Drugs, Health Technologies, Health Systems
Reimbursement Review
Obinutuzumab (Gazyva)
Sponsor: Hoffmann-La Roche Limited
Therapeutic area: Lupus nephritis
Summary
What Is Lupus Nephritis?
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease in which the immune system attacks healthy tissues throughout the body.1 Lupus nephritis is a serious kidney complication of SLE, resulting in inflammation and damage that can threaten kidney function.2
The estimated prevalence of SLE is about 1 in 2,000 individuals in Canada, with disease onset typically occurring in patients aged between 16 and 55 years.3,4 SLE is about 9 times more common in females than in males5 and approximately 50% of patients with SLE will develop lupus nephritis.2 Lupus nephritis is more common in patients who are Asian, Black, Hispanic, or Indigenous,6-8 and research suggests that patients who are Black or Hispanic are more likely to have their disease progress to kidney failure than patients who are white.2
What Are the Treatment Goals and Current Treatment Options for Lupus Nephritis?
The goals of lupus nephritis treatment include preserving kidney function, preventing disease flares, and reducing illness and deaths associated with chronic kidney disease, along with reducing treatment side effects and preserving fertility.
Patients seek treatments that are safe to take during pregnancy, cause fewer side effects, reduce fatigue, have simple administration, and provide better preservation of kidney function. Other important outcomes identified through clinician input include complete renal response (CRR) and reduction in steroid (prednisone) use.
Guidelines the European Alliance of Associations for Rheumatology released in 2025 recommend combination therapy for patients with active lupus nephritis, particularly for those with poor prognostic factors.9 Combination therapy includes a steroid (glucocorticoid) along with 1 of the following treatment options: a mycophenolic acid analogue (MPAA) or low-dose IV cyclophosphamide plus belimumab, an MPAA plus a calcineurin inhibitor (CNI), or an MPAA plus Gazyva.9
Guidelines recommend that patients with SLE additionally take hydroxychloroquine, supplements for their bone health (e.g., vitamin D, calcium, and antiresorptive agents), stay up to date with nonlive vaccines, and may use kidney-protective medicines such as ACE inhibitors.9,10
What Is Gazyva and Why Did Canada’s Drug Agency Conduct This Review?
Gazyva is a drug that is administered by IV infusion. Health Canada has approved Gazyva for the treatment of adult patients with active lupus nephritis who are receiving standard therapy.11
Canada’s Drug Agency (CDA-AMC) reviewed Gazyva to inform a recommendation to the participating public drug programs on whether it should be reimbursed for the treatment of adult patients with active lupus nephritis who are receiving standard therapy.
How Did CDA-AMC Evaluate Gazyva?
CDA-AMC reviewed the clinical evidence on the beneficial and harmful effects, as well as the economic evidence, of Gazyva versus other treatments used in Canada for the treatment of adult patients with active lupus nephritis who are receiving standard therapy (glucocorticoids and an MPAA). Low-dose IV cyclophosphamide, CNIs (i.e., tacrolimus, cyclosporine), and belimumab were considered relevant treatments to compare with Gazyva when reviewing the clinical evidence.
CDA-AMC identified equity and ethical considerations relevant to Gazyva and for the treatment of adult patients with active lupus nephritis who are receiving standard therapy.
The review was informed by materials submitted by the sponsor, which included clinical and economic evidence.
The review was also informed by 3 patient group submissions and 2 clinician group submissions in response to a call for input by CDA-AMC, and by input from the participating public drug programs around issues that may affect their ability to implement a recommendation.
One nephrologist and 1 rheumatologist with representation from Manitoba and Alberta were consulted as part of the review process.
What Were the Findings?
Clinical Evidence
CDA-AMC reviewed the following clinical evidence:
1 phase III randomized controlled trial (RCT), the REGENCY study, that compared Gazyva plus standard therapy with placebo plus standard therapy in 271 patients with lupus nephritis
1 network meta-analysis (NMA) of Gazyva plus standard therapy versus belimumab plus standard therapy.
Findings from the REGENCY study at week 76 showed that treatment with Gazyva plus standard therapy, compared with placebo plus standard therapy, likely results in:
a clinically important increase in the proportion of patients with CRR and in the proportion of patients with CRR with successful prednisone taper
a clinically important decrease in the proportions of patients with death or renal-related events
little to no difference in Functional Assessment of Chronic Illness Therapy–Fatigue scores
an increase in the proportion of patients with serious adverse events, although the clinical relevance of this increase was uncertain.
Indirect evidence from an NMA suggested that:
the effect of Gazyva plus standard therapy on CRR, partial renal response, overall response rate, change from baseline in estimated glomerular filtration rate, and serious adverse events compared to belimumab plus standard therapy was uncertain
the comparative estimates from the NMA were uncertain due to methodological limitations (i.e., risk of bias in the included studies and violation of the exchangeability assumption) and wide credible intervals, which made it unclear which treatment might be favoured.
Evidence gaps the clinical experts noted included that:
no evidence was available to show how Gazyva compares to CNIs (such as tacrolimus and cyclosporine) or cyclophosphamide, which the clinical experts noted are sometimes used in clinical practice in Canada
while single-drug immunosuppressive therapy with glucocorticoids and an MPAA or low-dose cyclophosphamide remains appropriate in selected patients or settings with limited access to combination immunosuppressive therapy, combination regimens are preferred and recommended when feasible
older CNIs such as tacrolimus and cyclosporine have less favourable safety profiles, require therapeutic drug monitoring, and have limited long-term evidence in populations other than patients who are Asian.9,12,13
Economic Evidence
Gazyva is available as a solution for infusion (25 mg/mL). At the submitted price of $5,751.73 per 40 mL vial, the annual cost of Gazyva is expected to be $23,007 per patient in the first year of treatment and $11,503 in subsequent years, based on the Health Canada–recommended dosage.
Key clinical efficacy in the economic analysis for Gazyva plus standard therapy (glucocorticoids plus mycophenolate mofetil) versus standard therapy alone was derived from the REGENCY trial. Evidence submitted by the sponsor indicated that Gazyva plus standard therapy is likely to increase the proportion of patients with CRR compared with standard therapy alone among patients with lupus nephritis, with moderate certainty. For Gazyva plus standard therapy versus belimumab plus standard therapy, clinical efficacy was informed by a sponsor-submitted indirect treatment comparison. According to the CDA-AMC clinical review, the relative effectiveness and safety of Gazyva plus standard therapy compared to belimumab plus standard therapy with respect to CRR and partial renal response are uncertain due to methodological limitations.
The results of the CDA-AMC base case suggest that:
Gazyva plus standard therapy is predicted to be associated with higher costs to the health care system than standard therapy alone (incremental costs = $40,437), primarily driven by increased costs associated with drug acquisition
Gazyva plus standard therapy is predicted to be associated with a gain of 0.56 life-years compared to standard therapy alone and may result in a gain of 0.51 quality-adjusted life-years compared to standard therapy alone
compared to standard therapy alone, the incremental cost-effectiveness ratio of Gazyva plus standard therapy was $79,492 per quality-adjusted life-years gained in the CDA-AMC base case; the estimated incremental cost-effectiveness ratio was highly sensitive to treatment duration and the length of treatment effect after discontinuation
there is no robust evidence to suggest that Gazyva plus standard therapy provides greater health benefit to patients than belimumab plus standard therapy; if there are no differences in health outcomes between Gazyva plus standard therapy and belimumab plus standard therapy, then the total cost of Gazyva plus standard therapy to the health system should not exceed that of belimumab plus standard therapy for the treatment of patients with lupus nephritis.
CDA-AMC estimates that the budget impact of reimbursing Gazyva plus standard therapy for the treatment of patients with active lupus nephritis will be approximately $6 million over the first 3 years of reimbursement compared to the amount currently spent on comparators, with an estimated expenditure of $14 million on Gazyva over this period. The actual budget impact of reimbursing Gazyva will depend on its uptake and how quickly it displaces standard therapy.
Abbreviations
AE
adverse event
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CNI
calcineurin inhibitor
CrI
credible interval
CRR
complete renal response
DIC
deviance information criterion
eGFR
estimated glomerular filtration rate
ESRD
end-stage renal disease
EULAR
European Alliance of Associations for Rheumatology
FACIT-F
Functional Assessment of Chronic Illness Therapy−Fatigue
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
ICE
intercurrent event
ICER
incremental cost-effectiveness ratio
ISN
International Society of Nephrology
ITC
indirect treatment comparison
MMF
mycophenolate mofetil
MPAA
mycophenolic acid analogue
NMA
network meta-analysis
OLT
open-label treatment
OR
odds ratio
PRR
partial renal response
QALY
quality-adjusted life-year
RCT
randomized controlled trial
RPS
Renal Pathology Society
SAE
serious adverse event
SC
subcutaneous
SE
standard error
SLE
systemic lupus erythematosus
SLEDAI-2K
Systemic Lupus Erythematosus Disease Activity Index 2000
SLR
systematic literature review
UPCR
urine protein creatinine ratio
Background
Introduction
The objectives of this report are as follows:
This report will review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of obinutuzumab, 25 mg/mL concentrate for solution for IV infusion, for the treatment of active lupus nephritis in adult patients who are receiving standard therapy. The focus will be placed on comparing obinutuzumab to relevant comparators in clinical practice in Canada and identifying gaps in the current evidence, as outlined in Table 1.
This report will review and critically appraise the economic information submitted by the sponsor, including a cost-effectiveness analysis and budget impact analysis. The focus of the economic review is aligned with the scope of the clinical review, unless otherwise stated. For most reviews, a Canada’s Drug Agency (CDA-AMC) base case is developed, informed by clinical expert input, the available clinical evidence, and the best interpretation of the economic evidence based on the information provided by the sponsor.
The application has received a Notice of Compliance from Health Canada. This report reflects the approved indication and recommended dosage for obinutuzumab.
