lab testing would also be important. Patients are typically assessed every 3 to 6 months, or more frequently in patients with advanced liver disease. The clinical experts expect that early treatment response would be evaluated in the first month or 2 after initiating odevixibat. The clinical experts noted that inadequate symptomatic response, intolerance to the drug (such as intolerable diarrhea), or disease progression necessitating a liver transplant would require discontinuation of the drug.
The clinical experts agreed that odevixibat should be prescribed and monitored by a pediatric gastroenterologist or hepatologist on an outpatient basis in a specialty pediatric gastroenterology or liver clinic. One of the clinical experts noted that patients living in remote areas could be managed by a general pediatrician in collaboration with a pediatric hepatologist or gastroenterologist.
Clinician Group Input
Input for this review was submitted by 1 clinician group, the Canadian Pediatric Hepatology Research Group (CPHRG), which is a committee of the Canadian Association for the Study of the Liver (CASL). CASL is the national professional organization for hepatology in Canada, which aims to eliminate liver disease through research, education, and advocacy. The CPHRG committee within CASL comprises representatives from all pediatric hepatology services in Canada. Information for the input submission was gathered through published literature, conferences, and the collective opinions of experts within the CPHRG, based on their experience in treating patients with ALGS.
The clinician group input was generally consistent with that provided by the consulted clinical experts regarding the limitations of currently available therapies for cholestatic pruritus. Both sources noted that existing pharmacologic options (i.e., antihistamines, cholestyramine, rifampin, naltrexone, and sertraline) are associated with limited efficacy and adverse effects and do not address the underlying pathophysiology of cholestatic pruritus. The clinician group also confirmed that, in the absence of effective symptom control, a liver transplant may be considered as a last resort, although this route has associated risks and a lifelong treatment burden.
With respect to the place in therapy, both the clinician group and clinical experts anticipated that odevixibat would be used as an adjunct to existing therapies rather than as a replacement. The clinician group added that some patients may be able to taper other medications after starting treatment. Both the experts and the clinician group identified patients with moderate to severe pruritus not adequately controlled on standard therapy as the population most likely to benefit from treatment with odevixibat. The clinician group also noted that those with moderate to severe pruritus and elevated bile acids are considered most likely to respond.
In terms of treatment monitoring, both the clinician group and the clinical experts highlighted improvements in pruritus and sleep as clinically meaningful indicators of treatment benefit. The clinician group expressed concerns regarding the practicality of implementing the pruritus assessment tools used in the odevixibat clinical trial (e.g., PRUCISION patient-reported outcome [PRO] and observer-reported outcome [ObsRO] instruments) in real-world practice settings, instead suggesting the Clinician Scratch Score, while the experts noted that similar domains are assessed routinely through clinical history. Both groups agreed that treatment with odevixibat should be discontinued in cases of liver disease progression, transplant, or intolerable adverse events (AEs). Both the experts and the clinician group indicated that odevixibat should be prescribed and monitored by a pediatric hepatologist or gastroenterologist within a specialized care setting.
Drug Program Input
Refer to Table 4 for a summary of the drug plan input.
Clinical Evidence
Systematic Review
Description of Studies
The systematic review included 1 phase III, double-blind, parallel-design, randomized controlled trial (RCT), ASSERT, that compared the efficacy and safety of odevixibat with placebo in patients with ALGS, elevated sBA, and significant pruritus. Patients were randomized to once-daily oral odevixibat 120 mcg/kg (N = 35) or a matching placebo (N = 17) for up to 24 weeks. During the trial, patients were permitted to continue with the standard antipruritic therapies they had been receiving before randomization. The primary end point was the change from baseline to month 6 in the PRUCISION ObsRO scratching severity score. Other key outcomes were the PRUCISION PRO itching severity score, the Pediatric Quality of Life Inventory (PedsQL), and sBA levels.
The mean age of patients enrolled was ███ years (standard deviation [SD] = ███; range, ███ to ████ years) in the odevixibat group and ███ years (SD = ███; range, ███ to ████) in the placebo group. The proportion of females was 40% and 64.7% and the proportion of males was 60% and 35.3% in the odevixibat and placebo groups, respectively. According to the National Cancer Institute Organ Dysfunction Working Group classification system, █████ and █████ of patients had mild hepatic impairment, █████ and █████ had moderate impairment, and █████ and █████ of patients had severe impairment at baseline in the odevixibat and placebo groups, respectively. The mean ObsRO scratching score was 2.80 points (SD = 0.52) and 3.01 points (SD = 0.64), and the mean sBA level was 237.4 µmol/L (SD = 114.9 µmol/L) and 246.1 µmol/L (SD = 120.5 µmol/L) in the odevixibat and placebo groups, respectively. Most patients were receiving UDCA, including 30 patients (85.7%) in the odevixibat group and 16 patients (94.1%) in the placebo group. All patients were on antipruritic medications at baseline, except for 1 patient randomized to the odevixibat group.
Efficacy Results
The primary outcome in the ASSERT trial was the change from baseline to month 6 (average of week 21 to week 24) in the scratching severity score (morning and evening scores combined) measured using the PRUCISION ObsRO questionnaire. Caregivers reported the observed scratching behaviours on a 0 to 4 scale (ranging from no scratching to worst possible scratching) twice daily covering the nighttime (p.m. score) and daytime (a.m. score) symptoms. The least squares (LS) mean difference in the change from baseline was −0.88 points (95% confidence interval [CI], −1.44 to −0.33; 1-sided P = 0.0012) favouring odevixibat versus placebo. The sponsor identified a 1.0-point to 1.5-point reduction as the clinically relevant within-patient threshold for change. The proportion of patients who achieved at least a 1.0-point or 1.5-point reduction in PRUCISION ObsRO scratching scores from baseline to month 6 also favoured odevixibat versus placebo, with point estimates of 44.7% and 36.6%, respectively, for the between-group difference.
Patients aged 8 years and older (N = 15) self-reported itching severity using the PRUCISION PRO questionnaire (0-point to 4-point scale). No statistical difference was detected between the odevixibat and placebo groups based on the change from baseline to month 6 in the itching severity score (LS mean difference of 0.18 points; 95% CI, −0.65 to 1.00; 1-sided P = 0.678).
Health-related quality of life (HRQoL) was assessed using the PedsQL questionnaire, a parent-reported instrument that includes 23 items and 4 domains: physical, emotional, social, and school.22 The PedsQL is scored on a 0 to 100 scale, with higher scores indicating improved HRQoL. The estimated within-group minimal important difference (MID) in children with chronic health conditions (asthma, attention-deficit/hyperactivity disorder [ADHD], depression, diabetes, or “other”) was 4.5 points for the parent-proxy report.22 For odevixibat versus placebo, the LS mean difference in the change from baseline to week 24 in the PedsQL total score was 2.78 points (95% CI, −6.31 to 11.87; 1-sided P = 0.27).
The change from baseline to month 6 in the sBA levels favoured odevixibat versus placebo, with the study reporting an LS mean difference of −112.7 µmol/L (95% CI, −178.8 µmol/L to −46.7 µmol/L; 1-sided P = 0.0006). For patients with ALGS, the MID for the change in sBA levels is unknown.
No deaths were reported, and no patients underwent a liver transplant during the 24-week study.
Harms Results
Overall, 26 (74.3%) of the 35 patients who received odevixibat experienced at least 1 treatment-emergent adverse event (TEAE), as did 12 (70.6%) of the 17 patients who received placebo. The most common TEAEs (≥ 10%) among patients who received odevixibat versus the corresponding frequency in patients who received placebo were diarrhea (28.6% versus 5.9%), pyrexia (22.9% versus 23.5%), COVID-19 (14.3% versus 23.5%), and abdominal pain (11.4% versus 5.9%). Treatment-emergent serious adverse events (SAEs) were reported in 5 patients (14.3%) who received odevixibat and in 2 patients (11.8%) who received placebo (proportion difference of 2.5% for odevixibat versus placebo; 95% CI, −23.0% to 21.6%).
No patients discontinued treatment due to a TEAE. Interruptions and dose reduction due to TEAEs were reported in 3 patients (8.6%) and 1 patient (2.9%), respectively, in the odevixibat group; none of the patients in the placebo group had an interruption in dosing or a dose reduction due to TEAEs.
Diarrhea TEAEs were reported in 10 patients (28.6%) in the odevixibat group and 1 patient (5.9%) in the placebo group (proportion difference of 22.7% for odevixibat versus placebo; 95% CI, −3.3% to 41.7%). No cases were rated as SAEs and none led to treatment discontinuation. ███ ███████ ████████ in the odevixibat group and | ███████ ██████ in the placebo group met the criteria for clinically significant diarrhea (defined as 1 of the following: diarrhea with a duration of 3 or more days without other etiology, diarrhea of grade 3 intensity or reported as an SAE, or diarrhea with concurrent dehydration requiring treatment with rehydration and/or other treatment intervention).
Critical Appraisal
The ASSERT study was a randomized, double-blind, placebo-controlled trial. No major sources of bias in the methods used to randomize and conceal the allocation of patients to treatment groups were identified by the CDA-AMC review team. However, due to the small sample size (N = 52), there is an increased risk that prognostic balance was not achieved, as evidenced by imbalances in patients’ baseline disease and demographic characteristics (age, sex, growth, medical and surgical history, time since diagnosis, extent of hepatic impairment, liver enzymes, and bilirubin levels). As such, it is possible that the observed effects were either overestimated or underestimated and may have been driven by prognostic differences between the 2 groups (i.e., may not be reflective of the true treatment effect). The extent of bias, however, is unclear. Although the trial was double blind, patients who experienced diarrhea, a known adverse effect of IBAT inhibitors, may have been able to infer which treatment group they were assigned to. Subjective outcomes, like pruritus, HRQoL, and harms, may be biased if treatment allocation is known or suspected. Given that diarrhea was reported more frequently in the odevixibat group versus the placebo group (29% versus 6%, respectively), the possibility of reporting bias cannot be ruled out.
No major issues in the statistical methods used in the study were identified by the CDA-AMC review team. A multiple testing procedure was employed for the change from baseline in the PRUCISION ObsRO scratching score and sBA levels. The study assessed pruritus and HRQoL, which were identified as important outcomes based on patient and clinician input. The PRUCISION instrument used to assess pruritus and sleep-related outcomes was developed by the sponsor for patients with cholestatic liver diseases, including ALGS.23 The CDA-AMC review team noted that the instrument has not been validated externally and has been assessed only within the sponsor’s own trials and in a small number of patients. The sponsor estimated that a 1.0-point to 1.5-point decrease in the ObsRO scratching score would represent a clinically important within-patient change.24,25 However, no data were provided on the MID for between-group differences. Thus, the clinical relevance of the mean differences between groups in the change in ObsRO scratching score was difficult to assess. No MIDs were available for the pruritus responder analyses or individual sleep-related outcome measures.
HRQoL was evaluated using the PedsQL instrument, which has evidence to support its validity, reliability, and responsiveness in pediatric patients with various chronic health conditions;26 however, the evidence in children with liver diseases was limited.27 The sponsor relied heavily on proxy reports to assess HRQoL. There is evidence supporting the validity of using this proxy approach for the PedsQL in pediatric populations.26 Of note, there may be a difference between what the proxy thinks and the child thinks with respect to HRQoL and symptoms. For the PedsQL total score analysis, data for 20% and 35% of patients were missing or excluded in the odevixibat and placebo groups, respectively. Given the magnitude of the missing data, the CDA-AMC review team considered this an important limitation of this outcome.
The change in sBA levels was a key secondary outcome in the trial. It has not yet been defined what sBA level or reduction can be associated with improved long-term outcomes in patients with ALGS. Thus, it is difficult to interpret the sBA level results, given that the surrogate threshold effect is unclear.
According to the clinical experts consulted by CDA-AMC, the patients enrolled in the ASSERT study were consistent with pediatric patients with ALGS in Canada. The experts noted that some patients excluded from the trial may be treated with odevixibat in practice, such as those with ascites or waiting for a liver transplant but, overall, the patients enrolled were generalizable. The dose of odevixibat used in the study matched the product monograph. During the trial, patients were allowed to continue the standard antipruritic medications they had been receiving before randomization, which aligns with the anticipated use of odevixibat in practice. The clinical experts confirmed the utilization of antipruritic cointerventions during the trial was consistent with clinical practice.
Limitations to the external validity include the relatively small sample size (N = 52) of the study, the focus on shorter-term end points, and the lack of head-to-head data with maralixibat. Liver transplants and mortality were identified as key clinical outcomes for patients with ALGS. While no deaths were reported and no patients underwent a liver transplant during the 24-week study, the study was not designed to test the comparative effectiveness of odevixibat versus placebo for these end points, and the lack of data for these key clinical outcomes is a limitation of the systematic review. The trial’s sample size and duration were likely insufficient to assess the impact of odevixibat on patients’ growth or on safety, except for the most common TEAEs.
GRADE Summary of Findings and Certainty of the Evidence
For the RCT identified in the sponsor’s systematic review, the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.28,29
Following the GRADE approach, evidence from the RCT started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. For the PRUCISION ObsRO and PRO, sBA levels, and harms, the presence or absence of any (non-null) effect was used and, for the PedsQL, the target of the certainty-of-evidence assessment (presence or absence of an important effect) was based on thresholds identified in the literature.
The selection of outcomes for the GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. Table 2 lists the key outcomes that were finalized in consultation with expert committee members.
Table 2: Summary of Findings for Odevixibat vs. Placebo for Patients With ALGS
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Odevixibat | Difference | |||||
Pruritus outcomes | |||||||
LS mean CFB in the PRUCISION ObsRO scratching severity scorea Follow-up: 24 weeks | 50 (1 RCT) | NA | −0.80 (SE = 0.23) | −1.69 (SE = 0.17) | −0.88 (95% CI, −1.44 to −0.33) | Lowb | Odevixibat may result in a reduction in the ObsRO scratching severity score when compared with placebo. |
LS mean CFB in the PRUCISION PRO itching severity scorea Follow-up: 24 weeks | 15 (1 RCT) | NA | −1.58 (SE = 0.31) | −1.40 (SE = 0.21) | 0.18 (95% CI to −0.65 to 1.00) | Lowc | Odevixibat may result in little to no difference in the PRO itching severity score when compared with placebo. |
HRQoL | |||||||
LS mean CFB in the PedsQL parent report total scored Follow-up: 24 weeks | 39 (1 RCT) | NA | 10.72 (SE = 3.96) | 13.50 (SE = 2.54) | 2.78 (95% CI, −6.31 to 11.87) | Very lowe | The evidence is very uncertain about the effect of odevixibat on the change in the PedsQL parent report total score when compared with placebo. |
sBA | |||||||
LS mean CFB in sBA (µmol/L)f Follow-up: 24 weeks | 52 (1 RCT) | NA | 22.4 (SE = 28.5) | −90.4 (SE = 21.3) | −112.7 (95% CI, −178.8 to −46.7) | Moderateg | Odevixibat likely results in a reduction in sBA levels when compared with placebo. The clinical importance of the decrease is unclear. |
Other efficacy outcomes | |||||||
Liver transplanth | NR | NR | NR | NR | NR | — | There is no evidence to assess the effect of odevixibat on the need for a liver transplant when compared with placebo. |
Mortalityh | NR | NR | NR | NR | NR | — | There is no evidence to assess the effect of odevixibat on mortality when compared with placebo. |
Harms | |||||||
Patients with any SAEs Follow-up: 24 weeks | 52 (1 RCT) | RR = 1.19 (95% CI, 0.26 to 5.45) | 118 per 1,000 | 143 per 1,000 | 25 more per 1,000 (230 fewer to 216 more per 1,000) | Very lowi | The evidence is very uncertain about the effect of odevixibat on SAEs when compared with placebo. |
Patients with diarrhea TEAEs Follow-up: 24 weeks | 52 (1 RCT) | RR = 4.71 (95% CI, 0.67 to 33.15) | 59 per 1,000 | 286 per 1,000 | 227 more per 1,000 (33 fewer to 417 more per 1,000) | Lowj | Odevixibat may result in an increase in diarrhea TEAEs when compared with placebo. The clinical importance of the increase is unclear. |
AE = adverse event; ALGS = Alagille syndrome; CDA-AMC = Canada’s Drug Agency; CFB = change from baseline; CI = confidence interval; HRQoL = health-related quality of life; LS = least squares; MID = minimal important difference; NA = not applicable; NR = not reported; ObsRO = observer-reported outcome; PedsQL = Pediatric Quality of Life Inventory; PRO = patient-reported outcome; RCT = randomized controlled trial; RR = relative risk; SAE = serious adverse event; sBA = serum bile acid; SE = standard error; TEAE = treatment-emergent adverse event; vs. = versus.
Note: Study limitations (which refer to internal validity or risk of bias), indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aPRUCISION ObsRO scratching scores and PRO itching scores range from 0 (no itching or scratching) to 4 (worst itching or scratching). The estimates were based on the a.m. and p.m. scores combined, with baseline levels calculated as the average scores for the 14 days before the start of treatment, and 24-week results based on the average of scores between week 21 and 24.
bThe ObsRO scratching score was rated down 2 levels for very serious concerns about imprecision. The sponsor identified a 1.0-point to 1.5-point decrease as the clinically important within-patient difference based on an analysis of data from the sponsor’s own trials; however, no data were submitted for the between-group MID and the clinical experts consulted by CDA-AMC were unable to estimate a between-group threshold for clinically important effects. Given the uncertainty in the between-group MID, the null was used. The small sample size raises concern for potential overestimation of the true effect and there is evidence of prognostic imbalance. The potential for reporting bias was identified by the CDA-AMC review team. Due to the differential frequency of known AEs potentially leading to patients being able to infer their assigned treatment, and the subjectivity of the outcome, the possibility of reporting bias could not be ruled out; however, the outcome was not downrated due to this risk of bias.
cThe PRO itching score was rated down 2 levels for very serious concerns about imprecision. No published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects; therefore, the null was used. The small sample size raises concern for potential overestimation of the true effect and there is evidence of prognostic imbalance. The potential for reporting bias was identified by the CDA-AMC review team. Due to the differential frequency of known AEs potentially leading to patients being able to infer their assigned treatment, and the subjectivity of the outcome, the possibility of reporting bias could not be ruled out; however, the outcome was not downrated due to this risk of bias.
dThe PedsQL is an HRQoL instrument that is scored from 0 to 100, with higher scores indicating better HRQoL. The data are based on parent-reported assessment.
eThe PedsQL outcome was rated down 1 level due to serious concerns with risk of bias due to missing data. In addition, the potential for reporting bias, due to the differential frequency of known AEs, could not be ruled out. The outcome was rated down 2 levels due to very serious imprecision. The estimated within-group MID was 4.5 points based on the literature (note, no between-group MID was submitted by the sponsor). Based on the available within-group threshold, the 95% CI includes the potential for improvement, little to no difference, and worse HRQoL.
fThe 24-week sBA levels were based on the average of the week 20 and week 24 results. The sBA levels were measured by a central laboratory and the results were concealed from study participants to maintain study blinding.
gThe sBA levels were rated down 1 level for serious concerns about imprecision. No published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects; therefore, the null was used. Although the point estimate and entire CI excluded the null, the small sample size raises concern for potential overestimation of the true effect and there is evidence of prognostic imbalance. Because the effect appeared plausible, the CDA-AMC review team rated it down only once.
hNo patients died or required a liver transplant in either treatment group during the study. However, the trial was not designed (neither in sample size nor in length of follow-up) to inform on these outcomes.
iThe risk of SAEs was rated down 2 levels due to very serious imprecision. No published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects; therefore, the null was used. Although the point estimate suggests an increase, the 95% CI includes the potential for both no difference and a decrease. The risk of SAEs was rated down 1 level due to serious indirectness. The follow-up duration and sample size were insufficient to evaluate the safety of the study drug except for the common AEs that occurred shortly after initiating treatment.30
jThe risk of diarrhea TEAEs was rated down 2 levels due to very serious imprecision. No published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects; therefore, the null was used. Although the point estimate suggests an increase, the 95% CI includes the potential for no difference and a decrease.
Sources: Clinical Study Report for the ASSERT study.31 Additional data supplied by the sponsor on April 28, 2025.32
Long-Term Extension Studies
Description of Studies
The ASSERT-EXT trial (N = 50) was a phase III, multicentre, open-label, long-term extension (LTE) study designed to evaluate the long-term safety and efficacy of odevixibat 120 mcg/kg once daily in patients with ALGS. The study consisted of a 72-week primary treatment period followed by a 4-week safety follow-up period. In the ASSERT-EXT trial, the eligibility criteria, intervention protocols, and evaluated outcomes were identical to those of the ASSERT RCT. Patients in the active treatment arm of the ASSERT RCT continued to be treated with odevixibat using the same dose and protocol (i.e., the continued-treatment group), and those receiving placebo were switched to odevixibat (i.e., the treatment-naive group). Like in the ASSERT RCT, standard of care (SOC) antipruritic treatments were permitted in both groups.
Efficacy Results
Observer-Reported Scratching Severity
At weeks 69 to 72, PRUCISION ObsRO scratching score data were available for ██ patients (███) and ██ patients (███) in the odevixibat continued-treatment and treatment-naive groups, respectively. The mean change from baseline in the ObsRO scratching severity score at weeks 69 to 72 in the continued-treatment group and treatment-naive group was █████ (SD = ████) and █████ (SD = ████), respectively.
From the baseline of the ASSERT trial, at weeks 69 to 72 of the ASSERT-EXT study, ███ of patients in the continued-treatment group had a reduction of 1.0 point or more in the scratching severity score and ███ had a reduction of 1.5 points or more.
From the ASSERT-EXT study baseline, at weeks 69 to 72, 77% of patients in the treatment-naive group had a reduction of 1.0 point or more in the scratching severity score and ███ had a reduction of 1.5 points or more; ███ of patients in the continued-treatment group had a reduction of 1.0 point or more in the scratching severity score and ███ had a reduction of 1.5 points or more.
Patient-Reported Itching Severity
At weeks 69 to 72, PRUCISION PRO itching score data were available for ██ patients (███) and ██ patients (███) in the odevixibat continued-treatment and treatment-naive groups, respectively. The mean change from baseline in itching severity score at weeks 69 to 72 in the odevixibat continued-treatment group and treatment-naive group was █████ (SD = ████) and █████ (SD = ████), respectively.
HRQoL Outcomes
For the PedsQL total score, data at week 72 were available for ██ patients (███) and ██ patients (███) in the continued-treatment and treatment-naive groups, respectively. ████ ████ █████████ ███ ██ █████ ████████ ███ ██ █████ ████████ ██ ███ █████████ █████████ ███ █████████ █████ ███████ ████████████ ███ ███ ██████ ██████ ██████ ██ ████ ███
The mean change from baseline to week 72 in the PedsQL total score was █████ (SD = ████) in the continued-treatment group and ████ (SD = █████) in the treatment-naive groups. ███ ███ ██████ ██████ ███████ ███ ████ ██████ ████ ████████ ██ ███ █████████ █████████ ███ █████████ █████ ███████ █████████████ ███ ████ ███ █ ██████ ███ ████ ███ █ ███████
sBA Levels
The mean change from baseline to week 72 in sBA levels was −36.0 µmol/L (SD = 90.76 µmol/L) in the continued-treatment group and −138.6 µmol/L (SD = 101.17 µmol/L) in the treatment-naive group. At 72 weeks, sBA data were available for 30 patients (91%) in the continued-treatment group and 13 patients (76%) in the treatment-naive group.
Harms Results
Overall, ███ of patients in the ASSERT-EXT study experienced at least 1 TEAE, including ███ of patients in the continued-treatment group and ███ patients in the treatment-naive group. Across both groups, the most common TEAEs were ███████████████ ██████ ████████ ██████ ███████ ██████ ███ ████████ ███ █████ ███████████ █████ █████████ █████. Most TEAEs were grade 1 or 2, with no grade 4 or 5 TEAEs. Treatment-emergent SAEs were reported in ███ of patients overall, including ███ of patients in the continued-treatment group and ███ of patients who were treatment-naive.
Because patients with ALGS are known to have multiple comorbidities, specific AEs of special interest included clinically significant diarrhea, new or worsening fat-soluble vitamin deficiency refractory to clinically recommended vitamin supplementation, potential sequelae of the deficiency, and hepatic events. Overall, ███ of patients had clinically significant diarrhea (most due to being more than 3 days in duration), although all were nonserious and assessed as grade 1 or 2; ██ had fat-soluble vitamin deficiency refractory to clinically recommended vitamin supplementation; ███ reported possible sequelae of fat-soluble vitamin deficiency; and ███ had liver-related TEAEs.
Overall, treatment discontinuation due to TEAEs (elevated bilirubin) occurred in | ███████, and | ████████ had dose reductions due to a TEAE. Treatment interruptions due to a TEAE occurred in ███ of patients in the continued-treatment group and ██ of patients in the treatment-naive group. There were no deaths during the study period.
Critical Appraisal
The ASSERT-EXT study was a phase III, multicentre, open-label study to evaluate the long-term safety and efficacy of odevixibat in patients with ALGS. This open-label design could bias the magnitude of treatment effect for subjective efficacy outcomes (pruritus and HRQoL) and the reporting of safety parameters due to unblinded exposure to the study drug. The direction and magnitude of these potential biases remain unclear. In addition, the absence of statistical hypothesis testing and a control group (i.e., no active comparator or placebo arm) limits the ability to draw definitive conclusions on whether the observed effects can be attributable to natural history, the placebo effect, or the effects of the drug.
The extension study consisted of patients who took part in the ASSERT trial; therefore, it is reasonable to expect that the same strengths and limitations related to generalizability apply to the extension period. There was considerable data missing at week 72 for most efficacy outcomes in the continued-treatment and treatment-naive groups, including caregiver-reported scratching severity score (███ and ███ of patients missing, respectively), patient-reported itching severity score (███ and ███), PedsQL total score (███ and ███), PedsQL Family Impact Module (███ and ███), and sBA levels (██ and ███). A complete case analysis was used, which assumes that the missing data were missing completely at random. Because this assumption is unlikely to be reasonable, there is a risk of bias due to missing outcomes data for these efficacy outcomes. The magnitude and direction of the bias cannot be predicted.
Indirect Comparisons
No indirect treatment comparisons were submitted by the sponsor for this review.
Studies Addressing Gaps in the Evidence From the Systematic Review
No studies in patients treated with odevixibat were submitted by the sponsor.
Conclusions
Based on a double-blind RCT in pediatric patients with ALGS, elevated sBA levels, and significant pruritus, odevixibat may result in a reduction in caregiver-reported pruritus severity at 6 months, compared with placebo, when used in addition to SOC treatments. The impact of odevixibat on patient-reported pruritus severity and HRQoL (measured using the PedsQL parent report total score) were of low and very low certainty, respectively. Odevixibat likely reduces sBA levels compared with placebo, but the clinical relevance of the sBA reduction observed was unclear because the change in sBA levels that results in an improvement in clinical outcomes has yet to be defined for patients with ALGS.
During the RCT and LTE study, no deaths were reported and 1 patient underwent a liver transplant; however, due to the limited sample size and treatment duration, no conclusions can be drawn on the impact of odevixibat on mortality or the need for a liver transplant.
While TEAEs were commonly reported by patients in both the odevixibat and placebo groups, no patients stopped therapy due to AEs. Diarrhea and SAEs were identified as important harms. Odevixibat may result in an increase in diarrhea TEAEs when compared with placebo; however, the clinical importance of the increase was unclear. The evidence was too uncertain to draw conclusions on the impact of odevixibat on the frequency of SAEs compared with placebo.
The evidence was limited to one 24-week placebo-controlled RCT (N = 52) and one 72-week open-label, uncontrolled LTE study (N = 50) and, thus, longer-term efficacy and safety are uncertain. No indirect evidence comparing odevixibat with maralixibat, the main comparator, was submitted because the sponsor-conducted feasibility assessment concluded that an indirect treatment comparison was unlikely to provide robust results. Therefore, the comparative efficacy and safety of odevixibat relative to maralixibat are unknown.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of odevixibat (Bylvay) oral capsules for the treatment of cholestatic pruritus in patients aged 12 months or older with ALGS.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
ALGS is a rare, life-threatening autosomal dominant genetic disorder that presents with a range of features, including cholestatic liver disease, failure to thrive, cardiovascular disease, skeletal abnormalities, ocular abnormalities, renal and vascular abnormalities, and distinct facial features. In most cases, the liver dysfunction associated with ALGS is an early and serious feature of this genetic condition and typically presents within the first 3 months of life.33,34 Elevated sBA levels and jaundice (elevated bilirubin) are hallmarks of ALGS and indicate the presence of impaired bile flow, also known as cholestasis. ALGS is caused by defects in components of the Notch signalling pathway, primarily characterized by hepatic involvement manifesting as cholestasis.35,36 Because Notch signalling plays a key role in determining the fate of biliary tract cells and directing the correct morphogenesis of the biliary tree, defects in this pathway may disrupt the differentiation of hepatocytes to bile duct cells and the formation of the bile ductal plates.35,36 Clinically, the hepatic manifestations may range from mild cholestasis to progressive liver failure and usually include severe pruritus and, later, progression to cirrhosis. Cholestatic pruritus may be debilitating and intractable, and have a considerable impact on sleep, growth, and school performance in children.5,6
Because ALGS clinical presentation is variable and detection has increased over time, the exact size of the ALGS population is somewhat uncertain.37 Prevalence is estimated to range from 1 in 30,000 to 1 in 100,000 individuals,7 and incidence is estimated to range from 1 in 30,000 to 1 in 100,000 births.8-11 The rate of premature mortality in ALGS is variable, with long-term follow-up studies over a 10-year to 40-year follow-up period reporting mortality rates of 7.5% to 35%, with the median age of death ranging from 2.3 to 4 years.2,3,12-18 The pivotal Global Alagille Alliance study, the largest known cohort of patients with ALGS, is a retrospective review of medical records of 1,433 patients with ALGS from 67 pediatric centres from 29 countries.38 During the study period from January 1997 to August 2019, 108 deaths were reported in the Global Alagille Alliance study. Notably, most deaths occurred during the first 5 years of life.38 Further evidence from the Global Alagille Alliance study showed that 570 patients presenting with ALGS-related neonatal cholestasis demonstrated 10-year and 18-year survival rates of 67.3% and 53.6%, respectively, among patients who had not undergone a liver transplant (native liver).39
The most commonly used definition for the diagnosis of ALGS requires that at least 3 of the following 5 criteria be fulfilled:5 cholestatic liver disease, congenital heart disease, skeletal anomalies, ocular abnormalities, characteristic facies. While genetic testing for JAG1 or NOTCH2 mutations supports diagnosis and is publicly funded in Canada, it is not required. It has been estimated that more than 95% of patients with ALGS who are tested will have these mutations.38 However, a small portion of patients fulfilling the classic criteria for ALGS do not appear to have a gene mutation or deletion, making genetic testing unreliable on its own.5 Current diagnostic criteria reflect a combination of clinical features and molecular diagnostic testing to account for clinical and genotypic variability.5
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
IBAT inhibitors are a novel class of drugs that inhibit the absorption of bile acids in the small intestine.20 The IBAT inhibitor maralixibat received a recommendation for reimbursement with conditions from CDA-AMC in April 2024 for the treatment of cholestatic pruritus in patients with ALGS.21 At the time of writing, maralixibat was undergoing negotiations with the pCPA and had not yet been reimbursed by public drug plans; on June 10, 2025, these negotiations concluded with a Letter of Intent.40
In Canada, several drugs are prescribed off label to manage symptoms of ALGS, although many of these treatments have limited or transient efficacy and may cause undesirable adverse effects.19 According to the clinical experts consulted by CDA-AMC, current treatments for cholestatic pruritus include UDCA, which is typically used early in cholestasis to promote bile excretion and may indirectly improve pruritus. Rifampin has been reported to improve pruritus in some patients with ALGS, according to the clinical experts, but may be poorly tolerated or contraindicated due to drug interactions. Cholestyramine, a bile acid–binding resin, may be considered, but its poor palatability and interference with the absorption of other drugs limit its use in clinical practice. Antihistamines are considered for mild cases, primarily for their sedative effects. Sertraline and naltrexone provide marginal additional benefit; however, according to the clinical experts, their use in pediatric patients is limited, and their efficacy in treating pruritus in children with ALGS is unclear.
Because existing pharmacological therapies are often ineffective in patients with ALGS, surgical alternatives are utilized, such as surgical biliary diversion procedures and liver transplants. Surgical biliary diversion procedures (partial internal biliary diversion and ileal exclusion) have been used in ALGS to ameliorate cholestasis, with variable results. They have short-term and long-term surgical and medical complications6,16,33,38,41 and are seldom used in Canada, according to the clinical experts consulted. Intractable pruritus or disfiguring xanthomas can be severe enough to warrant a liver transplant, even in the absence of liver failure.33,38,42 Most patients with ALGS will undergo a liver transplant in the first 2 decades of life, with only 40.3% of patients reaching adulthood with their original liver. Cholestatic pruritus is a leading cause of liver transplants in these patients.6,38 However, a liver transplant in ALGS carries an increased risk of complications, with studies reporting 1-year survival rates below 80%.2 Patients with ALGS who survive a liver transplant face lifelong immunosuppression and other long-term morbidities.33 Children who require transplant at a young age have poorer outcomes, particularly infants aged 1 year or younger, including higher mortality rates while on the transplant wait-list and have lower rates of graft survival.43 Of note, extrahepatic manifestations of ALGS, such as significant cardiac disease, are often a contraindication to a liver transplant, further limiting treatment options for these patients.44
In alignment with the clinical experts consulted by CDA-AMC, the sponsor stated that the management of cholestasis and related pruritus in patients with ALGS remains largely supportive, apart from the recent approval of maralixibat. Surgical treatment may be required for patients continuing to experience extensive morbidity. The clinical experts consulted by CDA-AMC emphasized that the current treatment focus is on improving symptoms, particularly the severely debilitating pruritus of ALGS, which significantly impacts patients’ quality of life. They also highlighted the lack of curative treatments.
Drug Under Review
Odevixibat was approved by Health Canada for the treatment of cholestatic pruritus in patients aged 12 months or older with ALGS.20 The sponsor has requested reimbursement as per the indication. Odevixibat is a reversible, selective inhibitor of the IBAT. It acts locally in the distal ileum to decrease the reuptake of bile acids from the terminal ileum and increase the clearance of bile acids through the colon, reducing the concentration of bile acids in the serum.
