Drugs, Health Technologies, Health Systems
Reimbursement Review
Roflumilast (Zoryve)
Sponsor: Arcutis Canada, Inc.
Therapeutic area: Atopic dermatitis
Summary
What Is Atopic Dermatitis?
Atopic dermatitis (AD) is a chronic, multifactorial, incurable inflammatory skin disease that is common in adults and children. AD is characterized by severe itch and recurrent eczematous lesions, which have a significant physical and psychosocial impact on patients and their families.
Canadian estimates suggest that the prevalence of AD is approximately 4% for adults and 15% for children, with most patients having mild to moderate AD.
What Are the Treatment Goals and Current Treatment Options for Atopic Dermatitis?
Treatment goals for AD include the control of signs (e.g., erythema, erosions, lichenification), reductions in symptoms (e.g., itching, burning, pain), secondary infections, flares, and improvements in health-related quality of life (HRQoL), sleep, and ability to carry out daily activities.
Important outcomes identified by patient groups included control of AD; reductions in symptoms, flares, and complications; relief from pain and discomfort; and improvement in HRQoL. In addition to these outcomes, input from clinical experts and clinician groups indicated it was important to assess the percentage of total body surface area affected by AD. The patient and clinician groups also expressed a need for topical treatments that allow for convenient application and have a favourable safety profile and tolerability.
After initial management strategies for AD (e.g., skin moisturizing practices, avoidance of triggers), topical treatments are considered the standard of care for patients with mild to moderate AD. Topical treatment options for mild to moderate AD include topical corticosteroids (TCSs), topical calcineurin inhibitors (TCIs), the phosphodiesterase type 4 inhibitor crisaborole (2% ointment), and the Janus kinase inhibitor ruxolitinib (1.5% cream). Of note, ruxolitinib (1.5% cream) is approved only for patients aged 12 years or older. All current topical treatments are approved for twice-daily application to affected areas.
What Is Zoryve and Why Did Canada’s Drug Agency Conduct This Review?
Zoryve is a topical cream of roflumilast 0.15% approved by Health Canada for the topical treatment of mild to moderate AD in patients aged 6 years and older. It is indicated to be applied once daily to affected areas.
Canada’s Drug Agency (CDA-AMC) reviewed Zoryve to inform a recommendation to the participating public drug programs on whether it should be reimbursed for the topical treatment of mild to moderate AD in patients aged 6 years and older with inadequate response, intolerance, or contraindications to TCSs.
How Did CDA-AMC Evaluate Zoryve?
CDA-AMC reviewed the clinical evidence on the beneficial and harmful effects, as well as the economic evidence, for Zoryve versus other treatments used in Canada for the topical treatment of mild to moderate AD in patients aged 6 years and older.
TCSs (e.g., desonide, triamcinolone acetonide, betamethasone valerate, hydrocortisone valerate, hydrocortisone acetate), TCIs (e.g., tacrolimus, pimecrolimus), ruxolitinib 1.5% cream, and crisaborole 2% ointment were considered to be relevant treatments to compare with Zoryve when reviewing the clinical evidence.
CDA-AMC identified equity and ethical considerations relevant to Zoryve and AD.
The review was informed by materials submitted by the sponsor that included clinical and economic evidence.
One patient group, 3 clinician groups, and the participating public drug programs made submissions in response to the CDA-AMC call for input for this review. The public drug programs submitted issues that may impact their ability to implement a recommendation.
CDA-AMC consulted 2 dermatologists from Ontario as part of the review process.
What Were the Findings?
Clinical Evidence
CDA-AMC reviewed the following clinical evidence:
Two phase III randomized controlled trials (the INTEGUMENT-1 and INTEGUMENT-2 trials) comparing Zoryve 0.15% cream with vehicle cream in a total of 1,337 patients with mild to moderate AD (INTEGUMENT-1 trial: Zoryve, n = 433 versus vehicle, n = 221; INTEGUMENT-2 trial: Zoryve, n = 451 versus vehicle, n = 232).
One long-term open-label extension study (INTEGUMENT-OLE) (Zoryve, n = 439 versus vehicle, n = 219).
One network meta-analysis of Zoryve versus other active therapies for mild to moderate AD (e.g., TCS, TCI, Janus kinase inhibitor or other phosphodiesterase type 4 inhibitor) consisting of 31 unique randomized controlled trials.
For the comparison of Zoryve 0.15% cream versus vehicle cream:
Evidence from the INTEGUMENT-1 and INTEGUMENT-2 trials demonstrated with high certainty that at week 4, Zoryve 0.15% cream results in a clinically important improvement in disease severity as indicated by the increase in the proportion of patients achieving a Validated Investigator Global Assessment for Atopic Dermatitis score of clear or almost clear plus a 2-grade or higher improvement from baseline at week 4 (success) and the proportion of patients achieving a 75% reduction in the Eczema Area and Severity Index when compared with vehicle cream.
Zoryve 0.15% cream likely results in a clinically important improvement in itch symptoms, as indicated by the proportion of patients achieving an improvement of 4 points or greater in their worst itch intensity (Worst Itch Numeric Rating Scale success) at week 4. However, Zoryve 0.15% cream may result in little to no important difference in general AD symptoms as represented by the least squares (LS) mean change in Patient-Oriented Eczema Measure score from baseline to week 4.
Zoryve 0.15% cream likely results in little to no meaningful improvement in HRQoL measured by the LS mean change in Dermatology Life Quality Index (DLQI) score from baseline to week 4 among adult patients. Zoryve 0.15% cream results in little to no meaningful improvement in HRQoL among pediatric patients, as represented by the LS mean change in Children’s Dermatology Life Quality Index (CDLQI) score from baseline to week 4.
During the open-label extension phase of the INTEGUMENT trials, the DLQI scores and the CDLQI scores at week 52 decreased from baseline, which may suggest an improvement in HRQoL. However, the long-term effect of Zoryve 0.15% cream on patients’ quality of life was uncertain due to the large amount of missing data in this extension study.
For the comparison of Zoryve 0.15% cream versus other active comparators for mild to moderate AD:
For the stringent success rate and rate for a 75% reduction in the Eczema Area and Severity Index, definite conclusions as to which treatment may be favoured could not be drawn when Zoryve 0.15% cream was compared to its comparators, except that treatment with Zoryve 0.15% cream may be associated with a lower rate of stringent treatment success or more severe disease at week 4 when compared to ruxolitinib.
For HRQoL measured by changes in the DLQI and CDLQI scores from baseline to week 4, definite conclusions as to which treatment may be favoured could not be drawn when Zoryve 0.15% cream was compared to all comparators.
Zoryve 0.15% cream was associated with a lower rate of all-cause discontinuation when compared with crisaborole ointment, whereas indirect evidence is insufficient to conclude whether Zoryve 0.15% cream differs from all other comparators in terms of all-cause discontinuation or withdrawals due to adverse events.
There was no evidence to inform how Zoryve compares with pimecrolimus.
No new safety signals were observed with treatment with Zoryve 0.15% cream in the INTEGUMENT-1 and INTEGUMENT-2 trials. The results for the interim analysis of the ongoing INTEGUMENT-OLE trial suggest that Zoryve 0.15% cream is safe for up to 52 weeks following the initial 4-week trials.
A key gap in the evidence is the lack of direct evidence comparing Zoryve 0.15% cream to other comparators for mild to moderate AD in Canadian clinical practice.
Economic Evidence
Zoryve is available as a 0.15% or 0.3% topical cream or a 0.3% topical foam, although only the 0.15% cream is indicated for the treatment of mild to moderate AD in patients aged 6 years and older. At the submitted price of $290.00 per 60 g tube, the annual cost of Zoryve 0.15% cream is expected to be $355 per patient, based on the Health Canada–recommended dosage, a mean of 18 administration days per year, and no wastage.
When considering the Health Canada indication (treatment of mild to moderate AD in patients aged 6 years and older), relevant comparators include the funded TCSs and TCIs. However, if funding is limited to the reimbursement request population (those who have also experienced an inadequate response, intolerance, or contraindications to TCSs), TCIs are the relevant funded comparators.
Based on the results of the CDA-AMC base case, Zoryve 0.15% cream is expected to be associated with higher costs to the health care system (including drug acquisition costs) compared to TCSs (incremental costs: $511 to $568 per patient in year 1) and to be within the range of costs for TCIs (incremental savings of $66 to incremental costs of $134 per patient in year 1), driven primarily by increased costs associated with drug acquisition and wastage. If no wastage is assumed, Zoryve 0.15% cream is still expected to be associated with higher costs compared to TCSs (incremental costs: $303 to $344 per patient per year), but lower costs compared to TCIs (incremental savings: $82 to $172 per patient per year).
CDA-AMC estimates that the budget impact of reimbursing Zoryve 0.15% cream for the treatment of the indicated population will be approximately $19.7 million over the first 3 years of reimbursement compared to the amount currently spent on TCIs and TCSs, with an estimated expenditure of $51.4 million on Zoryve over this period. If funding is limited to the reimbursement request population, CDA-AMC estimates that the budget impact of reimbursing Zoryve 0.15% cream would be approximately $871,000 over the first 3 years compared to the amount currently spent on TCIs, with an expected expenditure of $31.3 million on Zoryve. The actual budget impact of reimbursing Zoryve 0.15% cream will depend on the proportion of patients who will be reimbursed by the public drug plans, the potential wastage of excess product, the average amount of topicals used per patient-year, and the comparators being displaced. If reimbursement is limited to the reimbursement request population, the magnitude of uncertainty in the budget impact must be addressed to ensure the feasibility of adoption at the submitted price.
Abbreviations
AD
atopic dermatitis
AE
adverse event
BSA
body surface area
CDA-AMC
Canada’s Drug Agency
CDLQI
Children’s Dermatology Life Quality Index
CI
confidence interval
CrI
credible interval
DLQI
Dermatology Life Quality Index
EASI
Eczema Area and Severity Index
EASI-75
75% reduction in the Eczema Area and Severity Index
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
IIC-TCS
inadequate response, intolerance, or contraindications to topical corticosteroid
ITC
indirect treatment comparison
ITT
intention to treat
JAK
Janus kinase
LS
least squares
LTE
long-term extension
MID
minimal important difference
NMA
network meta-analysis
OLE
open-label extension
OR
odds ratio
PD
percent difference
PDE4
phosphodiesterase type 4
POEM
Patient-Oriented Eczema Measure
RCT
randomized controlled trial
SAE
serious adverse event
SCORAD
Scoring Atopic Dermatitis
TCI
topical calcineurin inhibitor
TCS
topical corticosteroid
TEAE
treatment-emergent adverse event
vIGA-AD
Validated Investigator Global Assessment for Atopic Dermatitis
WDAE
withdrawal due to adverse event
WI-NRS
Worst Itch Numeric Rating Scale
Background
Introduction
The objectives of this report are as follows:
Review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of roflumilast cream, 0.15% w/w, in the topical treatment of mild to moderate atopic dermatitis (AD) in patients aged 6 years and older. The focus will be placed on comparing roflumilast to relevant comparators in clinical practice in Canada and identifying gaps in the current evidence, and this focus is outlined in Table 1.
Review and critically appraise the economic information submitted by the sponsor, including a cost-effectiveness analysis and budget impact analysis. The focus of the Economic Review is aligned with the scope of the Clinical Review, unless otherwise stated. For most reviews, Canada’s Drug Agency (CDA-AMC) develops a base case informed by clinical expert input, the available clinical evidence, and the best interpretation of the economic evidence based on the information provided by the sponsor.
Table 1: Information on the Application Submitted for Review and on the CDA-AMC Review
Item | Description |
|---|---|
Information on the application submitted for review | |
Drug | Roflumilast (Zoryve), 0.15% w/w, topical cream |
Sponsor | Arcutis Canada, Inc. |
Health Canada indication | For topical treatment of mild to moderate AD in patients 6 years of age and older |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | March 13, 2025 |
Mechanism of action | Roflumilast and its active metabolite (roflumilast N-oxide) are inhibitors of PDE4 activity leading to accumulation of intracellular cyclic AMP and subsequent inhibition of inflammatory markers associated with plaque psoriasis, AD, or seborrheic dermatitis. |
Recommended dosage | Apply once daily to affected areas. For topical use only. Not for ophthalmic, oral, or intravaginal use. No dosage adjustment is required in geriatric patients, patients with renal impairment, or in patients with mild hepatic impairment (Child-Pugh A). |
Submission type | Initial |
Sponsor’s reimbursement request | For topical treatment of mild to moderate AD in patients 6 years of age and older, with inadequate response, intolerance, or contraindications to topical corticosteroids. |
Submitted price | $290.00 per 60 g tube |
Information on the CDA-AMC review | |
Review type | Standard |
Clinical review focusa | Population: As defined in the Health Canada indication Subgroups: IIC-TCS Intervention: Per recommended dosage Comparators:
Outcomes: vIGA-AD success, vIGA-AD score of clear or almost clear, EASI-75, WI-NRS, POEM, and DLQI or CDLQI |
AD = atopic dermatitis; AMP = adenosine monophosphate; CDA-AMC = Canada’s Drug Agency; CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; EASI-75 = 75% reduction in the Eczema Area and Severity Index; IIC-TCS = inadequate response, intolerance, or contraindications to topical corticosteroids; NOC = Notice of Compliance; PDE4 = phosphodiesterase type 4; POEM = Patient-Oriented Eczema Measure; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; vIGA-AD = Validated Investigator Global Assessment for Atopic Dermatitis; WI-NRS = Worst Itch Numeric Rating Scale.
aThe economic review aligns with the scope of the clinical review, unless otherwise stated.
Submission History for the Drug Under Review
CDA-AMC previously reviewed roflumilast 0.3% cream through the reimbursement review process for the topical treatment of plaque psoriasis, including treatment of psoriasis in the intertriginous areas, in patients aged 12 years and older, and issued a conditional recommendation of reimbursement.
Sources of Information
The contents of this Reimbursement Review report are informed by materials submitted by the sponsor, input received from interested parties (patient groups, clinician groups, and drug programs), and input from clinical experts consulted for this review.
Calls for patient group and clinician group input are issued for each Reimbursement Review. One patient group submission from the Eczema Society of Canada and 3 clinician group submissions, from the Canadian Dermatology Association, Dermatology Association of Ontario, and the Atlantic Dermatology Group, were received. The patient group gathered information through questionnaires; interviews with patients, caregivers, and health care professionals; and published literature. More than 3,000 people in Canada who live with eczema were surveyed by the patient group. The clinician groups gathered information from published literature, Canadian clinical triallists’ experience, and clinicians’ comments. In total, 17 clinicians contributed to the clinician group input. The full submissions received are available in the consolidated input document posted on the CDA-AMC project landing page. The drug programs provide input on each drug being reviewed through the reimbursement review process by identifying issues that may impact their ability to implement a recommendation.
Input from patient and clinician groups is considered throughout the review, including in the selection of outcomes to include in the clinical review and in the interpretation of the clinical and economic evidence. Relevant patient and clinician group input is summarized in the Disease Background, Current Management, and Unmet Needs and Existing Challenges sections of this report.
Each review team includes at least 1 clinical expert with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process. Two dermatologists from Ontario with expertise in the diagnosis and management of AD participated as part of the review team.
Disease Background
AD is a chronic, multifactorial, incurable inflammatory skin disease that is common in adults and children, but the majority of cases begin within the first 5 years of life.1-3 AD is characterized by severe itch and recurrent eczematous lesions, which have a significant physical and psychosocial impact on patients and their families.4
Canadian estimates suggest that the prevalence of AD is approximately 4% for adults and 15% for children, with most patients having mild to moderate AD.5-7 In 2021, a cross-sectional study of the CorEvitas Atopic Dermatitis Registry, which consisted of 53 sites based in Canada and the US, reported that, among the patients with AD enrolled in Canada, 2% were white or non-Hispanic, 11% were Asian or non-Hispanic, and 9% were Black or non-Hispanic. The study also reported facial involvement as the most common site for AD (62% among Asian patients, 49% among Hispanic patients, 43% among white patients, 42% among Black patients, and 49% among patients of other races).8 Of note, Indigenous populations in Canada experience a high burden related to AD. The true prevalence among these populations may be underestimated due to factors such as limited access to health care and specialists and underrepresentation in research.9 These populations often face numerous barriers to AD management, such as crowded housing and limited access to clean water and health care resources.9
According to the input from the AD patient groups, negative impacts on health-related quality of life (HRQoL) include significant suffering and discomfort due to itch, poor sleep quality, effects on intimacy and relationships, anxiety, and unpredictable patterns of flares. AD can also negatively impact mood, work, school, and social interactions. The burden of AD also extends to caregivers and family members, which can include financial burden and feelings of helplessness, guilt, and frustration.
Current Management
Treatment Goals
According to the clinical experts consulted by CDA-AMC, the goals of treatment for AD include improvements in HRQoL and control of signs (e.g., erythema, erosions, lichenification) and symptoms (e.g., itching, burning, and pain). One clinical expert indicated that the control of signs and symptoms is especially important to allow for adequate sleep, presence at work, and social activities. One clinical expert consulted by CDA-AMC also noted that reductions in superinfections and scarring were important goals of treatment for AD.
Similarly, the clinician groups that submitted input to CDA-AMC indicated that improvements in HRQoL and the control of signs and symptoms of AD were important goals of treatment for AD. These groups also noted that an ideal treatment for AD would result in both rapid itch resolution and skin clearance, have a favourable safety profile and tolerability, and would be accessible to all patients who would potentially benefit from treatment. They further indicated that ideal topical treatments would also have cosmetically appropriate bases that would be easy to use on any body area. The clinician groups noted that improvements in physician global assessment, body surface area (BSA) involvement and involvement of special sites (e.g., face, genitals, and folds) were important goals of treatment for AD.
According to the patient groups that submitted input to CDA-AMC, important treatment goals for AD among patients included improvements in disease control and reduced incidence of flares and symptoms (e.g., itch, dryness, flaking, inflammation, blistering, and cracked skin). Patients were also seeking a treatment that allows them to sleep better and provides relief from the pain, discomfort, and psychological burden they live with every day. Patients would like to have the ability to carry out simple daily activities, such as bathing, contributing at work, and exercising, as well as to feel comfortable in their skin, establish and maintain intimate relationships, and reduce or eliminate the potential complications and secondary infections that often arise as a result of living with uncontrolled forms of the disease.
Current Treatment Options
There are no curative treatments for AD. One clinical expert consulted by CDA-AMC stated that the initial management of AD consists of optimized skin moisturizing practices, which include daily bathing, the use of emollient moisturizers, and avoidance of symptom triggers. After these strategies, topical treatments are considered the standard of care for patients with mild to moderate AD. Treatment guidelines for AD recommend low to moderate potency topical corticosteroids (TCSs) as first-line therapy for mild to moderate AD that is refractory to moisturization alone. TCSs are available in various formulas and potencies, which range from class 1 (ultra-high potency) to class 6 or 7 (low potency).10 The choice of potency and formulation of TCS is based on the patient’s previous treatment history, site of application, accessibility, values, and preferences.11 However, treatment guidelines recommend that TCSs be limited to short-term use due to adverse events (AEs) (e.g., skin atrophy, infections, and other skin conditions), and that alternative treatment options should be considered if there is insufficient response after 4 weeks.6,11
Topical calcineurin inhibitors (TCIs) are approved for the second-line treatment of AD when TCSs are deemed inadvisable or ineffective. Pimecrolimus and tacrolimus are TCIs approved by Health Canada for the treatment of AD; the former is indicated for mild to moderate AD and the latter is indicated for moderate to severe AD. TCIs have a reduced adverse risk profile compared to TCSs and they can be used on sensitive skin areas, such as the face and skin folds.12 Moreover, treatment guidelines suggest that TCIs can be used as steroid-sparing drugs and reduce the need for TCS use over time.12 However, stinging and burning are common AEs of TCIs, which may affect patient experience with these drugs.6,11,12 Moreover, rare cases of malignancy have been reported in patients treated with TCIs, although a causal relationship has not been established. Other nonsteroidal topical treatments approved by Health Canada for mild to moderate AD are the phosphodiesterase type 4 (PDE4) inhibitor crisaborole (2% ointment) and the Janus kinase (JAK) inhibitor ruxolitinib 1.5% cream. Similar to TCIs, the most common AEs of crisaborole (2% ointment) are burning and stinging.6 Although guidelines suggest that treatment with ruxolitinib 1.5% cream is well tolerated, they note that it carries a boxed warning for serious infections, major cardiovascular events, and thrombosis.6,11
Treatment options for patients with moderate to severe AD that is refractory or who are intolerant to topical therapies include phototherapy and systemic therapies. Phototherapy is a safe and effective treatment for multiple skin conditions, including AD.11,13 However, most phototherapy regimens require multiple treatment sessions per week, which would require a significant time commitment from the patient.13 Moreover, due to a paucity of phototherapy centres across Canada,14 patients may have to travel long distances to receive treatment or may not view phototherapy as a viable option altogether. Systemic therapies for moderate to severe AD include immunosuppressants (methotrexate, cyclosporine, mycophenolate mofetil, and azathioprine), biologics (dupilumab, tralokinumab), and oral JAK inhibitors (upadacitinib, abrocitinib).11,13 One clinical expert consulted by CDA-AMC noted that dupilumab is indicated for patients aged 6 months and older, whereas tralokinumab, lebrikizumab, upadacitinib, and abrocitinib are indicated for patients aged 12 years and older.