Table 1: Information on the Application Submitted for Review and on the CDA-AMC Review
Item | Description |
|---|---|
Information on the application submitted for review | |
Drug | Obinutuzumab (Gazyva), 25 mg/mL, concentrate for solution for intravenous infusion |
Sponsor | Hoffmann-La Roche Limited |
Health Canada indication | For the treatment of adult patients with active lupus nephritis who are receiving standard therapy |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | January 22, 2026 |
Mechanism of action | Recombinant monoclonal humanized and glycoengineered Type II anti-CD20 antibody |
Recommended dosage | Dose 1: Initial infusion: 1,000 mg obinutuzumab administered at a rate of 50 mg/hr. The rate of infusion can be escalated in 50 mg/hr increments every 30 minutes to a maximum of 400 mg/hr. Dose 2 (two weeks after dose 1), dose 3 (week 24), dose 4 (week 26), dose 5a and thereafter (every 6 months): 1,000 mg obinutuzumab administered at a rate of 100 mg/hr. The rate of infusion can be escalated at a rate of 100 mg/hr every 30 minutes to a maximum of 400 mg/hr. |
Submission type | Initial |
Sponsor’s reimbursement request | Per indication |
Submitted price | $5,751.73 per 1,000 mg / 40 mL vial |
Information on the CDA-AMC review | |
Review type | Standard |
Clinical review focus | Population: As defined in the Health Canada indication Intervention: Per recommended dosage Comparators: MPAA, cyclophosphamide, CNIs (i.e., tacrolimus, cyclosporine), belimumab Outcomes: CRR, overall renal response, PRR, UPCR, change from baseline in eGFR, FACIT-F, AEs, SAEs, and notable harmsb (i.e., IRRs, neutropenia, serious infections, worsening of preexisting cardiac conditions) |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; CNI = calcineurin inhibitor; CRR = complete renal response; eGFR = estimated glomerular filtration rate; ESRD = end-stage renal disease; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; HRQoL = health-related quality of life; IRR = infusion-related reaction; LN = lupus nephritis; MMF = mycophenolate mofetil; MPAA = mycophenolate acid analogue; NMA = network meta-analysis; NOC = Notice of Compliance; PRR = partial renal response; RCT = randomized controlled trial; SAE = serious adverse event; SF-36 = Short Form (36) Health Survey; UPCR = urine protein creatinine ratio.
aDose 5 should be administered 6 months after dose 4.
bAs identified to be applicable to the lupus nephritis indication in the product monograph for obinutuzumab (Gazyva).11
Sources: The REGENCY trial Clinical Study Report and sponsor’s Summary of Clinical Evidence.14,15 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Submission History for the Drug Under Review
CDA-AMC has previously reviewed obinutuzumab through the reimbursement review process for several oncology indications; reimbursement was recommended for chronic lymphocytic leukemia,16 reimbursement with conditions was recommended for follicular lymphoma,17 and reimbursement was not recommended for previously untreated follicular lymphoma.18
Sources of Information
The contents of the reimbursement review report are informed by materials submitted by the sponsor, input received from interested parties (patient groups, clinician groups, and drug programs), and input from clinical experts consulted for this review.
Calls for patient group and clinician group input are issued for each reimbursement review report. Three patient group submissions and 2 clinician group submissions were received. Patient perspectives were collected through surveys and video interviews from Arthritis Consumer Experts (7 survey respondents); The Kidney Foundation of Canada and Lupus Canada (27 survey respondents); and a joint submission from Lupus Ontario, the Canadian Arthritis Patient Alliance, and Arthritis Society Canada (2 video interviews with 1 patient each and an unspecified number of survey respondents). Clinician group input was gathered from relevant scientific and medical literature and included feedback from the Canadian Network for Improved Outcomes in Systemic Lupus Erythematosus (23 clinicians) and the Toronto Lupus Program and Nephrology colleagues (10 clinicians). The full submissions received are available on the CDA-AMC project landing page in the consolidated input document. The drug programs provide input on each drug being reviewed through the reimbursement review process by identifying issues that may impact their ability to implement a recommendation.
Input from patient and clinician groups is considered throughout the review, including in the selection of outcomes to include in the clinical review and in the interpretation of the clinical and economic evidence. Relevant patient and clinician group input is summarized in the Disease Background, Current Management, and Unmet Needs and Existing Challenges sections.
Each review team includes at least 1 clinical expert with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process. One nephrologist and 1 rheumatologist with expertise in the diagnosis and management of lupus nephritis participated as part of the review team, with representation from the Prairies.
Disease Background
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease in which the immune system attacks healthy tissues throughout the body.1 Lupus nephritis is a serious renal manifestation of SLE, characterized by inflammation and damage that can lead to impaired kidney function.2 Clinically, lupus nephritis most commonly presents with proteinuria and/or hematuria, though manifestations range from asymptomatic (“silent”) disease to nephrotic-range proteinuria or acute kidney failure.19 Given this heterogeneity, kidney biopsy is often necessary for histological classification and to differentiate lupus nephritis from other renal pathologies in patients with SLE.19 According to the 2003 International Society of Nephrology/Renal Pathology Society (ISN/RPS) classification, lupus nephritis is categorized into 6 classes (I to VI), reflecting varying disease patterns and severities.20 Patients with class III, IV, or V lupus nephritis have the greatest risk of their disease progressing to chronic kidney disease.19
The estimated prevalence of SLE in people living in Canada, based on a 2016 publication, is approximately 1 in 2,000 individuals, with disease onset typically occurring between the ages of 16 and 55 years.3,4 SLE affects females about 9 times more frequently than males,5 and roughly 50% of individuals with SLE develop lupus nephritis.2 Lupus nephritis is more prevalent among Asian, Black, Hispanic, and Indigenous populations.6-8 Patients who are Black or Hispanic have a greater risk of their disease progressing to kidney failure compared to patients who are white.2
Patient group input: Patients groups noted that symptoms of lupus nephritis vary widely. Patients emphasized experiencing a range of difficult-to-manage symptoms, including fatigue, skin rashes, nausea, loss of appetite, joint pain, bruising, cognitive dysfunction (brain fog), back pain, and mental health challenges. Patient groups noted that lupus nephritis leads to chronic kidney inflammation and scarring, often progressing to kidney failure requiring dialysis or transplant. Patient groups highlighted that the disease and its treatment impose substantial physical, emotional, and socioeconomic burdens, leading to significant limitations in daily functioning, including household tasks, school or work activities, social engagement, personal hygiene, and cooking. Lupus-related fatigue, depression, and anxiety also negatively affect sexual health and intimacy. The burden of lupus nephritis extends beyond patients to their caregivers, who often face substantial emotional stress and logistical challenges. Caregivers frequently assist with daily activities, household responsibilities, and transportation to medical appointments, contributing to considerable ongoing strain.
Current Management
Treatment Goals
Patient group input: Treatment goals noted by patient groups include treatments that are affordable, have simpler administration, are safe to take during pregnancy, have fewer side effects, reduce fatigue, and provide preservation of kidney function.
Clinical experts consulted for this review: Treatment goals noted by the clinical experts consulted include maintaining kidney function, controlling disease activity, reducing mortality, and preserving health-related quality of life (HRQoL). The clinical experts noted that achieving remission, reflected in stepwise reductions in proteinuria over 3 to 12 months, is a key intermediary goal that is expected to predict long-term kidney stability. Additional priorities noted by the clinical experts include minimizing glucocorticoid exposure, preserving fertility, preventing relapses, and limiting treatment-related toxicity.
Clinician group input: Treatment goals noted by clinician groups include treatments that reduce disease activity, treatment toxicity, mortality, proteinuria, and glucocorticoid exposure; improve HRQoL; and prevent irreversible organ damage.
Current Treatment Options
According to the clinical experts consulted, patients in Canada with active lupus nephritis receive initial therapy in combination with glucocorticoids to induce remission, followed by a maintenance phase to sustain remission and prevent relapse. The maintenance phase usually begins 6 to 12 months after initial therapy and lasts 3 to 5 years if no relapse occurs. The clinical experts noted that initial therapy options for patients with active lupus nephritis in Canada align with current American College of Rheumatology 2024 and European Alliance of Associations for Rheumatology (EULAR) 2025 guidelines, which recommend combination therapy as the standard initial therapy.9,21,22 This includes glucocorticoids combined with 1 of the following multitargeted approaches:
a mycophenolic acid analogue (MPAA) or low-dose cyclophosphamide plus belimumab
an MPAA plus a calcineurin inhibitor (CNI)
an MPAA plus obinutuzumab (per EULAR 2025 guidance).
According to the clinical experts, single-drug immunosuppressive therapy with glucocorticoids and an MPAA or low-dose cyclophosphamide should only be considered in specific situations, such as when combination therapy is unavailable, not tolerated, or declined by the patient. The 2025 EULAR guidelines state that treatment with single-drug immunosuppressive therapies of glucocorticoids and an MPAA or cyclophosphamide remain appropriate for patients without poor prognostic factors or in settings with limited access to newer treatments.9 According to the clinical experts, about 20% of patients with active lupus nephritis who are eligible in clinical practice in Canada currently receive single-drug immunosuppressive therapy; however, the clinical experts noted that combination therapy has been demonstrated to be superior to single-drug immunosuppressive therapy and should be offered to all patients who are eligible whenever feasible. The clinical experts noted that treatment decisions should consider individual patient preferences and involve shared decision-making between patient and physician.
The clinical experts noted that voclosporin, a guideline-recommended CNI, is not available in Canada. Therefore, clinicians rely on older CNIs such as cyclosporine and tacrolimus. The 2025 EULAR guidelines state that cyclosporine is less suitable for long-term treatment due to its higher nephrotoxic potential.9 The clinical experts noted that both tacrolimus and cyclosporine have less favourable safety profiles than voclosporin and require the need for therapeutic drug monitoring. Evidence supporting their use with MPAAs in patients living outside Asian with lupus nephritis is scarce, and evidence supporting the use of tacrolimus in the indicated patient population beyond 2 years is limited.9,12,13
The clinical experts also noted that hydroxychloroquine is recommended for all patients, alongside initial therapy. Adjuvant treatments may also include bone protection through vitamin D, calcium, and antiresorptive agents; immunizations with nonlive vaccines; and lifestyle measures to reduce cardiovascular risk. Renal protection can be provided with ACE inhibitors or angiotensin receptor blockades, and with sodium-glucose cotransporter-2 inhibitors. Additional considerations may include treatment for latent tuberculosis and prophylaxis against pneumocystis.
Key characteristics of obinutuzumab are summarized with other treatments available for lupus nephritis in Table 1 in Appendix 1 in the Supplemental Material document (available on the CDA-AMC project landing page).
Unmet Needs and Existing Challenges
Patient group input: Unmet needs reported by patient groups include access to affordable treatments that may be used during pregnancy, have fewer side effects, offer simpler administration, and better protection of kidney function. Patient groups also want to reduce their use of corticosteroids because these drugs are associated with significant adverse events (AEs) when taken for longer durations and at higher doses, including increased bone density loss, weight gain, and mood changes. Current treatment challenges include difficulty working full-time because of infusion schedules, refrigeration requirements for self-injectors, long travel distances, limited specialist access, long wait times, and a lack of culturally appropriate care. Out-of-pocket costs for medications, travel, and allied health services further exacerbate the burden, underscoring the need for improved support and access.
Clinical experts consulted for this review: The clinical experts consulted for this review noted that current treatments for lupus nephritis often fail to achieve adequate disease control, with remission rates less than 50% in clinical trials (e.g., the BLISS-LN trial evaluating belimumab and the AURORA 1 trial evaluating voclosporin)23,24 and that patients experience substantial relapse rates over time. The clinical experts stated that combination therapy options with CNIs or belimumab have limitations, including nephrotoxicity, frequent monitoring, and burdensome administration, which can be especially challenging for patients in rural or remote areas. Rituximab is usually reserved for refractory patients according to the clinical experts, as B-cell depletion is often incomplete, limiting its utility. Many therapies are associated with significant short- and long-term AEs, and few options are safe during pregnancy, according to the clinical experts. Overall, they note that there is a clear need for more effective, safer, and accessible treatments that provide sustained disease control and reduce treatment burden.
Clinician group input: Clinician groups generally agreed with the clinical experts consulted for this review.