The recommended dose of odevixibat is 120 mcg/kg administered orally once daily in the morning with a meal.20 A temporary dose reduction to 40 mcg/kg per day may be considered if tolerability issues occur in the absence of other causes. Odevixibat is available as 200 mcg, 400 mcg, 600 mcg, and 1,200 mcg capsules that may be swallowed whole, opened and sprinkled on soft food, or added to a liquid.
Odevixibat received a recommendation for reimbursement from CDA-AMC on February 13, 2024, for the treatment of cholestatic pruritus in patients with progressive familial intrahepatic cholestasis.45
The key characteristics of odevixibat and other treatments available for cholestatic pruritus are summarized in Table 3.
Table 3: Key Characteristics of Drugs Used to Treat Cholestatic Pruritus
Characteristic | Odevixibat | Maralixibat | UDCA | Rifampin | Antihistamines (hydroxyzine)a |
|---|---|---|---|---|---|
Mechanism of action | An IBAT inhibitor that decreases reabsorption of bile acids from the terminal ileum to improve pruritus. | Same as odevixibat, i.e., IBAT inhibitor that decreases reabsorption of bile acids from the terminal ileum to improve pruritus. | Replaces or displaces toxic concentrations of endogenous hydrophobic bile acids that tend to accumulate in cholestatic liver disease. | Inhibits DNA-dependent RNA polymerase activity in susceptible cells. | Antihistamine that blocks H1 receptors, with anticholinergic, antiemetic, and sedative properties. |
Indicationb | For the treatment of cholestatic pruritus in patients aged 12 months or older with ALGS. | For the treatment of cholestatic pruritus in patients with ALGS. | Not approved for cholestatic pruritus associated with ALGS. For the management of cholestatic liver diseases, such as primary biliary cirrhosis. | Not approved for cholestatic pruritus associated with ALGS. | Not approved for cholestatic pruritus associated with ALGS. |
Route of administration | Oral | Oral | Oral | Oral | Oral |
Recommended dose | 120 mcg/kg once daily in the morning. Dose reduction to 40 mcg/kg/day may be considered if tolerability issues occur in the absence of other causes. Once the tolerability issues stabilize, the dose may be increased to 120 mcg/kg/day again. | Maintenance dose: 380 mcg/kg taken once daily in the morning Initial dose: 190 mcg/kg once daily on days 1 to 7. | 10 mg/kg to 20 mg/kg per day. | 10 mg/kg per day. | In children up to 40 kg in weight, the maximum daily dose is 2 mg/kg per day. In children weighing more than 40 kg and in adults, the maximum daily dose is 100 mg per day, given in divided doses. |
Serious adverse effects or safety issues | Diarrhea, increased levels of liver enzymes and bilirubin, FSV deficiency. | Diarrhea, serum ALT elevations, FSV deficiency. | Leukopenia, rash, abdominal pain. | Hypersensitivity reactions, thrombocytopenia, hemolytic anemia, renal failure, hepatitis, altered metabolism of some other drugs. | QTc prolongation, torsade de pointes, cardiac arrest, sudden death (rare). |
ALGS = Alagille syndrome; ALT = alanine transaminase; FSV = fat-soluble vitamin; IBAT = ileal bile acid transporter; QTc = corrected QT interval; RNA = ribonucleic acid; sBA = serum bile acid; UDCA = ursodeoxycholic acid.
aOther antihistamines include diphenhydramine, cetirizine, and so forth.
bHealth Canada–approved indication.
Sources: Product monographs for odevixibat,20 maralixibat,46 ursodiol (UDCA),47 rifampin,48 and hydroxyzine,49 and an article on ALGS published by UpToDate.50
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the CDA-AMC website.
Patient Group Input
This section was prepared by the review team based on the input provided by a patient group.
Input for this review was submitted by 1 patient group, ALGSA, a global nonprofit organization that supports the global ALGS community through resources, programs, and research and support initiatives. ALGSA gathered information for this submission through direct interviews with patients and caregivers and responses to an international September 2020 survey that included respondents living in Canada. Through interactions with patients and family members in international ALGSA support networks, some of whom were living in Canada, the patient group provided input received from those with firsthand experience with odevixibat.
The patient group highlighted ALGS as a complex, multisystem genetic disorder that profoundly impacts the day-to-day lives and emotional well-being of patients and caregivers. The most burdensome symptom of ALGS is chronic liver disease, resulting in symptoms such as jaundice, severe itch (pruritus), loss of sleep, discomfort, and malabsorption. The itch is described as “relentless, consuming, and life-altering,” and results in open wounds from scratching, sleep disruption, difficulty concentrating, and mental health challenges. Patients often face lengthy diagnostic journeys involving invasive diagnostic procedures and misdiagnoses. Beyond liver complications, ALGS can affect the heart, kidney, bones, eyes, and thyroid, sometimes requiring invasive interventions such as open-heart surgery, renal dialysis, and cardiac catheterizations. Nutritional challenges, including malabsorption, poor growth, and feeding issues, introduce further emotional and time burdens on patients and caregivers. The patient group noted that families often feel trapped in “survival mode,” managing complex medical regimens, frequent appointments, and financial stress.
Before maralixibat and odevixibat were approved in the US, itch was typically managed through the systemic antibiotic rifampicin and combined with medications like hydroxyzine, cholestyramine, naltrexone, or UDCA, alongside nonprescription remedies such as oatmeal baths, melatonin, and diphenhydramine. However, patients indicated that these approaches rarely provided significant relief from itch, and the risks of the long-term use of antibiotics remain unclear. The patient group noted that the Health Canada approval of maralixibat improved treatment options for ALGS, and that the approval of odevixibat would offer patients greater flexibility in administration methods and serve as an alternative treatment option if treatment efficacy is lost over time.
According to the patient group, an “ideal treatment” would provide meaningful and consistent relief from severe itch, reduce the need for multiple medications, avoid major adverse effects (particularly sedation, “liver strain,” and serious infection), minimize disruption to daily life, and offer a convenient administration method. Patients noted they would be willing to accept mild adverse effects if the treatment significantly improves itching and quality of life, while treatments with more substantial risks would be considered only as a last resort.
Overall, families who have had experience with odevixibat report substantially reduced or eliminated pruritus, improved appetite and weight gain, and reduced stress, anxiety, and depression. Reported adverse effects were related primarily to mild to moderate gastrointestinal discomfort, including cramping and diarrhea, which typically improved within 1 to 2 weeks of treatment. Families indicated that the benefit of itch relief offered by odevixibat outweighed the initial adverse effects. Families who discontinued odevixibat reported a rapid return of pruritus, which they felt emphasized its importance for maintaining quality of life.
Clinician Input
Input From the Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of ALGS.
Unmet Needs
The clinical experts identified the main treatment goals for a patient with cholestasis, which are to reduce the severity of symptoms (specifically pruritus), support normal growth and development, delay disease progression, and delay the need for a liver transplant.
The clinical experts emphasized the importance of pruritus to patients, which impacts sleep, school attendance and performance, and the overall quality of life of patients and their families. According to the clinical experts, the current standard treatment options for pruritus (sedatives, antihistamines, cholestyramine, rifampicin, naltrexone and, rarely, sertraline) are of limited effectiveness, have adverse effects, and do not address the mechanism of cholestatic pruritus. Given the lack of effective pruritus treatments, a liver transplant may be performed to manage symptoms in patients with severe pruritus. In such cases, a liver transplant may be performed before the onset of liver failure.
The clinical experts noted that partial external biliary diversion surgery may be performed to help control the condition, but it also has limitations. Patients will have a stoma in the front of the abdomen and must wear a bag over it, which may be challenging to manage at school. In addition, dehydration may be problematic for patients who have undergone partial external biliary diversion, and the procedure may pose a higher risk for patients with ALGS than for those with other chronic cholestasis disorders.
Maralixibat has recently been approved for the management of cholestatic pruritus in patients with ALGS; however, the majority of patients lack access due to cost and reliance on inconsistent private insurance coverage. According to the clinical experts, there are currently no specific treatments that have been demonstrated to modify the natural history of ALGS, although they noted that longer-term data for IBAT inhibitors is still developing.
Place in Therapy
The clinical experts stated that odevixibat would be used in combination with standard treatment options to control cholestatic pruritus. Most patients would also receive UDCA, which is not primarily indicated for management of pruritus. The clinical experts added that patients with mild pruritus, whose symptoms may be controlled with standard treatments, were unlikely to receive odevixibat. In the experts’ opinion, it would be preferable to use odevixibat early in the care of a patient with ALGS with cholestatic pruritus rather than as a last resort, due to its efficacy and its mechanism of action, which has the potential to alter the course of the disease.
One of the clinical experts anticipates that IBAT inhibitors, such as odevixibat, may replace partial external biliary diversion (PEBD) or, alternatively, may function as an adjunctive therapy for patients who experience a clinical response to PEBD that is suboptimal.51
One clinical expert suggested that odevixibat would be used as second-line or third-line therapy, after starting UDCA, which may improve cholestasis, and a trial of rifampin to manage pruritus. They noted that drug interactions are a limitation with rifampin. The other clinical expert identified specific cases where a first-line IBAT inhibitor may be warranted, such as an infant who is suffering with pruritus and whose development may be slowed at this key stage of neurodevelopment.
Patient Population
The patients who are best suited to treatment with odevixibat are those with significant or severe pruritus and consequent impairment in patient and family quality of life, according to the clinical experts consulted by CDA-AMC. These patients would be identified based on the clinical history provided by the patient and/or their family. Other criteria may include high sBA levels. Patients with ALGS and any of the genetic mutations involved (JAG1 or NOTCH2) would be suitable for odevixibat therapy.
When asked, both clinical experts were of the opinion that odevixibat could be continued beyond the age of 18, as long as patients were still deriving benefit from the drug and had not received a liver transplant.
Assessing the Response to Treatment
In clinical practice, the experts identified improvement in pruritus, sleep, and quality of life and improved growth as the most important outcomes for assessing response to therapy. Improved liver function on lab testing would also be important. Patients are typically assessed every 3 to 6 months, or more frequently in patients with advanced liver disease. The clinical experts expect that early treatment response would be evaluated a month or 2 after initiating odevixibat. The experts indicated that monitoring of sBA levels is not routine in clinical practice and would not be required to assess response to therapy. Testing of sBA levels may not be reimbursed in all jurisdictions, according to the clinical experts.
One expert indicated that a formal numerical pruritus assessment tool or HRQoL tools are not commonly used in clinical practice. However, routine clinical practice would include asking most of the questions that are assessed by these tools.
One clinical expert stated that a decrease in pruritus severity of at least 1 point on the PRUCISION instrument (or equivalent measurement tool for pruritus) would be clinically meaningful, although the other expert noted a partial improvement in pruritus would be an acceptable but less satisfactory outcome.
Discontinuing Treatment
The clinical experts noted that inadequate symptomatic response, intolerance to the drug (such as intolerable diarrhea) or disease progression necessitating a liver transplant would require discontinuation of the drug.
Prescribing Considerations
The clinical experts agreed that odevixibat should be prescribed and monitored by a pediatric gastroenterologist or hepatologist on an outpatient basis in a specialty pediatric gastroenterology or liver clinic. One of the clinical experts noted that patients living in remote areas could be managed by a general pediatrician in collaboration with a pediatric hepatologist or gastroenterologist.
Clinician Group Input
This section was prepared by the review team based on the input provided by a clinician group.
Input for this review was submitted by 1 clinician group, the CPHRG committee of the CASL. The CASL is the national professional organization for hepatology in Canada, which aims to eliminate liver disease through research, education, and advocacy. The CPHRG committee comprises representatives from all pediatric hepatology services in Canada. Information was gathered for the input submission through published literature, conferences, and the collective opinions of experts within the CPHRG, based on their experience managing patients with ALGS.
The clinician group input was generally consistent with that provided by the consulted clinical experts regarding the limitations of currently available therapies for cholestatic pruritus. Both sources noted that existing pharmacologic options (i.e., antihistamines, cholestyramine, rifampin, naltrexone, and sertraline) are associated with limited efficacy and adverse effects and do not address the underlying pathophysiology of cholestatic pruritus. The clinician group also confirmed that, in the absence of effective symptom control, a liver transplant may be considered as a last resort, although this route has associated risks and a lifelong treatment burden.
With respect to the place in therapy, both the clinician group and clinical experts anticipated that odevixibat would be used as an adjunct to existing therapies rather than as a replacement. The clinician group added that some patients may be able to taper other medications after starting treatment. Both the experts and clinician group identified patients with moderate to severe pruritus not adequately controlled on standard therapy as the population most likely to benefit from treatment with odevixibat. The clinician group also noted that those with moderate to severe pruritus and elevated bile acids are considered most likely to respond.
In terms of treatment monitoring, both the clinician group and the clinical experts highlighted improvements in pruritus and sleep as clinically meaningful indicators of treatment benefit. The clinician group expressed concerns regarding the practicality of implementing the pruritus assessment tools used in the odevixibat clinical trial (e.g., PRUCISION ObsRO and PRO) in real-world practice settings, instead suggesting the Clinician Scratch Score, while the experts noted that similar domains are assessed routinely through clinical history. Both groups agreed that treatment with odevixibat should be discontinued in cases of liver disease progression, transplant, or intolerable AEs. Both the experts and the clinician group indicated that odevixibat should be prescribed and monitored by a pediatric hepatologist or gastroenterologist within a specialized care setting.
Drug Program Input
The drug programs provide input on each drug being reviewed through CDA-AMC Reimbursement Review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The ASSERT study is a phase III, double-blind, randomized, placebo-controlled trial of odevixibat in children with ALGS. No comparative evidence was submitted by the sponsor for odevixibat vs. maralixibat. Maralixibat is indicated for the treatment of cholestatic pruritus in patients with ALGS and received a CDEC recommendation to reimburse with conditions. | The clinical experts consulted by CDA-AMC agreed that SOC (e.g., UDCA, rifampin, and antihistamines) as well as maralixibat were relevant comparators for odevixibat. |
Considerations for initiation of therapy | |
The instruments used to assess pruritus in the maralixibat and odevixibat clinical trials were different. The ICONIC trial (and CDEC recommendation) uses the ItchRO or the Clinician Scratch Scale for maralixibat. The ASSERT trial for odevixibat uses the PRUCISION ObsRO scratching score. Question: Is there a way to harmonize itch scoring tools across both drugs for ALGS so that clinicians would need to be versed in only 1 tool? | The ItchRO and PRUCISION ObsRO pruritus instruments are similar, with scores ranging from 0 (no itching or scratching) to 4 (the “worst” or “very severe” itching or scratching). The experts did not have an opinion on whether the pruritus scales could be considered interchangeable in clinical practice because they were not aware of evidence comparing the 2 instruments. |
If possible, consider aligning with the maralixibat initiation criteria regarding evidence of cholestasis, i.e., must include at least 1 of the following criteria:
| The experts noted that the ASSERT study inclusion criteria were different from the criteria used for the pivotal maralixibat trial and did not require patients to meet the bilirubin, vitamin deficiency, or GGT criteria listed. In addition, the sBA criteria in the ASSERT trial was above the ULN, not 3 times the ULN. Despite the differences in the criteria, the clinical experts anticipated that most patients with ALGS and moderate to severe pruritus would meet the reimbursement initiation criteria listed based on bilirubin levels or intractable pruritus. The experts expect that odevixibat and maralixibat would be used in the same patient population and emphasized the need for more than 1 IBAT inhibitor because not all patients will show an adequate response from the initial IBAT received. |
Consider aligning the prerequisite medications with the maralixibat recommendation, which specifies that patients must have undergone an adequate trial of 1 to 3 months with appropriate dosing of a systemic treatment for pruritus based on usual care. Such treatment may include:
| The clinical experts stated that most patients with moderate to severe pruritus will have been treated with 1 of the drugs listed, and it would be reasonable to apply these criteria to the reimbursement of odevixibat. They noted that SOC treatment options may control symptoms in some patients with mild pruritus. For patients with severe pruritus, the clinical experts noted that a requirement to use off-label antipruritic treatments before odevixibat would delay many patients from receiving an effective therapy. |
Can patients switch between maralixibat and odevixibat? | The experts stated that patients may switch between odevixibat and maralixibat if they had an inadequate response to either of these drugs. |
Considerations for continuation or renewal of therapy | |
Consider aligning with maralixibat renewal criteria, if possible, given that the submitted studies used different pruritus grading tools. | The clinical experts stated that the response threshold specified in the maralixibat renewal criteria (i.e., an improvement in pruritus to minimal or no itch [a score of 1 or less] on the ItchRO or Clinician Scratch Scale) was too stringent and suggested that a 1-point improvement on a 0 to 4 pruritus severity scale would be clinically important. In practice, patients would continue to receive therapy if they report any subjective improvement in pruritus and a partial response may be clinically important to patients, according to the experts. The clinical experts also noted that numerical rating scales are not commonly used in clinical practice to assess pruritus symptoms, but these instruments could be adopted if required for reimbursement. |
Considerations for prescribing of therapy | |
Given orally at a dose of 120 mcg/kg once daily in the morning. May decrease to 40 mcg/kg per day if tolerability issues occur in the absence of other causes. | This is a comment from the drug plans to inform CDEC deliberations. |
Odevixibat and maralixibat have the same mechanism of action. Question: Would the 2 drugs be prescribed together? | The clinical experts did not anticipate that these 2 IBAT inhibitors would be prescribed concurrently. |
Consider aligning prescribing criteria with maralixibat. | The clinical experts stated that odevixibat should be prescribed and monitored by a pediatric gastroenterologist or hepatologist on an outpatient basis in a specialty pediatric gastroenterology or liver clinic. One of the clinical experts noted that patients living in remote areas could be managed by a general pediatrician in collaboration with a pediatric hepatologist or gastroenterologist. |
System and economic issues | |
The sponsor’s base-case analysis assumed the following market share distribution:
The budget impact assessment resulted in varying annual cost estimates for odevixibat, depending on dosing assumptions. Over the first 3 years after the introduction of odevixibat, the total budget impact was estimated to range from −$15,063,908 to $11,713,715. The cost per capsule of odevixibat ranges from $175.92 for the 200 mcg capsule to $1,055.55 for the 1,200 mcg capsule. The annual cost by body weight ranges from $192,768 for less than 4 kg to $2,313,238 for 55.5 kg or higher. | This is a comment from the drug plans to inform CDEC deliberations. |
ALGS = Alagille syndrome; CDA-AMC = Canada’s Drug Agency; CDEC = Canadian Drug Expert Committee; GGT = gamma-glutamyl transferase; IBAT = ileal bile acid transporter; ItchRO = Itch Reported Outcome; ObsRO = observer-reported outcome; sBA = serum bile acid; SOC = standard of care; UDCA = ursodeoxycholic acid; ULN = upper limit of normal; vs. = versus.
Clinical Evidence
The objective of this Clinical Review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of odevixibat (Bylvay) 200 mcg, 400 mcg, 600 mcg, and 1,200 mcg oral capsules for the treatment of cholestatic pruritus in patients aged 12 months or older with ALGS. The focus will be placed on comparing odevixibat with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of odevixibat is presented in 2 sections, with the CDA-AMC critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes an RCT that was selected according to the sponsor’s systematic review protocol. Our assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes a sponsor-submitted LTE study.
No indirect evidence was submitted by the sponsor. Based on the feasibility assessment it conducted, the sponsor determined that an indirect treatment comparison for odevixibat versus maralixibat was not feasible.52
The sponsor submitted 2 studies intended to address gaps in the systematic review evidence.53,54 Because neither of these studies informed on the benefits or harms of the drug under review, the CDA-AMC review team did not summarize or appraise them in this report.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
1 pivotal RCT identified in the systematic review (the ASSERT trial)
1 LTE study (the ASSERT-EXT trial).
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
Characteristics of the included study are summarized in Table 5.
Table 5: Details of Study Included in the Systematic Review
Detail | ASSERT study |
|---|---|
Designs and populations | |
Study design | Phase III, double-blind, placebo-controlled RCT |
Locations | 21 sites: Belgium, France, Germany, Israel, Italy, Malaysia, Netherlands, Poland, Türkiye, UK, US |
Patient enrolment dates | Start date: February 26, 2021 End date: September 9, 2022 |
Randomized (N) | Total N = 52:
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Odevixibat oral capsules, 120 mcg/kg once daily for 24 weeks (concomitant SOCb therapy was permitted). |
Comparator(s) | Matching placebo (concomitant SOCb therapy was permitted). |
Study duration | |
Screening phase | Up to 56 days. |
Treatment phase | 24 weeks. |
Follow-up phase | 28-day safety follow-up (not required for patients who entered the 72-week LTE study). |
Outcomes | |
Primary end point | Change from baseline to month 6 (weeks 21 to 24) in the average morning and evening scratching severity score (PRUCISION ObsRO caregiver instrument). |
Secondary and exploratory end points | Key secondary:
Secondary:
Change from baseline to week 24 in the following:
Exploratory:
|
Publication status | |
Publications | |
ALGS = Alagille syndrome; ALT = alanine aminotransferase; AST = aspartate aminotransferase; GGT = gamma-glutamyl transferase; GIC = Global Impression of Change; GIS = Global Impression of Symptoms; INR = international normalized ratio; LTE = long-term extension; ObsRO = observer-reported outcome; PedsQL = Pediatric Quality of Life Inventory; PFIC = progressive familial intrahepatic cholestasis; PRO = patient-reported outcome; RCT = randomized controlled trial; SOC = standard of care; UDCA = ursodeoxycholic acid; ULN = upper limit of normal.
aMean weekly score for morning ObsRO and evening ObsRO were averaged to determine the overall ObsRO score. A minimum of 4 of the 7 expected scores were required to be reported.
bMedications for pruritus, including UDCA, rifampicin, and antihistamines, were allowed if patients were on a stable dosage for at least 4 weeks before enrolment, with no planned dosage changes. The use of topical treatments was unrestricted.
Sources: ASSERT Clinical Study Report31 and the sponsor’s summary of clinical evidence.52
The ASSERT study was a 24-week double-blind, randomized, placebo-controlled, multicentre, phase III study to evaluate the efficacy and safety of odevixibat versus placebo in patients with ALGS and significant pruritus (Table 5). Eligible patients were randomized (2:1) to receive oral odevixibat 120 mcg/kg once daily or placebo. An interactive web response system was used to assign patients to treatment based on a block size of 6 that was stratified by age group (< 10 years or 10 to < 18 years). A total of 52 patients were enrolled between February 26, 2021, and September 9, 2022, from 21 sites in Europe, Malaysia, and the US. The primary end point was the change in pruritus severity from baseline to month 6 (weeks 21 to 24) as measured using the caregiver-reported PRUCISION ObsRO scratching score. The design of the ASSERT study is provided in Figure 1.
In both groups, concomitant SOC antipruritic therapy was permitted; thus, the odevixibat group reflects odevixibat plus SOC and the placebo group reflects placebo plus SOC. However, in this report, the treatment groups will be referred to as odevixibat and placebo.
At the end of the randomized treatment period (week 24), patients had the option to enter the 72-week extension study and receive open-label odevixibat.

ALGS = Alagille syndrome; ObsRO = observer-reported outcome; R = randomization.
Source: Sponsor’s summary of clinical evidence.52
Populations
Inclusion and Exclusion Criteria
The ASSERT study included male and female patients of any age with a genetically confirmed diagnosis of ALGS, a history of significant pruritus, and elevated sBA levels (exceeding the upper limit of normal). Significant pruritus was defined as a caregiver-reported observed scratching score or a patient-reported itching score at an average of at least 2 points (on a 0 to 4 scale), as measured by the PRUCISION ObsRO instrument (for patients < 18 years) or the PRO instrument (for patients ≥ 18 years) in the 14 days before randomization. Exclusion criteria included a history of other liver diseases (e.g., biliary atresia, progressive familial intrahepatic cholestasis, liver cancer); conditions affecting drug absorption, distribution, metabolism, and excretion (e.g., inflammatory bowel disease); biliary diversion surgery within 6 months of screening; prior or planned liver transplant; and decompensated liver disease with ascites, variceal hemorrhage, or encephalopathy (Table 5).
Interventions
Patients were administered odevixibat orally once daily at a dose of 120 mcg/kg per day or a matching placebo for up to 24 weeks. The study drug was supplied as identical-looking capsules that contained pellets of placebo or odevixibat (200 mcg, 600 mcg, or 1,200 mcg). The capsules could be opened and the contents sprinkled on food or mixed in infant formula or breast milk, if needed. The number of capsules dispensed at each study visit was adjusted depending on the patient’s body weight to maintain the 120 mcg/kg daily dose during the trial. To manage potential AEs, dose modifications could be conducted by the principal investigator, in which case the patient continued taking the study drug at a reduced dose level of 40 mcg/kg per day. A total of 2 dose reductions were permitted during the study.
Concomitant use of SOC medications for pruritus, including UDCA, rifampicin, and antihistamines, was allowed if patients were on a stable dosage for at least 4 weeks before enrolment, with no planned dosage changes. The use of topical treatments was unrestricted. During the study, drugs affecting bile acid levels or gastrointestinal motility were prohibited. However, other products with possible impacts on gastrointestinal motility (e.g., selective serotonin reuptake inhibitors, tetracyclic antidepressants, fibre supplements, yogourt variants) were permitted if used at a stable dose for at least 4 weeks before enrolment.
To maintain blinding to treatment allocation over the course of the trial, sBA levels were measured by a central laboratory and the investigator, patient, caregiver, and sponsor were blinded to the results.
Outcomes
A list of efficacy end points assessed in this Clinical Review is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review, according to the clinical experts consulted, and input from the patient and clinician groups and public drug plans. Using the same considerations, the CDA-AMC review team selected end points that were considered the most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized key efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE. Supplementary end points have been summarized in this report but not assessed using GRADE.
Pruritus was identified by patients and clinicians as the most important symptom that has the greatest impact on HRQoL in patients with ALGS. Thus, both the observer-reported (primary study end point) and patient-reported pruritus outcomes were selected as key efficacy measures. Two HRQoL measures were selected, including the PedsQL total score (key outcome) and the PedsQL Family Impact Module score (supplementary end point). The clinical experts consulted by CDA-AMC agreed that sBA was a key surrogate end point and that SAEs and diarrhea were the most relevant harms to assess tolerance. Other supplementary end points included growth parameters (height, weight, and body mass index [BMI] z scores), sleep impacts, and pruritus responders, which were selected based on input from patients, clinicians, and expert committee members. A post hoc pruritus responder analysis that was used to inform the pharmacoeconomic model was also summarized as a supplementary end point.
Two other key outcomes, liver transplant and mortality, were identified based on the patient and clinician input; however, there were no data to inform these end points in the study submitted by the sponsor.
Table 6: Outcomes Summarized From the Study Included in the Systematic Review
Outcome measure | Time point | ASSERT study |
|---|---|---|
Key outcomes (assessed using GRADE) | ||
PRUCISION ObsRO scratching severity score (average of a.m. and p.m. scores)a | Change from baseline to month 6 (weeks 21 to 24) | Primary |
PRUCISION PRO itching severity score (average of a.m. and p.m. scores) | Change from baseline to month 6 (weeks 21 to 24) | Secondary |
PedsQL parent report total score | Change from baseline to week 24 | Secondary |
Serum bile acid levels (μmol/L)a | Change from baseline to the average of week 20 and week 24 | Key secondary |
Supplementary outcomes (not assessed using GRADE) | ||
Pruritus responders (≥ 1.0-point and ≥ 1.5-point change) as measured by the PRUCISION ObsRO a.m. and p.m. scores combined | At month 6 (week 21 to 24) | Secondary |
Pruritus responders (≥ 1.0-point change) as measured by the PRUCISION ObsRO a.m. scores | At week 24 (day 162 to 169) | Post hoc analysis used to inform the PE model |
PedsQL Family Impact Module | Change from baseline to week 24 | Secondary |
Growth parameters (height, weight, and BMI z scores) | Change from baseline to week 24 | Exploratory |
Sleep-related end points measured by the PRUCISION ObsRO | Change from baseline to month 6 (weeks 21 to 24) | Secondary |
Important outcomes not assessed in the clinical trial | ||
Liver transplant | NA | NA |
Mortality | NA | NA |
BMI = body mass index; GRADE = Grading of Recommendations Assessment, Development and Evaluation; NA = not applicable; ObsRO = observer-reported outcome; PE = pharmacoeconomic; PedsQL = Pediatric Quality of Life Inventory; PRO = patient-reported outcome.
aStatistical testing for these end points was adjusted for multiple testing.
Sources: ASSERT Clinical Study Report31 and sponsor’s summary of clinical evidence.52
PRUCISION ObsRO and PRO
The PRUCISION ObsRO and PRO instruments focus on the key symptoms of pruritus, sleep disturbance, and associated tiredness. The ObsRO includes 9 items and the PRO includes 7 items, which have a mix of response formats (Table 7). Each item is scored separately, with higher scores indicating a greater amount of scratching or itching, sleep disturbance, and tiredness.
All patients and/or their caregivers were asked to complete the ObsRO or PRO questions in the morning and evening of each day using an e-diary. The morning diary was to be completed shortly after the patient woke up and was used to assess nighttime symptoms, and the evening diary was to be completed just before the patient went to bed to report symptoms that occurred during the day. Patients aged 8 years or older completed the PRO questionnaire. For children aged 8 to 12 years, the caregiver was to read the PRO items along with the child and record the child’s response. Caregivers reported the ObsRO measures for patients of all ages and, ideally, the same caregiver completed the e-diary for a given patient throughout the day. Patients and caregivers were provided training on the e-diary at the screening visits.
The primary end point in the ASSERT study was the change from baseline in the PRUCISION ObsRO scratching score (morning and evening scores combined) to month 6 (the average of weeks 21 to 24). Secondary outcomes were the proportion of pruritus responders at month 6 (week 21 to 24), defined as patients with at least a 1.5-point decrease (primary analysis) or a 1.0-point decrease from baseline in the PRUCISION ObsRO scratching score (a.m. and p.m. scores combined). The sponsor also conducted a post hoc pruritus responder analysis that was used to inform the pharmacoeconomic model. This was the proportion of patients with at least a 1.0-point decrease in the PRUCISION ObsRO scratching score (a.m. only) at week 24 (day 162 to day 169).
The change from baseline to month 6 (week 21 to 24) in the PRUCISION PRO itching score (a.m. and p.m. scores combined) was a secondary outcome, based on the subgroup of patients who were 8 years and older.
Sleep-related end points measured using the PRUCISION ObsRO were secondary end points. These were reported as the change from baseline to month 6 for the number of awakenings (range, 0 to 99), tiredness score (range, 0 to 4), or the proportion of patients who showed sleep-related impacts (i.e., answered yes to binary sleep-related questions on the e-diary) (Table 7).
The PRUCISION instruments were developed by the sponsor for use in the odevixibat clinical trial program. The psychometric properties of the instrument were examined based on patients with ALGS enrolled in the ASSERT study (N = 52), and those with progressive familial intrahepatic cholestasis enrolled in the PEDFIC study (N = 62).23,24 These analyses showed some evidence to support the validity, reliability, and responsiveness of items in the ObsRO instrument; however, data were limited for the PRO questionnaire due to the small number of patients who completed the PRUCISION PRO. The sponsor estimated a 1.0-point to 1.5-point decrease in the ObsRO scratching score as the clinically meaningful threshold for a within-patient change.23,24
Table 7: PRUCISION PRO and ObsRO Diary Questions
Question | Response |
|---|---|
ObsRO morning diary | |
How bad was your child’s worst scratching since he/she went to bed last night?a | 0 = no scratching, 1 = a little scratching, 2 = medium scratching, 3 = a lot of scratching, 4 = worst possible scratching |
Since your child went to bed last night, did you see blood due to scratching? | Yes, no |
Did your child need a caregiver to help him/her fall asleep last night due to his/her itching? | Yes, no |
Did your child need a caregiver to soothe him/her at some time during the night last night due to his/her itching? | Yes, no |
Did your child need a caregiver to sleep with him/her at some time during the night last night due to his/her itching? | Yes, no |
How many times did you notice that your child woke up last night? | Numeric (0 to 99) |
Did your child take any prescribed or over-the-counter medicines before going to bed last night that may have made him/her sleepy? | Yes, no |
ObsRO evening diary | |
How bad was your child’s worst scratching since he/she woke up this morning?a | 0 = no scratching, 1 = a little scratching, 2 = medium scratching, 3 = a lot of scratching, 4 = worst possible scratching |
How tired did your child seem to be today?a | 0 = not tired at all, 1 = a little tired, 2 = medium tired, 3 = very tired, 4 = very, very tired |
PRO morning diary | |
How bad was your worst itching since you went to bed last night?a | 0 = no itching, 1 = a little itching, 2 = medium itching, 3 = a lot of itching, 4 = the worst itching |
How hard was it to fall asleep last night because of your itching?a | 0 = not hard at all, 1 = a little hard, 2 = medium hard, 3 = very hard, 4 = very, very hard |
How hard was it to stay asleep last night because of your itching?a | 0 = not hard at all, 1 = a little hard, 2 = medium hard, 3 = very hard, 4 = very, very hard |
Did you wake up last night because of itching? | Yes, no |
How tired do you feel this morning?a | 0 = not tired at all, 1 = a little tired, 2 = medium tired, 3 = very tired, 4 = very, very tired |
PRO evening diary | |
How bad was your worst itching since you woke up this morning?a | 0 = no itching, 1 = a little itching, 2 = medium itching, 3 = a lot of itching, 4 = the worst itching |
How tired were you since you woke up this morning?a | 0 = not tired at all, 1 = a little tired, 2 = medium tired, 3 = very tired, 4 = very, very tired |
ObsRO = observer-reported outcome; PRO = patient-reported outcome.
aScore based on 0 to 4 pictorial response scales in which each response was distinguished by a unique facial expression, verbal anchor, number, and colour code.
Source: ASSERT Clinical Study Report.31
Pediatric Quality of Life Inventory
Caregivers and patients (if applicable) were to complete the PedsQL (version 4.0), an instrument designed to assess HRQoL in children and adolescents. The PedsQL examines 4 functional domains: physical, emotional, social, and school.22 Different versions of the PedsQL were used, depending on the age of the patient: child and parent report core modules for patients aged 5 to 7 years, 8 to 12 years, and 13 to 18 years, and a parent report core module for toddlers (aged 2 to 4 years).
The caregiver was also asked to complete the PedsQL Family Impact Module (8 domains: physical, emotional, social, cognitive, communication, worry, daily activities, family relationships) designed to measure the impact of pediatric chronic health conditions on parents and the family.
