Drugs, Health Technologies, Health Systems
Reimbursement Review
Risankizumab (Skyrizi)
Sponsor: AbbVie Corporation
Therapeutic area: Ulcerative colitis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
AEOSI
adverse event of special interest
AGA
American Gastroenterological Association
AT
advanced therapy
AT-IR
inadequate response or intolerance to advanced therapy
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CrI
credible interval
ECCO
European Crohn’s and Colitis Organisation
GI
gastrointestinal
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HEMI
histologic, endoscopic, and mucosal improvement
HEMR
histologic, endoscopic, and mucosal remission
HRQoL
health-related quality of life
IBD
inflammatory bowel disease
IBDQ
Inflammatory Bowel Disease Questionnaire
ICE
intercurrent event
IL
interleukin
ITC
indirect treatment comparison
ITT
intention to treat
MID
minimal important difference
NICE
National Institute for Health and Care Excellence
NMA
network meta-analysis
OR
odds ratio
pCPA
pan-Canadian Pharmaceutical Alliance
PICOS
population, intervention, comparators, outcomes, study design
RCT
randomized controlled trial
RR
rerandomized responder
S1P
sphingosine-1-phosphate
SAE
serious adverse event
SC
subcutaneous
SD
standard deviation
Stride II
Selecting Therapeutic Targets in Inflammatory Bowel Disease-II
TEAE
treatment-emergent adverse event
TNF
tumour necrosis factor
TT
treat through
UC
ulcerative colitis
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Skyrizi (Risankizumab):
|
Sponsor | AbbVie Corporation |
Indication | For the treatment of adult patients with moderately to severely active UC who have had an inadequate response, loss of response, or were intolerant to conventional therapy, a biologic treatment, or a JAK inhibitor. |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Supplement to a New Drug Submission |
NOC date | October 10, 2024 |
Recommended dose | The recommended dose is 1,200 mg administered by IV infusion at week 0, week 4, and week 8, followed by 180 mg or 360 mg administered by subcutaneous injection at week 12, and every 8 weeks thereafter. Use the lowest effective dosage needed to maintain therapeutic response. |
NOC = Notice of Compliance; UC = ulcerative colitis.
Introduction
Ulcerative colitis (UC) is a chronic disease characterized by inflammation, predominantly of the mucosal layer of the large intestine (colon) most often involving the rectum and frequently extends continuously into the proximal colon.1 The cause of UC remains uncertain, but a combination of genetic and environmental factors contributes to immune dysregulation and up-regulation in response to micro-organisms in the gastrointestinal (GI) tract.2 UC is characterized by blood in the stool with mucus, frequent diarrhea, urgency, loss of appetite, and tenesmus (severe rectal cramp or spasm).3 Although UC principally affects the GI tract, extraintestinal manifestations, such as arthritis, may also occur.4 While most patients have a mild to moderate disease course, about 10% to 15% experience an aggressive course.5 Relapse is common, with the cumulative risk of relapse being 70% to 80% at 10 years.5 The chronic nature of UC has a considerable impact on patients’ health-related quality of life (HRQoL), including psychological, physical, sexual, and social domains of HRQoL, due to chronic symptoms such as urgency, frequency, and incontinence.6,7
The prevalence of UC in Canada was estimated to be 414 per 100,000 in 2023.8 It is estimated that 32% to 46% of Canadians with UC have moderate disease, and 13% to 14% have severe disease.9
The general goal of pharmacotherapy is to achieve complete remission, defined as both symptomatic and endoscopic remission without corticosteroid therapy; to preserve HRQoL; and to prevent disability.10,11 Guidelines generally recommend a standard step-up approach to the medical management of moderate to severe UC. It is further recommended that for maintenance of remission, the same drug should be used in patients whose disease has responded to induction therapy with that drug.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of risankizumab in the treatment of adult patients with moderately to severely active UC who have experienced an inadequate response, loss of response, or were intolerant to conventional therapy, a biologic treatment, or a JAK inhibitor.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to our call for input and from clinical experts consulted for the purpose of this review.
Patient Input
Canada’s Drug Agency (CDA-AMC) received input from 2 patient groups, the Gastrointestinal (GI) Society and Crohn’s and Colitis Canada. Both advocate for patients with UC and their caregivers. The GI Society gathered information from online surveys and round tables, and through a one-to-one conversation with a patient living with UC and receiving risankizumab. Crohn’s and Colitis Canada gathered information from a survey, from its 2023 Impact of Inflammatory Bowel Disease in Canada report, and through interviews with patients who have had experience with risankizumab.
Respondents reported that sustained remission or treatment response is more important than relieving any 1 symptom and the constant concern that there will be future flares, possibly worse than the last, at unpredictable times, can disastrously disrupt patients’ lives. Patients also noted that they need treatments that improve quality of life, not cause more symptoms, pain, frustration, or hardship. They also emphasized that improved outcomes included improved quality of life; access to different treatment options; access to a treatment that works, results in less suffering, and involves less unnecessary usage of health care resources (hospital stay, surgeries, and diagnostic procedures); access to a treatment with no side effects; reduction of symptoms; and ease of administration (fewer injections, self-injection, or oral administration).
The GI Society interviewed a patient with UC who started taking risankizumab through a clinical trial in December 2022. After 2 to 3 weeks, the patient noticed a significant improvement. The patient’s injections were every 8 weeks, and the patient noticed that, by about week 6, they could feel that they needed the medication again.
Clinician Input
Input From Clinical Experts Consulted for This Review
According to the clinical experts consulted by CDA-AMC, more effective and safer treatments are welcomed, as the currently available options are limited. The clinical experts noted that treating UC can be challenging and the goal is to control both symptoms (e.g., frequency, urgency) and signs of UC (e.g., clinical and endoscopic remission) with AT as quickly as possible. There are many unmet needs in patients with UC because UC responds to the available treatments only in some patients, and UC may become refractory to current treatment options in some patients. In addition, patients need new treatments that are better tolerated, with rapid onset and enhanced safety profiles. Furthermore, there have been few SC options for treatment of UC to date.
The clinical experts noted that risankizumab should be available as a first-line option for treatment of moderate to severe UC and that previous treatment failure should not be required to begin this drug. The only previous therapy that they would consider as a reasonable option would be corticosteroids, but these are not an option for long-term maintenance therapy.
The clinical experts noted that any patient with moderate to severe UC would be a suitable candidate for risankizumab. Patients unable to complete a self-injection may require support administering the medication. With regards to the diagnosis, and staging of UC, both the diagnosis and treatment need to be made by a gastroenterologist. Misdiagnosis is unlikely, as endoscopy is needed to establish the diagnosis.
The clinical experts noted that the important outcomes can be categorized as follows:
improvement in symptoms, including decreasing stool frequency, urgency, and bleeding; these have a direct impact on patients’ HRQoL and important end points, such as work or school participation, fatigue (by hindering patients from sleeping through the night), and overall function or activities of daily living
sustained clinical remission, avoiding the need to add steroids (e.g., steroid-sparing and steroid-free remission with ongoing clinical remission)
endoscopic bowel healing, which is the best marker to ensure that adverse outcomes such as surgery and hospitalization are avoided.
In practice, they noted that clinical scoring systems (e.g., Mayo and partial Mayo scores, rectal bleeding score), endoscopic outcomes, and histopathological evaluation are used to evaluate response to treatment. In addition, biomarkers (e.g., fecal calprotectin) and corticosteroid-free remission are used to monitor ongoing treatment response and during maintenance therapy. From the patient’s perspective, improvement in HRQoL is prudent to monitor.
The main issue with scoring systems is that there can be a level of subjectivity to them. Hard end points would include Mayo endoscopic subscore and histopathological evaluation. Following the Selecting Therapeutic Targets in Inflammatory Bowel Disease-II (Stride II) guidelines is imperative to ensuring appropriate ongoing clinical management.
The clinical experts noted that treatment discontinuation can be organized into 3 main categories:
primary nonresponse during induction treatment, with ongoing evidence of objective disease, despite a robust attempt at induction treatment (and possible reinduction) and no meaningful improvement with symptom persistence.
early improvement in symptoms and objective disease, followed by relapse with persistent symptoms and disease activity requiring corticosteroid treatment, either persistently or recurrently
serious adverse events (SAEs) (e.g., anaphylactic reactions).
The clinical experts noted that, following diagnosis and staging of UC by a gastroenterologist, treatment can be prescribed by a gastroenterologist or general internist with experience and expertise in treatment of inflammatory bowel disease (IBD). Treatment can occur in any clinical setting, but most patients will be initiated in the outpatient setting.
Clinician Group Input
CDA-AMC received input from 1 clinician group, the Canadian IBD Physicians Group, which consists of 25 clinicians who met through videoconference in order to contribute to this submission.
Regarding the place in therapy, the clinician group believed that risankizumab should be used as the first choice in patients with moderately to severely active UC in whom conventional therapies have failed, who are not candidates for other conventional therapy, or who have failed 1 or more ATs. They also noted that UC patients with current or previous mild to moderate active disease, whose remission can be adequately maintained with conventional treatments, are least suitable for treatment with risankizumab.
Drug Program Input
The drug plans identified jurisdictional implementation issues related to relevant comparators, considerations for initiation of therapy, considerations for prescribing of therapy, generalizability, care provision issues, and system and economic issues. The clinical experts consulted by CDA-AMC weighed evidence from the INSPIRE and COMMAND trials and other clinical considerations to provide responses to the drug plans’ questions.
Clinical Evidence
Systematic Review
Description of Studies
The systematic review included 2 pivotal trials (INSPIRE and COMMAND). The INSPIRE trial was a phase III, randomized, placebo-controlled, double-blind, parallel-group induction study in adult patients with moderately to severely active UC who experienced an inadequate response or intolerance to conventional therapies or other ATs. Patients in the INSPIRE trial were randomized to risankizumab 1,200 mg IV (650 patients) or placebo (325 patients). Patients who did not achieve clinical response at week 12 were eligible to enter another blinded risankizumab treatment period (i.e., Induction Period 2, which lasted for an additional 12 weeks).
The COMMAND trial was a phase III, randomized, placebo-controlled, double-blind, parallel-group maintenance study in adult patients with UC previously enrolled in the INSPIRE induction trial whose disease had an adequate response to risankizumab. Patients were randomized to risankizumab 180 mg subcutaneously (SC) (179 patients), risankizumab 360 mg SC (186 patients), or placebo (183 patients). The placebo group were patients who discontinued risankizumab after the induction phase (risankizumab withdrawal). Combined, the 2 studies lasted up to 81 weeks and included a screening period that lasted up to 35 days, a 12-week double-blind induction period, and a 52-week maintenance period. Randomization was stratified by history of inadequate response to advanced therapy (AT) (yes, no), last risankizumab induction dose (IV 600 mg, 1,200 mg, or 1,800 mg), and clinical remission status (per local evaluation) at the last visit of the induction trial (yes, no). Patients who experienced inadequate response were eligible for risankizumab rescue therapy starting at week 16 of maintenance (1 dose of 1,200 mg or 1,800 mg followed by 360 mg every 8 weeks). Loss of response or inadequate response was based on clinical and endoscopic symptoms (rectal bleeding score at least 1 point greater than the week 0 value or endoscopic subscore of 2 or 3). Final patient follow-up occurred in April 2023.
The following efficacy outcomes were assessed to be most relevant to inform expert committee deliberations and finalized in consultation with members of the expert committee: clinical remission per adapted Mayo score; clinical response per adapted Mayo score; endoscopic improvement; histologic, endoscopic, and mucosal improvement (HEMI); histologic, endoscopic, and mucosal remission (HEMR); discontinuation of corticosteroid use in patients taking steroids at baseline (maintenance only); UC-related hospitalization; and the patient-reported outcome Inflammatory Bowel Disease Questionnaire (IBDQ). Select notable harms outcomes considered important were treatment-emergent adverse events (TEAEs), SAEs, and adverse events of special interest (AEOSIs).
The baseline characteristics of patients in the risankizumab and placebo treatment arms in the INSPIRE trial were generally well balanced. UC was highly refractory in the entire study population. This is shown by the proportion of patients who had extensive UC or pancolitis at baseline, in addition to a high proportion in whom ATs had previously failed (close to 51%). Furthermore, this study population had a high proportion of patients with a baseline adapted Mayo score greater than 7, and most patients had a baseline endoscopic subscore of 3.
Similarly, in the population in the maintenance study (COMMAND), UC was also highly refractory, as evinced by severe disease characteristics: extensive UC or pancolitis, mean disease duration, and previous failure of an AT (close to 75%), including failure of more than 2 ATs (around 25%) and failure of JAK inhibitors (around 15%).
Efficacy Results
In the induction trial (INSPIRE), all efficacy outcomes were analyzed using all randomized patients who received at least 1 dose of study drug, according to the treatment that they were randomized to. Similarly, in the maintenance trial (COMMAND), all efficacy outcomes were analyzed using all randomized patients who received at least 1 dose of study drug after receiving IV risankizumab for only 1 period of 12 weeks in the induction trial, according to the treatment that they were randomized to.
The primary end point of the INSPIRE trial was clinical remission at 12 weeks using the adapted Mayo score. The risk difference was 14.0% (95% CI, 10.0% to 18.0%) for risankizumab compared to placebo. Similarly, in the COMMAND trial, the risk difference at 52 weeks was 16.3% (95% CI, 7.4% to 25.3%) for risankizumab 180 mg and 14.2% (95% CI, 5.3% to 23.2%) for risankizumab 360 mg compared to placebo (risankizumab withdrawal).
The secondary outcomes were in line with the primary end point. In the INSPIRE trial, the risk difference for clinical response was 28.6% (95% CI, 22.3% to 34.8%) for risankizumab compared to placebo. In the COMMAND trial, the risk difference was 17.1% (95% CI, 7.5% to 26.6%) and 11.5% (95% CI, 1.7% to 21.2%) for risankizumab 180 mg SC and risankizumab 360 mg SC, respectively, compared to placebo (risankizumab withdrawal).
For endoscopic improvement, in the INSPIRE trial, the risk difference was 24.3% (95% CI, 19.3% to 29.4%) for risankizumab compared to placebo. In the COMMAND trial, the risk difference was 20.1% (95% CI, 10.6% to 29.6%) and 17.4% (95% CI, 7.9% to 26.9%) for risankizumab 180 mg SC and risankizumab 360 mg SC, respectively, compared to placebo (risankizumab withdrawal).
For HEMI, in the INSPIRE trial, the risk difference was 16.6% (95% CI, 12.3% to 21.0%) for risankizumab compared to placebo. In the COMMAND trial, the risk difference was 20.2% (95% CI, 11.2% to 29.2%) and 19.8% (95% CI, 10.8% to 28.8%) for risankizumab 180 mg SC and risankizumab 360 mg SC, respectively, compared to placebo (risankizumab withdrawal).
For HEMR, in the INSPIRE trial, the risk difference was 5.6% (95% CI, 3.5% to 7.7%) for risankizumab compared to placebo. In the COMMAND trial, the risk difference was 4.0% (95% CI, −2.2% to 10.3%) and 6.1% (95% CI, −0.3% to 12.5%) for risankizumab 180 mg SC and risankizumab 360 mg SC, respectively, compared to placebo (risankizumab withdrawal).
For discontinuation of corticosteroid use in patients taking steroids at baseline, which was measured only in the COMMAND trial, the risk difference was 28.4% (95% CI, 14.0% to 42.8%) and 20.7% (95% CI, 4.9% to 36.6%) for risankizumab 180 mg SC and risankizumab 360 mg SC, respectively, compared to placebo (risankizumab withdrawal).
Total IBDQ score ranges between 32 and 224, with higher scores representing better HRQoL. Using the IBDQ scale, the mean difference was 18.3 (95% CI, 13.38 to 23.25) points for risankizumab compared to placebo in the INSPIRE trial. In the COMMAND trial, the mean difference was 17.5 (95% CI, 8.01 to 27.06) and 15.2 (95% CI, 5.18 to 25.31) points for risankizumab 180 mg SC and risankizumab 360 mg SC, respectively, compared to placebo (risankizumab withdrawal).
In the INSPIRE trial, the risk difference for UC-related hospitalization was −4.8% (95% CI, −7.3% to −2.2%) for risankizumab compared to placebo. In the COMMAND trial, the incidence rate difference was −2.5 per 100 patient-years (95% CI, −5.4 to 0.4) and −1.8 per 100 patient-years (95% CI, −5.0 to 1.3) for risankizumab 180 mg SC and risankizumab 360 mg SC, respectively, compared to placebo (risankizumab withdrawal).
Harms
The safety analyses in both trials included all randomized patients who received at least 1 dose of study drug, analyzed according to the treatment that they received. In the INSPIRE trial, the number of patients with at least 1 TEAE was similar between the risankizumab (42.4%) and placebo (49.4%) groups. The most common TEAE in the risankizumab group was COVID-19 (4.8%), and the most common TEAE in the placebo group was UC (refers to the worsening of the underlying disease) (10.2%). The proportion of patients with at least 1 SAE was higher in the placebo group (10.2%) than the risankizumab group (2.3%). The most common serious TEAEs in the risankizumab group were anemia, UC, and pulmonary embolism (2 patients [0.3%] each). In the placebo group, the most common serious TEAEs were UC (4.9%), anemia (0.6%), and anal fistula (0.6%). A higher proportion of patients in the placebo group (3.7%) discontinued treatment due to a TEAE than in the risankizumab group (0.6%). One patient in the risankizumab group died due to COVID-19. The most frequently occurring AEOSIs in both arms were hypersensitivity (risankizumab: 3.8%; placebo: 2.2%) and hepatic events (risankizumab: 1.5%; placebo: 4.0%).
In the COMMAND trial, the number of patients with at least 1 TEAE were similar between the risankizumab 180 mg (72.5%), risankizumab 360 mg (70.8%), and placebo (76.5%) groups. The proportion of patients with at least 1 SAE was similar across groups (5.2% with risankizumab 180 mg, 5.1% with risankizumab 360 mg, and 8.2% with placebo). In the risankizumab 180 mg group, the only serious TEAE occurring in more than 1 patient was appendicitis (2 patients, 1.0%). In the risankizumab 360 mg group, no serious TEAE occurred in more than 1 patient. In the placebo (risankizumab withdrawal) group, the most common serious TEAE was UC (2.6%); no other serious TEAE occurred in more than 1 patient. The proportions of patients who discontinued treatment due to a TEAE were highest in those taking risankizumab 360 mg (2.6%) compared to risankizumab 180 mg (1.6%) and placebo (1.5%) groups. Regarding AEOSIs, the risankizumab 180 mg group had the highest proportion of patients with hypersensitivity (10.4%). The risankizumab 360 mg group had the highest proportion of patients with hepatic events (6.7%).
Critical Appraisal
Internal Validity
INSPIRE trial: Randomization in the INSPIRE trial took place using a 2:1 computer-generated block randomization via interactive response technology. This is an adequate form of randomization and allocation concealment. Participants were stratified for major confounding factors (e.g., corticosteroid use, baseline Mayo score, and prior AT exposure). The baseline characteristics of the randomized groups were balanced, reflecting a proper randomization procedure. The trial was double-blind and placebo-controlled, with a detailed protocol including which concomitant medications were allowed and which changes in dosage were allowed after randomization. This decreases the probability of any deviations from the intended interventions during the trial. Concomitant medications were balanced across the treatment groups and were not expected to impact the efficacy results. The primary and secondary efficacy end points in INSPIRE were tested using a graphical multiple-testing procedure to ensure control of family-wise type I error at a significance level of 0.05 (2-sided), which was deemed adequate.
The sponsor considered the occurrence of appropriate intercurrent events (ICEs) that affect the interpretation of the effect of interest within the trial estimands. Data for patients who discontinued prematurely continued to be collected (i.e., treatment policy) while initiation or escalation of UC-related corticosteroids and the occurrence or UC-related surgery were considered as a nonresponse for binary end points and return to baseline for continuous end points. Regulatory authorities consider this approach adequate when the therapeutic intent is to induce remission in the short term.12,13
With regard to missing outcome data, the sponsor was unable to provide information requested by the review team about ICEs before study discontinuation or sporadic missing data for reasons other than study discontinuation (e.g., missing visit). Therefore, it was not possible to perform a complete appraisal of the potential for bias due to missing data arising from reasons that are incompatible with the nonresponder imputation approach used by the sponsor for binary end points (and missing at random for continuous end points). There was a higher discontinuation rate in the placebo group (8.3%) than in the risankizumab group (2%), which can introduce attrition bias when nonresponder imputation is used and the reasons for study discontinuation (and possibly missing data) are incompatible with lack of efficacy. More than half of patients in the placebo group who withdrew did so because of lack of efficacy or AEs, suggesting that the assumption of nonresponse is acceptable to account for withdrawals. Sensitivity analyses were conducted for the primary end point, including a tipping-point analysis which supported that the statistical significance of the results would be maintained across a range of assumptions about the missing data. The outcome of UC-related hospitalization used an as-observed analysis; this approach considers that data are missing completely at random, which is unlikely to be reasonable and may introduce bias if there is an increased proportion of missing data (which could not be assessed).
To minimize detection bias, the trial used central endoscopic review with blinded assessors for objective outcomes, but it is important to note that this is only 1 component of the adapted Mayo score. For subjective outcomes, such as patient-reported outcomes (e.g., other components of the adapted Mayo score, IBDQ) and safety outcomes, these results are unlikely to be biased because the trial was double-blind. A prespecified protocol, published on ClinicalTrials.gov (NCT03398148), minimized selective outcome and analysis reporting bias. Overall, the INSPIRE trial is at low risk of bias for most end points, although uncertainty remains about potential attrition bias, which could not be fully assessed due to insufficient reporting.
The collection of information on harms included worsening UC as well as UC-related symptoms. Given that these are likely to be increased in the placebo group, the interpretation of the difference between groups is challenging. Further, it is implausible that there were significantly more SAEs reported in the placebo group than in the active intervention group, which would imply that the placebo effect is associated with SAEs.
With regard to the subgroup analyses, randomization in the INSPIRE trial was stratified by the presence of baseline corticosteroid use (yes, no), baseline adapted Mayo score (≤ 7, > 7), and a history of intolerance or inadequate response to ATs (0, 1, > 1 treatment). All other subgroup analyses may have broken randomization and therefore could have been affected by biases, including selection bias and confounding due to unadjusted differences in baseline characteristics. The purpose of these analyses was to demonstrate consistency, and they cannot be used to draw credible conclusions about effect modification.
COMMAND trial: The trial protocol was publicly available (NCT03398135) and the randomization and allocation concealment methods were similar to those in the INSPIRE trial, but with a 1:1:1 allocation ratio stratified by prior advanced therapy response and induction dose. The baseline characteristics suggested imbalances in some disease characteristics, though these did not appear to systematically favour any treatment group. Therefore, they are not expected to have an important impact on the results.
While the trial was double-blind and open-label, rescue with risankizumab 1,200 mg IV was allowed after week 16. Unblinded therapy may have introduced performance bias in the collection of data related to harms. For efficacy end points, a patient needing rescue therapy was assumed to be a nonresponder, which is appropriate and would not introduce bias. Concomitant medications were balanced across the treatment groups and are not expected to impact the efficacy results. In the COMMAND trial, the testing began with testing the primary end point at the prespecified significance level of 0.025 (2-sided) for each risankizumab dosage group compared to placebo. This was deemed to be an adequate approach.
The sponsor considered the occurrence of appropriate ICEs that affect the interpretation of the effect of interest within the trial estimands. Data for patients who discontinued prematurely continued to be collected (i.e., treatment policy) while initiation or escalation of UC-related corticosteroids, the occurrence or UC-related surgery, and the need for rescue treatment were considered as a nonresponse for binary end points and return to baseline for continuous end points. Given that the intent of the COMMAND trial is to demonstrate maintenance of treatment efficacy in the long term, considering treatment discontinuations as nonresponse may be a preferred approach.13 Nonetheless, a supplemental analysis using the nonresponder imputation approach for treatment discontinuations showed similar results to the primary analysis on the primary and all secondary end points.
It was not possible to perform a comprehensive appraisal of the risk of bias due to missing outcome data as a result of incomplete reporting in the COMMAND study report. The concern for potential unreported missing data may be higher for the COMMAND trial than for the INSPIRE trial, resulting from the longer period of follow-up. There was a higher discontinuation rate in all groups compared to the INSPIRE trial, with 9.8% of patients in the placebo group discontinuing, and 7.8% to 12.4% of patients in the risankizumab groups discontinuing. This high discontinuation rate can introduce attrition bias when nonresponder imputation is used and the reasons for study discontinuation are incompatible with lack of efficacy (if these patients’ data are considered missing). The potential for bias cannot be appraised because it is unclear how many patients were already imputed as nonresponders before study discontinuation (due to ICE, which appeared to be common; for example 19% to 43% of patients received rescue medication). On request from the review team, the sponsor was unable to provide this information. Sensitivity analyses were conducted for the primary end point, including a tipping-point analysis indicating that the statistical significance of the results would be maintained across a range of assumptions about the missing data. The outcome of UC-related hospitalization used an as-observed analysis, which considers data to be missing completely at random, which is unlikely to be reasonable and may introduce bias if there is an increased proportion of missing data (which could not be assessed).
In addition, the outcome “discontinuation of corticosteroid use at week 52 in patients taking steroids at baseline” was not assessed in the full cohort of randomized participants, and it is unclear whether the baseline characteristics in the patients are similar between the groups. Overall, it is considered that bias is possible in the harms data due to open-label rescue treatment and in the corticosteroid discontinuation end point due to possible loss of randomization. The risk of bias due to missing outcome data could be increased because of attrition and lack of information on the proportion of data missing. This concern was mitigated for the primary outcome via sensitivity analysis.
Information on harms included worsening UC as well as UC-related symptoms. Given that these are likely to be increased in the placebo group, the interpretation of the difference between groups is challenging. Additionally, the comparison may not reflect a true placebo group due to the potential for risankizumab rescue therapy, which was received by 43% of patients in the placebo group.
In the COMMAND trial, randomization was stratified by history of inadequate response to advanced therapy (yes, no), last risankizumab induction dose (IV 600 mg, 1,200 mg, or 1,800 mg), and clinical remission status (per local evaluation) at the last visit of the induction trial (yes, no). All other subgroup analyses may have broken randomization and therefore could have been affected by biases, including selection bias and confounding due to unadjusted differences in baseline characteristics.
It is also important to note that the placebo group in the COMMAND trial received risankizumab during the induction phase (risankizumab withdrawal). As such, there could have been a carry-over effect due to an inadequate washout.
External Validity
The populations of both trials had a history of disease that was highly refractory to previous UC treatments, and the clinical experts consulted by the review team confirmed that patients in the newer UC trials are expected to have UC that is more refractory to treatment. In addition, the trials excluded patients with a history of IL-23 inhibitor use. With regard to co-interventions, in the INSPIRE trial, initiating or increasing the dose of comedications was prohibited, while in the COMMAND trial, patients undergoing corticosteroid therapy were required to taper by week 8. With regard to the comparator, no direct comparison to an active intervention was available (only compared to placebo). Furthermore, the main primary and secondary outcomes used the Mayo scoring system, while this score is not commonly used in clinical practice to inform decision-making. In the subgroup analyses, several subgroups, including patients in North America, showed less clinically significant results. As a result, the results may not be generalizable and credible conclusions on effect modification cannot be drawn. These specific trial characteristics and results may impact generalizability and implementation in a real-world setting.
GRADE Summary of Findings and Certainty of the Evidence
The selection of outcomes for Grading of Recommendations Assessment, Development and Evaluation (GRADE) assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members.
INSPIRE trial:
Clinical remission
Clinical response
Endoscopic improvement
HEMI
HEMR
IBDQ
SAEs
COMMAND trial:
Clinical remission
Clinical response
Endoscopic improvement
HEMI
HEMR
Discontinuation of corticosteroid use in patients taking steroids at baseline
IBDQ
SAEs
Table 2: Summary of Findings for Risankizumab vs. Placebo for Patients With UC (INSPIRE)
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Risankizumab 1,200 mg IV | Absolute difference (95% CI) | |||||
Efficacy | |||||||
Proportion of patients with clinical remission per the adapted Mayo score Follow-up: 12 weeks | 975 (1 RCT) | NR | 62 per 1,000 | 203 per 1,000 (172 to 234 per 1,000) | 140 more per 1,000 (100 more to 180 more) | Moderatea | Risankizumab likely results in a clinically important improvement in clinical remission compared to placebo. |
Proportion of patients with clinical response per the adapted Mayo score Follow-up: 12 weeks | 975 (1 RCT) | NR | 357 per 1,000 | 643 per 1,000 (606 to 679 per 1,000) | 286 more per 1,000 (223 more to 348 more) | Highb | Risankizumab results in a clinically important improvement in clinical response compared to placebo. |
Proportion of patients with endoscopic improvement Follow-up: 12 weeks | 975 (1 RCT) | NR | 121 per 1,000 | 365 per 1,000 (328 to 402 per 1,000) | 243 more per 1,000 (193 more to 294 more) | Highc | Risankizumab results in a clinically important improvement in endoscopic improvement compared to placebo. |
Proportion of patients with HEMI Follow-up: 12 weeks | 975 (1 RCT) | NR | 77 per 1,000 | 245 per 1,000 (212 to 278 per 1,000) | 166 more per 1,000 (123 more to 210 more) | Highd | Risankizumab results in an improvement in HEMI compared to placebo. The clinical importance is uncertain. |
Proportion of patients with HEMR Follow-up: 12 weeks | 975 (1 RCT) | NR | 6 per 1,000 | 63 per 1,000 (44 to 82 per 1,000) | 56 more per 1,000 (35 more to 77 more) | Highd | Risankizumab results in an improvement in HEMR compared to placebo. The clinical importance is uncertain. |
Patient-reported outcomes | |||||||
Change from baseline in IBDQ total score, points (range: 32 [worst] to 224 [best]) Follow-up: 12 weeks | 975 (1 RCT) | NR | 24.3 | 42.6 points (39.72 to 45.57) | MD 18.3 higher (13.38 higher to 23.25 higher) | Moderatee | Risankizumab likely results in a clinically important improvement in IBDQ scores compared to placebo. |
Harms | |||||||
Proportion of patients with serious adverse events Follow-up: 12 weeks | 975 (1 RCT) | NR | 102 per 1,000 | 23 per 1,000 (NR to NR) | 79 less per 1,000 (114 less to 44 less) | Lowf | Risankizumab may result in little to no clinically important difference in SAEs compared to placebo. |
CI = confidence interval; HEMI = histologic, endoscopic, and mucosal improvement; HEMR = histologic, endoscopic, and mucosal remission; IBDQ = Inflammatory Bowel Disease Questionnaire; MD = mean difference; MID = minimal important difference; NR = not reported; RCT = randomized controlled trial; vs. = versus.
Note: Study limitations (which refer to internal validity or risk of bias), indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes. The proportion of missing data was not reported and therefore cannot be fully appraised.
aAn empirically derived MID of 11% was identified in the literature14 for the between-group difference for this outcome. Rated down 1 level for imprecision (as the confidence intervals [CIs] show both potential benefit and little to no clinically important difference between risankizumab and placebo).
bAn empirically derived MID of 14% was identified in the literature14 for the between-group difference for this outcome.
cAn empirically derived MID of 12.5% was identified in the literature14 for the between-group difference for this outcome.
dAn empirically derived MID was not identified for the between-group difference for this outcome; effects were appraised using the null.
eAn empirically derived improvement of 15 points or more greater than placebo was considered be the MID for this outcome, as identified in the literature.15,16 Rated down 1 level for imprecision, as the CIs show both potential benefit and little to no clinically important difference between risankizumab and placebo.
fAn empirically derived MID of 6% was identified in the literature14 for the between-group difference for this outcome. Rated down 1 level for imprecision (as the CIs show both potential benefit and little to no clinically important difference between risankizumab and placebo). Rated down 1 level for serious indirectness, as the SAEs include symptoms of worsening UC, which complicates interpretation. The effect was judged as not different from placebo, given that a reduction in SAEs compared to placebo is implausible.
Source: INSPIRE Clinical Study Report.17 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 3: Summary of Findings for Risankizumab 180 mg SC vs. Placebo (Risankizumab Withdrawal) for Patients With UC (COMMAND)
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Risankizumab 180 mg SC | Absolute difference (95% CI) | |||||
Efficacy | |||||||
Proportion of patients with clinical remission per the adapted Mayo score Follow-up: 52 weeks | 362 (1 RCT) | NR | 251 per 1,000 | 402 per 1,000 (330 to 474 per 1,000) | 163 more per 1,000 (74 more to 253 more) | Moderatea | Risankizumab likely results in a clinically important improvement in clinical remission compared to placebo. |
Proportion of patients with clinical response per the adapted Mayo score Follow-up: 52 weeks | 362 (1 RCT) | NR | 519 per 1,000 | 682 per 1,000 (613 to 750 per 1,000) | 171 more per 1,000 (75 more to 266 more) | Moderateb | Risankizumab likely results in a clinically important improvement in clinical response compared to placebo. |
Proportion of patients with endoscopic improvement Follow-up: 52 weeks | 362 (1 RCT) | NR | 317 per 1,000 | 508 per 1,000 (435 to 581 per 1,000) | 201 more per 1,000 (106 more to 296 more) | Moderatec | Risankizumab likely results in a clinically improvement in endoscopic improvement compared to placebo. |
Proportion of patients with HEMI Follow-up: 52 weeks | 362 (1 RCT) | NR | 235 per 1,000 | 428 per 1,000 (356 to 501 per 1,000) | 202 more per 1,000 (112 more to 292 more) | Highd | Risankizumab results in an improvement in HEMI compared to placebo. The clinical importance is uncertain. |
Proportion of patients with HEMR Follow-up: 52 weeks | 362 (1 RCT) | NR | 98 per 1,000 | 129 per 1,000 (80 to 179 per 1,000) | 40 more per 1,000 (22 less to 103 more) | Moderatee | Risankizumab likely results in an improvement in HEMR compared to placebo. The clinical importance is uncertain. |
Proportion of patients with discontinuation of corticosteroid use in patients taking steroids at baseline Follow-up: 52 weeks | 142 (1 RCT) | NR | 368 per 1,000 | 649 per 1,000 (540 to 757 per 1,000) | 284 more per 1,000 (140 more to 428 more) | Lowf | Risankizumab may result in a clinically important improvement in discontinuation of corticosteroid use compared to placebo. |
Patient-reported outcomes | |||||||
Change in baseline in IBDQ total score, points (range: 32 [worst] to 224 [best]) Follow-up: 52 weeks | 362 (1 RCT) | NR | 35.0 | 52.6 (44.93 to 60.20) | MD 17.5 higher (8.01 higher to 27.06 higher) | Moderateg | Risankizumab likely results in a clinically important improvement in IBDQ scores compared to placebo. |
Harms | |||||||
Proportion of patients with serious adverse events Follow-up: 52 weeks | 389 (1 RCT) | NR | 82 per 1,000 | 52 per 1,000 (NR to NR) | 30 less per 1,000 (79 less to 20 more) | Lowh | Risankizumab may result in little to no clinically important difference in SAEs compared to placebo. |
CI = confidence interval; HEMI = histologic, endoscopic, and mucosal improvement; HEMR = histologic, endoscopic, and mucosal remission; IBDQ = Inflammatory Bowel Disease Questionnaire; MD = mean difference; MID = minimal important difference; NR = not reported; RCT = randomized controlled trial; vs. = versus.
Note: Study limitations (which refers to internal validity or risk of bias), indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes. The proportion of missing data was not reported and therefore cannot be fully appraised.
aAn empirically derived MID of 11% was identified in the literature14 for the between-group difference for this outcome. Rated down 1 level for serious imprecision; the point estimate suggests clinically important benefit while the lower bound of the CI suggests little to no difference.
bAn empirically derived MID of 14% was identified in the literature14 for the between-group difference for this outcome. Rated down 1 level of serious imprecision; the CI includes the potential for benefit and little to no difference.
cAn empirically derived MID of 12.5% was identified in the literature14 for the between-group difference for this outcome. Rated down 1 level for imprecision, as the CIs shows both potential benefit and little to no difference between risankizumab and placebo.
dAn empirically derived MID was not identified for the between-group difference for this outcome; effects were appraised using the null.
eAn empirically derived MID was not identified for the between-group difference for this outcome; effects were appraised using the null. Rated down 1 level for imprecision, as the CIs show both potential benefit and little to no difference between risankizumab and placebo.
fNo threshold of clinical importance could be established from the literature. An empirically derived MID was not identified for the between-group difference for this outcome. A difference of 15% between the groups was identified by the clinical experts consulted by CDA-AMC as a threshold of clinical importance for this outcome. Rated down 1 level for serious study limitations; the randomization was not stratified by baseline corticosteroid use; therefore, randomization in this population might not be upheld. Rated down 1 level for imprecision, as the CIs show both potential benefit and equivalence between risankizumab and placebo.
gAn empirically derived improvement of 15 points or more greater than placebo was considered be the MID for this outcome. Rated down 1 level for imprecision, as the CIs show both potential benefit and little to no clinically important difference between risankizumab and placebo.
hAn empirically derived MID of 6% was identified in the literature14 for the between-group difference for this outcome. Rated down 1 level for imprecision, as the CIs show both potential benefit and little to no clinically important difference between risankizumab and placebo. Rated down 1 level for serious indirectness, as SAEs include symptoms of worsening UC, which complicates interpretation.
Source: COMMAND Clinical Study Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 4: Summary of Findings for Risankizumab 360 mg SC vs. Placebo (Risankizumab Withdrawal) for Patients With UC (COMMAND)
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Risankizumab 360 mg SC | Absolute difference (95% CI) | |||||
Efficacy | |||||||
Proportion of patients with clinical remission per the adapted Mayo score Follow-up: 52 weeks | 362 (1 RCT) | NR | 251 per 1,000 | 376 per 1,000 (307 to 446 per 1,000) | 142 more per 1,000 (53 more to 232 more) | Moderatea | Risankizumab likely results in a clinically important improvement in clinical remission compared to placebo. |
Proportion of patients with clinical response per the adapted Mayo score Follow-up: 52 weeks | 362 (1 RCT) | NR | 519 per 1,000 | 623 per 1,000 (554 to 693 per 1,000) | 115 more per 1,000 (17 more to 212 more) | Moderateb | Risankizumab likely results in little to no clinically important difference in clinical response compared to placebo. |
Proportion of patients with endoscopic improvement Follow-up: 52 weeks | 362 (1 RCT) | NR | 317 per 1,000 | 483 per 1,000 (411 to 555 per 1,000) | 174 more per 1,000 (79 more to 269 more) | Moderatec | Risankizumab likely results in a clinically important improvement in endoscopic improvement compared to placebo. |
Proportion of patients with HEMI Follow-up: 52 weeks | 362 (1 RCT) | NR | 235 per 1,000 | 422 per 1,000 (351 to 494 per 1,000) | 198 more per 1,000 (108 more to 288 more) | Highd | Risankizumab results in an improvement in HEMI compared to placebo. The clinical importance is uncertain. |
Proportion of patients with HEMR Follow-up: 52 weeks | 362 (1 RCT) | NR | 98 per 1,000 | 156 per 1,000 (104 to 209 per 1,000) | 61 more per 1,000 (3 less to 125 more) | Moderatee | Risankizumab results in an improvement in HEMR compared to placebo. The clinical importance is uncertain. |
Proportion of patients with discontinuation of corticosteroid use in patients taking steroids at baseline Follow-up: 52 weeks | 142 (1 RCT) | NR | 368 per 1,000 | 542 per 1,000 (415 to 669 per 1,000) | 207 more per 1,000 (49 more to 366 more) | Lowf | Risankizumab may result in a clinically important improvement in discontinuation of corticosteroid use compared to placebo. |
Patient-reported outcomes | |||||||
Change from baseline in IBDQ total score, points (range: 32 [worst] to 224 [best]) Follow-up: 52 weeks | 362 (1 RCT) | NR | 35.0 | 50.3 (42.20 to 58.36) | MD 15.2 higher (5.18 higher to 25.31 higher) | Moderateg | Risankizumab likely results in a clinically important improvement in IBDQ scores compared to placebo. |
Harms | |||||||
Proportion of patients with serious adverse events Follow-up: 52 weeks | 389 (1 RCT) | NR | 82 per 1,000 (NR to NR) | 51 per 1,000 (NR to NR) | 30 less per 1,000 (80 less to 19 more) | Lowh | Risankizumab may result in little to no difference in SAEs compared to placebo. |
CI = confidence interval; HEMI = histologic, endoscopic, and mucosal improvement; HEMR = histologic, endoscopic, and mucosal remission; IBDQ = Inflammatory Bowel Disease Questionnaire; MD = mean difference; MID = minimal important difference; NR = not reported; RCT = randomized controlled trial; vs. = versus.
Note: Study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes. The proportion of missing data was not reported and therefore cannot be fully appraised.
aAn empirically derived MID of 11% was identified in the literature14 for the between-group difference for this outcome. Rated down 1 level for serious imprecision; the point estimate suggests clinically important benefit while the lower bound of the CI suggests little to no difference.
bAn empirically derived MID of 14% was identified in the literature14 for the between-group difference for this outcome. Rated down 1 level of serious imprecision; the CI includes the potential for benefit and little to no difference.
cAn empirically derived MID of 12.5% was identified in the literature14 for the between-group difference for this outcome. Rated down 1 level for imprecision, as the CIs show both potential benefit and equivalence between risankizumab and placebo.
dAn empirically derived MID was not identified for the between-group difference for this outcome; effects were appraised using the null.
eAn empirically derived MID was not identified for the between-group difference for this outcome; effects were appraised using the null. Rated down 1 level for imprecision, as the CIs show both potential benefit and little to no difference between risankizumab and placebo.
fNo threshold of clinical importance could be established from the literature. An empirically derived MID was not identified for the between-group difference for this outcome. A difference of 15% between the groups was identified by the clinical expert consulted by CDA-AMC as a threshold of clinical importance for this outcome. Rated down 1 level for serious study limitations; the randomization was not stratified by baseline corticosteroid use, therefore randomization in this population might not be upheld. Rated down 1 level for imprecision, as the CIs show both potential benefit and equivalence between risankizumab and placebo.
gAn empirically derived improvement of 15 points or more greater than placebo was considered be the MID for this outcome. Rated down 1 level for imprecision, as the CIs show both potential benefit and little to no clinically important difference between risankizumab and placebo.
hAn empirically derived MID of 6% was identified in the literature14 for the between-group difference for this outcome. Rated down 1 level for imprecision, as the CIs show both potential benefit and little to no clinically important difference between risankizumab and placebo). Rated down 1 level for serious indirectness, as SAEs include symptoms of worsening UC, which complicates interpretation.
Source: COMMAND Clinical Study Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Long-Term Extension Studies
The sponsor submitted an abstract19 for the COMMAND open-label extension study. Because of the lack of access to complete methods and results, the summary of this study is not reported in this section.
Indirect Evidence
Description of Studies
No direct comparison exists between risankizumab and other publicly reimbursed ATs or those recommended for reimbursement in Canada for moderately to severely active UC. In the absence of head-to-head randomized controlled trials (RCTs), the sponsor submitted an indirect treatment comparison (ITC). A network meta-analysis (NMA) was used to compare the efficacy and safety of risankizumab to other publicly reimbursed ATs or those recommended for reimbursement in Canada for moderately to severely active UC: adalimumab, infliximab, golimumab, vedolizumab, tofacitinib, ustekinumab, ozanimod, upadacitinib, mirikizumab, and etrasimod. The NMA included 28 phase III or higher randomized, double-blind trials. The NMA reported on clinical remission, clinical response, endoscopic improvement, adverse events (AEs), SAEs, and serious infections.
Induction
Efficacy: Results from the indirect comparison showed that, for the induction phase, risankizumab 1,200 mg was favoured over adalimumab (odds ratio [OR] █████ ███ ███ ████ ██ ████) and mirikizumab (OR █████ ███ ███ ████ ██ ████) for clinical response and to adalimumab (OR █████ ███ ███ ████ ██ ████), golimumab (OR █████ ███ ███ ████ ██ ████), mirikizumab (OR █████ ███ ███ ████ ██ ████), tofacitinib (OR █████ ███ ███ ████ ██ ████), and ustekinumab (OR █████ ███ ███ ████ ██ ████), with more patients in the risankizumab group achieving endoscopic improvement in the patient population who had not previously received AT (non-AT). Upadacitinib was favoured over risankizumab 1,200 mg for clinical remission (OR █████ ███ ███ ████ ██ ████), clinical response (OR █████ ███ ███ ████ ██ ████) and endoscopic improvement (OR █████ ███ ███ ████ ██ ████) in the population with previous inadequate response or intolerance to AT (AT-IR). There was no evidence of difference between risankizumab 1,200 mg and the other interventions in terms of efficacy during the induction period.
Safety: Risankizumab 1,200 mg was favoured over adalimumab (OR █████ ███ ███ ████ ██ ████), etrasimod (OR █████ ███ ███ ████ ██ ████), ozanimod (OR █████ ███ ███ ████ ██ ████), and tofacitinib (OR █████ ███ ███ ████ ██ ████), with fewer patients in the risankizumab 1,200 mg group having SAEs during the induction period. Etrasimod was favoured over risankizumab 1,200 mg (██ █████████ ███ ███ █████ ██ ████████) with regard to serious infections. There was no evidence of difference between risankizumab 1,200 mg and the other interventions in terms of safety during the induction phase.
Maintenance
Efficacy: For the maintenance phase, in the population with no previous inadequate response or intolerance to AT (non–AT-IR), upadacitinib 30 mg was favoured over risankizumab 180 mg (██ █████ ███ ███ ████ ██ ████) and risankizumab 360 mg (██ █████ ███ ███ ████ ██ ████), with fewer patients in the risankizumab groups having a clinical response in the non–AT-IR population during the maintenance period. There was no evidence of a difference between risankizumab and the other interventions in terms of efficacy during the maintenance phase.
For the maintenance phase, in AT-IR populations, risankizumab 180 mg was ████ ████████ ████████ ██ ████████████ █████████████ ███ ██ ██████ ███ ███ ████ ██ ██████ ████████████ ███ ██ █████ ███ ███ ████ ██ █████ ███ ███████████ ████████████ ███████████ ██ █████ ███ ███ ████ ██ ██████ ███████████ ███████ ██ █████ ███ ███ ████ ██ █████ ███ ████████ ██████████ ███████████ ███ █████ ███ ███ ████ ██ █████ ███ ████████████ █████████████ ███ ██ █████ ███ ███ ████ ██ █████ ████████████ ███ ██ █████ ███ ███ ████ ██ █████ ███ ████████ █████████ ███ ████████████ █████████████ ███ ██ █████ ███ ███ ████ ██ █████ ████████████ ███ ██ █████ ███ ███ ████ ██ █████ ███ ███████████ ███ █████ █████ ████ ██ █████ ███ ██████████ █████████. There was no evidence of a difference between risankizumab 180 mg and the other interventions in terms of efficacy during the maintenance phase.