Considerations for Using the Drug Under Review
Contents within this section have been informed by input from the clinical experts consulted for the purpose of this review and from clinician groups, as well as the reimbursement conditions proposed by the sponsor (e.g., refer to Table 2 in Appendix 1 in the Supplemental Material document). The implementation questions from the public drug programs and corresponding responses from the clinical experts consulted for this review are summarized in Table 3 in Appendix 1 in the Supplemental Material document. The following has been summarized by the review team.
Place in Therapy
According to the clinical experts consulted for this review, obinutuzumab represents a potential change in the lupus nephritis treatment paradigm, serving as an initial combination therapy option in combination with glucocorticoids and an MPAA for patients with active lupus nephritis.
The clinical experts noted that obinutuzumab would be a suitable treatment option for patients whose disease has not responded adequately to other combination or single-drug therapies. They emphasized that patients should not be required to receive such combination therapies without their disease responding before being eligible to receive obinutuzumab.
The clinical experts noted that obinutuzumab is the first anti-CD20 therapy that acts by depleting B cells in both blood and tissues. Given the role of B cells in lupus nephritis pathogenesis, the clinical experts noted that obinutuzumab provides a distinct therapeutic approach among currently available options.
Patient Population
The clinical experts were aligned with the initiation conditions proposed by the sponsor in terms of their appropriateness for identifying the suitable patient population and that these conditions could be easily implemented. The clinical experts indicated that eligibility for treatment with obinutuzumab should include any adult with active lupus nephritis, without specifying histologic class. In the REGENCY study,14 patients with mixed class III and V and IV and V disease demonstrated treatment response, suggesting the potential benefit in patients with pure class III, IV, or V disease, despite the absence of patients with pure class V disease in the study and the limited treatment options available for this population. This approach is aligned with the most recent EULAR guidelines, which reflect a shift away from class-based treatment definitions.9 The clinical experts suggested that current trial data do not clearly indicate which subgroup of patients are most likely to respond to obinutuzumab. They noted that patients with lupus nephritis are typically identified through findings such as hematuria, proteinuria, or reduced estimated glomerular filtration rate (eGFR), supported by laboratory tests including complement levels (C3 or C4) and anti–double-stranded DNA antibodies. However, these indicators are not specific to lupus nephritis, and a kidney biopsy is used for definitive diagnosis. The clinical experts noted that biopsies are generally performed in centres with nephrology services, which can delay diagnosis for patients living in rural or remote areas because of limited access to nephrologists and rheumatologists with specialized training in lupus nephritis. One clinician group noted that diagnosis is based on clinical and laboratory findings when a biopsy is not feasible, either due to contraindications or patient refusal, which happen in about 10% to 20% of cases. The clinician group suggested that obinutuzumab may be considered on an individualized basis in these cases to improve renal outcomes and reduce glucocorticoid use.
Assessing the Response to Treatment
According to the clinical experts, response to treatment in lupus nephritis is primarily evaluated using laboratory and clinical parameters that reflect kidney function and disease activity based on clinical guideline recommendations.9,10,21 The clinical experts stated that achieving these targets is associated with a lower risk of chronic kidney disease progression and provides useful benchmarks for assessing response. The targets include:
at least a 25% reduction in proteinuria after 3 months
at least a 50% reduction in proteinuria after 6 months
proteinuria between 0.5 to 0.8 g/day after 6 to 12 months (the response time can be delayed to 18 to 24 months if baseline proteinuria is in the nephrotic range, which is greater than 3.5 g/day)
stable eGFR (within 10% to 20% of baseline and > 60 mL/min)
reduction in glucocorticoid dose to less than or equal to 5 mg/day after 6 months.
The clinical experts noted that secondary indicators of response include:
improvements in serologic markers (e.g., higher complement levels, lower anti–double-stranded DNA)
reduced relapse rates or longer time to relapse
improvement in fatigue, HRQoL, and extrarenal lupus symptoms
better histologic activity on repeat biopsy, when performed.
According to the clinical experts, although the recommended targets help frame expectations for response, a lack of complete target achievement, particularly in patients with high baseline proteinuria or chronic damage, should not be taken on its own as evidence that the treatment is ineffective or warrants discontinuation.
The clinical experts noted that repeat biopsies are infrequently conducted in clinical practice if laboratory parameters show adequate improvement. Physicians experienced in the care of patients with lupus nephritis would consider these targets and secondary indicators to be reliable measures of renal response, with little variation across physicians.
The clinical experts agreed with the sponsor-proposed renewal criteria, including renewal at 12 months posttreatment and renewal conditions based on the clinician’s assessment of benefit as per appropriate treatment guidelines, without using fixed numeric thresholds as reimbursement requirements.
Discontinuing Treatment
The clinical experts were aligned with the discontinuation conditions proposed by the sponsor and noted that these conditions could be easily implemented. The clinical experts noted that consideration could be given to treatment discontinuation in cases of serious AEs (SAEs) (such as anaphylaxis, serious infection, or drug-induced neutropenia), lack of clinical improvement (defined as the treatment failing to achieve a meaningful reduction in proteinuria or stabilization of renal function after 12 months of therapy), or an inability to taper corticosteroids despite ongoing treatment. Discontinuation is also appropriate if a patient becomes pregnant, a disease relapse occurs, or following sustained remission (typically maintained for at least 3 to 5 years), although the optimal duration of therapy to prevent relapse remains uncertain according to the clinical experts. The Kidney Disease Improving Global Outcomes 2024 guidelines note that after complete or partial remission has been achieved, a relapse of active lupus nephritis should be treated with the same initial therapy used to achieve the original response or an alternative recommended therapy.25
Prescribing Considerations
The clinical experts were aligned with the prescribing conditions proposed by the sponsor that treatment with obinutuzumab should be initiated and supervised by a qualified physician experienced in the diagnosis and treatment of lupus nephritis and that these conditions could be easily implemented. One clinical expert noted that patients living in remote or less specialized settings may have delayed access to appropriate diagnosis and initiation of therapy.
Clinical Review
Methods
The review team considered trials in the sponsor’s systematic review (pivotal trials and randomized controlled trials [RCTs]), sponsor-submitted long-term extensions, indirect treatment comparisons (ITCs), and studies addressing gaps in the evidence for inclusion. Eligible studies for the systematic review included published and unpublished pivotal studies and phase III and IV RCTs. Relevant patients and interventions were defined by the indication and the recommended dosage in the product monograph. Relevant comparators were drugs used in clinical practice in Canada to treat patients described in the indication under review. These included glucocorticoids combined with 1 of the following: an MPAA, low-dose cyclophosphamide, an MPAA or low-dose cyclophosphamide plus belimumab, or an MPAA plus a CNI. ITCs and studies addressing gaps submitted by the sponsor were included when they filled an identified gap in the systematic review evidence (e.g., missing comparator, longer follow-up time).
The review team selected outcomes and follow-up times for review, considering the sponsor’s Summary of Clinical Evidence, clinical expert input, as well as patient and clinician group input. Included outcomes are those considered relevant to expert committee deliberations, and they were selected in consultation with committee members. Evidence from the systematic review for the most important outcomes was assessed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach. The outcomes assessed using GRADE, alongside the rationale for inclusion, were as follows:
complete renal response (CRR): serves as a surrogate outcome used to predict progression to end-stage renal disease (ESRD), sustained and significant decline in renal function, or death
CRR with successful prednisone taper: reflects reduced prednisone use, which was highlighted as important to patient and clinician groups; according to the clinical experts, it provides confirmation that disease control is being achieved by the immunosuppressant
death or renal-related events: represents outcomes important to patient and clinician groups and was used to inform the sponsor’s pharmacoeconomic model
change in the Functional Assessment of Chronic Illness Therapy–Fatigue (FACIT-F) scale: captures fatigue, a concern cited by the patient groups
SAEs: address safety outcomes of importance to clinician groups and the clinical experts consulted for this review.
Some outcomes were presented in the report but not appraised using GRADE, including proteinuric response, mean change from baseline in eGFR, partial renal response (PRR), and AEs. These outcomes were considered supportive to the review or were used to inform the pharmacoeconomic model.
Methods for data extraction, risk of bias appraisal, and certainty of evidence assessment are provided in Appendix 2 in the Supplemental Material document.
Clinical Evidence
In this report, the following sources of evidence submitted by the sponsor are reviewed and appraised:
1 pivotal trial (the REGENCY study) included in the systematic review
1 ITC.
Systematic Review
Description of Studies
Study Characteristics
Characteristics of the included study are summarized in Table 2. Details pertaining to the intervention and comparators, prohibited and permitted treatments, and relevant outcome measures are summarized in Appendix 3 in the Supplemental Material document.
The REGENCY Study
The REGENCY study is an ongoing, placebo-controlled phase III RCT evaluating the efficacy and safety of obinutuzumab in patients with class III or IV (with or without class V) lupus nephritis. The study is being conducted in 72 centres across 15 countries with no sites in Canada. Patients who were eligible were randomized to receive obinutuzumab or placebo in a 1:1 ratio, stratified by geographic region (Canada and the US versus Latin America and the Caribbean versus other) and race (Black versus other [wording of original source]). Patients randomized to receive obinutuzumab were further randomized in a 1:1 ratio to receive 1 of 2 obinutuzumab dosing schedules: patients in the 2-2-2 regimen received a blinded IV infusion of 1,000 mg obinutuzumab on day 1 and on weeks 2, 24, 26, 50, and 52; patients in the 2-2-1 regimen received the same regimen except that instead of obinutuzumab, they received a blinded placebo infusion at the week 50 visit. Efficacy results were presented with both obinutuzumab dosing groups combined.
All patients also received standard therapy of mycophenolate mofetil (MMF) and glucocorticoids (hereafter referred to as standard therapy). MMF was administered from baseline, titrated by week 4 to a target dose of 2.0 g to 2.5 g per day, and maintained through week 80 with dose adjustments allowed for intolerance or AEs. Before the first infusion, all patients were required to have received at least 1 dose of IV pulse methylprednisolone (250 mg to 1,000 mg) or equivalent. Patients then received oral prednisone using a standardized taper through week 80, with higher doses allowed when clinically necessary for renal or extrarenal flares. However, the use of pulse steroids or sustained high-dose corticosteroids from week 64 onward was considered corticosteroid rescue therapy and classified the disease in these patients as having not responded for subsequent efficacy assessments.
The REGENCY study consisted of the following study periods: screening, blinded treatment, open-label treatment (OLT) for patients meeting the criteria, and study follow-up. The primary efficacy end point was assessed at week 76. Efficacy and safety assessments were performed at scheduled visits throughout treatment, with safety follow-up continuing for at least 12 months after the last dose. The most recent analysis was based on a prespecified sponsor-defined clinical cut-off date of August 15, 2024 (with an estimated study completion date in 2028). The analysis was performed when all data were available for all patients who completed or discontinued before the week 76 visit. The combined safety data after week 76 from both the blinded treatment and OLT periods include 39 patients who began on placebo and later switched to obinutuzumab during the OLT period. While the REGENCY study is ongoing for long-term follow-up, the primary analysis has been completed, providing pivotal efficacy and safety data. The safety profile reported reflects these primary results.