The PedsQL is scored based on the questionnaire used (i.e., the report for toddlers is scored differently than the report for young children). PedsQL is scored on a 0 to 100 scale, with higher scores indicating improved HRQoL. The evidence to support its psychometric properties are summarized in Table 8. No MID was identified for patients with ALGS or other liver diseases, but in children with chronic health conditions (asthma, ADHD, depression, diabetes, or “other”), the estimated MID for the PedsQL total score for the child self-report was 4.4 points, and for the parent-proxy report was 4.5 points for within-group differences.22
Serum Bile Acid
The key secondary outcome was the change in sBA levels from baseline (defined as the average of the last 2 values before the first dose) to the average of week 20 and 24 (defined as the average of all nonmissing values during this period).
Blood samples for analysis of sBA were drawn at all visits (day 1 and weeks 4, 8, 12, 16, 20, and 24). Patients were required to fast (water intake only was permissible) for at least 4 hours before the collection of samples. Exceptions could be made for infants younger than 12 months if they were unable to fast for the full 4 hours. All postbaseline sBA results during the treatment period and at follow-up were blinded. A central laboratory performed the quantitative assessment of the sBA levels, utilizing a validated commercial enzyme cycling assay.
In other pediatric cholestatic diseases (progressive familial intrahepatic cholestasis and biliary atresia), sBA has been shown to have prognostic implications, with patients with higher sBA levels being more likely to progress to end-stage liver disease or to undergo a liver transplant.57,58 It has not yet been defined what sBA level or reduction can be associated with an improved long-term outcome in patients with ALGS.
Growth Parameters
Growth was measured as height (or length, depending on age) based on SOC, and weight was evaluated using a certified weight scale at various time points. BMI was calculated as weight (kg) divided by height (m).2 Change in growth parameters was assessed using linear growth (weight, height, and BMI for age) compared with a standard growth curve (z score, SD from the 50th percentile).41,59 No MID was identified in the literature.
Table 8: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
PRUCISION ObsRO | The ObsRO instrument focuses on the symptoms of pruritus, sleep disturbance, and associated tiredness. The ObsRO instrument asks caregivers about the patient’s scratching and other related behaviours observed during the daytime and nighttime hours. Items on the ObsRO consisted of a 9-item questionnaire with a mix of response formats, including binary (no, yes), rating scales (0 = no scratching, 1 = a little scratching, 2 = medium scratching, 3 = a lot of scratching, 4 = worst possible scratching), and numeric (0 to 99). Higher scores indicated a greater amount of scratching, sleep disturbance, and tiredness. Each item on the ObsRO is scored separately, with no total or domain scores calculated. | The PRUCISION ObsRO has shown evidence of validity, reliability, and responsiveness when assessed in caregivers of pediatric patients with ALGS from the ASSERT study (N = 52) and PFIC from the PEDFIC study (N = 62).23,24 Validity: Construct validity was assessed through analyses demonstrating cross-sectional relationships between the ObsRO, PRO, PedsQL, and GIS criterion measures. Moderate correlations (r ≥ 0.5) were observed between the ObsRO scratching scores and the CaGIS and CGIS scratching scores, supporting the validity of the scratching scores. Similar findings were observed in the sleep disturbance domain, with correlations ≥ 0.4 between the ObsRO sleep disturbance domain and CaGIS and CGIS sleep. Several scales on the PedsQL self-reported measure and Family Impact Module also correlated (r ≥ 0.3) with scratching scores. In contrast, mostly low correlations were observed between the sleep disturbance domain and PedsQL scores, except in the self-report scores and Family Impact Module, where correlations were ≥ 0.3. Reliability: Test–retest reliability was assessed with the ICC. Comparisons of first and second week ObsRO scores all showed reliability between acceptable (ICC > 0.60) and excellent (ICC > 0.90) for sleep disturbance items and scratching items. Responsiveness: Sensitivity to change was assessed through analyses demonstrating longitudinal relationships between the ObsRO and PRO and other measures, as well as correlations between changes in ObsRO and PRO scores and changes in PedsQL and GIS scores, and between changes in PRO and ObsRO scores and GIC scores. Mean differences were significant, as was the expected direction for all scales at week 24 when groups were identified with the CaGIC, except for taking medication, and the CGIC, except for taking medication and awakenings. An appropriate indication of improvement was observed with the GIC. Moderate correlations were observed between scratching and sleep disturbance domain scores and CaGIS and CGIS at week 24. PedsQL self-reported and parent-reported (total score, physical health, emotional functioning, and school functioning) score correlations and several PedsQL Family Impact Module score correlations (r ≥ 0.3) were observed between the scratching and sleep disturbance domain scores and at week 24. | Using anchor-based and distribution-based and ROC analyses on data from pediatric patients with ALGS, the clinically meaningful threshold for within-patient change in the ObsRO scratching severity score in ALGS was estimated to be a decrease of between 1.0 and 1.5 points.24 For the ObsRO sleep disturbance domain, a decrease in the sleep disturbance score of 26 to 42 points reflects a substantial numeric decrease in the average percent of nights where some type of support was provided by the caregiver. No MID was estimated for the ObsRO tiredness score.24 |
PRUCISION PRO | The PRO instrument focuses on the symptoms of pruritus, sleep disturbance, and associated tiredness. The PRUCISION PRO instrument (for patients aged ≥ 8 years) asked patients about their itching during the day and nighttime hours. Items on the PRO consisted of 7-item questionnaires with a mix of response formats including binary (i.e., no, yes) and rating scales (0 = no itching, 1 = a little itching, 2 = medium itching, 3 = a lot of itching, 4 = the worst itching). Higher scores indicated a greater amount of itching, sleep disturbance, and tiredness. Each item on the PRO is scored separately, with no total or domain scores calculated. | The PRUCISION PRO has shown some evidence of validity, reliability, and responsiveness when assessed in pediatric patients with ALGS from the ASSERT study (N = 17) and PFIC from the PEDFIC study (N = 10), though data were limited due to the small number of patients aged 8 years or older who could be included in the analysis.23,24 Validity: Construct validity was assessed through analyses demonstrating cross-sectional relationships between the ObsRO, PRO, PedsQL, and GIS criterion measures. Low to moderate positive correlations (r = 0.220 to 0.546) were observed between PRO itching items and the PGIS and the CGIS items, and low correlation with the CaGIS (r = 0.087 to 0.277). Also, low to high correlations were observed between sleep disturbance and tiredness items and the PGIS (r = 0.352 to 0.711) and CaGIS sleep items (r = 0.199 to 0.498). Reliability: Test–retest reliability was assessed with the ICC. The PRO itching, sleep disturbance, and tiredness items showed good test–retest reliability (ICC > 0.6) based on the ICC calculated between the first week and second week scores. Responsiveness: Sensitivity to change was assessed through analyses demonstrating longitudinal relationships between the ObsRO and PRO measures and other COA measures, as well as correlations between changes in ObsRO and PRO scores and changes in PedsQL and GIS scores, and between changes in PRO and ObsRO scores and GIC scores. Mean differences between groups for all itching measures, sleep disturbance, and tiredness were in the expected direction, with greater (though not significant) reductions observed for the “improved” group. Moderate correlations at week 24 were observed in itching, sleep disturbance, fall asleep and stay asleep, and tiredness items, which demonstrate sensitivity to detect change over time. Low to moderate correlations were observed between PRO scores and several PedsQL scales. | No MID was identified. |
PedsQL (total score and Family Impact Module) | The PedsQL is a 23-item tool that assesses HRQoL in children and adolescents, integrating both generic core scales and disease-specific modules into 1 measurement. The tool can be completed by the patient (self-report) or by caregivers (proxy report). The 4 functional domains include: physical (8 items), emotional (5 items), social (5 items), and school (5 items).22 Different versions of the PedsQL are used, depending on the age of the patient, and include the child and parent report core modules for patients aged 5 to 7 years, 8 to 12 years, and 13 to 18 years, and a parent report core module for toddlers (aged 2 to 4 years). The PedsQL is scored on a 0 to 100 scale, with higher scores indicating improved quality of life. The total score is calculated as the mean of all items. The PedsQL Family Impact Module is a parent-reported instrument that measures the impact of chronic medical conditions on parent or caregiver HRQoL. It has 36 items, with 6 subscales measuring parent-reported self-functioning (physical functioning, emotional functioning, social functioning, cognitive functioning, communication, and worry), and 2 subscales measuring parent-reported family functioning (daily activities and family relationships). Each item is scored using a 5-point Likert scale, ranging from “never” to “almost always.” The PedsQL module is scored on a 0 to 100 scale. The total and summary scores are calculated by summing all items and dividing by the number of questions answered. Higher scores indicate better functioning. | Validity: In a study of pediatric patients in Iran with chronic liver disease, the PedsQL demonstrated significant decreases in HRQoL in children and adolescents with chronic liver disease compared with healthy controls. There was also strong correlation between the child self-reported and parent-reported scores on all 4 scales (r = 0.528 to 0.740).27 In a study of 23 families of pediatric patients with chronic health conditions, a known-groups analysis of the PedsQL Family Impact Module demonstrated significantly decreased function in families with medically fragile children living at home compared with those living at a residential facility.60 Reliability: In a study of pediatric patients (N = 5,991) with acute or chronic health conditions and their caregivers (N = 7,021), the PedsQL total score demonstrated high internal consistency (alpha = 0.89) for the child self-report and the caregiver proxy report scores (alpha = 0.92).22 In a study of 23 families of pediatric patients with chronic health conditions, the PedsQL Family Impact Module showed high internal consistency, with most scales approaching or exceeding an alpha coefficient of 0.90, and all exceeding 0.70.60 Responsiveness: In a study of the caregivers of 4,500 hospitalized pediatric patients aged from 1 month to 18 years and 350 patients aged from 13 to 18 years, the PedsQL was found to be sensitive to clinical changes, with significant improvements in scores from admission to follow-up.61 | No MIDs were identified in pediatric patients with liver diseases. However, a PedsQL total score within-patient MID of 4.4 for the child self-report and 4.5 for the parent-proxy report has been estimated in children with chronic health conditions (asthma, ADHD, depression, diabetes, or “other”).22 |
ADHD = attention-deficit/hyperactivity disorder; ALGS = Alagille syndrome; CaGIC = Caregiver Global Impression of Change; CaGIS = Caregiver Global Impression of Symptoms; CGIC = Clinician Global Impression of Change; CGIS = Clinician Global Impression of Symptoms; COA = clinical outcome assessment; GIC = Global Impression of Change; GIS = Global Impression of Symptoms; HRQoL = health-related quality of life; ICC = intraclass correlation coefficient; MID = minimal important difference; ObsRO = observer-reported outcome; PedsQL = Pediatric Quality of Life Inventory; PFIC = progressive familial intrahepatic cholestasis; PGIS = Patient Global Impression of Symptoms; PRO = patient-reported outcome; ROC = receiver operating characteristic.
Statistical Analysis
Statistical Test or Model
The primary efficacy end point in the ASSERT study was the change in scratching severity score from baseline to month 6 (weeks 21 to 24) based on the combined average scratching score for the PRUCISION ObsRO from the morning and evening assessments. The primary efficacy analysis was based on a mixed model for repeated measures (MMRM) modelling change from baseline for each 4-week average morning and evening scratching score. The model included the baseline age stratification, average morning and evening scratching scores, and direct bilirubin; treatment group; time (in months as a categorical variable); and a treatment-by-time interaction. The analysis of the key secondary end point (change in sBA levels) used methods similar to those used for the primary end point. Table 9 describes the analysis methods for other secondary and exploratory end points and the sensitivity and supplemental analyses conducted.
For all efficacy end point analysis, the estimand strategy included all data collected through the end of study following the intercurrent event of premature discontinuation of treatment before week 24; data following intercurrent events of biliary diversion surgery or liver transplant were excluded. The Clinical Study Report states that because no intercurrent events occurred during the study, no data were excluded from the analysis of efficacy.
Multiple Testing Procedure
Statistical testing for the primary end point was performed using a 1-sided alpha of 0.025. A hierarchical testing strategy was used, and the key secondary efficacy end point was assessed only if the primary end point met its success criterion. Other secondary end points were assessed descriptively and provided supportive efficacy data without alpha adjustments for multiple comparisons.
Sample Size and Power Calculation
The study initially planned that 45 patients younger than 18 years of age would be randomized at an experimental-to-control allocation of 2 to 1 to obtain approximately 36 completers, assuming an approximate dropout rate of 20%. At a 1-sided significance alpha level of 0.025, assuming a pooled SD of 1.0 and a difference between the treatment groups of 1.2 in mean change in pruritus score favouring the experimental arm, the power of the study would be 0.909, using the exact method. The key secondary end point was also powered for a standardized treatment effect (mean change) of 1.2.
The study protocol included a noncomparative interim analysis of blinded data to determine whether the pooled SD was consistent with that used in the initial power calculations and to assess whether a larger sample size was needed to achieve adequate study power. The planned sample size re-estimation was conducted based on 17 patients having nonmissing outcomes at weeks 13 to 16, and 12 patients having nonmissing outcomes at weeks 21 to 24. Based on blinded pooled data, the observed SD at week 16 and week 24 was 1.11. Accordingly, the sample size was increased to target 48 completers (i.e., approximately 32 and 16 in the odevixibat and placebo groups, respectively, based on 2:1 randomization). Given the low actual dropout rate at that time, 7 patients were added to the study. A total of 52 patients were enrolled.
Subgroup Analyses
Efficacy analyses for the primary and key secondary efficacy end points were performed for the following patient subgroups: age category, region, sex, race, ethnicity, baseline sBA level (< median, ≥ median), baseline direct bilirubin (≤ 3 mg/dL, > 3 mg/dL), genetic mutation (JAG1, NOTCH2), use of UDCA and use of antipruritic medications (yes, no), Child-Pugh classification, hepatic impairment classification (no impairment; mild, moderate, or severe impairment).
The analyses used the MMRM model with baseline average a.m. and p.m. scratching score (for the primary end point) and baseline sBA level (for secondary efficacy end point), treatment group, time, and treatment-by-time interaction. A statistical analysis was performed only when the sample size was at least 5 patients in each treatment group. If the sample size was less than 5 in either treatment group, only summary statistics were provided and no P value was reported.
Table 9: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity and supplementary analyses |
|---|---|---|---|---|
PRUCISION ObsRO worse scratching score (average of a.m. and p.m. scores) change from baseline to month 6 (week 21 to 24)a | MMRM 1-sided alpha of 0.025 | Baseline age stratification, baseline average a.m. and p.m. scratching score, baseline direct bilirubin, treatment group, time in months (as a categorical variable: 0 to 4 weeks, 5 to 8 weeks, 9 to 12 weeks, 13 to 16 weeks, 17 to 20 weeks, and 21 to 24 weeks), and treatment-by-time interaction | No imputation of missing values | Sensitivity analyses: 1. Control-based multiple imputation method for missing average a.m. and p.m. monthly scratching scores 2. Tipping point analysis 3. MMRM based on worst weekly scratching score for baseline (14-day period) and each month (28-day period) 4. Baseline score redefined as the average of the 4 baseline weekly average a.m. and p.m. scores in the 28 days preceding the start of treatment for whom it was available 5. MMRM analysis of monthly scratching score at month 6 (weeks 21 to 24) (i.e., not change from baseline) Supplementary analysis: 6. Per-protocol population |
PRUCISION PRO worst itching score (average of a.m. and p.m. scores) change from baseline to month 6 (week 21 to 24)a | MMRM | Baseline age stratification, baseline average a.m. and p.m. scratching score, baseline direct bilirubin, treatment group, time in months (as a categorical variable: 0 to 4 weeks, 5 to 8 weeks, 9 to 12 weeks, 13 to 16 weeks, 17 to 20 weeks, and 21 to 24 weeks), and a treatment-by-time interaction | Not reported | Not reported |
Change in sBA levels (μmol/L) from baseline to the average of week 20 and week 24b | MMRM | Baseline age stratification, baseline sBA level, treatment group, visits (weeks 4, 8, 12, 16, 20, and 24), and treatment-by-visit interaction | Not reported | Sensitivity analyses: 1. Control-based multiple imputation method 2. Tipping point analysis Supplementary analyses: 3. Rank ANCOVA model with baseline age stratification, baseline bile acid level, and treatment group 4. MMRM with per-protocol population |
Change from baseline to week 24 in PedsQL parent report total score and Family Impact Module | ANCOVA | Baseline score, age category based on the age groups defined for the PedsQL (5 to < 8, ≥ 8 to < 13, ≥ 13 to < 18), and treatment | Not reported | Not reported |
Proportion of patients with a 1.5-point decrease in scratching score at month 6 | Cocran-Mantel-Haenszel test | Baseline age stratification | Patients with missing data imputed as nonresponders |
|
Proportion of patients with 1.0-point decrease in scratching score at week 24 (post hoc a.m. score only) | Exact CI and difference in proportions | None | Observed data (patients with missing data excluded) | Not reported |
Change from baseline in height, weight, and BMI z scores | MMRM | Baseline value, baseline age stratification, treatment group, visit (in weeks), and treatment-by-visit interaction | Not reported | Not reported |
PRUCISION ObsRO sleep-related end pointsa | MMRM | Baseline score, baseline age stratification, treatment group, time (in months), and treatment-by-time interaction | Not reported | Not reported |
ANCOVA = analysis of covariance; BMI = body mass index; CI = confidence interval; MMRM = mixed model for repeated measures; ObsRO = observer-reported outcome; PedsQL = Pediatric Quality of Life Inventory; PRO = patient-reported outcome; sBA = serum bile acid.
aThe baseline ObsRO value was calculated by averaging the 2 baseline weekly scores in the 14 days before the start of treatment.
bThe baseline sBA level was defined as the average of the last 2 values before the first dose of the study drug. If only 1 baseline value was available, that value was used.
Source: ASSERT Clinical Study Report.31
Analysis Populations
In the ASSERT trial, the full analysis set (FAS) was used for all efficacy analyses. The FAS included all randomized patients who received at least 1 dose of the study drug. Patients were analyzed according to the treatment group assigned at randomization. The safety analysis set, which was used for all safety analyses, included all randomized patients who received at least 1 dose of the study drug. Patients were analyzed according to the treatment received at first dose.
Supplementary efficacy analyses were conducted using the per-protocol population, which included patients in the FAS who did not have any important protocol deviations, met treatment adherence criteria of at least 70% of doses, and had a treatment duration of at least 50% of the days with e-diary entries during weeks 21 to 24.
Results
Patient Disposition
A total of 52 patients were screened and randomized in the ASSERT trial, including 35 patients in the odevixibat group and 17 patients in the placebo group. All patients completed the study and were included in the FAS and safety populations.
Table 10: Summary of Patient Disposition — ASSERT Study
Patient disposition | Odevixibat (N = 35) | Placebo (N = 17) |
|---|---|---|
Screened, N | 35 | 17 |
Randomized, N (%) | 35 (100%) | 17 (100%) |
Discontinued from study, n (%) | 0 | 0 |
FAS, N | 35 (100%) | 17 (100%) |
PPS, N | 31 (88.6%) | 16 (94.1%) |
Safety, N | 35 (100%) | 17 (100%) |
FAS = full analysis set; PPS = per-protocol set.
Source: ASSERT Clinical Study Report.31
Baseline Characteristics
The baseline characteristics outlined in Table 11 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results. There were some notable differences in the baseline characteristics between the treatment groups.
The mean age of patients enrolled was ███ years (SD = ███; range, ███ to ████ years) in the odevixibat group and ███ years (SD = ███; range, ███ to ████) in the placebo group. In the odevixibat and placebo groups, respectively, ████ and █████ of patients were younger than 2 years, █████ and █████ of patients were aged 2 years to younger than 12 years, and █████ and ████ of patients were aged 12 years to younger than 18 years. No adult patients were enrolled. The proportion of females was 40% and 64.7% and the proportion of males was 60% and 35.3% in the odevixibat and placebo groups, respectively. Patients in the odevixibat group had ███████ growth deficits at baseline (median z score █████ for height and █████ for weight) than patients in the placebo group (median z score █████ for height and █████ for weight).
On average, the patients in the odevixibat group had been diagnosed with ALGS for ███ years (SD = ███) compared with ███ years (SD = ███) in the placebo group. Most patients in both groups had the JAG1 gene mutation (91.4% and 94.1% in the odevixibat and placebo groups, respectively), while the remainder had the NOTCH2 gene mutation. According to the National Cancer Institute Organ Dysfunction Working Group classification system, █████ and █████ of patients had mild hepatic impairment, █████ and █████ had moderate impairment, and █████ and █████ of patients had severe impairment at baseline in the odevixibat and placebo groups, respectively. The mean ObsRO scratching score was 2.80 points (SD = 0.52) and 3.01 points (SD = 0.64), and the mean sBA level was 237.4 µmol/L (SD = 114.9 µmol/L) and 246.1 µmol/L (SD = 120.5 µmol/L) in the odevixibat and placebo groups, respectively. Most patients were receiving UDCA, including 30 patients (85.7%) in the odevixibat group and 16 patients (94.1%) in the placebo group. All patients but 1 in the odevixibat group were on antipruritic medications at baseline.
A summary of patients’ medical and surgical history is provided in Table 12.
Table 11: Summary of Baseline Characteristics — ASSERT Study (FAS)
Characteristic | Odevixibat (N = 35) | Placebo (N = 17) |
|---|---|---|
Age (years) | ||
Mean (SD) | ████ ███████ | ████ ███████ |
Median (range) | 6.10 (1.7 to 15.5) | 4.20 (0.5 to 14.3) |
Age category, n (%) | ||
< 2 years | | █████ | | ██████ |
≥ 2 to < 12 years | ██ ██████ | ██ ██████ |
≥ 12 to < 18 years | | ██████ | | █████ |
Sex, n (%) | ||
Female | 14 (40.0) | 11 (64.7) |
Male | 21 (60.0) | 6 (35.3) |
Race, n (%) | ||
Asian | 2 (5.7) | 1 (5.9) |
Black or African American | 2 (5.7) | 2 (11.8) |
White | 30 (85.7) | 13 (76.5) |
Other | 1 (2.9) | 1 (5.9) |
Height (cm) | ||
Mean (SD) | ██████ ████████ | ██████ ████████ |
Median (range) | ████ ██████ ██████ | ████ ██████ ██████ |
Height z scores | ||
Mean (SD) | █████ ███████ | █████ ███████ |
Median (range) | █████ ██████ ████ | █████ ██████ ████ |
Weight (kg) | ||
Mean (SD) | █████ ███████ | █████ ████████ |
Median | ████ █████ █████ | ████ █████ █████ |
Weight z scores | ||
Mean (SD) | █████ ███████ | █████ ███████ |
Median (range) | █████ ██████ ████ | █████ ██████ ████ |
BMI (kg/m2) | ||
Mean (SD) | █████ ███████ | █████ ███████ |
Median (range) | █████ ██████ █████ | █████ ██████ █████ |
BMI z scores | ||
Mean (SD) | █████ ███████ | █████ ███████ |
Median (range) | █████ ██████ ████ | █████ ██████ ████ |
Genetic mutation detected, n (%) | ||
JAG1 gene | 32 (91.4) | 16 (94.1) |
NOTCH2 gene | 3 (8.6) | 1 (5.9) |
Time since diagnosis (years) | ||
Mean (SD) | ████ ███████ | ████ ███████ |
Median (range) | ████ █████ █████ | ████ █████ █████ |
Scratching score (morning and evening combined), PRUCISION ObsRO | ||
Mean (SD) | 2.80 (0.520) | 3.01 (0.636) |
Median (range) | ████ █████ ████ | ████ █████ ████ |
sBA level (µmol/L) | ||
Mean (SD) | 237.4 (114.88) | 246.1 (120.53) |
Median (range) | █████ ████ ████ | █████ ████ ████ |
Prior medications, n (%) | ||
Antipruritic medications | 34 (97.1) | 17 (100.0) |
UDCA | 30 (85.7) | 16 (94.1) |
Rifampin | ██ ██████ | ██ ██████ |
Hepatic impairment classification, n (%)b | ||
Mild | | ██████ | | ██████ |
Moderate | ██ ██████ | | ██████ |
Severe | ██ ██████ | | ██████ |
Laboratory values | ||
ALT (U/L), mean (SD) | 185.6 (83.20) | 149.1 (84.15) |
AST (U/L), mean (SD) | 170.0 (80.51) | 160.8 (90.80) |
Total bilirubin (µmol/L), mean (SD) | 51.99 (43.38) | 61.62 (57.02) |
Direct bilirubin (µmol/L), mean (SD) | █████ ███████ | █████ ███████ |
GGT (U/L), mean (SD) | 366.3 (211.31) | 535.5 (345.34) |
eGFRc (mL/min/1.73 m2), mean (SD) | ██████ ███████ | ██████ ███████ |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; BMI = body mass index; eGFR = estimated glomerular filtration rate; FAS = full analysis set; GGT = gamma-glutamyl transferase; ObsRO = observer-reported outcome; sBA = serum bile acid; SD = standard deviation; UDCA = ursodeoxycholic acid.
aBased on the median baseline sBA levels across all 52 patients.
bBased on National Cancer Institute Organ Dysfunction Working Group classification system.
cCalculated by the modified Schwartz equation. Results were based on 30 patients and 12 patients in the odevixibat and placebo groups respectively.
Source: ASSERT Clinical Study Report.31
Table 12: Summary of Medical and Surgical History — ASSERT Study (FAS)
Characteristic | Odevixibat (N = 35) | Placebo (N = 17) |
|---|---|---|
Patients with any medical or surgical history, n (%)a | ██ █████ | ██ █████ |
Pruritus | ██ ██████ | ██ ██████ |
Pulmonary artery stenosis | ██ ██████ | | ██████ |
Pulmonary valve stenosis | | █████ | | ██████ |
Cardiac murmur | | ██████ | | ██████ |
Atrial septal defect | | █████ | | ██████ |
Aortic stenosis | | █████ | | █████ |
Ventricular septal defect | | █████ | | ██████ |
Cholestasis | ██ ██████ | | ██████ |
Jaundice | | █████ | | ██████ |
Portal hypertension | | █████ | | ██████ |
Gastrostomy | | █████ | | ██████ |
Splenomegaly | ██ | | ██████ |
Abdominal pain | | █████ | | █████ |
Cholangiogram | | █████ | | █████ |
Congenital musculoskeletal disorder of spine | | █████ | ██ |
Failure to thrive | | ██████ | | ██████ |
Anemia | | █████ | | ██████ |
Astigmatism | | █████ | ██ |
Vitamin D deficiency | ██ ██████ | | ██████ |
Vitamin E deficiency | | ██████ | | █████ |
Vitamin A deficiency | | █████ | | █████ |
FAS = full analysis set.
aEvents reported in at least 5% of patients overall.
Source: ASSERT Clinical Study Report.31
Exposure to Study Treatments
The mean treatment duration was ████ weeks (SD = ████) and ████ weeks (SD = ████) in the odevixibat and placebo groups, respectively (Table 13). All 52 patients took at least 1 concomitant medication, which included UDCA (85.7% and 94.1%), rifampin (65.7% and 70.6%), and hydroxyzine (17.1% and 5.9% of patients in the odevixibat and placebo groups, respectively). The use of vitamin supplements was also common in both treatment groups (Table 14).
Table 13: Summary of Patient Exposure — ASSERT Study (Safety Population)
Exposure | Odevixibat (N = 35) | Placebo (N = 17) |
|---|---|---|
Duration of exposure (weeks) | ||
Mean (SD) | ████ ██████ | ████ ██████ |
Median (range) | ████ ███ ██ ████ | ████ ██████ ████ |
Adherence, % | ||
Mean (SD) | ████ ██████ | █████ ██████ |
Median (range) | ███ ████ ██ █████ | ████ ███ ██ ████ |
SD = standard deviation.
Source: ASSERT Clinical Study Report.31
Table 14: Summary of Concomitant Medication — ASSERT Study (Safety Population)
Medication | Odevixibat (N = 35) | Placebo (N = 17) |
|---|---|---|
Patients with any concomitant medication, n (%) | ||
UDCA | 30 (85.7) | 16 (94.1) |
Rifampin | 23 (65.7) | 12 (70.6) |
Bile acid sequestrants (cholestyramine) | 1 (2.9) | 0 |
Naltrexone | 2 (5.7) | 1 (5.9) |
Antihistamines | ||
Hydroxyzine | 6 (17.1) | 1 (5.9) |
Diphenhydramine hydrochloride | 1 (2.9) | 1 (5.9) |
Cetirizine | 1 (2.9) | 1 (5.9) |
Cetirizine hydrochloride | 1 (2.9) | 0 |
Oxatomide | 1 (2.9) | 0 |
Loratadine | 1 (2.9) | 0 |
Vitamin preparations | ||
Vitamin D | 29 (82.9) | 12 (70.6) |
Vitamin A and D in combination | 2 (5.7) | 1 (5.9) |
Vitamin A, plain | 6 (17.1) | 4 (23.5) |
Vitamin K | 20 (57.1) | 12 (70.6) |
Other plain vitamin preparations | 18 (51.4) | 5 (29.4) |
Multivitamins with minerals | 2 (5.7) | 4 (23.5) |
Multivitamins, plain | 4 (11.4) | 1 (5.9) |
Multivitamins, other combinations | 1 (2.9) | 2 (11.8) |
Combination of vitamins | 2 (5.7) | 3 (17.6) |
Vitamins, other combinations | 1 (2.9) | 2 (11.8) |
UDCA = ursodeoxycholic acid.
Source: ASSERT Clinical Study Report.31
Efficacy
Observer-Reported Scratching Severity
The primary outcome was the change from baseline to month 6 (week 21 to week 24) in the scratching severity score (morning and evening scores combined) measured using the PRUCISION ObsRO. The results reported included 34 of 35 patients (97%) from the odevixibat group and 16 of 17 patients (94%) in the placebo group (Table 15).
From the baseline mean scores of 2.80 (SD = 0.52) and 3.01 (SD = 0.64) in the odevixibat and placebo groups, respectively, the LS mean change from baseline in the 6-month scratching severity score was −1.69 (standard error [SE] = 0.17) in the odevixibat group compared with −0.80 (SE = 0.23) in the placebo group. Based on the MMRM, the LS mean difference between groups was −0.88 (95% CI, −1.44 to −0.33; 1-sided P = 0.0012). Figure 2 shows the LS mean (95% CI) changes from baseline in scratching severity score over time by 4-week intervals based on the MMRM. The sensitivity analyses (multiple imputation, worst case scenario analysis, 4-week baseline score, and absolute difference at month 6) and the supplementary analysis (per-protocol population) conducted showed results that were consistent with the primary analysis. The change from baseline to month 6 in the ObsRO a.m. scratching score showed an LS mean difference of −0.90 (95% CI, −1.46 to −0.33) and the p.m. score reported an LS mean difference of −0.86 points (95% CI, −1.42 to −0.30) for the odevixibat versus placebo group, respectively. Due to the small number of patients in some subgroups, not all of the planned subgroup analyses could be conducted. Based on the subgroup data available, the treatment effects were generally consistent with the results in the overall study population. ████████ ███ ████████ ██ ████████ ████ ██████ ███████ ██████████ ██ █ ████ ███ ██████ █████████ ███████ ████ ████ ██████ ██ ████████ ██ █ ███ ██████ █████ █████████ ████ ███ ████ ███ ███████████ ███ ████ ███ ████████ ███ █████
Pruritus responder analyses were also conducted for the proportion of patients with at least a 1.0-point or 1.5-point decrease in the PRUCISION ObsRO scratching score (a.m. and p.m. scores combined) at 24 weeks (Appendix 1, Table 24). The proportion of patients with at least a 1.5-point decrease at 24 weeks was 54.3% versus 17.6%, with a between-group difference of 36.6% (95% CI, 5.1% to 58.7%) for the odevixibat versus placebo groups, respectively. Twenty-eight patients (80.0%) in the odevixibat group and 6 patients (35.3%) in the placebo group had at least a 1.0-point decrease in the scratching scores, with a between-group difference of 44.7% (95% CI, 10.3% to 68.6%) favouring odevixibat.
███ ███████ █████████ █ ████ ███ █████████ ████████ █████ ██ ██████████ ██ ████████ ████ ██ █████ █ █████████ ████████ ██ ███ ███████ █████████ █████ ██████████ █████ ██ ██ █████ ████████ ██ ██████ ████ ███ ███ ██ ███ █████ ████ ████ ███ ████ ██ ██████ ███ ████████████████ █████████ ███ ████ █████████ ██ ██ ██ ████████ ███████ ██ ███ ██████████ █████ ██████ █ ██ ██ ████████ ███████ ██ ███ ███████ █████ ███ ███ ████████ ████████ ███ █ ███████ █████ ██████████ ██ █████ ████ ███ ████ ██ ███████ ████ ████████ ███ █████ ██ ████████ ████ ███ ██████████ ███ ███████ █████ ███ ████████ █ ████ ███████ ██ ███ ██████████ █████ ███ █ █████ ████████ ██ ███ ███████ █████ ███ ███ ███████ ████ ██ ████ ███
Figure 2: Estimated Mean CFB in Monthly PRUCISION ObsRO Scratching Severity Score — ASSERT Study (FAS)
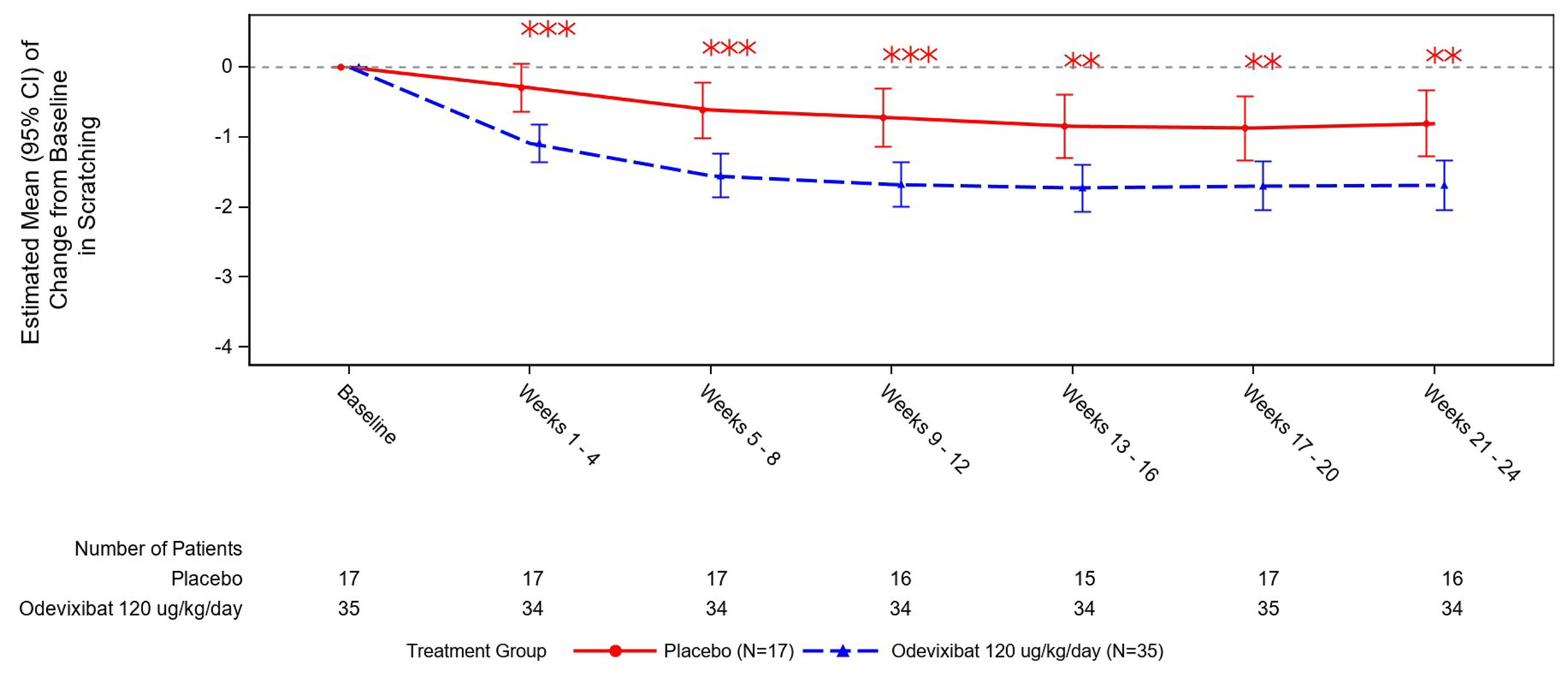
CFB = change from baseline; CI = confidence interval; FAS = full analysis set; MMRM = mixed model for repeated measures; ObsRO = observer-reported outcome.