Key characteristics of roflumilast are summarized with other treatments available for AD in the Supplemental Material document (available on the CDA-AMC project landing page) in the Key Characteristics table in Appendix 1.
Unmet Needs and Existing Challenges
According to the clinical experts consulted by CDA-AMC, there are patients with AD that is refractory or who are intolerant to existing treatment options for AD. The clinical experts also indicated safety concerns with existing treatment options for mild to moderate AD. These concerns range from potential absorption and tachyphylaxis associated with TCSs to intolerable burning and stinging associated with TCIs and crisaborole ointment. Moreover, it was noted that only a few of these treatment options are considered safe for the face and skin folds. One clinical expert consulted by CDA-AMC also noted that some preparations of current treatments contain high-frequency allergens (e.g., propylene glycol). One clinical expert noted that continued access to treatments for AD may be restricted after the end of coverage by public drug plans. One clinical expert noted that ruxolitinib was a costly treatment and available for patients aged 12 years or older. Moreover, 1 clinical expert consulted by CDA-AMC noted that current treatment options for mild to moderate AD require twice-daily applications, whereas a once-daily application would be preferred. The clinical experts consulted by CDA-AMC also indicated that specific populations are disproportionately affected by AD, which include pediatric patients, older adults, Indigenous populations, and racialized populations. One clinical expert suggested that AD is more common among Black, Asian, and South Asian populations, who may disproportionately rely on publicly funded programs and may be less likely to have access to private insurance.
The clinician groups described unmet needs that were similar to those noted by the clinical experts consulted by CDA-AMC, which included limited efficacy and tolerability to treatment options for AD, especially for patients with facial or intertriginous involvement. The clinician groups also indicated several barriers to treatment success for AD, which included fears of steroid-related side effects, low adherence observed with twice-daily regimens, availability of effective nonsteroidal treatments (e.g., crisaborole), and restriction of newer drugs (e.g., ruxolitinib) due to boxed safety warnings and BSA limitations. The clinician groups indicated that many patients require a topical drug that is safe, effective, well tolerated, and acceptable for long-term use.
The input from patient groups indicated that topical treatments work well to manage flares among some patients with AD. However, other patients may find that the currently available topical treatments are often inadequate, and there remains a significant unmet need among current treatment options. Patient groups noted that AD flares can be extremely itchy and painful, can lead to psychological distress, and can negatively impact patients and their families. They stated that patients reported frustration with the trial-and-error process of cycling through currently available treatments. The patient group input added that certain areas of the body (such as the face, eyelids, neck, genitals, and joint areas) are particularly challenging to treat due to constant use, friction, and exposure. These sensitive areas exacerbate physical discomfort and significantly impact daily functionality and confidence.
Considerations for Using the Drug Under Review
Contents within this section have been informed by input from the clinical experts consulted for the purpose of this review and from clinician groups, and by the reimbursement conditions proposed by the sponsor (refer to the Initiation, Renewal, Discontinuation, and Prescribing Conditions Proposed by the Sponsor table in Appendix 1 in the Supplemental Material document available on the CDA-AMC project landing page). The implementation questions from the public drug programs and corresponding responses from the clinical experts consulted for this review are summarized in the Supplemental Material document in the Summary of Drug Program Input and Clinical Expert Responses table in Appendix 1. The following has been summarized by the review team.
Place in Therapy
One clinical expert consulted by CDA-AMC noted that roflumilast cream would be an additional treatment option for mild to moderate AD and would be accepted as a first-line treatment for AD affecting the face or skin folds. One clinical expert consulted by CDA-AMC indicated that roflumilast cream would be an appropriate second-line treatment for patients with mild to moderate AD in which TCSs are not effective or advisable. Although they noted that roflumilast is not expected to shift the treatment paradigm for AD, the clinical expert believed it would be a more effective treatment option with fewer side effects compared to other second-line drugs.
Two of the 3 clinician groups that submitted input to CDA-AMC indicated that roflumilast would be best suited as a first-line topical treatment for mild to moderate AD. Of note, 1 group indicated that roflumilast would be used as a second-line or adjunctive topical treatment for mild to moderate AD because TCSs are considered the standard first-line treatment for most patients. However, all 3 clinician groups that submitted input to CDA-AMC agreed that roflumilast addresses several limitations of existing therapies for mild to moderate AD, such as efficacy, tolerability, suitability for sensitive areas, and convenience (due to its once-daily application).
Patient Population
One clinical expert consulted by CDA-AMC stated that roflumilast is best suited for patients with mild to moderate AD affecting the face or skin folds. The other clinical expert consulted by CDA-AMC indicated that all patients with AD would benefit from treatment with roflumilast, especially those who have experienced an inadequate response or intolerance to a TCS or TCI. One clinical expert consulted by CDA-AMC also indicated that roflumilast may especially benefit patients with concerns about long-term steroid use, particularly around sensitive areas of the body (e.g., face and skin folds). The experts consulted by CDA-AMC stated these patients with AD would be identified based on a clinical exam and assessment of treatment history. The clinical experts indicated that the misdiagnosis of AD is not common in clinical practice.
The sponsor proposed that eligibility for roflumilast should be based on the criteria used by each of the public drug programs for the reimbursement of tacrolimus and pimecrolimus for the treatment of mild to moderate AD after conventional therapies or as a second-line drug. The sponsor also proposed that eligibility for roflumilast should be for the treatment of mild to moderate AD in patients aged 6 years or older who have experienced an inadequate response, intolerance, or contraindications to topical corticosteroids (IIC-TCS). The sponsor’s rationale for these criteria was to ensure consistency with the real-world use of existing nonsteroidal treatments, and that Canadian consensus guidelines recommend the use of TCIs and roflumilast after a TCS when another TCS is not desired due to potential risks, inadequate control of AD, or intolerance.15,16 The clinical experts consulted by CDA-AMC agreed with the conditions for the initiation of roflumilast proposed by the sponsor and the underlying rationale. However, 1 clinical expert suggested that the failure of a TCS, TCI, and/or other topical PDE4 inhibitor should be an additional criterion for initiation of roflumilast.
Of the 3 clinician groups that submitted input to CDA-AMC, 2 indicated that roflumilast is best suited for patients aged 6 years and older with mild to moderate AD who experience an inadequate response to emollients. However, all 3 groups indicated that roflumilast would be suitable for patients aged 6 years and older with mild to moderate AD who have not achieved adequate control with, are intolerant to, or have contraindications to TCIs and TCSs. All 3 clinician groups indicated that roflumilast would be an ideal treatment option for AD that is affecting sensitive areas such as the face, skin folds, and intertriginous areas. One clinician group added that roflumilast would highly benefit pediatric patients and older adult patients with thinner skin that is prone to steroid-induced atrophy and patients with a darker skin phenotype that is prone to TCS-induced hypopigmentation. One clinician group indicated that the patients who are least suitable for treatment with roflumilast include those with moderate to severe liver impairment, those with severe disease requiring systemic therapy, and those with known hypersensitivity to any component of the formulation. All 3 clinician groups agreed that appropriate candidates for roflumilast are identified based on clinical diagnosis, as well as assessment of other factors such as disease severity and distribution, prior treatment response, and lifestyle factors. They also agreed that laboratory or companion diagnostic tests are not required to identify patients suitable for roflumilast.
Assessing the Response to Treatment
The clinical experts consulted by CDA-AMC noted that outcomes assessed for AD in clinical practice include visual improvement in eczema (e.g., decrease in scaling, redness, thickness, and erosions) and reduction in the frequency and duration of flares. The clinician groups that submitted input to CDA-AMC agreed that the outcomes assessed for AD in clinical practice involve a visual assessment of improvement in symptoms, although some groups also cited that the percentage of BSA affected and extent of patient-reported symptom relief (e.g., itching) would be assessed in clinical practice. The clinical experts consulted by CDA-AMC indicated that clinically meaningfully responses to treatment include a decrease in the Investigator’s Global Assessment score (1 to 2 points), reduction in the Eczema Area and Severity Index (EASI) score (50% to 75%), reduction in itch (e.g., 2- to 4-point drop in Worst Itch Numeric Rating Scale [WI-NRS]), and reduction in the number of flares requiring additional therapy. In addition to these measures, the clinician groups noted that a clinically meaningful response to treatment would be a reduction in the signs and symptoms of disease within 4 to 8 weeks of therapy. The clinical experts consulted by CDA-AMC stated that response to treatment should initially be assessed every 1 to 2 months, followed by every 3 to 6 months. One clinician group indicated that follow-up after initiation of topical treatment is variable and often driven by patient needs and access to care. If patients do not experience an improvement in symptoms, they are advised to follow up with a care provider as needed for reassessment or escalation of care.
Discontinuing Treatment
The clinical experts consulted by CDA-AMC indicated that factors for discontinuing treatment with roflumilast include the duration of treatment and whether improvements in clinical and patient-reported outcomes were observed. The sponsor proposed no conditions for discontinuation, with the rationale that roflumilast was associated with low rates of withdrawal due to adverse events (WDAEs) and serious adverse events (SAEs) in the pivotal INTEGUMENT-1 and INTEGUMENT-2 trials. The sponsor also noted that the rates of treatment-emergent adverse events (TEAEs) among patients who received roflumilast were similar to the rates among the patients who received vehicle cream in the pivotal trials. The sponsor did not provide additional rationale for not proposing conditions for discontinuation based on lack of response. The clinical experts consulted by CDA-AMC agreed with the sponsor’s rationale for proposing no conditions for discontinuation.
The clinician groups that responded to the CDA-AMC call for input indicated that treatment with roflumilast cream should be discontinued in cases of intolerable AEs, lack of meaningful efficacy after an adequate trial (e.g., 4 to 8 weeks), disease progression, pregnancy, new contraindications, or conflicts with patient preference.
Prescribing Considerations
The clinical experts consulted by CDA-AMC agreed that roflumilast can be prescribed by dermatologists and general practitioners. One clinical expert added that although general practitioners may prescribe roflumilast, initial prescriptions should be restricted to dermatologists to ensure an accurate diagnosis of AD and a sufficient trial of a TCS. The sponsor proposed no conditions for prescribing because the Canadian product monograph was not expected to restrict the prescribing of roflumilast 0.15% cream to specific health care professionals.17 It was also noted that the physicians surveyed by the sponsor suggested that roflumilast would be prescribed by both general practitioners and dermatologists. The clinical experts consulted by CDA-AMC agreed with the sponsor’s rationale for proposing no prescribing conditions.
The clinician groups that submitted input to CDA-AMC indicated that roflumilast can also be prescribed by any licensed health care provider with the appropriate training to diagnose and manage AD, which may include nurse practitioners, pediatricians, and allergy and immunology specialists. Moreover, the clinician groups also indicated that roflumilast can be prescribed in community care or outpatient settings. Lastly, the clinician groups noted that roflumilast cream is intended for application at home by patients or caregivers and does not require special administration techniques, pharmacy compounding, or clinic-based application.
Clinical Review
Methods
The review team considered studies in the sponsor’s systematic review (pivotal studies and randomized controlled trials [RCTs]), sponsor-submitted long-term extensions (LTEs), indirect treatment comparisons (ITCs), and studies addressing gaps in the evidence for inclusion in the clinical review. Eligible studies for the systematic review included published and unpublished pivotal studies and phase III RCTs and phase IV trials. The relevant patients and interventions were defined by the indication and the dosage recommended in the product monograph. Subgroups that were considered to be potentially important for informing the reimbursement recommendation included patients with a prior IIC-TCS, patients stratified by severity (e.g., mild, moderate), and patients stratified by age (e.g., children, adults). Relevant comparators were topical treatments used in clinical practice in Canada to treat patients with mild to moderate AD. These included TCSs and TCIs. Systemic treatment options were not considered relevant comparators for this review because their place in therapy and their target patient populations are distinct from those of topical therapies. Moreover, crisaborole and ruxolitinib were also excluded from the review because they are not currently reimbursed in Canada for mild to moderate AD.
LTEs of the included pivotal studies and RCTs were included, regardless of whether there was a comparison group. ITCs and studies submitted by the sponsor addressing gaps were included when they filled an identified gap in the systematic review evidence (e.g., relative efficacy and safety for roflumilast versus currently available active treatments, longer follow-up time).
The review team selected outcomes (and follow-up times) for review, considering the sponsor’s Summary of Clinical Evidence, clinical expert input, and the patient and clinician group input. The CDA-AMC review team consulted with committee members to select outcomes considered relevant to expert committee deliberations (Table 2). Evidence from the systematic review for the most important outcomes was assessed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach.
The following outcomes were considered important based on consultation with clinical experts and patient and clinician groups (Table 2).
Table 2: Summary of Outcomes Selected for GRADE Assessment
Outcome | Rationale |
|---|---|
vIGA-AD success at week 4 Proportion of patients in EASI-75 at week 4 | vIGA-AD and EASI are relevant measures of AD disease severity. vIGA-AD success and EASI-75 at week 4 are derived end points representing improvement in AD disease severity. |
Proportion of patients with WI-NRS success at week 4 | WI-NRS is a relevant measure of the highest itch intensity in the last 24 hours related to AD. WI-NRS success refers to a reduction in itch intensity. |
Change in POEM score from baseline to week 4 | POEM is used to assess AD-specific symptoms (e.g., dryness, itching, bleeding). A change in POEM score from baseline to week 4 is a change in symptom severity. |
Change in DLQI from baseline to week 4 | A desired treatment goal for AD is an improvement in HRQoL. DLQI is a relevant measure of AD-specific HRQoL in adults. A changes in DLQI from baseline to week 4 can represent changes in HRQoL among adults with AD. |
Change in CDLQI from baseline to week 4 | A desired treatment goal for AD is an improvement in HRQoL. CDLQI is a relevant measure of AD-specific HRQoL in children. A change in CDLQI from baseline to week 4 can represent changes in HRQoL among children with AD. |
Proportion of patients with SAEs | Reduction in side effects related to treatment for AD is considered to be an important treatment goal. |
AD = atopic dermatitis; CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; GRADE = Grading of Recommendations Assessment, Development and Evaluation; EASI = Eczema Area and Severity Index; EASI-75 = 75% reduction in the Eczema Area and Severity Index; POEM = Patient-Oriented Eczema Measure; SAE = serious adverse event; vIGA-AD = Validated Investigator Global Assessment for Atopic Dermatitis; WI-NRS = Worst Itch Numeric Rating Scale.
The methods for conducting the data extraction, risk of bias appraisal, and certainty of evidence assessment are in the Supplemental Material document in Appendix 2. A full description of outcomes is in the Supplemental Material document in Appendix 3.
Clinical Evidence
In this report, the following sources of evidence submitted by the sponsor are reviewed and appraised:
the 2 double-blind, vehicle-controlled, parallel-group, multicentre phase III RCTs included in the systematic review (the INTEGUMENT-1 and INTEGUMENT-2 trials)
1 LTE study (the INTEGUMENT-OLE study)
1 ITC (network meta-analysis [NMA]).
Systematic Review
Description of Studies
Study Characteristics
The characteristics of the INTEGUMENT-1 and INTEGUMENT-2 trials are summarized in Table 3. Details on the eligibility criteria, interventions and comparators, and relevant outcome measures are in the Supplemental Material document in Appendix 3.
The INTEGUMENT-1 and INTEGUMENT-2 trials have similar designs and characteristics: double-blind, vehicle-controlled, parallel-group, multicentre phase III RCTs that evaluated the efficacy and safety of roflumilast 0.15% cream compared with vehicle cream among patients with mild to moderate AD. The trials were conducted in study sites across Canada, Poland, and the US (INTEGUMENT-1 trial: 65 sites, 7 in Canada; INTEGUMENT-2 trial: 88 sites, 9 in Canada). Both trials enrolled patients who had a clinical diagnosis of mild to moderate AD of at least 3 months’ duration. Patients were also required to have 3% or higher BSA involvement (excluding scalp, palms, and soles), a Validated Investigator Global Assessment for Atopic Dermatitis (vIGA-AD) score of mild to moderate at baseline, and an EASI score of 5 or higher.
In both trials, patients were randomized in a 2:1 ratio to receive either roflumilast 0.15% cream or vehicle cream, which was applied once daily for 4 weeks. Randomization was performed according to a computer-generated randomization list, with stratification according to baseline vIGA-AD (i.e., mild versus moderate) and by study site. For both trials, patients were assessed at the screening phase and at baseline before the treatment period, and at week 1, week 2, and week 4 of the treatment period. Patients who completed 4 weeks of treatment in either trial were eligible to enrol in a separate open-label extension (OLE) study (the INTEGUMENT-OLE study)18 and receive up to 12 months of treatment. An overview of the study design for both trials is presented in Figure 1. There were approximately 650 patients planned for each of the INTEGUMENT-1 and INTEGUMENT-2 trials.