Table 2: Characteristics of the Study Included in the Systematic Review
Study name, design, and sample size | Key inclusion criteria | Key exclusion criteria | Intervention and comparator | Relevant end points |
|---|---|---|---|---|
The REGENCY trial Phase III, multicentre, double-blind RCT Total N = 271 |
|
| During blinded treatment (to week 76): Intervention:
Comparator:
Beyond week 76: Patients whose disease responded continued to receive blinded infusions every 6 months, starting at week 80 and continuing until study unblinding. Patients whose disease did not respond were eligible for open-label treatment.c | Primary end point:
Key secondary end points:
|
ACR = American College of Rheumatology; ANA = antinuclear antibody; CNI = calcineurin inhibitor; CRR = complete renal response; eGFR = estimated glomerular filtration rate; ESRD = end-stage renal disease; EULAR = European Alliance of Associations for Rheumatology; FACIT‑F = Functional Assessment of Chronic Illness Therapy–Fatigue; ISN/RPS = International Society of Nephrology/Renal Pathology Society; OLT = open-label treatment; RCT = randomized controlled trial; SLE = systemic lupus erythematosus; UPCR = urine protein to creatinine ratio.
aIncluding any anti-CD20 therapy (in the 9 months before or during screening); cyclophosphamide, tacrolimus, cyclosporin, or voclosporin (in the 2 months before or during screening); or a biologic B-cell–targeted therapy other than anti-CD20 (in the 2 months before or during screening).
bDefined by the presence of crescent formation in at least 50% of glomeruli assessed on renal biopsy or sustained doubling of serum creatinine during the 2 months before or during screening or investigator’s opinion that the patient has rapidly progressive glomerulonephritis.
cOLT followed the initial obinutuzumab treatment schedule with infusions on OLT day 1 and on OLT weeks 2, 24, 26, 52, and every 6 months thereafter. Patients who discontinued OLT entered safety follow-up.
Sources: REGENCY Clinical Study Report and sponsor’s Summary of Clinical Evidence.14,15 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Statistical Testing and Analysis Populations
The sample size determination for the REGENCY study was based on the phase II NOBILITY study.26 It was estimated that the addition of obinutuzumab to MMF would induce an overall CRR rate of 50% at week 76. Based on the planned sample size of 252 patients randomized in a 1:1 ratio (126 patients in each of the obinutuzumab and placebo groups), 90% power would be achieved to compare the combined obinutuzumab treatment group with the placebo group at the 2-sided type I error of 0.05, assuming the same CRR proportions across the strata.
To control the overall type I error, a fallback method maintaining a fixed sequence for testing was used. The sequence for end point testing was the primary end point, followed by key secondary end points: CRR with prednisone taper, proteinuric response, and change in eGFR at week 76. If the primary end point was significant at the 0.05 level, the first secondary was tested at the same level, and significance allowed the alpha to be split (i.e., 0.04 for proteinuric response and 0.01 for eGFR) and then reallocated based on results. Testing was to be continued sequentially through additional secondary end points (death or renal-related events, overall renal response, and FACIT-F) until the first nonsignificant result.
The efficacy analyses were based on all randomized patients according to their randomized treatment (efficacy-evaluable population). The safety analyses were based on patients who received any part of blinded infusion of obinutuzumab or placebo, grouped according to the actual treatment received. The primary analysis had a clinical cut-off date of August 15, 2024.
Patient Disposition
Patient disposition for each study is summarized in Appendix 4 in the Supplemental Material document.
Of 513 patients screened, 271 patients (52.8%) were randomized into the REGENCY study. Of the 242 patients who did not meet screening criteria, the main reasons were not meeting the urine protein creatinine ratio (UPCR) requirement (39.7%) and being ineligible or having unconfirmed class III or IV proliferative lupus nephritis (16.5%). A total of 135 patients (49.8%) were assigned to the obinutuzumab group and 136 patients (50.2%) to the placebo group, comprising the efficacy-evaluable population. Patients in the obinutuzumab group were further randomized 1:1 to 1 of 2 dosing schedules: 69 patients (51.1%) to the 2-2-2 regimen and 66 patients (48.9%) to the 2-2-1 regimen; however, the 2 regimens were combined for all outcome assessments. Three patients in the placebo group (2.2%) who did not receive study treatment were excluded from the safety population, and 1 patient assigned to placebo (0.7%) who inadvertently received obinutuzumab was included in the obinutuzumab safety group. The resulting safety-evaluable population included 268 patients: 136 patients in the obinutuzumab group (50.7%) and 132 patients in the placebo group (49.3%).
As of week 76, a total of 29 patients (10.7%) had discontinued the study: 13 patients (9.6%) in the obinutuzumab group and 16 patients (11.8%) in the placebo group. The most common reason for study discontinuation was because of withdrawal by the patient, with 9 patients (6.7%) in the obinutuzumab group and 6 patients (4.4%) in the placebo group. No patient in the obinutuzumab group discontinued because of a physician’s decision, compared to 5 patients (3.7%) in the placebo group.
As of week 76, totals of 29 patients (21.3%) in the obinutuzumab group and 38 patients (28.8%) in the placebo group had discontinued treatment. Common reasons for treatment discontinuation with obinutuzumab versus placebo were AEs (5.1% versus 1.5%, respectively), lack of efficacy (4.4% versus 16.7%, respectively), patient withdrawal (3.7% versus 3.8%, respectively), and physician decision (2.9% versus 4.5%, respectively).
Major protocol deviations were generally balanced between treatment groups, with 95 deviations experienced by 59 patients (43.7%) in the obinutuzumab group and 94 deviations experienced by 50 patients (36.8%) in the placebo group. The most common deviations, reported in at least 5% of patients, were receiving incorrect study medication (11.1% in the obinutuzumab group versus 11.0% in the placebo group), failure to follow approved study procedures (8.1% in the obinutuzumab group versus 9.6% in the placebo group), failure to report SAEs within 24 hours (7.4% in the obinutuzumab group versus 5.9% in the placebo group), and failure to document study drug administration (5.2% in the obinutuzumab group versus 5.1% in the placebo group).
Baseline Characteristics
Baseline characteristics of patients in the REGENCY study are summarized in Table 3.
Table 3: Summary of Baseline Characteristics From the REGENCY Study Included in the Systematic Review
Characteristic | REGENCY study | |
|---|---|---|
Obinutuzumab N = 135 | Placebo N = 136 | |
Age (years), median (range) | 30.0 (18 to 64) | 31.0 (18 to 72) |
Sex, n (%) | ||
Female | 114 (84.4) | 115 (84.6) |
Male | 21 (15.6) | 21 (15.4) |
Race, n (%) | ||
American Indian or Alaskan Native | 25 (18.5) | 26 (19.1) |
Asian | 9 (6.7) | 7 (5.1) |
Black or African American | 20 (14.8) | 20 (14.7) |
Multiple | 11 (8.1) | 9 (6.6) |
White | 65 (48.1) | 64 (47.1) |
Not reported | 1 (0.7) | 4 (2.9) |
Unknown or other | 4 (3.0) | 6 (4.4) |
Ethnicity, n (%) | ||
Hispanic or Latino | 71 (52.6) | 85 (62.5) |
Not Hispanic or Latino | 54 (40.0) | 48 (35.3) |
Not reported | 9 (6.7) | 1 (0.7) |
Unknown | 1 (0.7) | 2 (1.5) |
Serum creatinine (µmol/L) | ||
Serum creatinine, mean (SD) | 73.8 (34.1) | 77.5 (42.2) |
eGFR (mL/min per 1.73m2) | ||
Mean (SD) | 102.8 (29.3) | 101.9 (32.2) |
24-hour UPCR (mg/mg) | ||
n | 134 | 136 |
Mean (SD) | 3.14 (2.99) | 3.53 (2.76) |
Baseline lupus nephritis class, n (%) | ||
Class III | 56 (41.5) | 51 (37.5) |
Class IV | 79 (58.5) | 85 (62.5) |
Baseline lupus nephritis concomitant class V, n (%) | ||
Yes | 47 (34.8) | 38 (27.9) |
No | 88 (65.2) | 98 (72.1) |
Prior history of lupus nephritis, n (%) | ||
Yes | 81 (60.0) | 76 (55.9) |
No | 54 (40.0) | 60 (44.1) |
Duration of lupus nephritis for patients who had prior history of lupus nephritis (months) | ||
n | 81 | 76 |
Mean (SD) | 65.62 (78.64) | 59.33 (64.70) |
eGFR = estimated glomerular filtration rate; SD = standard deviation; UPCR = urine protein creatinine ratio.
Note: Racial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
Sources: REGENCY Clinical Study Report and the sponsor’s Summary of Clinical Evidence.14,15 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Treatment Exposure and Concomitant Medications
Details regarding treatment exposure, treatment adherence, use of concomitant medications, and use of rescue therapies in the included study are in Appendix 4 in the Supplemental Material document.
In the REGENCY study, at the clinical cut-off date of August 15, 2024, 43.4% of patients had received 5 infusions of obinutuzumab and 43.4% had received 6 infusions. Exposure to MMF and glucocorticoids was comparable between treatment groups. The median MMF duration to week 76 was 558 days in both treatment groups, with median cumulative doses of 1,099 g in the obinutuzumab group and 1,113 g in the placebo group. In the obinutuzumab group, the median glucocorticoid exposure was 519 days, with a median dose of 4,294 mg. In the placebo group, the median glucocorticoid exposure was 494 days, with a median dose of 4,366 mg.
Critical Appraisal
Internal Validity
In the REGENCY study, randomization sequences were generated centrally using an interactive voice or web response system (IxRS), confirming adequately concealed treatment allocation. Patients were stratified by known prognostic factors of race and region to ensure a balance in these characteristics across groups. In the REGENCY study, a higher proportion of patients in the obinutuzumab group (34.8%) had concomitant class V disease compared with the placebo group (27.9%); this indicates a potentially worse baseline prognosis that could bias the treatment effect, although the extent of potential bias is unknown. The clinical experts consulted noted that patients with concomitant class V lupus nephritis may require a longer duration to achieve a treatment response.
Patients randomized to obinutuzumab in the REGENCY study were further randomized to 1 of 2 dosing schedules to collect more pharmacokinetic and pharmacodynamic data. The schedules differed only in whether patients received obinutuzumab or placebo at week 50. Both regimens were projected to maintain adequate B-cell depletion through week 76; therefore, the 2 groups were combined for efficacy comparisons against the placebo group. The clinical experts suggested that the differences in dosing schedules were unlikely to impact efficacy outcomes. Additionally, the sponsor performed exploratory analyses of each obinutuzumab dosing schedule compared to placebo for the primary and key secondary end points. These analyses suggested that there were no major differences in effect estimates across the 2 doses that would impact the efficacy conclusions.