Risankizumab 360 mg was ████ ████████ ████████ ██ ███████████ ███ █████ ███ ███ ████ ██ ██████ ████████████ █████████████ ███ ██ █████ ███ ███ ████ ██ █████ ████████████ ███ ██ █████ ███ ███ ████ ██ █████ ███ ███████████ ████████████ ███████████ ██ █████ ███ ███ ████ ██ ██████ ███████████ █████ ███ █████ ███ ███ ████ ██ ██████ ███████████ ██████ ███ █████ ███ ███ ████ ██ █████ ███ ████████ ██████████ ███████████ ███ █████ ███ ███ ████ ██ █████ ███ ████████████ █████████████ ███ ██ █ ███ ███ ███ ████ ██ █████ ████████████ ███ ██ █████ ███ ███ ████ ██ █████ ███ ████████ █████████ ███ █████████ ███ █████ ███ ███ ████ ██ ██████ ████████████ █████████████ ███ ██ █████ ███ ███ ████ ██ █████ ████████████ ███ ██ █████ ███ ███ ████ ██ █████ ███ ███████████ ████████████ ███████ ██ █████ ███ ███ ████ ██ ████ ███ ███████████ ███████ ██ █████ ███ ███ ████ ██ █████ ███ ██████████ █████████. There was no evidence of a difference between risankizumab 360 mg and the other interventions in terms of efficacy during the maintenance phase.
Safety: Risankizumab 360 mg was favoured over golimumab (██ █████ ███ ███ ████ ██ ████), with fewer patients with AEs in the risankizumab group during the maintenance phase. There was no evidence of a difference between risankizumab 180 mg or risankizumab 360 mg and the other interventions in terms of safety during the maintenance phase.
Critical Appraisal of the NMA
In the sponsor-submitted ITC, relevant RCTs were identified using a systematic review produced following accepted methodological guidance and based on an a priori protocol that outlined the inclusion and exclusion criteria. The search is 2 years old, and it is not known whether more recent studies are available, nor what impact that might have on the results. Selection, data extraction, and risk-of-bias assessments were conducted in duplicate by independent researchers, which is considered adequate. Risk of bias of the included studies was conducted using the Cochrane Risk-of-Bias tool. The overall bias of all included studies was deemed to be low. The assessments occurred at the study level, and not the outcome level, as suggested by Cochrane, and bias concerns may vary by effect estimate. The appraisal may not be applicable across all outcomes. There was no assessment of the risk of publication bias; therefore, its presence or absence cannot be confirmed.
The methods used for the NMA were considered to be appropriate, including the use of Bayesian framework, as its conduct adhered to relevant guidance. However, there were no sensitivity analyses to understand the potential impact of the chosen priors for between-study standard deviation. Overall, the patient populations, interventions, comparators, and outcomes (PICO) of the included studies were consistent with the overall review’s objective. The clinical experts consulted by the review team did not expect any major issues regarding the representativeness of the study populations enrolled in the RCTs that were included in the ITC in relation to the populations in Canada that may be eligible for treatment with risankizumab.
As is common in NMAs of treatments for UC, 1 overall concern was the heterogeneity in the patient populations, which included their baseline characteristics, average disease severity (and how this was defined across trials), disease duration, and the extent of disease. In addition, the lengths of the induction and maintenance periods were not uniform across the trials. There were some differences in the AT-IR definitions used across studies, and due to changes in the standard of care and available treatments over time, the number and type of prior treatments to which patients were exposed likely varied. There was also heterogeneity in the use of concurrent immunomodulators and corticosteroids. Additionally, the timing of the outcome assessments varied substantially, and the definitions of the efficacy end points of interest were not always consistent. Some relevant evidence was excluded due to misalignment in the assessment time points; while this was necessary to reduce heterogeneity, the impact on results is not known.
There were additional sources of heterogeneity in the maintenance phase. In treat-through (TT) studies, patients were randomized to treatment or placebo at baseline and outcomes were measured at the end of an induction phase and again at the end of a maintenance phase. In rerandomization studies (e.g., risankizumab studies), patients were randomized to induction treatment or placebo at baseline, with outcomes measured at the end of the induction phase; induction responders were then rerandomized to maintenance treatment or placebo, with outcomes measured at the end of the maintenance phase strictly among induction responders. Adjustments were applied to include both TT and rerandomized trial designs in the same NMA. This was necessary and has been applied to other NMAs for UC but relies on assumptions that cannot be fully validated. Placebo was a common comparator in the NMAs, although the placebo groups across studies are likely not equivalent, due to carry-over effects from different induction treatments. Differences in the corticosteroid tapering strategy (and whether this was included) may also differ across the studies. For both induction and maintenance phases, the information needed to comprehensively assess the level of heterogeneity was not always available for all studies. The notable clinical and methodological heterogeneity raises concern for intransitivity, which undermines the validity of the indirect comparisons, as it indicates that the assumption of exchangeability may not hold.
Relevant efforts were made to reduce or account for heterogeneity across the NMAs, including stratification of the results by AT-IR status, baseline risk meta-regression, and sensitivity analyses. The stratification of analyses by AT-IR and non–AT-IR populations for efficacy analyses is appropriate but limits generalizability of the results of either analysis to the overall indicated population. The interpretation of these subgroups faces some limitations. The baseline characteristics within subgroups were not always fully known (i.e., the full population was used for comparison), making it difficult to comprehensively assess comparability. It is not clear whether randomization was stratified by AT-IR status across all trials. Confounding is possible, as the effect estimates might not be arising from fully randomized groups. None of the baseline risk-adjusted models converged; therefore, it became impossible to apply this adjustment. Sensitivity analyses of some sources of heterogeneity were not possible.
The networks were sparsely populated, with several nodes centred around a single connection (placebo) in a star geometry, with only 1 to 2 trials against placebo per treatment and only a few head-to-head trials (i.e., within-study comparisons of different doses of the same treatment). This reduces the robustness of the NMA and makes most comparisons underpowered by the lack of direct evidence. Further, all evidence for risankizumab was against placebo, increasing uncertainty in the estimates for each outcome, and the consistency assumption could not be assessed. The heterogeneity and network sparsity are reflected in imprecise credible intervals (CrIs). Fixed-effects models were chosen for a small number of outcomes based on model fit; when it was a better fit than the random-effects model. However, given the expected heterogeneity, the fixed-effects model has the potential to underestimate the uncertainty in the results (i.e., width of the CrIs). There was poor model fit for the analysis of serious infections in the induction phase. Last, based on the ITC feasibility assessment, 1 study was not included in the NMA because the drug is not approved in Canada.
Eight outcomes of interest to this review were reported: clinical remission, clinical response, endoscopic improvement, AEs, SAEs, and serious infections. Interpretation of harms outcomes was complicated by inclusion of UC worsening and related symptoms as harms in the placebo groups, and potential differences in how harms were collected across studies. Some outcomes of importance to patients, such as corticosteroid-free clinical remission and HRQoL, were not included.
Studies Addressing Gaps in the Evidence From the Systematic Review
Description of Studies
This study by Panaccione et al. (2025).20 was post hoc analysis of the pivotal INSPIRE and COMMAND trials specifically for AT-IR status. The objective of study was to assess the efficacy and safety of risankizumab induction and maintenance therapy in patients with moderately to severely active UC based on prior inadequate response or intolerance to AT (i.e., AT-IR status). For the efficacy analysis, the study included 472 patients in the non–AT-IR population and 503 patients in the AT-IR population for the induction phase, and 137 patients in the non–AT-IR population and 411 patients in the AT-IR population for the maintenance phase.
Induction Phase Efficacy Outcomes
During the induction phase, clinical outcomes at week 12 showed higher proportion of patients responding in the risankizumab 1,200 mg group than in the placebo group across both AT-IR and non–AT-IR subgroups. In the AT-IR subgroup, clinical remission rates were 11.4% for risankizumab and 4.3% for placebo (difference = 7.2%), while in the non–AT-IR subgroup, clinical remission rates were higher, at 29.7% and 8.4% (difference = 21.3%), respectively. Clinical response rates followed a similar trend, with 55.2% in the AT-IR subgroup and 73.8% in the non–AT-IR subgroup for risankizumab, compared to 31.2% and 40.6% for placebo (difference = 24.0% for AT-IR and 33.2% for non–AT‑IR subgroups).
Endoscopic outcomes also were higher in the risankizumab group. Endoscopic improvement at week 12 was observed in 25.9% of the AT‑IR subgroup and 47.6% of the non–AT‑IR subgroup receiving risankizumab, compared to 10.1% and 14.2% in the placebo groups (difference = 15.8% for AT‑IR and 33.2% for non–AT‑IR subgroups). Endoscopic remission rates were 4.8% (AT‑IR subgroup) and 16.7% (non–AT‑IR subgroup) for risankizumab, versus 3.0% and 3.9% for placebo (difference = 1.8% for AT‑IR and 12.8% for non–AT‑IR subgroups). Additionally, HEMI rates were 16.0% in the AT‑IR subgroup and 33.4% in the non–AT‑IR subgroup for risankizumab, compared to 7.1% and 8.4% for placebo, respectively (difference = 8.9% for AT‑IR and 25.1% for non–AT‑IR subgroups).
Maintenance Phase Efficacy Outcomes
During the maintenance period at week 52, the differences in clinical remission rates in the AT-IR subgroup versus placebo were 6.3% for risankizumab 360 mg and 13.4% for risankizumab 180 mg. In the non–AT-IR subgroup, remission rates versus placebo were 30.6% for risankizumab 360 mg and 19.8% for risankizumab 180 mg. Clinical response rates in the AT-IR subgroup versus placebo were 11.2% for risankizumab 360 mg and 17.8% for risankizumab 180 mg, while in the non–AT-IR subgroup, they were 7.5% and 11.1%, respectively.
Endoscopic outcomes were also higher in the risankizumab groups. Endoscopic improvement in the AT-IR subgroup versus placebo was observed in 8.4% for risankizumab 360 mg and 17.3% for risankizumab 180 mg, while in the non–AT-IR subgroup, rates were 40.6% and 24.2%, respectively. Endoscopic remission rates in the AT-IR subgroup versus placebo were 2.1% for risankizumab 360 mg and 5.6% for risankizumab 180 mg, and in the non–AT-IR subgroup were 31.6% for risankizumab 360 mg and 16.6% for risankizumab 180 mg. HEMI rates in the AT-IR subgroup versus placebo were 11.4% for risankizumab 360 mg and 17.1% for risankizumab 180 mg, and in the non–AT-IR subgroup were 40.4% for risankizumab 360 mg and 25.9% for risankizumab 180 mg.
Safety Outcomes
The proportions of patients with any TEAE in the AT-IR subgroup in the induction study was 44.1% and 53.5% in the risankizumab 1,200 mg and placebo groups, respectively.
The proportions of patients with any TEAE in the non–AT-IR subgroup in the induction study was 39.9% and 45.5% in the risankizumab 1,200 mg and placebo groups, respectively.
The proportions of patients with any TEAE in the AT-IR subgroup in the maintenance study was 69.4%, 74.7%, and 76.5% in risankizumab 360 mg, risankizumab 180 mg, and placebo groups, respectively.
The proportions of patients with any TEAE in the non–AT-IR subgroup in the maintenance study, was 75.0%, 66.0%, and 76.6% in risankizumab 360 mg, risankizumab 180 mg, and placebo groups, respectively.
Critical Appraisal of the Studies Addressing Gaps in the Evidence From the Systematic Review
This is a post hoc analysis. There is no type I error control, which increases the risk of type I error. Conversely, subgroups might not be powered to find significant differences. There is a risk of randomization being broken for some of the subgroups (those not included as stratification factors). Previous ATs were stratification factors, but classified differently in the post hoc analysis. The analysis could not distinguish whether observed differences across groups were due to AT-IR status or other differences between the groups. There are no tests for subgroup differences. Based on the previously noted reasons, credible conclusions about effect modification cannot be drawn, but the analysis can be used for hypothesis generation. In several cases the analysis suggests consistency in the direction of effect with the overall population in the trials, although magnitudes of effects differed.
Conclusions
Two pivotal phase III, randomized, placebo-controlled, double-blind, parallel-group trials for induction (INSPIRE) and maintenance (COMMMAND) phases in adult patients with moderately to severely active UC who experienced an inadequate response or intolerance to conventional therapies or other ATs were included in the CDA-AMC review. During the induction phase, moderate- to high-certainty evidence favoured risankizumab with regard to clinical response, clinical remission, endoscopic improvement, HEMI, and HEMR. Further, there was moderate-certainty evidence favouring risankizumab with regard to IBDQ and low-certainty evidence for little to no difference in SAEs. During the maintenance phase, there was high-certainty evidence favouring risankizumab for endoscopic improvement and HEMI. Further, there was moderate-certainty evidence favouring risankizumab for clinical response (for the 180 mg dose only, whereas the 360 mg dose showed little to no difference), clinical remission, HEMR, and IBDQ. Lastly, there was low-certainty evidence favouring risankizumab for discontinuation of corticosteroid use in patients taking steroids at baseline, and for little to no difference in SAEs.
There was some concern resulting from the inability to comprehensively appraise for risk of bias due to missing data, as this was not reported in the submitted materials. While the results of most subgroup analyses were similar in direction to the main analyses, they should be viewed as exploratory for hypothesis generation because the trial was not stratified by these subgroups. The occurrence of harms was relatively limited and aligned with the known safety profile of risankizumab. A study addressing gaps submitted by the sponsor suggested similar directions of effect for patients in the AT-IR and non–AT-IR groups, but the magnitude varied. Magnitudes of benefit appeared smaller for patients in the AT-IR group compared to those in the non–AT-IR groups in both the induction and maintenance phases. Conclusions on effect modification were not possible to draw from this study.
The sponsor-submitted NMA comparing risankizumab to other relevant comparators was affected by several limitations, including likely violation of the underlying assumptions, network sparsity, imprecision, and inconsistency of the results across outcomes. As a result, the relative treatment effects of risankizumab versus relevant comparators are subject to uncertainty and there is no conclusive evidence of which treatment may provide the preferred balance of benefits and harms.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of risankizumab:
600 mg in 10 mL (60 mg/mL) by IV infusion
180 mg in 1.2 mL (150 mg/mL) by SC injection
360 mg in 2.4 mL (150 mg/mL) by SC injection.
The indication is for the treatment of adult patients with moderately to severely active UC who experienced an inadequate response, loss of response, or were intolerant to conventional therapy, a biologic treatment, or a JAK inhibitor.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
Overview of the Condition
IBD is a group of diseases characterized by chronic recurrent, progressive inflammation of the GI tract.21 There are 2 main types of IBD: Crohn disease and UC. UC is a chronic disease characterized by inflammation, predominantly of the mucosal layer of the large intestine (colon) most often involving the rectum and frequently extends continuously into the proximal colon.1 The cause of UC remains uncertain, but a combination of genetic and environmental factors contributes to immune dysregulation and up-regulation in response to micro-organisms in the GI tract.2 UC is characterized by blood in the stool with mucus, frequent diarrhea, urgency, loss of appetite, and tenesmus (severe rectal cramp or spasm).3 Although UC principally affects the GI tract, extraintestinal manifestations may also occur, such as arthritis.4 There is no notable difference in frequency of UC among males and females.22 Although the risk of mortality from UC itself is low, the disease is associated with increased risk of other complications (e.g., respiratory diseases, colorectal cancer, lymphoma, and skin cancer) that result in higher premature mortality compared to the general population.23 About 30% to 60% of UC patients first present with isolated proctitis (involvement is limited to the rectum).24,25 Patients with proctitis are prone to proximal extension, associated with more colon involved in active disease, higher colectomy rates, increased need for AT, and higher hospitalization rates than patients who start with extensive colitis.24,26,27 Among patients with isolated proctitis who are untreated for 1 year, the relapse rate is between 47% and 86%.28 Resource utilization and direct and indirect costs are higher among patients with UC compared with matched controls, and in patients with moderate to severe UC the reported excess burden has been greatest.29
There is no single test used to diagnose UC, and diagnosis is based on clinical examination, laboratory results, endoscopic findings, histopathology results, and exclusion of alternative diagnoses.30
While most patients have a mild to moderate disease course, about 10% to 15% experience an aggressive course.5 Relapse is common, with the cumulative risk of relapse being 70% to 80% at 10 years.5 Achieving endoscopic healing earlier may be associated with reduced risk of future colectomy.5 The chronic nature of UC has a considerable impact on patients’ HRQoL, including psychological, physical, sexual, and social domains, due to chronic symptoms such as urgency, frequency, and incontinence.6,7 The medical and surgical treatments for UC (e.g., colectomies) and their potential accompanying complications can also negatively impact HRQoL and productivity.31-35 Individuals with UC are at greater risk of comorbid anxiety, depression, and impaired social interactions.6,7,36,37 Patients with UC frequently report fatigue and sleep disturbance, as well as an inability to perform regular daily routines such as jobs or domestic chores.32,38-40 Furthermore, the disease can impact the patients’ caregivers, family, workplace, and community.23
Estimated Disease Prevalence
The prevalence of UC in Canada was estimated to be 414 per 100,000 in 2023.8 It is estimated that 32% to 46% of Canadians with UC have moderate disease, and 13% to 14% have severe disease.9
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
Pharmacotherapy regimens for UC are evolving rapidly, but the general goal is to accomplish complete remission, defined as both symptomatic and endoscopic remission, without corticosteroid therapy; to preserve HRQoL; and to prevent disability.10,11 The most recent clinical practice guidelines used in Canada for the medical management of UC are the 2015 Toronto Consensus for patients with UC who are not hospitalized,10 the 2021 European Crohn’s and Colitis Organisation (ECCO)11 guideline, and the 2024 American Gastroenterological Association (AGA) guideline for the pharmacological management of moderate to severe UC.41 One of the main differences of the AGA guidelines is that they made recommendations specific to individual drugs rather than to classes, as they noted there are emerging data suggesting differences in efficacy even within a therapeutic class.
The guidelines generally recommend a standard step-up approach to the medical management of moderate to severe UC. The Toronto and ECCO guidelines recommend that patients should undergo a combination of oral corticosteroid therapy, 5-aminosalicylates, and/ or anti–tumour necrosis factor (TNF) agents (i.e., infliximab, adalimumab, and golimumab). Thiopurine, methotrexate, vedolizumab, tofacitinib, or ustekinumab may also be considered for induction of remission. It is further recommended that for maintenance of remission, the same drug should be used in patients who have responded to induction therapy with that drug. The AGA guidelines classified:
infliximab, vedolizumab, ozanimod, etrasimod, upadacitinib, risankizumab, and guselkumab as high-efficacy medications
golimumab, ustekinumab, tofacitinib, filgotinib, and mirikizumab as moderate-efficacy medications
adalimumab as having lower efficacy.
In Canada, the following drugs are available for patients with moderate to severe UC:
5-aminosalicylates (e.g., mesalamine, sulfasalazine, olsalazine)
corticosteroids
immunosuppressive therapy (e.g., azathioprine, 6-mercaptopurine)
anti-TNF therapies
anti-integrin therapies (e.g., vedolizumab)
anti–IL-12 and anti–IL-23 therapies (e.g., ustekinumab)
other targeted therapies, such as JAK inhibitors (e.g., tofacitinib and upadacitinib) and sphingosine‑1‑phosphate (S1P) receptor agonists (e.g., ozanimod).42,43
Mirikizumab, an anti–IL-23 therapy, received a Notice of Compliance in July 2023 and a recommendation for reimbursement with clinical criteria and/or conditions by CDA-AMC in November 2023.44,45 The pan-Canadian Pharmaceutical Alliance (pCPA) negotiations for mirikizumab concluded in 2024, and it is currently being reimbursed in some jurisdictions.46 Additionally, another S1P receptor modulator, etrasimod, received a Notice of Compliance in January 2024 and a final CDA-AMC recommendation in August 2024 to reimburse with clinical criteria and/or conditions, and it is currently undergoing pCPA negotiations.47,48
Drug Under Review
Key characteristics of risankizumab are summarized in Table 5 with other treatments available for the treatment of adult patients with moderately to severely active UC who experienced an inadequate response, loss of response, or were intolerant to conventional therapy, a biologic treatment, or a JAK inhibitor.
The recommended dose of risankizumab is 1,200 mg administered by IV infusion at week 0, week 4, and week 8, followed by 180 mg or 360 mg administered by SC injection at week 12, and every 8 weeks thereafter.
Other indications include the treatment of adult patients with:
moderate to severe plaque psoriasis
active psoriatic arthritis
moderately to severely active Crohn disease.
Risankizumab has been previously reviewed by CDA-AMC and received a recommendation to reimburse with clinical criteria and/or conditions for the following indications:
the treatment of moderate to severe plaque psoriasis in adults
the treatment of adults with moderately to severely active Crohn disease who experienced an inadequate response, intolerance, or a demonstrated dependence to corticosteroids; or an inadequate response, intolerance, or loss of response to immunomodulators or biologic therapies.
Risankizumab is a humanized immunoglobulin G1 (IgG1) monoclonal antibody that selectively binds with high affinity to the p19 subunit of human interleukin 23 (IL-23) cytokine and inhibits IL-23 signalling in cell-based assays, including the release of the proinflammatory cytokine IL-17. IL-23 is a naturally occurring cytokine that is involved in inflammatory and immune responses.
The sponsor’s reimbursement request is per the Health Canada–approved indication, for the treatment of adult patients with moderately to severely active UC who experienced an inadequate response, loss of response, or were intolerant to conventional therapy, a biologic treatment, or a JAK inhibitor. Risankizumab underwent review by Health Canada through a standard review pathway, and a Notice of Compliance was granted on October 10, 2024.
Table 5: Key Characteristics of Risankizumab and Main Comparators
Drug name (brand) | Mechanism of action | Indicationa | Route of administration and recommended dose | Serious adverse effects and/or safety issues |
|---|---|---|---|---|
Drug under review | ||||
Risankizumab (Skyrizi)49 | Humanized immunoglobulin G1 (IgG1) monoclonal antibody that selectively binds with high affinity to the p19 subunit of human interleukin 23 (IL-23) cytokine and inhibits IL‑23 signalling in cell‑based assays, including the release of the proinflammatory cytokine IL‑17 | For the treatment of adult patients with moderately to severely active UC who have had an inadequate response, loss of response, or were intolerant to conventional therapy, a biologic treatment, or a JAK inhibitor | 1,200 mg administered by IV infusion at week 0, week 4, and week 8, followed by 180 mg or 360 mg administered by SC injection at week 12, and every 8 weeks thereafter |
|
Comparators | ||||
S1P receptor modulators | ||||
Etrasimod (Velsipity)50 | Selective S1P receptor modulator; may reduce lymphocyte migration into inflammation sites and reduce cytokine response | For the treatment of adult patients with moderately to severely active UC who have had an inadequate response, lost response, or were intolerant to either conventional therapy or an advanced treatment | 2 mg orally once daily |
|
Ozanimod (Zeposia)51 | S1P receptor modulator; binds to the S1P1 receptors on lymphocytes preventing egress from lymph nodes; may reduce lymphocyte migration into the CNS and intestine | For the treatment of adult patients with moderately to severely active UC who had an inadequate response loss of response or were intolerant to either conventional therapy or biologic drugs | Initiation: 0.23 mg orally once daily on days 1 to 4, then 0.46 mg orally once daily on days 5 to 7 Maintenance: 0.92 mg orally once daily | Malignancies, particularly of the skin; initiation of ozanimod may result in transient reductions in heart rate and atrioventricular delays |
Anti-TNF biologics | ||||
Adalimumab (Humira)52 | Anti-TNF human IgG1 monoclonal antibody; binds and blocks TNF alpha and its interactions with p55 and p75 cell-surface TNF receptors |
|
| Serious infections, malignancies, and neurologic events. The most common adverse reaction in patients with rheumatoid arthritis treated with Humira was injection-site reactions. |
Adalimumab biosimilars: Abrilada, Amgevita, Hulio, Hyrimoz)53-56 | Anti-TNF human IgG1 monoclonal antibody; binds and blocks TNF alpha and its interactions with p55 and p75 cell-surface TNF receptors |
|
| Serious infections (pneumonia), malignancies, and neurologic events |
Adalimumab (biosimilars: Hadlima, Idacio, Simlandi, Yuflyma)57-60 | Anti-TNF human IgG1 monoclonal antibody; binds and blocks TNF alpha and its interactions with p55 and p75 cell-surface TNF receptors | For the treatment of adult patients with moderately to severely active UC who have had an inadequate response to conventional therapy, including corticosteroids and/or azathioprine or 6-MP, or who are intolerant to such therapies | SC administration of 160 mg at week 0, 80 mg at week 2, then 40 mg every other week thereafter as monotherapy or in combination with conventional therapies | Serious infections (pneumonia), malignancies, and neurologic events |
Golimumab (Simponi)61 | Anti-TNF human monoclonal antibody that binds with p55 or p75 human TNF receptors | For the treatment of adult patients with moderately to severely active UC who have had an inadequate response to, or have medical contraindications for conventional therapy, including corticosteroids, aminosalicylates, azathioprine, or 6-MP, for inducing and maintaining clinical response, inducing clinical remission, achieving sustained clinical remission in induction responders, or improving endoscopic appearance of the mucosa during induction | 200 mg administered by SC injection at week 0, followed by 100 mg at week 2 and then 50 mg every 4 weeks thereafter. For maintenance, 100 mg every 4 weeks can be considered at the discretion of the treating physician. | Upper respiratory tract infection |
Infliximab (Remicade)62 | Anti-TNF IgG1k monoclonal antibody that neutralizes the biological activity of TNF alpha by specifically binding to its receptors |
| IV infusion of 5 mg/kg at 0, 2 and 6 weeks, followed by 5 mg/kg every 8 weeks thereafter, for the treatment of adult and pediatric patients (≥ 6 years of age). Doses up to 10 mg/kg may be used in some adult patients. | Infections and malignancies |
Anti-TNF IgG1k monoclonal antibody that neutralizes the biological activity of TNF alpha by specifically binding to its receptors |
| Adults and pediatric patients (≥ 6 years of age): IV infusion of 5 mg/kg at 0, 2, and 6 weeks, followed by 5 mg/kg every 8 weeks thereafter. Doses up to 10 mg/kg may be used. | Infections and malignancies | |
Anti-integrin | ||||
Vedolizumab (Entyvio)66 | IgG1 monoclonal antibody; binds to the human alpha4beta7 integrin, acting as a gut-selective anti-inflammatory biologic | For the treatment of adult patients with moderately to severely active UC who have had an inadequate response to, loss of response to, or were intolerant to either conventional therapy or infliximab, a TNF alpha antagonist | 300 mg administered by IV infusion at 0, 2 and 6 weeks and then every 8 weeks thereafter. The SC maintenance dose is 108 mg every 8 weeks. | Infections and malignancies |
Interleukin inhibitors | ||||
Mirikizumab (Omvoh)45 | Humanized IgG4 monoclonal antibody that binds with high affinity and specificity to the p19 subunit of human IL‑23 cytokine to inhibit its interaction with the IL‑23 receptor | For the treatment of adult patients with moderately to severely active ulcerative colitis who have had an inadequate response, loss of response, or were intolerant to conventional therapy, a biologic treatment, or a JAK inhibitor | Induction: 300 mg IV for at least 30 minutes at week 0, week 4, and week 8. If patients do not have adequate therapeutic response at week 12, extended inducted dosing of 300 mg IV at weeks 12, 16, and 20 can be considered. Maintenance: 200 mg (given as 2 consecutive SC injections of 100 mg each) every 4 weeks after completion of induction dosing. | Upper respiratory tract infection, headache, and site injection reactions (e.g., rashes, including maculo‑papular, papular, and pruritic rashes). |
Ustekinumab (Stelara)67 | Human IgG1 monoclonal antibody; neutralizes cellular responses mediated by IL‑12 and IL‑23 | For the treatment of adult patients with moderately to severely active UC who have had an inadequate response with, lost response to, or were intolerant to either conventional therapy or a biologic or have medical contraindications to such therapies | Single, weight-based IV infusion (approximating 6 mg/kg) followed by a 90 mg SC dose 8 weeks later, then 90 mg SC every 8 weeks thereafter for maintenance. In patients with low inflammatory burden, a single IV dose followed 8 weeks later by 90 mg SC, then every 12 weeks thereafter may be considered at the discretion of the treating physician. | Immunomodulating drugs have the potential to increase the risk of infections and malignancy. |
JAK inhibitors | ||||
Selective JAK inhibitor; blocks several cytokine pathways and lymphocyte activation | For the treatment of adult patients with moderately to severely active UC with an inadequate response, loss of response, or intolerance to either conventional UC therapy or a TNF alpha inhibitor | 10 mg orally b.i.d. for induction for at least 8 weeks and 5 mg b.i.d. for maintenance. Depending on therapeutic response, 10 mg b.i.d. may also be used for maintenance in some patients. However, the lowest effective dose possible should be used for maintenance therapy to minimize adverse effects. | An HC warning indicated an increased risk of thrombosis (pulmonary and deep vein thrombosis) and death and increased risk of serious infections, including herpes zoster infections. Of note, tofacitinib is not recommended in combination with biological UC therapies or with potent immunosuppressants, such as azathioprine and cyclosporine | |
Upadacitinib (Rinvoq)72 | Selective JAK inhibitor; demonstrates activity against JAK1, JAK2, JAK3, and TYK2 | For the treatment of adult patients with moderately to severely active UC who have demonstrated prior treatment failure, i.e., an inadequate response to, loss of response to, or intolerance to at least 1 of conventional, and/or biologic therapy | Induction: 45 mg orally once daily for 8 weeks Maintenance: 15 mg orally once daily. For some patients, such as those with refractory, severe, or extensive disease, a maintenance dose of 30 mg once daily may be appropriate. The lowest effective dose needed to maintain response should be used. For patients ≥ 65 years of age, the only recommended maintenance dose is 15 mg once daily. | Upper respiratory tract infection. Of note, upadacitinib should not be used in combination with other JAK inhibitors, immunomodulating biologics (e.g., biologic DMARDs), or with potent immunosuppressants such as azathioprine, 6‑MP, and cyclosporine. |
6-MP = 6-mercaptopurine; AE = adverse event; b.i.d.= twice a day; CNS = central nervous system; CV = cardiovascular; DMARD = disease-modifying antirheumatic drug; HC = Health Canada; IgG1 = immunoglobulin G1; IgG1k = immunoglobulin G1-kappa; IL = interleukin; PRES = posterior reversible encephalopathy syndrome; S1P = sphingosine 1-phosphate; S1P1 = sphingosine 1-phosphate receptor subtype 1; SC = subcutaneous; TNF = tumour necrosis factor; TYK2 = tyrosine kinase 2; UC = ulcerative colitis; vs. = versus.
aHealth Canada–approved indication.
Note: All the comparators in this table were included in ITC as well as pharmacoeconomic analyses. Adalimumab was included in the ITC, but it was not broken down by adalimumab biosimilars vs. reference biologic drug. Infliximab was included in the ITC, but it was not broken down by infliximab biosimilars vs. reference biologic drug. Tofacitinib was included in the ITC, but it was not broken down by tofacitinib generic vs. reference biologic drug.
Source: Product monographs.45,50-72
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
CDA-AMC received 2 patient group inputs from GI Society and Crohn’s and Colitis Canada. The GI Society is a national charity committed to improving the lives of people with GI and liver conditions, supporting research, advocating for appropriate patient access to health care, and promoting GI and liver health. Crohn’s and Colitis Canada is a national health charity focused on finding cures for Crohn disease and UC through research, advocacy, and increasing awareness.
The GI Society gathered information through 1-to-1 conversation with a patient who is living with UC and is receiving the drug under review; a round table with gastroenterologists, patients, and patient groups across Canada in 2025; an online survey about the unmet needs of individuals living with IBD, with 514 respondents from Canada in 2024; a follow-up survey focusing on opinions regarding biologics and biosimilars with 55 respondents; interviews with 7 individuals living with IBD in 2023; a survey about the IBD patient journey with 54 respondents from Canada in 2022; a focus group with several patients with IBD in 2022; and 2 surveys regarding the unmet needs of patients with IBD in 2018 and 2020, with 432 and 579 respondents, respectively.
Crohn’s and Colitis Canada gathered information from Crohn and Colitis Canada’s report through its 2023 Impact of Inflammatory Bowel Disease in Canada report,- as well as through a survey conducted in 2022, in which 354 out of 1,706 survey respondents were patients with moderate to severe UC, and through interviews with patients who have had experience with the drug under review.
In terms of disease experience, the GI Society explained that diarrhea, pain, rectal bleeding, and loss of bowel control are common recurring symptoms of UC and anemia can result from blood loss due to ulcerations in the intestine and from general malnutrition due to decreased nutrient absorption. Some patients have extra-intestinal manifestations, including fever, inflammation of the eyes (uveitis) or joints (arthritis), ulcers of the mouth or skin, tender and inflamed nodules on the shins, and numerous other conditions. Anxiety, stress, suicidal thoughts, and poor mental health are major factors. UC often has a profound effect on an individual’s life — physically, emotionally, and socially — both at home and at school or in the workplace. Symptoms can be relentless, embarrassing, and scary. The severity of the disease can fluctuate, making it necessary to go through routine testing, reassessments, and medication changes. It is particularly difficult for children and young adults, because it often affects a person’s sense of self. According to the input, patients reported that sustained remission or treatment response is more important than relieving any single symptom. The constant concern about future flares, possibly worse than the last, at unpredictable times, can disastrously disrupt patients’ lives.
The GI Society noted that patients need treatments that improve quality of life, rather than cause more symptoms, pain, frustration, or hardship.
Regarding the currently available treatments, the patient group noted that patients with IBD still have a lot of difficulty achieving remission or adequate symptom relief. In the 2020 survey, 33% of respondents did not believe that their IBD was well-controlled by their current medications. In the 2024 survey, this number was 29%, compared with 38% who found it well-controlled and 33% who were unsure. When asked how concerned they were about running out of treatment options, 82% were at least somewhat concerned. Patients are still suffering, and each person living with IBD has a different experience. This is why it is so important that patients living with IBD have access to various effective treatments.
According to the patient group input, improved outcomes included improved quality of life; access to different treatment options; access to a treatment that works, which results in less patient suffering and involves less unnecessary usage of health care resources (hospital stay, surgeries, and diagnostic procedures); access to a treatment with no side effects; reduction of symptoms; and ease of administration (fewer injections, self-injection, or oral administration).
The GI Society had an interview with 1 patient with UC who started taking risankizumab through a clinical trial in December 2022. After 2 to 3 weeks, the patient noticed a significant improvement. The patient’s injections are every 8 weeks, and the patient noticed that, by about week 6, they could feel that they needed the medication again.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of UC.
Unmet Needs
The clinical experts noted that treating UC can be challenging. The goal is to control symptoms (e.g., frequency, urgency) and signs of UC (e.g., clinical and endoscopic remission) with AT as quickly as possible. There are many unmet needs in patients with UC because UC responds to the available treatments in only some patients and UC may become refractory to current treatment options in some patients. In addition, patients need new treatments that are better tolerated, with rapid onset and enhanced safety profiles. Furthermore, there have been few SC options for treatment of UC to date.
Place in Therapy
The clinical experts noted that risankizumab should be available as a first-line option for treatment of moderate to severe UC and that previous treatment failure should not be required to begin this drug. The only previous therapy that they would consider as a reasonable option would be corticosteroids, but these are not an option for long-term maintenance therapy.
Patient Population
The clinical experts noted that any patient with moderate to severe UC would be a suitable candidate for risankizumab. Patients unable to self-inject the drug may require support to administer the medication.
Both diagnosis and treatment need to be made by a gastroenterologist. Misdiagnosis is unlikely, as endoscopy is needed to establish the diagnosis.
Assessing the Response Treatment
The clinical experts noted the following important outcomes:
improvement in symptoms, including decreasing stool frequency, urgency, and bleeding; these have a direct impact on patients’ HRQoL and important end points, such as work or school participation, fatigue (by hindering patients from sleeping through the night), and overall function or activities of daily living
sustained clinical remission, avoiding the need to add steroids (e.g., steroid-sparing and steroid-free remission with ongoing clinical remission)
endoscopic bowel healing, which is the best marker to ensure that adverse outcomes such as surgery and hospitalization are avoided.
In practice, they noted that clinical scoring systems (e.g., Mayo and partial Mayo scores, rectal bleeding score), endoscopic outcomes, and histopathological evaluation are used to evaluate response to treatment. In addition, biomarkers (e.g., fecal calprotectin) and corticosteroid-free remission are used to monitor ongoing treatment response, including during maintenance therapy. From the patient’s perspective, it is prudent to monitor improvement in HRQoL.
The main issue with scoring systems is that they may be somewhat subjective. Hard end points include the Mayo endoscopic subscore and histopathological evaluation. Clinicians must follow the Stride II guidelines to ensure appropriate ongoing clinical management.
Discontinuing Treatment
The clinical experts noted that treatment may be discontinued for 3 main reasons:
primary nonresponse during induction treatment, with ongoing evidence of objective disease and no meaningful improvement of symptoms, despite a robust attempt at induction treatment (and possible repeated attempts)
early improvement in symptoms and objective disease, followed by relapse and with persistent symptoms and disease activity requiring corticosteroid treatment, either persistently or recurrently
SAEs (e.g., anaphylactic reactions).
Prescribing Considerations
The clinical experts noted that, following a diagnosis and staging of UC by a gastroenterologist, treatment can be prescribed by a gastroenterologist or general internist with experience and expertise in treatment of IBD. Treatment can be provided in any clinical setting, but, in most patients, it will be initiated in the outpatient setting.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
CDA-AMC received 1 clinician group input from Canadian Inflammatory Bowel Disease (IBD) Physicians Group. This group consists of more than 30 gastroenterologists in Canada with specific interest and expertise in IBD and with broad representation from across Canada. Twenty-five clinicians who met through videoconference contributed to this submission.
The clinician group strongly endorsed induction of remission with advanced targeted agents in patients with moderately to severely active UC. These ATs include anti-TNF drugs, alpha4beta7 integrin inhibitor vedolizumab, the JAK inhibitors tofacitinib and upadacitinib, the IL-12 and IL-23 inhibitor ustekinumab, sphingosine-1-phosphate receptor modulators ozanimod or etrasimod, or the IL-23 p19 inhibitor mirikizumab. The clinician group added that ATs were indicated in patients who were intolerant or had an inadequate response to conventional therapies (corticosteroids and immunomodulators such as thiopurines, azathioprine, or methotrexate).
According to the clinician group input, histologic healing is a potential future therapeutic target. The clinician group discussed that histologic remission, in conjunction with endoscopic remission (histo-endoscopic mucosal improvement and remission), has become increasingly important for new drugs to achieve. These measures are associated with a decreased likelihood of clinical relapse. The clinician group believes that rapid symptom relief, durability of treatment, and safety of treatment are important treatment goals, which is similar to patients’ wishes.
In terms of treatment gaps, the clinician group noted that, for moderately to severely active UC, conventional treatments for UC lack sustained efficacy and have long-term safety concerns. While ATs with varying mechanisms of action are available and have improved efficacy, a considerable proportion of patients (up to 20% to 30%) have disease that does not respond to induction therapy or loses response over time. As a result, there remains a significant medical need for additional efficacious, durable, and safe treatment options that provide symptom control and mucosal healing, improve quality of life, and reduce the risk of hospitalization and surgery in the long term for patients with moderately to severely active UC who have experienced inadequate response or intolerance to conventional therapies or ATs.
Regarding the place in therapy for patients with moderately to severely active UC, the clinician group believed that risankizumab could be used as the first choice in such patients in whom conventional therapy has failed, who are not candidates for other conventional therapy, or in whom 1 or more ATs have failed. The clinician group added that risankizumab is best suited to treat any such adult patient who have experienced an inadequate response, loss of response, or were intolerant to conventional therapy, a biologic treatment, or a JAK inhibitor. These patients are in most need of intervention, as they have active symptoms, lack long-term treatment options, and are at high risk of disease progression. On the other hand, UC patients with current or previous mild to moderate active disease, whose remission can be adequately maintained with conventional treatments, are least suitable level for treatment with risankizumab. The outcomes used to determine whether a patient is responding to treatment in clinical trials included clinical remission, clinical response, disease activity, patient-reported symptoms, stringent endoscopic and histologic outcomes that included mucosal improvement and healing, and HRQoL outcomes.
The Canadian IBD Physicians Group recommends discontinuing treatment with risankizumab if symptoms worsen or if there is an inadequate response, but this is not broadly anticipated. Available clinical data demonstrate that a large proportion of patients will respond to therapy during the first 6 months.
The clinician group noted that, during the induction phase, risankizumab needs to be administered in a clinic by a trained health care professional who is experienced in the management of patients with UC or is supervised by a health care professional with this experience.
Drug Program Input
The drug programs provide input on each drug being reviewed through the Reimbursement Review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 6.
Table 6: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Given that risankizumab was compared to placebo (and not an active drug comparator) in the submitted studies, how do the efficacy outcomes for risankizumab compare to the outcomes for other drugs for UC, in particular, in comparison to drugs with the same mechanism of action (IL-23 inhibitors or IL-12 and IL-23 inhibitors)? | The clinical experts noted that there is no strong evidence of a difference between the efficacy outcomes for risankizumab compared to other drugs for UC. |
Given that there are 3 other IL-23 inhibitors (mirikizumab, guselkumab, ustekinumab) with a similar mechanism of action, are there currently any unmet needs in the treatment of UC that risankizumab would address? | Not all drugs in the same class have the same molecular composition, so the mode of action may be slightly different between them. The choice for use will be made on a case-by-case basis, with factors including the patient’s history with drugs with similar mechanisms of action. |
Considerations for initiation of therapy | |
The INSPIRE study included participants with intolerance or inadequate response to conventional therapy, tofacitinib (nonbiologic), or 1 or more biologic therapies. However, the efficacy results were more notable in patients who are naive to ATs. What place in therapy would risankizumab have in patients with inadequate response to other ATs (i.e., biologics or JAK inhibitors)? | The clinical experts noted that, based on the available evidence from the studies and the indirect treatment comparison, the place in therapy of risankizumab would be similar to other ATs. |
The sponsor claims that risankizumab “is the first AT to demonstrate improvements in patient-reported outcomes such as tenesmus, fecal incontinence, sleep disturbance, and urgent bowel movements.” Would patients experiencing these symptoms be considered specifically for eligibility of risankizumab as opposed to other AT? | The clinical experts noted that these patient-reported outcomes are expected to be improved with all ATs and not just with risankizumab. |
Considerations for prescribing of therapy | |
Is there a possibility for the off-label use of a 24-week induction treatment in clinical practice? If appropriate, please comment on the utility and/or efficacy of using an extended induction | The clinical experts noted that it would depend on the patient history (e.g., patients with naive vs. refractory disease, corticosteroid response) and the decision would be made on a case-by-case basis. In the INSPIRE trial, patients who did not experience clinical response of risankizumab 1,200 mg IV at week 12 continued treatment with risankizumab up to week 24. At week 24, the proportion of patients who achieved clinical remission was 12.7% for those who received risankizumab 180 mg, 15.7% for those who received risankizumab 360 mg SC, and 8.8% for those who received risankizumab 1,200 mg IV. |
AT = advanced therapy; IL = interleukin; UC = ulcerative colitis; vs. = versus.
Clinical Evidence
The objective of this clinical review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of risankizumab:
600 mg in 10 mL (60 mg/mL) by IV infusion
180 mg in 1.2 mL (150 mg/mL) by SC injection
360 mg in 2.4 mL (150 mg/mL) by SC injection.
The indication is for the treatment of adult patients with moderately to severely active UC who have experienced an inadequate response, loss of response, or were intolerant to conventional therapy, a biologic treatment, or a JAK inhibitor. The focus is on comparing risankizumab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of risankizumab is presented in 4 sections with the CDA-AMC critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs selected according to the sponsor’s systematic review protocol. The CDA-AMC assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes sponsor-submitted long-term extension studies; however, none were available. The third section includes indirect evidence from the sponsor. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
2 pivotal studies or RCTs identified in systematic review
1 NMA
1 additional study addressing gaps in evidence.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
Description of Studies
Two pivotal trials, INSPIRE and COMMAND, were included in the systematic review. Characteristics of the included studies are summarized in Table 7.
The objective of these trials was to evaluate the efficacy and safety of risankizumab when administered as an induction (INSPIRE) or maintenance (COMMAND) therapy for patients with UC. The INSPIRE trial was a phase III, randomized, placebo-controlled, double-blind, parallel-group induction study in adult patients with moderately to severely active UC who experienced an inadequate response or intolerance to conventional therapies or other ATs. Patients in the INSPIRE trial who did not achieve clinical response at week 12 were eligible to enter another blinded risankizumab treatment period (i.e., Induction Period 2, which lasted for an additional 12 weeks). The COMMAND trial was a phase III, randomized, placebo-controlled, double-blind, parallel-group maintenance study in adult patients with UC previously enrolled in the INSPIRE induction trial who experienced an adequate response to risankizumab. Combined, the 2 studies lasted up to 81 weeks and included a screening period that lasted up to 35 days, a 12-week double-blind induction period, and a 52-week maintenance period. In both trials, randomization was performed using a web-based interactive response technology with block randomization methods.
During the INSPIRE trial, 977 patients were randomized in a 2:1 ratio to receive risankizumab or placebo across 261 sites in 41 countries, with 11 sites in Canada. Randomization was stratified by the presence of baseline corticosteroid use (yes, no), baseline adapted Mayo score (≤ 7, > 7), and a history of intolerance or inadequate response to ATs (0, 1, > 1 treatment). Study follow-up visits were scheduled at 4, 8, 12, and 24 weeks, and the final patient follow-up occurred in May 2023.
During the COMMAND trial, 584 patients were randomized in a 1:1:1 ratio to receive risankizumab 180 mg, risankizumab 360 mg, or placebo (risankizumab withdrawal) across 238 sites in 37 countries, with 11 sites in Canada. Randomization was stratified by history of inadequate response to advanced therapy (yes, no), last risankizumab induction dose (IV 600 mg, 1,200 mg, or 1,800 mg), and clinical remission status (per local evaluation) at the last visit of the induction trial (yes, no). Study follow-up visits were scheduled at 8, 16, 24, 32, 40, 48, and 52 weeks, and the final patient follow-up occurred in April 2023.