Note: Results were based on the combined morning and evening ObsRO scratching score, analyzed using an MMRM with baseline score as a covariate, and baseline age stratification, baseline direct bilirubin, treatment group, time (in months), and treatment-by-time interaction as fixed effects.
Double asterisks in the figure correspond to P > 0.0005 and ≤ 0.005. Triple asterisks in the figure correspond to P ≤ 0.0005, 1-sided, indicating superiority of odevixibat over placebo.
Source: ASSERT Clinical Study Report.31
Patient-Reported Itching Severity
Patient-reported itching severity was assessed in patients aged 8 years or older who were able to complete the PRUCISION PRO instrument. The change from baseline to month 6 (21 to 24 weeks) in the itching severity score (morning and evening scores combined) was a secondary end point in the ASSERT trial. Of the 17 patients who were aged 8 years or older, 16 patients completed the PRUCISION PRO instrument, and results for changes from baseline to month 6 in the itching severity score were available for 10 and 5 patients in the odevixibat and placebo groups, respectively (data from 1 patient [3%] were missing in the odevixibat group).
The mean itching severity score at baseline was 2.66 points (SD = 0.55) in the odevixibat group and 2.14 points (SD = 0.31) in the placebo group, and the LS mean change from baseline of −1.40 points (SE = 0.21) in the odevixibat group compared with −1.58 points (SE = 0.31) in the placebo group. The LS mean difference in the change from baseline to month 6 in the itching severity score was 0.18 points (95% CI, −0.65 to 1.00; 1-sided P = 0.678) for the odevixibat group versus the placebo group (Table 15). The mean (and SE) changes during the time on treatment in the average monthly itching severity scores are provided in Figure 3.
Figure 3: Mean CFB in Monthly PRUCISION PRO Itching Severity Score — ASSERT Study (FAS)
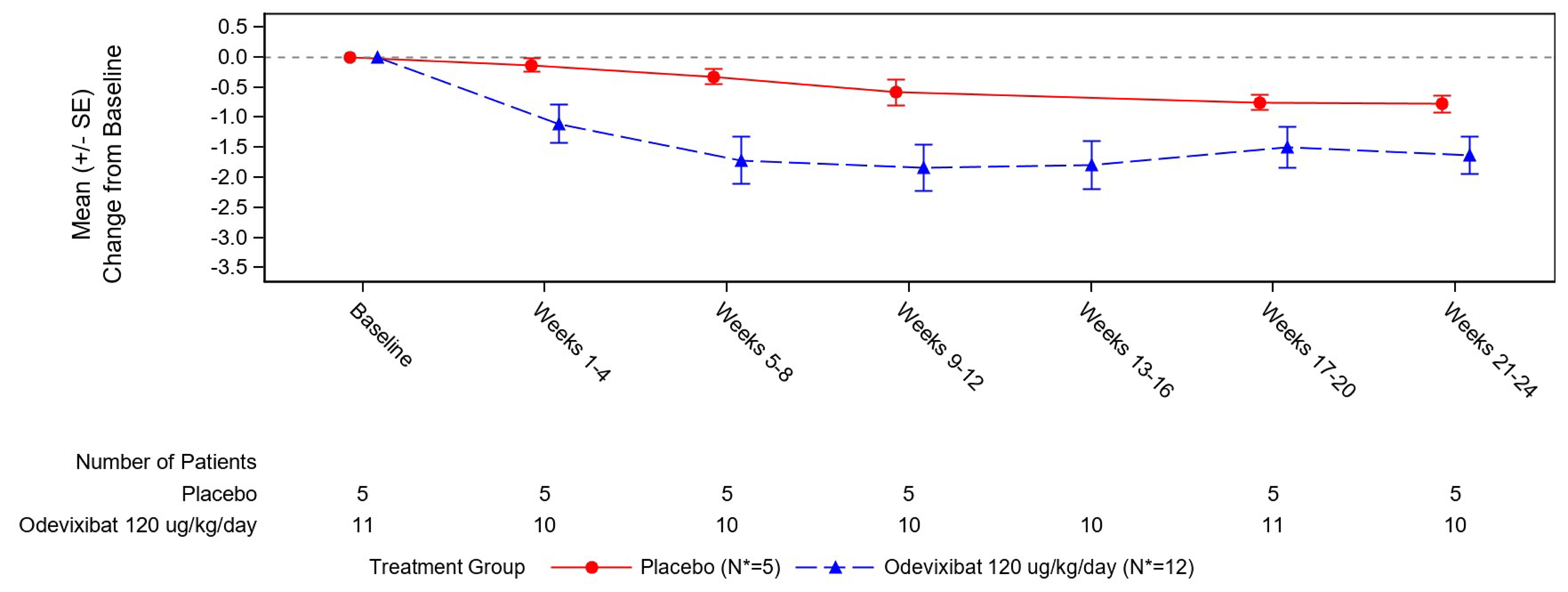
CFB = change from baseline; FAS = full analysis set; PRO = patient-reported outcome; SE = standard error.
Source: ASSERT Clinical Study Report.31
Table 15: Summary of Pruritus Severity Results — ASSERT Study (FAS)
Variable | Odevixibat (N = 35) | Placebo (N = 17) |
|---|---|---|
CFB to month 6 (21 to 24 weeks) in the scratching severity score (a.m. and p.m. scores combined; measured by PRUCISION ObsRO) | ||
Number of patients contributing to the analysis, n (%) | 34 | 16 |
Baseline, mean (SD) | 2.80 (0.52) | 3.01 (0.64) |
Average of week 21 to 24, mean (SD) | 1.14 (0.91) | 2.18 (0.98) |
Change from baseline, mean (SD) | −1.66 (0.97) | −0.76 (0.82) |
LS mean change from baseline (SE)a | −1.69 (0.17) | −0.80 (0.23) |
LS mean difference (95% CI) (odevixibat vs. placebo)a | −0.88 (−1.44 to −0.33) | |
One-sided P valuea | 0.0012 | |
CFB to month 6 (21 to 24 weeks) in the itching severity score (a.m. and p.m. scores combined; measured by PRUCISION PRO) | ||
Number of patients contributing to the analysis | 10 | 5 |
Baseline, mean (SD) | 2.66 (0.55) | 2.14 (0.31) |
Average of weeks 21 to 24, mean (SD) | 1.11 (0.77) | 1.36 (0.59) |
Change from baseline, mean (SD) | −1.63 (0.98) | −0.78 (0.32) |
LS mean change from baseline (SE)a | −1.40 (0.21) | −1.58 (0.31) |
LS mean difference (95% CI) (odevixibat vs. placebo)a | 0.18 (−0.65 to 1.00) | |
One-sided P valuea | 0.678 | |
CFB = change from baseline; CI = confidence interval; FAS = full analysis set; LS = least squares; MID = minimal important difference; MMRM = mixed model for repeated measures; ObsRO = observer-reported outcome; PRO = patient-reported outcome; SD = standard deviation; SE = standard error; vs. = versus.
Note: The pruritus severity score ranges from 0 (none) to 4 (worst possible itching or scratching). No estimate of the between-group MID was identified; however, the sponsor estimated a 1.0-point to 1.5-point within-patient change as clinically relevant, based on the ObsRO scratching score.
aThe analysis is based on an MMRM using a restricted maximum likelihood with baseline score as a covariate, and baseline age stratification, baseline direct bilirubin, treatment group, time (in months), and treatment-by-time interaction as fixed effects.
Source: ASSERT Clinical Study Report.31
HRQoL Outcomes
The analysis of caregiver-reported data for the PedsQL total score (patients aged 2 years and older) and the Family Impact Module (all patients) is summarized in Table 16. For the PedsQL total score, data were missing for 7 patients (20%) and 6 patients (35%) in the odevixibat and placebo groups, respectively. Data were missing for | █████ patients and | ████ patient in the odevixibat and placebo groups, respectively, for the Family Impact Module.
The change from baseline to week 24 in the PedsQL total score estimated an LS mean difference of 2.78 points (95% CI, −6.31 to 11.87) for odevixibat versus placebo. For the Family Impact Module, the LS mean difference in the change from baseline was ████ points (95% CI, █████ to █████) for odevixibat versus placebo.
Table 16: Summary of PedsQL Results — ASSERT Study (FAS)
Variable | Odevixibat (N = 35) | Placebo (N = 17) |
|---|---|---|
CFB to week 24 in PedsQL total score (patients ≥ 2 years) | ||
Number of patients contributing to the analysisa | 28 | 11 |
Baseline, mean (SD) | 67.75 (16.27) | 67.11 (13.49) |
Change from baseline, mean (SD) | 12.75 (17.81) | 9.47 (9.28) |
LS mean (SE)b | 13.50 (2.54) | 10.72 (3.96) |
LS mean difference (95% CI) (odevixibat vs. placebo)b | 2.78 (−6.31 to 11.87) | |
One-sided P valueb | 0.2691 | |
CFB to week 24 in PedsQL Family Impact Module | ||
Number of patients contributing to the analysisc | ██ | ██ |
Baseline, mean (SD) | █████ ███████ | █████ ███████ |
Change from baseline, mean (SD) | █████ ███████ | █████ ███████ |
LS mean (SE)b | █████ ██████ | ████ ██████ |
LS mean difference (95% CI) (odevixibat vs. placebo)b | ████ ██████ ██ ██████ | |
One-sided P valueb | ██████ | |
ANCOVA = analysis of covariance; CFB = change from baseline; CI = confidence interval; FAS = full analysis set; HRQoL = health-related quality of life; LS = least squares; MID = minimal important difference; PedsQL = Pediatric Quality of Life Inventory; SD = standard deviation; SE = standard error; vs. = versus.
Note: The PedsQL is scored from 0 to 100 with higher scores indicating better HRQoL. An MID of 4.5 points for the parent-proxy report has been estimated in the literature.
aThe PedsQL total score was reported by caregivers for patients aged 2 years or older. A total of 32 patients in the odevixibat group and 12 patients in the placebo group were aged at least 2 years. Data were missing for 7 patients (20%) and 6 patients (35%) in the odevixibat and placebo groups, respectively.
bThe analysis was based on an ANCOVA model at each visit, with baseline score as a covariate and treatment group and age category (< 5 years, 5 to 7 years, 8 to 12 years, 13 to 18 years) as fixed effects.
cData were missing for 5 patients (14%) and 1 patient (6%) in the odevixibat and placebo groups, respectively.
Source: ASSERT Clinical Study Report.31
sBA Levels
The key secondary outcome in the ASSERT trial was the change from baseline to the average of weeks 20 and 24 in sBA levels (µmol/L). This end point was controlled for multiple testing and the results were based on all 52 patients enrolled.
The LS mean change from baseline in sBA levels was −90.35 µmol/L (SE = 21.34 µmol/L) in the odevixibat group and 22.39 µmol/L (SE = 28.46 µmol/L) in the placebo group, for an LS mean difference of −112.74 µmol/L (95% CI, −178.78 to −46.69; 1-sided P = 0.0006) favouring odevixibat (Table 17).
Figure 4 shows the LS mean (95% CI) changes from baseline in sBA levels (µmol/L) over time by visit, based on the MMRM.
Figure 4: Estimated Mean Change From Baseline in sBA Levels (µmol/L) Through Week 24 (FAS)
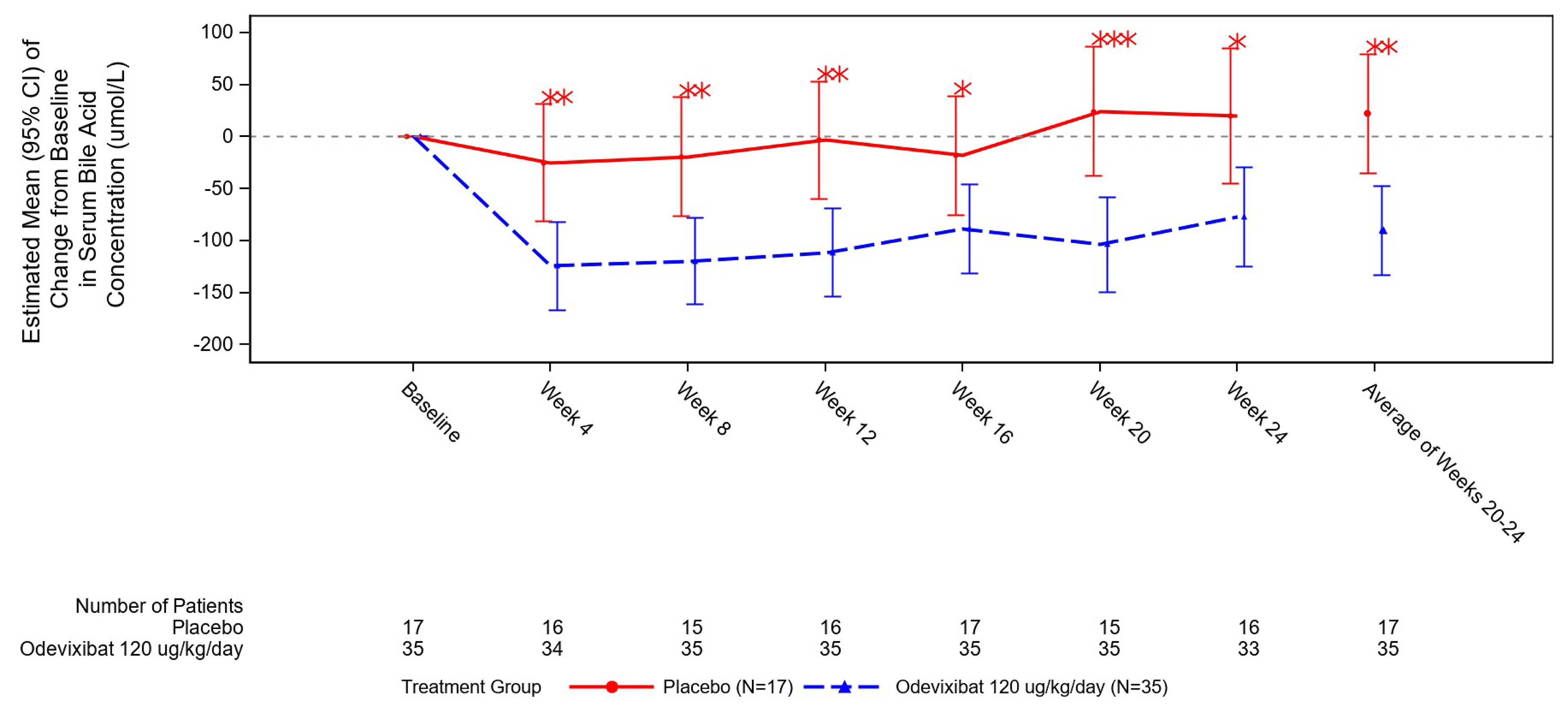
CI = confidence interval; FAS = full analysis set; LS = least squares; MMRM = mixed model for repeated measures; sBA = serum bile acid.
Note: The analysis is based on an MMRM with baseline sBA level as a covariate, and baseline age stratification, treatment group, visits (weeks 4, 8, 12, 16, 20, and 24), and treatment-by-time interaction as fixed effects. The comparison of treatment difference in change from baseline to the average of weeks 20 and 24 was estimated and tested using contrast.
The single asterisks in the figure correspond to P > 0.005 and ≤ 0.025; the double asterisks in the figure correspond to P > 0.0005 and ≤ 0.005; and the triple asterisks in the figure correspond to P ≤ 0.0005, 1-sided P value, indicating superiority of odevixibat over placebo.
Source: ASSERT Clinical Study Report.31
Table 17: Change From Baseline in sBA Levels — ASSERT Study (FAS)
Variable | Odevixibat (N = 35) | Placebo (N = 17) |
|---|---|---|
CFB to average of weeks 20 and 24 in sBA (µmol/L) | ||
Number of patients contributing to the analysis | 35 | 17 |
Baseline, mean (SD) | 237.4 (114.88) | 246.1 (120.53) |
Average of week 20 and 24, mean (SD) | 149.0 (102.30) | 270.7 (166.93) |
Change from baseline, mean (SD) | −88.4 (120.27) | 24.6 (131.63) |
LS mean change from baseline (SE)a | −90.35 (21.34) | 22.39 (28.46) |
LS mean difference (95% CI) (odevixibat vs. placebo)a | −112.74 (−178.78 to −46.69) | |
One-sided P valuea | 0.0006 | |
CFB = change from baseline; CI = confidence interval; FAS = full analysis set; LS = least squares; MMRM = mixed model for repeated measures; sBA = serum bile acid; SD = standard deviation; SE = standard error; vs. = versus.
aThe analysis is based on an MMRM using a restricted maximum likelihood with baseline sBA level as a covariate, and baseline age stratification, treatment group, visits (weeks 4, 8, 12, 16, 20, and 24), and treatment-by-time interaction as fixed effects. The comparison of treatment difference in change from baseline to the average of weeks 20 and 24 was estimated and tested using contrast.
Source: ASSERT Clinical Study Report.31
Growth Parameters
███████ ██ ███████ ██████ ███ ███ ████ ████████ ██ ███████████ ███ ██████ ██ ███ ██████ ██████ █████ ██ █████ ██ █████████ ███ ██ ████ ██████████ ██ ███ ██████ ████ ████████ ██ ██████ ███████ ███ ████ ████ ███ ████ ██ █████ ███ ██████████ ██████ ████████ ███ ██ ████ ██████████ ██ ███ ██████ ████ ████████ ██ ███ ██████ ███████ ███ █████ ████ ███ █████ ██ ██████ ███ ███ ██ ████ ██████████ ██ ███ ██████ ██ ███ ███ ███████ ███ █████ ████ ███ █████ ██ ██████ █████████ ██ █████ ████
Sleep-Related Parameters
A summary of the sleep-related outcomes based on the PRUCISION ObsRO questionnaire is provided in Appendix 1, Table 26. These outcomes were not controlled for multiple testing and were missing for 1 or 2 patients in the odevixibat group (3% or 6%), and 1 patient in the placebo group (6%).
The LS mean difference in the change from baseline to month 6 in the daytime tiredness score (range, 0 [best] to 4 [worst]) was −0.60 points (95% CI, −1.06 to −0.14), and the number of awakenings per night was −2.89 (95% CI, −6.12 to 0.34), for the odevixibat versus placebo groups. The other sleep outcomes were binary (yes, no) responses, reported as the percent of days with a sleep disturbance. The results favoured the odevixibat group versus the placebo group for the percent of days needing help falling asleep, requiring soothing during the night, and sleeping with a caregiver. No statistical difference between groups was detected for the percent of days seeing blood due to scratching or taking medication to induce sleep.
Mortality or Liver Transplant
No deaths were reported during the 24-week study, and no patients underwent a liver transplant or biliary diversion surgery.
Harms
Refer to Table 18 for harms data.
Adverse Events
Overall, 26 (74.3%) of the 35 patients who received odevixibat experienced at least 1 TEAE, as did 12 (70.6%) of the 17 patients who received placebo. Overall, infections and infestations were the most common AE type, which occurred with a similar frequency in the odevixibat and placebo groups (51.4% versus 58.8%, respectively). More patients in the odevixibat group experienced gastrointestinal-related AEs than patients who received placebo (34.3% versus 11.8%). The most common TEAEs (≥ 10%) among patients who received odevixibat versus the corresponding frequency in patients who received placebo, were diarrhea (28.6% versus 5.9%), pyrexia (22.9% versus 23.5%), COVID-19 (14.3% versus 23.5%), and abdominal pain (11.4% versus 5.9%).
Serious AEs
Treatment-emergent SAEs were reported in 5 patients (14.3%) who received odevixibat and in 2 patients (11.8%) who received placebo (proportion difference of 0.025%; 95% CI, −0.230% to 0.216%). None of the SAEs led to treatment discontinuation or dose reductions. One SAE of rhinovirus infection led to an interruption of odevixibat treatment.
Withdrawals Due to AEs
No patients discontinued treatment due to a TEAE. Dose interruptions and reductions due to TEAEs were reported in 3 patients (8.6%) and 1 patient (2.9%) in the odevixibat group, respectively; none of the patients in the placebo group had an interruption in dosing or a dose reduction due to TEAEs.
Mortality
There were no deaths during the study.
Notable Harms
Diarrhea TEAEs were reported in 10 patients (28.6%) in the odevixibat group and 1 patient (5.9%) in the placebo group (proportion difference of 0.227%; 95% CI, −0.033% to 0.417%). No cases were rated as SAEs and none led to treatment discontinuation. ███ ███████ ████████ in the odevixibat group and | ███████ ██████ in the placebo group met the criteria for clinically significant diarrhea (defined as 1 of the following: diarrhea with a duration of 3 or more days without other etiology, diarrhea of grade 3 intensity or reported as an SAE, or diarrhea with concurrent dehydration requiring treatment with rehydration and/or other treatment intervention). Of these clinically significant diarrhea cases, | ███████ in the odevixibat group required oral rehydration treatments. All events were grade 1 in severity, and none required a dose reduction. The median duration of clinically significant diarrhea was ███ ████ (range, | ██ ██ ████).
Three patients in each treatment group had a fat-soluble vitamin deficiency TEAE, of which 1 patient per group (2.9% versus 5.9% in the odevixibat and placebo groups, respectively) had vitamin D deficiency that was refractory to supplementation. For both patients, the deficiency resolved during the treatment period without interruption of the study drug.
Overall, 8 of the 52 patients had TEAEs that were possible sequelae of fat-soluble vitamin deficiency: 5 patients (14.3%) in the odevixibat group and 3 patients (17.6%) in the placebo group. The most common sequelae of fat-soluble vitamin deficiency were hematoma, reported in 3 patients (8.6%) in the odevixibat group; INR increased, reported in 1 patient (2.9%) in the odevixibat group and 2 (11.8%) in the placebo group; and epistaxis reported in 2 patients (11.8%) in the placebo group. Coagulopathy, hematemesis, and contusion were each reported in 1 patient in the odevixibat group. One patient in the odevixibat group had an SAE of hematemesis and increased INR that was considered a possible sequela of fat-soluble vitamin deficiency.
There were no liver decompensation events reported during the study. Further, none of the patients reported new or worsening portal hypertension or hepatic cirrhosis or TEAEs of ascites, hepatic encephalopathy, or variceal hemorrhage in either treatment group. Four patients (11.4%) in the odevixibat group and 2 patients (11.8%) in the placebo group reported a liver-related TEAE, which were all laboratory abnormalities, except for 1 patient in the odevixibat group who experienced jaundice.
Table 18: Summary of Harms Results — ASSERT Study (Safety Population)
Adverse event | Odevixibat (N = 35) | Placebo (N = 17) |
|---|---|---|
Any adverse event | ||
Patients with ≥ 1 TEAE, n (%)a | 26 (74.3) | 12 (70.6) |
Diarrhea | 10 (28.6) | 1 (5.9) |
Pyrexia | 8 (22.9) | 4 (23.5) |
COVID-19 | 5 (14.3) | 4 (23.5) |
Abdominal pain | 4 (11.4)b | 1 (5.9) |
Upper respiratory tract infection | 3 (8.6) | 2 (11.8) |
Respiratory tract infection | 3 (8.6) | 1 (5.9) |
Cough | 3 (8.6) | 1 (5.9) |
Bronchitis | 3 (8.6) | 0 |
Hematoma | 3 (8.6) | 0 |
Nasopharyngitis | 2 (5.7) | 1 (5.9) |
Conjunctivitis | 2 (5.7) | 0 |
Gastroenteritis | 2 (5.7) | 0 |
Vomiting | 2 (5.7) | 1 (5.9) |
Asthenia | 2 (5.7) | 0 |
Weight decreased | 2 (5.7) | 0 |
INR increased | 1 (2.9) | 2 (11.8) |
Epistaxis | 0 | 2 (11.8) |
SAEs | ||
Patients with ≥ 1 SAE, n (%) | 5 (14.3) | 2 (11.8) |
Description (number of patients) | Infections (3), hematemesis and increased INR (1), abdominal pain and constipation (1) | Infection (1), pyrexia (1) |
Treatment discontinuation | ||
Patients who stopped treatment, n | 0 | 0 |
Mortality | ||
Patients who died, n | 0 | 0 |
Adverse events of interest, n (%) | ||
Any clinically significant diarrhea TEAEc | | ██████ | | █████ |
Any fat-soluble vitamin deficiency TEAE | 3 (8.6)d | 3 (17.6) |
Any fat-soluble vitamin deficiency refractory to clinically recommended vitamin supplementation TEAEs | 1 (2.9) | 1 (5.9) |
Any possible sequelae of fat-soluble vitamin deficiency TEAEs | 5 (14.3) | 3 (17.6) |
Any liver-related TEAEse | | ██████ | | ██████ |
Any liver decompensation | 0 | 0 |
INR = international normalized ratio; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aTEAEs reported in 2 or more patients in either group.
bOne additional patient in the odevixibat group reported upper abdominal pain.
cClinically significant diarrhea defined as follows: diarrhea with a duration of 3 or more days without other etiology, diarrhea of grade 3 intensity or reported as an SAE, or diarrhea with concurrent dehydration requiring treatment with rehydration and/or other treatment intervention.
dOne patient in the odevixibat group had both vitamin A and vitamin E decrease reported.
eLiver-related TEAEs include the following Standardised Medical Dictionary for Regulatory Activities Queries: Drug-related hepatic disorders — comprehensive search (narrow and broad), biliary tract disorders, gallbladder-related disorders, and gallstone-related disorders.
Source: ASSERT Clinical Study Report.31
Critical Appraisal
Internal Validity
In the ASSERT study, no major sources of bias in the methods used to randomize and conceal the allocation of patients to treatment groups were identified by the CDA-AMC review team. ASSERT was a small study, with 52 patients enrolled overall and 35 patients assigned to the odevixibat group. Given that ALGS is an uncommon disorder, a small sample size was not unexpected. However, due to the small sample size, there is an increased risk that prognostic balance was not achieved, as evidenced by imbalances in patients’ baseline disease and demographic characteristics (age, sex, growth, medical and surgical history, time since diagnosis, extent of hepatic impairment, liver enzymes, and bilirubin levels). As such, it is possible that the observed effects were either overestimated or underestimated and may have been driven by prognostic differences between the 2 groups (i.e., may not be reflective of the true treatment effect).62 The extent of bias, however, is unclear.
The trial was double blinded, and the investigators took steps to prevent inadvertent unblinding, such as by concealing sBA levels from the study participants. Diarrhea is a known adverse effect of IBAT inhibitors; therefore, it is possible that patients who experienced diarrhea may have inferred they had been assigned to the active treatment group. This assumption may have biased the assessment of subjective patient-reported (and caregiver-reported) outcomes such as pruritus, HRQoL, and harms. Diarrhea AEs were reported more frequently in the odevixibat group than the placebo group (29% versus 6%); thus, any potential reporting bias may have favoured the odevixibat group.
No major issues in the statistical methods used in the study were identified by the CDA-AMC review team. A multiple testing procedure was employed; however, it included only the primary and key secondary outcome (sBA levels). Therefore, there was no multiplicity control, aside from a single pruritus outcome (change from baseline in pruritus severity score) and the change from baseline in sBA levels, meaning other outcomes reporting P values of less than 0.05 were at risk of type I error (false-positive results) and should be interpreted as supportive data only.
Although no patients withdrew from the study, outcome data were missing for some efficacy end points. For the PedsQL total score analysis, ███ and ███ of patients were missing or excluded in the odevixibat and placebo groups, respectively; for the PedsQL Family Impact Module, ███ and ██ of patients were missing or excluded. There were no methods used to impute the missing data for these end points, which were based on a complete case analysis and for which the assumption of “missing completely at random” was unrealistic. In addition, no sensitivity analyses were conducted to investigate other assumptions about the missingness. Given the magnitude of the missing data, the CDA-AMC review team considered this an important limitation of the HRQoL outcomes.
For the primary end point, ██ and ██ of patients in the odevixibat and placebo groups, respectively, were excluded from the 24-week analysis. Sensitivity analyses were conducted to explore the impact of the missing data, and these results were aligned with the results of the primary analysis. Based on the available data, there were no major concerns regarding missing data for the change from baseline in the PRUCISION ObsRO scratching score.
The study assessed pruritus and HRQoL, which were identified as important outcomes based on patient and clinician input. HRQoL was evaluated using the PedsQL instrument, which has evidence to support its validity, reliability, and responsiveness in pediatric patients with various chronic health conditions;26 however, the evidence in children with liver diseases was limited.27 The sponsor relied heavily on proxy reports to assess HRQoL. There is evidence supporting the validity of using this proxy approach with the PedsQL in the pediatric population.26 Of note, there may be a difference between what the proxy thinks and the child thinks with respect to HRQoL and symptoms. In children with chronic health conditions (asthma, ADHD, depression, diabetes, or “other”), the estimated within-group MID for the PedsQL total score for the child self-report and parent-proxy report was 4.4 and 4.5 points, respectively.22 No between-group MID was submitted by the sponsor; therefore, the within-group MID was used to interpret the clinical relevance of the results. The PRUCISION instrument that was used to assess pruritus and sleep-related outcomes was developed by the sponsor for patients with cholestatic liver diseases, including ALGS.23 The instrument’s psychometric properties were assessed in patients with progressive familial intrahepatic cholestasis and ALGS using data from the sponsor’s studies of odevixibat (PEDFIC and ASSERT trials).24,25 Due to the small number of patients completing the PRO instrument, validity, reliability, and responsiveness data were mainly reported for the ObsRO instrument.24,25 The CDA-AMC review team noted that the instrument has not been validated externally and has been assessed only within the sponsor’s own trials in a small number of patients. The sponsor estimated that a 1.0-point to 1.5-point decrease in the ObsRO scratching score would represent a clinically important within-patient change.24,25 However, no data were provided on the MID for between-group differences. Thus, the clinical relevance of the mean differences between groups in the change in ObsRO scratching score was difficult to assess. No MIDs were available for the pruritus responder analyses or the individual sleep-related outcome measures.
The change in sBA levels was a key secondary outcome in the trial. In other pediatric cholestatic diseases (progressive familial intrahepatic cholestasis and biliary atresia), sBA has prognostic implications, with patients with higher sBA levels being more likely to progress to end-stage liver disease or to undergo liver transplant.57,58 It has not yet been defined what sBA level or reduction can be associated with improved longer-term clinical outcomes in patients with ALGS. Thus, it is difficult to interpret the sBA level results, given that the surrogate threshold effect is unclear. ALGS may impact patients’ growth; thus, z scores for height, weight, and BMI were included in this report as supplementary end points. The Clinical Study Report states that in infants and toddlers, height, length, and weight measurements are sometimes challenging due to positioning, variable weight of diapers and clothing, or other confounding factors; thus, measurement errors may have occurred during the ASSERT trial. No MID for a change in z scores was submitted by the sponsor.
Numerous subgroup analyses were planned for the ASSERT study, but due to the small sample size of some groups, many subgroups could not be analyzed. The available subgroup data were generally consistent with the findings of the overall study population, except for patients with more severe liver impairment, for whom the point estimate for the between-group difference was near the null (no difference). The results raise uncertainty as to whether the estimated effect in the overall study population may be generalized to patients with more severe liver impairment. However, the small sample sizes in each subgroup, lack of predefined hypotheses regarding subgroup effects, and lack of testing for subgroup by treatment interactions preclude credible conclusions of effect modification. Because many subgroups could not be analyzed, these analyses were also insufficient to infer consistency of effects across all relevant subpopulations.
External Validity
The patients enrolled in the ASSERT study ranged in age from 6 months to 15.5 years, with the majority (███) aged 2 years to younger than 12 years, and the minority were either younger than 2 years (███) or were aged 12 years or older (███). The patients had been diagnosed with ALGS for a median of ███ years; half had sBA levels of 212.5 µmol/L or higher, and all patients but 1 had received antipruritic medications in the past. All patients had evidence of hepatic impairment, with ███, ███, and ███ classified as having mild, moderate, or severe impairment, respectively. According to the clinical experts consulted by CDA-AMC, these patients were consistent with pediatric patients with ALGS in Canada. The experts noted that some patients excluded from trial may be treated with odevixibat in practice, such as those with ascites or waiting for a liver transplant, but overall, the patients enrolled were generalizable to those in clinical practice.
The dose of odevixibat used in the study matched the product monograph and, during the trial, patients were allowed to continue the standard antipruritic medications they had been receiving before randomization. The clinical experts confirmed the use of antipruritic cointerventions during the trial was consistent with clinical practice. During the ASSERT study, most patients received UDCA (88%) and/or rifampin (67%).