All laboratory studies of blood specimens with unblinding potential were performed by a central laboratory. Results in the REGENCY study were presented through week 76, where the sponsor, patients, and investigators remained blinded to treatment assignment. After the primary analysis, the sponsor was unblinded, while investigators and patients remained blinded until all patients who were eligible had completed 1 year of follow-up beyond week 76. Although the study was double-blind and used matched infusions, there were more treatment-specific safety signals in the obinutuzumab group (notably infusion-related reactions, drug-related neutropenia, and higher rates of grade 3 to 5 infections), any of which could have signalled treatment assignment to investigators or patients. This may have introduced a risk of performance or detection bias; however, there is limited evidence to indicate that such a bias would exist. Common concomitant medications, including immunosuppressants, agents acting on the renin-angiotensin system, and antiprotozoals, were generally balanced between treatment groups. Corticosteroid-tapering protocols were prespecified and comparable across treatment groups. The rate of treatment discontinuation was high (21.3% with obinutuzumab and 28.8% with placebo). Reasons were often because of AEs and lack of treatment efficacy, which may be as expected (i.e., not related to the trial context). Reasons such as physician decision and patient withdrawal are more ambiguous and could be influenced by unblinding; there was not an important imbalance in these reasons across groups. The impact on detection bias is likely limited because key efficacy and safety end points were evaluated by an independent data monitoring committee.
The intercurrent event (ICE) handling strategies in the trial had the potential to introduce bias. ICEs included rescue therapy use, treatment failure, study treatment discontinuation, or death before end point assessment. For the primary and most categorical end points, ICEs were considered as a nonresponse. While this is appropriate for most ICEs when the aim is to induce remission, study withdrawal might not always be consistent with a lack of response. A total of 6.7% of patients in the obinutuzumab group and 9.6% in the placebo group experienced this ICE. After considering reasons for study discontinuation likely compatible with treatment failure (e.g., death, AE), the proportion of patients imputed as nonresponse for this reason was relatively low, suggesting limited potential for bias. There were notable between-group differences in treatment failure (3.7% versus 17.6%) and rescue therapy (5.9% versus 17.6%). A post hoc review of UPCR and eGFR data by the sponsor indicated that these ICEs generally reflected accurate clinical worsening, and most events occurred after week 24, allowing patients to receive sufficient blinded treatment before classification. Together, these findings mitigate concerns that the evaluation of ICEs meaningfully compromises internal validity. ICEs for continuous variables were considered under the treatment policy strategy (except deaths, which were imputed as 0), which is considered appropriate. The use of rescue therapy could affect the results; however, this occurred among more patients in the placebo group (17.6%) than the obinutuzumab group (5.9%), indicating that any potential bias would not favour the intervention.
Regarding loss to follow-up, 10.7% of patients discontinued the study through week 76 (9.6% in the obinutuzumab group versus 11.8% in the placebo group), representing a relatively balanced but higher-than-ideal attrition rate between groups. Missing data for the primary and key secondary outcomes were addressed using multiple imputation in the REGENCY study. For the primary end point, there were limited missing data after accounting for ICE strategies. Sensitivity analysis using alternative missing-data imputation methods and supplementary analyses, in which early withdrawals without an ICE were treated as missing, were both consistent with the primary analysis results, supporting the robustness of the findings. For continuous secondary outcomes, the extent of missing data (around 10% at 76 weeks and balanced across groups) has some potential to introduce bias because the missing at random mechanism assumed by the imputation approach may not be plausible. Analysis of the Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K) had a higher extent of missing data, with approximately 27% of patients missing data at week 76 in the obinutuzumab group and 14% in the placebo group. The differential and higher level of missing data, which are not likely to be missing at random, introduce a high risk of bias.
In the REGENCY study, multiplicity was addressed using a fallback method maintaining a fixed testing sequence. The sequence began with the primary end point, followed by key secondary end points. Because the hierarchy failed for the outcome of change from baseline in eGFR, there is an increased risk of type I error for the following outcome of death and renal-related events.
External Validity
According to the clinical experts consulted, the REGENCY study population was generally considered representative of adult patients with active lupus nephritis seen in routine clinical practice in Canada, although some limitations to generalizability were identified. The study excluded patients with severe renal involvement or with pure class V disease; according to the clinical experts, these patients may be eligible for the treatment under review in clinical practice. The EULAR 2025 guidelines highlight a transition from class-specific treatment approaches to a broader strategy centred on active lupus nephritis.9 The clinical experts noted that this shift will likely be widely applied to the treatment of lupus nephritis in clinical practice, in which active lupus nephritis encompasses pure class III, IV, or V disease, as well as mixed class III and V and class IV and V presentations.
The clinical experts indicated that, in clinical practice, patients would be eligible for treatment regardless of their baseline proteinuria levels, despite the study requiring specific thresholds for inclusion. One clinical expert noted that their patient population consisted of more patients who were Asian and fewer patients who were Black than in the REGENCY study. However, the distribution of sex, mean age, baseline disease activity, concomitant medication, and rescue therapy used in the study were broadly aligned with the clinical experience of the experts consulted.
In the REGENCY study, patients assigned to the obinutuzumab regimen that included a week 50 dose received 6 doses over 52 weeks, exceeding the product monograph recommendation of 5 doses per year.11 This higher dosing intensity may somewhat limit the generalizability of the results to patients receiving the standard dosing schedule. However, the clinical experts consulted indicated that they do not expect the additional dose to meaningfully impact the study outcomes. The clinical experts also noted that 76 weeks of follow-up was sufficient to assess induction of remission, but they emphasized that a longer duration would be necessary to adequately evaluate the maintenance of remission.
Results
The key efficacy and harms results and findings from the GRADE assessment are presented in this section. Detailed efficacy and harms results can be found in Appendix 4 in the Supplemental Material document.
Efficacy
The key efficacy results are summarized by outcome.
Complete Renal Response
The proportion of patients who achieved a CRR at week 76 was 46.4% in the obinutuzumab group compared to 33.1% in the placebo group, with an adjusted between-group difference of 13.40% (95% confidence interval [CI], 1.95% to 24.84%; P = 0.0232; threshold for significance, P = 0.05). The proportion of patients who achieved each individual component of the composite was numerically higher in the obinutuzumab group compared to the placebo group (not tested statistically):
UPCR less than 0.5 g/g: 47.4% versus 36.0%
eGFR of at least 85% of the baseline value: 83.7% versus 75.7%
no occurrence of ICEs: 88.9% versus 75.0%.
CRR With Successful Prednisone Taper
At week 76 in the REGENCY study, 42.7% of patients in the obinutuzumab group achieved a CRR with a successful prednisone taper versus 30.9% in the placebo group, for a difference of 11.88% (95% CI, 0.57% to 23.18% of patients; P = 0.0421).
Proteinuric Response
At week 76 in the REGENCY study, 55.5% of patients in the obinutuzumab group achieved a proteinuric response compared to 41.9% in the placebo group, with an adjusted difference of 13.68% (95% CI, 2.01% to 25.36% of patients; P = 0.0227).
Mean Change From Baseline in eGFR
Through week 76 in the REGENCY study, the adjusted mean change from baseline in eGFR was 2.31 mL/min per 1.73 m2 in the obinutuzumab group and −1.54 mL/min per 1.73 m2 in the placebo group, resulting in a treatment difference of 3.84 mL/min per 1.73 m2 (95% CI, −1.83 mL/min per 1.73 m2 to 9.51 mL/min per 1.73 m2; P = 0.1842).
Death or Renal-Related Events
Through week 76 in the REGENCY study, 18.9% of patients in the obinutuzumab group and 35.6% in the placebo group experienced death or renal-related events, corresponding to a difference of −16.83% (95% CI, −27.42% to −6.23%). There was no formal statistical testing because the statistical hierarchy failed before this end point.
Overall Renal Response
Through week 50 in the REGENCY study, 59.1% of patients in the obinutuzumab group achieved an overall renal response compared with 50.7% in the placebo group, yielding a difference of 8.36% (95% CI, −3.41% to 20.12%). There was no formal statistical testing because the statistical hierarchy failed before this end point.
Mean Change From Baseline in SLEDAI-2K Score
The SLEDAI-2K total score ranges from 0 to 105 points, with lower scores indicating improvement of disease activity.
Through week 76 in the REGENCY study, the adjusted mean change from baseline in SLEDAI-2K was −5.63 (standard error [SE] = 1.46) in the obinutuzumab group versus −5.51 (SE = 1.43) in the placebo group, yielding a treatment difference of −0.12 (95% CI, −3.11 to 2.87).
Mean Change From Baseline in FACIT-F Score
The FACIT-F scale ranges from 0 to 52 points, with lower scores indicating worse fatigue.
Through week 76 in the REGENCY study, the adjusted mean change from baseline in the FACIT-F score was 1.76 (SE = 1.22) in the obinutuzumab group versus 3.11 (SE = 1.21) in the placebo group, yielding a treatment difference of −1.35 (95% CI, −3.89 to 1.20). There was no formal statistical testing because the statistical hierarchy failed before this end point.
Harms
Treatment-Emergent AEs
The proportions of patients who experienced at least 1 AE in the REGENCY study were 93% in the obinutuzumab group versus 89% in the placebo group.
The most frequent AEs in the REGENCY study experienced by at least 10% of patients in any group (obinutuzumab group versus placebo group) included:
COVID-19: 28% versus 24%
diarrhea: 20% versus 16%
urinary tract infection: 15% versus 12%
infusion-related reactions: 15% versus 11%
upper respiratory tract infection: 11% versus 10%
neutropenia: 11% versus 3%
bronchitis: 10% versus 8%.
Serious Adverse Events
The proportions of patients experiencing at least 1 SAE in the REGENCY study were 32% in the obinutuzumab group versus 18% in the placebo group.
The most frequent SAEs experienced by at least 2% of patients in any group (obinutuzumab group versus placebo group) included:
COVID-19 pneumonia: 5% versus 0%
pneumonia: 3% versus 2%
urinary tract infection: 3% versus 2%.
When excluding events of confirmed or suspected COVID-19, the proportions of patients experiencing at least 1 SAE were 27% in the obinutuzumab group versus 18% in the placebo group.
Treatment Discontinuation
The proportions of patients who discontinued treatment because of AEs in the REGENCY study were 9% in the obinutuzumab group versus 4% in the placebo group.
Deaths
Through the 76-week blinded treatment period in the REGENCY study, 3 patients (2%) died in the obinutuzumab group (2 because of COVID-19 pneumonia and 1 because of nephrotic syndrome) versus 1 patient (1%) in the placebo group because of COVID-19.
AEs of Special Interest
AEs of special interest (obinutuzumab group versus placebo group) included:
infusion reactions (15% versus 11%)
grade 3 to 5 infections (15% versus 7%)
neutropenia (13% versus 4%).
AEs During OLT
Available safety data (AEs and deaths) from the blinded treatment and OLT period combined were comparable to the results up to the week 76 analysis.
Summary of Findings and Certainty of the Evidence
In the absence of literature-based minimal important difference estimates, a threshold for the smallest clinically important between-group difference of 10% suggested by the clinical experts was used for each of the following outcomes: CRR, CRR with prednisone taper, and occurrence of death or renal-related events. In the absence of a known threshold, the certainty in the presence of a nonnull effect was rated for FACIT-F and SAEs.