Figure 1: Study Design for the INTEGUMENT-1 and INTEGUMENT-2 Trials
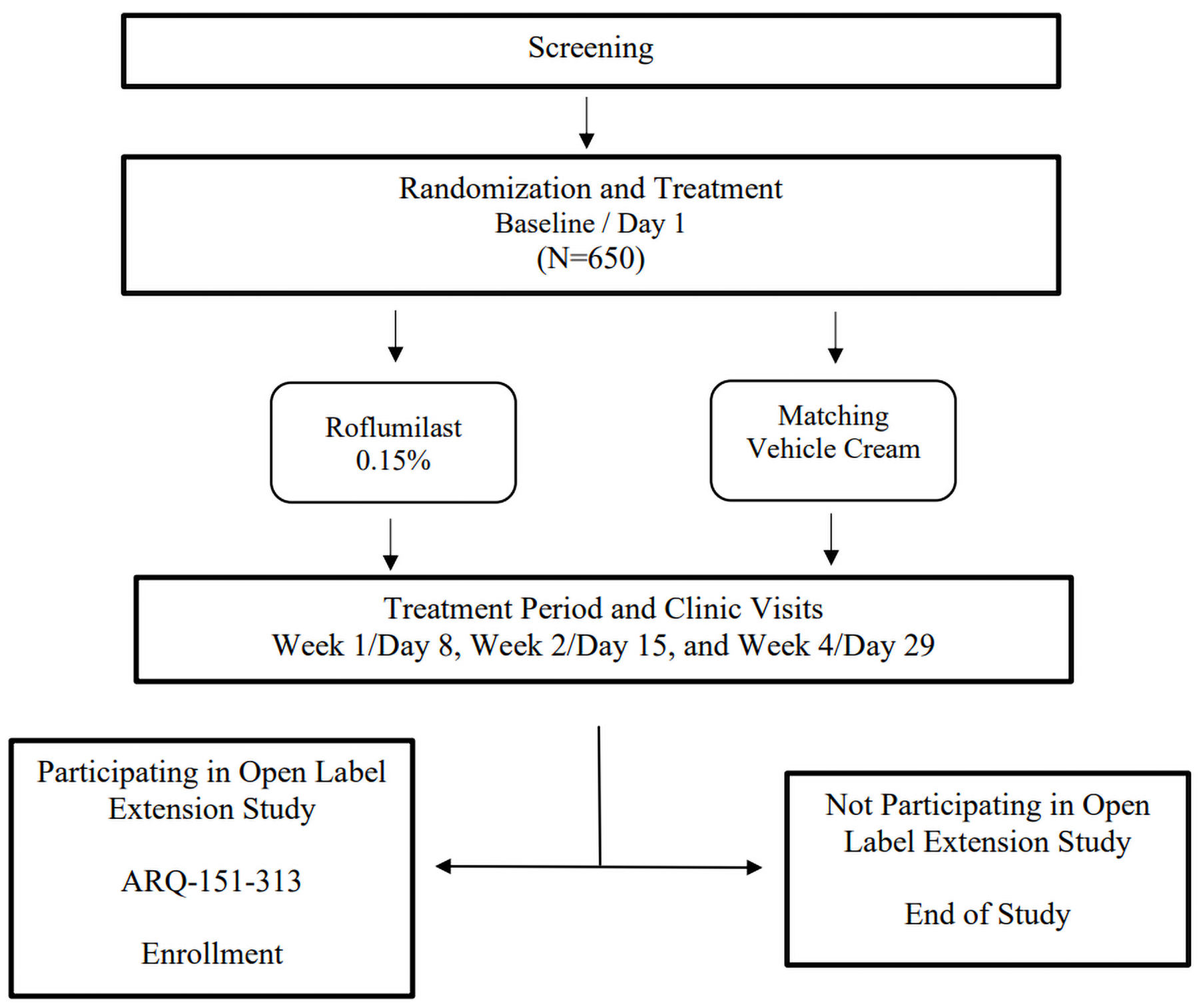
Sources: Statistical analysis plan for the INTEGUMENT-119 and INTEGUMENT-2 trials.20
The primary end point of both the INTEGUMENT-1 and INTEGUMENT-2 trials was vIGA-AD success, defined as a score of 0 (clear) or 1 (almost clear) plus a 2-grade or higher improvement from baseline at week 4. The secondary end points of the trials included vIGA-AD success at week 4 among patients with a vIGA-AD score of moderate at baseline; vIGA-AD success at weeks 1 and 2; vIGA-AD of clear or almost clear at weeks 1, 2, and 4; reduction in WI-NRS score of 4 or more points at weeks 1, 2, and 4 among patients aged 12 years or older with a baseline WI-NRS score of 4 or more; and the proportion of patients achieving at least a 75% reduction in the Eczema Area and Severity Index (EASI-75).
This review is based on the final analyses of the INTEGUMENT-1 (data cut-off: October 26, 2022) and INTEGUMENT-2 (data cut-off: November 21, 2022) trials. No interim analysis was planned for either trial. Both trials were funded by Arcutis Biotherapeutics, Inc.
Table 3: Characteristics of the INTEGUMENT-1 and INTEGUMENT-2 Trials
Study name, design, and sample size | Key inclusion criteria | Key exclusion criteria | Intervention and comparator | Relevant end points |
|---|---|---|---|---|
INTEGUMENT-1 trial Double-blind, vehicle-controlled, parallel-group, multicentre phase III RCT Total N = 654 INTEGUMENT-2 trial Double-blind, vehicle-controlled, parallel-group, multicentre phase III RCT Total N = 683 |
|
| Intervention: Roflumilast 0.15% cream administered topically once daily Comparator: Vehicle cream administered topically once daily | Primary end point:
Secondary end points:
Exploratory end points:
Safety end points:
|
AD = atopic dermatitis; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; DFI = Dermatitis Family Impact questionnaire; DLQI = Dermatology Life Quality Index; EASI-50 = 50% reduction in the Eczema Area and Severity Index; EASI-75 = 75% reduction in the Eczema Area and Severity Index; EASI-90 = 90% reduction in the Eczema Area and Severity Index; EASI-100 = 100% reduction in the Eczema Area and Severity Index; POEM = Patient-Oriented Eczema Measure; RCT = randomized controlled trial; SAE = serious adverse event; SCORAD = Scoring Atopic Dermatitis; TEAE = treatment-emergent adverse event; vIGA-AD = Validated Investigator Global Assessment for Atopic Dermatitis; WI-NRS = Worst Itch Numeric Rating Scale.
aThe basic features of the Hanifin criteria21 were pruritus, typical morphology and distribution (flexural lichenification in adults and facial and extensor eruptions in infants and children), chronic or chronically relapsing dermatitis, and personal or family history of atopy, in addition to 3 or more minor criteria.
Sources: Clinical Study Reports for INTEGUMENT-122 and INTEGUMENT-2 trials.23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Statistical Testing and Analysis Populations
There were approximately 650 patients planned for each of the INTEGUMENT-1 and INTEGUMENT-2 trials. This sample size provided approximately 95% power to detect an overall 15% difference between treatment groups on the primary end point (vIGA-AD success at week 4) at an alpha of 0.05 using a 2-sided stratified Cochran-Mantel-Haenszel test. The 15% treatment difference was estimated based on results of a phase II study comparing vIGA-AD success for roflumilast 0.15% cream versus vehicle cream for the treatment of plaque psoriasis.24
To control for a familywise type I error rate of 0.05, the secondary end point of vIGA-AD success at week 4 for patients with a vIGA-AD score of moderate at randomization was tested only if the primary end point was statistically significant. Moreover, if the primary and the selected secondary end points (vIGA-AD success at week 4 for patients with a vIGA-AD score of moderate at randomization) were statistically significant, the remaining secondary end points were inferentially tested using a hierarchical testing procedure by partitioning alpha. The remaining secondary end points were grouped into 2 families: secondary end point family 1 (WI-NRS success at week 4, week 2, and week 1) and secondary end point family 2 (EASI-75 at week 4, vIGA-AD of clear or almost clear at week 4, vIGA-AD success at week 2 and week 1, and vIGA-AD score of clear or almost clear at week 2 and week 1). An alpha level of 0.03 was used to test the end points in secondary end point family 1 sequentially, whereas an alpha level of 0.02 was used to test the end points in secondary end point family 2. A summary of the hierarchical testing procedure is presented in the Supplementary Material document, Appendix 3 (Figure 1).
Analyses for the primary end point and most secondary end points were based on the intention-to-treat (ITT) population, which consisted of all randomized patients. However, the analysis for the secondary end point vIGA-AD success at week 4 for patients with a vIGA-AD score of moderate at randomization was based on a subset of patients from the ITT population who had a vIGA-AD score of 3 at randomization. Moreover, the analyses for WI-NRS success were based on a subset of patients from the ITT population who were aged 12 years or older and had a baseline WI-NRS score of at least 4 points. The safety population consisted of all patients who received at least 1 application of the study intervention.
The primary end point was analyzed using the Cochran-Mantel-Haenszel test with a 2-sided significance level (alpha) of 0.05 and stratification by randomization factors (i.e., vIGA status and study site). The Mantel-Haenszel common odds ratio (OR) adjusted for randomization factors was reported alongside the associated 95% confidence intervals (CIs). Missing data for this end point was handled using multiple imputations based on the predictive mean matching method. The statistical tests and methods of handling the missing data that were used for the analysis of the secondary end points were similar to those used for the analysis of the primary end point. However, no adjustment was made for the randomized vIGA-AD score at baseline for the analysis for the secondary end point vIGA-AD success at week 4 for patients with a vIGA-AD score of moderate at randomization. Analyses for exploratory end points were performed using observed data from the ITT population. Exploratory end points evaluating change from baseline were analyzed using the analysis of covariance (ANCOVA) approach with stratification by randomization factors and the baseline value of the parameter under analysis as the covariate.
In addition to prespecified subgroup analyses performed for patients with a vIGA-AD score of moderate at baseline (i.e., the vIGA-AD success could be assessed) and patients aged 12 years or older with a WI-NRS score of 4 or higher at baseline (both of which were adjusted for multiplicity), additional subgroup analyses on the following subgroups were performed for the primary and secondary end points:
age group (6 to 11 years, 12 to 17 years, and 18 years or older)
sex (female, male)
race (Asian, Black or African American, white, other)
ethnicity (Hispanic or Latino, not Hispanic or Latino)
vIGA-AD score at randomization (mild, moderate)
actual baseline vIGA-AD score (mild, moderate)
baseline BSA affected by AD (less 10%; 10% or greater; and first, second, and third tertiles)
baseline EASI total score (7 or less; greater than 7; and first, second, and third tertiles)
Fitzpatrick skin type at screening (type I through III, type IV through VI)
prior IIC-TCS (no)
facial involvement (yes, no).
Of note, these additional subgroup analyses were not adjusted for multiplicity. Sensitivity analyses conducted for the primary end point included nonresponder imputation, a tipping point analysis, use of observed data on the ITT and per-protocol populations, and use of the modified ITT population (defined as the set of all randomized patients with the exception of patients who missed the week 4 vIGA-AD assessment specifically due to COVID-19 disruption). A similar set of sensitivity analyses (excluding the use of the modified ITT population) was performed for the secondary end point vIGA-AD success at week 4 based on the ITT population with a vIGA-AD score of moderate. If allowed by the hierarchical testing procedure, the sensitivity analyses performed on the other secondary end points were nonresponder imputation and the use of observed data in the ITT population. Additional analyses were conducted on the primary end point to assess the impact of the pooling of study sites.
Patient Disposition
Patient disposition for the INTEGUMENT-1 and INTEGUMENT-2 trials is summarized in the Supplemental Material, Appendix 4 (Table 5).
Of the 865 patients screened in the INTEGUMENT-1 trial, 433 patients and 221 patients were assigned to receive roflumilast 0.15% cream and vehicle cream, respectively. Of these, 404 patients (93.3%) who received roflumilast 0.15% cream and 208 patients (94.1%) who received vehicle cream completed the study. Of the 886 patients screened in the INTEGUMENT-2 trial, 451 patients and 232 patients were assigned to receive roflumilast 0.15% cream and vehicle cream, respectively. Of these, 410 patients (90.9%) who received roflumilast 0.15% cream and 211 patients (90.9%) who received vehicle cream completed the study.
In both trials, rates of study discontinuation were similar between the roflumilast 0.15% cream and vehicle cream groups (INTEGUMENT-1 trial: 6.7% for roflumilast 0.15% cream versus 5.9% for vehicle cream; INTEGUMENT-2 trial: 9.1% for both groups). In the INTEGUMENT-1 trial, the most common reason for study discontinuation was lost to follow-up (2.5%) among the roflumilast 0.15% cream group and withdrawal of consent among the vehicle cream group (2.3%). In the INTEGUMENT-2 trial, the most common reason for study discontinuation was withdrawal of consent for both the roflumilast 0.15% cream (3.3%) and vehicle cream (3.9%) groups.
In the safety population of the INTEGUMENT-1 trial, 24.9% of patients in the roflumilast 0.15% cream group and 21.7% of patients in the vehicle cream group had at least 1 important protocol deviation. In the safety population of the INTEGUMENT-2 trial, 27.4% of patients in the roflumilast 0.15% cream group and 21.7% of patients in the vehicle cream group had at least 1 important protocol deviation. Across both trials, the most common important protocol deviations were issues with investigational product administration (INTEGUMENT-1 trial: 8.1% for both roflumilast 0.15% cream and vehicle cream; INTEGUMENT-2 trial: 8.0% for roflumilast 0.15% cream versus 7.4% for vehicle cream), informed consent (INTEGUMENT-1 trial: 6.7% for roflumilast 0.15% cream versus 7.7% for vehicle cream; INTEGUMENT-2 trial: 5.5% for roflumilast 0.15% cream versus 6.5% for vehicle cream), and study visits (INTEGUMENT-1 trial: 4.8% for roflumilast 0.15% cream versus 2.7% for vehicle cream; INTEGUMENT-2 trial: 5.8% for roflumilast 0.15% cream versus 3.5% for vehicle cream).
Baseline Characteristics
The baseline characteristics for the INTEGUMENT-1 and INTEGUMENT-2 trials are summarized in Table 4.
Patient demographic and disease characteristics were similar across the roflumilast 0.15% cream and vehicle cream groups in both the INTEGUMENT-1 and INTEGUMENT-2 trials. Across both trials, most patients were adults (> 48%), female (> 54%), and white (> 58%). Across both trials, most patients had a vIGA-AD observed score of moderate (> 70%), were white (> 75%), and had AD affecting the face (> 41%). In the INTEGUMENT-1 trial, 285 patients (65.8%) treated with roflumilast 0.15% cream and 136 patients (61.5%) treated with vehicle cream had a prior IIC-TCS. In the INTEGUMENT-2 trial, 260 patients (57.6%) treated with roflumilast 0.15% cream and 132 patients (56.9%) treated with vehicle cream had a prior IIC-TCS.
Table 4: Summary of Baseline Characteristics for the INTEGUMENT-1 and INTEGUMENT-2 Trials (ITT Population)
Characteristic | INTEGUMENT-1 trial | INTEGUMENT-2 trial | ||
|---|---|---|---|---|
Roflumilast 0.15% cream | Vehicle | Roflumilast 0.15% cream | Vehicle | |
Demographic characteristics | ||||
Age (years) | ||||
Mean (SD) | 28.1 (19.14) | 28.5 (18.94) | 27.7 (19.60) | 26.2 (18.94) |
Median (range) | 20.0 (6 to 91) | 22.0 (6 to 84) | 20.0 (6 to 84) | 18.0 (6 to 79) |
Age category, N (%) | ||||
6 to 11 years | 88 (20.3) | 42 (19.0) | 126 (27.9) | 61 (26.3) |
12 to 17 years | 112 (25.9) | 54 (24.4) | 80 (17.7) | 52 (22.4) |
18 to 64 years | 209 (48.3) | 115 (52.0) | 225 (49.9) | 108 (46.6) |
≥ 65 years | 24 (5.5) | 10 (4.5) | 20 (4.4) | 11 (4.7) |
Sex at birth, N (%) | ||||
Female | 237 (54.7) | 129 (58.4) | 252 (55.9) | 143 (61.6) |
Male | 196 (45.3) | 92 (41.6) | 199 (44.1) | 89 (38.4) |
Ethnicity, N (%) | ||||
Hispanic or Latino | 99 (22.9) | 56 (25.3) | 51 (11.3) | 16 (6.9) |
Not Hispanic or Latino | 333 (76.9) | 164 (74.2) | 397 (88.0) | 213 (91.8) |
Not reported | 1 (0.2) | 1 (0.5) | 3 (0.7) | 3 (1.3) |
Race, N (%) | ||||
American Indian or Alaska Native | 2 (0.5) | 0 | 5 (1.1) | 1 (0.4) |
Asian | 63 (14.5) | 32 (14.5) | 51 (11.3) | 30 (12.9) |
Black or African American | 80 (18.5) | 46 (20.8) | 96 (21.3) | 50 (21.6) |
More than 1 race | 12 (2.8) | 6 (2.7) | 12 (2.7) | 8 (3.4) |
Native Hawaiian or other Pacific Islander | 1 (0.2) | 0 | 0 | 0 |
White | 261 (60.3) | 129 (58.4) | 268 (59.4) | 138 (59.5) |
Other | 14 (3.2) | 8 (3.6) | 19 (4.2) | 5 (2.2) |
Clinical characteristics | ||||
Height (cm) | ||||
N | 433 | 221 | 451 | 232 |
Mean (SD) | 160.9 (16.49) | 159.7 (17.49) | 158.1 (18.36) | 158.2 (18.63) |
Median (range) | 165.0 (110 to 196) | 163.0 (109 to 198) | 163.0 (108 to 196) | 162.0 (94 to 191) |
Baseline weight (kg) | ||||
N | 433 | 221 | 451 | 232 |
Mean (SD) | 69.4 (26.9) | 68.7 (27.8) | 68.4 (30.8) | 66.9 (30.4) |
Median (range) | 67.7 (19.4 to 150.5) | 67.8 (18.2 to 193.4) | 68.3 (15.0 to 157.7) | 64.9 (18.9 to 176.4) |
Baseline BMIa (kg/m2) | ||||
N | 233 | 125 | 245 | 119 |
Mean (SD) | 29.6 (7.1) | 29.5 (7.1) | 30.8 (8.1) | 29.8 (8.9) |
Median (range) | 28.0 (17.6 to 50.3) | 28.8 (18.0 to 56.5) | 28.9 (17.2 to 65.6) | 28.3 (17.2 to 66.4) |
BMI percentileb | ||||
N | 200 | 96 | 206 | 113 |
Mean | 69.5 | 67.1 | 68.3 | 67.8 |
SD | 29.7 | 27.8 | 27.1 | 28.9 |
Median (range) | 80.0 (0.0 to 99.7) | 73.1 (2.1 to 99.8) | 73.3 (0.0 to 99.8) | 76.6 (0.0 to 99.7) |
Fitzpatrick skin type, N (%) | ||||
Type I | 14 (3.2) | 13 (5.9) | 24 (5.3) | 10 (4.3) |
Type II | 97 (22.4) | 52 (23.5) | 112 (24.8) | 59 (25.4) |
Type III | 122 (28.2) | 47 (21.3) | 112 (24.8) | 57 (24.6) |
Type IV | 108 (24.9) | 50 (22.6) | 91 (20.2) | 58 (25.0) |
Type V | 53 (12.2) | 37 (16.7) | 65 (14.4) | 26 (11.2) |
Type VI | 39 (9.0) | 22 (10.0) | 47 (10.4) | 22 (9.5) |
Percent BSA affected by AD | ||||
N | 433 | 221 | 451 | 232 |
Mean (SD) | 13.4 (11.9) | 12.9 (11.1) | 13.7 (11.6) | 14.9 (11.3) |
Median (range) | 9.5 (3.0 to 87.0) | 9.0 (3.0 to 86.0) | 10.0 (3.0 to 88.0) | 11.0 (3.0 to 63.0) |
vIGA-AD observed, N (%) | ||||
Mild | 103 (23.8) | 59 (26.7) | 108 (23.9) | 53 (22.8) |
Moderate | 330 (76.2) | 162 (73.3) | 343 (76.1) | 179 (77.2) |
Average weekly WI-NRSc | ||||
N | 423 | 217 | 435 | 224 |
Mean (SD) | 5.9 (2.1) | 5.9 (2.4) | 6.2 (2.2) | 5.9 (2.1) |
Median (range) | 6.1 (0.0 to 10.0) | 6.0 (0.0 to 10) | 6.4 (0.0 to 10) | 6.1 (0.0 to 10) |
≥ 4, N (%) | 350 (80.8) | 168 (76.0) | 359 (79.6) | 181 (78.0) |
Baseline EASI | ||||
Mean (SD) | 9.9 (5.3) | 9.8 (5.1) | 10.3 (6.1) | 10.2 (5.3) |
Median (range) | 8.2 (4.4 to 47.4) | 8.2 (4.2 to 37.9) | 8.5 (4.9 to 52.5) | 8.4 (3.4 to 31.2) |
SCORAD | ||||
Mean (SD) | 44.9 (11.1) | 44.3 (10.3) | 46.0 (10.7) | 45.8 (10.9) |
Median (range) | 44.7 (18.9 to 77.9) | 43.6 (20.9 to 68.3) | 45.7 (18.2 to 81.5) | 44.1 (20.9 to 83.5) |
DLQId | ||||
N | 246 | 134 | 252 | 124 |
Mean (SD) | 7.8 (5.6) | 8.6 (6.8) | 9.4 (6.5) | 8.4 (6.0) |
Median (range) | 6.0 (0 to 28) | 7.0 (0 to 30) | 8.0 (1 to 30) | 7.0 (1 to 28) |
CDLQIe | ||||
N | 185 | 87 | 198 | 108 |
Mean (SD) | 7.6 (5.5) | 7.4 (5.8) | 8.0 (5.4) | 7.1 (5.3) |
Median (range) | 6.0 (0 to 28) | 6.0 (0 to 26) | 7.0 (0 to 23) | 6.0 (1 to 22) |
DFIf | ||||
N | 200 | 95 | 206 | 113 |
Mean (SD) | 6.6 (6.3) | 7.1 (6.8) | 6.3 (5.6) | 6.0 (5.7) |
Median (range) | 5.0 (0 to 27) | 5.0 (0 to 30) | 5.0 (0 to 24) | 4.0 (0 to 26) |
POEM | ||||
Mean (SD) | 15.6 (6.6) | 15.1 (6.8) | 16.0 (6.1) | 15.5 (6.1) |
Median (range) | 16.0 (0 to 28) | 15.0 (0 to 28) | 16.0 (2 to 28) | 15.0 (2 to 28) |
AD involvement location | ||||
Face, N (%) | 181 (41.8) | 98 (44.3) | 189 (41.9) | 99 (42.7) |
Eyelids, N (%) | 84 (19.4) | 43 (19.5) | 94 (20.8) | 56 (24.1) |
Prior inadequate response, intolerance, or contraindication to a TCS | ||||
TCS, N (%) | 285 (65.8) | 136 (61.5) | 260 (57.6) | 132 (56.9) |
TCI, N (%) | 77 (17.8) | 35 (15.8) | 84 (18.6) | 46 (19.8) |
Crisaborole, N (%) | 23 (5.3) | 14 (6.3) | 45 (10.0) | 16 (6.9) |
AD = atopic dermatitis; BMI = body mass index; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; DFI = Dermatitis Family Impact questionnaire; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity; ITT = intention to treat; POEM = Patient-Oriented Eczema Measure; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; vIGA-AD = Validated Investigator Global Assessment for Atopic Dermatitis; WI-NRS = Worst Itch Numeric Rating Scale.