Limitations to the external validity include the relatively small sample size (N = 52) of the study, the focus on shorter-term end points, and the lack of a head-to-head comparison with maralixibat. Liver transplants and mortality were identified as key clinical outcomes for patients with ALGS. While no deaths were reported and no patients underwent a liver transplant during the 24-week trial, the study was not designed to test the comparative effectiveness of odevixibat versus placebo for these longer-term end points. Given that this trial used a placebo control, there are likely ethical concerns with a longer treatment period; however, the lack of data for these key clinical outcomes is a limitation of the core study. The trial’s sample size and duration were likely insufficient to assess the impact of odevixibat on patients’ growth or on safety, except for the most common TEAEs.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations and a final certainty rating was determined, as outlined by the GRADE Working Group:28,29
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from the RCT started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. For the PRUCISION ObsRO and PRO, sBA levels, and harms, the presence or absence of any (non-null) effect was used and, for the PedsQL, the target of the certainty-of-evidence assessment (presence or absence of an important effect) was based on thresholds identified in the literature.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for odevixibat versus placebo.
Odevixibat may result in a reduction in the ObsRO scratching severity score when compared with placebo.
Odevixibat may result in little to no difference in the PRO itching severity score when compared with placebo.
The evidence is very uncertain about the effect of odevixibat on the change in PedsQL parent report total score when compared with placebo.
Odevixibat likely results in a reduction in sBA levels when compared with placebo. The clinical importance of the decrease is unclear.
There is no evidence to assess the effect of odevixibat on the need for liver transplant or on mortality, when compared with placebo.
The evidence is very uncertain about the effect of odevixibat on SAEs when compared with placebo.
Odevixibat may result in an increase in diarrhea TEAEs when compared with placebo. The clinical importance of the increase is unclear.
Long-Term Extension Studies
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
The ASSERT-EXT study (NCT05035030; N = 50) is a phase III, multicentre, open-label, LTE to the ASSERT RCT, which was designed to evaluate the long-term safety and efficacy of odevixibat (120 mcg/kg per day) in relieving pruritus in patients with ALGS. The duration of the primary treatment period was 72 weeks, followed by a 4-week safety follow-up period. The outcomes assessed in the ASSERT-EXT study were identical to those of the ASSERT RCT.
Patients were treated with the same odevixibat dose and protocol used in the active treatment arm of the ASSERT RCT. Those who received odevixibat in the RCT continued to receive odevixibat during the ASSERT-EXT study (i.e., the continued-treatment group), and those receiving placebo in the RCT were switched to odevixibat (i.e., the treatment-naive group). Patients who were not tolerating the 120 mcg/kg per day dose were down titrated to 40 mcg/kg per day dose after a minimum of 1 week of treatment and returned to the higher dose as soon as deemed appropriate.
Day 1 of the ASSERT-EXT study coincided with week 24 or clinic visit 9 of the ASSERT RCT; if not possible due to logistical reasons, patients could enrol in the extension study within 28 days of completing the RCT.
Statistical Analysis
No formal hypothesis testing was performed in the ASSERT-EXT study, and only descriptive statistics were used to summarize results. The FAS was the primary analysis set for all analyses unless otherwise specified and included all patients who received 1 dose of the study drug in the LTE.
Baseline 1 corresponded to the last value before treatment start in the ASSERT RCT, and baseline 2 corresponded to the last value before treatment start in the ASSERT-EXT study. Unless otherwise specified, baseline 2 was used in all analyses. For the purposes of safety analyses, AEs with missing start dates, but with stop dates either overlapping with the treatment period or missing, were considered TEAEs. A TEAE with missing severity was considered severe.
Baseline Characteristics
Refer to Table 19 for a summary of baseline clinical characteristics in the ASSERT-EXT study.
Most patient baseline clinical characteristics in the ASSERT-EXT study were similar to those of the ASSERT RCT, with a few notable differences. The mean scratching score (caregiver-reported) was lower in both groups compared with the ASSERT study baseline, at ████ points (SD = ████) in the continued-treatment group and ████ points (SD = ████) in the treatment-naive group. The mean sBA level in the continued-treatment group was █████ µmol/L (SD = ██████), which was lower than in the ASSERT study, and █████ µmol/L (SD = ██████) in the treatment-naive group, which was higher than in the ASSERT study. In the continued-treatment group, the mean alanine aminotransferase, aspartate aminotransferase, and gamma-glutamyl transferase levels were higher than in the ASSERT study, at 237.2 U/L (SD = 109.55 U/L), 206.9 U/L (SD = 114.43 U/L), and 456.3 U/L (SD = 254.35 U/L), respectively.
Table 19: Summary of Baseline Clinical Characteristics in the ASSERT-EXT Study (FAS)
Characteristic | Odevixibat to odevixibat (continued treatment) (N = 33) | Placebo to odevixibat (treatment naive) (N = 17) |
|---|---|---|
Height (cm) | ||
Mean (SD) | ██████ ████████ | ██████ ████████ |
Weight (kg) | ||
Mean (SD) | █████ ███████ | █████ ████████ |
Scratching score (morning and evening combined), PRUCISION ObsRO | ||
Mean (SD) | ████ ███████ | ████ ███████ |
Median (range) | ████ █████ ████ | ████ █████ ████ |
Serum bile acid level (µmol/L) | ||
Mean (SD) | █████ ████████ | █████ ████████ |
Median (range) | █████ ████ ████ | █████ ████ ████ |
Prior medications, n (%) | ||
Antipruritic medications | ██ ██████ | ██ █████ |
UDCA | ██ ██████ | ██ ██████ |
Hepatic impairment classification,b n (%) | ||
No Impairment | ██ | ██ |
Mild | | ██████ | | ██████ |
Moderate | ██ ██████ | ██ |
Severe | ██ ██████ | ██ ██████ |
Laboratory values | ||
ALT (U/L), mean (SD) | ██████ ████████ | █████ ███████ |
AST (U/L), mean (SD) | █████ ████████ | █████ ███████ |
Total bilirubin (µmol/L), mean (SD) | █████ ████████ | █████ ████████ |
Direct bilirubin (µmol/L), mean (SD) | █████ ████████ | █████ ████████ |
GGT (U/L), mean (SD) | █████ ████████ | █████ ████████ |
eGFRc (mL/min/1.73 m2), mean (SD) | ██████ ████████ | ██████ ████████ |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; BMI = body mass index; eGFR = estimated glomerular filtration rate; FAS = full analysis set; GGT = gamma-glutamyl transferase; ObsRO = observer-reported outcome; SD = standard deviation; UDCA = ursodeoxycholic acid.
aBaseline was the last value before treatment start in the ASSERT-EXT study.
bBased on National Cancer Institute Organ Dysfunction Working Group classification system.
cCalculated by the modified Schwartz equation.
Source: ASSERT-EXT Clinical Study Report.63
Results
Patient Disposition
Refer to Table 20 for a summary of the patient disposition in the ASSERT-EXT study. A total of 50 patients were enrolled and received treatment with odevixibat, including 33 who had received odevixibat in the ASSERT trial and 17 who had received placebo (i.e., were treatment-naive upon entry into the ASSERT-EXT study). Overall, 44 (88.0%) of the 50 patients completed the 72-week treatment period and 6 patients (4 in the treatment-naive group, and 2 in the continued-treatment group) discontinued treatment. Of all discontinuations, 1 was due to an AE (elevated bilirubin), 1 was due to withdrawal of consent, and 4 were for other reasons (1 patient whose caregiver elected to withdraw from the study because they did not think the study medication was working, 1 patient who discontinued to receive another medication, 1 patient who underwent a liver transplant, and 1 patient whose family withdrew consent due to patient’s itchiness).
Exposure to Study Treatments
Refer to Table 21 for a summary of patient exposure in the ASSERT-EXT study.
Table 20: Patient Disposition in the ASSERT-EXT Study (FAS)
Patient disposition | Odevixibat to odevixibat (continued treatment) (N = 33) | Placebo to odevixibat (treatment naive) (N = 17) |
|---|---|---|
Entered LTE, N | ██ | ██ |
Discontinued from study, n (%) | | █████ | | ██████ |
Reason for discontinuation, n (%) | ||
Adverse events | || | | █████ |
Withdrawal of consent | || | | █████ |
Other | | █████ | | ██████ |
FAS, n | ██ ███████ | ██ ███████ |
FAS = full analysis set; LTE = long-term extension; PPS = per-protocol set.
Sources: ASSERT-EXT Clinical Study Report63 and sponsor’s summary of clinical evidence.52
Table 21: Patient Exposure in the ASSERT-EXT Study (FAS)
Variable | Odevixibat to odevixibat (continued treatment) (N = 33) | Placebo to odevixibat (treatment naive) (N = 17) |
|---|---|---|
Duration of exposure (weeks)a | ||
Mean (SD) | █████ ████████ | █████ ████████ |
Median | █████ | █████ |
Adherence (%) | ||
Mean (SD) | █████ ███████ | █████ ███████ |
Median | █████ | █████ |
FAS = full analysis set; SD = standard deviation.
aDuration of exposure (in weeks) in the ASSERT-EXT study was calculated as: (date of last study drug intake minus date of first study drug intake plus 1) divided by 7.
Sources: ASSERT-EXT Clinical Study Report63 and sponsor’s summary of clinical evidence.52
Concomitant Medications
All 50 patients in the ASSERT-EXT study took at least 1 concomitant medication during the treatment period; most of these medications were for the treatment of pruritus. Overall, the most common types of concomitant medications were UDCA (███), vitamin D and analogues (███), rifampin (███), acetaminophen (███), vitamin K (███), ibuprofen (███), and other plain vitamin preparations (███). Concomitant use of “other antihistamines for systemic use” was higher in the continued-treatment group compared with the treatment-naive group, at ███ and ██, respectively; use of oral rehydration salt formulations was higher in the treatment-naive group compared with the continued-treatment group, at ███ and ███.
Efficacy
Refer to Table 22 for a summary of efficacy data from the ASSERT-EXT study.
Observer-Reported Scratching Severity
At weeks 69 to 72, PRUCISION ObsRO scratching severity data were available for ██ patients (███) and ███ patients (███) in the odevixibat continued-treatment and treatment-naive groups, respectively. The mean change from baseline in the ObsRO scratching severity score at weeks 69 to 72 in the continued-treatment group and treatment-naive group was █████ (SD = ████) and █████ (SD = ████), respectively.
From the baseline of the ASSERT RCT, at weeks 69 to 72 of the ASSERT-EXT study, 93% of patients in the continued-treatment group had a reduction of 1.0 point or more in the scratching severity score and 73% had a reduction of 1.5 points or more.
From the baseline of the ASSERT-EXT study to weeks 69 to 72, ███ of patients in the treatment-naive group had a reduction of 1.0 point or more in the scratching severity score and ███ had a reduction of 1.5 points or more; ███ of patients in the continued-treatment group had a reduction of 1.0 point or more in the scratching severity score and ███ had a reduction of 1.5 points or more.
Patient-Reported Itching Severity
The mean change from baseline in the PRUCISION PRO itching severity score at weeks 69 to 72 baseline in the odevixibat continued-treatment group and treatment-naive group was █████ (SD = █████) and █████ (SD = █████), respectively. At weeks 69 to 72, data were available for ██ patients (███) and ██ patients (███) in the odevixibat continued-treatment and treatment-naive groups, respectively.
HRQoL Outcomes
For the PedsQL total score, data at week 72 were available for ██ patients (███) and ██ patients (███) in the continued-treatment and treatment-naive groups, respectively. Data were available for 27 patients (82%) and ██ patients (███) in the continued-treatment and treatment-naive groups, respectively, for the Family Impact Module at week 72.
The mean change from baseline to week 72 in the PedsQL total score was █████ (SD = ████) in the continued-treatment group and ████ (SD = █████) in the treatment-naive groups. For the Family Impact Module, the mean change from baseline in the continued-treatment and treatment-naive groups, respectively, was ████ (SD = █████) and ████ (SD = █████).
sBA Levels
The mean change from baseline to week 72 in sBA levels (µmol/L) was ||||| (SD = |||||) in the continued-treatment group and ||||| (SD = ||||||) in the treatment-naive group. At 72 weeks, sBA data were available for || patients (|||) in the continued-treatment group and || patients (|||) in the treatment-naive group.
Sleep Parameters
In the ASSERT-EXT study, sleep-related outcomes were measured using the PRUCISION ObsRO questionnaire. Mean changes from baseline to weeks 69 to 72 in the continued-treatment group were ██████ (SD = ████) for percent of days with help falling asleep; ██████ (SD = ████) for percent of days with soothing; █████ (SD = █████) for percent of days sleeping with the caregiver; █████ (SD = ███) for mean daytime tiredness score; █████ (SD = ████) for percent of days seeing blood due to scratching, and ████ (SD = ███) for mean number of awakenings.
Mean changes from baseline to weeks 69 to 72, in the treatment-naive group were ██████ (SD = ████) for percent of days with help falling asleep, ██████ (SD = ████) for percent of days with soothing, ██████ (SD = ████) for percent of days sleeping with the caregiver, ████ (SD = ███) for mean daytime tiredness score, ██████ (SD = ████ for percent of days seeing blood due to scratching, and ████ (SD = ███) for mean number of awakenings.
Growth Parameters
███████ ██ ███████ ███████ ███ ███ ████ ███████████ ███ ██████ ██ ███████████ █████ ██ █████ ██ █████████ ██ ███ █████████ ██████ ███ ████ ██████ ████ ████████ ██ ███ ██████ ███████ ███ ████ ███ █ █████ ██ ███ █████████ █████████ █████ ███ ████ ███ █ █████ ██ ███ █████████ █████ ██████ ███ ████ ██████ ████ ████████ ██ ███ ██████ ███████ ███ █████ ███ █ █████ ██ ███ █████████ █████████ █████ ███ ████ ███ █ █████ ██ ███ █████████ █████ ██████ ███ ████ ██████ ████ ████████ ██ ███ ███ ███████ ███ █████ ███ █ █████ ██ ███ █████████ █████████ █████ ███ █████ ███ █ █████ ██ ███ █████████ █████ ██████
Mortality and Liver Transplant
No patients died or underwent biliary diversion surgery during the 72-week study. One patient received a liver transplant.
Table 22: Summary of Efficacy Results in the ASSERT-EXT Study (FAS)
Variable | Odevixibat to odevixibat (continued treatment) (N = 33) | Placebo to odevixibat (treatment naive) (N = 17) |
|---|---|---|
CFB to week 72 in the scratching severity score (a.m. and p.m. scores combined; measured by PRUCISION ObsRO) | ||
Baseline, n | ██ | ██ |
Mean (SD) | ████ ███████ | ████ ███████ |
Change from baseline to weeks 69 to 72, n | ██ | ██ |
Mean (SD) | █████ ███████ | █████ ███████ |
CFB to week 72 in the scratching severity score (a.m. and p.m. scores combined; measured by PRUCISION PRO) | ||
Baseline, n | ██ | ██ |
Mean (SD) | ████ ███████ | ████ ███████ |
Change from baseline to weeks 69 to 72, n | ██ | ██ |
Mean (SD) | █████ ███████ | █████ ███████ |
CFB to week 72 in PedsQL total score | ||
Baseline, n | ██ | ██ |
Mean (SD) | █████ ████████ | █████ ████████ |
Change from baseline to week 72, n | ██ | ██ |
Mean (SD) | █████ ███████ | ████ ████████ |
CFB to week 72 in PedsQL Family Impact Module | ||
Baseline, n | ██ | ██ |
Mean (SD) | █████ ████████ | █████ ████████ |
Change from baseline to week 72, n | ██ | ██ |
Mean (SD) | ████ ████████ | ████ ████████ |
CFB to week 72 in sBA level (µmol/L) | ||
Baseline, n | ██ | ██ |
Mean (SD) | █████ ████████ | █████ ████████ |
Change from baseline to week 72, n | ██ | ██ |
Mean (SD) | █████ ███████ | ██████ ████████ |
Percent of patients achieving a clinically meaningful reduction (≥ 1.5-point drop) from the ASSERT-EXT study baseline in the scratching score (measured by PRUCISION ObsRO) to week 72 | ||
Weeks 69 to 72 (monthly score), n | ██ | ██ |
Responders, n (%) | | ██████ | | ██████ |
Percent of patients achieving a clinically meaningful reduction (≥ 1.5-point drop) from ASSERT baseline in the scratching score (measured by PRUCISION ObsRO) to week 72 | ||
Weeks 69 to 72 (monthly score), n | ██ | ██ |
Responders, n (%) | ██ ██████ | ██ |
Percent of patients achieving a clinically meaningful reduction (≥ 1.0-point drop) from ASSERT-EXT study baseline in the scratching score (measured by PRUCISION ObsRO) to week 72 | ||
Weeks 69 to 72 (monthly score), n | ██ | ██ |
Responders, n (%) | | ██████ | ██ ██████ |
Percent of patients achieving a clinically meaningful reduction (≥ 1.0-point drop) from ASSERT baseline in the scratching score (measured by PRUCISION ObsRO) to week 72 | ||
Weeks 69 to 72 (monthly score), n | ██ | ██ |
Responders, n (%) | ██ ██████ | ██ |
BMI = body mass index; CFB = change from baseline; FAS = full analysis set; NA = not applicable; ObsRO = observer-reported outcome; PedsQL = Pediatric Quality of Life Inventory; PRO = patient-reported outcome; sBA = serum bile acid; SD = standard deviation.
Sources: ASSERT-EXT Clinical Study Report63 and sponsor’s summary of clinical evidence.52
Harms
Overall, ███ of patients in the ASSERT-EXT study experienced at least 1 TEAE, including ███ of patients in the continued-treatment group and ███ patients in the treatment-naive group (Table 23). Across both groups, the most common TEAEs were ███████████████ ██████ ████████ ██████ ███████ ██████ ███ ████████ ███ █████ ███████████ █████ █████████ ██████ Most TEAEs were grade 1 or 2 in intensity, with no grade 4 or 5 TEAEs. Treatment-emergent SAEs were reported in ███ of patients overall, including ███ of patients in the continued-treatment group and ███ of treatment-naive patients.
Because patients with ALGS are known to have multiple comorbidities, specific AEs of special interest included clinically significant diarrhea, new or worsening fat-soluble vitamin deficiency refractory to clinically recommended vitamin supplementation, potential sequelae of the deficiency, and hepatic events. Overall, ███ of patients had clinically significant diarrhea (most due to being more than 3 days in duration), though all were nonserious and assessed as grade 1 or 2, ██ had fat-soluble vitamin deficiency refractory to clinically recommended vitamin supplementation, ███ reported possible sequelae of fat-soluble vitamin deficiency, and ███ had liver-related TEAEs.
Overall, treatment discontinuation due to TEAEs (elevated bilirubin) occurred in | ███████ and | ████████ overall had dose reductions due to a TEAE. Treatment interruptions due to a TEAE occurred in ███ of patients in the continued-treatment group and ██ of patients in the treatment-naive group. There were no deaths during the study period.
Table 23: Summary of Harms Results From the ASSERT-EXT Study (FAS)
Harms | Odevixibat to odevixibat (continued treatment) (N = 33) | Placebo to odevixibat (treatment naive) (N = 17) |
|---|---|---|
TEAEs | ||
TEAE, n (%) | ██ ██████ | ██ █████ |
TEAEs by maximum intensity, n (%) | ||
Grade 1 | ██ ██████ | | ██████ |
Grade 2 | ██ ██████ | | ██████ |
Grade 3 | | ██████ | | ██████ |
Grade 4 | ██ | ██ |
Grade 5 | ██ | ██ |
Treatment-emergent SAEs, n (%) | | ██████ | | ██████ |
TEAEs leading to death, n (%) | ██ | ██ |
TEAEs leading to treatment discontinuation, n (%) | ██ | | █████ |
TEAEs leading to treatment interruption, n (%) | | ██████ | | █████ |
TEAEs leading to dose reduction, n (%) | | █████ | | █████ |
TEAEs reported in ≥ 15% of patients overall | ||
Patients with any TEAEs, n (%) | ██ ██████ | ██ █████ |
Nasopharyngitis | | ██████ | | ██████ |
COVID-19 | | ██████ | | ██████ |
Upper respiratory tract infection | | ██████ | | ██████ |
Otitis media | | ██████ | | ██████ |
Influenza | | ██████ | | █████ |
Bronchitis | | ██████ | ██ |
Viral infection | | █████ | | ██████ |
Diarrhea | | ██████ | | ██████ |
Cough | | ██████ | | ██████ |
Rhinorrhea | | █████ | | ██████ |
Vitamin D deficiency | | █████ | | ██████ |
Vitamin E deficiency | | █████ | | ██████ |
Pyrexia | | ██████ | | ██████ |
International normalized ratio increased | ██ | | ██████ |
Adverse events of special interest | ||
Any clinically significant diarrhea TEAEs | | ██████ | | ██████ |
Any FSV deficiency refractory to clinically recommended vitamin supplementation TEAEs | ██ | | ██████ |
Any possible sequelae of FSV deficiency TEAEs | | ██████ | | ██████ |
Any liver-related TEAEsa | | █████ | | ██████ |
FAS = full analysis set; FSV = fat-soluble vitamin; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aTEAEs reported in the Standardised Medical Dictionary for Regulatory Activities (MedDRA) queries of drug-related hepatic disorders — comprehensive search (narrow and broad), biliary tract disorders, gallbladder-related disorders, or gallstone-related disorders. Note this does not include 3 patients who underwent adjudication by the Hepatic Safety Adjudication Committee for elevated hepatic parameters because the elevations were not reported as TEAEs.
Sources: ASSERT-EXT Clinical Study Report and sponsor’s summary of clinical evidence.52
Critical Appraisal
The ASSERT-EXT study was a phase III, multicentre, open-label study to evaluate the long-term safety and efficacy of odevixibat in patients with ALGS. This open-label design could bias the magnitude of treatment effect for subjective efficacy outcomes, including pruritus, HRQoL, and sleep parameters, and the reporting of safety parameters due to unblinded exposure to the study drug. The direction and magnitude of these potential biases remain unclear. In addition, the absence of statistical hypothesis testing and a control group (i.e., no active comparator or placebo arm) limits the ability to draw definitive conclusions regarding the treatment effect (i.e., whether the observed effects could be attributable to natural history, the placebo effect, or the effects of the drug).
The extension study consisted of patients who took part in the ASSERT trial; therefore, it is reasonable to expect that the same strengths and limitations related to generalizability apply to the extension period. There were considerable data missing at week 72 for many efficacy outcomes in the continued-treatment and treatment-naive groups, including caregiver-reported scratching severity score (███ and ███ of patients missing, respectively), patient-reported scratching severity score (███ and ███), PedsQL total score (███ and ███), PedsQL Family Impact Module (███ and ███), and sBA levels (██ and ███). A complete case analysis was used, which assumes the missing data are missing completely at random. Because this assumption is unlikely to be reasonable, there is a risk of bias due to missing outcomes data for these efficacy outcomes. The magnitude and direction of the bias cannot be predicted.
Indirect Evidence
No indirect evidence was submitted by the sponsor for this review. The sponsor conducted a feasibility assessment to determine whether an indirect treatment comparison with maralixibat could be conducted. Based on the feasibility assessment, the sponsor concluded that an indirect treatment comparison was not feasible.52
The sponsor noted the differences in the trial design and outcome assessment between the ASSERT study and ICONIC study for maralixibat. Specifically, the ICONIC trial used a placebo withdrawal design, where all patients received maralixibat for 18 weeks, and then were randomized to continue maralixibat or switch to placebo for 4 weeks. After the 4-week randomized period, all patients received maralixibat. The primary end point was the change from week 18 (randomization) to week 22 in sBA, and the secondary outcome was the change in the pruritus severity score based on the Itch Reported Outcome instrument (range of 0 = no pruritus to 4 = very severe pruritus) among patients who met the treatment response criteria before randomization. The differences between the ASSERT and ICONIC trials in the pruritus severity assessment added heterogeneity to the comparison. Based on the differences in the placebo groups, the treatment duration between studies, and the outcome measures, the sponsor stated that an anchored indirect comparison was not feasible.
The sponsor also noted differences in the patient characteristics between the 2 studies and, considering the small sample sizes (35 patients received odevixibat and 31 patients received maralixibat), any adjustment for baseline difference would reduce the effective sample size to the extent that meaningful analysis was not possible. As a result, an unanchored matching-adjusted indirect comparison was not feasible.
The CDA-AMC review team evaluated the ICONIC and ASSERT trials and agreed there was substantial heterogeneity between the studies and that an anchored or unanchored indirect comparison was unlikely to provide robust evidence on the comparative efficacy or safety of odevixibat versus maralixibat.
Studies Addressing Gaps in the Systematic Review Evidence
The sponsor submitted 2 studies intended to address gaps in the systematic review evidence.53,54 Sokol et al. (2023)53 evaluated the 6-year predictors of event-free survival and transplant-free survival in patients with ALGS who received maralixibat. Hansen et al. (2024)54 evaluated the long-term impact of maralixibat treatment on event-free survival in children with ALGS and cholestatic pruritus. Because neither of these studies informed the objective of this Clinical Review, which is to evaluate the benefits and harms of the drug under review, the CDA-AMC review team did not summarize or appraise them in this report.
Discussion
Summary of Available Evidence
The systematic review included 1 double-blind, parallel-design RCT that compared the efficacy and safety of odevixibat with placebo (with SOC therapies) in patients with ALGS, elevated sBA, and significant pruritus (ASSERT study). Patients were randomized to once-daily oral odevixibat 120 mcg/kg per day (N = 35) or a matching placebo (N = 17) for up to 24 weeks. The primary end point was the change from baseline to month 6 in the PRUCISION ObsRO scratching severity score.
The mean age of the patients enrolled was ███ years (SD = ███; range, ███ to ████ years) in the odevixibat group and ███ years (SD = ███; range, ███ to ████) in the placebo group. The proportion of females was 40% and 64.7% and the proportion of males was 60% and 35.3% in the odevixibat and placebo groups, respectively. According to the National Cancer Institute Organ Dysfunction Working Group classification system, █████ and █████ of patients had mild hepatic impairment, █████ and █████ had moderate impairment, and █████ and █████ of patients had severe impairment at baseline in the odevixibat and placebo groups, respectively. The mean scratching ObsRO score was 2.80 points (SD = 0.52) and 3.01 points (SD = 0.64), and the mean sBA level was 237.4 µmol/L (SD = 114.9 µmol/L) and 246.1 µmol/L (SD = 120.5 µmol/L) in the odevixibat and placebo groups, respectively. Most patients were receiving UDCA, including 30 patients (85.7%) in the odevixibat group and 16 patients (94.1%) in the placebo group. All patients were on antipruritic medications at baseline, except for 1 patient randomized to the odevixibat group.
Patients who completed the ASSERT trial were eligible to enrol in the ASSERT-EXT study (N = 50), a phase III, multicentre, open-label, LTE designed to evaluate the long-term safety and efficacy of odevixibat 120 mcg/kg per day in relieving pruritus in patients with ALGS. The extension study consisted of a 72-week primary treatment period followed by a 4-week safety follow-up period. In the ASSERT-EXT study, eligibility criteria, intervention protocols, and evaluated outcomes were identical to those of the ASSERT RCT. Patients who had been in the active treatment arm of the ASSERT trial continued to be treated in the ASSERT-EXT study with odevixibat using the same dose and protocol, while those who had received placebo were switched to odevixibat. Like in the ASSERT RCT, concurrent use of standard antipruritic medications was permitted for all patients.
No indirect evidence comparing odevixibat with maralixibat and no studies addressing gaps in the odevixibat evidence were submitted by the sponsor.
Interpretation of Results
Efficacy
ALGS is a rare, life-threatening, genetic disorder that presents with a range of features, including cholestatic liver disease, failure to thrive, cardiovascular disease, skeletal abnormalities, ocular abnormalities, renal and vascular abnormalities, and distinct facial features. Patients and clinicians identified the need for well tolerated and effective treatments to reduce or eliminate the pruritus due to cholestatic liver disease in patients with ALGS. The ASSERT study met its primary end point and showed a statistically significant reduction in the PRUCISION ObsRO scratching score from baseline to week 24 favouring odevixibat versus placebo (LS mean difference −0.88 points; 95% CI, −1.44 to −0.33). While the sponsor estimated at least a 1-point difference in the scratching score as the minimum clinically important within-patient change, there is uncertainty in the MID for a between-group difference. The clinical experts consulted by CDA-AMC agreed that a mean difference of −0.88 points favouring odevixibat was clinically relevant, but they noted that the lower bound of the 95% CI (−0.33 points) was unlikely to represent a meaningful difference versus placebo. The CDA-AMC review team noted that the small sample size of the study and potential prognostic imbalance at baseline raise concern for the potential overestimation of the true effect, although the extent of bias is unclear. Also, there was potential for reporting bias due to the differential frequency of known AEs, which could result in some patients being able to infer which treatment group they were assigned to. Given these uncertainties and possible sources of bias, the CDA-AMC review team assessed the change from baseline in observer-reported pruritus severity as low-certainty evidence.
Observer-reported pruritus severity data were also analyzed based on the proportion of patients who had at least a 1.0-point or 1.5-point improvement from baseline using both the preplanned analyses (based on the a.m. and p.m. scratching scores) and a post hoc analysis that was used to inform the pharmacoeconomic model (based on a.m. scores only). The between-group differences favoured odevixibat versus placebo, reporting point estimates ranging from 36.6% to 44.7%. While these analyses provide supportive data for the primary outcome, they were not controlled for multiple testing (so there is an increased risk of type I errors), and the minimum clinically important between-group difference is unclear.
The patient-reported pruritus severity score did not detect a statistical difference between groups; however, there is uncertainty in this result because it was based on only 15 patients and showed very serious imprecision.
For patients experiencing cholestatic pruritus, the itch is described as “relentless” and it impacts sleep, school attendance and performance, and the overall quality of life of patients and their families. In the ASSERT study, HRQoL was assessed using the PedsQL questionnaire, which has evidence to support its validity, reliability, and responsiveness in pediatric patients with chronic health conditions. No statistical difference between groups was detected in the ASSERT trial for the change from baseline to week 24 in the PedsQL total score or the Family Impact Module. These results were limited by the extent of missing data and showed very serious imprecision; thus, the impact of odevixibat compared with placebo on HRQoL is unclear.
The change in sBA levels was a key secondary outcome in the trial, and the results showed statistically significant differences between groups favouring odevixibat versus placebo (LS mean difference of −112.74 µmol/L; 95% CI, −178.78 to −46.69). However, it has not yet been defined what sBA level or reduction can be associated with improved longer-term clinical outcomes in patients with ALGS. Thus, it is difficult to interpret the sBA level results, given that the surrogate threshold effect is unclear.
Children with ALGS often exhibit poor growth and nutritional deficiencies as a result of their condition. With respect to the change in height, weight, and BMI z scores in the ASSERT study, either little to no difference was detected between groups, or the results suggested that the placebo group was favoured. The follow-up duration and sample size of the trial may have been insufficient to detect differences in growth rates, and imbalances between groups in the proportion of infants (who have higher growth rates than older children) may have contributed uncertainty to these results.
The impact of odevixibat on sleep was challenging to interpret because some of the PRUCISION ObsRO sleep end points did not detect differences between groups (i.e., number of awakenings per night, percent of days seeing blood due to scratching, or taking medication to induce sleep) and, for other sleep end points, the clinical relevance for the between-group differences was unclear. Moreover, the evidence to support the validity and reliability of the sleep-related items on the PRUCISION instrument was limited. Given that the sleep end points were not controlled for multiple testing, the results should be interpreted as supportive data.
The evidence was limited to a single RCT that was 24 weeks in duration, with a 72-week open-label, uncontrolled extension study (N = 50). Neither the RCT nor the LTE study were designed to assess the effect of odevixibat on long-term end points, including mortality and the need for a liver transplant, compared with placebo. Over the course of the 2 studies, no patients died, and 1 patient received a liver transplant (during the LTE). Given the sample size and follow-up duration, assessing the comparative effects for these end points was likely not feasible; however, the lack of comparative data for these key clinical outcomes is a limitation of this review. The longer-term efficacy data reported in the ASSERT-EXT study suggested that treatment response was largely maintained for patients who continued odevixibat treatment after the RCT. However, the interpretation of the findings was limited by the small sample sizes, a high proportion of missing data across several outcomes, and the lack of a relevant randomized active comparator. Overall, the available evidence for the longer-term efficacy of odevixibat is uncertain.
According to the clinical experts consulted by CDA-AMC, the patients enrolled in the RCT and LTE were consistent with pediatric patients with ALGS in Canada. The experts noted that some patients who had been excluded from these trials may be treated with odevixibat in practice, such as those with ascites or those waiting for a liver transplant but, overall, the patients enrolled were generalizable to those in clinical practice. Limitations to the external validity include the relatively small sample size (N = 52) of the study and the focus on shorter-term end points.
There was no direct or indirect evidence comparing odevixibat versus maralixibat, the main comparator for this review. Due to the small sample sizes and heterogeneity between the 2 drugs’ clinical trials, conducting an indirect comparison was not feasible. As a result, the comparative effects between the drugs are unknown.
Harms
TEAEs were common during the 24-week ASSERT trial and occurred with a similar frequency in both treatment groups (74.3% for odevixibat and 70.6% for the placebo group). No patients discontinued treatment due to a TEAE and dose interruptions and reductions due to TEAEs were infrequent (< 9%) in the odevixibat group. Diarrhea was reported in 28.6% versus 5.9% of patients in the odevixibat versus placebo groups, respectively, and the comparative data suggest odevixibat may result in an increase in diarrhea TEAEs when compared with placebo. However, given the lack of an MID for this TEAE, the clinical importance of the increase was unclear. No diarrhea TEAEs were rated as SAEs and none led to treatment discontinuation. ██████████ ████████ in the odevixibat group and || ███████ ██████ in the placebo group met the criteria for clinically significant diarrhea, which was defined as diarrhea lasting 3 or more days that was grade 3 in severity or required treatment for dehydration.
There were no deaths during the 24-week ASSERT study. Treatment-emergent SAEs were reported in 5 patients (14.3%) who received odevixibat and in 2 patients (11.8%) who received placebo (proportion difference of 2.5% for odevixibat versus placebo; 95% CI, −23.0% to 21.6%). The data were too uncertain to draw any conclusions regarding the effect of odevixibat on SAEs when compared with placebo.
The controlled trial data were limited to a single 24-week study in which 35 patients received odevixibat. The sponsor-submitted 72-week LTE study (N = 50) provided additional safety data. Overall, the safety profile of odevixibat in the ASSERT-EXT study was consistent with the randomized study, with similar safety signals observed. █████████████ ███ ████████████████ █████ ███ ███ █ ████████ ████████ ████ ██ ███ ███ ███ ██ ██████████ ███ ███ ███ ████ ██████ ████ █████ ████ ██████ ██ ██████████ ████ ███ █████ ████████ ██ █████ ██ ███████████████ ██ ███ ███ ████ █████ Rates of notable harms, as well as treatment interruptions and dose reductions, were comparable with the RCT. One patient discontinued treatment due to a TEAE of elevated bilirubin during the extension study. No patients died during the extension study.