Table 4: Summary of Findings for Obinutuzumab vs. Placebo for Patients With Lupus Nephritis
Outcome and follow-up | Patients, N (studies) | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Obinutuzumab | Difference | |||||
Renal outcomes | |||||||
Patients with CRR Follow-up: week 76 | 271 (1 RCT) | NR | 331 per 1,000 | 464 per 1,000 | 134 more per 1,000 (20 to 284 more per 1,000) | Moderatea (serious imprecision) | Obinutuzumab plus standard therapy likely results in a clinically important increase in the proportion of patients with CRR compared with placebo plus standard therapy. |
Patients with CRR with successful prednisone taper Follow-up: week 76 | 271 (1 RCT) | NR | 309 per 1,000 | 427 per 1,000 | 118.8 more per 1,000 (6 to 232 more per 1,000) | Moderateb (serious imprecision) | Obinutuzumab plus standard therapy likely results in a clinically important increase in the proportion of patients with CRR with successful prednisone taper compared with placebo plus standard therapy. |
Patients with death or renal-related events Follow-up: week 76 | 271 (1 RCT) | NR | 356 per 1,000 | 189 per 1,000 | 168.3 fewer per 1,000 (62 to 274 fewer per 1,000) | Moderatec (serious imprecision) | Obinutuzumab plus standard therapy likely results in a clinically important decrease in the proportion of patients with death or renal-related events compared with placebo plus standard therapy. |
Fatigue | |||||||
Change from baseline in FACIT-F scale (range, 0 [worst] to 52 [best]), points Follow-up: week 76 | 271 (1 RCT) | NA | Adjusted mean = 3.11 | Adjusted mean (SE) = 1.76 (1.22) | Mean difference = –1.35 (−3.89 to 1.20) | Moderated (serious imprecision) | Obinutuzumab plus standard therapy likely results in little to no difference in fatigue compared with placebo plus standard therapy. |
Harms | |||||||
Patients with ≥ 1 SAEs (safety set) Follow-up: week 76 | 268 (1 RCT) | RR = 1.8 (1.2 to 2.8) | 182 per 1,000 | 324 per 1,000 | 142 more per 1,000 (39.2 to 244.2 more per 1,000) | Moderatee (serious imprecision) | Obinutuzumab plus standard therapy results in an increase in the proportion of patients with SAEs compared with placebo plus standard therapy. The clinical importance of the increase is uncertain. |
CI = confidence interval; CRR = complete renal response; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; NA = not applicable; NR = not reported; RCT = randomized controlled trial; RR = risk ratio; SAE = serious adverse event; SE = standard error; vs. = versus.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 1 level for serious imprecision. The point estimate suggests a clinically important difference in CRR based on a between-group difference threshold of 10% (100 per 1,000) suggested by clinical experts; however, the lower bound of the 95% CI crosses this threshold and approaches the null.
bRated down 1 level for serious imprecision. The point estimate suggests a clinically important difference in CRR with successful prednisone taper based on a between-group difference threshold of 10% (100 per 1,000) suggested by clinical experts; however, the lower bound of the 95% CI crosses this threshold and approaches the null.
cRated down 1 level for serious imprecision. The point estimate suggests a clinically important difference in death or renal-related events on a between-group difference threshold of 10% (100 per 1,000) suggested by clinical experts; however, the lower bound of the 95% CI crosses this threshold.
dDid not rate down for study limitations, although there is some concern for risk of bias due to missing outcome data. Rated down 1 level for serious imprecision. The 95% CI includes the possibility of trivial effects and no difference. A between-group minimal important difference in patients who are adults with lupus nephritis was not identified; therefore, the target of certainty appraisal was any effect (i.e., the null was used as a threshold); hence, the clinical importance of the estimated effect is uncertain.
eRated down 1 level for serious imprecision; the effect is informed by a small number of events and may be unstable. The null was used as the threshold to inform the target of the certainty rating and the precision of the effect; hence, the clinical importance of the estimated effect is uncertain. The between-group difference was requested from the sponsor to aid in the interpretation of the results.
Sources: REGENCY Clinical Study Report and sponsor’s Summary of Clinical Evidence.14,15 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Long-Term Extension Studies
The submission did not include any long-term extension studies.
Indirect Evidence
In the absence of head-to-head RCTs comparing obinutuzumab with relevant comparators, the sponsor submitted indirect evidence to inform comparative efficacy and safety. Furthermore, an appraisal of the indirect evidence was needed because evidence from the network meta-analysis (NMA) was incorporated into the sponsor’s pharmacoeconomic model.
Description of the ITC
The objective of the ITC was to provide evidence on comparative efficacy and safety of obinutuzumab plus standard therapy (MMF plus corticosteroids) versus relevant comparators in the treatment of adults with class III or IV lupus nephritis (with or without class V). The comparators included in the NMAs were:
belimumab through IV administration, in combination with corticosteroids and with MMF induction and maintenance or cyclophosphamide induction and azathioprine maintenance
rituximab plus MMF plus corticosteroids
high-dose voclosporin plus MMF plus corticosteroids
low-dose voclosporin plus MMF plus corticosteroids.
However, voclosporin is not available in Canada, and according to the clinical experts consulted by the review team, rituximab is given off-label and generally reserved for patients with refractory disease. Therefore, results for voclosporin and rituximab are not included in this report.
Study Selection and Review Methods
A broad systematic literature review (SLR) was performed to identify published RCTs evaluating the efficacy and safety of lupus nephritis treatments. Searches were undertaken in relevant databases, including MEDLINE and Embase, conference proceedings, trial registries, and relevant websites, between April and November 2024. The search was updated between February and April 2025. Studies eligible for inclusion were RCTs (phase II to IV) and NMAs of adult patients with active or active and chronic class III or IV lupus nephritis (with or without class V disease), who were receiving any relevant treatment, alone or in combination. The efficacy outcomes of interest specific to the NMA were CRR, PRR, overall renal response, and change from baseline in eGFR. Details of selection criteria are included in Table 15 in Appendix 6 in the Supplemental Material document.
Study selection was performed independently by 2 reviewers. Data extraction was conducted by 1 reviewer using standardized data extraction forms. A second independent reviewer validated all extracted data for accuracy and correctness. The risk of bias of the included studies was assessed by 2 reviewers using the National Institute for Health and Care Excellence 8-domain checklist for randomized trials from its technologies user guide.27 Any discrepancies between reviewers were resolved through discussion or the intervention of a third reviewer.
Feasibility Assessment
The sponsor conducted a feasibility assessment to evaluate the clinical and methodological validity of conducting ITCs against all approved and commonly used regimens for lupus nephritis, which was conducted in 4 steps. In step 1, relevant RCTs identified in the SLR were prioritized based on key criteria such as population characteristics (e.g., ≥ 90% adults), disease classification (e.g., ≥ 80% with ISN/RPS class III or IV [with or without class V]), and alignment with recommended treatments guidelines. In step 2, the connectivity of the RCTs was explored, and a best-case evidence network (largest connected network) was constructed to identify studies that were relevant for indirect comparisons. Between-study heterogeneity was not considered when selecting the best-case network. Step 3 involved evaluating the comparability of trials in the network in terms of study and patient characteristics and determining whether any heterogeneity may affect the validity of an NMA. Finally, the data reported for each outcome-specific evidence network were reviewed to ensure that the necessary data were reported at sufficiently similar time points. Outcome definitions were reviewed for similarity across trials.
The potential effect modifiers of interest were outlined in the statistical analysis plan and included age, sex, race, region, baseline UPCR, baseline eGFR, a history of lupus nephritis, and previous use of antimalarials. Other effect modifiers considered in the ITC report included ISN/RPS class, previous use of MMF, and previous use of rituximab.
ITC Analysis Methods
The ITC analysis methods are detailed in Table 15 in the Supplemental Material document. Bayesian NMAs were conducted for CRR, PRR independent of CRR, overall renal response, mean change from baseline in eGFR, grade 3 or higher AEs, and SAEs. Each treatment was modelled as a distinct node within the NMA, except where pooling of standard therapies was applied in the base-case structure (e.g., MMF induction plus maintenance, and cyclophosphamide induction plus azathioprine maintenance for the BLISS-LN study). Summary measures included odds ratios (ORs) for binary outcomes and mean differences for continuous outcomes. Vague priors were applied for study-specific baselines and treatment effects, while informative priors were employed for between-study standard deviations in accordance with available guidance. Model fit was evaluated using the deviance information criterion (DIC), with random-effects models preferred when DIC values were comparable across competing models. Between-study heterogeneity was quantified using tau-squared, and where applicable, the node-splitting approach was implemented to assess the consistency assumption.
Sensitivity analyses were conducted to test the robustness of results to model assumptions, including analyses using a fixed-effect model and alternative priors for between-study standard deviation. Additional supplemental analyses included using MMF subgroup data for CRR from the BLISS-LN study without combining the 2 standard therapies (i.e., MMF induction and maintenance, and cyclophosphamide induction and azathioprine maintenance) into a single node, and using week 50 overall renal response data from the REGENCY study.
Summary of Included Studies
The network has a star-shaped structure connected by MMF plus corticosteroids, linking obinutuzumab (the NOBILITY and REGENCY studies) to belimumab (the BLISS-LN study), rituximab (the LUNAR study), low-dose voclosporin (2 studies: AURA-LV, AURORA 1), and high-dose voclosporin (the AURA-LV study), with all treatments, including obinutuzumab, administered with either MMF or cyclophosphamide plus steroids as standard care. This allowed potential comparisons of efficacy and/or safety between obinutuzumab plus MMF plus corticosteroids and each of the following: belimumab plus MMF plus corticosteroids; rituximab plus MMF plus corticosteroids; and low-dose or high-dose voclosporin plus MMF plus corticosteroids. All studies were placebo-controlled, double-blind, phase II or phase III multicentre trials in adults aged 18 years or older with lupus nephritis. There was some variability in study design, study countries, and the number of patients recruited. Patient characteristics were comparable for age and sex across studies, but variability remained across other characteristics such as race, the inclusion of patients with pure class V disease, time since diagnosis, prior history of lupus nephritis, and treatment history.
The sponsor appraised 5 of the studies in the best-case network as having a high risk of bias, primarily because of imbalances in dropouts between groups. Other concerns included potential selective reporting (the AURA-LV and BLISS-LN studies), a lack of appropriate methods to address missing data (the BLISS-LN study), and authors not clearly declaring conflicts of interest (the LUNAR study). The REGENCY trial was appraised as being at unclear risk of bias because of differences in baseline characteristics across groups and some concerns about unexpected imbalances in dropouts across groups, potential selective reporting, and a lack of clear conflict of interest declarations.