Table 7: Details of Studies Included in the Systematic Review
Details | INSPIRE | COMMAND |
|---|---|---|
Designs and populations | ||
Study design | Phase III, randomized, placebo-controlled, double-blind, parallel-group induction study | Phase III, randomized, placebo-controlled, double-blind, parallel-group maintenance study |
Locations | 261 centres in 41 countries (Africa, Asia, Europe, North America, South America); 11 sites in Canada | 238 centres in 37 countries (Africa, Asia, Europe, North America, South America); 11 sites in Canada |
Patient enrolment dates | Start date: November 5, 2020 End date: August 4, 2022 | Start date: August 28, 2018 End date: March 30, 2022 |
Randomized (N) | Total = 977 Risankizumab, 1,200 mg = 652 Placebo = 325 | Total = 584 Risankizumab, 180 mg = 193 Risankizumab, 360 mg = 195 Placebo (risankizumab withdrawal) = 196 |
Inclusion criteria |
|
|
Exclusion criteria |
|
|
Drugs | ||
Intervention | Risankizumab, 1,200 mg, administered IV at 0, 4, and 8 weeks | Risankizumab, 180 or 360 mg, administered SC every 8 weeks for 52 weeks |
Comparator(s) | Placebo administered IV at 0, 4, and 8 weeks | Placebo administered SC every 8 weeks for 52 weeks |
Study duration | ||
Screening phase | 35 days | Not applicable |
Treatment phase | 12 to 24 weeks | 52 weeks |
Follow-up phase | Final follow-up was 140 days from the last dose of study drug. | Final follow-up was 140 days from the last dose of study drug, and patients could enrol in an open-label long-term extension study. |
Outcomes | ||
Primary end point | Clinical remission, determined using the adapted Mayo score, at 12‑week follow-up | Clinical remission, determined using the adapted Mayo score, at 52‑week follow-up |
Secondary and exploratory end points | Ranked secondary outcomes:
Other key secondary outcomes:
Exploratory outcomes:
| Ranked secondary outcomes:
Other key secondary outcomes:
Exploratory outcomes:
|
Safety end points | Incidence of TEAEs, changes in vital signs, physical examination results, and clinical laboratory data | Incidence of TEAEs, changes in vital signs, physical examination results, and clinical laboratory data |
Publication status | ||
Publications | Louis E, Schreiber S, Panaccione R, et al. Risankizumab for Ulcerative Colitis: Two Randomized Clinical Trials. JAMA. 2024;332(11):881-897.73 doi:10.1001/jama.2024.12414 AbbVie Inc. M16-067 Abbreviated Clinical Study Report — Final: A Multicenter, Randomized, Double-Blind, Placebo-Controlled Induction Study to Evaluate the Efficacy and Safety of Risankizumab in Patients with Moderately to Severely Active Ulcerative Colitis. 2023.17 | Louis E, Schreiber S, Panaccione R, et al. Risankizumab for Ulcerative Colitis: Two Randomized Clinical Trials. JAMA. 2024;332(11):881-897.73 doi:10.1001/jama.2024.12414 AbbVie Inc. M16-066 Abbreviated Clinical Study Report — Substudy 2 Interim: A Multicenter, Randomized, Double-Blind, Placebo-Controlled 52-week Maintenance and an Open-Label Extension Study of the Efficacy and Safety of Risankizumab in Patients with Ulcerative Colitis. 2024.18 |
Trial registration | ClinicalTrials.gov Identifier: NCT0339814874 | ClinicalTrials.gov Identifier: NCT0339813575 |
AE = adverse event; AT = advanced therapy; CRP = C-reactive protein; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; HEMI = histologic, endoscopic, and mucosal improvement; IBDQ = Inflammatory Bowel Disease Questionnaire; SC = subcutaneously; SF-36 = Short Form (36) Health Survey; TEAE = treatment-emergent adverse event; UC = ulcerative colitis; WPAI-UC = Work Productivity and Activity Impairment Questionnaire-Ulcerative Colitis.
Sources: INSPIRE Clinical Study Report,17 COMMAND Clinical Study Report.18
Populations
Inclusion and Exclusion Criteria
The INSPIRE trial enrolled adult patients with moderately to severely active UC and a history of intolerance or inadequate response to 1 or more conventional or ATs for UC. Additionally, eligible patients had to have a diagnosis of UC for at least 3 months, an adapted Mayo score of 5 to 9 points, and an endoscopic subscore of 2 to 3 points. Exclusion criteria included prior exposure to p40 inhibitors (e.g., ustekinumab) or p19 inhibitors (e.g., risankizumab or mirikizumab).
The COMMAND trial included participants enrolled in a risankizumab induction trial who experienced adequate response to risankizumab at the 12- or 24-week follow-up. An adequate response was defined as a decrease of 30% or more from baseline and of 2 points or more on the adapted Mayo score, and a decrease of 1 or more in rectal bleeding score or an absolute rectal bleeding score of 1 or less.
Interventions
For the induction trial (INSPIRE), patients were randomized to receive 1,200 mg of IV risankizumab or matching placebo at weeks 0, 4, and 8. IV treatment was administered over 3 hours in an inpatient setting. Patients who failed to respond at the end of the 12-week period entered the Induction Period 2. In this phase, risankizumab dosing was based on treatment received during the initial induction period. Patients who received IV risankizumab during the first induction period were randomized to receive risankizumab 1,200 mg IV, risankizumab 180 mg SC, or risankizumab 360 mg SC. IV doses were administered at weeks 12, 16, and 20, and SC doses were administered at weeks 12 and 20. Patients who received placebo during the first induction period received risankizumab 1,200 mg IV at weeks 12, 16, and 20.
For the maintenance trial (COMMAND), patients were assigned to receive 180 or 360 mg of risankizumab or matching placebo administered SC every 8 weeks for 52 weeks.
In the induction trial, patients taking stable doses of corticosteroids, aminosalicylates, or immunomodulators at baseline continued these treatments throughout the trial. Initiating or increasing the dose of these medications was prohibited, except in the event of moderate to severe treatment-related toxicities. UC-related antibiotics could be discontinued in the blinded Induction Period 2; oral corticosteroids could also be tapered during this period but not increased above baseline. Rectal aminosalicylates were prohibited from 14 days before the screening period and for the entire duration of the study.
In the maintenance trial, patients taking stable doses of aminosalicylates and/or immunomodulators at baseline continued these treatments throughout the trial. Initiating or increasing the dose of these medications was prohibited, except in the event of moderate to severe treatment-related toxicities. Patients undergoing corticosteroid therapy were required to taper by week 8. Patients who lost satisfactory clinical response after starting to taper steroids could increase their corticosteroid dose again, up to the dose used at baseline of the induction study. Starting at week 16, patients in any treatment group could receive open-label risankizumab therapy (i.e., 1 single dose of risankizumab administered IV followed by 360 mg administered SC every 8 weeks) if they lost adequate clinical response (defined as a rectal bleeding score of ≥ 1 point greater than the week 0 value on 2 consecutive assessments 7 to 14 days apart or a Mayo endoscopic subscore of 2 or 3).
Outcomes
A list of efficacy end points assessed in this clinical review report is provided in Table 8, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review according to the clinical expert(s) consulted and input from patient and clinician groups and public drug plans. Using the same considerations, CDA-AMC selected end points considered to be most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms considered important for informing expert committee deliberations were also assessed using GRADE.
Table 8: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | INSPIRE | COMMAND |
|---|---|---|---|
Efficacy | |||
Clinical remission per adapted Mayo scorea | Week 12 for induction or Week 52 for maintenance | Primary end point | Primary end point |
Clinical response per adapted Mayo scorea | Week 12 for induction or Week 52 for maintenance | Secondary end point | Secondary end point |
Endoscopic improvementa | Week 12 for induction or Week 52 for maintenance | Secondary end point | Secondary end point |
HEMIa | Week 12 for induction or Week 52 for maintenance | Secondary end point | Secondary end point |
HEMRa | Week 12 for induction or Week 52 for maintenance | Secondary end point | Secondary end point |
Discontinuation of corticosteroid use in patients taking steroids at baseline (maintenance only) | Week 52 | Not applicable | Secondary end point |
UC-related hospitalizationa | Week 12 for induction or Week 52 for maintenance | Secondary end point | Secondary end point |
Patient-reported outcomes | |||
IBDQa | Week 12 for induction or Week 52 for maintenance | Secondary end point | Secondary end point |
Harms outcomes | |||
Serious TEAEs | Week 12 for induction or Week 52 for maintenance | Secondary end point | Secondary end point |
HEMI = histologic, endoscopic, and mucosal improvement; HEMR = histologic, endoscopic, and mucosal remission; IBDQ = Inflammatory Bowel Disease Questionnaire; TEAE = treatment-emergent adverse event; UC = ulcerative colitis.
aP value adjusted for multiple testing.
Sources: INSPIRE Clinical Study Report,17 COMMAND Clinical Study Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Adapted Mayo Score
The adapted Mayo score consists of stool frequency, rectal bleeding, and endoscopic subscores, and is the most used method for measuring disease activity in UC (Table 9). Each subscore is scored from 0 to 3, and the total score ranges from 0 to 9, with higher scores indicating greater UC severity.76,77 Stool frequency and rectal subscores were based on patient-derived assessments captured in their daily diaries, and the endoscopic subscore was centrally read based on endoscopic imaging assessments.
Clinical Remission per Adapted Mayo Score
Clinical remission was determined using the adapted Mayo score (stool frequency score ≤ 1 and not greater than baseline, rectal bleeding score of 0, and endoscopic subscore ≤ 1 without friability), which is consistent with regulatory guidance.12,78,79 Notably, this definition for clinical remission is more stringent than definitions used in earlier UC trials.80-83 No published estimate of the MID for the between-group difference has been identified for this outcome. Clinical experts consulted by the review team suggested that a 10% difference between groups would be considered clinically important.
In a recently published article that assessed the effect size thresholds to inform research, guidelines, and clinical decisions in patients with IBD, it was reported that the consensus results of thresholds for clinical remission are 11% ± 6% for trivial to small, 20% ± 8% for small to moderate, and 31% ± 13% for moderate to large.14
Clinical Response per Adapted Mayo Score
Clinical response on the adapted Mayo score was defined as a decrease of 30% or more and 2 points or more from baseline and a decrease in rectal bleeding score of 1 or more or an absolute rectal bleeding score of 1 or less (Table 9).12 No published estimate of the MID for the between-group difference has been identified for this outcome. Clinical experts consulted by the review team suggested that a 10% difference between groups would be considered clinically important.
In a recently published article that assessed the effect size thresholds to inform research, guidelines, and clinical decisions in patients with IBD, it was reported that the consensus results of thresholds for clinical response are 14% ± 7% for trivial to small, 24% ± 10% for small to moderate, and 36% ± 15% for moderate to large thresholds.14
Endoscopic Improvement
Endoscopic improvement was defined as an endoscopic subscore of 1 or less without friability on the adapted Mayo score.12 No published estimate of the MID for the between-group difference has been identified for this outcome.
In a recently published article that assessed the effect size thresholds to inform research, guidelines, and clinical decisions in patients with IBD, it was reported that the consensus results of thresholds for endoscopic response are 12.5% ± 7% for trivial to small, 22% ± 9% for small to moderate, and 33 ± 14% for moderate to large thresholds.14
Histologic, Endoscopic, and Mucosal Improvement
The Geboes score is the most commonly used histologic scoring system in UC. It classifies structural (architectural) changes as grade 0, chronic inflammatory infiltrate as grade 1, lamina propria neutrophils and eosinophils as grade 2, neutrophils in the epithelium as grade 3, crypt destruction as grade 4, and erosion as ulceration as grade 5. International consensus has defined histological improvement as a Geboes score ≤ 3.1.84-88 HEMI was defined as an endoscopic subscore of 1 or less without friability on the adapted Mayo score and a Geboes score of 3.1 or less. No published estimate of the MID for the between-group difference has been identified for this outcome.
Histologic, Endoscopic, and Mucosal Remission
There is currently no universal consensus on how to define mucosal healing.12 Mucosal healing was measured as HEMR, which was defined as achieving an endoscopic subscore of 0 on the adapted Mayo score and a Geboes score of less than 2.0. No published estimate of the MID for the between-group difference has been identified for this outcome.
Discontinuation of Corticosteroid Use at Week 52 in Patients Taking Steroids at Baseline
This end point involved discontinuation of corticosteroids at week 52 and for the entire 52-week maintenance period in patients taking corticosteroids at baseline in the induction study. No published estimate of the MID for the between-group difference has been identified for this outcome.
Inflammatory Bowel Disease Questionnaire
The IBDQ is a disease-specific, multidimensional, self-reported questionnaire comprising 32 Likert-scaled items (Table 9). The total score ranges from 32 to 224 points using 7-point response options, with higher scores indicating better HRQoL. The IBDQ contains 4 component subscales: bowel symptoms, systemic symptoms, emotional function, and social function. Each subscale can be computed, with total scores ranging from 10 to 70, 5 to 35, 12 to 84, and 5 to 35 points, respectively.89-91 The IBDQ has been validated in a variety of settings, countries, and languages, and an absolute score of 170 points or greater indicates clinical remission.89,92 An absolute score change of 16 to 30 points is considered the MID for the change from baseline and a score of 15 points or greater above the placebo score is considered the MID for the between-group difference.15,16
UC-Related Hospitalization
This outcome included any UC-related event that resulted in an admission to the hospital for any length of time. Events per 100 person-years were also calculated for the maintenance trial. No MID for the between-group difference has been identified for this outcome.
Safety Evaluation Plan
TEAEs were recorded using the Medical Dictionary for Drug Regulatory Activities (MedDRA) System Organ Classes (SOCs) and preferred terms. The safety end points included any TEAE, TEAEs related to COVID-19, serious TEAEs, TEAEs leading to study drug discontinuation, deaths, and AEOSIs. SAEs included any of the following:
death
life-threatening events
hospitalization or prolongation of hospitalization
congenital anomaly
persistent or significant disability or incapacity
important medical event requiring medical or surgical intervention to prevent serious outcome.
Table 9: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
Adapted Mayo score (also known as Modified Mayo score) | In the adapted version of the Mayo score, the definition of an ES of 1 no longer includes mucosal friability and the Physician’s Global Assessment (PGA) is excluded. The components of the adapted Mayo score include:
Scale components are scored on a 4-point scale from 0 to 3, with the score of 0 indicating normal and higher score indicating more severe symptoms or disease. The adapted Mayo score is a sum of the Mayo SFS, RBS, and ES, with a maximum score of 9. | Validity: In a cross-sectional survey of 2,608 patients with UC and their treating gastroenterologist, increases in the adapted Mayo score were associated in increased in odds of adverse outcomes, including a current flare (OR = 1.52, SE = 0.10), a higher number of flares in the past year (OR = 1.17, SE = 0.03), deterioration in clinical status (OR = 1.48, SE = 0.10), and patient-reported overall WPAI (score = 6.94, SE = 0.888).76 A 1-point increase in the adapted Mayo score was associated with a 0.02‑unit decrease in EQ-5D and a 2.73‑point decrease in the SIBDQ, suggesting that a change in score of > 4 is associated with a clinically meaningful reduction in HRQoL.76 Reliability and responsiveness: No studies of the reliability and responsiveness of the adapted Mayo score were identified. | Evidence of an MID for the adapted Mayo score in patients with UC was not identified. However, a value of ≤ 1 indicates remission for the adapted Mayo score. In the context of UC, clinical remission is marked by a total Mayo score of ≤ 2 points.93,94 A clinical response is indicated by a decrease of ≥ 3 points in the Mayo score or the partial Mayo score, demonstrating a meaningful reduction in disease severity.93 Furthermore, according to Naegeli et al. (2021), changes of ≥ 4 units in Mayo permutations may be associated with clinically meaningful reductions in HRQoL, as measured by EQ‑5D and SIBDQ.76 |
IBDQ | The IBDQ is a disease-specific questionnaire used to assess self-reported disease-specific HRQoL in patients with IBD.95 The IBDQ is a 32-item Likert-based questionnaire divided into 4 dimensions:
Responses are graded on a scale from 1 (worst situation) to 7 (best situation). Total IBDQ score ranges between 32 and 224, with higher scores representing better HRQoL. Scores ranging from 170 to 190 indicate remission. | Validity: The emotional function dimension of the IBDQ was found to be strongly correlated with the Rand questionnaire (r = 0.76, P < 0.001); the systemic symptoms dimension of the IBDQ was weakly correlated to change in the disease activity index (r = 0.036, P = 0.442); and patients’ global rating of change in emotional function was moderately correlated to the emotional function dimension (r = 0.52, P < 0.001) and the bowel symptom dimension (r = 0.42, P = 0.003) of the IBDQ.95 The IBDQ was found to detect changes in social and emotional state of patients.92 Reliability and responsiveness: The IBDQ was shown to be highly reliable through evaluation of internal consistency (Cronbach alpha 0.7) and test-retest assessment (ICC = 0.9 to 0.99 or Pearson’s r ≥ 0.8). The IBDQ was also shown to be responsive to change in patients with IBD (P < 0.05).96,97 | While some suggest that an increase of 15 to 32 points may be considered a clinically relevant improvement in HRQoL for patients with Crohn disease or UC, evidence from clinical trials suggests that a change of more than 30 points is associated with clinical benefits and an improvement of 15 points or greater above placebo is required among patients with IBD including those with UC.15,16,90,98-100 |
ES = endoscopic subscore; HRQoL = health-related quality of life; IBD = inflammatory bowel disease; IBDQ = Inflammatory Bowel Disease Questionnaire; ICC = intraclass correlation; MID = minimal important difference; OR = odds ratio; PGA = Physician’s Global Assessment; RBS = rectal bleeding subscore; SE = standard error; SFS = stool frequency subscore; SIBDQ = Short Inflammatory Bowel Disease Questionnaire; UC = ulcerative colitis; WPAI = Work Productivity and Activity Impairment Questionnaire.
Statistical Analysis
Sample Size and Power Calculation
In the INSPIRE trial, the sponsor calculated the sample size based on the expected proportion of patients who achieved clinical remission per the adapted Mayo score at week 12. A sample size of 966 patients (644 patients in the risankizumab group and 322 in placebo group) provided 90% power or greater to detect a treatment difference of 10% for the primary outcome, based on an assumption of 6% for clinical remission in the placebo group and 16% in the risankizumab group, using the 2-sided Miettinen and Nurminen test at a significance level of 0.05.80 The sample size was determined to provide adequate power for the primary end point and selected ranked secondary end points (clinical remission per full Mayo score at week 12, endoscopic response at week 12, endoscopic remission at week 12, clinical response per adapted Mayo score at week 12, and clinical response per partial adapted Mayo score at week 4) and adequate patients with response to meet the requirement for this study. The assumed remission rates were based on substudy 1 and on ustekinumab data.
In the COMMAND trial, the sponsor calculated the sample size based on the expected proportion of patients who would achieve clinical remission per the adapted Mayo score at week 52. Assuming a clinical remission rate of 22% in the placebo group and 42% in the risankizumab groups, a sample size of 573 patients (191 in each treatment group) provided 90% power or greater to detect a treatment difference of 20% for the primary outcome using the 2-sided Miettinen and Nurminen test at a significance level of 0.025, adjusting the 0.05 significance level for the 2 dose comparisons.80
Statistical and Analytic Plans
Estimands
A summary of statistical analysis methods for all efficacy end points in the INSPIRE and COMMAND trials is presented in Table 10. The INSPIRE study estimands for the primary and secondary end points considered the following ICEs: premature discontinuation of the study drug, initiation or dose escalation of UC-related corticosteroids, and UC-related surgery. For ICE1, data were collected regardless of premature discontinuation. For binary end points, patients with ICE2 and/or ICE3 were considered to have no response. For continuous end points, measurements on or after the date of the ICE were not used (considered returned to baseline).
The COMMAND study estimands for the primary and secondary end points considered the following ICEs: premature discontinuation of study drug, UC-related corticosteroid therapy, risankizumab rescue therapy, and UC-related surgery. For ICE1, data collected were used regardless of premature discontinuation in the primary analysis. In supplementary analyses (except for UC-related hospitalization), patients were considered to have no response at all visits after premature discontinuation. For ICEs 2, 3, and/or 4, patients were considered to have no response for binary end points; for continuous end points, measurements on or after the date of the ICE were not used (considered returned to baseline).
Analysis Methods
In the INSPIRE trial, categorical variables were analyzed using the Cochran-Mantel-Haenszel test for common risk difference, stratified by AT-IR status, baseline steroid use, and baseline adapted Mayo score. In the COMMAND trial, the same approach was used, but the stratification factors included AT-IR status, clinical remission status at week 0, and the last risankizumab induction dose. If there was a stratum for a treatment group that had no patient in it, a value of 0.1 was added to all cells in the corresponding table to prevent dividing by 0. Greenland and Robins’ variance estimator was used for the common risk difference.101 Values for all stratification factors used in Cochran-Mantel-Haenszel analyses were based on actual values. The results of binary outcomes were presented as risk differences with 95% CIs.
Continuous variables collected longitudinally (more than 1 post baseline visit) were analyzed using the mixed-effects model for repeated measurements. The results of continuous outcomes were presented as least squares mean differences with 95% CIs.
Missing Data Handling
Evaluations of patients with ICE were handled as mentioned previously in the Estimands section. Missing data could occur due to various reasons, including missing visits or assessments, early withdrawal from the clinical trials, COVID-19 infection or logistic restrictions, or geopolitical conflict in Ukraine and its surrounding areas.
In both the INSPIRE and COMMAND trials, for binary end points, a nonresponder imputation was used (except for UC-related hospitalization), with multiple imputation methods used for missing data due to COVID-19 or geopolitical conflict in Ukraine (assuming to be missing at random). For UC-related hospitalization, an “as-observed” analysis was undertaken with no imputation of missing values. Additionally, all values were included regardless of ICEs.
For continuous end points, “return to baseline” imputation was used for patients with ICEs 2, 3, or 4 (in the COMMAND trial only) in the primary analysis. Missing data were considered to be missing at random in the mixed-effects model for repeated measurements.
Sensitivity Analyses
Sensitivity analyses were conducted for the primary end point and continuous secondary end points, as described in Table 10.
Multiple-Testing Procedure
The primary and secondary efficacy end points in INSPIRE were tested using a graphical multiple-testing procedure to ensure control of family-wise type I error at a significance level alpha = 0.05 (2-sided).102 The secondary efficacy end points were divided into 2 groups. The first group included the first 10 secondary end points. The second group included all of the remaining 5 secondary end points, which were tested using the Holm procedure.103 If the primary end point achieved significance, continued testing of the secondary outcomes followed a prespecified weight of allocation. No type I error control was applied to other study end points. The outcomes presented in the report for this trial are the results of the final analysis.
In the COMMAND trial, the testing began with testing the primary end point at the prespecified significance level of 0.025 (2-sided) for each risankizumab dose group compared to placebo. The secondary efficacy end points were divided into 2 groups. The first group included the first 12 secondary end points, and the second group included all of the remaining 5 secondary end points, which were tested using the Holm procedure.103 If the primary end point achieved significance, continued testing of the secondary outcomes followed a prespecified weight of allocation as indicated in the graphical multiple-testing procedure. No type I error control was applied to other study end points. The outcomes presented in the report for this trial are the results of the final analysis.
Subgroup Analyses
In the INSPIRE trial, subgroup analyses were assessed using a chi-square test or Fisher's exact test if 20% or greater of the cells had an expected cell count less than 5. The nonresponder imputation method was used for primary analysis. In this method, clinical remission for patients with missing data at scheduled assessment visits was considered “not achieved.” No control for the type I error rate was applied. These analyses aimed to understand whether treatment effects were consistent across subgroups. Forest plots were used to graphically depict treatment effect estimates in the subgroups. No inferential statistics (P values) were produced. The prespecified subgroups included:
sex
age
race
baseline corticosteroid use
baseline immunosuppressant use
baseline adapted Mayo score
baseline partial adapted Mayo score
prior exposure to AT
prior response to AT
baseline weight
baseline presence of pancolitis
baseline disease duration
baseline high-sensitivity C-reactive protein (hs-CRP)
baseline albumin
baseline calprotectin
geographic region.
Subgroup analyses in the COMMAND trial were conducted using the same methods as in the INSPIRE trial. In addition to the subgroups presented in the INSPIRE trial, the COMMAND trial also included the following prespecified subgroups:
week 0 clinical remission status
last risankizumab induction dose.
Table 10: Statistical Analysis of Efficacy End Points
Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses | |
|---|---|---|---|---|
INSPIRE | ||||
Clinical remission | CMH test for common risk difference | Stratified by:
| Nonresponder imputation with multiple imputation (included the treatment group, stratification factors, baseline measurement, and, if applicable, postbaseline measurements in the model) for data missing due to COVID‑19 or geopolitical conflict (assumed missing at random) |
|
Clinical response | CMH test for common risk difference | Stratified by:
| Nonresponder imputation with multiple imputation (included the treatment group, stratification factors, baseline measurement, and, if applicable, postbaseline measurements in the model) for data missing due to COVID‑19 or geopolitical conflict (assumed missing at random) |
|
Endoscopic improvement | CMH test for common risk difference | Stratified by:
| Nonresponder imputation with multiple imputation (included the treatment group, stratification factors, baseline measurement, and, if applicable, postbaseline measurements in the model) for data missing due to COVID‑19 or geopolitical conflict (assumed missing at random) |
|
HEMI | CMH test for common risk difference | Stratified by:
| Nonresponder imputation with multiple imputation (included the treatment group, stratification factors, baseline measurement, and, if applicable, postbaseline measurements in the model) for data missing due to COVID‑19 or geopolitical conflict (assumed missing at random) |
|
HEMR | CMH test for common risk difference | Stratified by:
| Nonresponder imputation with multiple imputation (included the treatment group, stratification factors, baseline measurement, and, if applicable, postbaseline measurements in the model) for data missing due to COVID-19 or geopolitical conflict (assumed missing at random) |
|
IBDQ | Mixed-effects model for repeated measurements | Fixed effects of treatment, visit, and treatment-by-visit interaction, stratification factors, and the continuous fixed covariates of baseline measurements | Multiple imputation method (assuming missing at random) incorporating return to baseline for patients with ICE 2 (initiation or escalation of UC-related corticosteroids) and ICE 3 (UC-related surgery) | Considering unequal variances due to the unequal sample size in the 2 treatment groups (2:1 randomization), a sensitivity analysis was performed for continuous secondary end points with only a single postbaseline measure using an ANCOVA model that estimates variance by treatment group |
UC-related hospitalization | Mixed-effects model for repeated measurements | Fixed effects of treatment, visit, and treatment-by-visit interaction, stratification factors, and the continuous fixed covariates of baseline measurements |
| None |
COMMAND | ||||
Clinical remission | CMH test for common risk difference | Stratified by:
| Nonresponder imputation with multiple imputation (that included the treatment group, stratification factors, baseline measurement, and, if applicable, postbaseline measurements in the model) for data missing due to COVID‑19 or geopolitical conflict (assumed missing at random) |
|
Clinical response | CMH test for common risk difference | Stratified by:
| Nonresponder imputation with multiple imputation (that included the treatment group, stratification factors, baseline measurement, and, if applicable, postbaseline measurements in the model) for data missing due to COVID‑19 or geopolitical conflict (assumed missing at random) | Composite strategy (i.e., discontinuation imputed as nonresponse) for discontinuations |
Endoscopic improvement | CMH test for common risk difference | Stratified by:
| Nonresponder imputation with multiple imputation (that included the treatment group, stratification factors, baseline measurement, and, if applicable, postbaseline measurements in the model) for data missing due to COVID-19 or geopolitical conflict (assumed missing at random) | None |
HEMI | CMH test for common risk difference | Stratified by:
| Nonresponder imputation with multiple imputation (that included the treatment group, stratification factors, baseline measurement, and, if applicable, postbaseline measurements in the model) for data missing due to COVID‑19 or geopolitical conflict (assumed missing at random) | None |
HEMR | CMH test for common risk difference | Stratified by:
| Nonresponder imputation with multiple imputation (that included the treatment group, stratification factors, baseline measurement, and, if applicable, postbaseline measurements in the model) for data missing due to COVID‑19 or geopolitical conflict (assumed missing at random) | None |
Discontinuation of corticosteroid use in patients taking steroids at baseline | CMH test for common risk difference | Stratified by:
| As-observed analysis except that premature study discontinuations and patients with ICE are considered “not discontinued” | None |
IBDQ | Mixed-effects model for repeated measurements | Fixed effects of treatment, visit, and treatment‑by‑visit interaction, stratification factors, and the continuous fixed covariates of baseline measurements | Multiple imputation method (assuming missing at random) incorporating return to baseline for patients with ICE 2 (initiation or escalation of UC‑related corticosteroids) and ICE 3 (UC‑related surgery) | Considering unequal variances due to the unequal sample size in the 2 treatment groups (2:1 randomization), a sensitivity analysis was performed for continuous secondary end points with only a single postbaseline measure using an ANCOVA model that estimates variance by treatment group. |
UC-related hospitalization | Exposure‑adjusted UC–related hospitalization based on incidence rates | Fixed effects of treatment, visit, and treatment-by-visit interaction, stratification factors, and the continuous fixed covariates of baseline measurements |
| None |
ANCOVA = analysis of covariance; AT-IR = inadequate response or intolerance to advanced therapy; CMH = Cochran-Mantel-Haenszel; HEMI = histologic, endoscopic, and mucosal improvement; HEMR = histologic, endoscopic, and mucosal remission; IBDQ = Inflammatory Bowel Disease Questionnaire; ICE = intercurrent event; UC = ulcerative colitis; vs. = versus.
Sources: INSPIRE Statistical Analysis Plan,104 COMMAND Statistical Analysis Plan.105 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Analysis Populations
The analysis populations in the INSPIRE and COMMAND trials included an intention-to-treat (ITT) population and safety analysis set (Table 11). In both trials, the ITT population was used for all efficacy analyses. The ITT population used in INSPIRE was the ITT2 population, which included all randomized patients who received at least 1 dose of study drug. The ITT population used in COMMAND was the ITT1RN_A population, which included all randomized patients who received at least 1 dose of study drug after receiving IV risankizumab for only 1 period of 12 weeks in the induction trial. Additionally, an efficacy analysis was conducted in patients who entered Induction Period 2 of the INSPIRE trial, which included all patients who received at least 1 dose of risankizumab during this period (ITT2_P2). The COMMAND trial also included supplementary efficacy analyses in 2 subsets of patients. The first subset was those in the ITT1RN_A population who received risankizumab 1,200 mg IV in the INSPIRE trial (ITT1RN_B). The second subset was those who received at least 1 dose of study drug in the COMMAND trial after receiving risankizumab 180 mg or 360 mg SC during Induction Period 2 in the INSPIRE trial or placebo during Induction Period 1 in the INSPIRE trial (ITT1NRN).
The safety analysis sets used in the safety analyses were the SA2 population and SA1RN population in the INSPIRE and COMMAND trials, respectively. For both trials, this population included all randomized patients who received at least 1 dose of study drug.
Table 11: Analysis Populations of the INSPIRE and COMMAND Trials
Study | Population | Definition | Application |
|---|---|---|---|
INSPIRE | ITT2 | All randomized patients who received at least 1 dose of study drug, analyzed according to the treatment that they were randomized to | All efficacy analyses |
ITT2_P2 | Patients who entered Induction Period 2 of the INSPIRE trial and received at least 1 dose of risankizumab during this period, analyzed according to the treatment that they were randomized to | All efficacy analyses | |
SA2 | All randomized patients who received at least 1 dose of study drug, analyzed according to the treatment that they actually received | Safety analyses | |
COMMAND | ITT1RN_A | All randomized patients who received at least 1 dose of study drug after receiving IV risankizumab for only 1 period of 12 weeks in the INSPIRE trial, analyzed according to the treatment that they were randomized to | All efficacy analyses |
ITT1RN_B | Patients in the ITT1RN_A population who received risankizumab 1,200 mg IV in the INSPIRE trial, analyzed according to the treatment that they were randomized to | All efficacy analyses | |
ITT1NRN | All nonrandomized patients who received at least 1 dose of study drug in COMMAND after receiving risankizumab 180 mg or 360 mg SC during Induction Period 2 in the INSPIRE trial or placebo during Induction Period 1 in the INSPIRE trial | All efficacy analyses | |
SA1RN | All randomized patients who received at least 1 dose of study drug, analyzed according to the treatment that they actually received | Safety analyses |
ITT = intention to treat; SA = safety analysis.
Sources: INSPIRE Clinical Study Report,17 COMMAND Clinical Study Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results
Patient Disposition
A total of 1,430 patients were screened and 977 were randomized in the INSPIRE trial (Table 12). The reasons for screening failure in 453 participants (31.7%) were not reported. Two patients randomized to risankizumab did not receive the study drug; 1 patient was unable to receive IV treatment and the other withdrew from the study before treatment. The ITT population included 650 patients in the risankizumab group and 325 in the placebo group. By the end of the study, discontinuation of the study drug was higher in the placebo group (8.3%) than the risankizumab group (2.0%). The most common primary reasons for withdrawal in the placebo group were AEs, withdrawal of consent, and lack of efficacy; in the risankizumab group, these were AEs, withdrawal of consent, and “other.” A similar number of patients discontinued for similar reasons in the 2 groups.
In the COMMAND trial, 754 patients were screened and 584 were randomized. Of the 584 randomized patients, 14 patients in the risankizumab 180 mg group, 9 in the risankizumab 360 mg group, and 13 in the placebo (risankizumab withdrawal) group had a clinical response before 24 weeks during the induction trial and were excluded from the ITT population; as the ITT population was defined as all randomized patients who received at least 1 dose of study drug after receiving IV risankizumab for only 1 period of 12 weeks in the induction trial. As such, 548 patients were included in the ITT population: risankizumab 180 mg = 179 patients, risankizumab 360 mg = 186 patients, placebo (risankizumab withdrawal) = 183 patients. A similar proportion of patients discontinued the study drug across the treatment groups, including 6.7% in the risankizumab 180 mg group, 11.3% in the risankizumab 360 mg group, and 9.8% in the placebo group. The most common reasons for discontinuation were lack of efficacy and withdrawal of consent in the risankizumab groups, and withdrawal of consent and “other” in the placebo group. A similar number of patients discontinued for similar reasons among the groups. A higher proportion of patients in the placebo group (42.6%) compared to the risankizumab 180 mg (19.0%) and risankizumab 360 mg (36.3%) groups required rescue treatment during the study.
Baseline Characteristics
The baseline characteristics outlined in Table 13 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results. Across both risankizumab and placebo treatment arms in the INSPIRE trial, baseline characteristics were generally well balanced. The total study population had highly refractory UC. This is shown by the proportion of patients who had extensive UC or pancolitis at baseline, in addition to a high proportion in whom ATs had previously failed (close to 51%). Specifically, these patients had prior failure of anti-TNFs, vedolizumab, or JAK inhibitors. Furthermore, this study population had a high proportion of patients with a baseline adapted Mayo score greater than 7, and most patients had a baseline endoscopic subscore of 3. Similarly, the maintenance (COMMAND) study population also had highly refractory UC with severe disease characteristics, indicated by the proportion of patients with extensive UC or pancolitis, mean disease duration, and a large proportion of patients in whom ATs had previously failed (close to 75%), including the failure of more than 2 ATs (around 25%) and failure of JAK inhibitors (around 15%).
Table 12: Summary of Patient Disposition From the INSPIRE and COMMAND Trials
Patient disposition | INSPIRE | COMMAND | |||
|---|---|---|---|---|---|
Risankizumab 1,200 mg IV (N = 650) | Placebo (N = 325) | Risankizumab 180 mg SC (N = 179) | Risankizumab 360 mg SC (N = 186) | Placebo (risankizumab withdrawal) (N = 183) | |
Screened, N | 1,430 | 754 | |||
Reason for screening failure, N (%) | NR | NR | |||
Randomized, N (%) | 652 | 325 | 193 | 195 | 196 |
Randomized and treated with study drug, N (%) | 650 | 325 | 179 | 186 | 183 |
Discontinued from study drug, N (%) | 13 (2.0) | 27 (8.3) | 12 (6.7) | 21 (11.3) | 18 (9.8) |
Reason for discontinuation, n (%) | |||||
Adverse event | 2 (0.3) | 12 (3.7) | 3 (1.7) | 2 (1.1) | 1 (0.5) |
Lost to follow-up | 0 | 0 | 0 | 1 (0.5) | 0 |
Withdrew consent | 4 (0.6) | 6 (1.8) | 3 (1.7) | 7 (3.8) | 5 (2.7) |
Lack of efficacy | 1 (0.2) | 5 (1.5) | 5 (2.8) | 8 (4.3) | 5 (2.7) |
COVID-19 infection | 1 (0.2) | 0 | 0 | 0 | 0 |
COVID-19 logistical restrictions | 1 (0.2) | 0 | 0 | 0 | 0 |
Geopolitical restrictions | 0 | 0 | 0 | 1 (0.5) | 1 (0.5) |
Other | 4 (0.6) | 4 (1.2) | 1 (0.6) | 2 (1.1) | 6 (3.3) |
Received rescue medication, N (%) | NA | NA | 24 (19.0) | 49 (26.3) | 78 (42.6) |
Discontinued from study, N (%) | 13 (2.0) | 27 (8.3) | 14 (7.8) | 23 (12.4) | 19 (10.4) |
Reason for discontinuation, N (%) | |||||
Adverse events | 2 (0.3) | 12 (3.7) | 4 (2.2) | 24 (1.1) | 1 (0.5) |
Withdrew consent | 4 (0.6) | 6 (1.8) | 4 (2.2) | 7 (3.8) | 7 (3.8) |
Lack of efficacy | 1 (0.2) | 6 (1.8) | 5 (2.8) | 10 (5.4) | 4 (2.2) |
COVID-19 infection | 1 (0.2) | 0 | 0 | 0 | 0 |
COVID-19 logistical restrictions | 1 (0.2) | 0 | 0 | 0 | 0 |
Geopolitical restrictions | 0 | 0 | 0 | 1 (0.5) | 1 (0.5) |
Lost to follow-up | 0 | 0 | 0 | 1 (0.5) | 1 (0.5) |
Other reason | 4 (0.6) | 3 (0.9) | 1 (0.6) | 2 (1.1) | 7 (3.8) |
ITT, N | 650 | 325 | 179 | 186 | 183 |
Safety, N | 651 | 324 | 193 | 195 | 196 |
ITT = intention-to-treat; NA = not applicable; NR = not reported; SC = subcutaneous.
Sources: INSPIRE Clinical Study Report,17 COMMAND Clinical Study Report,18 Louis et al.73 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 13: Summary of Baseline Characteristics From the INSPIRE and COMMAND Trials
Baseline characteristics | INSPIRE | COMMAND | |||
|---|---|---|---|---|---|
Risankizumab 1,200 mg IV (N = 650) | Placebo (N = 325) | Risankizumab 180 mg SC (N = 179) | Risankizumab 360 mg SC (N = 186) | Placebo (risankizumab withdrawal) (N = 183) | |
Sex, n (%) | |||||
Female | 265 (40.8) | 124 (38.2) | 74 (41.3) | 79 (42.5) | 82 (44.8) |
Male | 385 (59.2) | 201 (61.8) | 105 (58.7) | 107 (57.5) | 101 (55.2) |
Age (years), mean (SD) | 41.8 (13.5) | 42.8 (14.30) | 40.9 (14.7) | 42.5 (12.9) | 39.2 (14.2) |
BMI (kg/m2)a | |||||
Mean (SD) | 24.7 (5.3) | 24.9 (5.2) | 24.9 (5.4) | 24.2 (4.9) | 24.2 (5.3) |
Disease duration (years), mean (SD) | 7.7 (6.9) | 8.1 (7.0) | 8.5 (7.4) | 9.3 (7.1) | 8.2 (7.2) |
Location and extent of disease, n (%) | |||||
Left side | 313 (48.2) | 150 (46.2) | 84 (46.9) | 92 (49.5) | 85 (46.4) |
Extensive or pancolitis | 334 (51.4) | 174 (53.5) | 94 (52.5) | 94 (50.5) | 98 (53.6) |
Limited to rectum | 3 (0.5) | 1 (0.3) | 1 (0.6) | 0 | 0 |
Adapted Mayo score, mean (SD)b | 7.1 (1.2) | 7.1 (1.3) | 7.2 (1.2) | 7.0 (1.3) | 7.2 (1.2) |
Score ≤ 7, n (%) | 376 (57.9) | 190 (58.5) | 102 (57.0) | 109 (58.6) | 92 (50.3) |
Score > 7, n (%) | 273 (42.1) | 135 (41.5) | 77 (43.0) | 77 (41.4) | 91 (49.7) |
Endoscopic subscore, mean (SD) | 2.7 (0.5) | 2.7 (0.5) | 2.7 (0.4) | 2.7 (0.5) | 2.7 (0.5) |
Score = 2, n (%) | 208 (32.0) | 94 (28.9) | 47 (26.3) | 63 (33.9) | 52 (28.4) |
Score = 3, n (%) | 442 (68.0) | 231 (71.1) | 132 (73.7) | 123 (66.1) | 131 (71.6) |
Fecal calprotectin (mcg/g), median (IQR)c | 1,530.0 (592.0 to 3,196.0) | 1,624.0 (601.0 to 3,493.0) | 1,605.0 (611.0 to 3,180.0) | 1,568.0 (499.0 to 3,884.0) | 1,514.5 (729.0 to 2,910.0) |
Medication use, n (%) | |||||
Immunosuppressants | 108 (16.6) | 53 (16.3) | 35 (19.6) | 32 (17.2) | 37 (20.2) |
Aminosalicylates | 475 (73.1) | 238 (73.2) | 119 (66.5) | 135 (72.6) | 117 (63.9) |
Corticosteroids | 236 (36.3) | 112 (34.5) | 74 (41.3) | 59 (31.7) | 68 (37.2) |
Treatment response history, n (%) | |||||
AT-IR | 333 (51.2) | 170 (52.3) | 134 (74.9) | 139 (74.7) | 138 (75.4) |
Non–AT-IR | 317 (48.8) | 155 (47.7) | 45 (25.1) | 47 (25.3) | 45 (24.6) |
Number of times patient experienced an inadequate response to AT, n (%) | |||||
0 | 317 (48.8) | 155 (47.7) | 45 (25.1) | 47 (25.3) | 45 (24.6) |
1 | 153 (23.5) | 80 (24.6) | 52 (29.1) | 55 (29.6) | 62 (33.9) |
2 | 112 (17.2) | 55 (16.9) | 44 (24.6) | 37 (19.9) | 40 (21.9) |
> 2 | 68 (10.5) | 35 (10.8) | 38 (21.2) | 47 (25.3) | 36 (19.7) |
Prior failure to anti-TNF therapy, n (%) | 285 (43.8) | 142 (43.7) | 123 (68.7) | 128 (68.8) | 131 (71.6) |
Prior failure to vedolizumab, n (%) | 196 (30.2) | 97 (29.8) | 68 (38.0) | 84 (45.2) | 70 (38.3) |
Prior failure to JAK inhibitor, n (%) | 61 (9.4) | 40 (12.3) | 23 (12.8) | 29 (15.6) | 32 (17.5) |
AT = advanced therapy; AT-IR = inadequate response or intolerance to advanced therapy; BMI = body mass index; IQR = interquartile range; SC = subcutaneous; SD = standard deviation; TNF = tumour necrosis factor.
Notes: Where indicated, study participants with missing data for a given characteristic were excluded from the calculation of summary statistics.
aN = 647 in the risankizumab 1,200 mg IV arm; N = 177 in the risankizumab 180 mg SC arm.
bN = 649 in the risankizumab 1,200 mg IV arm.
cN = 602 in the risankizumab 1,200 mg IV arm; N = 302 in the placebo arm (INSPIRE); N = 151 in the risankizumab 180 mg SC arm; N = 166 in the risankizumab 360 mg SC arm; N = 162 in the placebo arm (COMMAND).
Sources: INSPIRE Clinical Study Report,17 COMMAND Clinical Study Report,18 Louis et al.73 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Exposure to Study Treatments
Exposure and adherence to study treatments in the INSPIRE (SA2 population) and COMMAND trials (SA1RN population) are summarized in Table 14. Treatment adherence was high and exposure to study treatments was similar between treatment arms in both trials.
Table 14: Summary of Patient Exposure From the INSPIRE and COMMAND Trials
Exposure | INSPIRE | COMMAND | |||
|---|---|---|---|---|---|
Risankizumab 1,200 mg IV (N = 650) | Placebo (N = 325) | Risankizumab 180 mg SC (N = 179) | Risankizumab 360 mg SC (N = 186) | Placebo (risankizumab withdrawal) (N = 183) | |
Duration (days), mean (SD) | 82.8 (8.3) | 80.7 (11.2) | 342.5 (97.5) | 317.4 (115.4) | 318.5 (98.7) |
Duration (days), median (range) | 84.0 (28 to 173) | 84.0 (28 to 98) | 390.0 (56 to 438) | 388.0 (56 to 399) | 373.5 (56 to 428) |
Duration intervals for induction therapy (days), n (%) | |||||
1 to 28 | 8 (1.2) | 10 (3.1) | NA | NA | NA |
29 to 56 | 4 (0.6) | 10 (3.1) | NA | NA | NA |
57 to 84 | 484 (74.3) | 230 (71.0) | NA | NA | NA |
85 to 112 | 153 (23.5) | 74 (22.8) | NA | NA | NA |
> 112 | 2 (0.3) | 0 | NA | NA | NA |
Duration intervals for maintenance therapy (days), n (%) | |||||
1 to 56 | NA | NA | 3 (1.6) | 5 (2.6) | 4 (2.0) |
57 to 112 | NA | NA | 10 (5.2) | 15 (7.7) | 3 (1.5) |
113 to 168 | NA | NA | 10 (5.2) | 20 (10.3) | 18 (9.2) |
169 to 224 | NA | NA | 9 (4.7) | 6 (3.1) | 13 (6.6) |
225 to 280 | NA | NA | 5 (2.6) | 10 (5.1) | 18 (9.2) |
281 to 336 | NA | NA | 2 (1.0) | 1 (0.5) | 17 (8.7) |
337 to 392 | NA | NA | 122 (63.2) | 120 (61.5) | 97 (49.5) |
> 392 | NA | NA | 32 (16.6) | 18 (9.2) | 26 (13.3) |
Treatment adherence (%)a | |||||
Mean (SD) | 99.3 (4.7) | 99.8 (2.6) | 99.7 (1.9) | 99.5 (3.0) | 99.5 (2.8) |
Median (range) | 100 (50.0 to 100) | 100 (66.7 to 100) | 100 (86 to 100) | 100 (71 to 100) | 100 (80 to 100) |
NA = not applicable; SC = subcutaneous; SD = standard deviation.
aTreatment compliance defined as 100% × (1 – (absolute value of (n of kits received – n of kits planned)) / n of kits planned during induction period).
Sources: INSPIRE Clinical Study Report,17 COMMAND Clinical Study Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Concomitant Medications and Co-interventions
In the INSPIRE trial, most patients (99.7% in both treatment groups) used at least 1 concomitant medication (Table 15). Concomitant medications were generally well balanced between treatment groups.
In the COMMAND trial, patients undergoing corticosteroid therapy were required to taper by week 8. Starting at week 16, patients in any treatment group could receive open-label risankizumab rescue therapy (i.e., 1 single dose of IV risankizumab followed by 360 mg administered subcutaneously every 8 weeks) in the event of loss of adequate clinical response. Most patients (> 98% in each treatment group) used at least 1 concomitant medication (Table 15).