Note: Racial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
aPatients aged ≥ 18 years.
bPatients aged 6 to 17 years. BMI percentile for children and adolescents is calculated using data files and instructions provided by the Centers for Disease Control and Prevention.
cBaseline is defined as the average of all nonmissing scores reported during the last 7 days of the screening period.
dPatients aged ≥ 17 years.
ePatients aged 6 to 16 years.
fPatients aged ≤ 17 years.
Sources: Clinical Study Reports for INTEGUMENT-122 and INTEGUMENT-2 trials.23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Treatment Exposure and Concomitant Medications
Details of patients’ treatment exposure and adherence in each of the INTEGUMENT-1 and INTEGUMENT-2 trials are in the Supplemental Material document, Appendix 4.
The mean number of applications per patient during the study period was similar for both treatment groups across the INTEGUMENT-1 (27.5 applications for roflumilast 0.15% cream versus 27.4 applications for vehicle cream) and INTEGUMENT-2 (27.1 applications for roflumilast 0.15% cream versus 26.9 applications for vehicle cream) trials. In both trials, higher mean values were observed for the total weight of vehicle cream applied (INTEGUMENT-1 trial: 77.2 g; INTEGUMENT-2 trial: 80.1 g) versus the total weight of roflumilast 0.15% cream applied (INTEGUMENT-1 trial: 55.4 g; INTEGUMENT-2 trial: 75.0 g). Rates of treatment compliance were high and similar across the INTEGUMENT-1 (97.0% for roflumilast 0.15% cream versus 98.6% for vehicle cream) and INTEGUMENT-2 (98.9% for roflumilast 0.15% cream versus 98.3% for vehicle cream) trials.
Details of patients’ use of prior and concomitant medications in each of the INTEGUMENT-1 and INTEGUMENT-2 trials are in the Supplemental Material document, Appendix 4 (Table 6 and Table 7).
Most patients received at least 1 prior medication in both the INTEGUMENT-1 (86.6% for roflumilast 0.15% cream versus 83.7% for vehicle cream) and INTEGUMENT-2 (83.6% for roflumilast 0.15% cream versus 82.2% for vehicle cream) trials. Across both trials, the most common class of prior medication received among patients was corticosteroid (INTEGUMENT-1 trial: 75.5% for roflumilast 0.15% cream versus 71.5% for vehicle cream; INTEGUMENT-2 trial: 73.9% for roflumilast 0.15% cream versus 70.0% for vehicle cream).
Most patients received at least 1 concomitant medication in the INTEGUMENT-1 (67.9% for roflumilast 0.15% cream versus 66.5% for vehicle cream) and INTEGUMENT-2 (66.4% for roflumilast 0.15% cream versus 67.8% for vehicle cream) trials. The most common classes of concomitant medications used in both trials were adrenergic inhalants (INTEGUMENT-1 trial: 14.3% for roflumilast 0.15% cream versus 12.2% for vehicle cream; INTEGUMENT-2 trial: 13.3% for roflumilast 0.15% cream versus 14.3% for vehicle cream), antihistamines for systemic use (INTEGUMENT-1 trial: 14.5% for roflumilast 0.15% cream versus 17.2% for vehicle cream; INTEGUMENT-2 trial: 22.3% for roflumilast 0.15% cream versus 19.6% for vehicle cream), and emollients and protectives (INTEGUMENT-1 trial: 13.9% for roflumilast 0.15% cream versus 14.9% for vehicle cream; INTEGUMENT-2 trial: 11.7% for roflumilast 0.15% cream versus 14.8% for vehicle cream).
Critical Appraisal
Internal Validity
The INTEGUMENT-1 and INTEGUMENT-2 trials were identical, double-blind, vehicle-controlled RCTs. For both trials, randomization was stratified by vIGA-AD score at baseline and by study site according to a computer-generated list, and treatment allocation was performed using an internet-based response system, which was deemed appropriate by the CDA-AMC review team. The baseline and demographic characteristics were generally balanced between the 2 groups in both trials. Although differences between groups were noted for the sex distribution in the INTEGUMENT-2 trial and the Fitzpatrick skin type in the INTEGUMENT-1 trial, the clinical experts consulted by CDA-AMC indicated these differences would have little impact on the interpretation of the results. The CDA-AMC review team deemed the use of a double-blind design to be appropriate in both trials; however, patients who received roflumilast cream may have noticed improvements in their symptoms over the study period compared with those who received vehicle cream. Therefore, there is a risk that patients could have become aware of their treatment allocation, which could have introduced performance bias in the patient-reported outcomes.
Efficacy analyses for most outcomes in the INTEGUMENT-1 and INTEGUMENT-2 trials were conducted based on the ITT populations, which is considered the ideal approach for analyzing efficacy outcomes. Of note, the analysis for WI-NRS success was performed only on patients who were aged 12 years or older with a WI-NRS score of 4 or higher at baseline, which comprised 63.1% and 58.6% of the ITT populations in the INTEGUMENT-1 and INTEGUMENT-2 trials, respectively. Of note, the sponsor indicated that patients who were younger than age 12 were excluded from the analysis of WI-NRS success due to the cognitive demands of the scale and the lack of validation of the WI-NRS instrument in younger children. However, the sponsor indicated that alternative instruments, such as the Children’s Dermatology Life Quality Index (CLDQI), can be used to assess treatment effect on itch and related symptoms for patients excluded from the analysis.
In both the INTEGUMENT-1 and INTEGUMENT-2 trials, the rates of discontinuation of the study intervention were low and similar between the roflumilast 0.15% cream and vehicle cream groups (INTEGUMENT-1 trial: 6.7% for roflumilast 0.15% cream versus 5.9% for vehicle cream; INTEGUMENT-2 trial: 9.1% for both groups). In both trials, rates of compliance were high (i.e., above 97%) and similar between the roflumilast 0.15% cream and vehicle cream groups. Across the trials, the most common reasons for discontinuation were withdrawal by patient and lost to follow-up. Missing data for the primary and secondary end points of the trials were handled using multiple imputation, which was deemed to be appropriate by the CDA-AMC team. For the analysis of the primary end point and most secondary end points, the rates of missing data were low (less than 10%) and similar between the roflumilast 0.15% cream and vehicle cream groups in both trials. Of note, sensitivity analyses were conducted for the primary end point to assess different methods for handling missing data; the results of these analyses were consistent with those of the primary analysis. For both trials, only observed data were used in the analyses of exploratory end points and no methods of imputation were described.
In both trials, a hierarchical testing procedure was used to adjust for multiplicity in the primary and secondary end points. The primary end point was met in both trials. All 10 secondary end points of the INTEGUMENT-1 trial were met, whereas only 7 of the secondary end points were met in the INTEGUMENT-2 trial. Of note, exploratory end points (e.g., percent BSA affected, Patient-Oriented Eczema Measure [POEM], Scoring Atopic Dermatitis [SCORAD], CDLQI, Dermatology Life Quality Index [DLQI]) were not included in the hierarchical testing procedure and were not adjusted for multiplicity. Thus, a type I error (i.e., a false-positive result) for these outcomes cannot be ruled out. For the primary and secondary outcomes in both trials, the study protocol prespecified multiple subgroup analyses for several demographic and clinical characteristics, which included patients with a prior IIC-TCS (i.e., the sponsor’s reimbursement request population). However, these subgroup analyses were not formally adjusted for multiplicity and should be interpreted as supportive evidence.
The INTEGUMENT-1 and INTEGUMENT-2 trials measured outcomes important to patients and clinicians (e.g., control of signs and symptoms, HRQoL) using several instruments that have been validated in patients with AD. Such instruments included vIGA-AD, EASI, WI-NRS, POEM, SCORAD, DLQI, and CLDQI. These instruments have established measurement properties in patients with AD, with most outcomes having evidence of reliability, validity, and responsiveness; some also have minimal important differences (MIDs). The clinical experts consulted by CDA-AMC agreed with the definitions of success for vIGA-AD and WI-NRS used in the trials and considered these to be standard across clinical trials for AD. Moreover, the definitions of vIGA-AD success, WI-NRS success, and EASI-75 were aligned with the MID estimates for these measures found in the literature.
External Validity
The clinical experts consulted by CDA-AMC agreed that the eligibility criteria of the INTEGUMENT-1 and INTEGUMENT-2 trials were representative of patients with mild to moderate AD in clinical practice. Of note, the eligibility criteria of both trials did not restrict patients based on prior treatment with a TCS. Although the clinical experts consulted by CDA-AMC noted variations in patient demographics across clinics in real-world practice, they agreed that a variety of ages, disease severities, and ethnicities were well represented in the baseline characteristics of patients enrolled in the INTEGUMENT-1 and INTEGUMENT-2 trials. They also noted that prior and concomitant medications received by patients in the trials were reflective of those observed in clinical practice.
Patients in the INTEGUMENT-1 and INTEGUMENT-2 trials were instructed to apply roflumilast topically once daily to AD lesions affecting at least 3% of BSA over a 4-week treatment period. The dose and instructions for applying roflumilast outlined in the INTEGUMENT-1 and INTEGUMENT-2 trials were aligned with those outlined in the Health Canada indication for roflumilast.25 The clinical experts consulted by CDA-AMC also indicated that the instructions used in the trials are aligned with how they would instruct patients to apply roflumilast in clinical practice. In the trials, efficacy measurements for the scalp, soles, and palms were not included in the analysis. The clinical experts consulted by CDA-AMC agreed that this protocol would not be expected to affect the interpretation of the results of the trials, noting that the trials aimed to assess patients with generalized mild to moderate AD rather than patients with dermatitis specifically affecting the scalp, palms, and soles.
The INTEGUMENT-1 and INTEGUMENT-2 trials assessed roflumilast cream compared with vehicle cream. Because the trials did not compare roflumilast with other relevant comparators for mild to moderate AD, the results of the trial do not provide an assessment of the efficacy and safety of roflumilast compared to existing treatments for mild to moderate AD in Canadian clinical practice. To address this gap in evidence, the sponsor submitted an NMA that evaluated roflumilast 0.15% cream compared with relevant comparators for mild to moderate AD in Canadian clinical practice.
The INTEGUMENT-1 and INTEGUMENT-2 trials assessed outcomes that were relevant to patients and clinicians. According to input on AD from patient groups, important outcomes include control of AD signs, reductions in symptoms and flares, and improvement in HRQoL. The clinical experts consulted by CDA-AMC considered several measured outcomes of the INTEGUMENT-1 and INTEGUMENT-2 trials to be of clinical importance, such as vIGA-AD, EASI-75, WI-NRS, and HRQoL measures (e.g., DLQI). Of note, the experts also indicated that vIGA-AD was a practical and informative measure for assessing AD in clinical practice, whereas the use of EASI was less practical for assessing mild to moderate AD in clinical practice. Although measures such as POEM and SCORAD are commonly assessed in clinical trials for AD, the clinical experts noted that these outcomes are not commonly assessed in clinical practice.
The patients in the INTEGUMENT-1 and INTEGUMENT-2 trials were treated for 4 weeks, with the latest time point for follow-up being week 4. One clinical expert consulted by CDA-AMC agreed that patients would not only be assessed for outcomes after 4 weeks; they would also be assessed every 3 months thereafter. The other clinical expert consulted by CDA-AMC indicated that the length of follow-up used in the trial is suitable for capturing the speed of improvement in signs and symptoms; however, the expert noted that efficacy and safety should be assessed over a longer time frame (e.g., at least 6 months).
Results
The key efficacy and harms results and findings from the GRADE assessment (Table 5) are presented in this section. Detailed efficacy and harms results can be found in Appendix 4 in the Supplemental Material document (Table 9 and Table 10).
Efficacy
Key results (based on a data cut-off of October 26, 2022, for the INTEGUMENT-1 trial and November 21, 2022, for the INTEGUMENT-2 trial) include the following.
vIGA-AD Success
The primary end point, the proportion of patients in the ITT population achieving vIGA-AD success at week 4, was met in both trials. At week 4, a higher proportion of patients receiving roflumilast 0.15% cream achieved vIGA-AD success compared with the patients receiving vehicle cream in both the INTEGUMENT-1 (32.0% for roflumilast versus 15.2% for vehicle; percent difference [PD] = 17.4%; 95% CI, 11.09 to 23.75) and INTEGUMENT-2 (28.9% for roflumilast versus 12.0% for vehicle; PD = 16.5%; 95% CI, 10.61 to 22.42) trials (Supplemental Material, Appendix 4, Table 9). For both trials, the results of the sensitivity and subgroup analyses favoured the roflumilast 0.15% cream group, which was consistent with the primary analysis.
The proportion of patients achieving vIGA-AD success at week 4 among patients with a vIGA-AD score of moderate at randomization, as well as the proportions of patients achieving vIGA-AD success at weeks 1 and 2 were secondary end points in both trials. For all 3 outcomes, the results favoured the roflumilast 0.15% cream group in both trials, which was consistent with the primary outcome (Supplemental Material, Appendix 4, Table 9).
Reduction of 75% in the EASI
The proportion of patients who achieved EASI-75 at week 4 was a secondary end point in both trials. At week 4, a higher proportion of patients receiving roflumilast 0.15% cream achieved EASI-75 compared with the vehicle cream in both the INTEGUMENT-1 trial (43.2% for roflumilast versus 22.0% for vehicle; PD = 22.0%; 95% CI, 15.09 to 28.94) and INTEGUMENT-2 trial (42.0% for roflumilast versus 19.7% for vehicle; PD = 20.8%; 95% CI, 14.14 to 27.50) (Supplemental Material, Appendix 4, Table 9).
WI-NRS Success
The proportion of patients (who were aged at least 12 years and had a baseline WI-NRS score of 4 or more points) achieving WI-NRS success at weeks 1, 2, and 4 were secondary end points in both trials. At week 4, a higher proportion of patients receiving roflumilast 0.15% cream achieved WI-NRS success compared with the patients receiving vehicle cream in both the INTEGUMENT-1 trial (33.6% for roflumilast versus 20.7% for vehicle; PD = 13.3%; 95% CI, 4.01 to 22.60) and INTEGUMENT-2 trial (30.2% for roflumilast versus 12.4% for vehicle; PD = 15.0%; 95% CI, 6.52 to 23.54) (Supplemental Material, Appendix 4, Table 9). Similar results were observed for this outcome that were measured at weeks 1 and 2 (Supplemental Material, Appendix 4, Table 9).
Change in POEM Score From Baseline
Change in POEM score from baseline at week 4 was evaluated as an exploratory end point for both trials and was not tested for multiplicity. A decrease in POEM score indicates improvement in HRQoL. At week 4, the between-group least squares (LS) mean difference comparing roflumilast 0.15% cream and vehicle cream was █████ ████ ███ █████ ██ ██████ in the INTEGUMENT-1 trial and ████ ██████ ██████ ██ ██████ in the INTEGUMENT-2 trial (Supplemental Material, Appendix 4, Table 9).
Change in DLQI Score From Baseline
Change in DLQI score from baseline to week 4 was evaluated as an exploratory end point for both trials and was not tested for multiplicity. A decrease in DLQI score indicates improvement in HRQoL. At week 4, the between-group LS mean difference comparing roflumilast 0.15% cream and vehicle cream was −0.5 points (95% CI, −1.34 to 0.30) in the INTEGUMENT-1 trial and −1.6 points (−2.51 to −0.59) in the INTEGUMENT-2 trial (Supplemental Material, Appendix 4, Table 9).
Change in CDLQI Score From Baseline
Change in CDLQI score from baseline at week 4 was evaluated as an exploratory end point for both trials and was not tested for multiplicity. A decrease in CDLQI score indicates improvement in HRQoL. At week 4, the between-group LS mean difference comparing roflumilast 0.15% cream and vehicle cream was −2.1 points (95% CI, −3.04 to 1.22) in the INTEGUMENT-1 trial and −1.6 points (−2.50 to −0.65) in the INTEGUMENT-2 trial (Supplemental Material, Appendix 4, Table 9).
Summary of Other Findings
Score of Clear or Almost Clear in the vIGA-AD
The proportion of patients achieving vIGA-AD success at weeks 1, 2, and 4 were secondary end points in both trials. At week 4, a higher proportion of patients receiving roflumilast 0.15% cream achieved a vIGA-AD score of clear or almost clear compared with the vehicle cream in both the INTEGUMENT-1 trial (41.5% for roflumilast versus 25.2% for vehicle; PD = 17.6%; 95% CI, 10.69 to 24.55) and INTEGUMENT-2 trial (39.0% for roflumilast versus 16.9% for vehicle; PD = 21.0%; 95% CI, 14.58 to 27.50) (Supplemental Material, Appendix 4, Table 9). Similar results for this outcome were observed at weeks 1 and 2 (Supplemental Material, Appendix 4, Table 9).
Percent Change From Baseline in BSA Affected by AD
Percent change in BSA affected by AD from baseline to week 4 was evaluated as an exploratory end point for both trials and was not tested for multiplicity. At week 4, a larger decrease in percent BSA affected by AD was noted for the roflumilast group compared with the vehicle group in both the INTEGUMENT-1 trial (−54.96% for roflumilast versus −33.62% for vehicle; LS mean change = −21.34%; 95% CI, −28.44% to −14.23%) and INTEGUMENT-2 trial (−51.78% for roflumilast versus −23.06% for vehicle; LS mean change = −28.72%; 95% CI, −36.35% to −21.10%) (Supplemental Material, Appendix 4, Table 9).
Change in SCORAD Score From Baseline
The change in SCORAD score from baseline to week 4 was evaluated as an exploratory end point for both trials and was not tested for multiplicity. A decrease in SCORAD score indicates improvement in HRQoL. At week 4, the between-group LS mean difference in SCORAD score comparing roflumilast 0.15% cream and vehicle cream was █████ ██████ ████ ███ █████ ██ ██████ in the INTEGUMENT-1 trial and █████ ██████ ████ ███ ██████ ██ ██████ in the INTEGUMENT-2 trial (Supplemental Material, Appendix 4, Table 9).
Harms
Key results are presented in the Supplementary Material document, Appendix 4 (Table 10) and include the following:
TEAEs were more frequent among patients treated with roflumilast 0.15% cream compared with the vehicle cream in both the INTEGUMENT-1 (21.2% for roflumilast versus 15.8% for vehicle) and INTEGUMENT-2 (22.6% for roflumilast versus 13.0% for vehicle) trials (Supplemental Material, Appendix 4, Table 10).