No direct or indirect evidence comparing odevixibat with maralixibat was submitted by the sponsor; thus, the relative safety of the 2 drugs is unknown.
Conclusion
Based on a double-blind RCT in pediatric patients with ALGS, elevated sBA levels, and significant pruritus, odevixibat may result in a reduction in caregiver-reported pruritus severity at 6 months, compared with placebo, when used in addition to SOC treatments. The impact of odevixibat on patient-reported pruritus severity and HRQoL (measured using the PedsQL parent report total score), were of low and very low certainty, respectively. Odevixibat likely reduces sBA levels compared with placebo, but the clinical relevance of the sBA reduction observed was unclear because the change in sBA levels that results in an improvement in clinical outcomes has yet to be defined for patients with ALGS.
During the RCT and the LTE study, no deaths were reported and 1 patient underwent a liver transplant; however, due to the limited sample size and treatment duration, no conclusions can be drawn on the impact of odevixibat on mortality or the need for a liver transplant.
While TEAEs were commonly reported by patients in both the odevixibat and placebo groups, no patients stopped therapy due to AEs. Diarrhea and SAEs were identified as important harms. Odevixibat may result in an increase in diarrhea TEAEs when compared with placebo; however, the clinical importance of the increase was unclear. The evidence was too uncertain to draw conclusions on the impact of odevixibat on the frequency of SAEs compared with placebo.
The evidence was limited to one 24-week placebo-controlled RCT (N = 52) and one 72-week open-label, uncontrolled LTE study (N = 50) and, thus, longer-term efficacy and safety are uncertain. No indirect evidence comparing odevixibat with maralixibat, the main comparator, was submitted because the sponsor-conducted feasibility assessment concluded that an indirect treatment comparison was unlikely to provide robust results. Therefore, the comparative efficacy and safety of odevixibat relative to maralixibat are unknown.
References
1.Drug Reimbursement Review sponsor submission: odevixibat (Bylvay) 200 mcg, 400 mcg, 600 mcg and 1200 mcg oral capsules [internal sponsor's package]. Medison Pharma Canada Inc; March 17, 2025.
2.Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology. 1999;29(3):822-9. doi:10.1002/hep.510290331 PubMed
3.Hoffenberg EJ, Narkewicz MR, Sondheimer JM, Smith DJ, Silverman A, Sokol RJ. Outcome of syndromic paucity of interlobular bile ducts (Alagille syndrome) with onset of cholestasis in infancy. J Pediatr. 1995;127(2):220-4. doi:10.1016/s0022-3476(95)70298-9 PubMed
4.Subramaniam P, Knisely A, Portmann B, et al. Diagnosis of Alagille syndrome-25 years of experience at King's College Hospital. J Pediatr Gastroenterol Nutr. 2011;52(1):84-9. doi:10.1097/MPG.0b013e3181f1572d PubMed
5.Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet. 2012;20(3):251-7. doi:10.1038/ejhg.2011.181 PubMed
6.Kamath BM, Ye W, Goodrich NP, et al. Outcomes of Childhood Cholestasis in Alagille Syndrome: Results of a Multicenter Observational Study. Hepatol Commun. 2020;4(3):387-398. doi:10.1002/hep4.1468 PubMed
7.Diaz-Frias J, Kondamudi N. Alagille Syndrome [sponsor supplied reference]. Accessed November 15, 2024. https://www.ncbi.nlm.nih.gov/books/NBK507827/
8.Danks DM, Campbell PE, Jack I, Rogers J, Smith AL. Studies of the aetiology of neonatal hepatitis and biliary atresia. Arch Dis Child. 1977;52(5):360-7. doi:10.1136/adc.52.5.360 PubMed
9.Langlois PH, Scheuerle AE. Descriptive epidemiology of birth defects thought to arise by new mutation. Birth Defects Res A Clin Mol Teratol. 2015;103(11):913-27. doi:10.1002/bdra.23412 PubMed
10.Leonard LD, Chao G, Baker A, Loomes K, Spinner NB. Clinical utility gene card for: Alagille Syndrome (ALGS). Eur J Hum Genet. 2014;22(3)doi:10.1038/ejhg.2013.140 PubMed
11.Powell JE, Keffler S, Kelly DA, Green A. Population screening for neonatal liver disease: potential for a community-based programme. J Med Screen. 2003;10(3):112-6. doi:10.1177/096914130301000303 PubMed
12.Alagille D, Estrada A, Hadchouel M, Gautier M, Odièvre M, Dommergues JP. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): review of 80 cases. J Pediatr. 1987;110(2):195-200. doi:10.1016/s0022-3476(87)80153-1 PubMed
13.Deprettere A, Portmann B, Mowat AP. Syndromic paucity of the intrahepatic bile ducts: diagnostic difficulty; severe morbidity throughout early childhood. J Pediatr Gastroenterol Nutr. 1987;6(6):865-71. doi:10.1097/00005176-198711000-00008 PubMed
14.Kamath BM, Podkameni G, Hutchinson AL, et al. Renal anomalies in Alagille syndrome: a disease-defining feature. Am J Med Genet A. 2012;158a(1):85-9. doi:10.1002/ajmg.a.34369 PubMed
15.Kamath BM, Spinner NB, Emerick KM, et al. Vascular anomalies in Alagille syndrome: a significant cause of morbidity and mortality. Circulation. 2004;109(11):1354-8. doi:10.1161/01.Cir.0000121361.01862.A4 PubMed
16.Lykavieris P, Hadchouel M, Chardot C, Bernard O. Outcome of liver disease in children with Alagille syndrome: a study of 163 patients. Gut. 2001;49(3):431-5. doi:10.1136/gut.49.3.431 PubMed
17.Narula P, Gifford J, Steggall MA, et al. Visual loss and idiopathic intracranial hypertension in children with Alagille syndrome. J Pediatr Gastroenterol Nutr. 2006;43(3):348-52. doi:10.1097/01.mpg.0000221895.51748.44 PubMed
18.Quiros-Tejeira RE, Ament ME, Heyman MB, et al. Variable morbidity in alagille syndrome: a review of 43 cases. J Pediatr Gastroenterol Nutr. 1999;29(4):431-7. doi:10.1097/00005176-199910000-00011 PubMed
19.Dull MM, Kremer AE. Newer approaches to the management of pruritus in cholestatic liver disease. Curr Hepatol Rep. 2020;19:86-95.
20.Medison Pharma Canada Inc. Bylvay (odevixibat): 200 mcg, 400 mcg, 600 mcg and 1200 mcg, oral capsules [product monograph] [sponsor supplied reference]. 2024. Updated July 22, 2025. Accessed July 23, 2025.
21.CADTH Drug Reimbursement Expert Review Committee final recommendation: maralixibat (Livmarli - Mirum Pharmaceuticals Inc.) [sponsor supplied reference]. CADTH; May 9, 2024. https://www.cda-amc.ca/sites/default/files/DRR/2024/SR0780%20Livmarli_Rec.pdf
22.Varni JW, Burwinkle TM, Seid M, Skarr D. The PedsQL 4.0 as a pediatric population health measure: feasibility, reliability, and validity. Ambul Pediatr. 2003;3(6):329-41. doi:10.1367/1539-4409(2003)003<0329:tpaapp>2.0.co;2 PubMed
23.Gwaltney C, Ivanescu C, Karlsson L, Warholic N, Kjems L, Horn P. Validation of the PRUCISION Instruments in Pediatric Patients with Progressive Familial Intrahepatic Cholestasis. Adv Ther. 2022;39(11):5105-5125. doi:10.1007/s12325-022-02262-7 PubMed
24.Medison Pharma Canada response to April 17, 2025 CDA-AMC request for additional information regarding Bylvay (odevixibat) CDA-AMC review: Psychometric Analysis Report: Study A4250-012 [internal sponsor's report]. Albireo Pharma Inc; November 7, 2022.
25.Gwaltney C, Bean S, Venerus M, et al. Development of the Patient- and Observer-Reported PRUCISION Instruments to Assess Pruritus and Sleep Disturbance in Pediatric Patients with Cholestatic Liver Diseases. Adv Ther. 2022;39(11):5126-5143. doi:10.1007/s12325-022-02261-8 PubMed
26.Varni JW, Limbers CA, Burwinkle TM. Parent proxy-report of their children's health-related quality of life: an analysis of 13,878 parents' reliability and validity across age subgroups using the PedsQL 4.0 Generic Core Scales. Health Qual Life Outcomes. 2007;5:2. doi:10.1186/1477-7525-5-2 PubMed
27.Tehranian S, Jafari S, Yousofi J, et al. Health-related quality of life (HRQOL) in children with chronic liver disease in North East Iran using PedsQL™ 4.0. Electron Physician. 2015;7(4):1214-9. doi:10.14661/2015.1214-1219 PubMed
28.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi:10.1016/j.jclinepi.2010.07.015 PubMed
29.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi:10.1016/j.jclinepi.2019.10.014 PubMed
30.Qureshi R, Mayo-Wilson E, Li T. Harms in Systematic Reviews Paper 1: An introduction to research on harms. J Clin Epidemiol. 2022;143:186-196. doi:10.1016/j.jclinepi.2021.10.023 PubMed
31.Clinical Study Report: A4250-012. A Phase 3 Double-Blind, Randomized, Placebo-Controlled Study of the Safety and Efficacy of Odevixibat (A4250) in Patients with Alagille Syndrome (ASSERT) [internal sponsor's report]. Albireo; November 10, 2022.
32.Medison Pharma Canada response to April 17, 2025 CDA-AMC request for additional information regarding Bylvay (odevixibat) CDA-AMC review [internal sponsor's report]. Medison Pharma Canada Inc; April 28, 2025.
33.Kamath BM, Baker A, Houwen R, Todorova L, Kerkar N. Systematic Review: The Epidemiology, Natural History, and Burden of Alagille Syndrome. J Pediatr Gastroenterol Nutr. 2018;67(2):148-156. doi:10.1097/mpg.0000000000001958 PubMed
34.Krantz ID, Piccoli DA, Spinner NB. Alagille syndrome. J Med Genet. 1997;34(2):152-7. doi:10.1136/jmg.34.2.152 PubMed
35.Zhou B, Lin W, Long Y, et al. Notch signaling pathway: architecture, disease, and therapeutics. Signal Transduct Target Ther. 2022;7(1):95. doi:10.1038/s41392-022-00934-y PubMed
36.Zong Y, Panikkar A, Xu J, et al. Notch signaling controls liver development by regulating biliary differentiation. Development. 2009;136(10):1727-39. doi:10.1242/dev.029140 PubMed
37.Alagille Syndrome [sponsor supplied reference]. Accessed October 1, 2024. https://rarediseases.org/rare-diseases/alagille-syndrome/
38.Vandriel SM, Li LT, She H, et al. Natural history of liver disease in a large international cohort of children with Alagille syndrome: Results from the GALA study. Hepatology. 2023;77(2):512-529. doi:10.1002/hep.32761 PubMed
39.Murillo Perez CF, Vandriel SM, Wang JS, et al. Serum bile acids are associated with native liver survival in patients with Alagille Syndrome: Results from the GALA Study Group. presented at: The Liver Meeting; 2023; Boston, MA. Session Abstract No. 121.
40.Livmarli (maralixibat) [sponsor supplied reference]. pan-Canadian Pharmaceutical Alliance. Accessed July 24, 2025, https://www.pcpacanada.ca/negotiation/22506
41.Emerick KM, Whitington PF. Partial external biliary diversion for intractable pruritus and xanthomas in Alagille syndrome. Hepatology. 2002;35(6):1501-6. doi:10.1053/jhep.2002.33332 PubMed
42.Mattei P, von Allmen D, Piccoli D, Rand E. Relief of intractable pruritus in Alagille syndrome by partial external biliary diversion. J Pediatr Surg. 2006;41(1):104-7; discussion 104-7. doi:10.1016/j.jpedsurg.2005.10.014 PubMed
43.Kwong AJ, Ebel NH, Kim WR, et al. OPTN/SRTR 2020 Annual Data Report: Liver. Am J Transplant. 2022;22 Suppl 2:204-309. doi:10.1111/ajt.16978 PubMed
44.Shneider BL. Liver transplantation for Alagille syndrome: the jagged edge. Liver Transpl. 2012;18(8):878-80. doi:10.1002/lt.23472 PubMed
45.CADTH Drug Reimbursement Expert Review Committee final recommendation: odevixibat (Bylvay - Medison Pharma Canada Inc.) for progressive familial intrahepatic cholestasis [sponsor supplied reference]. CADTH; March 1, 2024.
46.Mirum Pharmaceuticals Inc. Livmarli (maralixibat): 9.5 mg/mL oral solution [product monograph]. 2023. Updated May 13, 2025. Accessed June 12, 2025. https://pdf.hres.ca/dpd_pm/00071756.PDF
47.Pharmascience Inc. pms-Ursodiol C (urodiol): 250 mg, 500 mg oral tablet [product monograph]. 2006. Updated September 11, 2023. Accessed May 28, 2025. https://pdf.hres.ca/dpd_pm/00072506.PDF
48.Bausch Health Canada Inc. Rofact (rifampin): 150 mg, 300 mg oral capsule [product monograph]. 2019. Updated June 28, 2019. Accessed May 28, 2025. https://pdf.hres.ca/dpd_pm/00051995.PDF
49.Searchlight Pharma Inc. Atarax (hydroxyaine hydrochloride): 10 mg/5 mL oral syrup [product monograph]. 2022. Updated October 11, 2022. Accessed May 29, 2025. https://pdf.hres.ca/dpd_pm/00067726.PDF
50.Kohut T, Loomes, KM. Alagille syndrome. In: Post TW, ed. UpToDate. UpToDate; 2025. Accessed April 5, 2025. www.uptodate.com
51.Diamond T, Kamath BM. Bile acid transport inhibitors in paediatric hepatology: more than just an itch. Nat Rev Gastroenterol Hepatol. 2024;21(12):825-826. doi:10.1038/s41575-024-01008-w PubMed
52.Sponsor Summary of Clinical Evidence - Bylvay - Alagille Syndrome (SR0884) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: odevixibat (Bylvay), 200 mcg, 400 mcg, 600 mcg and 1200 mcg, oral capsules. Medison Pharma Canada Inc; 2025.
53.Sokol RJ, Gonzales EM, Kamath BM, et al. Predictors of 6-year event-free survival in Alagille syndrome patients treated with maralixibat, an ileal bile acid transporter inhibitor. Hepatology. 2023;78(6):1698-1710. doi:10.1097/hep.0000000000000502 PubMed
54.Hansen BE, Vandriel SM, Vig P, et al. Event-free survival of maralixibat-treated patients with Alagille syndrome compared to a real-world cohort from GALA. Hepatology. 2024;79(6):1279-1292. doi:10.1097/hep.0000000000000727 PubMed
55.Ovchinsky N, Aumar M, Baker A, et al. Efficacy and safety of odevixibat in patients with Alagille syndrome (ASSERT): a phase 3, double-blind, randomised, placebo-controlled trial. Lancet Gastroenterol Hepatol. 2024;9(7):632-645. doi:10.1016/s2468-1253(24)00074-8 PubMed
56.Albireo. NCT04674761: Efficacy and Safety of Odevixibat in Patients With Alagille Syndrome (ASSERT) [sponsor supplied reference]. ClinicalTrials.gov. Accessed December 18, 2024. https://clinicaltrials.gov/study/NCT04674761
57.van Wessel DBE, Thompson RJ, Gonzales E, et al. Genotype correlates with the natural history of severe bile salt export pump deficiency. J Hepatol. 2020;73(1):84-93. doi:10.1016/j.jhep.2020.02.007 PubMed
58.Harpavat S, Hawthorne K, Setchell KDR, et al. Serum bile acids as a prognostic biomarker in biliary atresia following Kasai portoenterostomy. Hepatology. 2023;77(3):862-873. doi:10.1002/hep.32800 PubMed
59.World Health Organization. Child Growth Standards [sponsor supplied reference]. Accessed October 1, 2024. https://www.who.int/tools/child-growth-standards
60.Varni JW, Sherman SA, Burwinkle TM, Dickinson PE, Dixon P. The PedsQL Family Impact Module: preliminary reliability and validity. Health Qual Life Outcomes. 2004;2:55. doi:10.1186/1477-7525-2-55 PubMed
61.Desai AD, Zhou C, Stanford S, Haaland W, Varni JW, Mangione-Smith RM. Validity and responsiveness of the pediatric quality of life inventory (PedsQL) 4.0 generic core scales in the pediatric inpatient setting. JAMA Pediatr. 2014;168(12):1114-21. doi:10.1001/jamapediatrics.2014.1600 PubMed
62.Guyatt GH, Oxman AD, Kunz R, et al. GRADE guidelines 6. Rating the quality of evidence—imprecision. J Clin Epidemiol. 2011;64(12):1283-1293. doi:10.1016/j.jclinepi.2011.01.012 PubMed
63.Clinical Study Report: A4250-015. An Open Label Study to Evaluate the Long-term Safety and Efficacy of Odevixibat (A4250) in Patients with Alagille Syndrome (ASSERT-EXT) [internal sponsor's report]. Albireo; July 8, 2024.
64.Medison Pharma Canada response to April 3, 2025 CDA-AMC request for additional information regarding Bylvay (odevixibat) CDA-AMC review [internal sponsor's report]. Medison Pharma Canada Inc; April 8, 2025.
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Table 24: Summary of Pruritus Responder Analyses — ASSERT Study (FAS)
Variable | Odevixibat (N = 35) | Placebo (N = 17) |
|---|---|---|
Patients achieving at least a 1.5-point decrease in the a.m. and p.m. PRUCISION ObsRO scratching scores at month 6 (week 21 to 24) (primary analysis)a | ||
Number of patients contributing to the analysis | 35 | 17 |
Number of responders (%)b | 19 (54.3) | 3 (17.6) |
Proportion difference (95% CI) (odevixibat vs. placebo)b | 0.366 (0.0514 to 0.5874) | |
OR (95% CI) (odevixibat vs. placebo)c | 5.15 (1.17 to 34.16) | |
One-sided P valued | 0.0071 | |
Patients achieving at least a 1.0-point decrease in the (a.m. and p.m.) PRUCISION ObsRO scratching scores at month 6 (week 21 to 24) (sensitivity analysis)a | ||
Number of patients contributing to the analysis | 35 | 17 |
Number of responders (%)b | 28 (80.0) | 6 (35.3) |
Proportion difference (95% CI) (odevixibat vs. placebo)b | 0.447 (0.1025 to 0.6862) | |
OR (95% CI) (odevixibat vs. placebo)c | 8.31 (1.82 to 44.48) | |
One-sided P valued | 0.0006 | |
Patients achieving at least a 1.0-point decrease in the a.m. PRUCISION ObsRO scratching score at week 24 (post hoc analysis for pharmacoeconomic review)e | ||
Number of patients contributing to the analysis | ██ | ██ |
Number of responders (%) | ██ ██████ | | ██████ |
Proportion difference (95% CI) (odevixibat vs. placebo)b | █████ ███████ ██ ████████ | |
OR (95% CI) (odevixibat vs. placebo) | ████ █████ ██ ██████ | |
One-sided P valuef | ██████ | |
CI = confidence interval; FAS = full analysis set; ObsRO = observer-reported outcome; OR = odds ratio; vs. = versus.
aThe monthly responder analyses were calculated based on the total number of patients randomized, using the change from baseline at week 24 (weeks 21 to 24) for the a.m. and p.m. scratching scores of the PRUCISION ObsRO instrument against the threshold of either a 1.5-point or 1.0-point decrease from baseline. All data collected through the end of the study following an intercurrent event due to premature discontinuation of study treatment before week 24 (i.e., treatment policy strategy) was included, while data following intercurrent events of biliary diversion surgery or liver transplant (i.e., hypothetical strategy) were excluded. Patients with missing data at week 24 were classified as nonresponders.
bA Clopper-Pearson exact CI is reported for the percentage of responders, and the exact unconditional CI is reported for the proportion difference without adjusting stratification factors.
cThe exact CI is reported based on Vollset, Hirji, and Elashoff (1991) adjusting baseline age stratification factors.
dThe analysis is based on the Cochran Mantel Haenszel test adjusting baseline age stratification factors. One-sided P value tests that odevixibat is better than placebo. The P value was not controlled for multiple testing.
eThe post hoc analysis of PRUCISION ObsRO score at 24 weeks was based on the average of a.m. scores from day 162 to day 169 using observed data, with no imputation for missing data. Pruritus scores were analyzed using a treatment policy strategy following intercurrent events related to premature discontinuation of study treatment before week 24. Data following intercurrent events such as biliary diversion surgery or liver transplant were excluded, consistent with a hypothetical strategy.
fThe P value was not controlled for multiple testing.
Sources: ASSERT Clinical Study Report.31 Additional data submitted by sponsor April 8, 2025.64
Table 25: Growth-Related Outcomes — ASSERT Study (FAS)
Variable | Odevixibat (N = 35) | Placebo (N = 17) |
|---|---|---|
CFB to week 24 in height z score | ||
Number of patients contributing to the analysis | ██ | ██ |
Baseline, mean (SD) | █████ ███████ | █████ ██████ |
Change from baseline, mean (SD) | ████ ███████ | █████ ██████ |
LS mean (SE)a | ████ ███████ | █████ ██████ |
LS mean difference (95% CI) (odevixibat vs. placebo)a | ████ █████ ██ █████ | |
One-sided P valuea | ██████ | |
CFB to week 24 in weight z score | ||
Number of patients contributing to the analysis | ██ | ██ |
Baseline, mean (SD) | █████ ███████ | █████ ██████ |
Change from baseline, mean (SD) | █████ ███████ | ████ ███████ |
LS mean (SE)a | █████ ███████ | ████ ███████ |
LS mean difference (95% CI) (odevixibat vs. placebo)a | █████ ██████ ██ ██████ | |
One-sided P valuea | ██████ | |
CFB to week 24 in BMI z score | ||
Number of patients contributing to the analysis | ██ | ██ |
Baseline, mean (SD) | █████ ███████ | █████ ██████ |
Change from baseline, mean (SD) | █████ ███████ | ████ ███████ |
LS mean (SE)a | █████ ███████ | ████ ███████ |
LS mean difference (95% CI) (odevixibat vs. placebo)a | █████ ██████ ██ ██████ | |
One-sided P valuea | ██████ | |
BMI = body mass index; CFB = change from baseline; CI = confidence interval; FAS = full analysis set; LS = least squares; MMRM = mixed model for repeated measures; SD = standard deviation; SE = standard error; vs. = versus.
aThe analysis is based on an MMRM using a restricted maximum likelihood with baseline growth data as a covariate, and baseline age stratification, treatment group, visit (in weeks), and treatment-by-visit interaction as fixed effects. The comparison of treatment difference in change from baseline to week 24 was estimated and tested using contrast.
Source: ASSERT Clinical Study Report.31
Table 26: PRUCISION ObsRO Sleep-Related Outcomes — ASSERT Study (FAS)
PRUCISION ObsRO outcomea | Odevixibat (N = 35) | Placebo (N = 17) |
|---|---|---|
CFB to month 6 in daytime tirednessb | ||
Number of patients contributing to the analyses | 34 | 16 |
Baseline, mean (SD) | 2.14 (0.873) | 2.44 (0.865) |
LS mean change from baseline (SE)c | −1.13 (0.147) | −0.53 (0.196) |
LS mean difference (95% CI) (odevixibat vs. placebo)c | −0.60 (−1.06 to −0.14) | |
One-sided P valuec | 0.0062 | |
CFB to month 6 in number of awakenings per night | ||
Number of patients contributing to the analyses | 33 | 16 |
Baseline, mean (SD) | 2.89 (2.336) | 6.18 (8.728) |
LS mean change from baseline (SE)c | −2.70 (0.930) | 0.19 (1.315) |
LS mean difference (95% CI) (odevixibat vs. placebo)c | −2.89 (−6.12 to 0.34) | |
One-sided P valuec | 0.0390 | |
CFB to month 6 in percent of days requiring help falling asleep | ||
Number of patients contributing to the analyses | 33 | 16 |
Baseline, mean (SD) | 63.17 (42.901) | 53.99 (48.065) |
LS mean change from baseline (SE)c | −43.44 (6.739) | −10.09 (9.205) |
LS mean difference (95% CI) (odevixibat vs. placebo)c | −33.35 (−54.86 to −11.85) | |
One-sided P valuec | 0.0016 | |
CFB to month 6 in percent of days requiring soothing during the night | ||
Number of patients contributing to the analyses | 33 | 16 |
Baseline, mean (SD) | 70.74 (39.384) | 59.73 (48.108) |
LS mean change from baseline (SE)c | −46.71 (5.812) | −6.34 (7.929) |
LS mean difference (95% CI) (odevixibat vs. placebo)c | −40.37 (−58.77 to −21.96) | |
One-sided P valuec | < 0.0001 | |
CFB to month 6 in percent of days sleeping with caregiver | ||
Number of patients contributing to the analyses | 33 | 16 |
Baseline, mean (SD) | 44.83 (45.425) | 49.86 (45.055) |
LS mean change from baseline (SE)c | −34.52 (5.357) | −8.27 (7.152) |
LS mean difference (95% CI) (odevixibat vs. placebo)c | −26.25 (−43.27 to −9.23) | |
One-sided P valuec | 0.0017 | |
CFB to month 6 in percent of days seeing blood due to scratching | ||
Number of patients contributing to the analyses | 33 | 16 |
Baseline, mean (SD) | 37.48 (32.003) | 54.72 (35.778) |
LS mean change from baseline (SE)c | −27.95 (4.446) | −18.98 (5.954) |
LS mean difference (95% CI) (odevixibat vs. placebo)c | −8.97 (−23.33 to 5.39) | |
One-sided P valuec | 0.1075 | |
CFB to month 6 in percent of days taking medication to induce sleep | ||
Number of patients contributing to the analyses | 33 | 16 |
Baseline, mean (SD) | 28.74 (43.645) | 23.26 (39.037) |
LS mean change from baseline (SE)c | −2.79 (4.254) | 4.34 (5.782) |
LS mean difference (95% CI) (odevixibat vs. placebo)c | −7.12 (−20.98 to 6.73) | |
One-sided P valuec | 0.1530 | |
CFB = change from baseline; CI = confidence interval; FAS = full analysis set; ICE = intercurrent event; LS = least squares; MMRM = mixed model for repeated measures; ObsRO = observer-reported outcome; SD = standard deviation; SE = standard error; vs. = versus.
aBaseline score was calculated by averaging weekly scores in the 14 days preceding start of treatment. The 6-month (28-day) average score was calculated by averaging 4 weekly scores (week 21 to 24) within the 4-week interval. All data collected through the end of the study following an ICE due to premature discontinuation of study treatment before week 24 (i.e., treatment policy strategy), while excluding data following ICEs of biliary diversion surgery or liver transplant (i.e., hypothetical strategy).
bTiredness severity score: 0 = not tired at all, 1 = a little tired, 2 = medium tired, 3 = very tired, 4 = very, very tired.
cThe analysis was based on an MMRM using a restricted maximum likelihood with baseline score as a covariate, and baseline age stratification, treatment group, time (in months), and treatment-by-time interaction as fixed effects. One-sided P value tests that odevixibat is better than placebo. P values were not adjusted for multiple testing and should be viewed as supportive data.
Source: ASSERT Clinical Study Report.31
Pharmacoeconomic Review
Abbreviations
AE
adverse event
ALGS
Alagille syndrome
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
IBAT
ileal bile acid transporter
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
QALY
quality-adjusted life-year
SOC
standard of care
SSRI
selective serotonin reuptake inhibitor
UDCA
ursodeoxycholic acid
Economic Review
The objective of the economic review is to review and critically appraise the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of odevixibat plus standard of care (SOC) compared with maralixibat plus SOC and SOC alone for the treatment of cholestatic pruritus in patients with Alagille syndrome (ALGS). SOC consisted of a basket of treatments used to control pruritus symptoms such as ursodeoxycholic acid (UDCA), hydroxyzine, rifampicin, cholestyramine, naltrexone and selective serotonin reuptake inhibitors (SSRIs).
Item | Description |
|---|---|
Drug product | Odevixibat (Bylvay), 200 mcg, 400 mcg, 600 mcg, and 1,200 mcg oral capsules |
Indication | For the treatment of cholestatic pruritus in patients aged 12 months or older with ALGS |
Submitted price | Odevixibat:
|
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | July 22, 2025 |
Reimbursement request | As per indication |
Sponsor | Medison Pharma Canada Inc. |
Submission history | Previously reviewed: Yes Indication: For the treatment of pruritus in patients aged 6 months or older with progressive familial intrahepatic cholestasis Recommendation date: February 13, 2024 Recommendation: Reimburse with clinical criteria and/or conditions |
ALGS = Alagille syndrome; NOC = Notice of Compliance.
Summary
Odevixibat is available as 200 mcg, 400 mcg, 600 mcg, and 1,200 mcg oral capsules (swallowed whole or sprinkled on food). At the submitted price of $175.92 per 200 mcg, $351.85 per 400 mcg, $527.77 per 600 mcg, and $1,055.55 per 1,200 mcg capsule, the annual cost of odevixibat is expected to range from $192,769 to $2,313,233 per patient depending on the patient’s weight (i.e., 4.0 kg to > 55.5 kg) based on Health Canada–recommended dosages. The daily dose may be reduced to 40 mcg/kg per day if tolerability issues occur.
Clinical efficacy in the economic analysis for odevixibat plus SOC was derived from the ASSERT trial, which suggests that odevixibat plus SOC may result in a reduction in caregiver-reported pruritus severity at 6 months compared with SOC alone for patients with cholestatic pruritus and ALGS. No indirect evidence comparing odevixibat with maralixibat, the main comparator, was submitted because a feasibility assessment conducted by the sponsor concluded that an indirect treatment comparison (ITC) was unlikely to provide robust results. The sponsor undertook an unadjusted, naive comparison of odevixibat and maralixibat. Therefore, the comparative efficacy and safety of odevixibat plus SOC relative to maralixibat plus SOC are unknown.
The results of the Canada’s Drug Agency (CDA-AMC) base case suggest:
Odevixibat plus SOC will be associated with higher costs to the health care system than SOC alone (incremental costs = $14,171,485), primarily driven by increased drug acquisition costs associated with odevixibat.
Odevixibat plus SOC is predicted to be associated with a gain of 2.85 life-years compared with SOC alone. When the impact on health-related quality of life is also considered, odevixibat plus SOC is predicted to result in a gain of 2.50 quality-adjusted life-years (QALYs) compared with SOC alone.
The incremental cost-effectiveness ratio (ICER) of odevixibat plus SOC compared with SOC alone was $5,678,772 per QALY gained in the CDA-AMC base case. The estimated ICER was highly sensitive to dosing assumptions, such as the distribution of patients across different dose levels and the associated caregiver burden captured using disutility.
Given the lack of direct comparative evidence and limitations with the sponsor’s submitted naive comparison of odevixibat and maralixibat, there is insufficient evidence to justify a price premium for odevixibat plus SOC compared with maralixibat plus SOC.
CDA-AMC estimates that the budget impact of reimbursing odevixibat plus SOC for the treatment of cholestatic pruritus in patients with ALGS will be approximately $211 million over the first 3 years of reimbursement compared with the amount currently spent on SOC alone, with an estimated expenditure of $212 million on odevixibat plus SOC over this period. The actual budget impact of reimbursing odevixibat plus SOC will depend on the dosing of odevixibat in clinical practice and the reimbursement status of maralixibat because the CDA-AMC estimate is based on the current situation in which maralixibat is not funded. The magnitude of uncertainty in the budget impact must be addressed to ensure the feasibility of adoption, given the difference between the sponsor’s estimate and the CDA-AMC estimate.
At the time of writing, maralixibat was not listed by public drug plans in Canada and was under active pan-Canadian Pharmaceutical Alliance negotiations for the treatment of cholestatic pruritus in patients aged 12 months and older with ALGS.1
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of odevixibat plus SOC from the perspective of a public health care payer in Canada over a lifetime horizon (99 years).4 The modelled population was assumed to begin with patients aged 12 months and older with ALGS and cholestatic pruritus, which is aligned with the Health Canada indication. The sponsor’s base-case analysis included costs related to drug acquisition (submitted price for odevixibat and public list prices for comparators), liver transplant and complications, adverse events (AEs), and health care resource use. Additional information about the sponsor’s submission is summarized in Appendix 3.
In the sponsor’s base case, odevixibat plus SOC was associated with incremental costs of $10,171,947 and 4.23 incremental QALYs relative to SOC alone. This resulted in an ICER of $2,405,534 per QALY gained. Maralixibat plus SOC was more costly and less effective than odevixibat plus SOC.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 2; full details are provided in Appendix 4). A revised base case was therefore developed.
Table 2: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The comparative efficacy and safety of odevixibat plus SOC and maralixibat plus SOC are unknown. | There is no direct or robust indirect evidence comparing odevixibat plus SOC with maralixibat plus SOC for the treatment of patients aged 12 months and older with ALGS and cholestatic pruritus. The trials assessing odevixibat and maralixibat compared with SOC were notably different. As such, no definitive conclusions could be drawn regarding the comparative efficacy and safety of odevixibat plus SOC relative to maralixibat plus SOC. | This issue could not be addressed. Given limitations with the sponsor’s naive comparison, it is uncertain whether odevixibat plus SOC provides a net benefit over maralixibat plus SOC for the treatment of patients aged 12 months and older with ALGS and cholestatic pruritus. | No scenario analysis was conducted owing to a lack of robust clinical evidence. |
The long-term treatment efficacy of odevixibat plus SOC compared with SOC is uncertain. | Approximately 58% of the sponsor’s estimated incremental QALYs were derived after the maximum follow-up period (i.e., 24 weeks) of the randomized double-blinded phase of the ASSERT trial. Although the ASSERT-EXT study supported the durability of treatment effect, there were serious methodological concerns with the open-label design of the study that precluded drawing definitive conclusions about the long-term efficacy of odevixibat plus SOC. | This issue could not be addressed. Given the absence of long-term efficacy data, the estimated incremental QALYs and costs associated with odevixibat plus SOC compared with SOC alone are uncertain. | No scenario analysis was conducted owing to a lack of long-term comparative data. |
The dosing of odevixibat is uncertain. | Although odevixibat doses exceeding the standard dose were observed in the sponsor’s internally collected data from an expanded access program, the model was not programmed to allow higher doses. The modelled distribution of patients on a low dose of odevixibat did not align with the trial evidence that suggested fewer patients received low-dose odevixibat. | CDA-AMC aligned the distribution of patients on a low dose of odevixibat with that observed in the ASSERT trial. CDA-AMC could not explore the impact of incorporating doses greater than the standard dose of odevixibat due to limitations with the model structure. | No scenario analysis was conducted due to high uncertainty in dosing assumptions, which made it challenging to define alternative values, and due to limitations of the model’s structure. |
The caregiver disutility is not appropriately captured or applied. | The sponsor’s approach for calculating the disutility for caregivers is inappropriate. In addition, the caregiver disutility should not be captured in the publicly funded health care payer perspective, given the indication is for patients, and such impacts are to be captured in nonreference-case analyses. | CDA-AMC removed caregiver disutilities in reanalysis. | No scenario analysis was conducted because the CDA-AMC base case used the appropriate methods for the analysis. |
The model structure does not appropriately capture the treatment paradigm of ALGS. | According to expert input, the modelled treatment paradigm is highly simplified. It does not capture the progression of underlying liver disease and the complexity of transplant-related treatment decisions. | This issue could not be addressed. | No scenario analysis was conducted owing to the limitations in the model’s structure. |
ALGS = Alagille syndrome; CDA-AMC = Canada’s Drug Agency; QALY = quality-adjusted life-year; SOC = standard of care.