Table 15: Summary of Concomitant Treatment From the INSPIRE and COMMAND Trials
Concomitant medication | INSPIRE | COMMAND | |||
|---|---|---|---|---|---|
Risankizumab 1,200 mg IV (N = 650) | Placebo (N = 325) | Risankizumab 180 mg SC (N = 179) | Risankizumab 360 mg SC (N = 186) | Placebo (risankizumab withdrawal) (N = 183) | |
Patients who received any concomitant medication, n (%) | 648 (99.7) | 324 (99.7) | 178 (99.4) | 184 (98.9) | 182 (99.5) |
Concomitant medications used by ≥ 10% of patients in any group, n (%) | |||||
Ascorbic acid, macrogol 3,350, potassium chloride, sodium ascorbate, sodium chloride, sodium sulphate | 89 (13.7) | 43 (13.2) | 26 (14.5) | 32 (17.2) | 26 (14.2) |
Azathioprine | 90 (13.8) | 47 (14.5) | 30 (16.8) | 24 (12.9) | 31 (16.9) |
Citric acid, magnesium oxide, sodium picosulfate | 72 (11.1) | 35 (10.8) | 21 (11.7) | 15 (8.1) | 11 (6.0) |
Cholecalciferol | 53 (8.2) | 27 (8.3) | 18 (10.1) | 16 (8.6) | 20 (10.9) |
Fentanyl | 84 (12.9) | 33 (10.2) | 16 (8.9) | 17 (9.1) | 22 (12.0) |
Lidocaine | 32 (4.9) | 20 (6.2) | 12 (6.7) | 16 (8.6) | 20 (10.9) |
Mesalazine | 463 (71.2) | 225 (69.2) | 109 (60.9) | 125 (67.2) | 114 (62.3) |
Midazolam | 144 (22.2) | 65 (20.0) | 29 (16.2) | 39 (21.0) | 43 (23.5) |
Paracetamol | 76 (11.7) | 46 (14.2) | 40 (22.3) | 51 (27.4) | 54 (29.5) |
Pethidine hydrochloride | 42 (6.5) | 22 (6.8) | 18 (10.1) | 14 (7.5) | 13 (7.1) |
Prednisolone | 61 (9.4) | 35 (10.8) | 18 (10.1) | 12 (6.5) | 20 (10.9) |
Prednisone | 118 (18.2) | 63 (19.4) | 39 (21.8) | 27 (14.5) | 33 (18.0) |
Propofol | 239 (36.8) | 110 (33.8) | 56 (31.3) | 57 (30.6) | 66 (36.1) |
Tozinameran | 50 (7.7) | 23 (7.1) | 30 (16.8) | 35 (18.8) | 26 (14.2) |
SC = subcutaneous.
Sources: INSPIRE Clinical Study Report,17 COMMAND Clinical Study Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Efficacy
A summary of the key efficacy results are presented in Table 16.
Table 16: Summary of Key Efficacy Results From Studies Included in the Systematic Review
End point | INSPIRE | COMMAND | |||
|---|---|---|---|---|---|
Risankizumab 1,200 mg IV (N = 650) | Placebo (N = 325) | Risankizumab 180 mg SC (N = 179) | Risankizumab 360 mg SC (N = 186) | Placebo (risankizumab withdrawal) (N = 183) | |
Week 12 | Week 52 | ||||
Clinical remission per adapted Mayo score | |||||
n (%) | 132 (20.3) | 20 (6.2) | 72 (40.2) | 70 (37.6) | 46 (25.1) |
RD (95% CI) vs. placebo | 14.0 (10.0 to 18.0) | NA | 16.3 (7.4 to 25.3) | 14.2 (5.3 to 23.2) | NA |
P value vs. placebo | < 0.00001a | NA | 0.0004a | 0.0019a | NA |
Clinical response per adapted Mayo score | |||||
n (%) | 418 (64.3) | 116 (35.7) | 122 (68.2) | 116 (62.3) | 95 (51.9) |
RD (95% CI) vs. placebo | 28.6 (22.3 to 34.8) | NA | 17.1 (7.5 to 26.6) | 11.5 (1.7 to 21.2) | NA |
P value vs. placebo | < 0.00001a | NA | 0.00045a | 0.02119a | NA |
Endoscopic improvement | |||||
n (%) | 237 (36.5) | 39 (12.1) | 91 (50.8) | 90 (48.3) | 58 (31.7) |
RD (95% CI) vs. placebo | 24.3 (19.3 to 29.4) | NA | 20.1 (10.6 to 29.6) | 17.4 (7.9 to 26.9) | NA |
P value vs. placebo | < 0.00001a | NA | 0.00003a | 0.00032a | NA |
HEMI | |||||
n (%) | 159 (24.5) | 25 (7.7) | 77 (42.8) | 79 (42.2) | 43 (23.5) |
RD (95% CI) vs. placebo | 16.6 (12.3 to 21.0) | NA | 20.2 (11.2 to 29.2) | 19.8 (10.8 to 28.8) | NA |
P value vs. placebo | < 0.00001a | NA | 0.00001a | 0.00001a | NA |
HEMR | |||||
n (%) | 41 (6.3) | 2 (0.6) | 23 (12.9) | 29 (15.6) | 18 (9.8) |
RD (95% CI) vs. placebo | 5.6 (3.5 to 7.7) | NA | 4.0 (−2.2 to 10.3) | 6.1 (−0.3 to 12.5) | NA |
P value vs. placebo | < 0.00001a | NA | 0.20616a | 0.06176a | NA |
Clinical remission with no corticosteroid use for 90 days | |||||
n (%) | NA | NA | 71 (39.6) | 69 (37.1) | 46 (25.1) |
RD (95% CI) vs. placebo (risankizumab withdrawal) | NA | NA | 15.8 (6.9 to 24.8) | 13.7 (4.8 to 22.7) | NA |
P value vs. placebo (risankizumab withdrawal) | NA | NA | 0.00052a | 0.00268a | NA |
Discontinuation of corticosteroid use in patients taking steroids at baseline | |||||
Number of patients contributing to the analysis | NA | NA | 74 | 59 | 68 |
n (%) | NA | NA | 48 (64.9) | 32 (54.2) | 25 (36.8) |
RD (95% CI) vs. placebo | NA | NA | 28.4 (14.0 to 42.8) | 20.7 (4.9 to 36.6) | NA |
P value vs. placebo | NA | NA | 0.0001 | 0.0105 | NA |
IBDQ | |||||
Number of patients contributing to the analysis | 619 | 310 | 168 | 168 | 172 |
Change from baseline, LS mean (95% CI) | 42.6 (39.72 to 45.57) | 24.3 (20.19 to 28.46) | 52.6 (44.93 to 60.20) | 50.3 (42.20 to 58.36) | 35.0 (27.15 to 42.92) |
LS mean difference (95% CI) vs. placebo | 18.3 (13.38 to 23.25) | NA | 17.5 (8.01 to 27.06) | 15.2 (5.18 to 25.31) | NA |
P value vs. placebo | < 0.00001a | NA | 0.00032a | 0.00308a | NA |
UC-related hospitalization | |||||
n (%) | 5 (0.8) | 18 (5.5) | NA | NA | NA |
RD (95% CI) vs. placebo | −4.8 (−7.3 to −2.2) | NA | NA | NA | NA |
P value vs. placebo | < 0.00001a | NA | NA | NA | NA |
UC-related hospitalization | |||||
Events per 100 person-years | NA | NA | 0.6 | 1.2 | 3.1 |
Incidence rate difference (95% CI) vs. placebo (risankizumab withdrawal) | NA | NA | −2.5 (−5.4 to 0.4) | −1.8 (−5.0 to 1.3) | NA |
P value vs. placebo (risankizumab withdrawal) | NA | NA | 0.09485a | 0.25307a | NA |
CI = confidence interval; HEMI = histologic, endoscopic, and mucosal improvement; HEMR = histologic, endoscopic, and mucosal remission; IBDQ = Inflammatory Bowel Disease Questionnaire; LS = least squares; NA = not applicable; RD = risk difference; SC = subcutaneous; UC = ulcerative colitis; vs. = versus.
aP value adjusted for multiple testing.
Sources: INSPIRE Clinical Study Report,17 COMMAND Clinical Study Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Clinical Remission per Adapted Mayo Score
The primary end point of the INSPIRE trial was clinical remission at 12 weeks. The risk difference was 14.0% (95% CI, 10.0% to 18.0%) compared to placebo, favouring risankizumab. Patients who did not experience clinical response to risankizumab 1,200 mg IV at week 12 (ITT2_P2 population) continued treatment with risankizumab up to week 24. At week 24, the proportion of patients who achieved clinical remission were risankizumab 180 mg SC: 9 of 71 (12.7%), risankizumab 360 mg SC: 11 of 70 (15.7%), risankizumab 1,200 mg IV: 6 of 68 (8.8%). For patients who received placebo during induction and were then rerandomized to receive risankizumab 1,200 mg IV during extended induction, 22% (38 of 173) achieved clinical remission at week 24.17
In the COMMAND trial, the risk differences versus placebo at 52 weeks were 16.3% (95% CI, 7.4% to 25.3%), favouring risankizumab 180 mg and 14.2% (5.3% to 23.2%), favouring risankizumab 360 mg.
Subgroup analyses in the INSPIRE and COMMAND studies showed similar results to the main analyses, except for a few notable exceptions. In the INSPIRE trial, patients aged 65 years or older, patients in North America, patients with baseline immunosuppressant use, patients with a history of more than 1 AT, and patients with AT-IR (with or without baseline steroid use) had a lower magnitude of effect. In the COMMAND trial, the effect estimates were consistent in direction.
In both the INSPIRE and COMMAND studies, sensitivity analyses using as-observed data, with FDA rules for calculating stool frequency and rectal bleeding subscores, and excluding patients impacted by geopolitical conflict had similar results as the primary analyses. The tipping-point analyses showed that the results remained statistically significant across a range of assumed values for patients with missing data in the placebo and risankizumab groups (including the most extreme case). In COMMAND, a supplemental analysis using the composite strategy (i.e., patients who discontinued treatment were classified as nonresponders) also had similar results as the primary analysis.
Clinical Response per Adapted Mayo Score
In the INSPIRE trial, the risk difference for clinical response was 28.6% (95% CI, 22.3% to 34.8%) compared to placebo, favouring risankizumab. In the COMMAND trial, the risk difference was 17.1% (95% CI, 7.5% to 26.6%) and 11.5% (95% CI, 1.7% to 21.2%) for risankizumab 180 mg SC and risankizumab 360 mg SC, respectively, compared to placebo (risankizumab withdrawal), favouring risankizumab. A supplemental analysis using the composite strategy (i.e., patients who discontinued treatment were classified as nonresponders) had similar results as the primary analysis. Similar results were reported for the subgroup of patients who received risankizumab 1,200 mg induction therapy in the INSPIRE trial (ITT1RN_B population).18
Among patients who did not experience clinical response to risankizumab 1,200 mg IV at week 12 (ITT2_P2 population), treatment with risankizumab up to week 24 demonstrated potential benefit for clinical response after Induction Period 2 (Table 17). At week 24, the proportion of patients who achieved clinical response per adapted Mayo score were comparable across all risankizumab treatment arms. Furthermore, for subjects who received placebo during induction and were then rerandomized to receive risankizumab 1,200 mg IV during extended induction, the proportion of subjects who achieved clinical response per adapted Mayo score at week 24 was comparable to that in the risankizumab 1,200 mg IV treatment arm at week 12 of the induction phase.17
Table 17: Clinical Response Per Adapted Mayo Score in Patients Who Entered Induction Period 2 in the INSPIRE Trial (ITT2_P2 Population)
Clinical response per adapted Mayo score | Week 24 | ||||
|---|---|---|---|---|---|
Risankizumab 180 mg SC | Risankizumab 360 mg SC | Risankizumab 1,200 mg IV | Placebo IV to risankizumab 1,200 mg IV | ||
Number of patients contributing to the analysis | 71 | 70 | 68 | 173 | |
n (%) | 40 (56.3) | 40 (57.1) | 34 (50.0) | 122 (70.5) | |
SC = subcutaneous.
Sources: INSPIRE Clinical Study Report.17
Endoscopic Improvement
In the INSPIRE trial, the risk difference for endoscopic improvement was 24.3% (95% CI, 19.3% to 29.4%) compared to placebo, favouring risankizumab. In the COMMAND trial, the risk difference was 20.1% (95% CI, 10.6% to 29.6%) and 17.4% (95% CI, 7.9% to 26.9%) for risankizumab 180 mg SC and risankizumab 360 mg SC, respectively, compared to placebo (risankizumab withdrawal), favouring risankizumab. A supplemental analysis using the composite strategy (i.e., patients who discontinued treatment were classified as nonresponders) had similar results as the primary analysis. Similar results were reported for the subgroup of patients who received risankizumab 1,200 mg induction therapy in the INSPIRE trial (ITT1RN_B population).18
Histologic, Endoscopic, and Mucosal Improvement
In the INSPIRE trial, the risk difference for HEMI was 16.6% (95% CI, 12.3% to 21.0%) compared to placebo, favouring risankizumab. In the COMMAND trial, the risk difference was 20.2% (95% CI, 11.2% to 29.2%) and 19.8% (95% CI, 10.8% to 28.8%) for risankizumab 180 mg SC and risankizumab 360 mg SC, respectively, compared to placebo (risankizumab withdrawal), favouring risankizumab. A supplemental analysis using the composite strategy (i.e., patients who discontinued treatment were classified as nonresponders) had similar results as the primary analysis. Similar results were reported for the subgroup of patients who received risankizumab 1,200 mg induction therapy in the INSPIRE trial (ITT1RN_B population).18
Histologic, Endoscopic, and Mucosal Remission
In the INSPIRE trial, the risk difference for HEMR was 5.6% (95% CI, 3.5% to 7.7%) compared to placebo, favouring risankizumab. In the COMMAND trial, the evidence was not sufficient to show a benefit for either arm, with a risk difference of 4.0% (95% CI, −2.2% to 10.3%) and 6.1% (95% CI, −0.3% to 12.5%) for risankizumab 180 mg SC and risankizumab 360 mg SC, respectively, compared to placebo (risankizumab withdrawal). A supplemental analysis using the composite strategy (i.e., patients who discontinued treatment were classified as nonresponders) had similar results as the primary analysis. Similar results were reported for the subgroup of patients who received risankizumab 1,200 mg induction therapy in the INSPIRE trial (ITT1RN_B population).18
Discontinuation of Corticosteroid Use in Patients Taking Steroids at Baseline
In the COMMAND trial, the risk difference was 28.4% (95% CI, 14.0% to 42.8%) and 20.7% (95% CI, 4.9% to 36.6%) for risankizumab 180 mg SC and risankizumab 360 mg SC, respectively, compared to placebo (risankizumab withdrawal), favouring risankizumab.
Inflammatory Bowel Disease Questionnaire
In the INSPIRE trial, the mean difference in IBDQ scores was 18.3 points (95% CI, 13.38 to 23.25 points) compared to placebo, favouring risankizumab. In the COMMAND trial, this end point was not formally tested due to earlier failure of the statistical hierarchy (the comparison for HEMR was not significant). The mean difference was 17.5 points (95% CI, 8.01 to 27.06 points) and 15.2 points (95% CI, 5.18 to 25.31 points) for risankizumab 180 mg SC and risankizumab 360 mg SC, respectively, compared to placebo (risankizumab withdrawal), favouring risankizumab. Similar results were reported for the subgroup of patients who received risankizumab 1,200 mg induction therapy in the INSPIRE trial (ITT1RN_B population).18
UC-Related Hospitalization
In the INSPIRE trial, the risk difference was −4.8% (95% CI, −7.3% to −2.2%) compared to placebo, favouring risankizumab. In the COMMAND trial, the evidence was insufficient to show a difference in the hospitalization rate. The incidence rate difference was −2.5 (95% CI, −5.4 to 0.4) per 100 patient-years and −1.8 (95% CI, −5.0 to 1.3) per 100 patient-years for risankizumab 180 mg SC and risankizumab 360 mg SC, respectively, compared to placebo (risankizumab withdrawal).
Harms
In both trials, the safety analysis sets included all randomized patients who received at least 1 dose of study drug, analyzed according to the treatment that they received. A summary of the overall TEAEs experienced by patients is provided in Table 18.
Table 18: Summary of Harms Results From the INSPIRE and COMMAND Trials
Adverse events | INSPIRE | COMMAND | |||
|---|---|---|---|---|---|
Risankizumab 1,200 mg IV (N = 651) | Placebo (N = 324) | Risankizumab 180 mg SC (N = 193) | Risankizumab 360 mg SC (N = 195) | Placebo (risankizumab withdrawal) (N = 196) | |
Week 12 | Week 52 | ||||
Most common adverse events (≥ 5% in any treatment group), n (%) | |||||
≥ 1 adverse event | 276 (42.4) | 160 (49.4) | 140 (72.5) | 138 (70.8) | 150 (76.5) |
COVID-19 | 31 (4.8) | 19 (5.9) | 17 (8.8) | 26 (13.3) | 23 (11.7) |
Anemia | 22 (3.4) | 20 (6.2) | 1 (0.5) | 1 (0.5) | 2 (1.0) |
Arthralgia | 21 (3.2) | 5 (1.5) | 11 (5.7) | 18 (9.2) | 9 (4.6) |
Headache | 19 (2.9) | 7 (2.2) | 9 (4.7) | 8 (4.1) | 15 (7.7) |
Nasopharyngitis | 18 (2.8) | 8 (2.5) | 18 (9.3) | 12 (6.2) | 16 (8.2) |
UC | 11 (1.7) | 33 (10.2) | 25 (13.0) | 27 (13.8) | 29 (14.8) |
Serious adverse events, n (%) | |||||
Patients with ≥ 1 SAE | 15 (2.3) | 33 (10.2) | 10 (5.2) | 10 (5.1) | 16 (8.2) |
Anemia | 2 (0.3) | 2 (0.6) | 0 | 0 | 0 |
Anal fistula | 0 | 2 (0.6) | 0 | 0 | 0 |
UC | 2 (0.3) | 16 (4.9) | 1 (0.5) | 1 (0.5) | 5 (2.6) |
Appendicitis | 0 | 1 (0.3) | 2 (1.0) | 0 | 0 |
Pulmonary embolism | 2 (0.3) | 1 (0.3) | 0 | 0 | 1 (0.5) |
Patients who stopped treatment due to adverse events, n (%) | |||||
Patients who stopped | 4 (0.6) | 12 (3.7) | 3 (1.6) | 5 (2.6) | 3 (1.5) |
Adverse events of special interest, n (%) | |||||
Hypersensitivity | 25 (3.8) | 7 (2.2) | 20 (10.4) | 10 (5.1) | 10 (5.1) |
Hepatic events | 10 (1.5) | 13 (4.0) | 3 (1.6) | 13 (6.7) | 1 (0.5) |
Serious infections | 4 (0.6) | 4 (1.2) | 2 (1.0) | 1 (0.5) | 4 (2.0) |
Injection-site reactions | 4 (0.6) | 4 (1.2) | 7 (3.6) | 5 (2.6) | 2 (1.0) |
Herpes zoster | 2 (0.3) | 0 | 2 (1.0) | 1 (0.5) | 3 (1.5) |
Malignancies (all types) | 0 | 2 (0.6) | 0 | 2 (1.0) | 1 (0.5) |
SAE = serious adverse event; SC = subcutaneous; UC = ulcerative colitis.
Note: Outcomes that occurred in at least 2 participants are reported.
Sources: INSPIRE Clinical Study Report,17 COMMAND Clinical Study Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Treatment-Emergent Adverse Events
In the INSPIRE trial, the number of patients with at least 1 TEAE were similar between the risankizumab (42.4%) and placebo (49.4%) groups. The most common TEAE in the risankizumab group was COVID-19 (4.8%), and the most common TEAE in the placebo group was UC (10.2%). In the COMMAND trial, the number of patients with at least 1 TEAE were similar between the risankizumab 180 mg (72.5%), risankizumab 360 mg (70.8%), and placebo (76.5%) groups.
Serious Adverse Events
In the INSPIRE trial, the proportion of patients with at least 1 SAE was higher in the placebo group (10.2%) than the risankizumab group (2.3%). The most common serious TEAEs in the risankizumab group were anemia, UC, and pulmonary embolism (2 patients [0.3%] each). In the placebo group, the most common serious TEAEs were UC (4.9%), anemia (0.6%), and anal fistula (0.6%). In the COMMAND trial, the proportion of patients with at least 1 SAE was similar across groups (5.2% with risankizumab 180 mg, 5.1% with risankizumab 360 mg, and 8.2% with placebo). In the risankizumab 180 mg group, the only serious TEAE occurring in more than 1 patient was appendicitis (2 patients, 1.0%). In the risankizumab 360 mg group, no serious TEAE occurred in more than 1 patient. In the placebo (risankizumab withdrawal) group, the most common serious TEAE was UC (2.6%); no other serious TEAE occurred in more than 1 patient.
Withdrawals Due to AEs
In the INSPIRE trial, a higher proportion of patients in the placebo group (3.7%) discontinued treatment due to a TEAE than in the risankizumab group (0.6%) (Table 18). In the COMMAND trial, the proportions of patients who discontinued treatment due to a TEAE were highest in patients taking risankizumab 360 mg (2.6%) compared to risankizumab 180 mg (1.6%) and placebo (1.5%) groups (Table 18).
Deaths
One patient in the risankizumab group died from COVID-19 in the INSPIRE trial (Table 18). In the COMMAND trial, 1 patient (in the risankizumab 360 mg group) died during the trial from colon adenocarcinoma (Table 18).
Adverse Events of Special Interest
In the INSPIRE trial, the most frequently occurring AEOSIs in both arms were hypersensitivity (risankizumab: 3.8%; placebo: 2.2%) and hepatic events (risankizumab: 1.5%; placebo: 4.0%). (Table 18). In the COMMAND trial, the risankizumab 180 mg group had the highest proportion of patients with hypersensitivity (10.4%) (Table 18). The risankizumab 360 mg group had the highest proportion of patients with hepatic events (6.7%).
Critical Appraisal
Internal Validity
INSPIRE Trial
Randomization in the INSPIRE trial involved 2:1 computer-generated, block randomization via interactive response technology. This is an adequate form of randomization and allocation concealment. Participants were stratified for major confounding factors (e.g., corticosteroid use, baseline Mayo score, and prior AT exposure). The baseline characteristics of the randomized groups were balanced, reflecting a proper randomization procedure. The trial was double-blind and placebo-controlled, with a detailed protocol including what concomitant medications were allowed and what changes in dosing were allowed after randomization. This decreases the probability of any deviations from the intended interventions during the trial. Concomitant medications were balanced across the treatment groups and not expected to impact the efficacy results. The primary and secondary efficacy end points in INSPIRE were tested using a graphical multiple-testing procedure to ensure control of family-wise type I error at a significance level alpha = 0.05 (2-sided), which was deemed adequate.
The sponsor considered the occurrence of appropriate ICEs that impact the interpretation of the effect of interest within the trial estimands. Data for patients who prematurely discontinued continued to be collected (i.e., treatment policy). Starting or escalating corticosteroids due to UC and surgery related to UC were considered a nonresponse for binary end points and return to baseline for continuous end points. This approach is considered adequate by regulatory authorities when the therapeutic intent is to induce remission in the short term.12,13
With regards to missing outcome data, on request by the review team the sponsor was unable to provide information about the occurrence of ICEs before study discontinuation nor the potential for sporadic missing data for reasons other than study discontinuation (e.g., missing visit). Therefore, it was not possible to perform a complete appraisal of the potential for bias due to missing data arising from reasons that are incompatible with the nonresponder imputation approach used by the sponsor for binary end points (and missing at random for continuous end points). There was a higher discontinuation rate in the placebo group (8.3%) than in the risankizumab group (2%), which can introduce attrition bias when nonresponder imputation is used. The reasons for study discontinuation (and possibly missing data) are incompatible with lack of efficacy. More than half of patients in the placebo group who withdrew did so because of lack of efficacy or AEs, suggesting that the assumption that withdrawals were due to nonresponse is acceptable. Sensitivity analyses were conducted for the primary end point, including a tipping-point analysis indicating that the statistical significance of the results would be maintained across a range of assumptions about the missing data. The outcome of UC-related hospitalization used an as-observed analysis; this approach considers that data are missing completely at random, which is unlikely and may introduce bias if there is an increased proportion of missing data (which could not be assessed).
To minimize detection bias, the trial used central endoscopic review with blinded assessors for objective outcomes, but it is important to note that this is only 1 component of the adapted Mayo score. For subjective outcomes, like patient-reported outcomes (e.g., other components of the adapted Mayo score, IBDQ) and safety outcomes, as the trial was double-blinded then it is unlikely that these results would be biased. Selective outcome and analysis reporting bias was marginalized by the publication of a prespecified protocol in ClinicalTrials.gov (NCT03398148). Overall, the INSPIRE trial is at low risk of bias for most end points, although uncertainty remains about potential attrition bias, which could not be fully assessed due to insufficient reporting.
The collection of information on harms included worsening UC as well as UC-related symptoms. Given that these are likely to be increased in the placebo group, the interpretation of the difference between groups is challenging. Further, it is implausible that there were significantly more SAEs reported in the placebo group than in the active intervention group, which would imply that the placebo effect is associated with SAEs.
With regards to the subgroup analyses, randomization in the INSPIRE trial was stratified by the presence of baseline corticosteroid use (yes, no), baseline adapted Mayo score (≤ 7, > 7), and a history of intolerance or inadequate AT response (0, 1, > 1 treatment). All other subgroup analyses may have broken randomization and therefore could have been affected by biases, including selection bias and confounding, due to unadjusted differences in baseline characteristics. The purpose of these analyses was to demonstrate consistency, and they cannot be used to draw credible conclusions about effect modification.
COMMAND Trial
The trial protocol was publicly available (NCT03398135) and the randomization and allocation concealment methods were similar to those in the INSPIRE trial, except that the trial used a 1:1:1 allocation ratio stratified by prior AT response and induction dose. The baseline characteristics suggested imbalances in some disease characteristics, although these did not appear to systematically favour any treatment group. Therefore, they are not expected to have an important impact on the results.
While the trial was double-blind, open-label rescue with risankizumab 1,200 mg IV was allowed after week 16. Unblinded therapy may have introduced performance bias in the collection of data related to harms. For efficacy end points, the need for rescue therapy resulted in an assumption that the patient was a nonresponder, which is appropriate and would not introduce bias. Concomitant medications were balanced across the treatment groups and not expected to impact the efficacy results. In the COMMAND trial, the testing began with testing the primary end point at the prespecified significance level of 0.025 (2-sided) for each risankizumab dose group compared to placebo. This was deemed an adequate approach.
The sponsor considered the occurrence of appropriate ICEs that impact the interpretation of the effect of interest within the trial estimands. Data for patients who prematurely discontinued continued to be collected (i.e., treatment policy). Starting or escalating corticosteroids related to UC, surgery related to UC, and rescue treatment were considered as nonresponse for binary end points and return to baseline for continuous end points. Given that the intent of COMMAND is to demonstrate maintenance of treatment efficacy in the long term, considering treatment discontinuations as nonresponse may be a preferred approach.13 Nonetheless, a supplemental analysis using the nonresponder imputation approach for treatment discontinuations showed similar results to the primary analysis on the primary and all secondary end points.
It was not possible to perform a comprehensive appraisal of the risk of bias due to missing outcome data as a result of incomplete reporting in the COMMAND study report. The concern for potential unreported missing data may be higher for the COMMAND trial compared to the INSPIRE trial as a result of the longer period of follow-up. There was a higher discontinuation rate in all groups compared to the INSPIRE trial, with 9.8% of patients in the placebo group discontinuing, and discontinuations in the risankizumab groups ranging from 7.8% to 12.4%. This can introduce attrition bias when nonresponder imputation is used and the reasons for study discontinuation are incompatible with lack of efficacy (if these patients’ data are considered missing). It is unclear how many patients were already imputed as nonresponders before study discontinuation (due to ICE, which appeared to be common; for example, 19% to 43% received rescue medication). As a result, the appraisal of potential for bias cannot be completed with certainty. On request from the review team, the sponsor was unable to provide this information. Sensitivity analyses were conducted for the primary end point, including a tipping-point analysis, which supported that the statistical significance of the results would be maintained across a range of assumptions about the missing data. The outcome of UC-related hospitalization used an as-observed analysis that considers data to be missing completely at random, which is unlikely to be reasonable and may introduce bias if there is an increased proportion of missing data (which could not be assessed).
In addition, the outcome “discontinuation of corticosteroid use at week 52 in patients taking steroids at baseline” was not assessed in the full cohort of randomized participants, and it is unclear whether the baseline characteristics in these individuals are similar between the groups. Having said that, the outcome “clinical remission with no corticosteroid use for 90 days” was assessed in the full cohort of randomized participants, and the results favoured risankizumab. Overall, bias is possible in the harms data due to open-label rescue treatment and in the corticosteroid discontinuation end point due to possible loss of randomization. The risk of bias due to missing outcome data could be increased because of attrition and lack of information to understand the proportion of data that may be missing. This concern was mitigated for the primary outcome via sensitivity analysis.
The collection of information on harms included worsening UC as well as UC-related symptoms. Given that these are likely to be increased in the placebo group, the interpretation of the difference between groups is challenging. Additionally, the comparison may not reflect a true placebo group due to the potential for risankizumab rescue therapy, which was received by 43% of patients in the placebo group.
In the COMMAND trial, randomization was stratified by history of inadequate AT response (yes, no), last risankizumab induction dose (IV 600 mg, 1,200 mg, or 1,800 mg), and clinical remission status (per local evaluation) at the last visit of the induction trial (yes, no). All other subgroup analyses may have broken randomization and therefore could have been affected by biases, including selection bias and confounding due to unadjusted differences in baseline characteristics. It is also important to note that the placebo group in the COMMAND trial received risankizumab during the induction trial (risankizumab withdrawal). As such, there could have been a carry-over effect due to an inadequate washout.
External Validity
The populations of both trials had a history of UC that was highly refractory to previous UC treatments, and the clinical experts consulted by the review team confirmed that patients in the newer UC trials are expected to have UC that is more refractory to treatment. In addition, the trials excluded patients with a history of IL-23 inhibitor use. With regard to co-interventions, in the INSPIRE trial, initiating or increasing the dose of comedications was prohibited, while in the COMMAND trial, patients undergoing corticosteroid therapy were required to taper by week 8. With regard to the comparator, no direct comparison to an active intervention was available (risankizumab was only compared to placebo). Furthermore, the main primary and secondary outcomes used the Mayo scoring system, which is not commonly used in clinical practice to inform decision-making. In the subgroup analyses, several subgroups showed less clinically significant results, including patients in North America. This may impact generalizability, although credible conclusions on effect modification cannot be drawn. These specific trial characteristics and results may impact generalizability and implementation in a real-world setting.
GRADE Summary of Findings and Certainty of the Evidence
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
INSPIRE trial:
Clinical remission
Clinical response
Endoscopic improvement
HEMI
HEMR
IBDQ
SAEs
COMMAND trial:
Clinical remission
Clinical response
Endoscopic improvement
HEMI
HEMR
Discontinuation of corticosteroid use in patients taking steroids at baseline
IBDQ
SAEs
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:106,107
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
For RCTs: Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. Literature-based effect size thresholds were available for clinical response and remission, endoscopic improvement, and SAEs.14 A literature-based MID was available for IBDQ, with a change of more than 30 points considered to be associated with clinical benefits and an improvement of 15 points or greater above placebo required among patients with IBD, including those with UC (Table 7). For discontinuation of corticosteroid use in patients taking steroids at baseline, between-group differences were identified by the clinical expert consulted by the review team as a threshold of clinical importance for these outcomes. The certainty for all other outcomes was based on the presence or absence in any effect, as a threshold of clinical importance could not be established.
Results of GRADE Assessments
Risankizumab Versus Placebo
Table 2 presents the GRADE summary of findings for risankizumab versus placebo for patients with UC (INSPIRE). Table 3 presents the GRADE summary of findings for risankizumab 180 mg SC versus placebo for patients with UC (COMMAND). Table 4 presents the GRADE summary of findings for risankizumab 360 mg SC versus placebo for patients with UC (COMMAND).
Long-Term Extension Studies
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
The sponsor submitted an abstract19 for the COMMAND open-label extension study. Because of the lack of access to complete information of methods and results, the summary of this study is not reported in this section.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
Objectives for the Summary of Indirect Evidence
No direct comparison exists between risankizumab and other publicly reimbursed ATs or those recommended for reimbursement in Canada for moderately to severely active UC. In the absence of head-to-head RCTs, an NMA was used to compare the efficacy and safety of risankizumab to other publicly reimbursed ATs or those recommended for reimbursement in Canada for moderately to severely active UC: adalimumab, infliximab, golimumab, vedolizumab, tofacitinib, ustekinumab, ozanimod, upadacitinib, mirikizumab, and etrasimod.
Specifically, the objectives were the following:
to determine the comparative efficacy of risankizumab versus relevant AT comparators separately, as induction and maintenance therapies for adults with moderately to severely active UC, and also separately for patients with non–AT-IR and AT-IR, per the risankizumab approved indication
to determine the comparative safety of risankizumab versus relevant AT comparators separately, as induction and maintenance therapies for adults with moderately to severely active UC, in the combined sample of overall safety populations.
Description of Indirect Comparisons
Study Selection Methods
A systematic review was conducted as per standardized guidance from Cochrane, the Centre for Reviews and Dissemination, and the National Institute for Health and Care Excellence (NICE).108,109 A search for English-language RCTs was conducted in MEDLINE, Embase, and Cochrane Library (including Cochrane Database of Systematic Reviews, Cochrane Central Register of Controlled Trials, Database of Abstracts of Reviews of Effects, and health technology assessment database) reporting the clinical efficacy or safety of ATs in adult patients with moderately to severely active UC. Searches were from database inception to June 27, 2023. Search strategies included search terms (free text) and controlled vocabulary for UC and the interventions. Search terms from previously published systematic reviews and NICE guidance were used to comprehensively capture RCT search terms.110
In addition to the bibliographic database searches, grey literature searches included relevant conference proceedings (Digestive Disease Week, Crohn’s & Colitis Congress, ISPOR–The Professional Society for Health Economics and Outcomes Research, United European Gastroenterology Week, American College of Gastroenterology, ECCO) for the last 5 years and searching trial registries keyword searches of the annual proceedings of scientific meetings and clinical trial registers (ClinicalTrials.gov, Health Canada’s Clinical Trial Database, EU Clinical Trials Register, International Clinical Trials Registry Platform). Last, the following websites of the regulatory and health technology assessment authorities in UK (England [NICE] and Scotland [Scottish Medicines Consortium]), Canada (CDA-AMC), France (Haute Autorité de Santé), Australia (Pharmaceutical Benefits Advisory Committee), and Germany (Federal Joint Committee [Gemeinsamer Bundesausschuss]) were searched for additional studies.
Finally, the bibliographies of systematic reviews and meta-analyses identified through database searches were used to identify key studies. Bibliographies from selected key studies were also reviewed to ensure literature saturation.
Title and abstract screening were conducted by 2 blinded, independent researchers in parallel using the predefined population, intervention, comparators, outcomes, and study design (PICOS) criteria presented in Table 19. The study selection was performed by 2 independent reviewers and based on a 2-step approach according to a predefined set of eligibility criteria: screening of abstracts and titles, followed by in-depth review of full-text articles. Each reviewer independently assessed the studies for inclusion according to the criteria, structured using the PICOS framework. Discrepancies between researchers were resolved by a third independent researcher. Data from included studies were extracted into a predefined Excel template by a single analyst and all results were 100% quality checked.
The risk of bias of individual full-text studies included in the systematic review was assessed using the Cochrane Collaboration risk-of-bias tool for RCTs.111 The risk-of-bias assessment was performed independently by 2 reviewers, and any discrepancies were resolved through discussions to reach a consensus.
Table 19: Study Selection Criteria and Methods for ITCs Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Adults (≥ 16 years) with moderately to severely activea UC who have experienced inadequate response, loss of response, or were intolerant to conventional therapy or AT (e.g., biologic treatment, JAK inhibitor, and/or S1P receptor modulator) |
Intervention | Risankizumab, adalimumab, golimumab, infliximab, vedolizumab, ustekinumab, tofacitinib, upadacitinib, ozanimod, mirikizumab, etrasimod, and filgotiniba |
Comparator | Head-to-head comparisons and/or placebo-controlled |
Outcome | Reported relevant efficacy and/or safety outcomes after 6 to 12 weeks of induction treatment or after 40 to 54 weeks of maintenance treatment: Efficacy:
Safety:
|
Study designs | Phases III and higher randomized and double-blind studies (only outcomes during randomized, double-blind phases were assessed) |
Publication characteristics |
|
Exclusion criteria |
|
Databases searched |
|
Selection process | The study selection was performed by 2 independent reviewers and based on a 2-step approach according to a predefined set of eligibility criteria: screening of abstracts and titles; in-depth review of full-text articles. |
Data extraction process | Data from included studies were extracted into a predefined Excel template by a single analyst, and all results were 100% quality checked by a research associate. |
Risk-of-bias assessment | Cochrane Collaboration risk-of-bias tool for RCTs. Assessment was performed independently by 2 reviewers, and any discrepancies were resolved through discussions to reach a consensus. |
AE = adverse event; AT = advanced therapy; EU = European Union; RCT = randomized controlled trial; S1P = sphingosine 1-phosphate.
aWhile filgotinib was included in the global systematic review, it was excluded from the feasibility assessment, as it is not approved in Canada.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
ITC Design
ITC Analysis Methods
Feasibility Assessment
Network plots were created to demonstrate the network connectivity of all included induction and maintenance RCTs. Then, relevant study and patient characteristics (e.g., age, sex, weight, duration and extent of disease, baseline Mayo scores, and concurrent medications for UC) were reviewed across the included induction and maintenance RCTs to get a sense of their comparability and identify potential sources of cross-RCT heterogeneity. The potential effect modifiers were identified based on clinical opinion. The mean baseline (placebo) effects across the included induction and maintenance RCTs were also assessed by performing a meta-analysis of all placebo arms included in each network.
Model Specifications
For each feasible network, NMAs were conducted in a generalized linear model framework using Bayesian Markov chain Monte Carlo simulations and 3 chains with 100,000 runs each. The burn-in period was set to half of the convergence sequence, until convergence (minimum set size of 25,000).112-114 Convergence was assessed with the Brooks-Gelman-Rubin method using the potential scale reduction factor.
Per NICE Decision Support Unit Technical Support Document 2, all binary response outcomes were modelled with a binomial likelihood and logit link function.112,113 All posterior distributions for ORs were summarized by their medians and 95% CrIs.
By default, RCT-specific baselines were modelled as independent, such that an unrelated model parameter was specified for each one. However, in networks with 1 or more placebo arm(s) having a value of 0 (i.e., no events), an exchangeable baseline assumption with a half-normal (0, 0.322) prior for heterogeneity was used to aid parameter estimation and numerical stability or convergence.113
For all networks, both fixed-effects and random-effects models were tested.
Priors
Vague or flat prior distributions were given to the parameters to be estimated by default.112,113 For parameters assumed to be specified on a continuous scale, namely, the relative treatment effects (d), RCT-specific baselines (mu), and baseline adjustment regression term (beta) (for models with baseline risk adjustment), a normal (0, 1002) prior distribution was used. For the between-study standard deviation (SD) (for random-effects models), a uniform (0, 2) prior distribution was used. For the between-study heterogeneity SD (or sigma) associated with the random-effects model, when most (≥ 50%) interventions in a network are informed by a single study, a half-normal (0, 0.322) prior distribution, which gives a low prior weight to unfeasibly large SDs on the logit scale, was used.113,115
Baseline Risk Adjustment
For each fixed-effects and random-effects model tested, a baseline risk-adjusted version was also tested that adjusted for differences in mean placebo effects across studies using the code provided in NICE Decision Support Unit Technical Support Document 3.116 A common regression term (beta) was assumed for all adjustments (i.e., the relationship between the placebo response and the active treatment response was assumed not to depend on treatment). The model with the baseline risk covariate was selected if, because of its inclusion, the median posterior SD (for random-effects models) decreased and the 95% CrI of the regression term beta excluded 0.113
Model Fit
Models’ global fits were assessed and compared using their overall posterior mean residual deviance and deviance information criteria.112,113 All else being equal between fixed-effects and random-effects models, the random-effects model was selected to account for the expected heterogeneity in outcomes, study design, and study populations across included RCTs.
Heterogeneity
The relevant study and patient characteristics were compared (age, gender, weight, duration of disease, extent of disease, baseline Full Mayo score and Adapted Mayo score, and concurrent medications for UC). Any disparities were evaluated in sensitivity analyses.
The statistical assessment of homogeneity was based on the I2 statistic, derived from a direct head-to-head meta-analysis of the treatment comparisons in each network that were evaluated in more than 1 study.117
All comparisons with an I2 higher than 50% were investigated. Possible sources of heterogeneity were reviewed and, if applicable, studies introducing heterogeneity to the model were excluded via a sensitivity analysis where possible.
Consistency
To verify the consistency of the direct and indirect comparisons, when available, a node-splitting approach was considered. Whenever applicable, a sensitivity analysis was performed based on the inconsistency assessment to determine the impact of treatment comparisons with significant inconsistency between the direct and indirect evidence.
A list of all sensitivity analyses is provided in Table 20.
Included Treatments and Nodes
The interventions of interest for the NMA included ATs publicly reimbursed or recommended for reimbursement in Canada for moderately to severely active UC (adalimumab, golimumab, infliximab, vedolizumab, ustekinumab, tofacitinib, upadacitinib, ozanimod, mirikizumab, etrasimod), as well as risankizumab (Table 20). Of note, although filgotinib was included in the systematic review, it was excluded from the NMA results, as it is not approved in Canada. As previously mentioned, although not publicly reimbursed yet, etrasimod has been recommended by CDA-AMC and is currently subject to negotiations with the pCPA.48 Only RCTs identified by the clinical systematic review that reported useful relative efficacy and safety estimates for the licensed dose(s) were included. Where the drug licence allows for dose escalation during the maintenance phase, both the low and high doses were included if they had been assessed in the RCTs. In RCTs that assessed both licensed and unlicensed doses, arms using unlicensed doses were excluded from the networks. Of note, different doses of treatment were treated as separate treatment nodes in the NMA networks. Last, placebo was included as a common comparator in the NMA.
Following a feasibility assessment, the NMA was deemed feasible for the following efficacy and safety outcomes:
Efficacy (both induction and maintenance phases, both non–AT-IR and AT-IR populations):
Clinical remission
Clinical response
Endoscopic improvement
Safety (both induction and maintenance phases, overall population):
% of AEs
% of SAEs
% of serious infections
TT Versus Rerandomized Responder Design
Two maintenance study designs were employed across the UC RCTs: TT and rerandomized responder (RR). In TT studies, patients were randomized to treatment or placebo at baseline, and outcomes were measured at the end of an induction phase and again at the end of a maintenance phase. In RR studies (e.g., risankizumab studies), patients were randomized to induction treatment or placebo at baseline, with outcomes measured at the end of the induction phase; induction responders were then rerandomized to maintenance treatment or placebo, with outcomes measured at the end of the maintenance phase strictly among induction responders.
Because these 2 study designs are incompatible in a standard NMA, the sponsor re-calculated data from the TT RCTs to mimic a RR design to conduct the NMA.
Table 20: Indirect Comparison Analysis Methods
Methods | Description |
|---|---|
Analysis methods | An NMA was performed using a Bayesian framework. Two models were tested: fixed-effects and random-effects. |
Priors |
|
Assessment of model fit | Model fit was assessed with:
|
Assessment of consistency | Node-splitting was used to assess consistency. |
Assessment of convergence | The overall PSRF was verified to be under a threshold of 1.05. |
Outcomes |
|
Follow-up time points |
|
Construction of nodes | Each treatment and dose were its own node. All placebo groups were combined in 1 node. |
Planned sensitivity analyses | Induction: 1. Exclusion of predominant Asian RCTs (clinical remission, non–AT‑IR and AT‑IR) 2. Heterogeneity in the duration of disease: exclusion of Jiang et al. (2015) and NCT01551290 (clinical remission, non–AT‑IR) 3. Meta-regression of corticosteroids (clinical remission, non–AT-IR) 4. Statistical heterogeneity defined as I2 > 50% (placebo vs. vedolizumab): exclusion of NCT02039505 (clinical response, AT-IR) 5. Statistical heterogeneity defined as I2 > 50% (placebo vs. upadacitinib): exclusion of U-ACHIEVE Study 2 (all AEs, overall population) 6. Statistical heterogeneity defined as I2 > 50% (placebo vs. vedolizumab): exclusion of NCT02039505 (SAEs, overall population) Maintenance: 7. Exclusion of predominant Asian RCTs (clinical remission, non–AT-IR and AT‑IR) 8. Meta-regression of corticosteroids (clinical remission, non–AT‑IR) 9. Statistical heterogeneity defined as I2 > 50% (placebo vs. golimumab): exclusion of PURSUIT-J (clinical remission, non–AT‑IR) 10. Statistical heterogeneity defined as I2 > 50% (placebo vs. golimumab): exclusion of PURSUIT-J (endoscopic improvement, non–AT-IR) 11. Statistical heterogeneity defined as I2 > 50% (placebo vs. golimumab): exclusion of PURSUIT-J (all AEs, overall population) 12. Statistical heterogeneity defined as I2 > 50% (placebo vs. golimumab): exclusion of PURSUIT-J (serious AEs, overall population) |
Subgroup analysis |
|
Methods for pairwise meta-analysis |
|
AE = adverse event; AT = advanced therapy; AT-IR = inadequate response or intolerance to advanced therapy; DIC = deviance information criteria; NMA = network meta-analysis; PSRF = potential scale reduction factor; RCT = randomized controlled trial; SAE = serious adverse event; SD = standard deviation; vs. = versus.
Note: Not all the planned sensitivity analyses were possible due to lack of data.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results of the ITC
Summary of Included Studies
Thirty-nine studies (with 118 reports) with potential for NMA inclusion were selected for data extraction. Per the NMA PICOS criteria, all 21 induction RCTs randomized patients to relevant induction treatment(s) versus placebo for 6 to 12 weeks in a double-blind manner. Likewise, all 16 maintenance RCTs randomized patients to relevant maintenance treatment(s) versus placebo (except for SERENE-C118) for 40 to 54 weeks in a double-blind manner. A total of 28 original RCTs reported by 77 records were included in the NMA, 21 in the induction phase and 16 in the maintenance phase. Table 21 presents an overview of the included studies.
Induction
A network plot of the included induction RCTs is shown in Figure 1. The network is star-shaped, in that all treatments are anchored on placebo as the common comparator. The network plots for individual outcomes differed slightly, as not all outcomes were reported by all the included trials.