In both trials, the most common TEAEs among the roflumilast 0.15% cream group were headache (INTEGUMENT-1 trial: 2.3%; INTEGUMENT-2 trial: 3.5%), application-site pain (INTEGUMENT-1 trial: 2.1%; INTEGUMENT-2 trial: 0.9%), and nausea (INTEGUMENT-1 trial: 1.8%; INTEGUMENT-2 trial: 2.0%). The most common TEAEs among the vehicle cream group were COVID-19 (INTEGUMENT-1 trial: 2.3%; INTEGUMENT-2 trial: 1.3%) and headache (INTEGUMENT-1 trial: 1.4%; INTEGUMENT-2 trial: 0.4%) (Supplemental Material, Appendix 4, Table 10).
The incidence of SAEs was low for both treatment groups in both trials. However, SAEs were more frequent among patients treated with roflumilast 0.15% cream compared with the vehicle cream in both the INTEGUMENT-1 (0.9% for roflumilast versus 0% for vehicle) and INTEGUMENT-2 (0.9% for roflumilast versus 0% for vehicle) trials (Supplemental Material, Appendix 4, Table 10).
Rates of WDAEs were low and similar between both treatment groups in the INTEGUMENT-1 trial (1.4% for both groups) but were higher for the roflumilast 0.15% cream group in the INTEGUMENT-2 trial (1.8% for roflumilast versus 0.9% for vehicle cream) (Supplemental Material, Appendix 4, Table 10).
There were no deaths observed during either of the trials (Supplemental Material, Appendix 4, Table 10).
Similar proportions of patients who received roflumilast and vehicle cream had investigator-assessed dermal responses of 0 (no evidence of irritation) and 1 (minimal erythema, barely perceptible) in both the INTEGUMENT-1 (0 = no evidence of irritation: 97.5% for roflumilast versus 99.0% for vehicle cream; 1 = minimal erythema, barely perceptible: 2.2% for roflumilast versus 1.0% for vehicle cream), and INTEGUMENT-2 (0 = no evidence of irritation: 97.1% for roflumilast versus 95.7% for vehicle cream; 1 = minimal erythema, barely perceptible: 2.4% for roflumilast versus 3.3% for vehicle cream) trials (Supplemental Material, Appendix 4, Table 10).
Compared with vehicle cream, a higher proportion of patients who received roflumilast reported a sensation of 0 (none) in both the INTEGUMENT-1 (81.3% for roflumilast versus 74.5% for vehicle cream) and INTEGUMENT-2 (79.6% for roflumilast versus 70.7% for vehicle cream) trials (Supplemental Material, Appendix 4, Table 10).
Summary of Findings and Certainty of the Evidence
Literature-based MID estimates were used as the thresholds for the following outcomes: LS mean change in POEM score from baseline to 4 weeks (MID = 3.4 points), LS mean change in DLQI score from baseline to 4 weeks (MID = 4 points), and LS mean change in CDLQI score from baseline to 4 weeks (MID = 6 to 8 points). Of note, each of the MID estimates identified from the published literature pertained to a within-group change. Refer to the summary of outcome measures in Appendix 3 of the Supplemental Material document. In the absence of literature-based MID estimates, the clinical experts suggested a threshold of 10% for each of the following outcomes: proportion of patients achieving vIGA-AD success at week 4, proportion of patients achieving EASI-75 at week 4, proportion of patients achieving WI-NRS success, and proportion of patients with SAEs.
Table 5: Summary of Findings for Roflumilast 0.15% Cream vs. Vehicle Cream for Patients With Mild to Moderate AD
Outcome and follow-up | Patients, N (studies) | Effect (95% CI) | Certainty | What happens |
|---|---|---|---|---|
Extent and severity of disease | ||||
vIGA-AD score of 0 (clear) to 4 (severe AD) Proportion of patients achieving vIGA-AD success, i.e., score of 0 (clear) or 1 (almost clear) plus a 2-grade or higher improvement from baseline Follow-up: 4 weeks | 1,327 (2 RCTs) | INTEGUMENT-1 trial:
INTEGUMENT-2 trial:
| Higha,b | Roflumilast 0.15% cream results in a clinically important increase in the proportion of patients achieving vIGA-AD success at week 4 when compared with vehicle cream. |
EASI of 0 (clear) to 72 (very severe) Proportion of patients achieving EASI-75 (i.e., at least a 75% reduction in EASI score from baseline) Follow-up: 4 weeks | 1,327 (2 RCTs) | INTEGUMENT-1 trial:
INTEGUMENT-2 trial:
| Higha,b | Roflumilast 0.15% cream results in a clinically important increase in the proportion of patients achieving EASI-75 at week 4 when compared with vehicle cream. |
Symptom control | ||||
WI-NRS of 0 (no itch) to 10 (worst itch imaginable) Proportion of patients achieving WI-NRS success (i.e., at least a 4-point reduction in WI-NRS score among patients aged ≥ 12 years with a baseline WI-NRS score of 4 or higher) Follow-up: 4 weeks | 813 (2 RCTs) | INTEGUMENT-1 trial:
INTEGUMENT-2 trial:
| Moderateb,c | Roflumilast 0.15% cream likely results in a clinically important increase in the proportion of patients achieving WI-NRS success at week 4 when compared with vehicle cream. |
POEM score of 0 (clear) to 28 (very severe) LS mean change in POEM score from baseline to 4 weeks Follow-up: 4 weeks | 1,236 (2 RCTs) | INTEGUMENT-1 trial:
INTEGUMENT-2 trial:
| Lowb,d,e,f | Roflumilast 0.15% cream may result in little to no important difference in the LS mean change in POEM score from baseline to week 4 compared with vehicle cream. |
Health-related quality of life | ||||
DLQI score of 0 (best) to 30 (worst) LS mean change from baseline in score Follow-up: 4 weeks | 685 (2 RCTs) | INTEGUMENT-1 trial:
INTEGUMENT-2 trial:
| Moderateb,d,g,h | Roflumilast 0.15% cream likely results in little to no important difference in the LS mean change in DLQI score from baseline to week 4 when compared with vehicle cream. |
CDLQI score of 0 (best) to 30 (worst) LS mean change from baseline in score Follow-up: 4 weeks | 551 (2 RCTs) | INTEGUMENT-1 trial:
INTEGUMENT-2 trial:
| Highb,d,i | Roflumilast 0.15% cream results in little to no important difference in the LS mean change in CDLQI score from baseline to week 4 when compared with vehicle cream. |
Harms | ||||
Proportion of patients with SAEs Follow-up: 4 weeks | 1,336 (2 RCTs) | INTEGUMENT-1 trial:
INTEGUMENT-2 trial:
| Moderateb,d,j | Roflumilast 0.15% cream likely results in little to no important difference in the proportion of patients with 1 or more SAEs when compared with vehicle cream. |
AD = atopic dermatitis; CDA-AMC = Canada’s Drug Agency; CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI-75 = 75% reduction in the Eczema Area and Severity Index; LS = least squares; MID = minimal important difference; POEM = Patient-Oriented Eczema Measure; RCT = randomized controlled trial; SAE = serious adverse event; SE = standard error; vIGA-AD = Validated Investigator Global Assessment for Atopic Dermatitis; vs. = versus; WI-NRS = Worst Itch Numeric Rating Scale.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aAt the time of this review, there were no MIDs identified from published literature for this outcome. The clinical experts consulted by CDA-AMC indicated that a 10% between-group difference for this outcome could be clinically meaningful. In both the INTEGUMENT-1 and INTEGUMENT-2 trials, the point estimate and 95% CI for the between-group difference suggested a clinically important difference between roflumilast and vehicle cream.
bNot rated down for serious indirectness. The CDA-AMC review team noted that a duration of follow-up of 4 weeks may not be long enough to assess the efficacy and safety of roflumilast for AD. One clinical expert consulted by CDA-AMC agreed that patients would not only be assessed for outcomes after 4 weeks; they would also be assessed every 3 months thereafter. The other clinical expert consulted by CDA-AMC indicated that efficacy and safety should be assessed over a longer time frame than 4 weeks (e.g., at least 6 months) to inform reimbursement. However, Canadian treatment guidelines suggest that a minimum of 4 weeks is an appropriate length of follow-up to assess treatment response for AD.6
cRated down 1 level for serious imprecision. At the time of this review, there were no MIDs identified from published literature for this outcome. The clinical experts consulted by CDA-AMC indicated that a 10% between-group difference for this outcome could be clinically meaningful. In both the INTEGUMENT-1 and INTEGUMENT-2 trials, the point estimate and upper bound of the 95% CI for the between-group difference suggested a clinically important difference between roflumilast and vehicle cream, whereas the lower bound of the 95% CI suggested no clinically important difference between the 2 groups.
dThe statistical testing for this end point was not adjusted for multiplicity in the INTEGUMENT-1 and INTEGUMENT-2 trials and should be considered as supportive evidence.
eRated down 1 level for serious imprecision. An MID of 3.4 points was identified from published literature for this outcome. In the INTEGUMENT-1 trial, the point estimate and upper bound of the 95% CI for the between-group difference suggested no clinically important difference between roflumilast and vehicle cream, whereas the lower bound of the 95% CI suggested a clinically important difference between the 2 groups. In the INTEGUMENT-2 trial, the point estimate and lower bound of the 95% CI for the between-group difference suggested a clinically important difference between roflumilast and vehicle cream, whereas the upper bound of the 95% CI suggested no clinically important difference between the 2 groups.
fRated down 1 level for serious inconsistency. The results for this outcome in the INTEGUMENT-1 trial suggested a clinically important difference between roflumilast and vehicle cream. However, the results for this outcome in the INTEGUMENT-2 trial suggested no clinically important difference between the 2 groups.
gAn MID of 4 points was identified from published literature for this outcome. In both the INTEGUMENT-1 and INTEGUMENT-2 trials, the point estimate and 95% CI for the between-group difference did not suggest a clinically important difference between roflumilast and vehicle cream.
hRated down 1 level for study limitations. Although similar between the roflumilast and vehicle groups, the rates of dropout were large in both groups for this outcome (more than 10%) at week 4. Moreover, no methods for handling missing data were described for this outcome.
iAn MID of 6 to 8 points was identified from published literature for this outcome. In both the INTEGUMENT-1 and INTEGUMENT-2 trials, the point estimate and 95% CI for the between-group difference did not suggest a clinically important difference between roflumilast and vehicle cream.
jRated down 1 level for serious imprecision. At the time of this review, there was no MID identified from published literature for this outcome. The clinical experts consulted by CDA-AMC indicated that a 10% between-group difference for this outcome could be clinically meaningful. In both the INTEGUMENT-1 and INTEGUMENT-2 trials, the point estimate and lower bound of the 95% CI for the between-group difference suggested no clinically important difference between roflumilast and vehicle cream, whereas the upper bound of the 95% CI suggested a clinically important difference between the 2 groups.
Sources: Clinical Study Reports for INTEGUMENT-122 and INTEGUMENT-2 trials.23 The details included in the table are from the sponsor’s Summary of Clinical Evidence.
LTE Studies
Description of Studies
The INTEGUMENT-OLE trial (ARQ-151-313, NCT04804605) is an ongoing phase III, multicentre, open-label, LTE study of the INTEGUMENT-1, INTEGUMENT-2, and INTEGUMENT-PED trials. The INTEGUMENT-OLE trial is evaluating up to 52 weeks of treatment with roflumilast 0.15% cream in patients aged 6 years and older, or roflumilast 0.05% cream in patients aged 2 to 5 years who had mild to moderate AD in the parent studies. While the INTEGUMENT-OLE trial has not been completed, the data from interim analysis 1 (data cut-off: January 20, 2023) are available and include results for all patients who completed the INTEGUMENT-1 and INTEGUMENT-2 trials and rolled over to the INTEGUMENT-OLE trial (n = 658).26 The median duration of disease control with twice-weekly application was 281 days in the INTEGUMENT-OLE trial as of interim analysis 1.27 The results for roflumilast 0.05% cream in patients aged 2 to 5 years from the INTEGUMENT-PED trial are not included in this review. The INTEGUMENT-OLE trial aims to assess the long-term safety profile of roflumilast 0.15% cream in patients who had completed the previous 4-week studies for the INTEGUMENT-1 and INTEGUMENT-2 trials (cohort-week 52). In the INTEGUMENT-OLE trial, 1 group started with roflumilast in the INTEGUMENT-1 and INTEGUMENT-2 trials and continued with roflumilast in the OLE trial. A second group started with vehicle in the INTEGUMENT-1 and INTEGUMENT-2 trials and continued with roflumilast in the OLE trial.
The inclusion and exclusion criteria for patients reported in interim analysis 1 were consistent with those in the INTEGUMENT-1 and INTEGUMENT-2 trials. However, patients who experienced a TEAE or SAE that precluded further treatment with roflumilast 0.15% cream in the INTEGUMENT-1 and INTEGUMENT-2 trials were also excluded from the INTEGUMENT-OLE trial. An additional cohort comprising the patients in the INTEGUMENT-1 and INTEGUMENT-2 trials aged 6 to 17 years who were treated for up to 24 weeks with roflumilast 0.15% cream (cohort-week 24) was added during the INTEGUMENT-OLE trial, with the intention of gathering further long-term data for this patient group.
In the OLE study, roflumilast 0.15% cream was applied once daily for up to 52 weeks in patients with mild to moderate AD. Roflumilast 0.15% cream was applied to all currently affected areas (except the scalp) and any newly appearing AD lesions that arose during the study, as per the protocol for the preceding studies (the INTEGUMENT-1 and INTEGUMENT-2 trials). From the week 4 study visit, patients with AD lesions that had completely resolved (vIGA-AD score of 0 [clear]) switched to twice-weekly maintenance treatment. At subsequent scheduled or unscheduled clinic visits, patients on twice-weekly maintenance therapy would switch back to the original once-daily dosing regimens used in the INTEGUMENT-1 and INTEGUMENT-2 trials if the vIGA-AD score was 2 or greater or the AD was considered by an investigator to be inadequately controlled, while patients on once-daily maintenance therapy would switch to twice-weekly maintenance therapy if they achieved a vIGA-AD score of 0.
The definitions of the efficacy and safety outcomes in the INTEGUMENT-OLE trial were consistent with those used in the INTEGUMENT-1 and INTEGUMENT-2 trials. The 2 primary outcomes in the INTEGUMENT-OLE trial were occurrence of TEAEs and SAEs. The secondary outcomes evaluated in this clinical review were:
vIGA-AD value of 0 (clear) or 1 (almost clear) at each assessment
vIGA-AD success, defined as a vIGA-AD value of 0 (clear) or 1 (almost clear) plus a 2-grade improvement from baseline
WI-NRS success, defined as an achievement of a 4-point or greater improvement over time from a baseline score of 4 or higher
EASI score with a 75% reduction over time.
Change from baseline in the DLQI and CDLQI scores from baseline were the exploratory outcomes in the INTEGUMENT-OLE trial.
At the time of the interim analysis of the INTEGUMENT-OLE trial, there were 439 patients enrolled from the roflumilast treatment groups (considered the previously treated group) in the INTEGUMENT-1 and INTEGUMENT-2 trials, and 219 from the vehicle groups (considered the treatment-naive group).26 No formal statistical testing (i.e., multiple testing scheme) was conducted in the INTEGUMENT-OLE trial. An overview of the safety and efficacy results, including the HRQoL results, was provided using descriptive statistics. In the INTEGUMENT-OLE trial, 66.7% of the patients had experienced prior treatment failure with TCS at baseline (Appendix 5, Table 12). Subgroup analyses were performed for patients with a prior IIC-TCS. All analyses for AEs were based on the safety population (defined as all patients who were enrolled in the INTEGUMENT-OLE trial and received at least 1 confirmed dose of roflumilast 0.15% cream in the study). Secondary efficacy end points using the vIGA-AD score were reported for the full analysis population, which includes patients who withdrew prematurely from the study.
After patients were assigned to a prespecified analysis visit, missing data were estimated using multiple imputation with predictive mean matching sequential regression. For secondary end points using the WI-NRS pruritis score or EASI total score, descriptive statistics were accompanied with 2-sided 95% CIs. For efficacy end points investigating WI-NRS success and EASI-75, the results were presented with 95% CIs using the Wilson method.28 The safety population and full analysis population are nearly identical; however, 1 patient was not included in the safety population due to an early discontinuation from the study before any application of roflumilast 0.15% cream.
Patient Disposition
Patient disposition for the INTEGUMENT-OLE trial is summarized in the Supplemental Material document in Appendix 5, Table 11.
In the INTEGUMENT-OLE trial, a total of 327 patients (49.7%) completed the study, 214 patients (32.5%) withdrew, and the trial was still ongoing for 117 patients (17.8%) at interim analysis 1. The proportions of patients who completed, withdrew, and were still being assessed at the data cut-off point were similar between the patients who had been previously treated and the patients who were naive to treatment. In the overall safety population, the number of patients who discontinued treatment due to AEs was 2.7%, and AE-related treatment discontinuation was more common among patients who were naive to treatment (4.1%) compared to previously treated patients (2.1%). The rate of treatment discontinuation due to lack of efficacy was 5.8%, and was slightly more common in previously treated patients (6.2%) compared to patients who were naive to treatment (5.0%).
Baseline Characteristics
In the INTEGUMENT-OLE trial, most patients had a baseline vIGA-AD of almost clear (25.9%), mild (37.0%) or moderate (28.3%). The baseline mean WI-NRS score in the INTEGUMENT-OLE trial was 3.53, which is lower compared to the INTEGUMENT-1 and INTEGUMENT-2 trials (range, 5.85 to 6.21). Patients who were naive to treatment had a slightly higher mean WI-NRS baseline score (4.18) compared to previously treated patients (3.18). Total EASI score at baseline was lower in the INTEGUMENT-OLE trial (5.61) compared to the INTEGUMENT-1 and INTEGUMENT-2 trials (range, 9.77 to 10.31), and higher in patients who were naive to treatment (8.05) compared to previously treated patients (4.40). The mean percent BSA affected by AD was 9.07% in the INTEGUMENT-OLE trial, which was smaller than the baseline percentages in the INTEGUMENT-1 and INTEGUMENT-2 trials (range, 12.86% to 14.89%). Patients who were naive to treatment had a higher baseline mean percent BSA (12.88%) compared to previously treated patients (7.18%).
Exposure to Study Treatments
Details of patients’ treatment exposure and adherence and use of concomitant medications in the INTEGUMENT-OLE study are in the Supplemental Material document, Appendix 5 (Table 11).
Treatment compliance was high in the overall safety population (92.2%) and comparable between the patients who had been previously treated (92.3%) and the patients who were naive to treatment (92.2%). The proportion of patients who were compliant (applied at least 80% of the expected applications during the study drug application period and did not miss more than 3 consecutive doses while on the once-daily dosing schedule or more than 2 nonconsecutive days per week on the twice-weekly dosing schedule) was also high (83.0%) and comparable between the subgroups.
Of the 658 patients enrolled, 130 patients (19.8%) achieved a vIGA-AD score of 0 and switched to the twice-weekly maintenance treatment at least once after the 4-week trial. While the proportion of patients who returned to the daily dosing schedule once (32.8%) was noteworthy, repeat interruption of the twice-weekly maintenance treatment was rare (1.6%). The median duration of disease control with a twice-weekly application was 281 days (95% CI lower limit of 147; upper limit not estimable).27
In the safety population, the majority of patients had used concomitant medications (73.1%). Among the 657 patients in the safety population, the most commonly used medications were salbutamol (11.3%), cetirizine hydrochloride (8.5%), ibuprofen (7.8%), paracetamol (6.8%), and emollients and protectives (5.2%).
Critical Appraisal
Internal Validity
The interim analysis of the INTEGUMENT-OLE trial suggests that roflumilast is safe for up to 52 weeks following the initial 4-week duration of the INTEGUMENT-1 and INTEGUMENT-2 studies. The design of this OLE, with a primary end point of long-term safety rather than effectiveness (i.e., vIGA-AD success), is a change that may introduce selection bias because the patients who had previously experienced an AE or an SAE in the INTEGUMENT-1 and INTEGUMENT-2 trials were excluded. Enrolling only patients who tolerated and responded favourably to roflumilast potentially underestimates the harms and overestimates the benefits.