Critical Appraisal of ITC
The methods used to conduct the SLR and NMA were prespecified with a protocol and statistical analysis plan, and appropriate criteria were used to search databases, select studies, and extract data. The SLR appears to be generally well conducted and adequately designed to minimize the risk of bias and error in study identification. However, the risk of bias assessment was performed at the trial level rather than the outcome level; outcome-specific appraisal can provide a more nuanced understanding of study reliability. All included studies were assessed as having a high risk of bias, except for the REGENCY study, for which the overall risk of bias was unclear. Consequently, all cross-study comparisons within the network are considered at risk of bias, meaning that the reported between-group differences may differ systematically from the true effects.
The feasibility assessment steps were appropriate and considered relevant sources of heterogeneity across studies, such as outcome definitions and imbalances in effect modifiers. Following the feasibility appraisal, the sponsor concluded that an NMA should be undertaken but acknowledged that variability in effect modifiers could introduce bias. The statistical approach was appropriate, and sensitivity analyses exploring assumptions made in the analysis supported the robustness of the findings.
Some patient characteristics that were identified as potential effect modifiers were similar or had minor variability across trials, including age and sex distribution, baseline UPCR, and baseline eGFR. However, heterogeneity was observed for some other characteristics across the included trials. Specifically, there were differences in race or ethnicity, prior lupus nephritis history, time since diagnosis, the inclusion of patients with pure class V disease, and the use of prior treatments, which may introduce important heterogeneity. In some instances, the information was not available in the included studies to fully appraise the extent of possible heterogeneity. An additional concern was that the outcome definitions were inconsistent across the studies being compared; for example, the CRR criteria were variable and may not be directly comparable. The sponsor noted that eGFR data were reported differently across trials, with some reporting corrected eGFR capped at 90 mL/min per 1.73 m2 and others reporting uncorrected eGFR. Reporting time points also varied from 48 weeks to 104 weeks. These cross-trial differences raise the concern that the assumption of exchangeability is violated. This assumption needs to be met for the NMA to produce valid effect estimates; when it is violated, the results are at risk of bias (i.e., may differ systematically from the truth). Although alternative methods such as a matching-adjusted indirect comparison or simulated treatment comparison would have allowed some adjustment for cross-trial differences, the review team acknowledges that the data were not available from all included studies to perform a meaningful adjustment, and inconsistencies in outcome definitions could not have been addressed.
Most comparators within the evidence network were informed by a single study, resulting in a star-shaped network and limiting the ability to test for consistency between direct and indirect evidence. The sponsor assessed the consistency assumption within the NMA; however, this evaluation was constrained by the network structure. With only 1 closed loop available, there was insufficient power or information to formally assess consistency. The sparse network and key sources of heterogeneity contributed to wide credible intervals (CrIs) for all the analyses. Notwithstanding other limitations, the imprecision precludes meaningful conclusions from the NMA. The CrIs span a wide range of values and include the possibility that either obinutuzumab or the comparator treatment may be favoured. Additionally, the network is sparse and the relevant comparisons rely only on indirect information, which reduces confidence in the results.
Another limitation of the NMA was that it is missing some relevant comparators because of a lack of anchor to the network, differences in reporting time points, or both. These included glucocorticoids plus cyclophosphamide, and glucocorticoids plus MMF plus a CNI (tacrolimus or cyclosporine). The clinical SLR report submitted by the sponsor also indicated that an unanchored matching-adjusted indirect comparison would have been necessary to compare obinutuzumab plus MMF plus corticosteroids to tacrolimus plus MMF plus corticosteroids. However, the sponsor indicated that this approach may be unsuitable because of several challenges, including substantial patient heterogeneity that reduces overlap in key prognostic and effect-modifying variables, small sample size, and inconsistent outcome definitions that limit comparability. The CDA-AMC review team agrees that such an analysis would have important limitations to its validity and may not be feasible.
Although the HRQoL of patients with lupus nephritis was considered an important treatment goal by clinical experts, this outcome was not assessed in the NMA submitted by the sponsor. PRR independent of CRR is a novel outcome in the NMA. The redefinition, which removes patients with CRR who would have also met PRR criteria (i.e., patients doing well on treatment), makes it unclear whether the outcome of PRR without CRR should be conceptualized as a benefit or a harm. The outcome is therefore most relevant for economic modelling; for clinical interpretation, CRR and overall renal response are more relevant.
Results
Of the 82 RCTs included at the full-text screening stage, 28 studies (reported across 86 records) were selected for data extraction to inform the NMA feasibility assessment because these were considered most relevant to the decision problem based on the population criteria defined. Of these 28 studies, 20 were selected for inclusion in the NMA feasibility assessment. The NMA feasibility assessment resulted in 4 separate networks: 2 networks in studies with a primary analysis end point of less than 48 weeks, where there was no link to the treatment obinutuzumab; 1 network of 5 treatments, including obinutuzumab, with all studies having a primary analysis time point of 48 weeks or longer; and 1 single unconnected study with a primary analysis time point of 48 weeks or longer. This led to the selection of a best-case network of 6 studies connected by MMF plus corticosteroids with a time point of 48 weeks or longer. The best-case network refers to the set of studies the sponsor included in this scenario (the AURA-LV, AURORA-1, BLISS‑LN, LUNAR, NOBILITY, and REGENCY studies).
Efficacy
Results of the NMA are presented as ORs or mean differences with associated 95% CrIs. An OR greater than 1 indicates a better renal response (CRR, PRR independent of CRR, and overall renal response) with obinutuzumab plus MMF plus corticosteroids relative to other comparators. Similarly, a mean difference greater than 0 indicates a higher eGFR preservation with obinutuzumab plus MMF plus corticosteroids relative to other treatments. For all analyses, the model fit was similar between the base-case model and the 2 sensitivity analyses; therefore, it was the preferred model. The posterior median tau suggested moderate heterogeneity for all outcomes, but was estimated with substantial uncertainty, as demonstrated by the wide CrIs. The consistency assessment did not identify evidence of inconsistency in the networks (when a closed loop was available).
For CRR at ≥ 48 weeks or longer, the OR was █ ███ █ (95% CrI, ████ █████ ██ █████ ██████ ████████ █ ███ █ █████████████ ).
For PRR independent of CRR at 48 weeks or longer, the OR was ███ (95%CrI, ████ █████ ██ █████ ██████ ████████ █ ███ █ █████████████ ).
For overall renal response at 48 weeks or longer, the OR was ███ █ (95% CrI, ████ █████ ██ █████ █████ ████████ █ ███ █ ██████████████ ).
For change from baseline in eGFR at 48 weeks or longer, the mean difference was ████ (95% CrI, █████ ███████ ██ █████ ████████ █████ █ ████ ███████████).
Harms
Serious Adverse Events
For SAEs, the model fit was similar between the base-case model and the 2 sensitivity analyses (DIC differences less than 5 points); therefore, the base-case model was the preferred model. The posterior median tau suggested moderate heterogeneity for SAE. The wider CrI of tau indicates uncertainty in the heterogeneity.
Results of the NMA for SAEs are presented as ORs with associated 95% CrIs.
For SAEs at 48 weeks or longer, the OR was ███ (95% CrI, ████ ███ ███ ███ ██ ████ █████ ███████ █ ███ █ ███████████ ).
Studies Addressing Gaps in the Systematic Review Evidence
The submission did not include any studies addressing gaps in the systematic review evidence.
Discussion
This report summarizes the evidence for obinutuzumab plus standard therapy (MMF and corticosteroids) compared with placebo plus standard therapy for adult patients with active lupus nephritis. The evidence appraisal was based on the primary and key secondary end points from the REGENCY trial, a phase III, randomized, double-blind, placebo-controlled, multicentre, global study. Patients who were eligible for the REGENCY study had biopsy-proven class III or IV lupus nephritis (with or without class V) and were receiving standard therapy. Key outcomes of interest to patients and clinicians included CRR, CRR with successful prednisone taper, death or renal-related events, fatigue, and SAEs.
The report also included the sponsor-submitted NMA, which indirectly compared obinutuzumab to belimumab in the current absence of head-to-head comparative trials. Some comparators were not relevant to the clinical context in Canada (voclosporin and rituximab); therefore, they were excluded from this report. Outcomes of interest included CRR, PRR independent of CRR, overall renal response, eGFR, and SAEs.
In terms of the clinical landscape, the EULAR 2025 guidelines give a strong (grade A) recommendation for using obinutuzumab along with standard therapy in patients with active lupus nephritis. The guidelines highlight a transition from class-specific treatment approaches to a broader strategy centred on active lupus nephritis. The clinical experts consulted supported these recommendations, noting that this shift will likely be widely applied to the management of lupus nephritis in clinical practice, where active lupus nephritis encompasses pure class III, IV, or V disease, as well as mixed class III and V and class IV and V presentations.
Efficacy
Input from 3 patient groups received for this review highlighted the key unmet needs for patients, including the need for treatments that are affordable, have simpler administration, are safe to take during pregnancy, have fewer side effects, reduce fatigue, and preserve kidney function. The clinical experts consulted for this review identified key clinical efficacy outcomes from the REGENCY trial, including CRR, CRR with successful prednisone taper, and death or renal-related events. The clinical experts considered a 10% between-group difference to be the smallest clinically meaningful difference for each of these outcomes. Evidence from the REGENCY trial showed that obinutuzumab plus standard therapy likely results in a clinically important increase in the proportion of patients with CRR and CRR with successful prednisone taper through week 76 compared with placebo plus standard therapy. CRR is a composite end point combining a marked reduction in proteinuria with stable kidney function, which reflects remission in clinical practice. A successful prednisone taper also reflects treatment efficacy, in addition to reducing corticosteroid-related toxicities. Therefore, obinutuzumab may address patient-identified unmet needs for treatments that preserve kidney function and have fewer side effects. In addition, obinutuzumab likely results in a clinically important decrease in the proportion of patients with death or renal-related events, which was considered a supportive end point in the trial.
Findings from the REGENCY trial suggest that a higher proportion of patients in the obinutuzumab group achieved a proteinuric response. Additionally, the obinutuzumab group showed a greater mean change from baseline in eGFR at week 76 compared with the placebo group. Although this difference did not reach statistical significance, the clinical experts noted that the observed difference was clinically meaningful in the context of a 76-week trial. They noted that the magnitude of the eGFR change between treatment groups in the REGENCY study represented a clinically meaningful effect, while the wide CI reflected limitations in sample size and trial duration.
Studies have shown that having a modified primary efficacy renal response or modified CRR within 2 years of lupus nephritis diagnosis,28 as well as a proteinuric response at 12 months,29,30 is predictive of long-term kidney function. Across kidney diseases, progression to kidney failure requires a decrease in eGFR, supporting its use as a surrogate end point for chronic kidney disease progression.31 This evidence indicates that these biomarkers are prognostic of long-term outcomes. However, the best evidence for surrogacy validation would be consistent RCT evidence showing that the treatment effect on CRR reliably predicts the treatment effect on the hard kidney end point of ESRD. This evidence is not yet available. Persistent proteinuria despite immunologic remission may reflect chronic damage or delayed healing rather than active inflammation.32 In such cases, the EULAR 2025 guidelines recommend considering a repeat kidney biopsy to assess whether disease activity persists.9,33 Clinical experts noted that patients with extensive scarring may not achieve CRR or a proteinuric response, as defined in the REGENCY study, even when other clinically meaningful improvements are observed. Overall, while these surrogate end points are reasonably likely to predict long-term renal benefit, there remains a lack of RCT evidence demonstrating that treatment-driven improvements in CRR, proteinuria, or eGFR consistently translate into reductions in ESRD.