Figure 1: Induction Network Plot
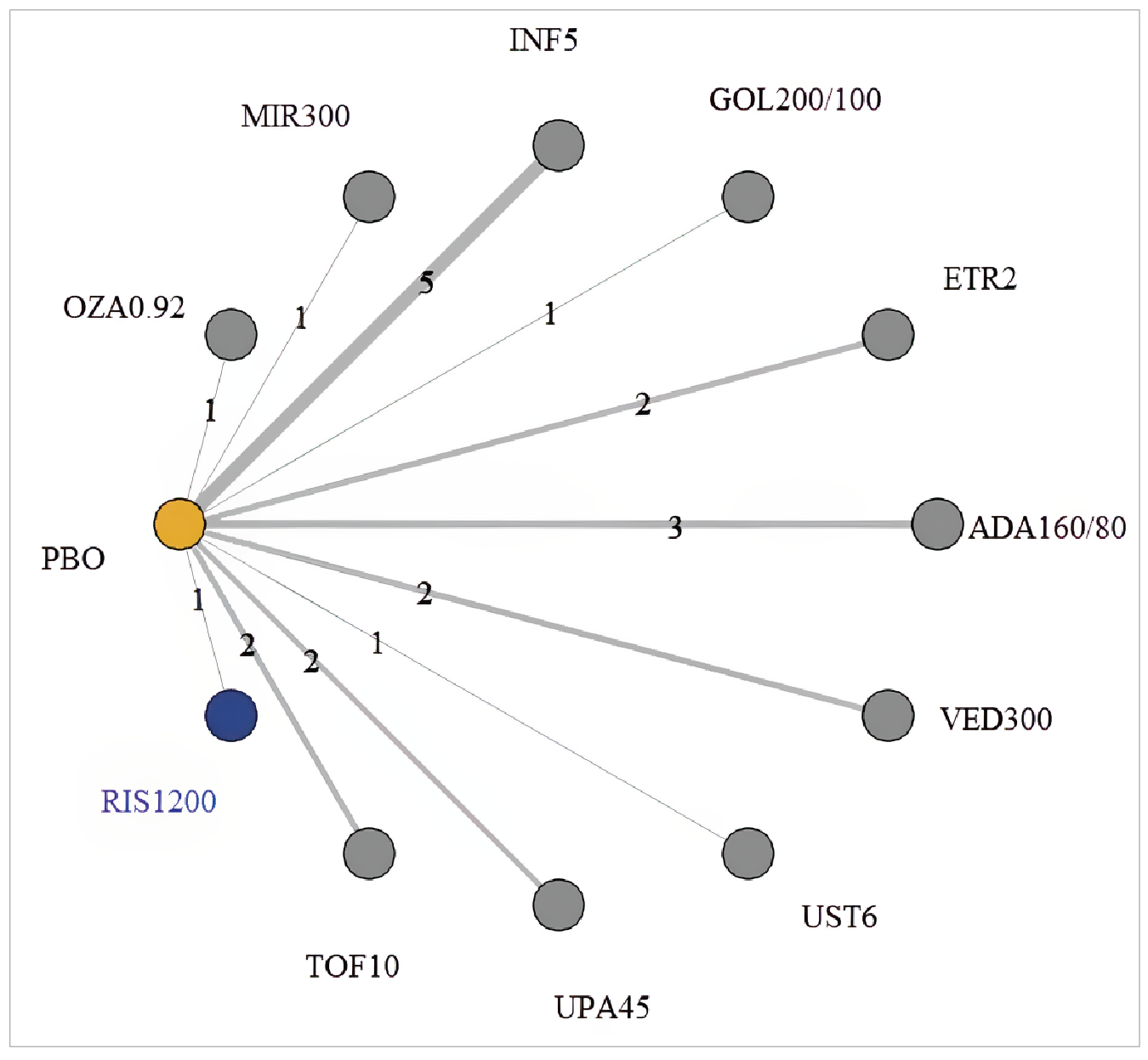
ADA = adalimumab; ETR = etrasimod; GOL = golimumab; INF = infliximab; MIR = mirikizumab; OZA = ozanimod; PBO = placebo; RIS = risankizumab; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab; VED = vedolizumab.
Notes: The nodes are weighted based on the number of participants. The lines are weighted based on the number of studies (numbers) with a direct comparison between treatments.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
The key findings from the feasibility assessment, along with the list of resulting sensitivity analyses, were as follows:
NMA connectivity was feasible for both non–AT-IR and AT-IR populations in induction studies.
A sensitivity analysis excluding predominantly Asian RCTs was planned to assess the impact on the clinical response of risankizumab relative to other treatments.
Due to the limited number of studies, no relevant sensitivity analyses based on outcome definitions could be performed. However, the sponsor considered the outcome definitions across studies to be sufficiently similar for inclusion in the NMA.
Table 22 outlines the key findings of the baseline characteristics’ heterogeneity and the approach taken to address them. The sponsor considered that baseline patient characteristics were comparable across the induction RCTs. However, some heterogeneity was observed.
No other sensitivity analyses were deemed necessary by the sponsor based on the feasibility assessment process.
Additionally, there are sources of heterogeneity that extend beyond the patient baseline characteristics (e.g., differences in the studies’ inclusion or exclusion criteria, outcome definitions, and time points). For example, the included studies used varying definitions of clinical remission and response (e.g., central endoscopy versus local investigator readings), durations of follow-up for outcome assessment (e.g., 6 to 12 weeks), differing definition of AT-IR, number and type of patients with AT were previously exposed differed (as the commercially available options changed over time), differences in how disease severity inclusion criteria was defined across the trials, differences in disease duration (e.g., ranging from 3.7 to 9.1 years), variation in extent of disease (e.g., from 32 to 80% with extensive colitis or pancolitis), heterogeneity in concurrent corticosteroid use (particularly for the non–AT-IR population), and wide variation in placebo risk.
Table 21: Overview of the RCTs Included In the Clinical Systematic Review
Study | Phase | UC severity (scores) | Bio-experience | Asian study population | Induction phase | Maintenance phase | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Duration (weeks) | Total, N | Included regimen(s) (+ placebo) | RCT design | Induction treatment(s) | Induction status | Duration (weeks) | Total N | Included regimen(s) (+ placebo) | |||||
ACT-1 (NCT00036439)82 | III | FMS 6 to 12 EMS ≥ 2 | Naive | No | 8 | 364 | INF5 | TT | INF10 INF5 placebo | All | 46 | 364 | INF10 INF5 |
ACT-2 (NCT00096655) | III | FMS 6 to 12 EMS ≥ 2 | Naive | No | 8 | 364 | INF5 | Excluded: Duration < 40 weeks | |||||
ELEVATE UC 12 (NCT03996369) | III | AMS 4 to 9 EMS ≥ 2 RBS ≥ 1 | Mixed | No | 12 | 354 | ETR2 | No maintenance | |||||
ELEVATE UC 52 (NCT03945188) | III | AMS 4 to 9 EMS ≥ 2 RBS ≥ 1 | Mixed | No | 12 | 433 | ETR2 | TT | ETR2 placebo | All | 40 | 433 | ETR2 |
GEMINI 1 (NCT00783718) | III | FMS 6 to 12 EMS ≥ 2 | Mixed | No | 6 | 374 | VED300 | RR | VED300 | FM responseb | 46 | 373 | VED300Q8W VED300Q4W |
Japic CTI‑060298 | III | FMS 6 to 12 EMS ≥ 2 | Naive | Yes | 8 | 208 | INF5 | Excluded: Duration < 40 weeks | |||||
Jiang et al. (2015)130 | NR | FMS 6 to 12 EMS ≥ 2 | Naive | Yes | 8 | 123 | INF5 (INF3.5 excluded) | Excluded: Duration < 40 weeks | |||||
LUCENT-1 (NCT03518086) | III | AMS 4 to 9 EMS ≥ 2 | Mixed | No | 12 | 1,281 | MIR300 | Maintenance in LUCENT-2 | |||||
III | AMS 4 to 9 EMS ≥ 2 | Mixed | No | Induction in LUCENT-1 | RR | MIR300 | AM responsea | 40 | 644 | MIR200 | |||
M10 to 447 (NCT00853099)141 | II/III | FMS 6 to 12 EMS ≥ 2 | Naive | Yes | 8 | 274 | ADA160/80 (ADA80/40 excluded) | TT | ADA160/80 ADA80/40 placebo | All | 44 | 274 | ADA40Q2W (ADA160/80 and ADA80/40 combined) |
M16 to 067 (NCT03398148) Substudy 2 Induction Period 1142 | III | AMS 5 to 9 EMS ≥ 2 | Mixed | No | 12 | 977 | RIS1200 | Maintenance in M16 to 066 | |||||
M16 to 066 (NCT03398135) Substudy 1143 | III | AMS 5 to 9 EMS ≥ 2 | Mixed | No | Induction in M16 to 067 | RR | RIS1200 RIS600 RIS1800 | AM responsea | 52 | 548 | RIS180 RIS360 | ||
NCT01551290144 | III | FMS 6 to12 EMS ≥ 2 | Naive | Yes | 8 | 99 | INF5 | Excluded: Duration < 40 weeks | |||||
III | FMS 6 to 12 EMS ≥ 2 | Mixed | Yes | 10 | 246 | VED300 | RR | VED300 | FM responseb | 50 | 83 | VED300Q8W | |
OCTAVE 1 (NCT01465763 | III | FMS 6 to 12 EMS ≥ 2 RBS ≥ 1 | Mixed | No | 8 | 598 | TOF10 | Maintenance in OCTAVE Sustain | |||||
OCTAVE 2 (NCT01458951 | III | FMS 6 to 12 EMS ≥ 2 RBS ≥ 1 | Mixed | No | 8 | 541 | TOF10 | Maintenance in OCTAVE Sustain | |||||
OCTAVE Sustain (NCT01458574) | III | FMS 6 to 12 EMS ≥ 2 RBS ≥ 1 | Mixed | No | Induction in OCTAVE 1 and OCTAVE 2 | RR | TOF10 TOF15 placebo | FM responseb | 52 | 593 | TOF10 TOF5 | ||
PURSUIT-J (NCT01863771)162 | III | FMS 6 to 12 EMS ≥ 2 | Naive | Yes | Excluded: Open-label | RR | GOL200/100 | FM responseb | 54 | 63 | GOL100 | ||
PURSUIT-M (NCT00488631)163 | III | FMS 6 to 12 EMS ≥ 2 | Naive | No | Induction in PURSUIT-SC | RR | GOL400/200 GOL200/100 | FM responseb | 54 | 464 | GOL100 GOL50 | ||
PURSUIT-SC (NCT00487539)164 | III | FMS 6 to 12 EMS ≥ 2 | Naive | No | 6 | 774 | GOL200/100 (GOL400/200 excluded) | Maintenance in PURSUIT-M | |||||
SERENE-UC (NCT02065622 | III | FMS 6 to 12 EMS ≥ 2 | Mixed | No | Excluded: Intervention ADA HIR | RR | ADA HIR ADA160/80 | All (efficacy evaluated in FM responders) | 44 | 371 | ADA40Q2W ADA40QW (ADA TDM excluded; no placebo) | ||
TRUE NORTH (NCT02435992) | III | FMS 6 to 12 | Mixed | No | 10 | 645 | OZA0.92 | RR | OZA0.92 | FM responseb | 42 | 457 | OZA0.92 |
U-ACCOMPLISH (Study M14 to 675; NCT03653026)173 | III | AMS 5 to 9 EMS ≥ 2 | Mixed | No | 8 | 522 | UPA45 | Induction in U-ACHIEVE Study 3 | |||||
U-ACHIEVE Study 2 and 3 (Study M14 to 234; NCT02819635)174 | III | AMS 5 to 9 EMS ≥ 2 | Mixed | No | 8 | 474 | UPA45 | RR | UPA45 | AM responsea | 52 | 451 | UPA30 UPA15 |
ULTRA-1 (NCT00385736)83 | III | FMS 6 to 12 EMS ≥ 2 | Naive | No | 8 | 390 | ADA160/80 (ADA80/40 excluded) | No maintenance | |||||
ULTRA-2 (NCT00408629) | III | FMS 6 to 12 EMS ≥ 2 | Mixed | No | 8 | 518 | ADA160/80 | TT | ADA160/80 placebo | All | 44 | 518 | ADA40Q2W |
UNIFI (NCT02407236) | III | FMS 6 to 12 EMS ≥ 2 | Mixed | No | 8 | 961 | UST6 (UST130 excluded) | RR | UST130 UST6 | FM responseb | 44 | 523 | UST90Q12W UST90Q8W |
VISIBLE 1 (NCT02611830)186 | III | FMS 6 to 12 EMS ≥ 2 | Mixed | No | Excluded: Open-label | RR | VED300 | FM responseb | 46 | 216 | VED300Q8W VED108Q2W (SC) | ||
ADA = adalimumab; AMS = adapted Mayo score; EMS = endoscopic Mayo subscore; ETR = etrasimod; FMS = full Mayo score; GOL = golimumab; HIR = higher induction dosing regimen; INF = infliximab; MIR = mirikizumab; NR = not reported; OZA = ozanimod; RBS = rectal bleeding subscore; RCT = randomized clinical trial; RIS = risankizumab; RR = rerandomized responder; TDM = therapeutic drug monitoring; TOF = tofacitinib; TT = treat through; UC = ulcerative colitis; UPA = upadacitinib; UST = ustekinumab; VED = vedolizumab; X = applicable.
aAM response = decrease in AMS ≥ 2 points and ≥ 30% from baseline, and a decrease in RBS ≥ 1 or an absolute RBS ≤ 1.
bFM response = decrease in FMS ≥ 3 points and ≥ 30% from baseline, and a decrease in RBS ≥ 1 or an absolute RBS ≤ 1.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 22: Key Findings from the Baseline Patient Characteristics Assessment (Induction Population)
Patient characteristics at baseline | Heterogeneity assessment | Approach to addressing Heterogeneity | ||
|---|---|---|---|---|
Non–AT-IR | AT-IR | Non–AT-IR | AT-IR | |
Age | Minimal | NA | ||
Gender | Minimal | NA | ||
Weight | Notable heterogeneity with lower values in Asian RCTs | NA Although most treatments are not weight dependent, and therefore testing would not be necessary, SA excluding predominantly Asian RCTs addressed this concern. | ||
Duration of disease | Notable heterogeneity with lower values in Jiang et al. (2015) (INF) and NCT01551290 (INF) | Minor | SA excluding Jiang et al. (2015) and NCT01551290 RCTs. | NA |
Extent of disease | Notable heterogeneity with higher values in Asian RCTs | Notable heterogeneity with lower values in ELEVATE UC 12/52 (ETR), LUCENT-1 (MIR), and TRUE NORTH (OZA) | NA SA excluding higher values was considered. However, the SA excluding predominantly Asian RCTs addressed this concern. | NA SA excluding lower values was considered. However, given the star-shaped structure of the network, this analysis would exclude treatments from those studies (ETR, MIR, and OZA) and would not affect the results. |
Baseline FMS | Minimal | NA | ||
Baseline AMS | Minimal | NA | ||
Concurrent immunomodulators | Notable heterogeneity with near-0%, largely due to study inclusion criteria for TRUE NORTH (OZA), U-ACCOMPLISH (UPA), and U‑ACHIEVE Study 2 (UPA) | NA SA excluding lower values was near-0%. However, given the star‑shaped structure of the network, this analysis would exclude treatments from those studies (UPA and OZA) and would not affect the results. | ||
Concurrent corticosteroids | Notable heterogeneity | Minimal | Meta-regression based on concurrent corticosteroid use | NA |
AMS = adapted Mayo score; AT-IR = inadequate response or intolerance to advanced therapy ETR = etrasimod; INF = infliximab; FMS = full Mayo score; MIR = mirikizumab; NA = not applicable; OZA = ozanimod; RCT = randomized clinical trial; SA = sensitivity analysis; UC = ulcerative colitis; UPA = upadacitinib.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Maintenance
A network plot of the included maintenance RCTs is shown in Figure 2. Like the induction network, the maintenance network is also star-shaped. All maintenance treatments are anchored on placebo as the common comparator.
Figure 2: Maintenance Network Plot

ADA = adalimumab; ETR = etrasimod; GOL = golimumab; INF = infliximab; MIR = mirikizumab; OZA = ozanimod; PBO = placebo; Q#W = every # weeks; RIS = risankizumab; SC = subcutaneous; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab; VED = vedolizumab.
Notes: The nodes are weighted based on the number of participants. The lines are weighted based on the number of studies (numbers) with a direct comparison between treatments.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
The key findings from the feasibility assessment, along with the list of resulting sensitivity analyses, were as follows:
NMA connectivity was feasible for both non–AT-IR and AT-IR populations in maintenance phase.
A sensitivity analysis excluding RCTs in predominantly Asian populations was planned to assess the impact on the clinical response of risankizumab relative to other treatments.
Two sensitivity analyses were planned for clinical response and endoscopic improvement due to a stricter time point requirement (i.e., excluding PURSUIT-J for clinical response and PURSUIT-M for endoscopic improvement). Due to the limited number of studies, no other relevant sensitivity analyses based on outcome definitions could be performed. However, the outcome definitions were considered sufficiently similar by the sponsor to being included in the NMA.
A sensitivity analysis was planned based on the study design, using TT data instead of RR for risankizumab for efficacy outcomes (true placebo analysis).
Table 23 outlines the key findings about the baseline characteristics’ heterogeneity and the approach taken to address them. In summary, the sponsor considered that baseline patient characteristics were comparable across the maintenance RCTs, which supports the feasibility of the NMA. However, some heterogeneity was observed.
No other sensitivity analyses were deemed necessary by the sponsor based on the feasibility assessment process.
Additionally, there are sources of heterogeneity that extend beyond the patient baseline characteristics (e.g., differences in the studies’ inclusion and exclusion criteria, outcome definitions, and time points). For example, the included studies used varying definitions of clinical remission and response (e.g., central endoscopy versus local investigator readings), durations of follow-up for outcome assessment (e.g., 40 to 54 weeks), differing definition of AT-IR, number and type of patients with AT were previously exposed differed (as the commercially available options changed over time), differences in how disease severity inclusion criteria was defined across the trials, differences in disease duration (e.g., ranging from 3.7 to 9.1 years), variation in extent of disease (e.g., from 32% to 80% with extensive colitis or pancolitis), heterogeneity in concurrent corticosteroid use (particularly for the non–AT-IR population), and wide variation in placebo risk. It is unclear how the effect of these variations was mitigated or could have biased the analyses.
Table 23: Key Findings from the Baseline Patient Characteristics Assessment (Maintenance Population)
Patient characteristics at baseline | Heterogeneity assessment | Approach to addressing heterogeneity | ||
|---|---|---|---|---|
Non–AT-IR | AT-IR | Non–AT-IR | AT-IR | |
Age | Minimal | NA | ||
Gender | Minimal | NA | ||
Weight | Notable heterogeneity with lower values in Asian RCTs | NA Although most treatments are not weight dependent, and therefore testing would not be necessary, SA excluding predominantly Asian RCTs addressed this concern | ||
Duration of disease | Minor | NA | NA | |
Extent of disease | Notable heterogeneity with higher values in M10 to 447 (ADA) and NCT02039505 (VED) | Notable heterogeneity with lower values in ELEVATE UC 52 (ETR) and LUCENT-2 (MIR) | NA SA excluding higher values was considered. However, the SA excluding predominantly Asian RCTs addressed this concern on ADA and VED. | NA SA excluding lower values was considered. However, given the star-shaped structure of the network, this analysis would exclude treatments from those studies (ETR and MIR) and would not affect the results. |
Baseline FMS | Minimal, exception of OCTAVE Sustain | NA No adjustment required as OCTAVE Sustain reported FMS for rerandomized responders after induction, while other studies reported FMS at true baseline. | ||
Baseline AMS | Minimal | NA | ||
Concurrent immunomodulators | Notable heterogeneity with near-0%, largely due to study inclusion criteria for TRUE NORTH (OZA), and U-ACHIEVE Study 3 (UPA) | NA SA excluding lower values was near-0%. However, given the star-shaped structure of the network, this analysis would exclude treatments from those studies (UPA and OZA) and would not affect the results. | ||
Concurrent corticosteroids | Notable heterogeneity | Minimal | Meta-regression based on concurrent corticosteroids use | NA |
ADA = adalimumab; AMS = adapted Mayo score; AT-IR = inadequate response or intolerance to advanced therapy; ETR = etrasimod; FMS = full Mayo score; MIR = mirikizumab; NA = not applicable; OZA = ozanimod; RCT = randomized clinical trial; SA = sensitivity analysis; UC = ulcerative colitis; UPA = upadacitinib; VED = vedolizumab.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Induction
Efficacy
Risankizumab 1,200 mg was favoured over adalimumab ███ █████ ███ ███ ████ ██ █████ and mirikizumab ███ █████ ███ ███ ████ ██ ████) for clinical response and to adalimumab (██ █████ ███ ███ ████ ██ ████), golimumab (██ █████ ███ ███ ████ ██ ██████ mirikizumab ███ █████ ███ ███ ████ ██ ██████ tofacitinib (██ █████ ███ ███ ████ ██ ████), and ustekinumab ███ █████ ███ ███ ████ ██ █████ for achieving endoscopic improvement in the non-AT population during induction (Table 24). Risankizumab 1,200 mg was less favoured compared to upadacitinib for clinical remission (██ █████ ███ ███ ████ ██ ████), clinical response (██ █████ ███ ███ ████ ██ █████ and endoscopic improvement (██ █████ ███ ███ ████ ██ ████) in the AT-IR population during induction (Table 24). There was no evidence of a significant difference between risankizumab 1,200 mg and the other interventions in the efficacy NMAs during the induction phase (Table 24).
Table 24: League Table of ORs for Clinical Remission, Clinical Response, Endoscopic Improvement in Non–AT-IR and AT-IR Induction NMAs
Drug | Clinical remission | Clinical response | Endoscopic improvement | |||
|---|---|---|---|---|---|---|
Non–AT-IR induction RIS1200 | AT-IR induction RIS1200 | Non–AT-IR induction RIS1200 | AT-IR induction RIS1200 | Non–AT-IR induction RIS1200 | AT-IR induction RIS1200 | |
ADA 160/80 | ████ | ████ | ████ | ████ | ████ | ████ |
ETR2 | ████ | ████ | ████ | ████ | ████ | ████ |
GOL 200/100 | ████ | ████ | ████ | ████ | ████ | ████ |
INF 5 | ████ | ████ | ████ | ████ | ████ | ████ |
MIR 300 | ████ | ████ | ████ | ████ | ████ | ████ |
OZA 0.92 | ████ | ████ | ████ | ████ | ████ | ████ |
TOF 10 | ████ | ████ | ████ | ████ | ████ | ████ |
UPA 45 | ████ | ████ | ████ | ████ | ████ | ████ |
UST 6 | ████ | ████ | ████ | ████ | ████ | ████ |
VED 300 | ████ | ████ | ████ | ████ | ████ | ████ |
ADA = adalimumab; AT-IR = inadequate response or intolerance to advanced therapyETR = etrasimod; GOL = golimumab; INF = infliximab; MIR = mirikizumab; OR = odds ratio; OZA = ozanimod; RIS = risankizumab; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab; VED = vedolizumab.
Note: All NMAs utilized a random-effects model except clinical response in the non–AT-IR induction NMA, which utilized a fixed-effects model. Asterisks (**) indicate significance (95% CrI does not cross 1). Results interpretation: OR > 1 favours risankizumab in the column.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Safety
Risankizumab 1,200 mg was favoured over adalimumab (██ █████ ███ ███ ████ ██ ██████ █████████ ███ █████ ███ ███ █████ ██████ ████████ ███ █████ ███ ███ ████ ██ ██████ ███ ███████████ ███ █████ ███ ███ ████ ██ ████) with regard to SAEs during the induction phase (Table 25). Risankizumab 1,200 mg was less favoured than etrasimod ███ █████████ ███ ███ █████ ██ █████████ with regard to serious infections during the induction phase (Table 25). There was no evidence of a difference between risankizumab 1,200 mg and the other interventions in the safety NMAs during the induction phase (Table 25).
Table 25: League Table of ORs for AEs, SAEs, and Serious Infections in Induction NMAs
Drug | AEs RIS1200 | SAEs RIS1200 | Serious infections RIS1200 |
|---|---|---|---|
ADA 160/80 | ████████████ | ████████████ | ████████████ |
ETR 2 | ████████████ | ████████████ | ████████████ |
GOL 200/100 | ████████████ | ████████████ | ████████████ |
INF 5 | ████████████ | ████████████ | ████████████ |
MIR 300 | ████████████ | ████████████ | ████████████ |
OZA 0.92 | ████████████ | ████████████ | ████████████ |
TOF 10 | ████████████ | ████████████ | ████████████ |
UPA 45 | ████████████ | ████████████ | ████████████ |
UST 6 | ████████████ | ████████████ | ████████████ |
VED 300 | ████████████ | ████████████ | ████████████ |
ADA = adalimumab; ETR = etrasimod; GOL = golimumab; INF = infliximab; MIR = mirikizumab; OR = odds ratio; OZA = ozanimod; RIS = risankizumab; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab; VED = vedolizumab.
Note: All NMAs utilized a random-effects model except serious infections in the overall induction NMA, which utilized a fixed-effects model. Asterisks (**) indicate significance (95% CrI do not cross 1). Results interpretation: OR < 1 favours risankizumab in the column.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Maintenance
Efficacy
For the maintenance phase, in non–AT-IR populations, risankizumab 180 mg ███ █████ ███ ███ ████ ██ ████) and risankizumab 360 mg ███ █████ ███ ███ ████ ██ █████ were less favoured than upadacitinib 30 mg for clinical response in the non–AT-IR population during the maintenance phase (Table 26). There was no evidence of a difference between risankizumab 1,200 mg and the other interventions in the efficacy NMAs during the maintenance phase (Table 26).
Table 26: League Table of ORs for Clinical Remission, Clinical Response, Endoscopic Improvement in Maintenance Non–AT-IR NMAs
Drug | Clinical remission | Clinical response | Endoscopic improvement | |||
|---|---|---|---|---|---|---|
RIS180 | RIS360 | RIS180 | RIS360 | RIS180 | RIS360 | |
ADA 40Q2W | ████ | ████ | ████ | ████ | ████ | ████ |
ETR 2 | ████ | ████ | ████ | ████ | ████ | ████ |
GOL 100 | ████ | ████ | ████ | ████ | ████ | ████ |
GOL 50 | ████ | ████ | ████ | ████ | ████ | ████ |
INF 10 | ████ | ████ | ████ | ████ | ████ | ████ |
INF 5 | ████ | ████ | ████ | ████ | ████ | ████ |
MIR 200 | ████ | ████ | ████ | ████ | ████ | ████ |
OZA 0.92 | ████ | ████ | ████ | ████ | ████ | ████ |
RIS 180 | ████ | ████ | ████ | ████ | ████ | ████ |
RIS 360 | ████ | ████ | ████ | ████ | ████ | ████ |
TOF 10 | ████ | ████ | ████ | ████ | ████ | ████ |
TOF 5 | ████ | ████ | ████ | ████ | ████ | ████ |
UPA 15 | ████ | ████ | ████ | ████ | ████ | ████ |
UPA 30 | ████ | ████ | ████ | ████ | ████ | ████ |
UST 90Q12W | ████ | ████ | ████ | ████ | ████ | ████ |
UST 90Q8W | ████ | ████ | ████ | ████ | ████ | ████ |
VED 108Q2W(SC) | ████ | ████ | ████ | ████ | ████ | ████ |
VED 300Q4W | ████ | ████ | ████ | ████ | ████ | ████ |
VED 300Q8W | ████ | ████ | ████ | ████ | ████ | ████ |
ADA = adalimumab; ETR = etrasimod; GOL = golimumab; INF = infliximab; MIR = mirikizumab; OR = odds ratio; OZA = ozanimod; RIS = risankizumab; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab; VED = vedolizumab.
Note: All NMAs utilized a random-effects model. Asterisks (**) indicate significance (95% CrI do not cross 1). Results interpretation: OR > 1 favours risankizumab in the column.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
For the maintenance phase, in AT-IR populations, risankizumab 180 mg was less favoured than upadacitinib (███████████████ ██ ██████ ███ ███ ████ ██ ██████ ████████████ ███ ██ █████ ███ ███ ████ ██ █████ ███ ███████████ ████████████ ███████████ ██ █████ ███ ███ ████ ██ ██████ ███████████ ███████ ██ █████ ███ ███ ████ ██ █████ ███ ████████ ██████████ ███████████ ████████████ ███ ██ █████ ███ ███ ████ ██ █████ ███ ████████ █████████ ███ ████████████ █████████████ ███ ██ █████ ███ ███ ████ ██ █████ ████████████ ███ ██ █████ ███ ███ ████ ██ █████ ███ ███████████ ███ █████ █████ ████ ██ █████ ███ ██████████ █████████ ██████ ██). There was no evidence of a difference between risankizumab 180 mg and the other interventions in the efficacy NMAs during the maintenance phase (Table 27).
Risankizumab ███ ██ ███ ████ ████████ ████████ ██ ███████████ ███ █████ ███ ███ ████ ██ ██████ ████████████ █████████████ ███ ██ █████ ███ ███ ████ ██ █████ ████████████ ███ ██ █████ ███ ███ ████ ██ █████ ███ ███████████ ████████████ ███████████ ██ █████ ███ ███ ████ ██ ██████ ███████████ █████ ██ █████ ███ ███ ████ ██ ██████ ███████████ ██████ ███ █████ ███ ███ ████ ██ █████ ███ ████████ ██████████ ███████████ ███ █████ ███ ███ ████ ██ ██ █████ ███ ███ ████ ██ █████ ███ ███████████ ███ █████ ███ ███ ████ ██ ████) for endoscopic remission (Table 27). There was no evidence of a difference between risankizumab 180 mg and the other interventions in the efficacy NMAs during the maintenance phase (Table 27). There was no evidence of a difference between risankizumab 180 mg and the other interventions in the efficacy NMAs during the maintenance phase (Table 27).
Table 27: League Table of ORs for Clinical Remission, Clinical Response, Endoscopic Improvement in Maintenance AT-IR NMAs
Drug | Clinical remission | Clinical response | Endoscopic improvement | |||
|---|---|---|---|---|---|---|
RIS180 | RIS360 | RIS180 | RIS360 | RIS180 | RIS360 | |
ADA 40Q2W | ████ | ████ | ████ | ████ | ████ | ████ |
ETR 2 | ████ | ████ | ████ | ████ | ████ | ████ |
GOL 100 | ████ | ████ | ████ | ████ | ████ | ████ |
GOL 50 | ████ | ████ | ████ | ████ | ████ | ████ |
INF 10 | ████ | ████ | ████ | ████ | ████ | ████ |
INF 5 | ████ | ████ | ████ | ████ | ████ | ████ |
MIR 200 | ████ | ████ | ████ | ████ | ████ | ████ |
OZA 0.92 | ████ | ████ | ████ | ████ | ████ | ████ |
RIS 180 | ████ | ████ | ████ | ████ | ████ | ████ |
RIS 360 | ████ | ████ | ████ | ████ | ████ | ████ |
TOF 10 | ████ | ████ | ████ | ████ | ████ | ████ |
TOF 5 | ████ | ████ | ████ | ████ | ████ | ████ |
UPA 15 | ████ | ████ | ████ | ████ | ████ | ████ |
UPA 30 | ████ | ████ | ████ | ████ | ████ | ████ |
UST 90Q12W | ████ | ████ | ████ | ████ | ████ | ████ |
UST 90Q8W | ████ | ████ | ████ | ████ | ████ | ████ |
VED 108Q2W(SC) | ████ | ████ | ████ | ████ | ████ | ████ |
VED 300Q4W | ████ | ████ | ████ | ████ | ████ | ████ |
VED 300Q8W | ████ | ████ | ████ | ████ | ████ | ████ |
ADA = adalimumab; ETR = etrasimod; GOL = golimumab; INF = infliximab; MIR = mirikizumab; OR = odds ratio; OZA = ozanimod; RIS = risankizumab; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab; VED = vedolizumab.
Notes: All NMAs utilized a random-effects model. Asterisks (**) indicate significance (95% CrI do not cross 1). Results interpretation: OR > 1 favours risankizumab in the column.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Safety
Risankizumab 360 mg was favoured over golimumab ███ █████ ███ ███ ████ ██ █████ with regard to AEs during the maintenance phase (Table 28). There was no evidence of a difference between risankizumab 180 mg or risankizumab 360 mg and the other interventions in the safety NMAs during the maintenance phase (Table 28).
Table 28: League Table of ORs for AEs, SAEs, and Serious Infections in Maintenance NMAs
Drug | AEs | SAEs | Serious infections | |||
|---|---|---|---|---|---|---|
RIS180 | RIS360 | RIS180 | RIS360 | RIS180 | RIS360 | |
ADA 40Q2W | ████ | ████ | ████ | ████ | ████ | ████ |
ADA 40QW | ████ | ████ | ████ | ████ | ████ | ████ |
ETR 2 | ████ | ████ | ████ | ████ | ████ | ████ |
GOL 100 | ████ | ████ | ████ | ████ | ████ | ████ |
GOL 50 | ████ | ████ | ████ | ████ | ████ | ████ |
INF 10 | ████ | ████ | ████ | ████ | ████ | ████ |
INF 5 | ████ | ████ | ████ | ████ | ████ | ████ |
MIR 200 | ████ | ████ | ████ | ████ | ████ | ████ |
OZA 0.92 | ████ | ████ | ████ | ████ | ████ | ████ |
RIS 180 | ████ | ████ | ████ | ████ | ████ | ████ |
RIS 360 | ████ | ████ | ████ | ████ | ████ | ████ |
TOF 10 | ████ | ████ | ████ | ████ | ████ | ████ |
TOF 5 | ████ | ████ | ████ | ████ | ████ | ████ |
UPA 15 | ████ | ████ | ████ | ████ | ████ | ████ |
UPA 30 | ████ | ████ | ████ | ████ | ████ | ████ |
UST 90Q12W | ████ | ████ | ████ | ████ | ████ | ████ |
UST 90Q8W | ████ | ████ | ████ | ████ | ████ | ████ |
VED 108Q2W(SC) | ████ | ████ | ████ | ████ | ████ | ████ |
VED 300Q4W | ████ | ████ | ████ | ████ | ████ | ████ |
VED 300Q8W | ████ | ████ | ████ | ████ | ████ | ████ |
ADA = adalimumab; ETR = etrasimod; GOL = golimumab; INF = infliximab; MIR = mirikizumab; OR = odds ratio; OZA = ozanimod; RIS = risankizumab; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab; VED = vedolizumab.
Notes: All NMAs utilized a random-effects model. Asterisks (**) indicate significance (95% CrI do not cross 1). Results interpretation: OR < 1 favours risankizumab in the column.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal of the NMA
In the sponsor-submitted ITC, relevant RCTs were identified using a systematic review produced following accepted methodological guidance and based on an a priori protocol that outlined the inclusion and exclusion criteria. The search is 2 years old, and it is not known whether more recent studies are available, nor what impact that might have on the results. Selection, data extraction, and risk-of-bias assessments were conducted in duplicate by independent researchers, which is considered adequate. Risk of bias of the included studies was assessed using the Cochrane Risk-of-Bias tool. The overall bias of all included studies was deemed to be low. The risk was assessed at the study level, not the outcome level, as suggested by Cochrane, and bias concerns may vary by effect estimate. The appraisal may not be universally applicable across all outcomes. There was no assessment of the risk of publication bias; therefore, its presence or absence cannot be confirmed.
The methods used for the NMA were considered appropriate, including the use of a Bayesian framework, as its conduct adhered to relevant guidance. However, there were no sensitivity analyses to understand the potential impact of the chosen priors for between-study standard deviation. Overall, the patient populations, interventions, comparators, and outcomes of the included studies were consistent with the overall review’s objective. The clinical experts consulted by the review team did not expect any major issues regarding the representativeness of the study populations enrolled in the RCTs that were included in the ITC in relation to the populations in Canada that may be eligible for treatment with risankizumab.
As is common in NMAs of treatments for UC, 1 overall concern was the heterogeneity in the patient populations, which included their baseline characteristics, average disease severity (and how this was defined across trials), disease duration, and the extent of disease. There were some differences in the AT-IR definitions used across studies, and due to changes in the standard of care and available treatments over time, the number and type of prior treatments to which patients were exposed likely varied. There was also heterogeneity in the use of concurrent immunomodulators and corticosteroids. Additionally, the timing of outcome assessments varied substantially, and the definitions of the efficacy end points of interest were not always consistent. Some relevant evidence was excluded due to misalignment in the assessment time points; while this was necessary to reduce heterogeneity, the impact on results is not known.
There were additional sources of heterogeneity in the maintenance phase. Adjustments were applied to include both TT and RR trial designs. This was necessary and has been applied to other NMAs for UC but relies on assumptions that cannot be fully validated. Placebo was a common comparator in the NMAs, though the placebo groups across studies are likely not equivalent, due to carry-over effects from different induction treatments. Differences in the corticosteroid tapering strategy (and whether this was included) may also differ across the studies. For both induction and maintenance phases, the information needed to comprehensively assess the level of heterogeneity was not always available for all studies. The notable clinical and methodological heterogeneity raises concern for intransitivity, which undermines the validity of the indirect comparisons, as it indicates that the assumption of exchangeability may not hold.
Relevant efforts were made to reduce or account for heterogeneity across the NMAs, including stratification of the results by AT-IR status, baseline risk meta-regression, and sensitivity analyses. The stratification of analyses by AT-IR and non–AT-IR populations for efficacy analyses is appropriate but limits generalizability of the results of either analysis to the overall population for this indication. The interpretation of these subgroups faces some limitations. The baseline characteristics within subgroups were not always fully known (i.e., the full population was used for comparison), challenging comprehensive assessments of comparability. It is not clear whether randomization was stratified by AT-IR status across all trials. Confounding is possible, as the effect estimates might not be arising from fully randomized groups. None of the baseline risk-adjusted models converged; therefore, it became not possible to apply this adjustment. Sensitivity analyses for some sources of heterogeneity were not possible.
The networks were sparsely populated, with several nodes centred around a single connection (placebo) in a star geometry, with only 1 to 2 trials against placebo per treatment and only a few head-to-head trials (i.e., within-study comparisons of different doses of the same treatment). This reduces the robustness of the NMA and makes most comparisons underpowered due to the lack of direct evidence. Further, all evidence for risankizumab was against placebo, increasing uncertainty in the estimates for each outcome, and the consistency assumption could not be assessed. The heterogeneity and network sparsity are reflected in imprecise CrIs. Fixed-effects models were chosen for a small number of outcomes based on model fit when it was a better fit than the random-effects model. However, given the expected heterogeneity, the fixed-effects model has the potential to underestimate the uncertainty in the results (i.e., width of the CrIs). There was poor model fit for the analysis of serious infections in the induction phase. Last, based on the ITC feasibility assessment, 1 study was not included in the NMA because the drug is not approved in Canada.
Eight outcomes of interest to this review were reported: clinical remission, clinical response, endoscopic improvement, AEs, SAEs, and serious infections. Interpretation of harms outcomes was complicated by inclusion of UC worsening and related symptoms as harms in the placebo groups, and potential differences in how harms were collected across studies. Some outcomes of importance to patients, such as corticosteroid-free clinical remission and HRQoL, were not included.
Summary
In the absence of direct comparisons between risankizumab and other available treatments in Canada, the sponsor submitted an NMA that included 28 (21 for induction and 16 for maintenance) phases III or higher randomized, double-blind trials. They reported on clinical remission, clinical response, endoscopic improvement, any AE, SAEs, and serious infections.
In the AT-IR population, the results of the NMA suggest that upadacitinib was favoured over risankizumab for clinical response and remission and endoscopic improvement in the induction and maintenance phases. For clinical remission and endoscopic improvement, the results also suggest that vedolizumab is often favoured over risankizumab during the maintenance phase. Other comparisons were less consistent across outcomes and/or had wide CrIs that included the possibility that either risankizumab or the comparator may be favoured.
In the non–AT-IR population, results of the NMA suggest that risankizumab was favoured over adalimumab and mirikizumab for clinical response and over adalimumab, mirikizumab, golimumab, tofacitinib, and ustekinumab for endoscopic improvement in the induction phase. Similar findings were not observed in the maintenance phase. Other comparisons were less consistent across outcomes and/or had wide CrIs that included the possibility that either risankizumab or the comparator may be favoured.
In the full population, the results suggest that risankizumab was favoured over adalimumab, etrasimod, ozanimod, and tofacitinib 10 for SAEs in the induction phase, and etrasimod was favoured over risankizumab for serious infections. Similar findings were not observed in the maintenance phase, where the CrIs were wide, suggesting uncertainty in whether risankizumab or the comparator may be favoured.
Limitations of the NMA included likely violation of the underlying assumptions, network sparsity, imprecision, and inconsistency of the results across outcomes. As a result, the relative treatment effects of risankizumab versus relevant comparators are subject to uncertainty and there is no conclusive evidence of which treatment may provide the preferred balance of benefits and harms.
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
Table 29: Summary of Study Addressing Gaps in the Systematic Review Evidence
Detail | Description |
|---|---|
Panaccione et al. (2025)20 | |
Title | Risankizumab efficacy and safety based on prior inadequate response or intolerance to advanced therapy: post hoc analysis of the INSPIRE and COMMAND phase 3 studies |
Evidence gap | Efficacy of risankizumab in subgroups based on AT-IR status, including the number and mechanism of action of prior ATs received |
Study design |
|
Population | The same as the INSPIRE and COMMAND trials (Table 11) |
Interventions | As this was a post hoc analysis of the pivotal trials, there are no additional interventions. The comparison is risankizumab vs. placebo. |
Key findings | For the following results, confidence intervals were unreported. Induction efficacy outcomes at week 12 with risankizumab 1,200 mg:
Maintenance efficacy outcomes at week 52 with risankizumab 360 mg and risankizumab 180 mg:
Safety outcomes:
|
Limitations |
|
AT = advanced therapy; AT-IR = inadequate response or intolerance to advanced therapy; HEMI = histologic, endoscopic, and mucosal improvement; TEAE = treatment-emergent adverse event; vs. = versus.
Sources: Panaccione et al. (2025).20 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Discussion
Summary of Available Evidence
The CDA-AMC reviewed a systematic review that included 2 pivotal trials (INSPIRE and COMMAND), 1 NMA, and 1 study addressing gaps in the systematic review evidence. The INSPIRE trial was a phase III, randomized, placebo-controlled, double-blind, parallel-group induction study in adult patients with moderately to severely active UC who experienced an inadequate response or intolerance to conventional therapies or other ATs. Patients in the INSPIRE trial were randomized to risankizumab 1,200 mg IV (ITT population N = 650) or placebo (ITT population N = 325). Patients who did not achieve clinical response at week 12 were eligible to enter another blinded risankizumab treatment period (i.e., Induction Period 2, which lasted for an additional 12 weeks). The main outcomes were:
proportion of patients with clinical remission per the adapted Mayo score
proportion of patients with clinical response per the adapted Mayo score
proportion of patients with endoscopic improvement
proportion of patients with HEMI
proportion of patients with HEMR
change from baseline in IBDQ total score
proportion of patients with SAEs.
The COMMAND trial was a phase III, randomized, placebo-controlled, double-blind, parallel-group maintenance study in adult patients with UC previously enrolled in the INSPIRE induction trial who experienced an adequate response to risankizumab. The ITT population was: risankizumab 180 mg = 179 patients, risankizumab 360 mg = 186 patients, and placebo (risankizumab withdrawal) = 183 patients. Combined, the 2 studies lasted up to 81 weeks and included a screening period that lasted up to 35 days, a 12-week double-blind induction period, and a 52-week maintenance period. Randomization was stratified by history of inadequate response to AT (yes, no), last risankizumab induction dose (IV 600 mg, 1,200 mg, or 1,800 mg), and clinical remission status (per local evaluation) at the last visit of the induction trial (yes, no). Final patient follow-up occurred in April 2023.
The main outcomes were:
proportion of patients with clinical remission per the adapted Mayo score
proportion of patients with clinical response per the adapted Mayo score
proportion of patients with endoscopic improvement
proportion of patients with HEMI
proportion of patients with HEMR
proportion of patients with discontinuation of corticosteroid use in patients taking steroids at baseline
change from baseline in IBDQ total score
proportion of patients with SAEs.
Additional reviewed patient-centric outcomes were fecal incontinence, abdominal pain, bowel urgency, nocturnal bowel movements, tenesmus, and sleep interrupted due to UC symptoms (Table 30).
In the absence of direct comparisons between risankizumab and other available treatments in Canada, the sponsor submitted an NMA that included 28 (21 for induction and 16 for maintenance) phases III or higher randomized, double-blind trials. They reported on clinical remission, clinical response, endoscopic improvement, AEs, SAEs, and serious infections.
The study addressing the gaps in the systematic review evidence reported on a post hoc analysis of the INSPIRE and COMMAND trials. The objective was to assess the efficacy and safety of risankizumab for induction and maintenance phases in patients with moderately to severely active UC based on prior inadequate response or intolerance to AT (i.e., AT-IR status), including the number and mechanism of action of prior ATs received.
Interpretation of Results
Efficacy
Patient and clinician input received by CDA-AMC for this review indicated that outcomes such as clinical remission and clinical response based on the adapted Mayo scale, endoscopic improvement, HEMI, HEMR, HRQoL, and safety outcomes are highly valued. The clinical experts consulted by the review team confirmed that the goal of treatment is generally to improve patient quality of life, achieve clinical and endoscopic remission as quickly as possible, and decrease AEs. The experts also concur that most patients will prioritize sustained remission and/or treatment response and note that not all patients have disease that responds to currently available therapies.
The clinical experts consulted by CDA-AMC and other interest holders considered the aforementioned outcomes as critical for decision-making. A recent consensus guideline for outcome effect size thresholds was developed by an international group of researchers;14 it recommend thresholds of small, moderate, and large effect sizes as follows: clinical remission, 11% ± 6%, 20% ± 8%, and 31% ± 13%; endoscopic remission, 9% ± 5%, 17% ± 9%, and 28% ± 14%; and SAEs 6% ± 6%, 11% ± 9%, and 17% ± 12%, respectively. Discussions with the clinical experts highlighted that the thresholds used may differ depending on how refractory the patient’s UC is. A higher threshold would be expected for patients with less refractory UC. Because the populations in the INSPIRE and COMMAND trials had highly refractory UC, the review team, in consultation with the clinical experts, decided to use the small threshold for the evaluation of evidence certainty. This aligns with the usual CDA-AMC approach to GRADE, which applies a minimally contextual approach and rates effect estimates versus a threshold for a small effect. If some of the more stringent expectations of benefit were used, the clinical implications would be more uncertain. Additionally, placebo is not a commonly used intervention in this patient population, with no evidence of comparative efficacy and safety against commonly prescribed treatments in Canada. Based on these thresholds, the INSPIRE trial showed that risankizumab 1,200 mg (induction phase) resulted in important improvements in clinical response based on the adapted Mayo scale, endoscopic improvement, HEMI, and HEMR as compared to placebo. In the COMMAND (maintenance phase) pivotal trial, there was a clinically important improvement in HEMR with both risankizumab 180 mg and 360 mg as compared to placebo. The results of the pivotal trials were supported by evidence from a post hoc subgroup analysis on the efficacy of risankizumab based on AT-IR status, including the number and mechanism of action of prior ATs received. These suggested that the magnitude of benefit might be smaller for the AT-IR group than the non–AT-IR group, although the design was not adequate to draw credible conclusions for effect modification. It is also important to note that the placebo group in the COMMAND trial received risankizumab during the induction phase (risankizumab withdrawal). As such, there could have been a carry-over effect due to an inadequate washout.
The additional patient-centric outcomes generally favoured risankizumab over placebo during both induction and maintenance phases. Further, fewer patients who received risankizumab during the induction phase were hospitalized, but there was no evidence of a difference in risk of hospitalization during the maintenance phase. Also, during the maintenance phase, the outcome “clinical remission with no corticosteroid use for 90 days,” which was assessed in the full cohort of randomized participants, favoured risankizumab.