In the interim analysis of the INTEGUMENT-OLE trial, 439 patients from the roflumilast treatment groups (previously treated) and 219 from the vehicle groups (naive to treatment) were enrolled from the INTEGUMENT-1 and INTEGUMENT-2 trials. At the baseline of this OLE study, the patients in the previously treated group compared with the treatment-naive group were more likely to have a smaller affected BSA (mean BSA affected by AD: 7.18% versus 12.88%), milder AD (mean vIGA-AD score: 1.7 versus 2.3), lower WI-NRS score (mean WI-NRS score: 3.18 versus 4.18), and lower EASI total score (mean EASI total score: 4.40 versus 8.05). Therefore, the imbalance in the severity of AD between the previously treated group and the treatment-naive group may have an impact on treatment response. Because the vIGA-AD and EASI scales have been validated for change in patient populations with moderate to severe AD but have decreased sensitivity for detecting changes in patients with milder disease,29,30 it may be difficult to detect a meaningful change in response to roflumilast in patients with milder disease, and this may underestimate the efficacy of roflumilast in that subgroup. The distributions of Fitzpatrick skin type were similar between the patients from the 2 INTEGUMENT trials and the INTEGUMENT-OLE trial.
There was inconsistency in reporting how missing data were handled in the INTEGUMENT-OLE trial. The sponsor’s statistical analysis plan for the INTEGUMENT-OLE trial states that multiple imputation was planned with predictive mean matching sequential regression. In the OLE publication,27 no imputation was performed for missing values. In the interim analysis of the ongoing OLE study, there was a risk of bias from outcome data missing at later time points (refer to Supplementary Material, Appendix 5, Figure 2). Considering the proportion of patients achieving a vIGA-AD score of 0 (clear) or 1 (almost clear), there were wider 95% CIs over time, representing greater uncertainty in estimating treatment effects. Long-term HRQoL results were available in the INTEGUMENT-OLE study; however, there was a large amount of missing data in the assessment of change from baseline to week 52 in the DLQI and CDLQI scores. The proportion of patients who completed the assessments at week 52 ranged from 23% to 55%. A definite conclusion regarding the treatment effect of roflumilast on patients’ longer-term quality of life cannot be drawn.
In the OLE study, all patients were treated with roflumilast without a comparator group. Descriptive statistics summarized the safety and efficacy results. However, in the absence of formal statistical testing and without a comparator group, CDA-AMC cannot quantify the magnitude of difference or draw conclusions about clinically meaningful differences between the groups. An ideal comparator would be an active treatment used for the same indication and place in therapy. CDA-AMC cannot rule out changes in AD severity that occur independent of roflumilast treatment.
Results of the subgroup analyses in patients with a prior IIC-TCS were generally consistent with those in the overall population; however, the 95% CIs for the point estimate were relatively wider (indicating greater uncertainty in the treatment-effect estimate), which was consistent with the effect of a smaller patient population, especially for patients who were naive to treatment.
External Validity
In the OLE study, patients’ demographic characteristics and Fitzpatrick skin type distribution were similar to those for the INTEGUMENT-1 and INTEGUMENT-2 trials. Those patients who had been treated previously had milder disease and improved symptoms compared to patients who were naive to treatment when they entered the extension phase. The clinical experts consulted for this review indicated that the study populations in the trials had a wide range of demographic characteristics (e.g., age, race) and disease characteristics (e.g., disease severity, Fitzpatrick skin type, prior treatments), and well represented the patients seen in their practice. The INTEGUMENT trials and the extension study included patients living in Canada, Poland, and the US, yet the proportion of patients recruited from each of these countries was not explicitly stated. In the OLE study, from the week 4 study visit, patients with AD lesions that had completely resolved (vIGA-AD score of 0 [clear]) switched to twice-weekly maintenance treatment. As per 1 of the clinical experts, use of roflumilast as an anti-inflammatory therapy and as twice-weekly maintenance is consistent with Canadian clinical practice guidelines.6 Therefore, the study findings appear generalizable to patients living in Canada with mild to moderate AD who would be treated with roflumilast cream.
Results
Efficacy
Detailed results for outcomes relevant to this review are in Table 14 and Table 15 in Appendix 5 in the Supplemental Material document.
Key results include the following:
vIGA-AD of Clear or Almost Clear
At the primary baseline, 40 patients (6.1%) had a vIGA-AD score of clear or almost clear. At weeks 4, 12, 24, 36, and 52, 38.9%, 40.6%, 44.3%, 46.3%, and 46.3% of the overall safety population achieved a vIGA-AD score of clear or almost clear, respectively. Approximately 20% of patients achieved a vIGA-AD score of clear and switched to a twice-weekly dosing schedule with a median duration of at least 147 days or approximately 5 months for the first twice-weekly dosing period, and 65.6% of patients continued twice-weekly dosing for the remainder of their time in the study. The median duration of disease control with twice-weekly application was 281 days in the INTEGUMENT-OLE trial.
vIGA-AD Success
Achievement of a vIGA-AD score of clear or almost clear plus a 2-grade improvement from primary baseline (vIGA-AD success) in the full analysis population was a secondary efficacy end point, while in the INTEGUMENT-1 and INTEGUMENT-2 trials, this was a primary efficacy end point.
At week 4, vIGA-AD success was achieved in 31.0% of patients (95% CI, 26.87% to 35.55%) in the previously treated group and 7.9% of patients (95% CI, 4.91% to 12.33%) in the treatment-naive group. Response rates for vIGA-AD success in both the previously treated and treatment-naive groups trended upward during the study. At week 52, 42.4% of patients (95% CI, 34.88% to 50.32%) in the previously treated group and 23.7% of patients (95% CI, 15.95% to 33.60%) in the treatment-naive group achieved vIGA-AD success.
Similarly, for the IIC-TCS population, vIGA-AD success was achieved in █████ ████ ███ ██████ ██████ of patients in the previously treated group and ████ ████ ███ █████ █████ of patients in the treatment-naive group at week 4. Response rates for vIGA-AD success in both the previously treated and treatment-naive groups trended upward during the study for patients with a prior IIC-TCS. At week 52, █████ ████ ███ ██████ █████ of patients in the treatment-naive group achieved vIGA-AD success.
Proportion of Patients With EASI-75
For the IIC-TCS population, at week 4, EASI-75 success was achieved in ████ of patients in the previously treated group and ████ of patients in the treatment-naive group. The percentage of patients achieving EASI-75 in both the previously treated and treatment-naive groups showed a general trend upward during the study. At week 52, ██ of patients in the previously treated group and ████ of patients in the treatment-naive group achieved EASI-75.
Health-Related Quality of Life
At week 52, the mean change from baseline in the DLQI score was ████ ███ ████ in the previously treated group and ███ ███ ████ in the treatment-naive group; the mean change from baseline in the CDLQI score was ████ ███ ████ in the previously treated group and ████ ███ ████ in the treatment-naive group.
Harms
The incidence of TEAEs and SAEs was the primary study end point for the OLE study. Detailed results for harms are presented in Appendix 5 of the Supplemental Material document (Table 15). Key results include the following:
A TEAE was defined as an AE that started at any time following application of roflumilast 0.15% cream through to study completion. The proportion of patients who experienced at least 1 TEAE during 52 weeks of treatment with roflumilast 0.15% cream in the INTEGUMENT-OLE trial was 33.5% in previously treated patients, 39.0% in patients who were naive to treatment, and 35.3% in the overall safety population. Most TEAEs were considered mild or moderate in severity, while only 1.2% of patients in the overall safety population experienced severe TEAEs (1.1% of previously treated patients and 1.4% of patients who were naive to treatment). One patient experienced a life-threatening TEAE with grade 4 pneumonia; the AE was considered unrelated to roflumilast 0.15% cream and the patient recovered from the event and went on to complete the study. No fatal TEAEs were reported in the study.
The most commonly reported TEAEs (≥ 1% of patients) were:
COVID-19 infection, which occurred in 30 patients (4.6%) overall: 18 patients (4.1%) who had been treated previously and 12 patients (5.5%) who were naive to treatment.
Nasopharyngitis, which occurred in 18 patients (2.7%) overall: 12 patients (2.7%) who had been treated previously and 6 patients (2.8%) who were naive to treatment.
Upper respiratory tract infection, which occurred in 17 patients (2.6%) overall: 8 patients (1.8%) who had been treated previously and 9 patients (4.1%) who were naive to treatment.
Headache, which occurred in 17 patients (2.6%) overall: 12 patients (2.7%) who had been treated previously and 5 patients (2.3%) who were naive to treatment.
A total of 8 patients (1.2%) reported 1 SAE: 5 patients (1.1%) in the previously treated group and 3 patients (1.4%) in the treatment-naive group. No individual SAE was reported by more than 1 patient. Details of the occurrence of SAEs are presented in Appendix 5 of the Supplemental Material document (Table 15).
A total of 19 patients (2.9%) discontinued roflumilast 0.15% cream: 10 patients (2.3%) who had been treated previously and 9 patients (4.1%) who were naive to treatment. Two patients withdrew from the study due to AEs: 1 patient withdrew due to an application-site infection that was unlikely related to roflumilast 0.15% cream, and 1 patient withdrew due to mild (grade 1) vomiting.
Indirect Evidence
A review of indirect evidence was undertaken and submitted by the sponsor31 because there was no direct evidence comparing roflumilast 0.15% cream versus other relevant comparators for the treatment of mild to moderate AD. A research protocol for this study is not available, although detailed selection criteria and methods of analyses were provided in the original ITC report.
The objective of this section is to summarize and critically appraise the sponsor-submitted ITC and to inform the pharmacoeconomic model.
Description of Indirect Comparisons
The objective of the submitted ITC report was to derive estimates of the relative efficacy and safety of roflumilast versus existing therapies for patients with mild to moderate AD using an NMA.
In the sponsor-submitted NMA, in addition to the patient population that was consistent with the Health Canada–approved indication (patients with mild to moderate AD), roflumilast was also compared to other active comparators in a subset of patients with a prior IIC-TCS or with prior exposure to a TCS not otherwise specified; however, this clinical review presents the results for the overall population.
Study Selection and Review Methods
█████████ ██ ███ ████████ ███ ███ ███ █████████ ██ ██████████ ████ ███ ████████ ████████ ███ ██████████ ███████ ██ ███████████████ ████████ ██ █████████ ████ ███ █████████ █████████ █████ ███ ██████████ ██████████ ███████ ███ █████████████ ████████ ██████████ █ ██████████ ██████████ ██████ █████ ███ █████████ ██ ████████ ████████ ██████ █████████ ████████ ███ ██████ ██ █████████████ ███ ████████ ████ ████ ██ ████████ ███ █████████ ████ ██ ████████ █ █████ ██ █████ ███ ████████ █████ █████████ ███ ████ ██ ████████ ██ ████ █████████ ██████████ ██ ████████ ████████ ███████████ █████ ██████ ███ ███ ████ ███████ ████████ █████ ███ ███████████ ███ █████ █████ ███████████ ████████ ███ ████ ████████ ████ ████████ ██ ████ ████ ████████ █████████ ███ █████ ██████████ ████ ████████ ███ ███████████ ████████ ████████ █████████ ██ ████ ██ ██████ ███ ██████████ █████████ █████████ ██ ████ ██ █████ ████ ████████ ██ █████ █████ █████████ ███ ████ ██████████ ████ █████████ ██ ███ █████████ ██████████████ █ █████ ████████ ███ ████████ ██ █████████ ███ █████████████ ███████ ███ █████ █ █████████ ██████ ███ ████████ ███████ ███████████ ████ █████████ ████ ███ ███████ ████████ ██ █ █████████████ █████████ ███████ ███████████ ██ ████████ █████ ██████ ██ █ ██████ ████████ ███ █████████ ██ █ ██████ █████████ ███ █████████████ ███████ ███ ███ █████████ ████ ████████ ██ █████████ ██ ████ ████████ ██ ███ ████████ ██ █ █████ ███████████ █████████. A summary of the study selection criteria and methodology used to conduct the systematic literature review contributing to the ITC is described in Appendix 6 of the Supplemental Material document (Table 16).
█████ ██ ███ ███ █████████ ███████ ██████████ ██ ███ ███ ████ ███████ █████████ ██ █ ███████████ ██████████ ██ █████████ ██ ███ ███████ █████ █████████████ ██████ ██████████ ████████████ ██ █████ ███████████████ ██████ █████ ███████ █████████ ███ █████████ █████████ ████████████ ███ ████████████ ████████ █████████ █████████ ██████ ████████████ ██ ████████ ███████ ███████████████ ██████ ███████████ ███████████████ ███ ███████ █████████████████ ███████ ████████████ ███ ███ ███████ ██ ███████████ ███ ███████ █████ ████ ██ ███ ████ ███████████ █████████ ██████ █████████ ████████ ████ ████ ███ ███████ ████████ ██████ █████████ ██ ████████
ITC Analysis Methods
The NMA was performed within a Bayesian framework, following the guidelines outlined in the National Institute for Health Care Excellence Evidence Synthesis Decision Support Unit Technical Support Document series.35 Both fixed- and random-effects models were considered, and random-effects models were prioritized to adequately account for between-trial heterogeneity. In the NMA, assessments of model fit and consistency were performed. In addition, subgroup analyses were conducted in patients with a prior IIC-TCS or prior exposure to a TCS not otherwise specified. Nodes were created by grouping the interventions according to drug product, drug strength, and treatment class as much as possible, assuming similar treatment effects within each node.
In this ITC report, the relative efficacy and harms of roflumilast cream versus existing therapies were estimated for patients with mild to moderate AD using an NMA for stringent treatment success rate, EASI-75, change in HRQoL (e.g., DLQI or CDLQI scores), and safety (all-cause discontinuation and WDAEs).
Details of the NMA methods are presented in the Supplementary Material, Appendix 6, Table 17.
Results of the NMA
Summary of Included Studies
The systematic literature review identified 3,458 citations. The grey literature searches identified 488 conference abstracts, 436 records from bibliographies of on-topic systematic literature reviews, and 362 records from a targeted search of other sources that were eligible for further screening. In total, 3,512 records were excluded. The full texts of the remaining 227 records were obtained, and 213 of those documents were assessed for eligibility. Ultimately, 181 records were excluded for various reasons. This process led to the inclusion of 32 records reporting on 31 unique RCTs. A list of all citations excluded at the full-text stage, along with the reasons for their exclusion, was provided in the sponsor’s ITC report. The 2 INTEGUMENT trials were the only studies evaluating the treatment effects of roflumilast 0.15% cream in patients with mild to moderate AD.
The risk of bias of the included RCTs was appraised at the study level using the Cochrane risk of bias tool36 for 16 of the 31 trials identified. The level of the overall risk of bias was determined to be high for 3 trials, unclear for 10 trials, and low for 2 trials. The 3 trials with a high risk of bias all had performance bias, with 1 trial having an open-label design and 2 having a single-blind design.
Assessment of Similarity
Most trials were similar in their design and population, facilitating valid comparisons. The acute treatment period was around 4 weeks, and outcome assessments occurred within a period of symptom resolution. There was overlap between trials in their eligibility criteria, such as age, disease duration, and level of BSA involvement, which are examples of similarity. Patient age was older than 6 years, and patient race, particularly the proportions of patients who are Black or African American, and the proportions of patients with moderate to severe disease were relatively balanced between groups. Outcome measures of treatment withdrawal, all-cause discontinuation, and WDAE were commonly reported and measured using comparable ordinal scales. These outcomes support similarity in efficacy and safety assessments.
There were notable differences between studies and in patient characteristics in concomitant medication use, vehicle composition and use, and treatment formulations. For example, Meurer (2004) allowed concomitant TCS use, which could confound results in the relative effect of pimecrolimus versus vehicle, and was excluded. NCT03539601 had an unbalanced vehicle control design, leading to exclusion of treatment groups (pimecrolimus and hydrocortisone butyrate) with crisaborole vehicle. Treatments not evaluated with a comparable vehicle in the same trial were removed to avoid bias. Differences in formulation and dosing (e.g., fluocinonide twice a day versus once a day) were addressed by combining treatments into potency-based nodes. Some trials used noncomparable definitions for stringent treatment success, leading to their exclusion. Additionally, poor reporting of certain eligibility data, such as duration of AD, limited full assessment of heterogeneity.
Several sources of similarities and differences across the 31 studies are summarized in the Supplementary Material, Appendix 6, Table 18.
Effect Modifiers
The authors of the NMA report identified several potentially important effect modifiers for mild to moderate AD, including age, race, and disease severity. Across the included RCTs, patients included both children and adults, with ages ranging from 6 to 44 years. Age was considered a treatment-effect modifier of moderate clinical importance, with the age of 6 years generally considered an important clinical cut-off, given that many patients tend to outgrow the disease by that age. The mean age in all trials was at least 6 years; all trials were included in the NMA regardless of mean age. The percentage of BSA involvement comprised a wide range (4.9% to 36.7%). Race or ethnicity and disease severity were considered to be important treatment-effect modifiers, with most study patients identified as white, and the proportions of patients with mild or moderate AD were generally balanced across studies.
Trial characteristics and patient characteristics of the RCTs included in the NMA are shown in Supplementary Material, Appendix 6, Table 19, Table 20, and Table 21.
Evidence Networks
Stringent treatment success was the only outcome whose network included at least 1 comparator from each treatment class of interest. Comparators within the TCI or TCS classes were less represented across other outcome networks. None of the networks for EASI-75, CDLQI, and DLQI included comparators from these 2 treatment classes. Generally, even in networks where TCI or TCS comparators were present, data from only 1 trial were available to inform each treatment node. Connectivity to both high-potency and low-to-medium potency TCS therapies was not possible in any of the networks. All eligible RCTs for the NMA were placebo-controlled; therefore, all networks were generally star-shaped with placebo as the central node. Closed loops were available only for the comparisons between various concentrations of ruxolitinib, and there were no closed loops between different active treatments.
The diagrams for the network of evidence for the efficacy and safety outcomes assessed at week 4 are presented in the Supplementary Material, Appendix 6, Figure 3 to Figure 8.
Critical Appraisal of the ITC
First, the sponsor did not provide a research protocol or statistical analysis plan for the ITC or NMA; therefore, it is unclear whether the analysis methods presented in the report were determined a priori. However, the sponsor noted that the ITC was performed in accordance with the Cochrane Handbook for Systematic Reviews of Interventions and was reportedly in alignment with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement. The risk of bias for the included studies was assessed using the Cochrane risk of bias tool. It was unclear whether the results of the risk of bias assessment were used in the NMA, such as in a sensitivity analysis, to examine the robustness of the study findings based on the quality of the clinical evidence.
Based on the reported information, the analysis methods of the NMA, such as model selection, model fit assessment, convergence, or assessment of similarity, were considered appropriate. Random-effects models were prioritized to adequately account for between-trial heterogeneity. The wider credible intervals (CrIs) increase the uncertainty in the effect size.
Age, race, and disease severity were identified by the sponsor as important effect modifiers for the treatments in the study population. According to the clinical experts consulted for this review, all relevant effect modifiers were included in the NMA, which helps adequately address the issues of heterogeneity.
In the sponsor’s NMA report, several potential threats to transitivity were identified, including differences in concomitant medication use, vehicle composition and use, and treatment formulations. These differences may influence treatment effects and introduce bias into indirect comparisons, increasing uncertainty in the estimated relative effectiveness.
The report also noted clinical and methodological heterogeneity across trials, such as variations in patient baseline characteristics (e.g., age, prior treatments) and trial features (e.g., treatment duration, outcome definitions). To address these differences, the sponsor applied strategies like combining brand name and generic TCIs at equivalent doses and aggregating vehicles with different formulations into a single node to maintain network connectivity. The clinical experts consulted for this review considered these approaches reasonable. However, for some individual RCTs, limited reporting of key variables — such as WI-NRS scores, disease severity, amount of BSA affected, or EASI scores — limits the ability to fully assess the distribution of potential effect modifiers. This lack of information further challenges the transitivity assumption, which is a requirement for the credibility of the indirect comparisons in the NMA.
The networks were sparse for several comparisons. Only a limited number of trials were included for the NMA of the change in the EASI-75 rate, change in DLQI or CDLQI score, risk of all-cause discontinuation, or risk of WDAEs at week 4. All evidence for the comparisons of interest was indirect. Thus, the outcome assessments were affected by substantial imprecision and the certainty of the effect estimates was greatly reduced, which was reflected by the very wide 95% CrIs of the effect estimates.