Note: Full details of the issues identified by CDA-AMC are provided in Appendix 3.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to Table 6), in consultation with clinical experts. Detailed information about the base case is provided in Appendix 4.
Impact on Health Care Costs
Odevixibat plus SOC is expected to be associated with additional health care costs compared with comparators (incremental costs versus SOC alone = $14,171,485). This increase in health care spending primarily results from drug acquisition costs associated with odevixibat plus SOC (refer to Figure 1).
Impact on Health
Relative to comparators, odevixibat plus SOC is predicted to increase the amount of time spent in the “Response” health state by approximately 8 years, increase time to liver transplant by approximately 10 years, and increase survival by approximately 3 years per patient over the lifetime horizon (refer to Figure 2). Considering the impact on both quality and length of life, odevixibat plus SOC is expected to result in 2.50 additional QALYs per patient compared with SOC alone.
Figure 1: Impact of Odevixibat Plus SOC vs. SOC on Health Care Costs
SOC = standard of care; vs. = versus.
Note: Other costs include costs related to liver transplant, post-transplant complications, and adverse events. A detailed cost breakdown for maralixibat plus SOC is provided in Appendix 4, Table 9.
Figure 2: Impact of Odevixibat Plus SOC vs. SOC on Patient Health
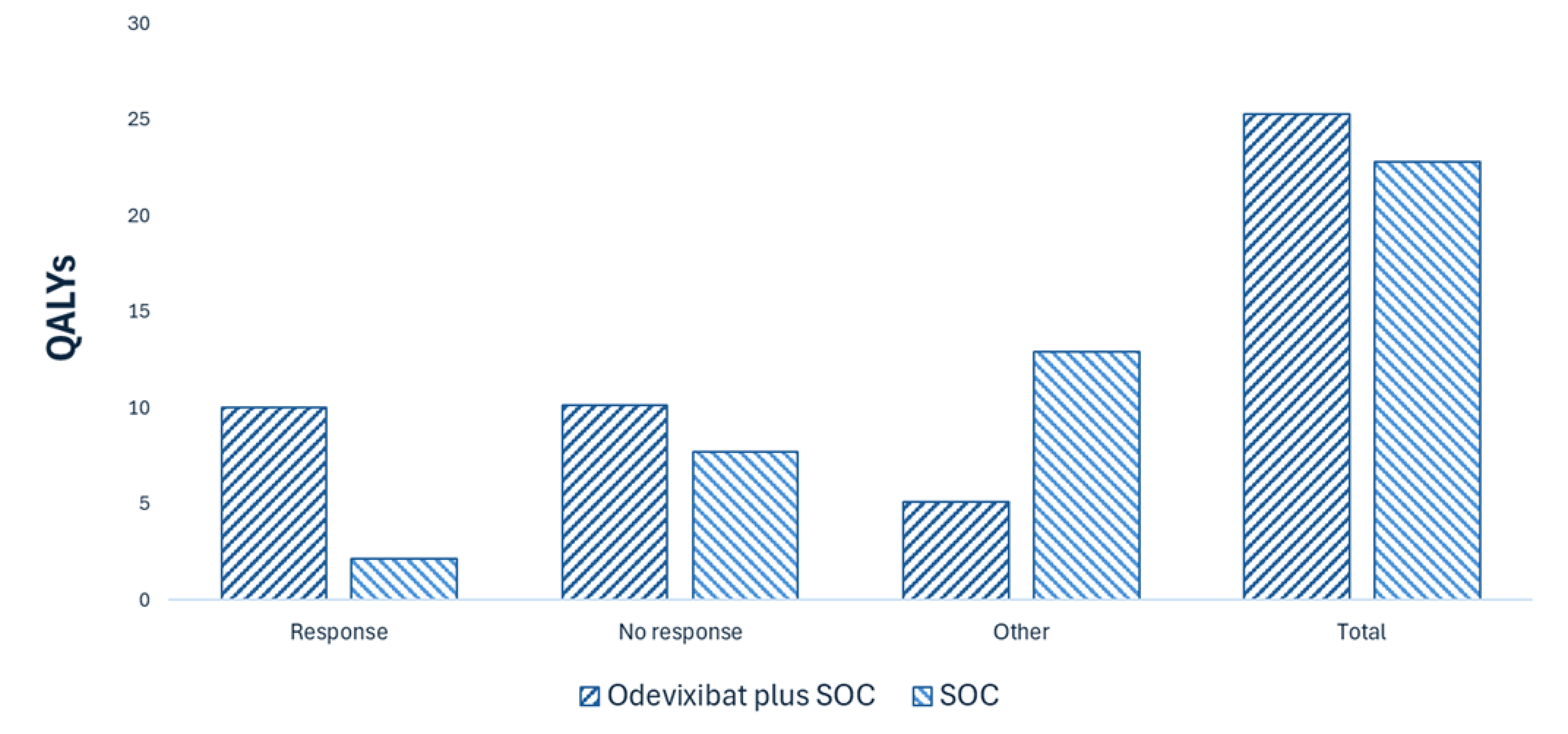
QALY = quality-adjusted life-year; SOC = standard of care; vs. = versus.
Note: “Other” health state QALYs include QALYs gained in health states characterized by liver transplant and post-transplant complications. A detailed QALY breakdown for maralixibat plus SOC is provided in Appendix 4, Table 9.
Overall Results
The results of the CDA-AMC base case suggest that odevixibat plus SOC is more effective (incremental QALYs: 2.50) and more costly (incremental costs: $14,171,485) compared with SOC alone, resulting in an ICER of $5,678,772 per QALY gained. Additional details on the CDA-AMC base case are available in Appendix 4.
Table 3: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
SOC | 291,707 | 22.77 | Reference |
Odevixibat plus SOC | 14,463,192 | 25.26 | 5,678,772 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Note: Publicly available list prices were used for all comparators.
Uncertainty and Sensitivity
Due to the lack of direct or robust indirect evidence, the comparative efficacy and safety of odevixibat with maralixibat are unknown. As a result, there is insufficient evidence to justify a price premium for odevixibat plus SOC compared with maralixibat plus SOC.
The long-term efficacy and safety of odevixibat plus SOC compared with SOC alone are uncertain due to the lack of long-term comparative data.
The model structure did not fully capture the current treatment paradigm for ALGS, particularly in relation to liver transplant–related treatment decisions. This may have led to an overestimation of the cost savings associated with transplant.
Summary of the Budget Impact
The sponsor submitted a budget impact analysis (BIA) to estimate the 3-year (2026 to 2028) budget impact of reimbursing odevixibat for use in the Health Canada–indicated population. The sponsor assumed that the payer would be the CDA-AMC participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The price of odevixibat was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on the publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in Appendix 5.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (Appendix 5). CDA-AMC estimated that 172 patients would be eligible for treatment with odevixibat over a 3-year period (year 1 = 164; year 2 = 168; year 3 = 172), 89 of whom are expected to receive odevixibat plus SOC (year 1 = 82; year 2 = 85; year 3 = 89). The estimated incremental budget impact of reimbursing odevixibat plus SOC is expected to be approximately $211 million over the first 3 years, with an expected expenditure of $212 million on odevixibat. The CDA-AMC estimate is based on the currently available situation in which maralixibat is not funded. The actual budget impact of reimbursing odevixibat plus SOC will depend on the dosing of odevixibat in clinical practice and the reimbursement status of maralixibat.
Conclusion
Based on the CDA-AMC base case, odevixibat plus SOC would be considered cost-effective at the submitted price if the public health care system were willing to pay at least $5,678,772 for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details are presented in Table 9). The estimated cost-effectiveness of odevixibat plus SOC compared with maralixibat plus SOC is uncertain due to the unknown comparative efficacy and safety. There is insufficient evidence to justify a price premium for odevixibat plus SOC compared with maralixibat plus SOC.
The budget impact on the public drug plans of reimbursing odevixibat plus SOC in the first 3 years is estimated to be approximately $211 million. The 3-year expenditure on odevixibat plus SOC (i.e., not accounting for current expenditures on comparators) is estimated to be $212 million. The estimated budget impact is uncertain due to uncertainty in the expected dosing of odevixibat in clinical practice and the reimbursement status of maralixibat.
Figure 3: Summary of the CDA-AMC Economic Analysis and Price Reduction
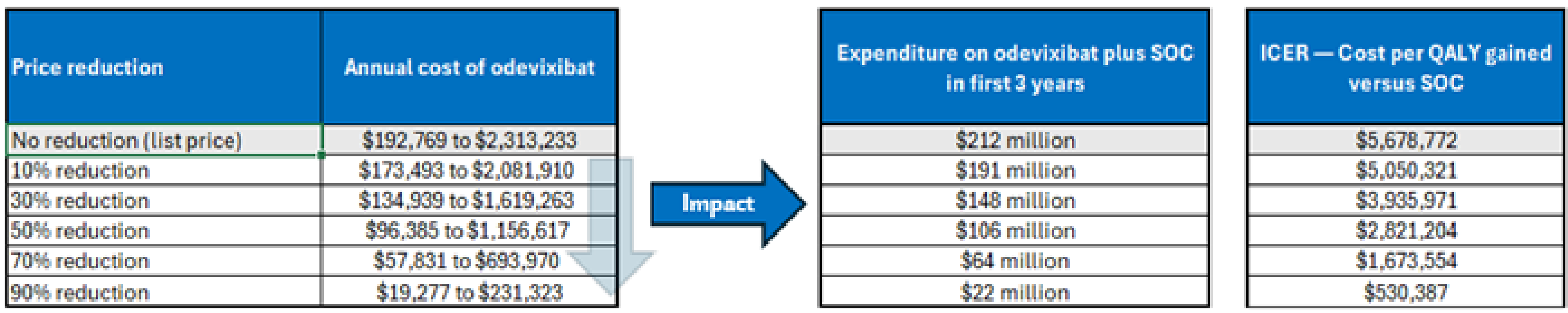
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Note: The annual cost of treatment in the cost table presents a range based on patient weight, while expenditure is derived from the budget impact analysis in which the proportion of patients within each weight category was based on data observed in the ASSERT-EXT study and was extrapolated over the time horizon, resulting in an annual cost of $800,000 to $850,000 with no price reduction. “Expenditure on odevixibat plus SOC” includes only the drug cost of odevixibat. Furthermore, there is no robust evidence to suggest that odevixibat should have a price premium over maralixibat.
References
1.Livmarli (maralixibat) [sponsor provided reference]. pan-Canadian Pharmaceutical Alliance. 2024. Accessed December 1, 2024. https://www.pcpacanada.ca/negotiation/22506
2.Bylvay (Odevixibat): Capsule; 200 mcg, 400 mcg, 600 mcg and 1200 mcg, Oral [Product Monograph] [sponsor provided reference]. Accessed August 15, 2024.
3.Mirum Pharmaceuticals Inc. Livmarli (maralixibat oral solution): solution, 9.5 mg/mL maralixibat (as maralixibat chloride), oral [product monograph]. 2023. Accessed May 13, 2025. https://pdf.hres.ca/dpd_pm/00071756.PDF
4.Medison Pharma Canada Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: odevixibat (Bylvay) 200 mcg, 400 mcg, 600 mcg and 1200 mcg oral capsules [internal sponsor's package]. 2025.
5.CADTH. Drug Reimbursement Review pharmacoeconomic report: Maralixibat (Livmarli) for ALGS. 2024. Accessed April 09, 2025. https://www.cda-amc.ca/sites/default/files/DRR/2024/SR0780-Livmarli-Combined-Report.pdf
6.Ontario Ministry of Health. Ontario drug benefit formulary/comparative drug index. Accessed May 20, 2025. https://www.formulary.health.gov.on.ca/formulary/
7.Pharmascience Inc. pms-Ursodiol C: 250 mg and 500 mg tablets [product monograph]. 2017. Accessed April 7, 2025. https://pdf.hres.ca/dpd_pm/00040988.PDF
8.Sanis Health Inc. Sertraline (sertraline hydrochloride): 25, 50, and 100 mg Capsules [product monograph]. 2010. Accessed April 7, 2025. https://pdf.hres.ca/dpd_pm/00009791.PDF
9.Teva Canada Limited. Revia (naltrexone hydrochloride): tablets, 50 mg [product monograph]. 2015. Accessed April 7, 2025. https://pdf.hres.ca/dpd_pm/00030323.PDF
10.Valeant Canada LP. Rofact (rifampin): capsules, 150 mg and 300 mg [product monograph]. 2018. Accessed April 7, 2025. https://pdf.hres.ca/dpd_pm/00046484.PDF
11.Odan Laboratories Ltd. Cholestyramine-Odan: Light powder, 4g per sachet [product monograph]. 2022. Updated April 07, 2025. https://pdf.hres.ca/dpd_pm/00068840.PDF
12.Grady J. Scratching the Itch: Management of pruritus in cholestatic liver disease. Liver Fellow Network. 2024. Accessed May 20, 2025. https://www.aasld.org/liver-fellow-network/core-series/clinical-pearls/scratching-itch-management-pruritus-cholestatic
13.Gregorio GV, Ball CS, Mowat AP, Mieli-Vergani G. Effect of rifampicin in the treatment of pruritus in hepatic cholestasis. Arch Dis Child. 1993;69(1):141-3. doi:10.1136/adc.69.1.141 PubMed
14.Lindor KD, Bowlus CL, Boyer J, Levy C, Mayo M. Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology. 2019;69(1):394-419. doi:10.1002/hep.30145 PubMed
15.Mayo Foundation for Medical Education and Research. Cholestyramine (oral route). Mayo Clinic. Accessed May 20, 2025. https://www.mayoclinic.org/drugs-supplements/cholestyramine-oral-route/description/drg-20068562
16.Sweigart AJ, Moore A, Kendsersky R, Shah AA, Zook J. Evaluation of the Use of Naltrexone for Cholestatic Pruritus. J Pediatr Pharmacol Ther. 2023;28(6):524-529. doi:10.5863/1551-6776-28.6.524 PubMed
17.Dietitians of Canada. WHO Growth Charts for Canada. 2025. Accessed April 7, 2025. https://www.dietitians.ca/growthcharts
18.BC PharmaCare. Drug shortages. 2025. Accessed May 20, 2025. https://www2.gov.bc.ca/gov/content/health/practitioner-professional-resources/pharmacare/pharmacies/drug-shortage-information
19.Government of Saskatchewan. Saskatchewan Drug Plan: search formulary. 2024. Accessed May 20, 2025. http://formulary.drugplan.ehealthsask.ca/SearchFormulary
20.Diaz-Frias J KNP. Alagille Syndrome [sponsor provided reference]. StatPearls Publishing. 2020. Accessed November 15, 2024. https://www.ncbi.nlm.nih.gov/books/NBK507827/
21.Canada's population estimates: Age and gender, July 1, 2024 [sponsor provided reference]. Accessed December 19, 2024. https://www150.statcan.gc.ca/n1/daily-quotidien/240925/dq240925a-eng.htm
22.Live births by birth weight [sponsor provided reference]. Accessed December 19, 2024. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310042201
23.Clinical Study Report: Protocol A4250-012. A Phase 3 Double-Blind, Randomized, Placebo-Controlled Study of the Safety and Efficacy of Odevixibat (A4250) in Patients with Alagille Syndrome (ASSERT) [Sponsor’s Internal Report] [sponsor provided reference]. 2022.
24.Week 72 Clinical Study Report: Protocol A4250-015. An Open Label Study to Evaluate the Long-term Safety and Efficacy of Odevixibat (A4250) in Patients with Alagille Syndrome (ASSERT-EXT) [Sponsor’s Internal Report] [sponsor supplied reference]. 2024.
25.Gonzales E, Hardikar W, Stormon M, et al. Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): a randomised phase 2 study. The Lancet. 2021;398(10311):1581-1592. PubMed
26.Vandriel SM, Li LT, She H, et al. Natural history of liver disease in a large international cohort of children with Alagille syndrome: Results from the GALA study. Hepatology. 2023;77(2):512-529. doi:10.1002/hep.32761 PubMed
27.Hansen BE, et al. Event-free survival of maralixibat-treated patients with Alagille syndrome compared to a real-world cohort from GALA Hepatology. 2023;9(6):1279-1292. doi:10.1097/HEP.00000000000727 PubMed
28.Kamath BM, Yin W, Miller H, et al. Outcomes of liver transplantation for patients with Alagille syndrome: the studies of pediatric liver transplantation experience. Liver Transpl. 2012;18(8):940-8. doi:10.1002/lt.23437 PubMed
29.Khan KA, Petrou S, Rivero-Arias O, Walters SJ, Boyle SE. Mapping EQ-5D utility scores from the PedsQL™ generic core scales. Pharmacoeconomics. 2014;32:693-706. PubMed
30.Kini SP, DeLong LK, Veledar E, McKenzie-Brown AM, Schaufele M, Chen SC. The impact of pruritus on quality of life: the skin equivalent of pain. Arch Dermatol. 2011;147(10):1153-1156. PubMed
31.Odevixibat for treating progressive familial intrahepatic cholestasis [HST17] Committee papers [sponsor provided reference]. Accessed December 8, 2024. https://www.nice.org.uk/guidance/hst17/resources/odevixibat-for-treating-progressive-familial-intrahepatic-cholestasis-pdf-50216263887301
32.Quadrados L, et al. Patient and caregiver burden associated with caring for a child with Alagille syndrome: Mixed-methods development of health state vignettes [sponsor supplied reference]. ISPOR conference. 2022;
33.Rare disease database: Alagille Syndrome [sponsor provided reference]. National Organization for Rare Disorders. 2024. Accessed December 8, 2024. https://rarediseases.org/rare-diseases/alagille-syndrome/
34.Kronsten V, Fitzpatrick E, Baker A. Management of cholestatic pruritus in paediatric patients with alagille syndrome: the King's College Hospital experience. J Pediatr Gastroenterol Nutr. 2013;57(2):149-54. doi:10.1097/MPG.0b013e318297e384 PubMed
35.Maralixibat (Livmarli): CADTH Reimbursement Recommendation: Indication: For the treatment of cholestatic pruritus in patients with Alagille syndrome [sponsor provided reference]. Canadian Agency for Drugs and Technologies in Health. 2024. Accessed December 1, 2024. https://www.cda-amc.ca/sites/default/files/DRR/2024/SR0780%20Livmarli_Rec.pdf
36.Ontario Drug Benefit Formulary/Comparative Drug Index [sponsor provided reference]. Accessed December 8, 2024. https://www.formulary.health.gov.on.ca/formulary/
37.Webb AN, Lester ELW, Shapiro AMJ, Eurich DT, Bigam DL. Cost-utility analysis of normothermic machine perfusion compared to static cold storage in liver transplantation in the Canadian setting. Am J Transplant. 2022;22(2):541-551. doi:10.1111/ajt.16797 PubMed
38.Interactive Costing Tool [sponsor provided reference]. Accessed December 8, 2024. https://www.nihr.ac.uk/documents/interactive-costing-tool-ict-getting-started/12170
39.Ontario Schedule of Benefits [sponsor provided reference]. 2024. Updated December 18, 2024. https://www.dr-bill.ca/ohip_billing_codes
40.Data on file: PICTURE study final results summary [Sponsor’s Internal Report] [sponsor provided reference]. 2021.
41.Home Care Costs in Ontario: A Complete Breakdown [sponsor provided reference]. 2024. Accessed December 8, 2024. https://www.closingthegap.ca/home-care-costs-in-ontario-a-complete-breakdown
42.Schedule of Benefits for Laboratory Services [sponsor provided reference]. Accessed December 8, 2024. https://www.ontario.ca/files/2024-01/moh-ohip-schedule-of-benefits-laboratory-services-2024-01-24.pdf
43.How Much Does Therapy Cost in Canada [sponsor provided reference]. 2024. Accessed December 8, 2024. https://www.pharecounselling.com/mental-health-blog/how-much-does-therapy-cost-in-canada
44.How Much Does a Dietitian Cost? [sponsor provided reference]. 2024. Accessed December 8, 2024. https://korunutrition.com/dietitian-cost/
45.Murphy G, Farah B, Wong W, et al. CADTH Therapeutic Reviews. Direct-Acting Antiviral Agents for Chronic Hepatitis C Genotype 1 [sponsor provided reference]. 2014.
46.CADTH. Guidelines for the economic evaluation of health technologies: Canada. 4th ed. 2017. Accessed May 15, 2025. https://www.cda-amc.ca/guidelines-economic-evaluation-health-technologies-canada-0
47.York Health Economics Consortium (YHEC). Disutility [online]. University of York. 2016. Accessed May 14, 2025. https://yhec.co.uk/glossary/disutility/
48.Medison Pharma Canada Inc. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: odevixibat (Bylvay) 200 mcg, 400 mcg, 600 mcg and 1200 mcg oral capsules [internal sponsor's package]. 2025.
49.Kamath BM, Ye W, Goodrich NP, et al. Outcomes of childhood cholestasis in Alagille syndrome: results of a multicenter observational study. Hepatology Communications. 2020;4(3):387-398. PubMed
50.Leonard LD, Chao G, Baker A, Loomes K, Spinner NB. Clinical utility gene card for: Alagille Syndrome (ALGS). Eur J Hum Genet. 2014;22(3)doi:10.1038/ejhg.2013.140 PubMed
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and the CDA-AMC participating public drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and, as such, the table may not represent the actual costs to public drug plans.
Table 4: Cost Comparison for Alagille Syndrome
Treatment | Strength and concentration | Form | Price | Recommended dosage | Daily cost | Annual cost |
|---|---|---|---|---|---|---|
Odevixibat (Bylvay) | 200 mcg (sprinkle) 400 mcg (swallow) 600 mcg (sprinkle) 1,200 mcg (swallow) | Oral capsule | $175.9247a $351.8493a $527.7740a $1,055.5480a | 120 mcg per kg administered once daily in the morning, although the PM also suggests dosing is rounded (refer to Table 1 in the PM). If tolerability issues occur in the absence of other causes, the dose may be reduced to 40 mcg per kg per day. Once the tolerability issues stabilize, the dose may be increased to 120 mcg per kg per day, with a maximum daily dose of 7,200 mcg.b | For patients weighing 4 kg to 55.5 kg or greater: $527.77 to $6,333.29b Low dose for patients weighing 4 kg to 55.5 kg or greater: $175.92 to $2,111.10b | For patients weighing 4 kg to 55.5 kg or greater: $192,769 to $2,313,233b Low dose for patients weighing 4 kg to 55.5 kg or greater: $64,256 to $771,078b |
Ileal bile acid transporter | ||||||
Maralixibat (Livmarli) | 9.5 mg/mL | 30 mL vial Solution for oral administration | $1,787.0000 per mLc or $188.1052 per mgc | Week 1: 190 mcg per kg daily Week 2 and onward: 380 mcg per kg daily up to 28.5 mg (or 3 mL) daily for patients weighing more than 70 kg | First year: $353.98 (5 kg patient) to $5,309.63 (75 kg patient)d Subsequent years: $357.40 (5 kg patient) to $5,361.00 (75 kg patient)d | First year: $129,289 (5 kg patient) to $1,939,342 (75 kg patient)d Subsequent years: $130,540 (5 kg patient) to $1,958,105 (75 kg patient)d |
PM = product monograph.
Note: Dosing of odevixibat and maralixibat was obtained from their respective PMs.2,3
aSponsor-submitted price.4
bTable 1 in the odevixibat PM caps the recommended weight-based dosage at 7,200 mcg daily for patients weighing 55.5 kg and greater.2 As such, the cost range is estimated for a patient with a maximum weight of at least 55.5 kg.
cPrice obtained from the CADTH Reimbursement Review of maralixibat (Livmarli).5
dThe cost range is estimated for a patient with a minimum weight of 5 kg and a maximum weight of 75 kg.
Table 5: Cost Comparison for Alagille Syndrome — Off-Label Treatments
Treatment | Strength | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Cholestyramine | 4,000 mg | Light powder in vial | 0.9217a | 4,000 to 24,000 mg daily | 0.92 to 5.53 | 337 to 2,020 |
Naltrexone (generic) | 50 mg | Tablet | 2.8075 | Pediatric patients: 0.25 to 5 mg per kg daily Adults: 12.5 mg to 50 mg daily | Pediatric patients: 2.81 to 11.23 Adults: 2.81 | Pediatric patients: 1,025 to 4,102 Adults: 1,025 |
Rifampin (Rofact) | 150 mg 300 mg | Capsule | 0.8514b 1.3403b | Pediatric patients: 4 mg to 10 mg per kg daily Adults: 150 mg to 300 mg twice daily | Pediatric patients: 0.85 to 2.19 Adults: 0.85 to 1.34 | Pediatric patients: 311 to 801 Adults: 311 to 490 |
Ursodeoxycholic acid (Ursodiol generics) | 250 mg 500 mg | Tablet | 0.3818 0.7242 | 10 mg to 20 mg per kg daily | 0.72 to 2.55 | 265 to 933 |
Selective serotonin reuptake inhibitor | ||||||
Sertraline (generic) | 25 mg 50 mg 100 mg | Capsule | 0.1516 0.3032 0.3303 | Pediatric patients: 1 mg to 4 mg per kg per day Adults: 75 mg to 100 mg daily | Pediatric patients: 0.30 to 0.63 Adults: 0.33 | Pediatric patients: 111 to 231 Adults: 121 |
Note: All prices are from the Ontario Drug Benefit Formulary (accessed May 20, 2025),6 unless otherwise indicated, and do not include dispensing fees. Formulation information was obtained from respective product monographs7-11 and the dosage was sourced from published literature.12-16 Annual costs assume 365.25 days. A weight of 31.5 kg and 76 kg was estimated for the pediatric and adult populations, respectively.17
According to the expert input received for this review, hydroxyzine and selective serotonin reuptake inhibitors are not commonly used to treat pruritus in clinical practice. Other selective serotonin reuptake inhibitors, such as citalopram, escitalopram, fluoxetine, paroxetine, and vortioxetine, were not identified as being used in clinical practice in Canada.
aDue to a drug shortage, US-labelled cholestyramine for oral suspension sachets have been temporarily added to regular benefit.18
bSaskatchewan drug formulary,19 accessed May 20, 2025. Coverage is restricted to treatment of pruritus in patients with cholestatic liver disease who do not experience a response to or are intolerant to cholestyramine.
cAccording to published literature and the expert input received for this review,12 ursodeoxycholic acid is the mainstay treatment for treating cholestasis rather than pruritus.
Appendix 2: Input Relevant to the Economic Review
Please note that this appendix has not been copy-edited.
This section is a summary of the input received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from Alagille Syndrome Alliance through surveys, focus groups, and interviews with patients and caregivers in Canada and worldwide. Patients with ALGS reported symptoms such as jaundice, severe pruritus (itching), and nutritional challenges (malabsorption and poor growth). Itch, in particular, was highlighted as a significant concern, often leading to sleep disturbances and fatigue. These challenges collectively had a negative impact on mental health and overall quality of life and made it difficult for patients and caregivers to carry out daily activities. Current treatments reported by patient input included rifampin, hydroxyzine, cholestyramine, naltrexone, ursodiol, nonprescription remedies (such as lotions and Benadryl), surgical interventions (internal or external biliary diversion and liver transplant), and maralixibat. Patient input indicated that odevixibat would be an alternative option for those who do not experience a response to, or lose response to, existing therapies. Treatment goals included meaningful and consistent relief from severe pruritus (i.e., reduction in itch intensity and frequency). Caregivers of 2 children (1 child was aged 3 years; the age of the other child was not reported) reported having experience with odevixibat. One of the caregivers noted a reduction in the number of medications for their child, and the other caregiver reported no side effects of odevixibat. Patient input also indicated that, based on their interactions with patients and families, odevixibat effectively reduced or eliminated pruritus (which led to uninterrupted sleep and restored energy levels), improved growth, and relieved the emotional toll of the disease. However, the impact of odevixibat was sustained only while patients remained on treatment. Symptoms such as pruritus returned upon treatment discontinuation. The side effects reported included mild to moderate gastrointestinal discomfort, cramping, and diarrhea.
Clinician input was received from the Canadian Pediatric Hepatology Research Group, a committee of the Canadian Association for the Study of the Liver, which was gathered through interviews with clinical experts and published literature. The current standard of treatment for ALGS aims to improve symptoms and delay the need for a liver transplant rather than target the underlying disease mechanism of bile duct paucity. The management of pruritus included off-label treatments such as antihistamines (used as first-line therapy), UDCA, cholestyramine, rifampicin, naltrexone, hydroxyzine, and SSRIs (such as sertraline). Clinicians anticipated the use of odevixibat, in addition to SOC, in patients with ALGS who have persistent pruritus on treatment with UDCA, antihistamines, and rifampin. Odevixibat would be considered for patients whose pruritus is persistent while on existing medications and may be considered in patients who were adequately controlled on existing medications. Treatment goals included reducing pruritus, improving long-term liver disease outcomes and delaying or preventing the need for liver transplant. Treatment response would be assessed using patient-reported reduction in severity of pruritus and sleep disturbance as well as examination of the skin for excoriations. Treatment should be discontinued if liver disease progresses, there is a liver transplant, or intolerable AEs. Odevixibat should be administered in a pediatric gastroenterologist or hepatologist in a specialty clinic setting.
Input from the CDA-AMC participating drug plans flagged that there is no comparison with maralixibat plus SOC, which received a positive recommendation. The input also questioned whether treatment may be switched between odevixibat and maralixibat. Additionally, the drug plans noted significant variability in the estimated budget impact, largely attributed to dosing assumptions.
Several of these concerns were addressed in the sponsor’s model:
Treatment response was assessed as a reduction in pruritus.
CDA-AMC addressed some of these concerns as follows:
The impact of uncertainty in odevixibat dosing on the cost-effectiveness and budget impact of odevixibat plus SOC was explored.
CDA-AMC was unable to address the following concerns:
The comparative efficacy and safety of odevixibat plus SOC and maralixibat plus SOC are unknown.
Appendix 3: Summary of the Sponsor’s Submission
Please note that this appendix has not been copy-edited.
Summary of the Sponsor’s Economic Evaluation
For the pharmaceutical reviews program, clinical and economic information is submitted to CDA-AMC by the sponsor. The CDA-AMC health economics team reviews the submitted economic information and appraises the information in collaboration with clinical experts and the clinical review team to evaluate key assumptions, influential parameters, and the overall rigour of the economic submission. Based on what the team learns through this process, adjustments may be made to the sponsor’s model to produce the CDA-AMC base case. The CDA-AMC base case represents the team’s current understanding of the clinical condition, clinical evidence currently available, and best interpretation of the economic evidence based on the information provided.
For the review of odevixibat plus SOC, the sponsor provided a cost-utility analysis and a BIA. The sponsor’s economic submission is summarized in Table 6.
Table 6: Key Components of the Sponsor’s Economic Evaluation
Component | Description |
|---|---|
Treatment information | |
Drug under review | Odevixibat (Bylvay), oral capsules (200 mcg, 400 mcg, 600 mcg, and 1,200 mcg)2 |
Submitted price of drug under review |
|
Regimen | The recommended dosage is 120 mcg/kg administered once daily in the morning with a meal. If tolerability issues occur in the absence of other causes, dose may be reduced to 40 mcg/kg/day. Once the tolerability issues stabilize, the dose may be increased to 120 mcg/kg/day.2 In the sponsor’s submission, the modelled total daily dosage ranged from 600 mcg to 7,200 mcg (maximum dosage) for a patient weight of 4.0 kg to 55.5 kg or greater. This was based on the total daily dose required for this patient weight at the Health Canada–recommended dosage of 120 mcg per kg per day. |
Annual cost of drug under review | $133,139 to $1,597,6734 |
Model information | |
Type of economic evaluation | Cost-utility analysis Markov model |
Treatment | Odevixibat plus SOC |
Included comparators |
|
Perspective | Publicly funded health care payer perspective |
Time horizon | Lifetime (99 years) |
Cycle length | Markov model: 1 year |
Modelled population | Patients aged 12 months and older with ALGS and cholestatic pruritus |
Characteristics of modelled population | Derived from epidemiology data on ALGS and population estimates for Canada (baseline age: 1 year; percentage male: 51.90%)20-22 |
Model health states | Markov model:
For additional information, refer to the Model Structure section and Figure 4. |
Data sources | |
Comparative efficacy | The probability of a pruritic response was derived from the following:
|
Natural history and/or clinical pathway |
AEs and complications:
|
Health-related utilities and disutilities |
|
Costs included in the model |
|
Summary of the submitted results | |
Base-case results |
|
Scenario analysis resultsa |
|
AE = adverse event; ALGS = Alagille syndrome; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; ITC = indirect treatment comparison; QALY = quality-adjusted life-year; SOC = standard of care; SSRI = selective serotonin reuptake inhibitor; UDCA = ursodeoxycholic acid.
aResults of scenario analyses that had a meaningful impact on the estimated ICER compared with the sponsor’s base case. Additional scenarios were submitted that had no meaningful impact on the estimated ICER, including adopting a shorter time horizon and alternative discount rates. The results of the sponsor’s model differed from the report; CDA-AMC used the results from the model. For the scenario assuming generic pricing, the results in the pharmacoeconomic report are reported.