The sponsor also submitted an NMA considered of relevance for the current CDA-AMC review. These analyses used data from published trials and Clinical Study Reports to compare risankizumab to active comparators. The analyzed outcomes were:
efficacy (both induction and maintenance phases, both non–AT-IR and AT-IR populations)
clinical remission
clinical response
endoscopic improvement
safety (both induction and maintenance phases, overall population)
percent of AEs
percent of SAEs
percent of serious infections.
In the AT-IR population, results of the NMA suggest that upadacitinib was favoured over risankizumab for clinical response and remission and for endoscopic improvement in the induction and maintenance phases. For clinical remission and endoscopic improvement, the results also suggest that vedolizumab is often favoured over risankizumab during maintenance. Other comparisons were less consistent across outcomes and/or had wide CrIs that included the possibility that either risankizumab or the comparator may be favoured.
In the non–AT-IR population, results of the NMA suggest that risankizumab was favoured over adalimumab and mirikizumab for clinical response and was favoured over adalimumab, mirikizumab, golimumab, tofacitinib, and ustekinumab for endoscopic improvement in the induction phase. Similar findings were not observed in the maintenance phase. Other comparisons were less consistent across outcomes and/or had wide CrIs that included the possibility that either risankizumab or the comparator may be favoured.
In the full population, the results suggest that risankizumab was favoured over adalimumab, etrasimod, ozanimod, and tofacitinib for SAEs in the induction phase, and etrasimod was favoured over risankizumab for serious infections. Similar findings were not observed in the maintenance phase, where the CrIs were wide, suggesting uncertainty as to whether risankizumab or the comparator may be favoured.
Hence, except for a few isolated cases, the results of the NMA generally did not provide evidence favouring 1 treatment over another, with wide CrIs crossing the null for most analyses. Furthermore, due to differences in study designs, inclusion criteria, patient populations, definitions of outcomes measures, and timing of assessment, there is uncertainty in the results because it is likely that the underlying assumption of exchangeability is violated.
Harms
In the INSPIRE trial, the number of patients with at least 1 TEAE were similar between the risankizumab (42.4%) and placebo (49.4%) groups. The most common TEAE in the risankizumab group was COVID-19 (4.8%), and the most common TEAE in the placebo group was UC (10.2%). The proportion of patients with at least 1 SAE was higher in the placebo group (10.2%) than in the risankizumab group (2.3%). The most common serious TEAEs in the risankizumab group were anemia, UC, and pulmonary embolism (2 patients [0.3%] each). In the placebo group, the most common serious TEAEs were UC (4.9%), anemia (0.6%), and anal fistula (0.6%). A higher proportion of patients in the placebo group (3.7%) discontinued treatment due to a TEAE than in the risankizumab group (0.6%). One patient in the risankizumab group died due to COVID-19. The most frequently occurring AEOSIs in both arms were hypersensitivity (risankizumab: 3.8%; placebo: 2.2%) and hepatic events (risankizumab: 1.5%; placebo: 4.0%).
In the COMMAND trial, the number of patients with at least 1 TEAE was similar between the risankizumab 180 mg (72.5%), risankizumab 360 mg (70.8%), and placebo (76.5%) groups. The proportion of patients with at least 1 SAE was similar across groups (5.2% with risankizumab 180 mg, 5.1% with risankizumab 360 mg, and 8.2% with placebo). In the risankizumab 180 mg group, the only serious TEAE occurring in more than 1 patient was appendicitis (2 patients [1.0%]). In the risankizumab 360 mg group, no serious TEAE occurred in more than 1 patient. In the placebo (risankizumab withdrawal) group, the most common serious TEAE was UC (2.6%); no other serious TEAE occurred in more than 1 patient. The proportions of patients who discontinued treatment due to a TEAE were highest in patients taking risankizumab 360 mg (2.6%) compared to risankizumab 180 mg (1.6%) and placebo (1.5%) groups. Regarding AEOSIs, the risankizumab 180 mg group had the highest proportion of patients with hypersensitivity (10.4%). The risankizumab 360 mg group had the highest proportion of patients with hepatic events (6.7%).
Based on these results, risankizumab 1,200 mg (induction phase) and risankizumab 180 mg and 360 mg (maintenance phase) was well tolerated by patients. Even so, it is unclear how patients taking placebo had more AEs and SAEs than those receiving risankizumab during the induction phase (risankizumab: 2.3%, placebo: 10.2%) and the maintenance phase (risankizumab 180 mg: 5.2%; risankizumab 360 mg: 5.1%; placebo: 8.2%). This brings into question the validity of the safety assessments and likely results from the lack of efficacy of the placebo (i.e., symptoms related to worsening disease). The evidence on the comparative safety of risankizumab from the NMA was unremarkable, with the results generally providing no evidence of superiority of 1 treatment over another, with wide CrIs crossing the null for most analyses. Furthermore, due to differences in study designs, inclusion criteria, patient populations, and length of follow-up, there is uncertainty in the results.
Conclusion
The CDA-AMC review included 2 pivotal phase III, randomized, placebo-controlled, double-blind, parallel-group trials for the induction phase (INSPIRE) and the maintenance phase (COMMMAND) in adult patients with moderately to severely active UC who experienced an inadequate response or intolerance to conventional or other ATs. During the induction phase, there was moderate- to high-certainty evidence favouring risankizumab with regard to clinical response, clinical remission, endoscopic improvement, HEMI, and HEMR. Further, there was moderate-certainty evidence favouring risankizumab with regard to IBDQ and low-certainty evidence for little to no difference in serious AEs. During the maintenance phase, there was high-certainty evidence favouring risankizumab for endoscopic improvement and HEMI. Further, there was moderate-certainty evidence favouring risankizumab for clinical response (180 mg only, the 360 mg dose showed likely little to no difference), clinical remission, HEMR, and IBDQ. Last, there was low-certainty evidence favouring risankizumab for discontinuation of corticosteroid use in patients taking steroids at baseline, and for little to no difference in SAEs.
There was some concern resulting from the inability to comprehensively appraise for risk of bias due to missing data, as this was not reported in the submitted materials. While the results of most subgroup analyses were similar in direction to the main analyses, they should be viewed as exploratory, for hypothesis generation only, because the trial was not stratified along these subgroups. The occurrence of harms was relatively limited and aligned with the known safety profile of risankizumab. A study addressing gaps submitted by the sponsor suggested similar directions of effect for AT-IR and non–AT-IR populations, but the magnitude of effects varied. Benefits appeared smaller for patients with AT-IR compared to those considered non–AT-IR in both the induction and maintenance phases. Conclusions on effect modification were not possible to draw from this study.
The sponsor submitted an NMA comparing risankizumab to other relevant comparators was affected by several limitations, which included likely violation of the underlying assumptions, network sparsity, imprecision, and inconsistency of the results across outcomes. As a result, the relative treatment effects of risankizumab versus relevant comparators are subject to uncertainty, and there is no conclusive evidence of which treatment may provide the preferred balance of benefits and harms.
References
1.Rubin DT, Ananthakrishnan AN, Siegel CA, Sauer BG, Long MD. ACG Clinical Guideline: Ulcerative Colitis in Adults. Am J Gastroenterol. 2019;114(3):384-413. doi:10.14309/ajg.0000000000000152 PubMed
2.Kaplan GG, Ng SC. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology. 2017;152(2):313-321 e2. doi:10.1053/j.gastro.2016.10.020 PubMed
3.Longo D, Fauci A, Kasper D, Hauser S, Jameson J, Loscalzo J. Harrison’s Principles of Internal Medicine. 18th ed. McGraw-Hill Education; 2011.
4.Greuter T, Vavricka SR. Extraintestinal manifestations in inflammatory bowel disease - epidemiology, genetics, and pathogenesis. Expert Rev Gastroenterol Hepatol. 2019;13(4):307-317. doi:10.1080/17474124.2019.1574569 PubMed
5.Fumery M, Singh S, Dulai PS, Gower-Rousseau C, Peyrin-Biroulet L, Sandborn WJ. Natural History of Adult Ulcerative Colitis in Population-based Cohorts: A Systematic Review. Clin Gastroenterol Hepatol. 2018;16(3):343-356 e3. doi:10.1016/j.cgh.2017.06.016 PubMed
6.Armuzzi A, Liguori G. Quality of life in patients with moderate to severe ulcerative colitis and the impact of treatment: A narrative review. Dig Liver Dis. 2021;53(7):803-808. doi:10.1016/j.dld.2021.03.002 PubMed
7.Benchimol EI, Bernstein CN, Bitton A, et al. The Impact of Inflammatory Bowel Disease in Canada 2018: A Scientific Report from the Canadian Gastro-Intestinal Epidemiology Consortium to Crohn's and Colitis Canada. J Can Assoc Gastroenterol. 2019;2(Suppl 1):S1-S5. doi:10.1093/jcag/gwy052 PubMed
8.Crohn’s and Colitis Canada (CCFC). 2023 Impact of inflammatory bowel disease in Canada [sponsor-supplied reference]. 2023. Accessed August 8, 2023. https://crohnsandcolitis.ca/Crohns_and_Colitis/documents/reports/2023-IBD-Report-English-LR.pdf
9.Schreiber S, Panés J, Louis E, Holley D, Buch M, Paridaens K. National differences in ulcerative colitis experience and management among patients from five European countries and Canada: An online survey. J Crohns Colitis. 2013;7(6):497-509. doi:10.1016/j.crohns.2012.07.027 PubMed
10.Bressler B, Marshall JK, Bernstein CN, et al. Clinical practice guidelines for the medical management of nonhospitalized ulcerative colitis: the Toronto consensus. Gastroenterology. 2015;148(5):1035-1058 e3. doi:10.1053/j.gastro.2015.03.001 PubMed
11.Raine T, Bonovas S, Burisch J, et al. ECCO Guidelines on Therapeutics in Ulcerative Colitis: Medical Treatment. J Crohns Colitis. 2022;16(1):2-17. doi:10.1093/ecco-jcc/jjab178 PubMed
12.U. S. Food Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research. Ulcerative Colitis: Developing Drugs for Treatment : Guidance for Industry. Draft Guidance [sponsor-supplied reference]. 2022. Accessed February 6, 2025. https://www.fda.gov/media/158016/download
13.European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP). Guideline on the development of new medicinal products for the treatment of ulcerative colitis : Revision 1. 2018. Accessed June 16, 2025. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-development-new-medicinal-products-treatment-ulcerative-colitis-revision-1_en.pdf
14.Gordon M, Shaban N, Sinopoulou V, et al. Developing IBD Outcome Effect Size Thresholds to Inform Research, Guidelines, and Clinical Decisions. Inflamm Bowel Dis. 2025:izaf085. doi:10.1093/ibd/izaf085 PubMed
15.Irvine EJ. Quality of life of patients with ulcerative colitis: past, present, and future. Inflamm Bowel Dis. 2008;14(4):554-65. doi:10.1002/ibd.20301 PubMed
16.Irvine EJ. Development and subsequent refinement of the inflammatory bowel disease questionnaire: a quality-of-life instrument for adult patients with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 1999;28(4):S23-7. doi:10.1097/00005176-199904001-00003 PubMed
17.AbbVie Inc. M16-067 Abbreviated Clinical Study Report - Final: A Multicenter, Randomized, Double-Blind, Placebo-Controlled Induction Study to Evaluate the Efficacy and Safety of Risankizumab in Subjects with Moderately to Severely Active Ulcerative Colitis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Skyrizi (risankizumab injection), 150 mg in 1 mL sterile solution (150 mg/mL) subcutaneous injection; 180 mg in 1.2 mL sterile solution (150 mg/mL) subcutaneous injection; 360 mg in 2.4 mL sterile solution (150 mg/mL) subcutaneous injection; 75 mg in 0.83 mL sterile solution (90 mg/mL) subcutaneous injection; 90 mg in 1 mL sterile solution (90 mg/mL) subcutaneous injection; 600 mg in 10 mL sterile solution (60 mg/mL) for intravenous infusion. October 30, 2023.
18.AbbVie Inc. M16-066 Abbreviated Clinical Study Report – Substudy 2 Interim: A Multicenter, Randomized, Double-Blind, Placebo-Controlled 52-Week Maintenance and an Open-Label Extension Study of the Efficacy and Safety of Risankizumab in Subjects with Ulcerative Colitis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Skyrizi (risankizumab injection), 150 mg in 1 mL sterile solution (150 mg/mL) subcutaneous injection; 180 mg in 1.2 mL sterile solution (150 mg/mL) subcutaneous injection; 360 mg in 2.4 mL sterile solution (150 mg/mL) subcutaneous injection; 75 mg in 0.83 mL sterile solution (90 mg/mL) subcutaneous injection; 90 mg in 1 mL sterile solution (90 mg/mL) subcutaneous injection; 600 mg in 10 mL sterile solution (60 mg/mL) for intravenous infusion. July 22, 2024.
19.Atreya R, Louis E, Loftus Jr EV, et al. DOP014 Efficacy and safety up to 3 years of risankizumab maintenance treatment in patients with moderately to severely active ulcerative colitis: interim results from phase 3 COMMAND open-label extension study. J Crohns Colitis. 2025;19(Supplement_1):i108-i110. doi:10.1093/ecco-jcc/jjae190.0053
20.Panaccione R, Louis E, Colombel JF, et al. Risankizumab efficacy and safety based on prior inadequate response or intolerance to advanced therapy: post hoc analysis of the INSPIRE and COMMAND phase 3 studies. J Crohns Colitis. 2025;19(1):jjaf005. doi:10.1093/ecco-jcc/jjaf005 PubMed
21.Hanauer SB. Inflammatory bowel disease: Epidemiology, pathogenesis, and therapeutic opportunities. Inflamm Bowel Dis. 2006;12 Suppl 1:S3-9. doi:10.1097/01.MIB.0000195385.19268.68 PubMed
22.Crohn’s and Colitis Foundation of Canada (CCFC). The impact of inflammatory bowel disease in Canada: 2012 Final Report and Recommendations [sponsor-supplied reference]. 2012. Accessed May 10, 2023. http://crohnsandcolitis.ca/Crohns_and_Colitis/documents/reports/ccfc-ibd-impact-report-2012.pdf
23.Crohn’s and Colitis Canada (CCFC). 2018 Impact of inflammatory bowel disease in Canada [sponsor-supplied reference]. 2018. Accessed October 5, 2022. https://crohnsandcolitis.ca/Crohns_and_Colitis/documents/reports/2018-Impact-Report-LR.pdf
24.Torres J, Billioud V, Sachar DB, Peyrin-Biroulet L, Colombel JF. Ulcerative colitis as a progressive disease: the forgotten evidence. Inflamm Bowel Dis. 2012;18(7):1356-63. doi:10.1002/ibd.22839 PubMed
25.Ungaro R, Mehandru S, Allen PB, Peyrin-Biroulet L, Colombel JF. Ulcerative colitis. Lancet. 2017;389(10080):1756-1770. doi:10.1016/S0140-6736(16)32126-2 PubMed
26.Roda G, Narula N, Pinotti R, et al. Systematic review with meta-analysis: proximal disease extension in limited ulcerative colitis. Aliment Pharmacol Ther. 2017;45(12):1481-1492. doi:10.1111/apt.14063 PubMed
27.Ungar B, Kopylov U. Long-term outcome of ulcerative proctitis. United Eur Gastroenterol J. 2020;8(8):847-848. doi:10.1177/2050640620948795 PubMed
28.Regueiro MD. Diagnosis and treatment of ulcerative proctitis. J Clin Gastroenterol. 2004;38(9):733-40. doi:10.1097/01.mcg.0000139178.33502.a3 PubMed
29.Cohen R, Skup M, Ozbay AB, et al. Direct and indirect healthcare resource utilization and costs associated with ulcerative colitis in a privately-insured employed population in the US. J Med Econ. 2015;18(6):447-56. doi:10.3111/13696998.2015.1021353 PubMed
30.Gajendran M, Loganathan P, Jimenez G, et al. A comprehensive review and update on ulcerative colitis. Dis Mon. 2019;65(12):100851. doi:10.1016/j.disamonth.2019.02.004 PubMed
31.Casellas F, Arenas JI, Baudet JS, et al. Impairment of health-related quality of life in patients with inflammatory bowel disease: a Spanish multicenter study. Inflamm Bowel Dis. 2005;11(5):488-96. doi:10.1097/01.mib.0000159661.55028.56 PubMed
32.Armuzzi A, Tarallo M, Lucas J, et al. The association between disease activity and patient-reported outcomes in patients with moderate-to-severe ulcerative colitis in the United States and Europe. BMC Gastroenterol. 2020;20(1):18. doi:10.1186/s12876-020-1164-0 PubMed
33.Sarid O, Slonim-Nevo V, Schwartz D, et al. Differing Relationship of Psycho-Social Variables with Active Ulcerative Colitis or Crohn's Disease. Int J Behav Med. 2018;25(3):341-350. doi:10.1007/s12529-018-9712-5 PubMed
34.Ghosh S, Sanchez Gonzalez Y, Zhou W, et al. Upadacitinib Treatment Improves Symptoms of Bowel Urgency and Abdominal Pain, and Correlates With Quality of Life Improvements in Patients With Moderate to Severe Ulcerative Colitis. J Crohns Colitis. 2021;15(12):2022-2030. doi:10.1093/ecco-jcc/jjab099 PubMed
35.Peyrin-Biroulet L, Germain A, Patel AS, Lindsay JO. Systematic review: outcomes and post-operative complications following colectomy for ulcerative colitis. Aliment Pharmacol Ther. 2016;44(8):807-16. doi:10.1111/apt.13763 PubMed
36.Rubin DT, Dubinsky MC, Panaccione R, et al. The impact of ulcerative colitis on patients' lives compared to other chronic diseases: a patient survey. Dig Dis Sci. 2010;55(4):1044-52. doi:10.1007/s10620-009-0953-7 PubMed
37.Becker HM, Grigat D, Ghosh S, et al. Living with inflammatory bowel disease: A Crohn's and Colitis Canada survey. Can J Gastroenterol Hepatol. 2015;29(2):77-84. doi:10.1155/2015/815820 PubMed
38.Sikirica M, Lynch J, Hoskin B, Kershaw J, Wild R, Lukanova R. P0412 Humanistic burden and impact of the disease in moderate to severe patients with Crohn’s disease (CD) and ulcerative colitis (UC): findings from a cross sectional study in the US and France, Germany, Italy, Spain, UK (EU5). United Eur Gastroenterol J. 2020;8(Supplement_1):348. doi:10.1177/2050640620927345
39.Burisch J, Zhao M, Odes S, et al. P860 The Costs of Inflammatory Bowel Diseases in High-Income Settings. J Crohns Colitis. 2023;17(Supplement_1):i983. doi:10.1093/ecco-jcc/jjac190.0990
40.Ruiz-Casas L, Evans J, Rose A, et al. The LUCID study: living with ulcerative colitis; identifying the socioeconomic burden in Europe. BMC Gastroenterol. 2021;21(1):456. doi:10.1186/s12876-021-02028-5 PubMed
41.Singh S, Loftus EV, Jr., Limketkai BN, et al. AGA Living Clinical Practice Guideline on Pharmacological Management of Moderate-to-Severe Ulcerative Colitis. Gastroenterology. 2024;167(7):1307-1343. doi:10.1053/j.gastro.2024.10.001 PubMed
42.CADTH. Drug Reimbursement Review: Ustekinumab (Stelara/Stelara I.V - Janssen Inc.). September 16, 2020. Accessed February 25, 2025. https://www.cda-amc.ca/ustekinumab-2
43.CADTH. Drug Reimbursement Review: Vedolizumab (Entyvio - Takeda Canada Inc.). July 14, 2020. Accessed February 25, 2025. https://www.cda-amc.ca/vedolizumab-1
44.Canada's Drug Agency (CDA). Reimbursement Review: Mirikizumab (Omvoh). February 9, 2024. Accessed February 7, 2025. https://www.cda-amc.ca/mirikizumab
45.Eli Lilly Canada, Inc. Omvoh (mirikizumab): 100 mg/mL solution for subcutaneous injection; 20 mg/mL solution for intravenous infusion [product monograph; sponsor supplied reference]. July 20, 2023. Accessed September 19, 2023. https://pdf.hres.ca/dpd_pm/00071771.PDF
46.pan-Canadian Pharmaceutical Alliance (pCPA). Omvoh (mirikizumab) for Ulcerative Colitis. 2024. Accessed February 25, 2025. https://www.pcpacanada.ca/negotiation/22481
47.pan-Canadian Pharmaceutical Alliance (pCPA). Velsipity (etrasimod) for Ulcerative Colitis. 2024. Accessed February 25, 2025. https://www.pcpacanada.ca/negotiation/22823
48.Canada's Drug Agency (CDA). Reimbursement Review: Etrasimod (Velsipity). December 9, 2024. Accessed February 25, 2025. https://www.cda-amc.ca/etrasimod
49.AbbVie Corporation. Skyrizi (risankizumab injection): 150 mg in 1 mL sterile solution (150 mg/mL) subcutaneous injection; 180 mg in 1.2 mL sterile solution (150 mg/mL) subcutaneous injection; 360 mg in 2.4 mL sterile solution (150 mg/mL) subcutaneous injection; 75 mg in 0.83 mL sterile solution (90 mg/mL) subcutaneous injection; 90 mg in 1 mL sterile solution (90 mg/mL) subcutaneous injection; 600 mg in 10 mL sterile solution (60 mg/mL) [product monograph; sponsor-supplied reference]. October 10, 2024.
50.Pfizer Canada ULC. Velsipity (etrasimod): 2 mg etrasimod (as etrasimod L-arginine) oral tablets [product monograph; sponsor-supplied reference]. January 30, 2024.
51.Bristol-Myers Squibb Canada. Zeposia (ozanimod as ozanimod hydrochloride): capsules, 0.23 mg, 0.46 mg and 0.92 mg, oral [product monograph; sponsor supplied reference]. December 19, 2023. Accessed August 11, 2023. https://www.bms.com/assets/bms/ca/documents/productmonograph/ZEPOSIA_EN_PM.pdf
52.AbbVie Corporation. Humira (adalimumab): 40 mg in 0.8 mL sterile solution (50 mg/mL) subcutaneous injection, 10 mg in 0.1 mL sterile solution (100 mg/mL) subcutaneous injection, 20 mg in 0.2 mL sterile solution (100 mg/mL) subcutaneous injection, 40 mg in 0.4 mL sterile solution (100 mg/mL) subcutaneous injection, 80 mg in 0.8 mL sterile solution (100 mg/mL) subcutaneous injection [product monograph; sponsor-supplied reference]. September 16, 2022. Accessed August 10, 2023. https://www.abbvie.ca/content/dam/abbvie-dotcom/ca/en/documents/products/HUMIRA_PM_EN.pdf
53.Amgen Canada Inc. Amgevita (adalimumab): 20 mg in 0.4 mL sterile solution (50 mg/mL), prefilled syringe for subcutaneous injection; 40 mg in 0.8 mL sterile solution (50 mg/mL), prefilled syringe for subcutaneous injection; 40 mg in 0.8 mL sterile solution (50 mg/mL), prefilled SureClick® autoinjector for subcutaneous injection [product monograph; sponsor-supplied reference]. September 9, 2022. Accessed August 17, 2023. https://pdf.hres.ca/dpd_pm/00067468.PDF
54.BGP Pharma ULC. Hulio (adalimumab): 20 mg in 0.4 mL sterile solution (50 mg/mL) subcutaneous injection; 40 mg in 0.8 mL sterile solution (50 mg/mL) subcutaneous injection [product monograph; sponsor supplied reference]. July 20, 2023. Accessed August 17, 2023. https://pdf.hres.ca/dpd_pm/00071858.PDF
55.Pfizer Canada ULC. Abrilada (adalimumab): 40 mg in 0.8 mL sterile solution (50 mg/mL), prefilled pen for subcutaneous injection; 10 mg in 0.2 mL sterile solution (50 mg/mL), prefilled glass syringe for subcutaneous injection; 20 mg in 0.4 mL sterile solution (50 mg/mL), prefilled glass syringe for subcutaneous injection; 40 mg in 0.8 mL sterile solution (50 mg/mL), prefilled glass syringe for subcutaneous injection; 40 mg in 0.8 mL sterile solution (50 mg/mL), glass vial for subcutaneous injection [product monograph; sponsor-supplied reference]. November 14, 2022. Accessed August 21, 2023. https://pdf.hres.ca/dpd_pm/00068228.PDF
56.Sandoz Canada Inc. Hyrimoz (adalimumab): 20 mg in 0.4 mL sterile solution (50 mg/mL) subcutaneous injection in pre-filled syringe; 40 mg in 0.8 mL sterile solution (50 mg/mL) subcutaneous injection in a pre-filled syringe; 40 mg in 0.8 mL sterile solution (50 mg/mL) subcutaneous injection in a pre-filled autoinjector [product monograph; sponsor-supplied reference]. October 11, 2022. Accessed August 21, 2023. https://pdf.hres.ca/dpd_pm/00067757.PDF
57.Merck Canada Inc. Hadlima (adalimumab): 40 mg in 0.8 mL sterile solution (50 mg/mL), pre-filled syringe for subcutaneous injection; Hadlima PushTouch (adalimumab): 40 mg in 0.8 mL sterile solution (50 mg/mL), auto-injector for subcutaneous injection [product monograph; sponsor-supplied reference]. November 26, 2020. Accessed August 21, 2023. https://crohnsandcolitis.ca/Crohns_and_Colitis/documents/HADLIMA-PM_E.pdf
58.Celltrion Healthcare Canada Limited. Yuflyma (adalimumab): 40 mg in 0.4 mL sterile solution (100 mg/mL), pre-filled syringe for subcutaneous injection; 40 mg in 0.4 mL sterile solution (100 mg/mL), pre-filled syringe with Needle guard for subcutaneous injection; 40 mg in 0.4 mL sterile solution (100 mg/mL), pre-filled pen (auto-injector) for subcutaneous injection; 80 mg in 0.8 mL sterile solution (100 mg/mL), pre-filled syringe for subcutaneous injection; 80 mg in 0.8 mL sterile solution (100 mg/mL), pre-filled syringe with Needle guard for subcutaneous injection; 80 mg in 0.8 mL sterile solution (100 mg/mL), pre-filled pen (auto-injector) for subcutaneous injection [product monograph; sponsor-supplied reference]. February 3, 2023. Accessed August 21, 2023. https://pdf.hres.ca/dpd_pm/00069429.PDF
59.Fresenius Kabi Canada Ltd. Idacio (adalimumab): sterile solution, 40 mg / 0.8 mL (50 mg/mL), subcutaneous injection [product monograph; sponsor-supplied reference]. October 30, 2020. Accessed August 21, 2023. https://pdf.hres.ca/dpd_pm/00058541.PDF
60.JAMP Pharma Corporation. Simlandi (adalimumab): 40 mg in 0.4 mL sterile solution (100 mg/mL) subcutaneous injection; 80 mg in 0.8 mL sterile solution (100 mg/mL) subcutaneous injection [product monograph; sponsor-supplied reference]. December 9, 2022. Accessed August 21, 2023. https://pdf.hres.ca/dpd_pm/00068814.PDF
61.Janssen Inc. Simponi (golimumab): 50 mg/0.5 mL, 100 mg/1.0 mL solution for injection; Simponi I.V (golimumab): 50 mg/4.0 mL solution for infusion [product monograph; sponsor-supplied reference]. January 15, 2018. Accessed August 11, 2023. https://pdf.hres.ca/dpd_pm/00043422.PDF
62.Janssen Inc. Remicade (infliximab): powder for solution, sterile, lyophilized, 100 mg/vial [product monograph; sponsor-supplied reference]. August 4, 2017. Accessed August 11, 2023. https://crohnsandcolitis.ca/Crohns_and_Colitis/images/living-with-crohns-colitis/REMICADE-MONOGRAPH.PDF
63.Amgen Canada Inc. Avsola (infliximab): powder for solution, sterile, lyophilized, 100 mg/vial, intravenous infusion [product monograph; sponsor-supplied reference]. May 17, 2022. Accessed August 17, 2023. https://pdf.hres.ca/dpd_pm/00065940.PDF
64.Organon Canada Inc. Renflexis (infliximab): powder for solution, sterile, lyophilized, 100 mg /vial, intravenous infusion [product monograph; sponsor-supplied reference]. February 9, 2023. Accessed August 21, 2023. https://pdf.hres.ca/dpd_pm/00069528.PDF
65.Pfizer Canada Inc. Inflectra (infliximab): powder for solution, sterile, lyophilized, 100 mg/vial [product monograph; sponsor-supplied reference]. November 7, 2017. Accessed August 11, 2023. https://pdf.hres.ca/dpd_pm/00042037.PDF
66.Takeda Canada Inc. Entyvio (vedolizumab): powder for concentrate for solution for infusion, 300 mg/vial [product monograph; sponsor-supplied reference]. March 22, 2016. Accessed August 15, 2023. https://pdf.hres.ca/dpd_pm/00034239.PDF
67.Janssen Inc. Stelara (ustekinumab): solution for subcutaneous injection, 45 mg/0.5 mL, 90 mg/1.0 mL; Stelara I.V. (ustekinumab): solution for intravenous infusion, 130 mg/26 mL (5 mg/mL) [product monograph; sponsor-supplied reference]. December 12, 2008. Accessed August 15, 2023. https://pdf.hres.ca/dpd_pm/00069002.PDF
68.Pfizer Canada ULC. Xeljanz (tofacitinib): tablets, oral, 5 mg tofacitinib (as tofacitinib citrate), 10 mg tofacitinib (as tofacitinib citrate); Xeljanz XR (tofacitinib extended-release): tablets, oral, 11 mg tofacitinib (as tofacitinib citrate) [product monograph; sponsor-supplied reference]. August 11, 2023. Accessed August 21, 2023. https://pdf.hres.ca/dpd_pm/00071982.PDF
69.Pharmascience Inc. pms-Tofacitinib (tofacitinib): tablets, 5 mg tofacitinib (as tofacitinib citrate), oral [product monograph; sponsor-supplied reference]. August 23, 2022. Accessed August 15, 2023. https://pdf.hres.ca/dpd_pm/00069222.PDF
70.Taro Pharmaceuticals Inc. Taro-tofacitinib (tofacitinib): tablets, oral, 5 mg tofacitinib (as tofacitinib citrate), 10 mg tofacitinib (as tofacitinib citrate) [product monograph; sponsor-supplied reference]. July 13, 2022. Accessed August 18, 2023. https://pdf.hres.ca/dpd_pm/00068405.PDF
71.Auro Pharma Inc. Auro-tofacitinib (tofacitinib): tablets, 5 mg tofacitinib (as tofacitinib citrate), 10 mg tofacitinib (as tofacitinib citrate) [product monograph; sponsor-supplied reference]. August 19, 2022. Accessed September 22, 2023. https://pdf.hres.ca/dpd_pm/00068455.PDF
72.AbbVie Corporation. Rinvoq (upadacitinib): extended-release tablets for oral use, 15 mg upadacitinib, 30 mg upadacitinib, 45 mg upadacitinib [product monograph; sponsor-supplied reference]. October 31, 2023. Accessed December 11, 2023. https://www.abbvie.ca/content/dam/abbvie-dotcom/ca/en/documents/products/RINVOQ_PM_EN.pdf
73.Louis E, Schreiber S, Panaccione R, et al. Risankizumab for Ulcerative Colitis: Two Randomized Clinical Trials. JAMA. 2024;332(11):881-897. doi:10.1001/jama.2024.12414 PubMed
74.AbbVie. NCT03398148: A Multicenter, Randomized, Double-Blind, Placebo Controlled Induction Study to Evaluate the Efficacy and Safety of Risankizumab in Subjects With Moderately to Severely Active Ulcerative Colitis [sponsor-supplied reference]. ClinicalTrials.gov; 2018. https://clinicaltrials.gov/study/NCT03398148
75.AbbVie. NCT03398135: A Multicenter, Randomized, Double-Blind, Placebo Controlled 52-Week Maintenance and an Open-Label Extension Study of the Efficacy and Safety of Risankizumab in Subjects With Ulcerative Colitis [sponsor-supplied reference]. ClinicalTrials.gov; 2018. https://clinicaltrials.gov/study/NCT03398135
76.Naegeli AN, Hunter T, Dong Y, et al. Full, Partial, and Modified Permutations of the Mayo Score: Characterizing Clinical and Patient-Reported Outcomes in Ulcerative Colitis Patients. Crohns Colitis 360. 2021;3(1):otab007. doi:10.1093/crocol/otab007 PubMed
77.Swenson A, Burke K, Womack K, Quinn M, Curhan G. Methods to Derive a Modified Mayo Score from Electronic Health Records Data. Inflamm Bowel Dis. 2023;29(Supplement_1):S21-S21. doi:10.1093/ibd/izac247.040
78.U.S. Food and Drug Administration. Ulcerative colitis: clinical trial endpoints: guidance for industry [sponsor-supplied reference]. August 2016. Accessed February 6, 2025. https://www.fda.gov/media/99526/download
79.European Medicines Agency. Guideline on the development of new medicinal products for the treatment of Ulcerative Colitis (CHMP/EWP/18463/2006) [sponsor-supplied reference]. 2008. Accessed February 6, 2025. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-development-new-medicinal-products-treatment-ulcerative-colitis_en.pdf
80.Sands BE, Sandborn WJ, Panaccione R, et al. Ustekinumab as Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med. 2019;381(13):1201-1214. doi:10.1056/NEJMoa1900750 PubMed
81.Sandborn WJ, van Assche G, Reinisch W, et al. Adalimumab induces and maintains clinical remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2012;142(2):257-65.e1-3. doi:10.1053/j.gastro.2011.10.032
82.Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353(23):2462-76. doi:10.1056/NEJMoa050516 PubMed
83.Reinisch W, Sandborn WJ, Hommes DW, et al. Adalimumab for induction of clinical remission in moderately to severely active ulcerative colitis: results of a randomised controlled trial. Gut. 2011;60(6):780-7. doi:10.1136/gut.2010.221127 PubMed
84.Jauregui-Amezaga A, Geerits A, Das Y, et al. A Simplified Geboes Score for Ulcerative Colitis. J Crohns Colitis. 2017;11(3):305-313. doi:10.1093/ecco-jcc/jjw154 PubMed
85.Geboes K, Riddell R, Ost A, Jensfelt B, Persson T, Löfberg R. A reproducible grading scale for histological assessment of inflammation in ulcerative colitis. Gut. 2000;47(3):404-9. doi:10.1136/gut.47.3.404 PubMed
86.Kim JE, Kim M, Kim MJ, et al. Histologic improvement predicts endoscopic remission in patients with ulcerative colitis. Sci Rep. 2024;14(1):19926. doi:10.1038/s41598-024-68372-0 PubMed
87.Cushing KC, Tan W, Alpers DH, Deshpande V, Ananthakrishnan AN. Complete histologic normalisation is associated with reduced risk of relapse among patients with ulcerative colitis in complete endoscopic remission. Aliment Pharmacol Ther. 2020;51(3):347-355. doi:10.1111/apt.15568 PubMed
88.Ma C, Sedano R, Almradi A, et al. An International Consensus to Standardize Integration of Histopathology in Ulcerative Colitis Clinical Trials. Gastroenterology. 2021;160(7):2291-2302. doi:10.1053/j.gastro.2021.02.035 PubMed
89.Feagan BG, Rochon J, Fedorak RN, et al. Methotrexate for the treatment of Crohn's disease. The North American Crohn's Study Group Investigators. N Engl J Med. 1995;332(5):292-7. doi:10.1056/nejm199502023320503 PubMed
90.Irvine EJ, Feagan B, Rochon J, et al. Quality of life: a valid and reliable measure of therapeutic efficacy in the treatment of inflammatory bowel disease. Canadian Crohn's Relapse Prevention Trial Study Group. Gastroenterology. 1994;106(2):287-96. doi:10.1016/0016-5085(94)90585-1 PubMed
91.Higgins PD, Schwartz M, Mapili J, Krokos I, Leung J, Zimmermann EM. Patient defined dichotomous end points for remission and clinical improvement in ulcerative colitis. Gut. 2005;54(6):782-8. doi:10.1136/gut.2004.056358 PubMed
92.Pallis AG, Mouzas IA, Vlachonikolis IG. The inflammatory bowel disease questionnaire: a review of its national validation studies. Inflamm Bowel Dis. 2004;10(3):261-9. doi:10.1097/00054725-200405000-00014 PubMed
93.Lewis JD, Chuai S, Nessel L, Lichtenstein GR, Aberra FN, Ellenberg JH. Use of the noninvasive components of the Mayo score to assess clinical response in ulcerative colitis. Inflamm Bowel Dis. 2008;14(12):1660-6. doi:10.1002/ibd.20520 PubMed
94.Higgins PD, Schwartz M, Mapili J, Zimmermann EM. Is endoscopy necessary for the measurement of disease activity in ulcerative colitis? Am J Gastroenterol. 2005;100(2):355-61. doi:10.1111/j.1572-0241.2005.40641.x. PubMed
95.Guyatt G, Mitchell A, Irvine EJ, et al. A new measure of health status for clinical trials in inflammatory bowel disease. Gastroenterology. 1989;96(3):804-10. doi:10.1016/0016-5085(89)90905-0 PubMed
96.Chen XL, Zhong LH, Wen Y, et al. Inflammatory bowel disease-specific health-related quality of life instruments: a systematic review of measurement properties. Health Qual Life Outcomes. 2017;15(1):177. doi:10.1186/s12955-017-0753-2 PubMed
97.Alrubaiy L, Rikaby I, Dodds P, Hutchings HA, Williams JG. Systematic review of health-related quality of life measures for inflammatory bowel disease. J Crohns Colitis. 2015;9(3):284-92. doi:10.1093/ecco-jcc/jjv002 PubMed
98.Pallis AG, Vlachonikolis IG, Mouzas IA. Assessing health-related quality of life in patients with inflammatory bowel disease, in Crete, Greece. BMC Gastroenterol. 2002;2:1. doi:10.1186/1471-230x-2-1 PubMed
99.Gregor JC, McDonald JW, Klar N, et al. An evaluation of utility measurement in Crohn's disease. Inflamm Bowel Dis. 1997;3(4):265-76. doi:10.1097/00054725-199712000-00004 PubMed
100.LeBlanc K, Mosli MH, Parker CE, MacDonald JK. The impact of biological interventions for ulcerative colitis on health-related quality of life. Cochrane Database Syst Rev. 2015;2015(9):Cd008655. doi:10.1002/14651858.CD008655.pub3 PubMed
101.Greenland S, Robins JM. Estimation of a common effect parameter from sparse follow-up data. Biometrics. 1985;41(1):55-68. PubMed
102.Bretz F, Maurer W, Brannath W, Posch M. A graphical approach to sequentially rejective multiple test procedures. Stat Med. 2009;28(4):586-604. doi:10.1002/sim.3495 PubMed
103.Holm S. A Simple Sequentially Rejective Multiple Test Procedure. Scandinavian Journal of Statistics. 1979;6(2):65-70.
104.AbbVie Inc. Study M16-067 – Statistical Analysis Plan for Sub-Study 2: A Multicenter, Randomized, Double-Blind, Placebo-Controlled Induction Study to Evaluate the Efficacy and Safety of Risankizumab in Subjects with Moderately to Severely Active Ulcerative Colitis. Version 2.0 [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Skyrizi (risankizumab injection), 150 mg in 1 mL sterile solution (150 mg/mL) subcutaneous injection; 180 mg in 1.2 mL sterile solution (150 mg/mL) subcutaneous injection; 360 mg in 2.4 mL sterile solution (150 mg/mL) subcutaneous injection; 75 mg in 0.83 mL sterile solution (90 mg/mL) subcutaneous injection; 90 mg in 1 mL sterile solution (90 mg/mL) subcutaneous injection; 600 mg in 10 mL sterile solution (60 mg/mL) for intravenous infusion. February 10, 2023.
105.AbbVie Inc. Study M16-066 – Statistical Analysis Plan for Sub-study 1: A Multicenter, Randomized, Double-Blind, Placebo Controlled 52-Week Maintenance and an Open-Label Extension Study of the Efficacy and Safety of Risankizumab in Subjects with Ulcerative Colitis. Version 6.0 [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Skyrizi (risankizumab injection), 150 mg in 1 mL sterile solution (150 mg/mL) subcutaneous injection; 180 mg in 1.2 mL sterile solution (150 mg/mL) subcutaneous injection; 360 mg in 2.4 mL sterile solution (150 mg/mL) subcutaneous injection; 75 mg in 0.83 mL sterile solution (90 mg/mL) subcutaneous injection; 90 mg in 1 mL sterile solution (90 mg/mL) subcutaneous injection; 600 mg in 10 mL sterile solution (60 mg/mL) for intravenous infusion. April 27, 2023.
106.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi:10.1016/j.jclinepi.2019.10.014 PubMed
107.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi:10.1016/j.jclinepi.2010.07.015 PubMed
108.Tacconelli E. Systematic reviews: CRD's guidance for undertaking reviews in health care. Lancet Infect Dis. 2010;10(4):226. doi:10.1016/S1473-3099(10)70065-7
109.National Institute for Health and Care Excellence. NICE Process and Methods Guides [sponsor-supplied reference]. 2012.
110.National Institute for Health and Care Excellence. Appendix A: Literature search strategies. In: Cirrhosis in over 16s: assessment and management (NICE guideline NG50) [sponsor-supplied reference]. 2016. https://www.nice.org.uk/guidance/ng50/documents/search-strategies
111.Higgins JPT, Green S, eds. Cochrane Handbook for Systematic Reviews of Interventions [sponsor-supplied reference]. 2022. http://handbook-5-1.cochrane.org/
112.Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 2: A Generalised Linear Modelling Framework for Pairwise and Network Meta-Analysis of Randomised Controlled Trials [sponsor-supplied reference]. National Institute for Health and Clinical Excellence; 2011.
113.Dias S, Ades AE, Welton NJ, Jansen JP, Sutton AJ. Network Meta-Analysis for Decision-Making. First ed. Statistics in Practice. John Wiley & Sons; 2018.
114.Gelman A, Rubin DB. Markov chain Monte Carlo methods in biostatistics. Stat Methods Med Res. 1996;5(4):339-55. doi:10.1177/096228029600500402 PubMed
115.Lambert PC, Sutton AJ, Burton PR, Abrams KR, Jones DR. How vague is vague? A simulation study of the impact of the use of vague prior distributions in MCMC using WinBUGS. Stat Med. 2005;24(15):2401-28. doi:10.1002/sim.2112 PubMed
116.Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 3: Heterogeneity—Subgroups, Meta-Regression, Bias, and Bias-Adjustment [sponsor-supplied reference]. National Institute for Health and Clinical Excellence; 2011. http://nicedsu.org.uk/wp-content/uploads/2016/03/TSD3-Heterogeneity.final-report.08.05.12.pdf
117.Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med. 2002;21(11):1539-58. doi:10.1002/sim.1186 PubMed
118.Colombel JF, Panés J, D’Haens G, et al. OP01 Higher vs. standard adalimumab maintenance regimens in patients with moderately to severely active ulcerative colitis: Results from the SERENE-UC maintenance study. J Crohns Colitis. 2020;14(Supplement_1):S001-S001. doi:10.1093/ecco-jcc/jjz203.000
119.Sandborn WJ, Vermeire S, Peyrin-Biroulet L, et al. Etrasimod as induction and maintenance therapy for ulcerative colitis (ELEVATE): two randomised, double-blind, placebo-controlled, phase 3 studies. Lancet. 2023;401(10383):1159-1171. doi:10.1016/S0140-6736(23)00061-2 PubMed
120.Magro F, Peyrin-Biroulet L, Sands BE, et al. P602 Achievement of stringent histologic and composite endpoints in subjects with moderately to severely active ulcerative colitis treated with etrasimod: a post hoc analysis of the phase 3 ELEVATE UC 52 and ELEVATE UC 12 trials. J Crohns Colitis. 2023;17(Supplement_1):i728-i729. doi:10.1093/ecco-jcc/jjac190.0732
121.Chaparro M, Armuzzi A, Irving PM, et al. DOP42 Efficacy of etrasimod on symptomatic relief in patients with ulcerative colitis: an analysis of the phase 3 ELEVATE UC 52 and ELEVATE UC 12 trials. J Crohns Colitis. 2023;17(Supplement_1):i107-i110. doi:10.1093/ecco-jcc/jjac190.0082
122.UEG Week 2022 Oral Presentations. United Eur Gastroenterol J. 2022;10(S8):9-184. doi:10.1002/ueg2.12293
123.UEG Week 2022 Moderated Posters. United Eur Gastroenterol J. 2022;10(S8):185-472. doi:10.1002/ueg2.12294
124.Sands BE, Rubin DT, Panés J, et al. DOP41 Achievement of corticosteroid-free clinical endpoints in subjects with ulcerative colitis: an analysis of the phase 3 ELEVATE UC 52 trial. J Crohns Colitis. 2023;17(Supplement_1):i106-i107. doi:10.1093/ecco-jcc/jjac190.0081
125.Vermeire S, Sands BE, Dubinsky MC, et al. P582 Efficacy of etrasimod at week 52 among subjects who reached clinical response at week 12: post hoc analysis of the phase 3 ELEVATE UC 52 trial. J Crohns Colitis. 2023;17(Supplement_1):i709-i711. doi:10.1093/ecco-jcc/jjac190.0712
126.Feagan BG, Rutgeerts P, Sands BE, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369(8):699-710. doi:10.1056/NEJMoa1215734 PubMed
127.Sandborn WJ, Colombel JF, Panaccione R, et al. Deep Remission With Vedolizumab in Patients With Moderately to Severely Active Ulcerative Colitis: A GEMINI 1 post hoc Analysis. J Crohns Colitis. 2019;13(2):172-181. doi:10.1093/ecco-jcc/jjy149 PubMed
128.Feagan BG, Rubin DT, Danese S, et al. Efficacy of Vedolizumab Induction and Maintenance Therapy in Patients With Ulcerative Colitis, Regardless of Prior Exposure to Tumor Necrosis Factor Antagonists. Clin Gastroenterol Hepatol. 2017;15(2):229-239 e5. doi:10.1016/j.cgh.2016.08.044 PubMed
129.Kobayashi T, Suzuki Y, Motoya S, et al. First trough level of infliximab at week 2 predicts future outcomes of induction therapy in ulcerative colitis-results from a multicenter prospective randomized controlled trial and its post hoc analysis. J Gastroenterol. 2016;51(3):241-51. doi:10.1007/s00535-015-1102-z PubMed
130.Jiang XL, Cui HF, Gao J, Fan H. Low-dose Infliximab for Induction and Maintenance Treatment in Chinese Patients With Moderate to Severe Active Ulcerative Colitis. J Clin Gastroenterol. 2015;49(7):582-8. doi:10.1097/mcg.0000000000000319 PubMed
131.D'Haens G, Dubinsky M, Kobayashi T, et al. Mirikizumab as Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med. 2023;388(26):2444-2455. doi:10.1056/NEJMoa2207940 PubMed
132.Dubinsky MC, Clemow DB, Hunter Gibble T, et al. Clinical Effect of Mirikizumab Treatment on Bowel Urgency in Patients with Moderately to Severely Active Ulcerative Colitis and the Clinical Relevance of Bowel Urgency Improvement for Disease Remission. Crohns Colitis 360. 2023;5(1):otac044. doi:10.1093/crocol/otac044 PubMed
133.UEG Week 2022 Poster Presentations. United Eur Gastroenterol J. 2022;10(S8):473-1092. doi:10.1002/ueg2.12295
134.D’Haens G, Kobayashi T, Morris N, et al. OP26 Efficacy and safety of mirikizumab as induction therapy in patients with moderately to severely active Ulcerative Colitis: Results from the Phase 3 LUCENT-1 study. J Crohns Colitis. 2022;16(Supplement_1):i028-i029. doi:10.1093/ecco-jcc/jjab232.025
135.Clemow D, Sapin C, Hibi T, et al. S765 Association of Ulcerative Colitis Bowel Urgency Improvement with Clinical Response and Remission. Am J Gastroenterol. 2022;117(10S):e543-e543. doi:10.14309/01.ajg.0000859700.09368.7e
136.Long M, Gisbert J, McGinnis K, et al. Improvement in Bowel Urgency Is Associated with Stool Frequency and Rectal Bleeding Remission at 12 and 52 Weeks in Patients with Moderately-to-Severely Active Ulcerative Colitis. Gastroenterology. 2023;164(4):S13-S14. doi:10.1053/j.gastro.2023.03.033
137.Travis S, Hibi T, Hisamatsu T, et al. S773 Effect of Mirikizumab on Bowel Urgency Clinically Meaningful Improvement and Remission: Results From the Phase 3 LUCENT Induction and Maintenance Studies. Am J Gastroenterol. 2022;117(10S):e549-e550. doi:10.14309/01.ajg.0000859732.42332.94
138.Navabi S, D’Haens G, Samaan KH, et al. P469 Effect of mirikizumab on clinical and endoscopic outcomes based on prior advanced therapy failure in patients with moderately to severely active ulcerative colitis. J Crohns Colitis. 2023;17(Supplement_1):i598-i598. doi:10.1093/ecco-jcc/jjac190.0599
139.D'Haens G, Higgins PDR, Peyrin-Biroulet L, et al. P554 Extended induction response in patients treated with mirikizumab with moderately to severely active ulcerative colitis in the LUCENT trials. J Crohns Colitis. 2023;17(Supplement_1):i682-i683. doi:10.1093/ecco-jcc/jjac190.0684
140.Dubinsky MC, Irving PM, Li X, et al. 867e: Efficacy and safety of mirikizumab as maintenance therapy in patients with moderately to severely active ulcerative colitis: Results from the phase 3 LUCENT-2 study. Gastroenterology. 2022;162(7):S-1393-S-1394. doi:10.1016/S0016-5085(22)64060-5
141.Suzuki Y, Motoya S, Hanai H, et al. Efficacy and safety of adalimumab in Japanese patients with moderately to severely active ulcerative colitis. J Gastroenterol. 2014;49(2):283-94. doi:10.1007/s00535-013-0922-y PubMed
142.AbbVie. Clinical Study Protocol M16-067 [sponsor-submitted reference]. 2022.