Roflumilast was not part of the closed loop in any of the networks, which limits the ability to assess the consistency assumption, which assumes that direct and indirect evidence for the treatment comparisons is in agreement. In addition, although ruxolitinib was included in all of the efficacy and HRQoL outcomes assessed, it was absent from the safety outcome assessment, which limits the comprehensiveness of the safety comparisons for this treatment.
For all the efficacy and safety outcomes assessed in the NMA, the results were available for week 4 only. The long-term benefit or harm of roflumilast cream versus currently available treatments for mild to moderate AD is unknown.
Efficacy and Harms Results of the ITC
Efficacy
Results for all efficacy outcomes favour the random-effects model. Key results of the NMA are in Table 6. Forest plots for the indirect comparisons between roflumilast 0.15% cream and the comparators are shown in the Supplementary Material, Appendix 6 (Figure 9 to Figure 12).
Stringent Treatment Success at Week 4
Roflumilast was statistically favoured over vehicle cream and numerically but not statistically favoured over crisaborole (OR [95% CrI] = █████ ██████ ██ ███████ and tacrolimus (OR [95% CrI] = █████ ██████ ██ ███████. Low and medium potency TCS treatments were numerically but not statistically favoured over roflumilast (OR [95% CrI] = █████ ██████ ██ ███████. Both strengths of ruxolitinib were statistically favoured over roflumilast (OR [95% CrI] = █████ ██████ ██ █████ and OR [95% CrI] = █████ ██████ ██ ██████ for the 1.5% and 0.75% strengths, respectively).
EASI-75 Reduction Rate at Week 4
Roflumilast was statistically favoured over vehicle cream and numerically but not statistically favoured over crisaborole (OR [95% CrI] = █████ ██████ ██ ███████. Both strengths of ruxolitinib were statistically favoured over roflumilast (OR [95% CrI] = █████ ██████ ██ ██████ ███ OR [95% CrI] = █████ ██████ ██ ██████ for the 1.5% and 0.75% strengths, respectively). No data on TCI or TCS treatments were available for comparison in the EASI-75 network.
DLQI Mean Change From Baseline at Week 4
Roflumilast was slightly numerically but not statistically favoured over crisaborole (mean difference [95% CrI] = ██████ ███████ ██ ██████ and vehicle. Both strengths of ruxolitinib were numerically but not statistically favoured over roflumilast (mean difference [95% CrI] = █████ ███████ ██ ████ and mean difference [95% CrI] = ████ ███████ ██ ███ for the 0.75% and 1.5% strengths, respectively). No data for TCI or TCS were available for comparison in the DLQI change from baseline network.
CDLQI Mean Change From Baseline at Week 4
Roflumilast was statistically favoured over vehicle and numerically but not statistically favoured over crisaborole (mean difference [95% CrI] = ██████ ███████ ██ ███████ Both strengths of ruxolitinib were numerically but not statistically favoured over roflumilast (mean difference [95% CrI] = ████ ███████ ██ ██████ and mean difference [95% CrI] = █████ ███████ ██ ██████ for the 1.5% and 0.75% strengths, respectively). No data for TCI or TCS treatments were available for comparison in the CDLQI change from baseline network.
Table 6: Summary of Efficacy Outcome Measures in the Sponsor-Submitted NMA, Roflumilast 0.15% vs. Relevant Comparators (Random-Effect Models) [Redacted]
██████████ | █████ ████ | █████ ████ | █████ ████ | █████ ████ |
|---|---|---|---|---|
████████ | ████████ | ████████ | ████████ | ████████ |
████████ | ||||
████████ | ████████ | ██ | ██ | ██ |
████████ | ██ | ██ | ██ | ██ |
████████ | ||||
████████ | ████████ | ██ | ██ | ██ |
████████ | ██ | ██ | ██ | ██ |
████████ | ||||
████████ | ████████ | ████████ | ████████ | ████████ |
████████ | ████████ | ████████ | ████████ | ████████ |
████████ | ||||
████████ | ████████ | ████████ | ████████ | ████████ |
█████ █ ██████████ ███████████ ████ ███████ ██████ ███ █ ████████ █████████ ████ █ ███████████ ████ ███████ ██████ ███████ █ ██████ ████ ███ ████████ █████ ███ ██ █ ███ ███████████ ███ █ ███████ ██████████████ ██ █ ████ ██████ ██ █ █████████ ██████████████ █████ ██ ██ █████████ █████████████ █████ █████ ██ ██ █████████ █████████
Harms
Results for both harm outcomes favoured the random-effects model. Key results of the ITCs are shown in Table 7. Forest plots for the indirect comparisons between roflumilast 0.15% cream and the comparators are shown in the Supplementary Material, Appendix 6 (Figure 13 and Figure 14).
All-Cause Discontinuation Rate at Week 4
Roflumilast was statistically favoured over crisaborole (OR [95% CrI] = █████ ██████ ██ ███████ and low- to medium- potency TCS treatments (OR [95% CrI] = █████ ██████ ██ ███████. Roflumilast was numerically but not statistically favoured over the vehicle cream and tacrolimus 0.3% (OR [95% CrI] = █████ ██████ ██ ███████.
WDAEs Rate at Week 4
Roflumilast was numerically favoured over all treatments; however, due to 0 WDAEs in some studies, the estimates are too uncertain to draw reliable conclusions.
Table 7: Summary of Harm Outcome Measures in the Sponsor-Submitted NMA, Roflumilast 0.15% vs. Relevant Comparators (Random-Effects Models) [Redacted]
██████████ | ███████ ████ | ███████ ████ |
|---|---|---|
███ ██ █████ | ███ ██ █████ | ███ ██ █████ |
███ ██ █████ | ██ | ██ |
███ ██ █████ | ███ ██ █████ | ███ ██ █████ |
███ ██ █████ | ██ | ███ ██ █████ |
███ ██ █████ | ██ | ██ |
███ ██ █████ | ██ | ██ |
███ ██ █████ | ███ ██ █████ | ███ ██ █████ |
███ ██ █████ | ███ ██ █████ | ███ ██ █████ |
███ █ ████████ █████████ ██ █ ███ ███████████ ██ █ ████ ██████ ███ █ ███████ █████████████ ████ █ ██████████ ███ ██ ███████ █████████████ █████ █████████ ███████████ █████████████████ ██
Studies Addressing Gaps in the Systematic Review Evidence
There were no studies addressing the gaps in the evidence submitted by the sponsor.
Discussion
Efficacy
Disease control was identified as an important goal of treatment for AD by the clinical experts consulted by CDA-AMC and by the patient and clinician groups. The INTEGUMENT-1 and INTEGUMENT-2 trials assessed several outcomes pertaining to disease control among patients aged 6 years and older with mild to moderate AD, which included vIGA-AD, EASI, and the percent BSA affected by AD. According to an assessment using GRADE, high-certainty evidence from the INTEGUMENT-1 and INTEGUMENT-2 trials demonstrated that at week 4, roflumilast 0.15% cream likely resulted in clinically meaningful improvements in the proportion of patients achieving vIGA-AD success and the proportion of patients achieving EASI-75 when compared to vehicle cream (Table 5). Although not formally included in the GRADE assessment, the results of both trials demonstrated that a higher proportion of patients who received roflumilast 0.15% cream achieved vIGA-AD success at week 1 and week 2 and a vIGA-AD score of clear or almost clear at weeks 1, 2, and 4. In both trials, patients in the roflumilast 0.15% cream group had a larger decrease in percent BSA affected by AD from baseline to week 4 compared with those in the vehicle cream group.
The clinical experts consulted by CDA-AMC indicated that reduction of the symptoms of AD was an important goal of treatment for AD, which was echoed by patient and clinician groups. In both trials, WI-NRS success was a secondary outcome that measured the extent of itching. For this outcome, moderate-certainty evidence demonstrated that when compared with vehicle cream, roflumilast 0.15% cream likely results in a clinically important increase in the proportion of patients achieving WI-NRS success at week 4 (Table 5). Conclusions for this outcome were limited by serious imprecision in the estimates. The INTEGUMENT-1 and INTEGUMENT-2 trials reported the LS mean change in POEM and SCORAD scores from baseline, which were both used to measure the extent of AD symptoms. For both measures, a decrease in score indicates improvement in AD symptoms. In both trials, patients in the roflumilast 0.15% cream group were associated with a larger LS mean decrease in POEM and SCORAD scores from baseline to week 4 compared with the vehicle cream group. According to the GRADE assessment, low-certainty evidence demonstrated that roflumilast 0.15% cream may result in little to no important difference in LS mean change in POEM score from baseline to week 4 compared with vehicle cream (Table 5). This was attributed to imprecision in the estimates and the inconsistency of results between the INTEGUMENT-1 and INTEGUMENT-2 trials based on the MID of 3.4 points identified from the literature. Of note, the POEM and SCORAD outcomes were not adjusted for multiplicity. Hence, there is a higher probability of statistical significance by chance alone, increasing the risk of type I error (i.e., a false-positive result).
Key HRQoL outcomes of the INTEGUMENT-1 and INTEGUMENT-2 trials were measured using the DLQI and CDLQI instruments, which have been validated in adults and children with AD. For these measures, a decrease in scores represents improved HRQoL. In the INTEGUMENT-1 and INTEGUMENT-2 trials, treatment with roflumilast was associated with larger LS mean decreases in DLQI and CDLQI scores from baseline to week 4 compared with vehicle cream. Based on the MID of 4 points identified from the literature, moderate-certainty evidence demonstrated that roflumilast 0.15% cream likely results in little to no important difference in the LS mean change in DLQI score from baseline to week 4 when compared with vehicle cream (Table 5). Conclusions were limited by large rates of dropout at week 4 (e.g., more than 10%), with no methods described for handling missing data for this outcome. Based on the MID of 6 to 8 points identified from the literature, high-certainty evidence demonstrated that roflumilast 0.15% cream results in little to no important difference in the LS mean change in CDLQI score from baseline to week 4 when compared with vehicle cream (Table 5). Of note, during the OLE phase of the INTEGUMENT trials, the DLQI scores and the CDLQI scores at week 52 decreased from baseline, which suggests an improvement in HRQoL. However, the long-term effect of roflumilast cream on the patients’ HRQoL was uncertain due to the large amount of missing data in the extension study.
For the primary and secondary end points, the INTEGUMENT-1 and INTEGUMENT-2 trials included subgroup analyses on patients with a prior IIC-TCS, which was the population for the reimbursement request outlined by the sponsor. Results of the subgroup analyses for this population demonstrated that the results for patients with a prior IIC-TCS were consistent with those of the overall population in both trials. Conclusions on the results for this subgroup are limited due to a lack of consideration for sample size and lack of adjustment for multiplicity for the subgroup analyses conducted in the trials. Moreover, data for exploratory outcomes (e.g., percent BSA affected, SCORAD, POEM, DLQI, CDLQI) were not measured for this subgroup in the trial. Of note, the sponsor submitted post hoc analyses of pooled data from the INTEGUMENT-1 and INTEGUMENT-2 trials that evaluated DLQI and CLDQI outcomes among patients with a prior IIC-TCS.
This CDA-AMC review highlighted a few considerations pertaining to the generalizability of the results of the INTEGUMENT-1 and INTEGUMENT-2 trials to clinical practice in Canada. The clinical experts consulted by CDA-AMC agreed that the eligibility criteria for the trials were representative of patients with AD in clinical practice. Although the clinical experts consulted by CDA-AMC noted variations in patient demographics across clinics, they agreed that a variety of ages, disease severities, and ethnicities were well represented in the baseline characteristics of the trials. One clinical expert noted that although the trial follow-up of 4 weeks was sufficient for capturing the speed of improvement in signs and symptoms, patients in clinical practice should be assessed over a longer time frame (e.g., at least 6 months). Lastly, because the trials did not compare roflumilast with other relevant comparators available in Canada for the treatment of mild to moderate AD, the stand-alone results of the trial may not provide a full assessment of the efficacy and safety of roflumilast compared to existing treatments in Canadian clinical practice for mild to moderate AD. However, the sponsor submitted an NMA that evaluated roflumilast 0.15% cream compared with relevant comparators in Canadian clinical practice for mild to moderate AD.
The INTEGUMENT-OLE trial is an ongoing phase III, multicentre, open-label LTE study assessing the long-term safety of roflumilast 0.15% cream in the patients who completed the initial 4-week INTEGUMENT-1 and INTEGUMENT-2 studies. At the interim analysis, 658 patients were enrolled, comprising 439 patients who had been previously treated with roflumilast and 219 patients who were naive to treatment.26 A total of 327 patients (49.7%) completed the study, 214 patients withdrew (32.5%), and the trial was still ongoing for 117 patients (17.8%) at interim analysis 1. The results of the interim analysis of the INTEGUMENT-OLE trial suggest that roflumilast is safe for up to 52 weeks following the initial 4-week trials. Of note, sources of risk of bias in the OLE trial included the open-label design, changing the primary end point from vIGA-AD success to long-term safety, and enrolling patients who tolerated and responded favourably to roflumilast. Compared to the treatment-naive group, patients in the previously treated group were more likely to have milder AD; therefore, the imbalance in the severity of AD between the previously treated group and treatment-naive group may have an impact on treatment response because patients with milder disease may respond better to roflumilast. In the interim analysis of the ongoing OLE study, there was a risk of bias due to outcome data missing at later time points. The absence of statistical testing and comparator groups limits conclusions about the magnitude of difference and limits the ability of the CDA-AMC review team to draw conclusions about clinically meaningful differences between the groups. The results of the subgroup analyses of patients with a prior IIC-TCS were generally consistent with those in the overall population. The wider 95% CIs for the point estimate were consistent with the effect of smaller patient populations, especially for patients who were naive to treatment.
In the OLE study, patients’ demographic characteristics and Fitzpatrick skin type distribution were similar to those for the INTEGUMENT-1 and INTEGUMENT-2 trials, while patients who had been treated previously had milder disease and improved symptoms when they entered the extension phase compared to those patients who were naive to treatment. The clinical experts consulted for this review indicated that the study populations in the trials had a wide range of demographic characteristics (e.g., age, race) and disease characteristics (e.g., disease severity, Fitzpatrick skin type, prior treatments) and well represented the patients seen in their practice. In the OLE study, from the week 4 study visit, patients with AD lesions that had completely resolved (vIGA-AD score of 0 [clear]) switched to twice-weekly maintenance treatment. As per the clinical experts, this approach is consistent with clinical practice, although this may not be standard practice based on Canadian guidelines; thus, the study findings can be generalized to patients in Canada with mild to moderate AD who would be treated with roflumilast cream.
One ITC report was submitted by the sponsor to compare the efficacy and safety of roflumilast 0.15% cream with other active therapies (e.g., TCSs, TCIs, JAK inhibitors, or other PDE4 inhibitors) for the treatment of mild to moderate AD. The comparative efficacy and harms of roflumilast versus the comparator treatments were evaluated based on evidence from 31 RCTs. Based on the results of the NMA, the indirect evidence is insufficient to conclude whether roflumilast differs from TCIs, TCSs, or other PDE4 inhibitors in terms of stringent treatment success rate and EASI-75 rate. Treatment with roflumilast may be associated with a lower stringent treatment success rate or more severe disease at week 4 when compared to a JAK inhibitor (ruxolitinib). For HRQoL measured with changes in DLQI or CDLQI scores from baseline to week 4, the indirect evidence is insufficient to make any conclusions on comparisons between roflumilast and all comparators. For harm assessments, roflumilast cream was associated with a lower rate of all-cause discontinuation when compared with crisaborole ointment, while the indirect evidence is insufficient to conclude whether roflumilast differs from all other comparators for the outcomes of all-cause discontinuation or WDAEs.
Definite conclusions on the comparative efficacy and safety of roflumilast cream versus other active treatments for mild to moderate AD cannot be made due to the limitations of the NMA, which include clinical and methodological heterogeneities noted in the patient baseline characteristics and trial characteristics. Various approaches were used to address the heterogeneity. Meanwhile, in some included studies, the information reported for patients’ disease severity at baseline was insufficient for judging the degree of heterogeneity in potential treatment-effect modifiers across the trials, which would challenge the transitivity assumption underlying the NMA. In addition, the networks were sparse for several comparisons, especially for the analyses of EASI-75 rate, change in DLQI or CDLQI score, risk of all-cause discontinuation, and risk of WDAEs at week 4. Thus, the outcome assessments were affected by substantial imprecision and the certainty of the effect estimates was greatly reduced, which was reflected by very wide 95% CrIs of the effect estimates. Roflumilast was not part of the closed loop in any of the networks. This means the consistency assumption, in which the relative treatment effects can be compared across studies, cannot be adequately assessed. For all of the efficacy and safety outcomes assessed in the NMA, the results were available for week 4 only. The long-term benefit or harm of roflumilast cream versus currently available treatments for mild to moderate AD is unknown.
Harms
In the INTEGUMENT-1 and INTEGUMENT-2 trials, TEAEs were reported more frequently among patients who received roflumilast 0.15% cream compared with the vehicle cream, with the most common TEAEs being headache, application-site pain, and nausea. Incidences of SAEs and WDAEs were low for both treatment groups, although slightly higher in the roflumilast 0.15% cream group. Moderate-certainty evidence from the trials suggested that roflumilast 0.15% cream likely results in little to no important difference in the proportion of patients with 1 or more SAEs when compared with vehicle cream at week 4 (Table 5). However, this conclusion was limited by imprecision in the estimates. Similar rates of high scores for investigator-assessed tolerability were observed among patients who received roflumilast 0.15% cream and vehicle cream at week 4, but high scores for patient-reported tolerability were reported among more patients who received the roflumilast 0.15% cream at week 4. The results of the INTEGUMENT-OLE trial identified no new safety signals associated with roflumilast 0.15% cream after 52 weeks of treatment. According to the results of the NMA, roflumilast 0.15% cream was associated with a lower rate of all-cause discontinuation when compared with crisaborole ointment, while the indirect evidence is not sufficient to conclude whether roflumilast differs from all other comparators in terms of all-cause discontinuation or WDAEs.
The clinical experts consulted by CDA-AMC indicated safety concerns with existing treatment options for mild to moderate AD, which were echoed by the patient and clinician groups that submitted input to CDA-AMC. Treatment with TCS is associated with steroid-related effects and dependence, whereas TCIs and crisaborole ointment are often associated with intolerable burning and stinging. The patient and clinician groups agreed that roflumilast 0.15% cream meets the need for a nonsteroidal topical treatment option for mild to moderate AD. Moreover, treatment with roflumilast 0.15% cream in the INTEGUMENT-1 and INTEGUMENT-2 trials was associated with low rates of administration-site conditions (i.e., less than 2.5%). Although rates of administration-site conditions were generally similar between roflumilast 0.15% cream and vehicle cream in the trials, the safety profile of roflumilast 0.15% cream compared to other relevant comparators for AD in Canada is unclear because the pivotal trials did not assess this, and the information from the ITC was insufficient for a definitive conclusion on safety. The patient and clinician groups also indicated a need for topical treatments that are safe for use on AD affecting sensitive areas (e.g., face, neck, skin folds, intertriginous areas). Although a subgroup analysis on facial involvement was prespecified for the primary and secondary outcomes in the INTEGUMENT-1 and INTEGUMENT-2 trials, safety data were not available for this subgroup.
Ethics and Equity Considerations
The clinical experts consulted by CDA-AMC highlighted several considerations for equity in the treatment of patients with AD. One clinical expert noted that AD is more common among Black and South Asian populations, and 1 clinical expert suggested that AD disproportionately affects Indigenous populations due to suboptimal access to moisturizers and clean water. Compared to previous trials for AD, the clinical experts noted that various ethnicities and skin types were well represented in the INTEGUMENT-1 and INTEGUMENT-2 trials, including among those patients who may be disproportionately affected by AD. One clinical expert also indicated that pediatric patients were disproportionately affected by AD due to a higher incidence of the disease compared with adults and limited treatment options. Subgroup data from the INTEGUMENT-1 and INTEGUMENT-2 trials showed that the results for children (aged 6 to 11 years) and adolescents (aged 12 to 17 years) were generally consistent with those of the overall population in the trials. Of note, 1 clinician group added that roflumilast would highly benefit pediatric and older adult patients with thinner skin that is prone to steroid-induced atrophy and patients with a darker skin phenotype that is prone to TCS-induced hypopigmentation.