Model Structure
The sponsor submitted a Markov state transition model (Figure 4) with annual cycles. Patients entered the Markov model, which simulated the movement of patients through 5 mutually exclusive health states (“Response,” “No Response,” “Liver Transplant,” “Post–Liver Transplant,” and “Death”) over the lifetime horizon. Responders entered the “Response” health state and continued with initial treatment. Nonresponders entered the “No Response” health state and switched to SOC. Responders could remain in their assigned health states or experience the possibility of transitioning to the “No Response” state in each cycle. Patients who did not experience a response to treatment or lost response were at risk of a liver transplant. Patients in the “Liver Transplant” health state discontinued SOC. Following surgical intervention, patients remained in the “Post–Liver Transplant” health state until death. Patients could transition to the “Death” state from all health states at any time.
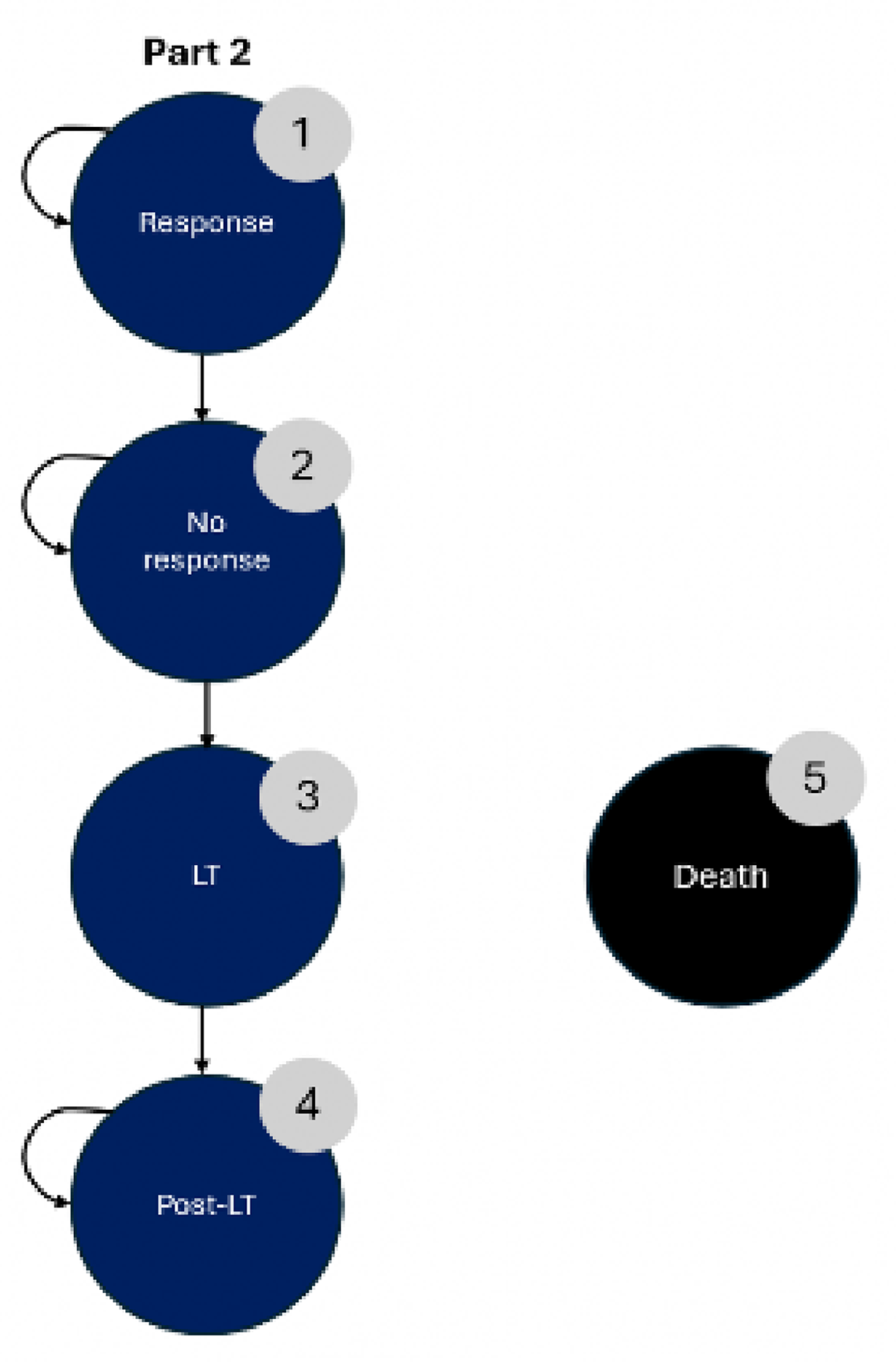
Table 7: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
SOC | 291,796 | 18.81 | Reference |
Odevixibat plus SOC | 10,463,743 | 23.04 | 2,405,534 |
Dominated | |||
Maralixibat plus SOC | 10,728,463 | 22.13 | Dominated by odevixibat plus SOC |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Note: The results of the sponsor’s model differed from the report; CDA-AMC used the results from the model.
Appendix 4: Additional Details of CDA-AMC Reanalyses
Please note that this appendix has not been copy-edited.
Clinical Data in the Economic Model
The CDA-AMC Clinical Review of the ASSERT trial found that odevixibat plus SOC may result in a reduction in the caregiver-reported pruritus severity score at 6 months compared with placebo, when used in addition to SOC treatments. The impact of odevixibat on patient-reported pruritus severity, and health-related quality of life were of low and very low certainty, respectively. The evidence was also too uncertain to draw conclusions on the impact of odevixibat plus SOC on the frequency of serious AEs compared with placebo in addition to SOC. The long-term comparative efficacy and safety of odevixibat plus SOC was also uncertain due to the open-label design of the ASSERT-EXT study. No conclusions could be drawn on the impact of odevixibat on mortality or the need for liver transplant due to limited sample size and treatment duration of the ASSERT and ASSERT-EXT studies. There was no robust indirect evidence comparing odevixibat plus SOC to maralixibat plus SOC, the main comparator. Therefore, the comparative efficacy and safety of odevixibat relative to maralixibat are unknown.
Estimates of relative efficacy for the economic evaluation were obtained from the ASSERT trial, ASSERT-EXT study, and ICONIC trial. The comparative efficacy of odevixibat plus SOC relative to SOC alone was based on data from the ASSERT and ASSERT-EXT. The sponsor incorporated data for maralixibat plus SOC based on a naive indirect comparison of ASSERT and ICONIC trials that did not adjust for baseline differences in patient characteristics and study design. The outcome of interest, as it relates to the economic evaluation, was the probability of pruritus response defined as the proportion of patients achieving a score decrease by 1 point or more on the observer-reported outcome tool based on morning scores only. The sponsor’s economic model predicted that odevixibat plus SOC provides a net benefit over SOC alone and maralixibat plus SOC for the treatment of patients aged 12 months and older with ALGS and cholestatic pruritus. The sponsor’s submitted base case also predicted that odevixibat plus SOC results in a survival advantage and reduced transplant events compared with SOC alone and maralixibat plus SOC.
Key Issues of the Submitted Economic Evaluation
CDA-AMC identified the following key issues with the sponsor’s analysis:
The comparative efficacy and safety of odevixibat plus SOC and maralixibat plus SOC are unknown: In the absence of a direct head-to-head comparison, the sponsor conducted an unadjusted naive ITC of the ASSERT and ICONIC trials to inform the comparative efficacy of odevixibat and maralixibat, respectively, in the pharmacoeconomic model.4,23,25 The sponsor performed a feasibility assessment and concluded that a more robust method, such as a matching-adjusted indirect comparison, was not feasible due to significant differences in study design. The sponsor acknowledged that differences in baseline pruritus severity and response rates for patients on placebo may confound the results of the submitted unadjusted naive ITC. The sponsor’s submitted naive ITC suggested that patients on odevixibat plus SOC may have higher treatment response rates and longer survival compared with those on maralixibat plus SOC. The Clinical Review agreed there was substantial heterogeneity between the ASSERT and ICONIC trials and that an anchored or unanchored indirect comparison was unlikely to provide robust comparative evidence. Overall, there was insufficient evidence to draw any robust conclusions regarding the comparative efficacy of odevixibat plus SOC and maralixibat plus SOC. The expert input received for this review echoed this uncertainty, noting that the lack of robust evidence precluded any definitive statements about their relative efficacy.
Due to the lack of direct comparative evidence and limitations with the sponsor’s submitted unadjusted naive ITC, it is uncertain whether odevixibat plus SOC provides a net benefit over maralixibat plus SOC for the treatment of patients aged 12 months and older with ALGS and cholestatic pruritus. CDA-AMC was unable to address this issue. The results of the sponsor’s submitted cost-utility analysis for odevixibat and maralixibat are subject to a high degree of uncertainty and should be interpreted with caution.
Long-term treatment efficacy of odevixibat plus SOC compared to SOC is uncertain: The CDA-AMC Clinical Review of the ASSERT trial found that odevixibat plus SOC may reduce the severity of caregiver-reported pruritus in children with ALGS compared with SOC alone.23 The randomized, double-blinded period of this study was 24 weeks. The CDA-AMC Clinical Review also found that while the ASSERT-EXT study supported the durability of the treatment effect,24 there were serious methodological concerns with the open-label design of the study that precluded drawing definitive conclusions about long-term treatment efficacy. Approximately 58% of the sponsor’s estimated incremental QALYs were derived after the maximum follow-up period (i.e., 24 weeks) of the ASSERT trial. The lack of strong evidence to support the duration of treatment response beyond the trial period, coupled with the model’s reliance on extrapolation and assumptions of treatment effectiveness beyond 24 weeks, adds uncertainty to the results of the economic analysis of odevixibat plus SOC compared with SOC alone.
This issue could not be addressed by CDA-AMC.
Dosing of odevixibat is uncertain: The sponsor estimated that █████████████ ████ ███████ of the patients receiving odevixibat remain on the lower dose (40 mcg per kg per day) over the time horizon based on internal data from an Expanded Access Program.4 CDA-AMC requested the sponsor to provide detailed information regarding the patient population and breakdown of doses in the cited internal data for validation, which was not shared by the sponsor. In response to this request, the sponsor indicated that most patients in this dataset were residing in Noth America or Europe and that, approximately ███ of patients received a dose greater than the standard dose (120 mcg per kg per day) of odevixibat. Despite internal data showing that some patients received a dose exceeding the standard dose, these high doses were not incorporated in the model. In addition, the product monograph of odevixibat does not clearly specify whether there is an upper limit to dosing, which makes it difficult to determine whether a cap on odevixibat dosing would be used in clinical practice. In the absence of access to the data supporting the modelled distribution of patients on odevixibat doses, CDA-AMC obtained clinical expert feedback which indicated that the proportion of patients on low dose of odevixibat was likely overestimated, because most patients are expected to initiate on standard dose in clinical practice and follow the dosing protocols outlined in the product monograph.
In a reanalysis, CDA-AMC aligned the distribution of patients on a low dose of odevixibat with that observed in the ASSERT trial (i.e., 2.9% of patients on a low dose of 40 mcg per kg per day).23 CDA-AMC could not explore the impact of incorporating a dose greater than the standard dose of odevixibat due or remove the cap on the odevixibat dosing due to limitations with the model structure. If the dose of odevixibat in clinical practice differs to those modelled, the treatment costs associated with odevixibat will not be accurate.
Caregiver disutility is not appropriately captured: The sponsor’s base case, conducted from the perspective of the public health care payer, incorporated caregiver disutilities to the “No Response” and liver transplant–related health states until patients in the model reached the age of 18 years. The sponsor derived these disutilities as the complement of utility values estimated for cholestasis in a poster presentation by Quadrado et al. (2022).32 The sponsor’s approach is problematic for several reasons. First, the disutilities associated with caregiving should not be calculated by subtracting utility values from 1.0. Disutilities should reflect the marginal difference in utility for caregivers of children with ALGS and cholestatic pruritus compared with caregivers of children in the general populatoin.46,47 The sponsor’s approach to estimating disutility implicitly implies that caregivers of children who respond to pruritus treatment and have not undergone a liver transplant have perfect health, which can lead to an overestimation of disutility associated with caregiving. Additionally, the sponsor incorporated caregiver disutilities in the submitted analysis conducted from the perspective of the public health care payer. As the target population of the economic evaluation was a patient population with ALGS and cholestatic pruritus, the analysis from the perspective of the health care payer should only include the health effects for the target population.32 While there may be impacts on informal caregivers, any associated impact beyond the targeted population(s), in terms of either costs or effects, should be addressed in a nonreference-case analysis.46 As such, the inclusion of caregiver utilities in the base-case public health care payer perspective was inappropriate. The sponsor’s approach of estimating and applying caregiver disutilities overestimated the impact of caregiver burden on health utility and inflated the incremental QALYs in favour of odevixibat plus SOC.
In a reanalysis, CDA-AMC removed caregiver disutilities. In the process, CDA-AMC also corrected caregiver disutility for the liver transplant health state, which was hard-coded to a value of 0.05 in the parameter tab.
The model structure does not appropriately capture the treatment paradigm of ALGS: The sponsor-submitted Markov model does not adequately capture the complexity of the treatment pathway. The model assumes a simplified treatment pathway in which patients with uncontrolled pruritus (i.e., those in “No Response” health state) are at risk of having a liver transplant. However, according to expert feedback received for this review, some patients with pruritus control may also undergo transplant due to progression of the underlying liver disease (i.e., cholestasis). At the same time, some ALGS patients with uncontrolled pruritus and progressive cholestasis may not have a liver transplant due to potential contraindications such as cardiac complications.5,32 These aspects of the treatment paradigm are not reflected in the modelled health states, which are characterized by pruritus response only and disregard the progression of the underlying disease. Because the model structure does not fully reflect the complexity of transplant-related treatment decisions, the estimated cost savings derived from reduced transplant events for patients on odevixibat plus SOC are uncertain and may have been overestimated.
This issue could not be addressed by CDA-AMC.
Additional issues were identified but were not considered to be key issues. These included the sponsor’s assumption that 100% of patients on ileal bile acid transporter (IBAT) inhibitors incurred the cost of odevixibat and maralixibat in the first year and a half, regardless of treatment response. This assumption effectively disconnected cost from response, which was reported as 89% and 72% for odevixibat plus SOC and maralixibat plus SOC, respectively. During this time, the sponsor also assumed that patients on IBAT inhibitors were not at risk of death or liver transplant, regardless of treatment response. Although this approach may have overestimated treatment costs, it also artificially inflated treatment benefit and cost savings by assuming no death and a liver transplant in the first year. Lastly, poor modelling practices were noted, including a calculation error and the use of multiple hard-coded inputs to inform the same parameter, which made it challenging to validate model outputs.
CDA-AMC Reanalysis of the Economic Evaluation
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts (refer to Table 8). The impact of these changes, individually and collectively, is presented in Table 9. Due to the lack of comparative efficacy data for odevixibat plus SOC relative to maralixibat plus SOC, maralixibat plus SOC was removed from the CDA-AMC base case. Results of a scenario analysis including maralixibat plus SOC are presented in Table 10. The CDA-AMC analyses are based on the publicly available prices of the comparator treatments.
Table 8: Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Proportion of patients on a low dose of odevixibat | █████ | 2.9% |
2. Caregiver disutility for no response and liver transplant–related health states | Included (caregiver disutility for the LT health state was set to 0.05) | Excluded (caregiver disutility for LT health state was set to 0) |
CDA-AMC base case (health care payer perspective) | ― | Reanalysis 1 + 2 |
CDA-AMC = Canada’s Drug Agency; LT = liver transplant; SOC = standard of care.
Note: CDA-AMC was unable to resolve the issues with the absence of robust evidence on the comparative efficacy and safety of odevixibat and maralixibat, uncertainty in long-term efficacy of odevixibat plus SOC and lack of face validity of the model structure.
Table 9: Summary of the Stepped Analysis
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor base case | SOC | 291,796 | 18.81 | Reference |
Odevixibat plus SOC | 10,463,743 | 23.04 | 2,405,534 | |
Dominated | ||||
Maralixibat plus SOC | 10,728,463 | 22.13 | Dominated by Odevixibat plus SOC | |
Sponsor base case, deterministic | SOC | 291,975 | 18.77 | Reference |
Odevixibat plus SOC | 10,130,599 | 22.98 | 2,335,387 | |
Dominated | ||||
Maralixibat plus SOC | 10,713,175 | 22.11 | Dominated by Odevixibat plus SOC | |
CDA-AMC reanalysis 1 | SOC | 291,975 | 18.77 | Reference |
Odevixibat plus SOC | 14,301,492 | 22.98 | 3,325,428 | |
CDA-AMC reanalysis 2 | SOC | 291,975 | 22.75 | Reference |
Odevixibat plus SOC | 10,130,599 | 25.25 | 3,930,757 | |
CDA-AMC base case Reanalysis 1 + 2 (deterministic) | SOC | 291,975 | 22.75 | Reference |
Odevixibat plus SOC | 14,301,492 | 25.25 | 5,597,125 | |
CDA-AMC base case Reanalysis 1 + 2 (probabilistic) | SOC | 291,707 | 22.77 | Reference |
Odevixibat plus SOC | 14,463,192 | 25.26 | 5,678,772 | |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Table 10: Summary of Scenario Analysis
Scenario analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
CDA-AMC base case Reanalysis 1 + 2 (deterministic) | SOC | 291,975 | 22.75 | Reference |
Maralixibat plus SOC | 10,713,175 | 24.93 | 4,787,566 | |
Odevixibat plus SOC | 14,301,492 | 25.25 | 10,998,251 | |
CDA-AMC base case Reanalysis 1 + 2 (probabilistic) | SOC | 291,707 | 22.77 | Reference |
Maralixibat plus SOC | 10,762,337 | 24.93 | 4,845,908 | |
Odevixibat plus SOC | 14,463,192 | 25.26 | 11,053,818 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Table 11: Disaggregated Results of the CDA-AMC Analyses
Parameter | Odevixibat plus SOC | SOC | Maralixibat plus SOC |
|---|---|---|---|
Discounted LYs | |||
Total | 33.09 | 30.24 | 32.95 |
By health state | |||
Response | 11.53 | 2.45 | 9.54 |
No Response | 14.53 | 10.63 | 15.72 |
LT | 0.29 | 0.66 | 0.32 |
Post-LT | 6.74 | 16.5 | 7.37 |
Discounted QALYs | |||
Total | 25.26 | 22.77 | 24.93 |
By health state | |||
Response | 10.04 | 2.14 | 8.31 |
No Response | 10.13 | 7.69 | 11.02 |
LT | 0.17 | 0.40 | 0.19 |
Post-LT | 4.92 | 12.54 | 5.41 |
Caregiver QALY decrement | 0 | 0 | 0 |
Discounted costs ($) | |||
Total | 14,463,192 | 291,707 | 10,762,337 |
Response | 14,290,122 | 4,405 | 10,576,683 |
No Response | 65,553 | 40,709 | 69,374 |
LT | 59,581 | 133,043 | 64,147 |
Post-LT | 47,922 | 113,548 | 52,121 |
Adverse events | 13 | 3 | 12 |
CDA-AMC = Canada’s Drug Agency; LY = life-year; LT = liver transplant; QALY = quality-adjusted life-year; SOC = standard of care.
Price Reduction Analysis
CDA-AMC conducted price reduction analyses using the sponsor’s base case and the CDA-AMC base case (refer to Table 12).
Table 12: Results of the Price Reduction Analysis
Price reduction | Unit drug costa ($) | Annual cost ($) | ICERs for odevixibat plus SOC vs. SOC ($/QALY) | |
|---|---|---|---|---|
Sponsor base case | CDA-AMC base case | |||
No price reduction | 0.88 | For patients weighing 4 kg to 55.5 kg: $192,769 to $2,313,233 | 2,405,534 | 5,677,442 |
10% | 0.79 | For patients weighing 4 kg to 55.5 kg: $173,493 to $2,081,910 | 2,150,114 | 5,050,321 |
20% | 0.70 | For patients weighing 4 kg to 55.5 kg: $154,216 to $1,850,587 | 1,916,688 | 4,477,503 |
30% | 0.62 | For patients weighing 4 kg to 55.5 kg: $134,939 to $1,619,263 | 1,687,201 | 3,935,971 |
40% | 0.53 | For patients weighing 4 kg to 55.5 kg: $115,662 to $1,387,940 | 1,433,207 | 3,359,349 |
50% | 0.44 | For patients weighing 4 kg to 55.5 kg: $96,385 to $1,156,617 | 1,200,050 | 2,821,204 |
60% | 0.35 | For patients weighing 4 kg to 55.5 kg: $77,108 to $925,293 | 940,889 | 2,244,701 |
70% | 0.26 | For patients weighing 4 kg to 55.5 kg: $57,831 to $693,970 | 705,304 | 1,673,554 |
80% | 0.18 | For patients weighing 4 kg to 55.5 kg: $38,554 to $462,647 | 464,147 | 1,094,183 |
90% | 0.09 | For patients weighing 4 kg to 55.5 kg: $19,277 to $231,323 | 220,520 | 530,387 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; SOC = standard of care; vs. = versus.
aSponsor’s submitted price for odevixibat.4 Although multiple dosage forms (i.e., 200 mcg, 400 mcg, 600 mcg, and 1,200 mcg) are available for odevixibat, the unit cost is the same for all forms. As such, a cost per mcg is presented in the table.
Issues for Consideration
The availability of maralixibat for the treatment of cholestatic pruritus in patients with ALGS is uncertain. Maralixibat for the treatment of cholestatic pruritus in patients aged 12 months and older with ALGS received a positive reimbursement recommendation35 and is currently in active pan-Canadian Pharmaceutical Alliance negotiations.1
Appendix 5: Budget Impact Analysis
Please note that this appendix has not been copy-edited.
Summary of the Submitted BIA
The sponsor submitted a BIA that estimated the expected incremental budgetary impact of reimbursing odevixibat plus SOC for the treatment of cholestatic pruritus in patients with ALGS.48
The BIA was conducted from the perspective of public drug plan payers over a 3-year time horizon (2026 to 2028), with 2025 as the base year. The sponsor’s estimate reflects the aggregated results from the jurisdictional provincial budgets (excluding Quebec) as well as the Non-Insured Health Benefits Program. The sponsor estimated the eligible population using an epidemiological approach. The sponsor’s base case included drug acquisition costs of odevixibat, maralixibat, and SOC (consisting of UDCA, cholestyramine, rifampicin, naltrexone, hydroxyzine, and SSRIs). The market uptake for odevixibat was estimated using internal market research by Pharmalytics Group. The key inputs to the BIA are documented in Table 13.
The sponsor estimated the 3-year incremental budgetary savings associated with reimbursing odevixibat for the treatment of cholestatic pruritus in patients with ALGS would be $9,457,383 (year 1 = $2,883,475, year 2 = $3,187,069, year 3 = $3,386,840).
Table 13: Key Model Parameters
Parameter | Sponsor’s estimate |
|---|---|
Target population | |
Prevalent cohort | |
Total population | 32,256,749 |
Prevalence of ALGS | 0.00154% |
Percentage of diagnosed patients | 100% |
Percentage of medically eligible patients | 80% |
Percentage of pretransplant patients | 30% |
Number of patients eligible for drug under review | 119 / 0 / 0 |
Incident cohort | |
Live births | 274,906 |
Incidence of ALGS | 0.003% |
Percentage of diagnosed patients | 100% |
Percentage of medically eligible patients | 30% |
Percentage of pretransplant patients | 100% |
Number of patients eligible for drug under review | 2 / 2 / 2 |
Market shares (reference scenario) | |
Odevixibat | ██ █ ██ █ ██ |
Maralixibat | ███ █ ███ █ ███ |
SOC | ███ █ ███ █ ███ |
Market shares (new-drug scenario) | |
Odevixibat | ███ █ ███ █ ███ |
Maralixibat | ███ █ ███ █ ███ |
SOC | ███ █ ███ █ ███ |
Cost of treatment (per patient per year)a | |
Odevixibat | |
Year 1 | $559,390 |
Year 2 | $586,431 |
Year 3 | $597,761 |
Maralixibat | |
Year 1 | $718,044 |
Year 2 | $756,953 |
Year 3 | $774,109 |
SOC | |
Year 1 | $1,635 |
Year 2 | $1,661 |
Year 3 | $1,665 |
ALGS = Alagille syndrome; SOC = standard of care.
aTreatment costs were weight-based, with dosing adjusted by weight categories ranging from less than 4 kg to 55 kg or greater. The proportion of patients within each weight category was based on data observed in the ASSERT-EXT study and was extrapolated over the time horizon.
Key Issues of the Submitted BIA
CDA-AMC identified several key issues for the sponsor’s analysis that have notable implications on the results of the BIA:
The size of the population is uncertain: The sponsor restricted treatment eligibility to those ALGS patients with cholestasis and pruritus. The sponsor assumed that only 30% of the patients in the incident cohort met this criterion. However, this estimate lacked face validity. According to expert feedback received for this review, a majority of patients would be expected to exhibit pruritus at presentation. The sponsor’s assumption likely underestimated the number of newly diagnosed patients eligible for treatment. The also sponsor estimated 30% of medically eligible ALGS patients are pretransplant patients based on a combination of literature and sponsor-sought expert feedback.4,26,49 The study by Vandriel et al. (2023) indicated that approximately 40.3% of patients retain their native liver by adulthood.26 The study by Kamath et al. (2020) is a longitudinal study that enrolled pretransplant patients only and reported that only 20% of patients (58 out of 293) had had a transplant, indicating that 80% of patients did not have a transplant.49 This shows there is a wide range of estimates for the proportion of pretransplant patients, which is highly uncertain. Furthermore, although the model distributed patients across various weight categories, the sponsor does not estimate transplant rates by age group, which would have been a more appropriate approach given the structure of the model. Compared with the published literature cited by the sponsor, the sponsor’s adopted value is a much lower estimate, potentially underestimating the number of eligible patients and, consequently, the budget impact.
In a reanalysis, CDA-AMC increased the proportion of incident patients deemed medically eligible to 60% based on expert input.
In a reanalysis, CDA-AMC increased the proportion of pretransplant patients to 40.3% to align with the results of the GALA study. It was noted that the highest proportion of patients were in the 17.5 to 25.5 age category in the sponsor’s BIA model. CDA-AMC explored the impact of assuming 80% of patients are pretransplant in a scenario analysis.
In a scenario analysis, CDA-AMC used an alternative estimate of incidence and prevalence (i.e., 1 in 30,000 persons).
Treatment cost of SOC is inappropriately captured: In the submitted BIA model, the sponsor attributed the cost of SOC when used in combination with either maralixibat or odevixibat to SOC alone. This is inappropriate because the costs associated with SOC for patients on maralixibat and odevixibat should be accounted for in the total cost calculation of maralixibat plus SOC and odevixibat plus SOC, respectively, rather than SOC alone.
In a reanalysis, the cost of SOC for patients receiving it in addition to maralixibat or odevixibat was accounted for in estimating the total treatment cost for maralixibat plus SOC and odevixibat plus SOC.
The proportion of patients on a low dose of odevixibat was overestimated: As noted in the Key Issues of the Submitted Economic Evaluation section, dosing of odevixibat is highly uncertain. The sponsor’s estimate of the proportion of patients on a low dose of odevixibat could not be validated. According to expert feedback, the dosing patterns observed in the ASSERT trial are more likely to reflect clinical practice.
In a reanalysis, CDA-AMC aligned the distribution of patients on a low dose of odevixibat with that observed in the ASSERT trial. This resulted in an increase in the annual cost of odevixibat to $800,000 to $850,000 per patient per year from year 1 to year 3 of the BIA.
Market share of maralixibat is uncertain: The sponsor assumed that ███ of patients eligible for treatment are on maralixibat plus SOC in the reference scenario. However, this estimate is highly uncertain and lacks face validity. Although maralixibat has received a positive recommendation for the treatment of cholestatic pruritus in patients with ALGS, pan-Canadian Pharmaceutical Alliance negotiations are still active, and it is uncertain whether maralixibat will be reimbursed by jurisdictions for ALGS.1
In a reanalysis, the market share of maralixibat was decreased to 0% in the reference scenario.
In a scenario analysis, CDA-AMC assumed maralixibat received a Letter of Intent and market share.
Market uptake of odevixibat plus SOC and maralixibat plus SOC may have been underestimated: The sponsor assumed that both maralixibat and odevixibat would have █████ market share by year 3 and, together, they would occupy ███ of the market share. According to expert feedback, if IBAT inhibitors are reimbursed, they would likely be a first-line treatment and widely adopted in clinical practice.
In a reanalysis, the market share of odevixibat plus SOC was increased to 80% in years 2 and 3 based on expert input. CDA-AMC explored the impact of assuming that maralixibat is publicly reimbursed, in which case the sponsor’s assumptions regarding market share distribution between maralixibat plus SOC and odevixibat plus SOC were retained.
Prevalence of ALGS is uncertain: The sponsor calculated the prevalence and incidence of ALGS as a midpoint between 2 extreme values reported in published literature.20,50 However, the cited sources indicate a wide range of plausible estimates, suggesting considerable uncertainty in the true prevalence and incidence estimates. The sponsor’s approach of relying on the midpoint may underestimate the actual number of patients with ALGS if the true prevalence and incidence is closer to the upper bound. This may have underestimated the number of patients with ALGS.
In scenario analysis, CDA-AMC adopted the upper bound of the prevalence and incidence range (i.e., 1 in 30,000) to explore its impact on budget impact.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analyses by making changes in model parameter values and assumptions, in consultation with clinical experts, as outlined in Table 14.
The results of the CDA-AMC stepwise reanalysis are presented in summary format in Table 15, and a more detailed breakdown is presented in Table 16. In the CDA-AMC base case, the 3-year budget impact of reimbursing odevixibat plus SOC for cholestatic pruritus in patients with ALGS was $211,497,590 (year 1 = $65,176,495, year 2 = $71,074,001, year 3 = $75,247,093).
Table 14: Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Percentage of patients deemed medically eligible in incident cohort | 30% | 60% |
2. Percentage of pretransplant patients in prevalent cohort | 30% | 40.3% |
3. Treatment cost of SOC for patients on maralixibat and odevixibat | Included in estimating total cost of SOC | Included in estimating total cost of maralixibat and odevixibat |
4. Proportion of patients on a low dose of odevixibat | █████ | 2.9% |
5. Market uptake of maralixibat and changes in IBAT use (year 1 / year 2 / year 3) | Reference scenario: Maralixibat: ███ █ ███ █ ███ SOC: ███ █ ███ █ ███ New-drug scenario: Odevixibat: ███ █ ███ █ ███ Maralixibat: ███ █ ███ █ ███ SOC: ███ █ ███ █ ███ | Reference scenario: Maralixibat: 0% / 0% / 0% SOC: 100% / 100% / 100% New-drug scenario: Odevixibat: 50% / 80% / 80% Maralixibat: 0% / 0% / 0% SOC: 50% / 20% / 20% |
CDA-AMC base case | ― | Reanalysis 1 + 2 + 3 + 4 + 5 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; IBAT = ileal bile acid transporter; SOC = standard of care.
Table 15: Summary of the Stepped Analysis of the CDA-AMC Base Case
Stepped analysis | Three-year total |
|---|---|
Submitted base case | –$9,457,383 |
CDA-AMC reanalysis 1 | –$9,883,419 |
CDA-AMC reanalysis 2 | –$12,558,146 |
CDA-AMC reanalysis 3 | –$9,457,383 |
CDA-AMC reanalysis 4 | $4,233,903 |
CDA-AMC reanalysis 5 | $108,568,515 |
CDA-AMC base case (reanalysis 1 + 2 + 3 + 4 + 5 + 6) | $211,497,590 |
CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments.
CDA-AMC used the CDA-AMC base case to conduct the following scenario analyses to explore uncertainty in the estimated budget impact of reimbursing odevixibat plus SOC. The results are provided in Table 16.
Assuming maralixibat is reimbursed by public drug plans in the new-drug scenario. In year 1, the market share was 35% for maralixibat and 15% for odevixibat. In years 2 and 3, the market share was assumed to be split equally (i.e., 40%) between the 2 IBAT inhibitors.
Assuming a higher incidence and prevalence rate (1 in 30,000 persons).
Table 16: Disaggregated Summary of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference total | 37,419,349 | 43,698,469 | 46,842,571 | 48,701,605 | 139,242,645 |
Odevixibat plus SOC | 0 | 0 | 0 | 0 | 0 | |
All other comparators | 37,419,349 | 43,698,469 | 46,842,571 | 48,701,605 | 139,242,645 | |
New-drug total | 37,419,349 | 40,814,995 | 43,655,502 | 45,314,764 | 129,785,261 | |
Odevixibat plus SOC | 0 | 10,166,651 | 10,960,385 | 11,480,258 | 32,607,294 | |
All other comparators | 37,419,349 | 30,648,344 | 32,695,116 | 33,834,506 | 97,177,967 | |
Budget impact | 0 | –2,883,475 | –3,187,069 | –3,386,840 | –9,457,383 | |
CDA-AMC base case | Reference total | 249,585 | 268,262 | 279,366 | 287,045 | 834,673 |
Odevixibat plus SOC | 0 | 0 | 0 | 0 | 0 | |
All other comparators | 249,585 | 268,262 | 279,366 | 287,045 | 834,673 | |
New-drug total | 249,585 | 65,444,757 | 71,353,367 | 75,534,138 | 212,332,263 | |
Odevixibat plus SOC | 0 | 65,310,626 | 71,215,738 | 75,394,736 | 211,921,101 | |
All other comparators | 249,585 | 134,131 | 137,629 | 139,402 | 411,162 | |
Budget impact | 0 | 65,176,495 | 71,074,001 | 75,247,093 | 211,497,590 | |
CDA-AMC scenario analyses | ||||||
Scenario 1: Assuming maralixibat is reimbursed by public drug plans | Reference total | 249,585 | 59,189,908 | 64,890,942 | 68,916,629 | 192,997,478 |
New-drug total | 249,585 | 61,066,363 | 66,879,621 | 71,000,376 | 198,946,360 | |
Budget impact | 0 | 1,876,455 | 1,988,679 | 2,083,747 | 5,948,881 | |
Scenario 2: Assuming prevalence and incidence is 1 in 30,000 persons | Reference total | 540,768 | 573,371 | 589,316 | 597,896 | 1,760,582 |
New-drug total | 540,768 | 139,878,556 | 149,375,415 | 155,058,508 | 444,312,478 | |
Budget impact | 0 | 139,305,185 | 148,786,099 | 154,460,612 | 442,551,896 | |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; SOC = standard of care.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.