143.AbbVie. Clinical Study Protocol M16-066 [sponsor-submitted reference]. 2023.
144.Xi'an Janssen Pharmaceutical Ltd. Clinical Study Report Synopsis: REMICADEUCO3001. A Phase 3, Multicenter, Randomized, Double-Blind, Placebo-Controlled Study Evaluating the Efficacy and Safety of Infliximab in Chinese Subjects With Active Ulcerative Colitis [sponsor-supplied reference]. 2014. https://yoda.yale.edu/wp-content/uploads/2023/02/nct01551290.pdf
145.Motoya S, Watanabe K, Ogata H, et al. Vedolizumab in Japanese patients with ulcerative colitis: A Phase 3, randomized, double-blind, placebo-controlled study. PLoS One. 2019;14(2):e0212989. doi:10.1371/journal.pone.0212989 PubMed
146.Nagahori M, Watanabe K, Motoya S, et al. Week 2 Symptomatic Response with Vedolizumab as a Predictive Factor in Japanese Anti-TNFα-Naive Patients with Ulcerative Colitis: A post hoc Analysis of a Randomized, Placebo-Controlled Phase 3 Trial. Digestion. 2021;102(5):742-752. doi:10.1159/000512235 PubMed
147.Sandborn WJ, Su C, Sands BE, et al. Tofacitinib as Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med. 2017;376(18):1723-1736. doi:10.1056/NEJMoa1606910 PubMed
148.Lichtenstein GR, Moore GT, Soonasra A, et al. P479 Analysis of haematological changes in tofacitinib-treated patients with ulcerative colitis across Phase 3 induction and maintenance studies. J Crohns Colitis. 2019;13(Supplement_1):S351-S352. doi:10.1093/ecco-jcc/jjy222.603
149.Dubinsky MC, Peyrin-Biroulet L, Melmed GY, et al. Efficacy of Tofacitinib in Patients With Ulcerative Colitis by Prior Tumor Necrosis Factor Inhibitor Treatment Status: Results From OCTAVE Induction and Maintenance Studies: 640. Am J Gastroenterol. 2017;112:S354.
150.Sandborn W, Sands B, Steinwurz F, et al. P113 Evaluation of the Efficacy of Tofacitinib in Patients with Ulcerative Colitis Utilizing the Modified Mayo Score: Data from the Octave Program. Inflamm Bowel Dis. 2020;26(Supplement_1):S17-S18. doi:10.1093/ibd/zaa010.041
151.Sandborn WJ, Peyrin-Biroulet L, Sharara AI, et al. Efficacy and Safety of Tofacitinib in Ulcerative Colitis Based on Prior Tumor Necrosis Factor Inhibitor Failure Status. Clin Gastroenterol Hepatol. 2022;20(3):591-601 e8. doi:10.1016/j.cgh.2021.02.043 PubMed
152.D'Haens GR, Sands BE, Sandborn WJ, et al. Tofacitinib has induction efficacy in moderately to severely active ulcerative colitis, regardless of prior TNF inhibitor therapy. United Eur Gastroenterol J. 2016;4(5 Supplement 1):A45-A46. doi:10.1177/2050640616663688
153.Hanauer S, Panaccione R, Danese S, et al. Tofacitinib Induction Therapy Reduces Symptoms Within 3 Days for Patients With Ulcerative Colitis. Clin Gastroenterol Hepatol. 2019;17(1):139-147. doi:10.1016/j.cgh.2018.07.009 PubMed
154.Chiorean M, Rubin DT, Löwenberg M, et al. Improvement in Physicianʼs Global Assessment Within 2 Weeks in Patients With Ulcerative Colitis Treated With Tofacitinib. Am J Gastroenterol. 2018;113(Supplement):S414. doi:10.14309/00000434-201810001-00734
155.Danese S, Sandborn WJ, Panes J, et al. OC.06.1: Onset of Efficacy of Tofacitinib for Induction Therapy in Patients with Active Ulcerative Colitis in two Multinational, Phase 3 Clinical Trials. Dig Liver Dis. 2017;49:e90-e91. doi:10.1016/s1590-8658(17)30328-6
156.Sands BE, Long MD, Reinisch W, et al. Tofacitinib for the Treatment of Ulcerative Colitis: Analysis of Nonmelanoma Skin Cancer Rates From the Ulcerative Colitis Clinical Program. Inflamm Bowel Dis. 2022;28(2):234-245. doi:10.1093/ibd/izab056 PubMed
157.Vavricka SR, Greuter T, Cohen BL, et al. DOP85 Corticosteroid-free efficacy and safety outcomes in patients receiving tofacitinib in the OCTAVE Sustain maintenance study. J Crohns Colitis. 2021;15(Supplement_1):S116-S117. doi:10.1093/ecco-jcc/jjab073.124
158.Reinisch W, Osterman M, Doherty G, et al. Tu1720 – Efficacy of Tofacitinib Maintenance Therapy for Ulcerative Colitis in Remitting Patients Vs Patients with Clinical Response After 8 Weeks of Induction Treatment. Gastroenterology. 2019;156:S-1098. doi:10.1016/S0016-5085(19)39706-9
159.Feagan BG, Vermeire S, Sandborn WJ, et al. Tofacitinib for Maintenance Therapy in Patients With Active Ulcerative Colitis in the Phase 3 OCTAVE Sustain Trial: Results by Local and Central Endoscopic Assessments: 607. Am J Gastroenterol. 2017;112:S329-S330.
160.Gary L, Benjamin C, Laurent P-B, et al. P036 Impact of prior immunosuppressant and tumor necrosis factor inhibitor therapies on tofacitinib efficacy and safety in patients with ulcerative colitis. Am J Gastroenterol. 2019;114:S10. doi:10.14309/01.ajg.0000578216.94201.78
161.Hudesman D, Torres J, Salese L, et al. Patient-reported outcome improvement with tofacitinib in the ulcerative colitis octave clinical program. Am J Gastroenterol. 2021;116(SUPPL):S404-S405. doi:10.14309/01.ajg.0000776996.06251.75
162.Hibi T, Imai Y, Senoo A, Ohta K, Ukyo Y. Efficacy and safety of golimumab 52-week maintenance therapy in Japanese patients with moderate to severely active ulcerative colitis: a phase 3, double-blind, randomized, placebo-controlled study-(PURSUIT-J study). J Gastroenterol. 2017;52(10):1101-1111. doi:10.1007/s00535-017-1326-1 PubMed
163.Sandborn WJ, Feagan BG, Marano C, et al. Subcutaneous golimumab maintains clinical response in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2014;146(1):96-109.e1. doi:10.1053/j.gastro.2013.06.010 PubMed
164.Sandborn WJ, Feagan BG, Marano C, et al. Subcutaneous golimumab induces clinical response and remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2014;146(1):85-95; quiz e14-5. doi:10.1053/j.gastro.2013.05.048 PubMed
165.Panes J, Colombel JF, D'Haens GR, et al. High versus standard adalimumab induction dosing regime ns in patients with moderately to severely active ulcerative colitis: Results from the SERENE-UC induction study. United Eur Gastroenterol J. 2019;7(8 Supplement):118. doi:10.1177/2050640619854670
166.Sandborn WJ, Feagan BG, D'Haens G, et al. Ozanimod as Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med. 2021;385(14):1280-1291. doi:10.1056/NEJMoa2033617 PubMed
167.William S, Geert D, Doug W, et al. P025 Ozanimod Efficacy, Safety, and Histology in Patients with Moderate-to-Severe Ulcerative Colitis During Induction in the Phase 3 True North Study. Am J Gastroenterol. 2020;115(Suppl 1):S6-s7. doi:10.14309/01.ajg.0000722896.32651.d6 PubMed
168.Silvio D, Brian F, Stephen H, et al. P030 Ozanimod Efficacy, Safety, and Histology in Patients with Moderate-to-Severe Ulcerative Colitis During Maintenance in the Phase 3 True North Study. Am J Gastroenterol. 2020;115(Suppl 1):S8. doi:10.14309/01.ajg.0000722916.98351.89 PubMed
169.Subrata G, Geert D, Vipul J, et al. P012 Ozanimod Reduced Fecal Calprotectin Levels in Patients with Ulcerative Colitis in the Phase 3 True North Study. Am J Gastroenterol. 2020;115(Suppl 1):S3. doi:10.14309/01.ajg.0000722844.18503.37 PubMed
170.Schreiber S, Armuzzi A, Dignass A, et al. Corticosteroid-Free Remission In Patients With Moderately To Severely Active Ulcerative Colitis Treated With Ozanimod: Results From The Maintenance Phase Of True North. Gastroenterology. 2021;160(6 Supplement):S-83. doi:10.1016/S0016-5085(21)00944-6
171.Sands BE, Nguyen D, Pondel M, et al. Impact of prior biologic exposure on patient response to ozanimod for moderate-to-severe ulcerative colitis in the phase 3 true north study. Am J Gastroenterol. 2021;116(SUPPL):S313-S314. doi:10.14309/01.ajg.0000776304.52681.bf PubMed
172.Osterman MT, Longman R, Sninsky C, et al. Rapid Induction Effects of Ozanimod on Clinical Symptoms and Inflammatory Biomarkers in Patients with Moderately to Severely Active Ulcerative Colitis: Results from the Induction Phase of True North. Gastroenterology. 2021;160(6 Supplement):S-93-S-94. doi:10.1016/S0016-5085(21)00965-3
173.AbbVie. Clinical Study Protocol M14-675 [sponsor-submitted reference]. 2020.
174.AbbVie. Clinical Study Protocol M14-234 [sponsor-submitted reference]. 2020.
175.Ghosh S, Wolf D, Sandborn W, et al. P568 Sustained efficacy in patients with ulcerative colitis treated with adalimumab: results from ULTRA 2. J Crohns Colitis. 2013;7(Supplement_1):S238-S238. doi:10.1016/s1873-9946(13)60589-9
176.Colombel JF, Plevy S, Sandborn W, et al. Time to remission and response in adalimumabtreated patients with moderately to severely active ulcerative colitis from ultra 2. United Eur Gastroenterol J. 2013;1(1 Suppl):A219-A220. doi:10.1177/2050640613502900
177.Sandborn WJ, Wolf DC, Van Assche G, et al. Rapid onset of adalimumab and long-term efficacy among week-8 responders in adults with moderate to severe active Ulcerative Colitis: O-7. Inflamm Bowel Dis. 2011;17(Suppl_2):S4-S4. doi:10.1097/00054725-201112002-00009
178.D'Haens G, Van Assche G, Wolf D, et al. Mucosal healing in ulcerative colitis patients with week 8 response to adalimumab: Subanalysis of ultra 2. Am J Gastroenterol. 2012;107(1):S610-S611.
179.Sandborn WJ, Colombel JF, D'Haens G, et al. One-year maintenance outcomes among patients with moderately-to-severely active ulcerative colitis who responded to induction therapy with adalimumab: subgroup analyses from ULTRA 2. Aliment Pharmacol Ther. 2013;37(2):204-13. doi:10.1111/apt.12145 PubMed
180.Panaccione R, Colombel J-F, Sandborn W, et al. Sa1229 Durable Clinical Remission and Response in Adalimumab-Treated Patients With Ulcerative Colitis. Gastroenterology. 2015;148(4):S-264. doi:10.1016/s0016-5085(15)30869-6
181.van Assche G, Targan SR, Baker T, et al. Sustained Remission in Patients with Moderate to Severe Ulcerative Colitis: Results from the Phase 3 UNIFI Maintenance Study. Z Gastroenterol. 2019;57(05):P05. doi:10.1055/s-0039-1691865
182.Sands B, Peyrin-Biroulet L, Marano C, et al. Efficacy in Biologic Failure and Nonbiologic-Failure Populations in a Phase 3 Study of Ustekinumab in Moderate-Severe Ulcerative Colitis: UNIFI. Z Gastroenterol. 2019;57(05):e136-e137. doi:10.1055/s-0039-1691869
183.Alcalá M, Sulleiro S, Hernando T, García I. P0516 Efficacy of Ustekinumab At The End of Maintenance in Ulcerative Colitis Patients Receiving 6 Mg/ Kg Induction Posology. Data From Unifi Trial (Ueg Week 2020 Poster Presentations). United Eur Gastroenterol J. 2020;8(S8):144-887. doi:10.1177/2050640620927345
184.Danese S, Sands BE, Sandborn WJ, et al. LB07 Efficacy of ustekinumab subcutaneous maintenance treatment by induction-dose subgroup in the unifi study of patients with ulcerative colitis. United Eur Gastroenterol J. 2019;7(10):1415. doi:10.1177/2050640619888859
185.Panaccione R, Peyrin-Biroulet L, Danese S, et al. Impact of Response and Inflammatory Burden at Start of Maintenance Therapy on Clinical Efficacy of Ustekinumab Dosing Regimen in UC: Week 44 Results From UNIFI: 842. Am J Gastroenterol. 2019;114:S487-S488. doi:10.14309/01.ajg.0000592904.87773.5d
186.Sandborn WJ, Baert F, Danese S, et al. Efficacy and Safety of Vedolizumab Subcutaneous Formulation in a Randomized Trial of Patients With Ulcerative Colitis. Gastroenterology. 2020;158(3):562-572 e12. doi:10.1053/j.gastro.2019.08.027 PubMed
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Table 30: Summary of Additional Efficacy Results From Studies Included in the Systematic Review
End point | INSPIRE | COMMAND | |||
|---|---|---|---|---|---|
RIS 1,200 mg IV (N = 650) | Placebo (N = 325) | RIS 180 mg SC (N = 179) | RIS 360 mg SC (N = 186) | Placebo (RIS withdrawal) (N = 183) | |
Week 12 | Week 52 | ||||
No bowel urgency | |||||
n (%) | 287 (44.1) | 287 (44.1) | 96 (53.6) | 92 (49.4) | 57 (31.1) |
RD (95% CI) vs. placebo | 16.3 (10.3, 22.4) | — | 22.6 (13.1, 32.2) | 18.4 (8.8, 28.0) | — |
P value vs. placebo | < 0.00001a | — | < 0.00001a | 0.00018a | — |
No abdominal pain | |||||
n (%) | 232 (35.8) | 86 (26.5) | 84 (46.9) | 70 (37.8) | 54 (29.5) |
RD (95% CI) vs. placebo | 9.3 (3.4, 15.3) | — | 17.0 (7.4, 26.7) | 8.2 (−1.3, 17.7) | — |
P value vs. placebo | 0.00213a | — | 0.00053a | 0.08954a | — |
No nocturnal bowel movements | |||||
n (%) | 437 (67.3) | 140 (43.1) | 75 (41.9) | 81 (43.5) | 55 (30.1) |
RD (95% CI) vs. placebo | 24.2 (17.9, 30.5) | — | 12.0 (3.3, 20.6) | 14.8 (6.1, 23.5) | — |
P value vs. placebo | < 0.00001a | — | 0.00691a | 0.00090a | — |
No tenesmus | |||||
n (%) | 317 (48.7) | 98 (30.2) | 66 (36.9) | 68 (36.8) | 43 (23.5) |
RD (95% CI) vs. placebo | 18.6 (12.4, 24.8) | — | 13.1 (4.6, 21.7) | 14.4 (5.7, 23.0) | — |
P value vs. placebo | < 0.00001a | — | 0.00258a | 0.00110a | — |
Fecal incontinence (episodes per week) | |||||
Number of patients contributing to the analysis | 602 | 288 | 66 | 70 | 69 |
Change from baseline, LS mean (95% CI) | −3.84 (−4.27, −3.41) | −2.21 (−2.85, −1.57) | −3.44 (−4.72, −2.16) | −2.87 (−4.28, −1.46) | −2.77 (−4.19, −1.34) |
LS mean difference (95% CI) vs. placebo | −1.63 (−2.38, −0.87) | — | −0.68 (−2.56, 1.21) | −0.10 [-2.13, 1.92] | — |
P value vs. placebo | 0.00003a | — | 0.48140a | 0.92009a | — |
Sleep interrupted due to UC symptoms (nights per week) | |||||
Number of patients contributing to the analysis | 602 | 288 | 66 | 70 | 69 |
Change from baseline, LS mean (95% CI) | −2.49 (−2.69, −2.28) | −1.51 (−1.80, −1.21) | −2.57 (−3.12, −2.02) | −2.48 (−3.01, −1.95) | −1.77 (−2.30, −1.25) |
LS mean difference (95% CI) vs. placebo | −0.98 (−1.33, −0.63) | — | −0.80 (−1.56, −0.04) | −0.71 (−1.44, 0.02) | — |
P value vs. placebo | < 0.00001a | — | 0.03896a | 0.05788a | — |
vs. = versus.
Pharmacoeconomic Review
Abbreviations
5-ASA
5-aminosalicylic acid
AT
advanced therapy
AT-IR
inadequate response or intolerance to advanced therapies
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
CT
conventional therapy
GI
gastrointestinal
IR
inadequate response or intolerance
NMA
network meta-analysis
S1P
sphingosine 1-phosphate
SC
subcutaneous
UC
ulcerative colitis
Economic Review
The objective of the economic review undertaken by Canada’s Drug Agency (CDA-AMC) is to review and critically appraise the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of risankizumab compared to other advanced therapies (ATs) for moderate to severely active ulcerative colitis (UC) in adults who have had an inadequate response, loss of response, or were intolerant to conventional therapy (CT), a biologic treatment, or a JAK inhibitor. The sponsor submitted subgroup analyses that assessed the cost-effectiveness of risankizumab among the population with an inadequate response or intolerance (IR) to ATs, including biologics, JAK inhibitors, or sphingosine 1-phosphate (S1P) receptor modulators (the AT-IR population) and in the population with an inadequate response or intolerance to CT (the non–AT-IR population).
Item | Description |
|---|---|
Drug product | Risankizumab (Skyrizi):
|
Indication | For the treatment of adult patients with moderately to severely active ulcerative colitis who have had an inadequate response, loss of response, or were intolerant to conventional therapy, a biologic treatment, or a JAK inhibitor |
Submitted price | $4,593.14 per 600 mg vial for IV infusion $4,593.14 per 180 mg or 360 mg cartridge for SC injection |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | October 10, 2024 |
Reimbursement request | Per indication |
Sponsor | AbbVie Corporation |
Submission history | Previously reviewed: Yes Indication: Plaque psoriasis Recommendation date: May 24, 2019 Recommendation: Reimburse with clinical criteria and/or conditions Indication: Crohn disease Recommendation date: May 12, 2023 Recommendation: Reimburse with clinical criteria and/or conditions |
NOC = Notice of Compliance; SC = subcutaneous.
Summary
Risankizumab is available as 600 mg vials for IV infusion as well as 180 mg and 360 mg cartridges for subcutaneous injection. At the submitted price of $4,593.14 per vial or cartridge, the annual cost of risankizumab is expected to be $50,627 per patient in the first year of treatment and $29,958 in subsequent years, based on the Health Canada–recommended dosage for UC.
Clinical efficacy in the economic analysis was derived from a sponsor-submitted network meta-analysis (NMA), with the efficacy of risankizumab informed by the INSPIRE and COMMAND trials. Indirect evidence submitted by the sponsor was affected by several methodological limitations identified by CDA-AMC, which included likely violations of the underlying assumptions, network sparsity, imprecision, and inconsistency of results across outcomes. As a result, the relative treatment effects of risankizumab versus relevant comparators are uncertain. There is no conclusive evidence concerning which treatment may provide the preferred balance of benefits and harms.
In the sponsor’s base case, risankizumab was dominated (less effective, more costly) by multiple comparators in both populations. This finding is uncertain due to limitations in the submitted NMA. Thus, it is uncertain if, and to what extent, risankizumab provides a net benefit relative to other therapies currently funded.
Given the limitations of the sponsor-submitted indirect evidence, there is insufficient evidence to suggest risankizumab should be priced higher than other ATs for the treatment of UC. Further price reductions may be required to ensure cost-effectiveness.
CDA-AMC estimates that the budget impact of reimbursing risankizumab for the treatment of the indicated population will be approximately $13.8 million over the first 3 years of reimbursement compared to the amount currently spent on other ATs funded for the treatment of UC, with an estimated expenditure of $41.7 million on risankizumab over this period. The actual budget impact of reimbursing risankizumab will depend on the uptake of risankizumab, the comparators displaced, the underlying growth in market size of patients receiving ATs for UC, and the potential confidential pricing of comparators.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of risankizumab from the perspective of a public health care payer in Canada over a lifetime horizon (58 years). The modelled populations consisted of adults with moderately to severely active UC who were non–AT-IR or AT-IR. When both populations are considered together, they are aligned with the full Health Canada indication. Clinical data were based on the participants in the INSPIRE and COMMAND trials. The sponsor’s base-case analysis included costs related to drug acquisition, administration, conventional therapy, adverse events, surgery, and use of other health care resources.
In the sponsor’s base case, risankizumab was dominated (more costly and less effective) by multiple comparators in both patient populations. In the sponsor’s analysis of the non–AT-IR population, the following treatments were on the cost-effectiveness frontier: adalimumab, etrasimod, tofacitinib, and upadacitinib. In the sponsor’s analysis of the AT-IR population, the following treatments were on the cost-effectiveness frontier: adalimumab and upadacitinib. Additional information about the sponsor’s submission is summarized in Appendix 3.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 2; full details are provided in Appendix 4).
Table 2: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The comparative clinical efficacy and safety of risankizumab vs. other ATs is uncertain. | There is a lack of head-to-head evidence comparing risankizumab to other ATs for the treatment of UC. An NMA submitted by the sponsor reported wide CrIs for most comparisons. It was associated with several important methodological limitations, leading to inconclusive evidence concerning a preferred treatment. | CDA-AMC could not address this issue in the base case due to uncertainty in the comparative evidence. Risankizumab was dominated (more costly, less effective) by several other comparators in both populations of the sponsor’s submitted pharmacoeconomic analyses. | No scenario analysis was conducted. |
The proportion of patients receiving high and low doses of comparators is uncertain. | The proportion of patients requiring higher doses of comparators was informed by IQVIA GPM data, the literature, and the sponsor’s market research. It is unclear whether these proportions are consistent with clinical practice in Canada. | CDA-AMC could not address this issue due to a lack of alternative data on the proportions of patients with UC receiving each funded dose of the comparators. | No scenario analyses were conducted. |
AT = advanced therapy; CDA-AMC = Canada’s Drug Agency; CrI = credible interval; UC = ulcerative colitis; vs. = versus.
Note: Full details of the CDA-AMC identified issues are provided in Appendix 3.
CDA-AMC Assessment of Cost-Effectiveness
Based on the CDA-AMC clinical review, risankizumab demonstrated improvements for most outcomes when compared to placebo in the INSPIRE induction and COMMAND maintenance trials. However, there was an absence of direct evidence comparing it with active comparators. The sponsor conducted NMAs in both the non–AT-IR and AT-IR populations to inform parameters for each comparator in the economic model. These parameters included the proportion of patients achieving clinical remission or response without remission during the induction phase and maintaining it through the first year of treatment, as well as the proportion experiencing a serious infection during the maintenance phase. The CDA-AMC clinical review noted that the NMA was affected by several limitations, including likely violation of the underlying assumptions, network sparsity, imprecision, and inconsistency of results across outcomes. As a result, the relative treatment effects of risankizumab versus relevant comparators are uncertain. There is no conclusive evidence concerning which treatment may provide the preferred balance of benefits and harms. Due to this uncertainty, no reanalyses were conducted.
Summary of the Budget Impact
The sponsor submitted a budget impact analysis (BIA) to estimate the 3-year (2026 to 2028) budget impact of reimbursing risankizumab for the Health Canada–indicated population. The sponsor assumed that the payer would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an claims-based approach. The price of risankizumab was aligned with the price in the sponsor’s economic evaluation, while the prices of comparators were based on the publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in Appendix 5.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (Appendix 5). CDA-AMC estimated that 16,808 patients would be eligible for treatment with risankizumab over a 3-year period. The estimated incremental budget impact of reimbursing risankizumab is predicted to be approximately $13.8 million over the first 3 years, with an expected expenditure of $41.7 million on risankizumab. The actual budget impact of reimbursing risankizumab will depend on the uptake of risankizumab, the comparators displaced, the underlying growth in the market of patients receiving ATs for UC, and the potential confidential pricing of comparators.
Conclusion
Based on the sponsor’s submitted base case, risankizumab would be considered dominated (i.e., more costly and less effective) by multiple comparators for both populations. The estimated cost-effectiveness of risankizumab compared to other ATs is uncertain, due to limitations and inconsistency in the submitted indirect evidence comparing it with other active comparators and in the proportion of patients with UC using each funded dose of each comparator. Overall, there is insufficient evidence to justify a price premium for risankizumab compared to other ATs for the treatment of UC.
The budget impact of reimbursing risankizumab to the public drug plans in the first 3 years is estimated to be approximately $13.8 million. The 3-year expenditure on risankizumab (i.e., not accounting for current expenditure on comparators) is estimated to be $41.7 million.
References
1.Ontario Ministry of Health. Ontario drug benefit formulary/comparative drug index. 2025. Accessed May 8, 2025. https://www.formulary.health.gov.on.ca/formulary/
2.AbbVie Corporation. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Skyrizi (risankizumab injection), 150 mg in 1 mL sterile solution (150 mg/mL) subcutaneous injection; 180 mg in 1.2 mL sterile solution (150 mg/mL) subcutaneous injection; 360 mg in 2.4 mL sterile solution (150 mg/mL) subcutaneous injection; 75 mg in 0.83 mL sterile solution (90 mg/mL) subcutaneous injection; 90 mg in 1 mL sterile solution (90 mg/mL) subcutaneous injection; 600 mg in 10 mL sterile solution (60 mg/mL) for intravenous infusion. March 25, 2025.
3.Ontario Ministry of Health. Exceptional Access Program (EAP). 2025. Accessed May 8, 2025. http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx
4.AbbVie Corporation. Skyrizi (risankizumab injection): 150 mg in 1 mL sterile solution (150 mg/mL) subcutaneous injection; 180 mg in 1.2 mL sterile solution (150 mg/mL) subcutaneous injection; 360 mg in 2.4 mL sterile solution (150 mg/mL) subcutaneous injection; 75 mg in 0.83 mL sterile solution (90 mg/mL) subcutaneous injection; 90 mg in 1 mL sterile solution (90 mg/mL) subcutaneous injection; 600 mg in 10 mL sterile solution (60 mg/mL) for intravenous infusion [product monograph]. October 10, 2024.
5.AbbVie Corporation. The Relative Efficacy and Safety of Risankizumab vs. Relevant Comparators for Treating Patients with Moderately-to-Severely Active Ulcerative Colitis: A Bayesian Network Meta-analysis (NMA) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Skyrizi (risankizumab injection), 150 mg in 1 mL sterile solution (150 mg/mL) subcutaneous injection; 180 mg in 1.2 mL sterile solution (150 mg/mL) subcutaneous injection; 360 mg in 2.4 mL sterile solution (150 mg/mL) subcutaneous injection; 75 mg in 0.83 mL sterile solution (90 mg/mL) subcutaneous injection; 90 mg in 1 mL sterile solution (90 mg/mL) subcutaneous injection; 600 mg in 10 mL sterile solution (60 mg/mL) for intravenous infusion. February 21, 2025.
6.Ferrante M, Declerck S, De Hertogh G, et al. Outcome after proctocolectomy with ileal pouch-anal anastomosis for ulcerative colitis. Inflamm Bowel Dis. 2008;14(1):20-8. doi:10.1002/ibd.20278 PubMed
7.Gonzalez R, Pereyra L, Omodeo M, et al. P359 Risk factors for pouchitis after laparoscopic ileal-pouch-anal anastomosis in patients with ulcerative colitis. J Crohns Colitis. 2014;8(Supplement_1):S215-S215. doi:10.1016/s1873-9946(14)60479-7
8.Misra R, Askari A, Faiz O, Arebi N. Colectomy Rates for Ulcerative Colitis Differ between Ethnic Groups: Results from a 15-Year Nationwide Cohort Study. Can J Gastroenterol Hepatol. 2016;2016:8723949. doi:10.1155/2016/8723949 PubMed
9.Segal JP, McLaughlin SD, Faiz OD, Hart AL, Clark SK. Incidence and Long-term Implications of Prepouch Ileitis: An Observational Study. Dis Colon Rectum. 2018;61(4):472-475. doi:10.1097/dcr.0000000000000978 PubMed
10.Suzuki H, Ogawa H, Shibata C, et al. The long-term clinical course of pouchitis after total proctocolectomy and IPAA for ulcerative colitis. Dis Colon Rectum. 2012;55(3):330-6. doi:10.1097/DCR.0b013e3182417358 PubMed
11.Statistics Canada. Life expectancy and other elements of the complete life table, three-year estimates, Canada, all provinces except Prince Edward Island [sponsor supplied reference]. 2025. Accessed March 27, 2025. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401&pickMembers%5B0%5D=1.1&pickMembers%5B1%5D=3.2&pickMembers%5B2%5D=4.3&cubeTimeFrame.startYear=2013+%2F+2015&cubeTimeFrame.endYear=2017+%2F+2019&referencePeriods=20130101%2C20170101
12.Woehl A, Hawthorne AB, McEwan P. The relation between disease activity, quality of life and health utility in patients with ulcerative colitis. Gut. 2008;57(Suppl 1):A153.
13.Arseneau KO, Sultan S, Provenzale DT, et al. Do patient preferences influence decisions on treatment for patients with steroid-refractory ulcerative colitis? Clin Gastroenterol Hepatol. 2006;4(9):1135-42. doi:10.1016/j.cgh.2006.05.003 PubMed
14.Stevenson M, Archer R, Tosh J, et al. Adalimumab, etanercept, infliximab, certolizumab pegol, golimumab, tocilizumab and abatacept for the treatment of rheumatoid arthritis not previously treated with disease-modifying antirheumatic drugs and after the failure of conventional disease-modifying antirheumatic drugs only: systematic review and economic evaluation. Health Technol Assess. 2016;20(35):1-610. doi:10.3310/hta20350 PubMed
15.IQVIA. DeltaPA [sponsor supplied reference]. 2024.
16.Canada's Drug Agency. Reimbursement Review: Etrasimod (Velsipity) [sponsor supplied reference]. 2024.
17.Biogen Genentech U. S. A. Inc. Prescribing Information: Rituxan Hycela [sponsor supplied reference]. 2017.
18.Government of Canada. Job Bank. Wages for Registered nurses and registered psychiatric nurses [sponsor supplied reference]. 2024. Accessed February 26, 2022. https://www.jobbank.gc.ca/wagereport/occupation/993
19.MaRS Discovery District. Employee benefits and benefits packages: What Ontario employers should know [sponsor supplied reference]. 2015. Accessed February 1, 2022. https://www.marsdd.com/mars-library/employee-benefits-and-benefits-packages-what-ontario-employers-should-know/
20.Statistics Canada. Consumer Price Index, health and personal care, by province (monthly) [sponsor supplied reference]. 2025. Accessed March 27, 2025. http://www.statcan.gc.ca/tables-tableaux/sum-som/l01/cst01/cpis13a-eng.htm
21.Tam VC, Ko YJ, Mittmann N, et al. Cost-effectiveness of systemic therapies for metastatic pancreatic cancer. Curr Oncol. 2013;20(2):e90-e106. doi:10.3747/co.20.1223 PubMed
22.Government of Canada. Job Bank. Wages: Pharmacist in Canada [sponsor supplied reference]. 2024. Accessed February 26, 2022. https://www.jobbank.gc.ca/marketreport/wages-occupation/18196/ca
23.Millar DR, Corrie P, Hill M, Pulfer A. PCN74 A service evaluation to compare secondary care resource use between Xelox and Folfox-6 regimens in the treatment of metastatic colorectal cancer (MCRC) from a UK National Health Service (NHS) perspective. Value in Health. 2008;6(11):A483. doi:10.1016/S1098-3015(10)66610-7
24.Ontario Ministry of Health and Long Term Care. Schedule of benefits for laboratory services: (effective July 24, 2023) [sponsor supplied reference]. 2023. https://www.ontario.ca/files/2024-01/moh-ohip-schedule-of-benefits-laboratory-services-2024-01-24.pdf
25.Ontario Ministry of Health and Long Term Care. Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 24, 2023) [sponsor supplied reference]. 2024. Accessed September 1, 2024. https://www.ontario.ca/files/2024-08/moh-schedule-benefit-2024-08-30.pdf
26.PeriPharm Inc. Canadian Costing of SKYRIZI (Risankizumab) for the Treatment of Ulcerative Colitis [sponsor supplied reference]. 2024.
27.Ontario Ministry of Health and Long Term Care. Ontario Case Costing Inititative-OCCI Costing Analysis Tool [sponsor supplied reference]. 2018. Accessed May 1, 2017. https://hsim.health.gov.on.ca/hdbportal/
28.Tsai HH, Punekar YS, Morris J, Fortun P. A model of the long-term cost effectiveness of scheduled maintenance treatment with infliximab for moderate-to-severe ulcerative colitis. Aliment Pharmacol Ther. 2008;28(10):1230-9. doi:10.1111/j.1365-2036.2008.03839.x PubMed
29.I. M. S. Health Quintiles via IQVIA. Geographic Prescription Monitoring (GPM) Database [sponsor supplied reference]. 2025.
30.pan-Canadian Pharmaceutical Alliance. Abrilada (adalimumab) for rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, hidradenitis suppurativa, plaque psoriasis, uveitis. 2022. Accessed May 07, 2025. https://www.pcpacanada.ca/negotiation/21637
31.pan-Canadian Pharmaceutical Alliance. Velsipity (etrasimod) for Ulcerative Colitis. 2025. Accessed May 07, 2025. https://www.pcpacanada.ca/negotiation/22823
32.pan-Canadian Pharmaceutical Alliance. Avsola (infliximab) for Ankylosing spondylitis, plaque psoriasis, psoriatic arthritis, rheumatoid arthritis, Crohn’s disease and Ulcerative Colitis. 2020. Accessed May 07, 2025. https://www.pcpacanada.ca/negotiation/21207
33.pan-Canadian Pharmaceutical Alliance. Omvoh (mirikizumab) for Ulcerative Colitis. 2024. Accessed May 07, 2025. https://www.pcpacanada.ca/negotiation/22481
34.pan-Canadian Pharmaceutical Alliance. Zeposia (ozanimod) for Ulcerative Colitis. 2023. Accessed May 07, 2025. https://www.pcpacanada.ca/negotiation/22096
35.pan-Canadian Pharmaceutical Alliance. Xeljanz (tofacitinib) for Ulcerative Colitis. 2020. Accessed May 07, 2025. https://www.pcpacanada.ca/negotiation/21103
36.pan-Canadian Pharmaceutical Alliance. Rinvoq (upadacitinib) for Ulcerative Colitis. 2024. Accessed May 07, 2025. https://www.pcpacanada.ca/negotiation/22462
37.pan-Canadian Pharmaceutical Alliance. Wezlana (ustekinumab) for Multiple Indications. 2024. Accessed May 07, 2025. https://www.pcpacanada.ca/negotiation/22413
38.pan-Canadian Pharmaceutical Alliance. Entyvio (vedolizumab) for Ulcerative Colitis. 2017. Accessed May 07, 2025. https://www.pcpacanada.ca/negotiation/20913
39.pan-Canadian Pharmaceutical Alliance. Entyvio SC (vedolizumab) for Ulcerative Colitis. 2021. Accessed May 07, 2025. https://www.pcpacanada.ca/negotiation/21228
40.Louis E, Panaccione R, Parkes G, et al. S845 Risankizumab Induction Therapy in Patients With Moderately to Severely Active Ulcerative Colitis: Efficacy and Safety in the Randomized Phase 3 INSPIRE Study. Am J Gastroenterol. 2023;118(10S):S624-S625. doi:10.14309/01.ajg.0000953020.05891.05
41.AbbVie Corporation. Skyrizi (risankizumab injection): 150 mg in 1 mL sterile solution (150 mg/mL) subcutaneous injection; 180 mg in 1.2 mL sterile solution (150 mg/mL) subcutaneous injection; 360 mg in 2.4 mL sterile solution (150 mg/mL) subcutaneous injection; 75 mg in 0.83 mL sterile solution (90 mg/mL) subcutaneous injection; 90 mg in 1 mL sterile solution (90 mg/mL) subcutaneous injection; 600 mg in 10 mL sterile solution (60 mg/mL) for intravenous infusion [product monograph; sponsor supplied reference]. June 04, 2025. https://pdf.hres.ca/dpd_pm/00080731.PDF
42.Canada's Drug Agency. Reimbursement Review: Guselkumab (Tremfya) for Ulcerative Colitis. 2025. Accessed June 10, 2025. https://www.cda-amc.ca/guselkumab-1
43.AbbVie Corporation. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Skyrizi (risankizumab injection), 150 mg in 1 mL sterile solution (150 mg/mL) subcutaneous injection; 180 mg in 1.2 mL sterile solution (150 mg/mL) subcutaneous injection; 360 mg in 2.4 mL sterile solution (150 mg/mL) subcutaneous injection; 75 mg in 0.83 mL sterile solution (90 mg/mL) subcutaneous injection; 90 mg in 1 mL sterile solution (90 mg/mL) subcutaneous injection; 600 mg in 10 mL sterile solution (60 mg/mL) for intravenous infusion. March 26, 2025.
44.AbbVie Corporation. NCT06880744: A Study to Assess the Change in Disease Activity in Adult Participants With Moderate to Severe Ulcerative Colitis Treated With Risankizumab Compared to Vedolizumab (REVAMP). ClinicalTrials.gov; 2025. Accessed June 06, 2025. https://clinicaltrials.gov/study/NCT06880744
45.Das R, Steinhart AH. Choosing Therapies in Ulcerative Colitis. J Can Assoc Gastroenterol. 2024;7(1):9-21. doi:10.1093/jcag/gwad025 PubMed
46.Panaccione R, Lee WJ, Clark R, et al. Dose Escalation Patterns of Advanced Therapies in Crohn's Disease and Ulcerative Colitis: A Systematic Literature Review. Adv Ther. 2023;40(5):2051-2081. doi:10.1007/s12325-023-02457-6 PubMed
47.IQVIA. PharmaStat. 2025. Accessed June 11, 2025. https://www.iqvia.com/
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and CDA-AMC–participating public drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans
Table 3: CDA-AMC Cost Comparison Table for Advanced Therapies for Moderately to Severely Active Ulcerative Colitis
Drug/ Comparator | Strength | Dosage Form | Price ($) | Recommended Dosage | Average Daily Cost ($) | Average Annual Cost ($) |
|---|---|---|---|---|---|---|
Risankizumab (Skyrizi) | 180 mg/1.2 mL 360 mg/2.4 mL 600 mg/10 mL | SC injection SC injection IV infusion | 4,593.1400a 4,593.1400a 4,593.1400a | 1,200 mg IV infusion weeks 0, 4, and 8, then 180 mg or 360 mg SC week 12 and every 8 weeks thereafter | Year 1: 138.61 Thereafter: 82.02 | Year 1: 50,627 Thereafter: 29,958 |
Comparators | ||||||
Adalimumab (Humira) | 40 mg/0.8 mL | Prefilled syringe or autoinjector for SC injection | 794.1000b | 160 mg at week 0, 80 mg at week 2, then 40 mg every other week thereafter | Year 1: 66.51 Thereafter: 56.72 | Year 1: 24,291 Thereafter: 20,718 |
Adalimumab (SEBs) | 40 mg/0.8 mL | Prefilled syringe or autoinjector for SC injection | 471.2700 | 160 mg at week 0, 80 mg at week 2, then 40 mg every other week thereafter | Year 1: 39.47 Thereafter: 33.66 | Year 1: 14,180 Thereafter: 12,295 |
Etrasimod (Velsipity) | 2 mg | Tablet | 43.1000 | 2 mg daily | 43.10 | 15,742 |
Golimumab (Simponi) | 50 mg/0.5 mL 100 mg/1 mL | Prefilled syringe or autoinjector for SC injection | 1,555.1700b 1,555.1700b | 200 mg at week 0, 100 mg at week 2, then 50 mg every 4 weeks thereafter. A maintenance dose of 100 mg every few weeks can be considered. | Year 1: 65.12 Thereafter: 55.54 | Year 1: 23,786 Thereafter: 20,287 |
Infliximab (Remicade) | 100 mg | Vial for IV infusion | $987.5600b | 5 mg/kg at week 0, 2, and 6, then every 8 weeks thereafter. Dose may be adjusted up to 10 mg/kg to sustain response and remission. | Year 1: 86.76 to 141.08c Thereafter: 70.54 to 141.08 | Year 1: 31,690 to 51,529c Thereafter: 25,765 to 51,529 |
Infliximab (SEBs: Avsola, Ixifi, Remdantry, Renflexis) | 100 mg | Vial for IV infusion | $493.0000 | Year 1: 43.31 to 70.43c Thereafter: 35.21 to 70.43 | Year 1: 15,820 to 25,724c Thereafter: 12,862 to 25,724 | |
Infliximab (SEB: Inflectra) | 100 mg | Vial for IV infusion | $525.0000 | Year 1: 46.12 to 75.00c Thereafter: 37.50 to 75.00 | Year 1: 16,847 to 27,394c Thereafter: 13,697 to 27,394 | |
Infliximab (Remsima SC) | 120 mg | Prefilled syringe or pen for SC injection | 474.5100 | 5 mg/kg IV at week 0, 2, and 6, then 120 mg SC every 2 weeks starting 4 weeks after the induction regimen is completed. | Year 1:43.60d Thereafter: 33.89 | Year 1: 15,923d Thereafter: 12,380 |
Mirikizumab (Omvoh) | 100 mg 300 mg | Prefilled syringe or pen Vial for IV infusion | 1,268.0700 2,536.1400 | 300 mg IV at week 0, 4, and 8, then 200 mg SC every 4 weeks. At 12 weeks, patients without adequate response may extend induction with 300 mg IV at weeks 12, 16, and 20, then 200 mg SC every 4 weeks. | Year 1: 90.58 Thereafter: 90.58 | Year 1: 33,083 Thereafter: 33,083 |
Ozanimod (Zeposia) | 0.23 mg 0.46 mg 0.92 mg | Capsule | 78.2776 78.2776 68.4932 | 0.23 mg daily days 1 to 4, then 0.46 mg daily days 5 to 7, then 0.92 mg daily thereafter | Year 1: 68.68 Thereafter: 68.49 | Year 1: 25,086 Thereafter: 25,017 |
Tofacitinib (Xeljanz, generics) | 5 mg 10 mg | Tablet | 5.9897 21.1718 | 10 mg twice daily for at least 8 weeks, then 5 mg twice daily thereafter. 10 mg twice daily may be used for maintenance for some patients | Year 1: 11.98 to 23.96e Thereafter: 13.82 to 23.96e | Year 1: 5,046 to 8,751e Thereafter: 4,375 to 8,751e |
Upadacitinib (Rinvoq) | 15 mg 30 mg 45 mg | Tablet | 51.6810 76.9600 101.8100 | 45 mg once daily for 8 weeks, then 15 mg or 30 mg once daily thereafter | Year 1: 59.37 to 80.77 Thereafter: 51.68 to 76.96 | Year 1: 21,684 to 29,501 Thereafter: 18,876 to 28,110 |
Ustekinumab (Stelara) | 130 mg/26 mL 90 mg/mL | Vial for IV infusion Prefilled Syringe for SC injection | 2,080.0000 4,593.1400 | IV induction at week 0: 260 mg if ≤ 55 kg, 390 mg if > 55 kg to ≤ 85 kg, 520 mg if > 85 kg. Then 90 mg SC every 8 weeks. A maintenance dose of 90 mg SC every 12 weeks after an initial SC dose at week 8 may be considered for some patients. | Year 1: 67.57 to 92.82 Thereafter: 54.68 to 82.02 | Year 1: 24,681 to 33,901 Thereafter: 19,972 to 29,958 |
Ustekinumab (SEBs) | 130 mg/26 mL 90 mg/mL | Vial for IV infusion Prefilled Syringe for SC injection | 1,248.0000 2,755.8840 | Year 1: 40.54 to 55.69 Thereafter: 32.81 to 49.21 | Year 1: 14,809 to 20,341 Thereafter: 11,983 to 17,975 | |
Vedolizumab (Entyvio) (IV) | 300 mg | Vial for IV infusion | 3,711.2500b | 300 mg at week 0, 2, 6, then every 8 weeks thereafter. Some patients may benefit from 300 mg every 4 weeks if response decreases. | Year 1: 81.51 to 142.71 Thereafter: 66.27 to 132.54 | Year 1: 29,773 to 52,123 Thereafter: 24,206 to 48,412 |
Vedolizumab (Entyvio) (SC) | 108 mg/ 0.68 mL | Prefilled syringe or pen for SC injection | 927.8050b | 300 mg IV infusions at weeks 0 and 2, then 108 mg SC starting week 6 and every 2 weeks thereafter | Year 1: 81.51 Thereafter: 66.27 | Year 1: 29,773 Thereafter: 24,206 |
SC = subcutaneous; SEB = subsequent entry biologic.