The input from the patient and clinician groups noted that many treatments for AD require frequent or complex administration, which can conflict with patient and caregiver preferences and negatively affect treatment adherence. The clinical experts consulted by CDA-AMC and the patient and clinician groups indicated that the once-daily schedule of roflumilast cream is more convenient compared to other treatments for AD, which can address the unmet need for more convenient treatment options for AD and appeal to patient and caregiver preferences for treatment. Of note, the advantage of once-daily formulations on adherence is further supported by expert panel recommendations and adherence studies in other conditions requiring topical therapy,37,38 as well as in AD.39
Conclusion
The evidence base for this review included 2 phase III double-blind RCTs (the INTEGUMENT-1 and INTEGUMENT-2 trials), 1 LTE study (the INTEGUMENT-OLE study), and 1 sponsor-submitted NMA. The INTEGUMENT-1 and INTEGUMENT-2 trials evaluated the efficacy and safety of roflumilast in patients aged 6 years or older with mild to moderate AD compared with vehicle cream. The outcomes evaluated in these trials were considered important to patients and clinicians, which included assessment of signs and symptoms of AD and HRQoL. High-certainty evidence from the INTEGUMENT-1 and INTEGUMENT-2 trials demonstrated that at week 4, roflumilast 0.15% cream results in a clinically important increase in the proportion of patients achieving vIGA-AD success at week 4 and proportion of patients achieving EASI-75 when compared with vehicle cream. The results of the trials also suggest that when compared with vehicle cream, roflumilast 0.15% cream likely results in a clinically important increase in the proportion of patients achieving WI-NRS success at week 4, whereas roflumilast 0.15% cream may result in little to no important difference in LS mean change in POEM score from baseline to week 4. Moreover, the GRADE assessment for LS mean change from baseline to week 4 in DLQI and CLDQI scores suggests that roflumilast likely results in little to no meaningful improvement in HRQoL among adult patients and results in little to no meaningful improvement in HRQoL in pediatric patients. Overall, no new safety signals were observed with treatment with roflumilast 0.15% cream in the INTEGUMENT-1 and INTEGUMENT-2 trials. The results for the interim analysis of the ongoing INTEGUMENT-OLE trial suggest that roflumilast maintained its safety profile through 52 weeks following the initial 4-week trials. Of note, sources of risk of bias in the OLE trial included the open-label design, enrolling patients who tolerated and responded favourably to roflumilast, and outcome data missing at later time points. Based on the sponsor’s submitted NMA, the indirect evidence is insufficient to conclude whether roflumilast differs from its comparators with respect to stringent treatment success rate and EASI-75 rate. Treatment with roflumilast may be associated with a lower stringent treatment success rate or more severe disease at week 4 when compared to ruxolitinib. The indirect evidence is insufficient to draw any conclusions for comparisons between roflumilast and all comparators in terms of HRQoL measured by changes in the DLQI and CDLQI scores from baseline to week 4. Roflumilast cream was associated with a lower rate of all-cause discontinuation when compared with crisaborole ointment, whereas indirect evidence is insufficient to conclude whether roflumilast differs from all other comparators in terms of all-cause discontinuation or WDAEs.
Economic Review
Methods
The review team appraised the pharmacoeconomic evidence submitted by the sponsor on the cost and budget impact of roflumilast 0.15% cream compared to TCIs and TCSs for the treatment of mild to moderate AD. For the review of roflumilast, the sponsor provided a cost-minimization analysis and a budget impact analysis. The sponsor’s economic evaluation is summarized in the Supplemental Material document, Appendix 9.
The sponsor’s cost-minimization analysis compared roflumilast to TCIs and TCSs from the perspective of a public drug plan payer in Canada over 1-, 3-, and 5-year time horizons. The modelled population comprised patients aged 6 years and older with mild to moderate AD, which is aligned with the Health Canada indication and was based on the participants in the INTEGUMENT-1 and INTEGUMENT-2 trials. The sponsor’s base-case analysis included costs related to drug acquisition only (submitted price for roflumilast 0.15% cream and public list prices for comparators).
In the sponsor’s base case, roflumilast 0.15% cream was associated with incremental costs of $511 to $563 per patient per year relative to TCSs while, for TCIs, the incremental costs associated with roflumilast 0.15% ranged from $45 per patient per year relative to pimecrolimus to a savings of $66 per patient per year relative to tacrolimus 0.1%.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 8; full details are provided in the Supplemental Material document, Appendix 10). A revised base case was therefore developed.
Table 8: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The clinical similarity of roflumilast 0.15% cream to relevant comparators is highly uncertain. | There have been no head-to-head trials comparing roflumilast 0.15% cream to relevant TCI and TCS comparators. The CDA-AMC review of the submitted NMA indicated that definitive conclusions of the comparative efficacy and safety of roflumilast vs. its funded comparators could not be made. | CDA-AMC could not address uncertainty in the comparative clinical evidence. | No scenario analysis was conducted. |
The average annual amount of the topical comparators needed to treat mild to moderate AD is uncertain. | The sponsor assumed the mean numbers of flares, days of treatment per flare, amount of BSA affected, and amount of product used per application per unit of BSA from the literature. The clinical expert opinion obtained by CDA-AMC deemed the sponsor’s assumptions reasonable but uncertain. | CDA-AMC did not address uncertainty in the number of treatment days or the amount of product required per application. Any increases in these assumptions would lead to corresponding increases in cost differences between comparators. | No scenario analysis was conducted. |
Wastage of excess product was overestimated. | The sponsor overestimated the amount of product that would be wasted for some comparators by not considering the availability of smaller package sizes. Additionally, the assumption that all products not used at the end of every year of therapy is wasted is unlikely to represent clinical practice over multiple years, especially when assuming the product had not expired. | CDA-AMC could not address uncertainty in the comparative clinical evidence. | To represent the average cost per year of therapy, a scenario was conducted that assumed no wastage because any excess medication dispensed in a year would be balanced out by its use in the next. |
AD = atopic dermatitis; BSA = body surface area; CDA-AMC = Canada’s Drug Agency; NMA = network meta-analysis; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; vs. = versus.
Note: Full details of the issues identified by CDA-AMC are provided in the Supplemental Material document, Appendix 10.
CDA-AMC Assessment of Cost
The CDA-AMC base case included drug acquisition only. The CDA-AMC base case was derived by making changes to model parameter values and assumptions, in consultation with clinical experts. Detailed information about the base case is provided in the Supplemental Material document, Appendix 10. When considering the Health Canada–indicated population, relevant comparators include TCSs and TCIs reimbursed for the treatment of mild to moderate AD. When considering the reimbursement request population of patients with mild to moderate AD with a prior IIC-TCS, TCIs would be the relevant comparators.
The acquisition cost of roflumilast 0.15% cream is expected to be $580 per patient in year 1 compared to a year 1 cost of $17 to $69 per patient for TCS comparators and $446 to $646 for TCI comparators over the same time period (Table 9). The use of roflumilast 0.15% cream is expected to result in incremental costs of $511 to $568 per patient when compared to TCSs and range from providing incremental savings of $66 to incurring incremental costs of $134 when compared to TCIs. This increase in health care spending results from the higher drug acquisition costs with roflumilast 0.15% cream and wastage assumptions. When wastage is not assumed, roflumilast 0.15% cream is associated with $303 to $344 per patient per year in incremental costs compared to the TCS comparators, but provides $82 to $172 per year in incremental savings compared to the TCI comparators (Supplemental Material, Appendix 10).
Table 9: Summary of CDA-AMC Economic Evaluation Results
Drug | Drug acquisition costs, year 1 ($) |
|---|---|
Betamethasone valerate 0.05% (TCS) | 12 |
Hydrocortisone 0.05% (TCS) | 17 |
Hydrocortisone valerate 0.02% (TCS) | 40 |
Triamcinolone acetonide 0.1% (TCS) | 49 |
Desonide 0.05% (TCS) | 69 |
Pimecrolimus 1% (TCI) | 446 |
Roflumilast 0.15% | 580 |
Tacrolimus 0.03% (TCI) | 604 |
Tacrolimus 0.1% (TCI) | 646 |
CDA-AMC = Canada’s Drug Agency; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid.
Summary of the Budget Impact
The sponsor submitted a budget impact analysis to estimate the 3-year (2026 to 2028) budget impact of reimbursing roflumilast 0.15% cream for use in the Health Canada–indicated population. The sponsor assumed that the payer would be the CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic-based approach. The price of roflumilast 0.15% cream was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on publicly available list prices. Additional information on the sponsor’s submission is provided in the Supplemental Material document, Appendix 11.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (Supplemental Material, Appendix 11). CDA-AMC estimated that 299,584 patients would be eligible for treatment with roflumilast 0.15% cream by the third year of reimbursement, of whom 44,938 would be expected to receive roflumilast 0.15% cream. The estimated incremental budget impact of reimbursing roflumilast 0.15% cream for the Health Canada–indicated population is expected to be approximately $19.7 million over the first 3 years, with an expected expenditure of $51.4 million on roflumilast. If funded for the sponsor’s narrower reimbursement request population, 182,147 patients would be eligible for roflumilast 0.15% cream by year 3 of reimbursement, with 27,322 expected to receive it, at an estimated incremental budget impact of $871,000 over the first 3 years and an expected expenditure of $31.3 million on roflumilast. The actual budget impact will depend on the population eligible for roflumilast, the proportion of patients who will be reimbursed by the public drug plans, the potential wastage of excess product, the average amount of topicals used per patient-year, and the comparators being displaced.
Conclusions
At the submitted price, roflumilast 0.15% cream is expected to be more costly than TCSs and within the range of the costs for the TCIs used for the treatment of patients aged 6 years and older with mild to moderate AD. However, confidential pricing agreements exist for comparators, and a price reduction for roflumilast 0.15% cream may be required so that no additional costs are incurred by the health care system. Results are highly sensitive to the volume of comparator treatments used per year, and to the amount of wastage associated with each comparator.
The budget impact on the public drug plans of reimbursing roflumilast 0.15% cream in the first 3 years is estimated to be approximately $19.7 million if funded for its full indication, and $871,000 if funded for its reimbursement request population. The 3-year expenditure on roflumilast 0.15% cream (i.e., not accounting for current expenditure on comparators) is estimated to be $51.4 million in the Health Canada–indicated population and $31.3 million in the reimbursement request population. The estimated budget impact is uncertain due to uncertainties in the proportion of patients who will be reimbursed by the public drug plans, the potential wastage of excess product, the average amount of topicals used per patient-year, and the comparators being displaced.
References
1.Association CD. Eczema [sponsor supplied reference]. Accessed March 31, 2024. https://dermatology.ca/public-patients/skin/eczema/
2.Kapur S, Watson W, Carr S. Atopic dermatitis. Allergy Asthma Clin Immunol. 2018;14(Suppl 2):52. doi:10.1186/s13223-018-0281-6 PubMed
3.Vakharia PP, Chopra R, Sacotte R, et al. Burden of skin pain in atopic dermatitis. Ann Allergy Asthma Immunol. 2017;119(6):548-552 e3. doi:10.1016/j.anai.2017.09.076 PubMed
4.Langan SM, Irvine AD, Weidinger S. Atopic dermatitis. Lancet. 2020;396(10247):345-360. doi:10.1016/S0140-6736(20)31286-1 PubMed
5.Barbarot S, Auziere S, Gadkari A, et al. Epidemiology of atopic dermatitis in adults: Results from an international survey. Allergy. 2018;73(6):1284-1293. doi:10.1111/all.13401 PubMed
6.Gooderham MJ, Hong HC, Lynde C, et al. Canadian Consensus Guidelines for the Management of Atopic Dermatitis with Topical Therapies. Dermatol Ther (Heidelb). 2025;15:1467–1485. doi:10.1007/s13555-025-01386-2 PubMed
7.Silverberg JI, Barbarot S, Gadkari A, et al. Atopic dermatitis in the pediatric population: A cross-sectional, international epidemiologic study. Ann Allergy Asthma Immunol. 2021;126(4):417-428 e2. doi:10.1016/j.anai.2020.12.020 PubMed
8.Silverberg JI, Shi VY, Alexis A, et al. Racial and Ethnic Differences in Sociodemographic, Clinical, and Treatment Characteristics Among Patients with Atopic Dermatitis in the United States and Canada: Real-World Data from the CorEvitas Atopic Dermatitis Registry. Dermatol Ther (Heidelb). 2023;13(9):2045-2061. doi:10.1007/s13555-023-00980-6 PubMed
9.Asiniwasis RN, Chu DK. Atopic Dermatitis and Canadian Indigenous Peoples: Burdens, Barriers, and Potential for Solutions. Canadian Allergy & Immunology Today. 2022;2(3):26-30.
10.Butala S, Paller AS. Optimizing topical management of atopic dermatitis. (1534-4436 (Electronic))
11.Panel AAJADG, Chu DK, Schneider L, et al. Atopic dermatitis (eczema) guidelines: 2023 American Academy of Allergy, Asthma and Immunology/American College of Allergy, Asthma and Immunology Joint Task Force on Practice Parameters GRADE- and Institute of Medicine-based recommendations. Ann Allergy Asthma Immunol. 2024;132(3):274-312. doi:10.1016/j.anai.2023.11.009 PubMed
12.Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2. Management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71(1):116-32. doi:10.1016/j.jaad.2014.03.023 PubMed
13.Davis DMR, Drucker AM, Alikhan A, et al. Guidelines of care for the management of atopic dermatitis in adults with phototherapy and systemic therapies. J Am Acad Dermatol. 2024;90(2):e43-e56. doi:10.1016/j.jaad.2023.08.102 PubMed
14.Kim WB, Jerome D, Yeung J. Diagnosis and management of psoriasis. (1715-5258 (Electronic))
15.LEO Pharma Inc. Protopic (tacrolimus ointment 0.03% and 0.1% (w/w)). Thornhill (ON)2020.
16.Valeant Canada LP. Elidel (Pimecrolimus Cream, 1%). Laval (QB)2014.
17.Arcutis Biotherapeutics. ZORYVE (roflumilast cream 0.15%). 2024.
18.ARQ-151-313: A Phase 3, Multicenter, Open-Label Extension Study of the Long-Term Safety of ARQ-151 Cream 0.15% and ARQ-151 Cream 0.05% in Subjects with Atopic Dermatitis, (2023).
19.Arcutis Biotherapeutics, Inc. Statistical Analysis Plan. ARQ-151-311. A Phase 3, 4-Week, Parallel Group, Double Blind, Vehicle-Controlled Study of the Safety and Efficacy of ARQ-151 Cream 0.15% Administered QD in Subjects with Atopic Dermatitis [internal sponsor's report]. Oct 25, 2022.
20.Arcutis Biotherapeutics, Inc. Statistical Analysis Plan: ARQ-151-312. A Phase 3, 4-Week, Parallel Group, Double Blind, Vehicle-Controlled Study of the Safety and Efficacy of ARQ-151 Cream 0.15% Administered QD in Subjects with Atopic Dermatitis [internal sponsor's report]. November 10, 2022.
21.Hanifin JM, Thurston M Fau - Omoto M, Omoto M Fau - Cherill R, Cherill R Fau - Tofte SJ, Tofte Sj Fau - Graeber M, Graeber M. The eczema area and severity index (EASI): assessment of reliability in atopic dermatitis. EASI Evaluator Group. (0906-6705 (Print))
22.ARQ-151-311: A Phase 3, 4-Week, Parallel Group, Double Blind, Vehicle-Controlled Study of the Safety and Efficacy of ARQ-151 Cream 0.15% Administered QD in Subjects with Atopic Dermatitis, (2023). Accessed March 31, 2024.
23.ARQ-151-312: A Phase 3, 4-Week, Parallel Group, Double Blind, Vehicle-Controlled Study of the Safety and Efficacy of ARQ-151 Cream 0.15% Administered QD in Subjects with Atopic Dermatitis, (2023). Accessed March 31, 2024.
24.Gooderham M, Kircik L, Zirwas M, et al. The Safety and Efficacy of Roflumilast Cream 0.15% and 0.05% in Patients With Atopic Dermatitis: Randomized, Double-Blind, Phase 2 Proof of Concept Study. J Drugs Dermatol. 2023;22(2):139-147. doi:10.36849/JDD.7295 PubMed
25.Arcutis Canada, Inc. Zoryve (roflumilast): roflumilast cream, 0.15% and 0.3% w/w, topical; roflumilast foam, 0.3% w/w, topical [product monograph]. November 7, 2023. Updated March 13, 2025.
26.ARQ-151-313: A Phase 3, Multicenter, Open-Label Extension Study of the Long-Term Safety of ARQ-151 Cream 0.15% and ARQ-151 Cream 0.05% in Subjects with Atopic Dermatitis [sponsor supplied reference]. 2023. Accessed July 26, 2023.
27.Simpson EL, Eichenfield LF, Papp KA, et al. Long-Term Safety and Efficacy with Roflumilast Cream 0.15% in Patients Aged ≥6 Years with Atopic Dermatitis: A Phase 3 Open-Label Extension Trial. Dermatitis. 2025;doi:10.1089/derm.2024.0418 PubMed
28.Lott A. Wilson Confidence Intervals for Binomial Proportions With Multiple Imputation for Missing Data. The American Statistician. 2020;74(2):6. doi:10.1080/00031305.2018.1473796
29.Simpson EL, Bissonnette R, Paller AS, et al. The Validated Investigator Global Assessment for Atopic Dermatitis (vIGA-AD): a clinical outcome measure for the severity of atopic dermatitis. Br J Dermatol. 2022;187(4):531-538. doi:10.1111/bjd.21615 PubMed
30.Tian J, Shi L, Kang S, et al. Evaluation of outcome indicators in trials for atopic dermatitis. Mol Biomed. 2025;6(1):33. doi:10.1186/s43556-025-00273-8 PubMed
31.Eversana. Systematic literature review and network meta-analysis of clinical evidence for the acute topical treatment of mild to moderate atopic dermatitis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Zoryve (roflumilast cream 0.15% for topical treatment). North York (ON): Arcutis Canada Inc.; September 24, 2024. 2024. Accessed May 26, 2025.
32.Higgins JPT, Thomas J, Chandler J, et al. Cochrane Handbook for Systematic Reviews of Interventions [sponsor supplied reference]. Cochrane; 2023. www.training.cochrane.org/handbook
33.Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. doi:10.1136/bmj.n71 PubMed
34.Page MJ, Moher D, Bossuyt PM, et al. PRISMA 2020 explanation and elaboration: updated guidance and exemplars for reporting systematic reviews. BMJ. 2021;372:n160. doi:10.1136/bmj.n160 PubMed
35.Dias S, Sutton AJ, Ades AE, Welton NJ. Evidence synthesis for decision making 2: a generalized linear modeling framework for pairwise and network meta-analysis of randomized controlled trials. Med Decis Making. 2013;33(5):607-17. doi:10.1177/0272989x12458724 PubMed
36.Lunny C, Higgins JPT, White IR, et al. Risk of Bias in Network Meta-Analysis (RoB NMA) tool. BMJ. 2025;388:e079839. doi:10.1136/bmj-2024-079839 PubMed
37.Puig L, Carrascosa JM, Belinchon I, et al. Adherence and patient satisfaction with topical treatment in psoriasis, and the use, and organoleptic properties of such treatments: a Delphi study with an expert panel and members of the Psoriasis Group of the Spanish Academy of Dermatology and Venereology. Actas Dermosifiliogr. 2013;104(6):488-96. doi:10.1016/j.ad.2012.12.005 PubMed
38.Yentzer BA, Ade RA, Fountain JM, et al. Simplifying regimens promotes greater adherence and outcomes with topical acne medications: a randomized controlled trial. Cutis. 2010;86(2):103-8. PubMed
39.Tier HL, Balogh EA, Bashyam AM, et al. Tolerability of and Adherence to Topical Treatments in Atopic Dermatitis: A Narrative Review. Dermatol Ther (Heidelb). 2021;11(2):415-431. doi:10.1007/s13555-021-00500-4 PubMed
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.