Notes: All prices are from the Ontario Drug Benefit Formulary (accessed May 2025),1 unless otherwise indicated, and do not include dispensing fees. Patients are assumed to weigh 75 kg. Only strengths relevant to dosing for ulcerative colitis have been included. A year was defined as 365.25 days. To avoid overly penalizing medications with an injection scheduled on the 365th day of the year relative to those with schedules avoiding that day, the annual cost of the first year is calculated assuming a 364 day year, with an additional 1.25 days of the mean daily maintenance cost added. E.g., risankizumab year 1 cost assumes 3 * 1,200 mg doses ($27,558.84), 4 * 180 mg or 360 mg maintenance doses ($22,965.70), and an additional 1.25 days of maintenance dosing ($4,593.14 / 56 * 1.25 = $102.53). Adalimumab (Humira) year 1 cost assumes 1 * 160 mg dose ($3,176.40), 1 * 80 mg dose ($1,588.20), 24 * 40 mg dose ($19,058.40), and 1.25 days of maintenance dosing ($794.10 / 14 * 1.25 = $70.90)
aSponsor’s submitted price.2
bOntario Drug Benefit Exceptional Access Program list price.3
cInduction dosing for infliximab is assumed to be 5 mg/kg, and maintenance is assumed to be 5 or 10 mg/kg.
dAssumes the least expensive infliximab IV product has been used for induction (e.g., Avsola).
eAssumes patients receiving the 10 mg dose are using the less expensive option of 2x5 mg tablets.
Appendix 2: Input Relevant to the Economic Review
Please note that this appendix has not been copy-edited.
This section is a summary of the input received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient group input was received from the Gastrointestinal (GI) Society and from Crohn and Colitis Canada. The GI Society gathered information through online surveys, interviews, roundtables and focus groups with clinicians and patients, and one-to-one conversations, including 1 with a patient receiving risankizumab for UC. Crohn and Colitis Canada gathered information from a survey on unmet needs which included patients in Canada with moderate to severe UC), interviews with patients experienced with risankizumab, and from their own 2023 report (Impact of Inflammatory Bowel Disease in Canada). Patient groups reported patients having experience with systemic steroids, sulfasalazine and 5-aminosalicylic acid (5-ASA), corticosteroids, immunomodulators, antibiotics, biologics, JAK inhibitors, and S1P modulators. Respondents reported that sustained remission/treatment response is more important than relieving any 1 symptom and the constant concern of future flares disrupts their lives, and that systemic steroids are a burden in their UC management. A respondent noted the long time required to determine whether conventional therapies worked, and the GI Society highlighted that a tiered and patchwork approach to treatment is used across Canada, where conventional therapy must be trialled before funding access to advanced therapy, except in Quebec. The interviewed patient with risankizumab experience reported significant improvement within weeks of starting it, with the patient’s symptoms improving from “having no quality of life” to “mild and bearable,” although the patient was unable to lower their dose from 360 mg due to a flare up.
Clinician group input was received from the Canadian Inflammatory Bowel Disease (IBD) Physicians Group. The group noted that uncontrolled inflammation in UC leads to progressive bowel damage that can result in mucosal fibrosis, functional bowel issues, structure formation, and critically, a significantly increased long-term risk of colorectal cancer. For moderately to severely active UC, the clinician group indicated that initial induction of remission with systemic corticosteroids is recommended, but that steroid therapy cannot be used for maintenance given long-term safety concerns and the burden of exposure on patients. Guidelines recommend that maintenance of remission should continue with the same drug in which remission was induced. Therefore, the clinician group endorsed the induction of remission with advanced agents including biologics, JAK inhibitors, or S1P modulators, noting that patients with moderately to severely active UC may not respond to 5-ASA therapy, and that immunomodulators such as azathioprine or methotrexate are not recommended for induction of remission due to lack of efficacy. While noting that advanced therapies have improved efficacy compared to conventional therapy, all are associated with unique efficacy and safety concerns, and 20% to 30% of patients do not respond to therapy or lose response over time. The group therefore notes significant need remains for additional efficacious, durable, and safe options, and believes risankizumab should be used as the first choice in patients with moderately to severely active UC who have failed or are not candidates for conventional therapy, or 1 or more advanced therapies.
Input from CDA-AMC–participating drug plans noted that 3 other IL-23 inhibitors are available, wondering what unmet needs risankizumab might fill. The plans also noted that an extended induction to 24 weeks was permitted in the trial but not indicated by Health Canada.
Several of these concerns were addressed in the sponsor’s model:
The submitted model included both clinical remission and response without remission as health states based on Mayo score improvement, with the impact on patient quality of life captured in utility values.
CDA-AMC was unable to address the following concerns:
Owing to uncertainty in the submitted indirect comparative evidence, CDA-AMC was unable to address whether risankizumab resolves the treatment gaps identified by patient and clinician groups.
Appendix 3: Summary of the Sponsor’s Submission
Please note that this appendix has not been copy-edited.
Summary of the Sponsor’s Economic Evaluation
For the pharmaceutical reviews program, clinical and economic information is submitted to CDA-AMC by the sponsor. The CDA-AMC health economics team reviews the submitted economic information and appraises the information in collaboration with clinical experts and the clinical review team to evaluate key assumptions, influential parameters, and the overall rigour of the economic submission. Based on what the team learns through this process, adjustments may be made to the sponsor’s model to produce the CDA-AMC base case. The CDA-AMC base case represents the team’s current understanding of the clinical condition, clinical evidence currently available, and best interpretation of the economic evidence based on the information provided.
For the review of risankizumab, the sponsor provided a cost-utility analysis and a BIA. The sponsor’s economic submission is summarized in Table 4.
Table 4: Key Components of the Sponsor’s Economic Evaluation
Component | Description |
|---|---|
Treatment information | |
Drug under review | Risankizumab (Skyrizi)
Note: other sizes of risankizumab exist but are not relevant to this review. |
Submitted price of drug under review | $4,593.14 per 600 mg vial for IV infusion $4,593.14 per 180 mg or 360 mg cartridge for SC injection |
Regimen | Risankizumab 1,200 mg IV at weeks 0, 4, and 8, then 180 mg or 360 mg SC every 8 weeks4 |
Annual cost of drug under review | $27,559 per patient per 12-week induction period, then $29,855 per patient per year (note: a modelled year was 364 days [52 weeks]) |
Model information | |
Type of economic evaluation | Cost-utility analysis Decision tree followed by Markov model |
Treatment | Risankizumab 1,200 mg IV at weeks 0, 4, and 8, then SC 180 mg or 360 mg every 8 weeks |
Included comparators |
|
Perspective | Publicly funded health care payer perspective |
Time horizon | Lifetime (58 years) |
Cycle length | The decision tree is 12 weeks, followed by the Markov model with 4-week cycles. |
Modelled populations | Adult patients with moderately to severely active UC who have had: 1. an inadequate response or intolerance to CT (Non-AT-IR) 2. an inadequate response or intolerance to advanced therapy, including biologics, JAKs and/or S1P receptor modulators (AT-IR) |
Characteristics of modelled population | Age and gender ratios were derived from the M16 to 066 and M16 to 067 trials, while body weight was based on the weighted average weights of all trials in the NMA. Non-AT-IR population mean age: 42.5 years (62% male, 38% female, mean weight 73.5kg) AT-IR population mean age: 41.8 years (58% male, 42% female, mean weight 73.5 kg) |
Model health states | Decision tree: death, nonresponder and responder (with responders achieving either remission or response without remission). Markov model: remission, response without remission, active UC, first surgery, post first surgery remission, post first surgery complications, second surgery, post second surgery remission, and death. For additional information, refer to Model Structure (Figure 4 and Figure 5). |
Data sources | |
Comparative efficacy |
|
Natural history and/or clinical pathway |
|
Health-related utilities and disutilities |
|
Costs |
|
Summary of the submitted results | |
Base-case results | Risankizumab was dominated (being more costly and less effective) by multiple comparators for both populations. The following treatments were on the cost-effectiveness frontier:
|
Scenario analysis results |
|
AE = adverse event; AT = advanced therapy; CT = conventional therapy; ICER = incremental cost-effectiveness ratio; IR = intolerant or inadequate response; NMA = network meta-analysis; QALY = quality-adjusted life-years; S1P = sphingosine-1-phosphate; SC = subcutaneous; UC = ulcerative colitis.
aThese scenarios included varying the discount rate, time horizon, or proportion of patients on high-dose maintenance treatment, assuming a societal perspective, including patients who remained on placebo in the maintenance phase, extending the induction period to 24 weeks, removing treatment sequencing, including the cost of branded adalimumab, and adding extra utility benefits for risankizumab.
Model Structure
The sponsor submitted a decision tree followed by a Markov model.2 The decision tree (Figure 4) for the base case consisted of patients who achieved either remission or response without remission, or who were nonresponders, or who died. For the scenario where an extended induction was possible, patients who were initially nonresponders could then achieve remission, response without remission, or remain in active UC. In the base case, the decision tree lasted 12 weeks, while in the extended induction scenario, it lasted up to 24 weeks.
Responding patients then entered the Markov model (Figure 5) in either the remission or response without remission health states and were assumed to continue on maintenance dosing of their original therapy until loss of response or death. The Markov had 4-week long cycles. Patients not achieving a response or who later lost response were assumed to receive an additional line of advanced therapy (i.e., treatment sequencing), before entering the active UC or later health states. This second line advanced therapy (AT) was assumed to be tofacitinib for all patients except those initially treated with tofacitinib, who were assumed to receive vedolizumab. Patients not responding to this second line of AT were assumed to enter the active UC health state on CT alone. Patients in active UC could transition to surgery states, starting with a first surgery, after which they could be in post first surgery remission or post first surgery complications, with those in the latter state able to have a second surgery and achieve post second surgery remission. Patients in the surgery health states were assumed to stop all drug treatments including CT for the remainder of the time horizon.
Table 5: Summary of the Sponsor’s Economic Evaluation Results — Non–AT-IR Population
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/ QALY) |
|---|---|---|---|
Adalimumab 160/80 | 145,053 | 12.2823 | Reference |
Etrasimod 2 | 156,700 | 12.5926 | 37,536 |
Tofacitinib 10 | 170,002 | 12.8025 | 63,374 |
Upadacitinib 45 | 242,965 | 13.6855 | 82,627 |
Dominated Treatments | |||
Ozanimod 0.92 | 157,997 | 12.2627 | Dominated by adalimumab 160/80 |
Golimumab 200/100 | 160,717 | 12.4213 | Dominated by etrasimod 2 |
Ustekinumab 6 | 161,410 | 12.5264 | Dominated by etrasimod 2 |
Infliximab 5 | 166,364 | 12.4237 | Dominated by etrasimod 2 |
Vedolizumab 108 | 185,094 | 12.7122 | Dominated by tofacitinib 10 |
Vedolizumab 300 | 199,347 | 12.6110 | Dominated by tofacitinib 10 |
Risankizumab 1,200 | 200,531 | 12.4675 | Dominated by tofacitinib 10 |
Mirikizumab 300 | 206,809 | 12.6452 | Dominated by tofacitinib 10 |
AT = advanced therapy; ICER = incremental cost-effectiveness ratio; IR = intolerant or inadequate response; QALY = quality-adjusted life-year.
Note: CDA-AMC has omitted listing comparators as dominating others when the comparator is itself dominated or extendedly dominated.
Source: Sponsor’s pharmacoeconomic submission.2
Figure 1: Decision Tree Model Structure
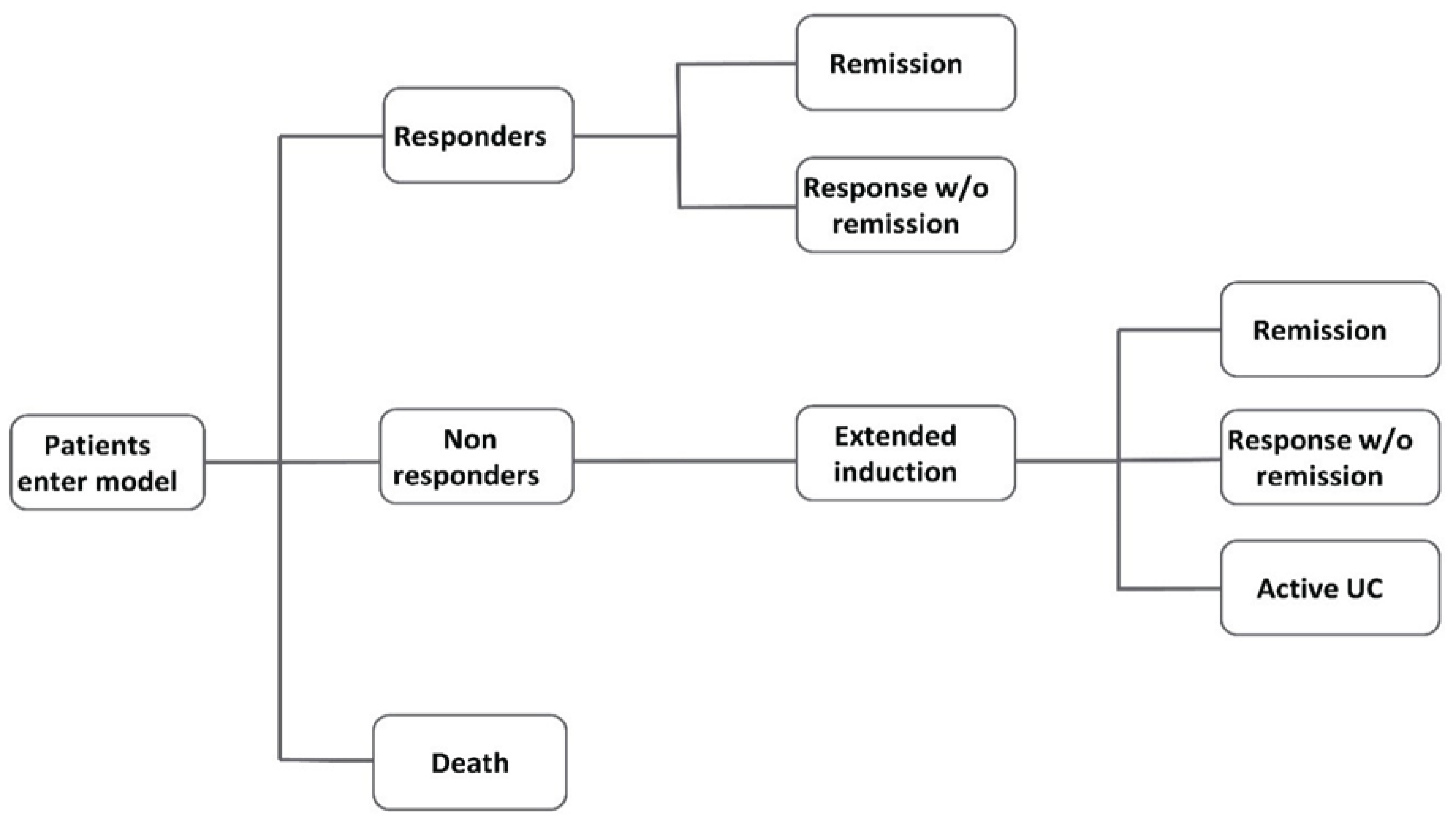
UC = ulcerative colitis.
Note: the extended induction section of the decision tree only applies to the sponsor’s extended duration scenario. In the sponsor’s base case, nonresponders received an additional line of AT before entering the Markov model.
Source: Sponsor’s pharmacoeconomic submission.2
Table 6: Summary of the Sponsor’s Economic Evaluation Results — AT-IR Population
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/ QALY) |
|---|---|---|---|
Adalimumab 160/80 | 150,759 | 12.5791 | Reference |
Upadacitinib 45 | 232,813 | 13.7044 | 72,915 |
Dominated Treatments | |||
Ustekinumab 6 | 153,919 | 12.3941 | Dominated by adalimumab 80/160 |
Etrasimod 2 | 155,093 | 12.5435 | Dominated by adalimumab 80/160 |
Ozanimod 0.92 | 163,518 | 12.4229 | Dominated by adalimumab 80/160 |
Tofacitinib 10 | 167,246 | 12.6528 | Extendedly dominated through adalimumab and upadacitinib |
Vedolizumab 108 | 178,161 | 12.6705 | Extendedly dominated through adalimumab and upadacitinib |
Mirikizumab 300 | 179,284 | 12.4393 | Dominated by adalimumab 80/160 |
Risankizumab 1,200 | 187,054 | 12.3282 | Dominated by adalimumab 80/160 |
Vedolizumab 300 | 190,468 | 12.5444 | Dominated by adalimumab 80/160 |
AT = advanced therapy; ICER = incremental cost-effectiveness ratio; IR = intolerant or inadequate response; QALY = quality-adjusted life-year.
Note: CDA-AMC has omitted listing comparators as dominating others when the comparator is itself dominated or extendedly dominated.
Source: Sponsor’s pharmacoeconomic submission.2
Figure 2: Markov Model Structure
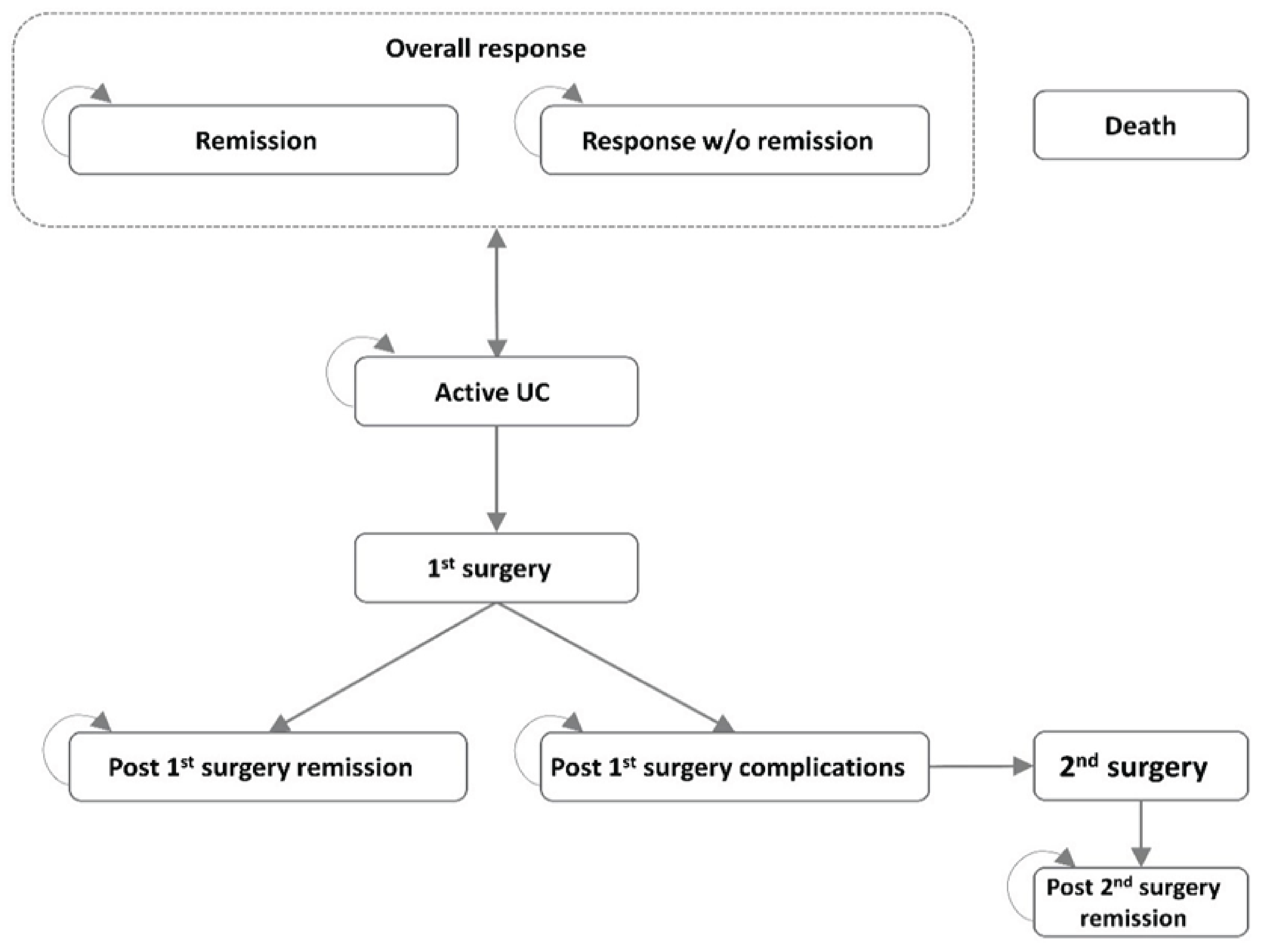
UC = ulcerative colitis.
Source: Sponsor’s pharmacoeconomic submission.2
Appendix 4: Additional Details of CDA-AMC Reanalyses
Please note that this appendix has not been copy-edited.
Clinical Data in the Economic Model
The CDA-AMC clinical review found that risankizumab demonstrated improvements for most outcomes when compared to placebo in the INSPIRE induction and COMMAND maintenance trials. However, there was an absence of direct evidence against active comparators. The sponsor conducted NMAs in both the non-AT-IR and AT-IR populations to inform parameters for each comparator in the economic model, including the proportion of patients achieving clinical remission or response without remission (equal to the proportion in response minus the proportion in remission) in the induction phase and maintaining it through the first year of treatment, as well as the proportion experiencing a serious infection during the maintenance phase. The CDA-AMC clinical review noted the NMA was affected by several limitations, which included likely violation of the underlying assumptions, network sparsity, imprecision, and inconsistency of results across outcomes. As a result, the relative treatment effects of risankizumab versus relevant comparators is subject to uncertainty and there is no conclusive evidence of which treatment may provide the preferred balance of benefits and harms. Consequently, the estimates of the probability of treatment remission and response, as well as the maintenance of such improvements used in the model are highly uncertain.
Owning to this uncertainty, no reanalyses were conducted.
Key Issues of the Submitted Economic Evaluation
CDA-AMC identified the following key issues with the sponsor’s analysis:
The comparative clinical efficacy and safety of risankizumab is uncertain: There was no head-to-head evidence comparing risankizumab to other advanced therapies used to treat UC in Canada. To inform comparative efficacy, the sponsor conducted NMAs that assessed the short-term comparative efficacy of risankizumab versus other advanced therapies in the non-AT-IR and AT-IR populations separately, for the induction period and maintenance period (generally ending 1 year after treatment initiation). According to the CDA-AMC clinical review, for the AT-IR population, upadacitinib was favoured over risankizumab for clinical response, remission, and endoscopic improvement in the induction and maintenance phases, with vedolizumab also favoured over risankizumab for clinical response and endoscopic remission. For the non-AT-IR population, risankizumab was favoured over adalimumab and mirikizumab for clinical response and endoscopic improvement in induction, however similar findings were not maintained in the maintenance phase. In the full population, risankizumab was favoured over several comparators (adalimumab, etrasimod, ozanimod, and tofacitinib 10) for serious adverse events in the induction phase, while etrasimod was favoured over risankizumab for serious infections, again with similar findings not observed in the maintenance phase. All other comparisons were less consistent across outcomes and/or had wide CrI that included the possibility of either risankizumab or the comparator being favoured. The NMA comparing risankizumab to other relevant comparators was affected by several limitations, which included likely violation of the underlying assumptions, network sparsity, imprecision, and inconsistency of results across outcomes. As a result, the relative treatment effects of risankizumab versus relevant comparators are are subject to uncertainty and there is no conclusive evidence of which treatment may provide the preferred balance of benefits and harms.
Additionally, as assumed in the sponsor’s BIA, some comparators are used more frequently than stipulated in their product monographs for some patients in Canada. As such, these comparators may be associated with both increased costs and increased efficacy compared to how they are represented in the submitted model and the trials included in the NMA. Thus, there is further uncertainty in the cost-effectiveness of risankizumab compared to other advanced therapies as currently reimbursed in Canada.
Given the lack of direct evidence, limitations with the sponsor’s NMAs and lack of clinical evidence to inform variations in clinical practice that differ from what was used in clinical trials, the comparative efficacy and cost-effectiveness of risankizumab relative to other advanced therapies reimbursed for the treatment of moderately to severely active UC remains highly uncertain in both the non-AT-IR and AT-IR populations. As a result, it is uncertain if, and to what extent, risankizumab provides a net benefit relative to other therapies currently funded. CDA-AMC was unable to address this limitation in reanalysis.
The proportion of patients receiving high and low dosing of comparators is uncertain: The sponsor estimated varying proportions of patients would require the highest doses recommended in the product monograph and funded by public drug plans, when such options were available: ██% for risankizumab, ██% for golimumab, ██% for infliximab, ██% for tofacitinib, ██% for upadacitinib, 100% for ustekinumab, and 37% to 49% for vedolizumab. The proportion of patients requiring these higher doses was based on assumption for risankizumab, while proportions for other comparators were reportedly based on IQVIA GPM data,29 the literature, or the sponsor’s market research.2 It is unclear whether the proportions of patients using each funded dose of each comparator are consistent with clinical practice in Canada.
CDA-AMC did not address this limitation in reanalysis, as it would not resolve the uncertainty in the comparative efficacy, safety, and cost-effectiveness of risankizumab noted in the previous limitation.
Additional issues were identified but were not considered to be key issues:
The sponsor’s model had additional uncertainty: During the review process, additional issues were identified with the sponsor’s model but were not assessed in detail or addressed in reanalysis given the uncertainty in the comparative clinical evidence. For example, these issues include the lack of long-term evidence available, uncertainty in utility values derived from non-Canadian sources, the assumption of no vial sharing, and uncertainty in the choice, efficacy, safety, and number of subsequent therapies.
Addressing these limitations would not resolve the uncertainty in the clinical evidence and thus would not improve conclusions regarding the comparative efficacy, safety, and cost-effectiveness of risankizumab versus its comparators.
Issues for Consideration
The sponsor’s analyses rely on publicly accessible list prices and do not reflect existing confidential prices negotiated by public plans. Given that multiple subsequent entry adalimumab products (e.g., Abrilada30), etrasimod,31 golimumab, multiple subsequent entry infliximab products (e.g., Avsola32), mirikizumab,33 ozanimod,34 tofacitinib,35 upadacitinib,36 multiple subsequent entry ustekinumab products (e.g., Wezlana37), and vedolizumab38,39 have successfully undergone price negotiations for the treatment of ulcerative colitis, it is likely that the current costs paid by public drug plans for these treatments are lower than the public list prices.
In the INSPIRE induction trial,40 some patients randomized to risankizumab who had not responded by week 12 continued induction up to week 24. However, in the product monograph,41 the recommended induction period for risankizumab is 12 weeks. The cost-effectiveness of risankizumab when this off-label extended induction of up to 24 weeks is used was not reviewed.
The sponsor emphasized that endoscopic improvement was an important outcome, indicating that risankizumab should be considered as a first-line AT due to endoscopic improvement demonstrated in the pivotal trials against placebo.2 However, endoscopic improvement was not included within the submitted economic model and potential benefit to endoscopic improvement reported for risankizumab versus some of the comparators in the sponsor’s submitted NMA during the induction period for the non-AT-IR population were not detected in the NMA for the maintenance period.5 Refer to CDA-AMC clinical report.
Guselkumab is currently under review by CDA-AMC for the treatment of adults with moderately to severely active UC.42 Should guselkumab become funded, it would be a comparator to risankizumab with an unknown relative cost-effectiveness.
Appendix 5: Budget Impact Analysis
Please note that this appendix has not been copy-edited.
Summary of the Submitted BIA
The sponsor submitted a BIA43 that estimated the expected incremental budgetary impact of reimbursing risankizumab for the treatment of adult patients with moderately to severely active UC who have had an inadequate response, loss of response, or were intolerant to CT or advanced therapies.
The BIA was conducted from the perspective of public drug plan payers over a 3-year time horizon (2026 to 2028), with 2025 as the base year. The sponsor’s estimate reflects the aggregated results from the jurisdictional provincial budgets (excluding Quebec) as well as the Non-Insured Health Benefits Program. The sponsor estimated the eligible population using a claims-based approach. The sponsor used IQVIA GPM data from 2023 to 2024 to estimate the market size, projecting the number of patients receiving each treatment using a simple exponential smoothing forecast, and supplementing with IQVIA Pharmastat data (Q4 2024) to inform public coverage proportions. Market share for mirikizumab and etrasimod, which are new enough that forecasts from previous years were not feasible, were based on the sponsor’s internal research and assumed to have the same public funding proportions as upadacitinib. The sponsor’s base case included drug acquisition costs, dispensing fees, markups, and assumed wastage of excess medication in vials, where applicable. The market uptake for risankizumab was estimated using the sponsor’s internal projections, with ████% of uptake projected to displace the anti-integrins, ████% for the anti-TNFs, ████% from the anti-IL-12/23s and ███% from the JAK inhibitors and S1P receptor modulators. The key inputs to the BIA are documented in Table 7.
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Number of publicly funded patients using advanced therapies for UC | 14,153 / 15,533 / 16,808 |
Number of patients eligible for drug under review | 14,153 / 15,533 / 16,808 |
Market shares (reference scenario) | |
Risankizumab (Skyrizi) | 0% / 0% / 0% |
Adalimumab (Humira, biosimilars) | ███% / ███% / ███% |
Etrasimod (Velsipity) | ███% / ███% / ███% |
Golimumab (Simponi) | ███% / ███% / ███% |
Infliximab (Remicade, biosimilars) | ███% / ███% / ███% |
Mirikizumab (Omvoh) | ███% / ███% / ███% |
Ozanimod (Zeposia) | ███% / ███% / ███% |
Tofacitinib (Xeljanz, biosimilars) | ███% / ███% / ███% |
Upadacitinib (Rinvoq) | ███% / ███% / ███% |
Ustekinumab (Stelara, biosimilars) | ███% / ███% / ███% |
Vedolizumab (Entyvio) | ███% / ███% / ███% |
Market shares (new drug scenario)b | |
Risankizumab (Skyrizi) | ███% / ███% / ███% |
Adalimumab (Humira, biosimilars) | ███% / ███% / ███% |
Etrasimod (Velsipity) | ███% / ███% / ███% |
Golimumab (Simponi) | ███% / ███% / ███% |
Infliximab (Remicade, biosimilars) | ███% / ███% / ███% |
Mirikizumab (Omvoh) | ███% / ███% / ███% |
Ozanimod (Zeposia) | ███% / ███% / ███% |
Tofacitinib (Xeljanz, biosimilars) | ███% / ███% / ███% |
Upadacitinib (Rinvoq) | ███% / ███% / ███% |
Ustekinumab (Stelara, biosimilars) | ███% / ███% / ███% |
Vedolizumab (Entyvio) | ███% / ███% / ███% |
Cost of treatment per patient per year (induction Year / maintenance year) | |
Risankizumab (50% high dose) | $50,607 / $29,937 |
Adalimumab (39% weekly dosing) | $18,596 / $17,078 |
Etrasimod | $15,732 / $15,732 |
Golimumab (47% high dose) | $23,772 / $20,273 |
Infliximab (71% high dose, 6% regular dose q6w, 2% regular dose q.4.w.) | $28,921 / $21,652 |
Mirikizumab | $33,060 / $33,060 |
Ozanimod | $25,000 / $25,000 |
Tofacitinib (Xeljanz, 28% high dose)a | $23,484 / $22,018 |
Tofacitinib (generics, 28% high dose)a | $8,712 / $7,514 |
Upadacitinib (73% high dose) | $27,389 / $25,618 |
Ustekinumab (100% high dose) | $26,706 / $26,706 |
Vedolizumab (43% high dose, 15% get SC) | $38,342 / $31,778 |
q = every; SC = subcutaneous; UC = ulcerative colitis; w = weeks.
aThe sponsor has assumed that all biologic products which no longer have market exclusivity are funded at the price of the least expensive biosimilar. However, for tofacitinib, the sponsor assumed public plans would fund the full price of Xeljanz for patients receiving that brand.
b50% of the uptake of risankizumab was projected to come from the anti-integrins (vedolizumab), 30% from the anti-TNFs (adalimumab, golimumab, infliximab), 15% from the anti-IL-12/23s (ustekinumab, mirikizumab), and 5% from the JAK inhibitors and S1PR modulators (ozanimod, tofacitinib, upadacitinib, estrasimod).
The sponsor estimated the 3-year incremental budget impact associated with reimbursing risankizumab would be $13,441,343 (year 1 = $4,882,332; year 2 = $4,617,932; year 3 = $3,941,079).
Key Issues of the Submitted BIA
CDA-AMC identified several key issues to the sponsor’s analysis that have notable implications on the results of the BIA:
Use of claims-based approach to estimate market size is uncertain: The sponsor estimated the market size of patients using advanced therapies for UC by using IQVIA GPM data on the number of users of each therapy in Canada (excluding Quebec) from January 2023 to December 2024 to project the number of patients on each therapy through to December 2028. The method used by the sponsor to determine which patients were using the comparators for UC rather than for other indications (e.g., Crohn disease) was not specified. The number of patients was then converted to the number of publicly reimbursed patients using proportions derived from IQVIA Pharmastat data (Q4 2024), which does not allow for filtering by indication. Forecasts were based on simple exponential smoothing for all comparators with the exception of biosimilar adalimumab, which was assumed to hold steady, as well as mirikizumab and estrasimod, both of which were deemed too new for exponential smoothing and were instead based on internal research and upadacitinib’s public coverage rates in Q4 2024. The method is associated with uncertainty in the proportion of patients who are publicly reimbursed specifically for UC, the proportion who will use each comparator in future, and the total number of patients being publicly reimbursed for advanced therapies for UC. For example, in forecasting from 2023 to 2024 public claims data, the sponsor assumes that upadacitinib uptake will continue to grow at the same rate through 2028 as it did when entering the publicly reimbursed market in 2023. This assumption leads to the sponsor’s model projecting an average increase of 112 patients per month using any AT for UC during the forecasted period, with upadacitinib itself gaining 117 new patients per month. Between 2025 and 2028, the model projects the overall UC AT market to increase by 32% (16,808/12,725). While clinical expert opinion obtained by CDA-AMC agreed that the overall market for advanced therapies for UC is increasing due to patients spending less time trialing 5-ASA, having easier access to advanced therapies, and more aggressive treatment paradigms, a 32% increase in 3 years was deemed implausible, as was such a steep and sustained estimation of upadacitinib’s uptake. Of note, using the patient number override provided within the submitted BIA overrides patient numbers for all jurisdictions, breaking the model’s ability to report meaningful pan-Canadian results.
CDA-AMC could not adjust for this limitation in reanalyses due to a lack of data on the future uptake of upadacitinib and due to the structure and programming of the submitted model making the testing of alternative assumptions unfeasible within the time frame of this review. Therefore, number of eligible patients in reanalyses may continue to be overestimated.
The comparators being displaced are uncertain: The sponsor assumed, based on internal projections, that ████% of the uptake of risankizumab would be displacing anti-integrins (vedolizumab), ████% from the anti-TNFs (adalimumab, golimumab, infliximab), ████% from the anti-IL-12/23s (ustekinumab, mirikizumab), and ████% from the JAK inhibitors and S1P modulators (ozanimod, tofacitinib, upadacitinib, estrasimod). The sponsor’s rationale for such a large displacement of vedolizumab was based on vedolizumab’s underlying market share, and the sponsor’s estimation that risankizumab has an advantage over vedolizumab in the non-AT-IR population, and a planned head-to-head trial comparing risankizumab to vedolizumab. CDA-AMC notes that this trial is anticipated to begin in June 2025 and reach primary completion in September 2028,44 thus finding are unlikely to impact market uptake within the time horizon of the current BIA. Clinical expert input obtained by CDA-AMC disagreed that such a large proportion of risankizumab’s uptake would come from vedolizumab, indicating vedolizumab is often preferred due to its gut-specific mechanism of action and its association with safety.45 The clinical expert input indicated that more uptake from anti-TNF comparators would be expected instead.
In reanalysis, CDA-AMC assumed that 40% of risankizumab’s uptake would come from vedolizumab (the only anti-integrin) and 40% would come from the anti-TNF comparators.
The proportions of patients using high or off-label escalated dosing are uncertain: The submitted BIA includes comparators with multiple recommended dosing regimens (i.e., risankizumab, golimumab, infliximab, tofacitinib, ustekinumab, upadacitinib, and vedolizumab), and also considers off-label administration frequency for some comparators (i.e., adalimumab, infliximab, ustekinumab). Clinical expert input obtained by CDA-AMC considered most of these assumptions to be appropriate; however, noted that the proportion of patients using infliximab 10 mg every 8 weeks (████% in the sponsor’s base case) may be overestimated, while the proportion of patients using upadacitinib 30 mg daily (████%) was likely underestimated. Clinical expert input estimated that approximately 30% of patients were likely using the 5 mg/kg every 8 week dosing, while the remainder would use either 10 mg/kg every 8 weeks, or 5 mg/kg every 4 to 6 weeks. CDA-AMC noted that a systematic review of dose escalation patterns in advanced therapies in Crohn and UC46 reported that 57% of patients in the US and Canada using infliximab ended follow-up on a dose of 10 mg/kg every 8 weeks. For upadacitinib, clinical expert input obtained by CDA-AMC indicated that approximately 85% of patients are using the 30 mg daily dose, rather than ████%. CDA-AMC notes that the relative safety and efficacy between comparators reported in the NMA was based on their dosing in clinical trials, which is generally not consistent with off-label increases in dosing frequency. Thus the relative cost-effectiveness of the comparators when including off-label dosing is unknown. CDA-AMC also notes that risankizumab is flat-priced such that the cost of the 180 mg and 360 mg every 8 week regimens are the same and thus assumptions altering the proportion of patients using each dose do not change the anticipated budgetary impact. However, any off-label increases in frequency of risankizumab administration would increase its budgetary cost.
In reanalysis. CDA-AMC assumed 57% of patients using infliximab were receiving the 10 mg/kg every 8 weeks dose which, due to further calculations in the submitted model, resulted in approximately 32% of patients receiving the 5 mg/kg every 8 week dose, 9% receiving 5 mg/kg every 6 weeks, and 3% every 4 weeks. CDA-AMC also assumed that 85% of patients using ustekinumab would receive 30 mg daily. For consistency with the submitted pharmacoeconomic analysis and the dosing used to inform relative safety and efficacy in the submitted NMA, CDA-AMC also included a scenario removing off-label frequency increases.
The cost of tofacitinib is likely overestimated: While the sponsor’s cost-utility analysis assumed that all use of molecules which have lost exclusivity was at the price of the least expensive generic or biosimilar product, the sponsor’s BIA only makes this assumption for biologic products; patients receiving the Xeljanz brand of tofacitinib are assumed to be reimbursed for the full cost of Xeljanz, despite generic tofacitinib being interchangeable. The Ontario Drug Benefit formulary, for example, only reimburses originator brand products for the price of their interchangeable generics, and thus the cost to the drug plan for Xeljanz is the same as that of generic tofacitinib.1 Additionally, at $21 per tablet, the publicly listed unit price of 10 mg generic tofacitinib is more than double the unit price of 5 mg tofacitinib ($6 per tablet). It is likely that many patients are using 2 5 mg tablets to make up a 10 mg dose of tofacitinib. This assumption is supported by data from the IQVIA Pharmastat database, which indicated that more than 95% of publicly funded tofacitinib 5 mg and 10 mg units reimbursed in Q1 of 2025 were 5 mg tablets.47
In reanalysis, CDA-AMC assumed that tofacitinib would be reimbursed at the price of the generic product, using 2 5 mg tablets.
The price of drugs paid by public drug plans is uncertain: Both the sponsor’s analyses and those conducted by CDA-AMC are based on publicly available list prices for all comparators. Confidential negotiated prices are available for most comparators.30,31,33-39 Thus, the actual costs paid by the public drug plans for advanced therapies for UC are unknown. Depending on the negotiated prices, reimbursing risankizumab may lead to a higher budgetary impact than estimated.
CDA-AMC was unable to incorporate the presence of confidential negotiated prices in the reanalysis.
Poor modelling practices were employed. The sponsor’s submitted model included numerous IFERROR statements, which lead to situations in which the parameter value is overwritten with an alternative value without alerting the user to the automatized overwriting. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impractical. It remains unclear whether the model is running inappropriately by overriding errors.
CDA-AMC was unable to address this limitation.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analyses by making changes in model parameter values and assumptions, in consultation with clinical experts, as outlined in Table 8. Additionally, CDA-AMC removed dispensing fees and markups from the base-case reanalysis.
Table 8: Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Remove dispensing fees and markups | Included | Excluded |
2. Displacement of comparators (% risankizumab share coming from each class) | Anti-Integrins: ████% Anti-TNFs: ████% Anti-IL-12/23s: ████% JAKs/S1Ps: █████% | Anti-Integrins: 40% Anti-TNFs: 40% Anti-IL-12/23s: 15% JAKs/S1Ps: 5% |
3. Proportion patients on higher dose of infliximab and upadacitinib | Infliximab: ████% use 10 mg/kg q.8.w., ███% 5 mg/kg q.6.w., ████% using 5 mg/kg q.4.w.a Upadacitinib: ████% using 30 mg daily | Infliximab: 57% using 10 mg/kg q.8.w., 8.6% 5 mg/kg q.6.w., 2.58% using 5 mg/kg q.4.w., 31.8% 5 mg/kg q.8.w.a Upadacitinib: 85% use 30 mg daily |
4. Tofacitinib pricing | Xeljanz unit price for patients using Xeljanz 10 mg tablets to achieve 10 mg doses | Generic tofacitinib unit price used for all patients using 2x5 mg tablets to achieve 10 mg doses |
CDA-AMC base case | ― | Reanalysis 1 + 2 + 3 + 4 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; q.#.w. = every # weeks.
Note: CDA-AMC was unable to resolve the issues with uncertainty in the market shares and growth in market size derived from the sponsor’s claims-based approach, nor with issues regarding confidential pricing of comparators.
aThe sponsor’s model assumes that 20% of patients receiving infliximab who are not using 10 mg q.8.w. are using 5 mg/kg q.6.w. and 6% of them are using 5 mg/kg q.4.w. For example, in the sponsor’s base case, this results in 71.4% of patients using 10 mg/kg q.8.w., 5.7% using 5 mg/kg q.6.w. ([100 to 71.4]*20%), 1.7% using 5 mg/kg q.4.w. ([100% to 71.4%]*6%), and the remaining 21% using 5 mg/kg q.8.w. Altering the proportion of patients receiving 10 mg/kg q.8.w. dosing also alters the proportion using the other 3 dosing schedules.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 9 and a more detailed breakdown is presented in Table 10. In the CDA-AMC base case, the 3-year budget impact of reimbursing risankizumab for the treatment of adult patients with moderately to severely active UC who have had an inadequate response, loss of response, or were intolerant to CT or advanced therapies was $13,802,682 (year 1: $4,630,458, year 2: $4,749,029, year 3: $4,423,195). CDA-AMC estimated that 16,808 patients would be eligible for treatment with risankizumab by year 3. Although this may be an overestimate of the number of eligible patients due to uncertainty in the sponsor’s projected claims data, it is unclear how the budgetary impact of reimbursing risankizumab would be impacted due to differences in comparator pricing.
Table 9: Summary of the Stepped Analysis of the CDA-AMC Base Case
Stepped analysis | Three-year total ($) |
|---|---|
Sponsor’s submitted base case | 13,441,343 |
CDA-AMC reanalysis 1: dispensing fees and markups | 12,207,087 |
CDA-AMC reanalysis 2: displacement of comparators | 14,561,455 |
CDA-AMC reanalysis 3: proportion high-dose infliximab and upadacitinib | 13,793,498 |
CDA-AMC reanalysis 4: tofacitinib costs | 13,506,838 |
CDA-AMC base case: Reanalysis 1 + 2 + 3 + 4 | 13,802,682 |
CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments
CDA-AMC used the CDA-AMC base case to conduct scenario analyses to explore uncertainty in the estimated budget impact of reimbursing risankizumab. The results are provided in Table 10.
Off-label dose frequency increases were removed for consistency with the pharmacoeconomic analysis and with dosing in the NMA.
Dispensing fees and markups were included.
Table 10: Disaggregated Summary of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Sponsor’s submitted base case | Reference total | 349,726,613 | 387,426,439 | 420,682,638 | 452,687,185 | 1,260,796,262 |
Risankizumab | 0 | 0 | 0 | 0 | 0 | |
All other comparators | 349,726,613 | 387,426,439 | 420,682,638 | 452,687,185 | 1,260,796,262 | |
New drug total | 349,726,613 | 392,308,770 | 425,300,570 | 456,628,265 | 1,274,237,605 | |
Risankizumab | 0 | 10,868,457 | 15,481,676 | 18,422,823 | 44,772,957 | |
All other comparators | 349,726,613 | 381,440,313 | 409,818,893 | 438,205,442 | 1,229,464,648 | |
Budget Impact | 0 | 4,882,332 | 4,617,932 | 3,941,079 | 13,441,343 | |
CDA-AMC base case | Reference total | 317,407,427 | 356,134,422 | 390,446,524 | 422,517,830 | 1,169,098,775 |
Risankizumab | 0 | 0 | 0 | 0 | 0 | |
All other comparators | 317,407,427 | 356,134,422 | 390,446,524 | 422,517,830 | 1,169,098,775 | |
New drug total | 317,407,427 | 360,764,880 | 395,195,553 | 426,941,025 | 1,182,901,457 | |
Risankizumab | 0 | 9,955,572 | 14,406,152 | 17,293,425 | 41,655,148 | |
All other comparators | 317,407,427 | 350,809,308 | 380,789,401 | 409,647,600 | 1,141,246,309 | |
Budget Impact | 0 | 4,630,458 | 4,749,029 | 4,423,195 | 13,802,682 | |
CDA-AMC scenario analyses | ||||||
Scenario 1: Off-label frequencies removed | Reference total | 307,009,753 | 345,723,807 | 379,880,666 | 411,764,604 | 1,137,369,077 |
New drug total | 307,009,753 | 350,709,044 | 385,297,901 | 417,089,994 | 1,153,096,939 | |
Budget Impact | 0 | 4,985,237 | 5,417,235 | 5,325,390 | 15,727,862 | |
Scenario 2: Dispensing fees and markups included | Reference total | 337,378,635 | 378,108,760 | 414,126,460 | 447,745,050 | 1,239,980,270 |
New drug total | 337,378,635 | 383,296,472 | 419,322,359 | 452,470,936 | 1,255,089,768 | |
Budget Impact | 0 | 5,187,712 | 5,195,899 | 4,725,887 | 15,109,498 | |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@cda-amc.ca.