Obinutuzumab likely results in little to no difference in FACIT-F scores, a HRQoL measure that captures fatigue. However, there was some uncertainty in the findings because of imprecision. The null value was used to assess the certainty of the evidence for FACIT‑F because there was no established minimal important difference identified in patients with active lupus nephritis and the questionnaire is not used in clinical practice. Although patient input highlighted fatigue as an important unmet need, the clinical experts noted that the lack of variation in FACIT-F scores between treatment groups did not preclude the potential for significant differences in other clinically relevant efficacy outcomes because fatigue is influenced by many nonrenal factors and the FACIT-F scale may not be sensitive to kidney-specific treatment effects.
A key limitation of the REGENCY study is the absence of direct comparative evidence against treatments commonly used in clinical practice in Canada. To address this gap, the sponsor submitted an NMA comparing obinutuzumab with other treatments, of which belimumab, a B-lymphocyte stimulator inhibitor, is considered to be the most relevant comparator; however, imprecision and a high risk of bias rendered the comparative efficacy uncertain. Notwithstanding other limitations, the wide CrIs crossed the null for all efficacy outcomes, indicating that it is possible that obinutuzumab plus standard therapy could offer similar, better, or worse efficacy compared to belimumab plus standard therapy. Interpretation of the NMA is further limited by a high risk of bias in most of the included studies as well as several important sources of heterogeneity across the studies, including differences in trial design, patient eligibility criteria, baseline characteristics, and outcome definitions. These limitations suggest violation of the underlying exchangeability assumption, leading to a risk that effect estimates are biased (i.e., systematically different from the truth). The clinical experts consulted for this review noted that maintaining HRQoL is a key treatment goal, which was not evaluated in the NMA submitted by the sponsor. The NMA also did not include indirect comparisons to other therapies, such as tacrolimus, cyclosporine, or cyclophosphamide, which clinical experts noted are sometimes used in Canada for this patient population. The absence of evidence leaves the relative efficacy and safety of obinutuzumab versus these other treatment options uncertain.
Harms
The overall frequency of AEs in the REGENCY study was similar between treatment groups. Discontinuations because of AEs were uncommon in both treatment groups, which is suggestive of good tolerability. According to the experts consulted, the safety findings through week 76 showed that obinutuzumab had an overall AE profile consistent with what is already known for this drug class.
Using the null value as a threshold, treatment with obinutuzumab plus standard therapy likely resulted in an increase in the proportion of patients with SAEs compared with placebo plus standard therapy, although the clinical relevance of this increase is uncertain. SAEs occurred more frequently with obinutuzumab than with placebo, driven largely by serious infections, including COVID-19. The REGENCY study began enrolling patients before the availability of robust vaccination and antiviral therapy for COVID-19. The sponsor indicated that COVID-19–related SAEs occurred earlier in the study, with none reported in the latter half of the trial. When COVID-19–related events were excluded, the imbalance in SAEs persisted but was of a smaller magnitude. Deaths were infrequent in both treatment groups. Infusion-related reactions, drug-related neutropenia, and higher-grade infections were observed more frequently in the obinutuzumab group than in the placebo group. These patterns align with the known mechanistic effects of B-cell depletion according to the experts consulted.
Exploratory comparisons of the 2 obinutuzumab dosing regimens showed similar overall proportions of patients with AEs, although SAEs were more common in the group that did not receive the additional week 50 dose. A post hoc analysis found that these differences were already present before the dosing schedules diverged, suggesting that they are unlikely to be attributable to the dosing regimen itself.
Indirect evidence from the sponsor-submitted NMA suggested that comparative harms, assessed as SAEs, of obinutuzumab plus standard therapy versus belimumab plus standard therapy are uncertain. The NMA of SAEs was affected by limitations similar to those of the efficacy NMAs, including a high risk of bias in individual studies and violation of exchangeability. Additionally, wide CrIs leave it uncertain whether obinutuzumab plus standard therapy offers a similar, better, or worse incidence of SAEs compared to belimumab plus standard therapy.
Ethics and Equity Considerations
SLE is about 9 times more common in females than in males,5 and approximately 50% of patients with SLE will develop lupus nephritis.2 Lupus nephritis is more common in patients who are Asian, Black, Hispanic, or Indigenous,6-8 and research suggests that patients who are Black or Hispanic are more likely to progress to kidney failure than patients who are white.2 Patient group input noted that these disparities arise not only from biological factors but also from structural barriers such as delayed diagnosis, reduced access to specialists, and socioeconomic inequities related to income, education, and systemic racism.
Patient groups reported a wide range of debilitating symptoms and significant impacts on daily functioning, emotional well-being, and socioeconomic stability, with caregivers also experiencing substantial emotional and logistical strain in supporting daily needs and medical care. Current treatment challenges reported by patient groups include difficulty working full-time because of infusion schedules, refrigeration requirements for self-injectors, long travel distances, limited specialist access, long wait times, and a lack of culturally appropriate care. Out-of-pocket costs for medications, travel, and allied health services further exacerbate the burden, underscoring the need for improved support and access. The clinical experts also noted that kidney biopsies, which are necessary for diagnosis and treatment guidance, are typically performed at specialized centres; this can delay care for patients with limited local access.
Conclusion
One phase III, randomized, double-blind, placebo-controlled trial (the REGENCY study) provided evidence for the efficacy and safety of obinutuzumab in combination with standard therapy in patients with lupus nephritis. According to the clinical experts, the study population was broadly representative of patients seen in clinical practice in Canada. Evidence from the REGENCY study demonstrated that obinutuzumab plus standard therapy likely results in a clinically important increase in the proportion of patients with a CRR and CRR with a successful prednisone taper through week 76 compared with placebo plus standard therapy. These surrogate biomarkers are considered to be prognostic of long-term clinical outcomes, based on observational evidence. Therefore, obinutuzumab may address patient-identified unmet needs for treatments that preserve kidney function. In addition, obinutuzumab likely results in a clinically important decrease in the proportion of patients with death or renal-related events and likely leads to little to no difference in fatigue based on FACIT-F scores. Obinutuzumab likely results in an increase in the proportion of patients with SAEs, the clinical importance of which is uncertain. Patients in the obinutuzumab group experienced numerically more SAEs and grade 3 to 5 AEs than patients in the placebo group; however, according to the experts consulted, harms were considered manageable in clinical practice and consistent with the known safety profile of B-cell–targeted therapy.
Indirect evidence from an NMA submitted by the sponsor suggested that obinutuzumab plus standard therapy has an uncertain effect on renal response outcomes and SAEs compared with belimumab plus standard therapy. The limitations that rendered the results uncertain were a high risk of bias in the included studies and between-trial heterogeneity that likely violates the underlying exchangeability assumption. Together, these factors introduce a risk of bias in the network, which may cause the effect estimates to systematically differ from the true differences between treatments. Irrespective of methodological concerns, wide CrIs included the possibility that obinutuzumab plus standard therapy has better, worse, or similar efficacy and harms compared to belimumab plus standard therapy. No evidence was available to inform the comparative effectiveness of obinutuzumab versus other relevant therapies that the clinical experts noted are sometimes used in clinical practice, such as CNIs (tacrolimus and cyclosporine) and cyclophosphamide.
Economic Review
Methods
The review team appraised the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of obinutuzumab plus standard therapy, defined as MMF in combination with glucocorticoids (i.e., methylprednisolone, and prednisone), compared to standard therapy alone, IV belimumab plus standard therapy, and subcutaneous (SC) belimumab plus standard therapy for adults with active lupus nephritis.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of obinutuzumab plus standard therapy from the perspective of a public health care payer in Canada over a lifetime horizon of 67 years. The modelled population consisted of adults with class III or IV lupus nephritis, with or without class V disease, which is narrower than the Health Canada indication and was based on the participants in the REGENCY trial. The sponsor’s base-case analysis included costs related to drug acquisition and administration, AEs, supportive care, kidney transplant, rescue therapy, and end-of-life care.
In the sponsor’s base case, obinutuzumab plus standard therapy was associated with incremental costs of $38,595 and 0.83 incremental quality-adjusted life-years (QALYs) gained relative to standard therapy alone. This resulted in an incremental cost-effectiveness ratio (ICER) of $46,752 per QALY gained. Of the incremental benefit compared to standard therapy alone (0.83 incremental QALYs gained), approximately 98% of the benefit was predicted to be accrued after the observation period of the REGENCY trial (76 weeks). SC belimumab was also on the frontier. The ICER of SC belimumab plus standard therapy compared to obinutuzumab plus standard therapy was $605,632 per QALY gained. Additional information about the sponsor’s submission is summarized in Appendix 10 in the Supplemental Material document.
CDA-AMC identified several key issues with the sponsor’s analysis (e.g., refer to Table 5; full details are provided in Appendix 11 in the Supplemental Material document).
Table 5: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
Comparative clinical efficacy and safety of obinutuzumab plus standard therapy vs. belimumab plus standard therapy is highly uncertain. | Conclusions of the sponsor’s NMA are uncertain due to risk of bias in the included studies, inability to test consistency assumption, and a violation of the exchangeability assumption. Comparative efficacy for obinutuzumab vs. belimumab is highly uncertain. | CDA-AMC could not address this issue. | No scenario analysis was conducted. |
Relevant comparators were excluded. | Glucocorticoids in combination with mycophenolate and tacrolimus, glucocorticoids in combination with cyclophosphamide, and glucocorticoids in combination with mycophenolate and rituximab — all therapies recommended by EULAR and used in clinical practice in Canada — were not included in the economic model. | CDA-AMC could not address this issue because comparative efficacy between obinutuzumab plus standard therapy and the missing comparators is unknown. | No scenario analysis was conducted. |
The sponsor assumed that treatment response after discontinuation was maintained indefinitely. | No evidence was presented to suggest that patients who discontinue treatment sustain their treatment response indefinitely. | CDA-AMC assumed that the full treatment response was maintained for 3 years after treatment discontinuation. | The length of response after treatment discontinuation is highly uncertain. CDA-AMC explored the impact in a scenario analysis. |
The sponsor assumed that all patients received obinutuzumab for 36 months. | The KDIGO guidelines recommend that a subset of patients with active lupus nephritis receive treatment for at least 36 months. | CDA-AMC could not address this issue. | Given uncertainty in obinutuzumab treatment duration, CDA-AMC explored the impact of a 6-year treatment duration in a scenario analysis. |
Transition probabilities used in the probabilistic analysis were not aligned with the sponsor’s submitted report. | The transition probabilities applied by the sponsor in the probabilistic analysis were not aligned with the sponsor’s report and generated results that were very different from the deterministic analysis. | CDA-AMC could not address this issue and used the deterministic analysis throughout the report. | No scenario analysis was conducted. |
CDA-AMC = Canada’s Drug Agency; EULAR = European Alliance of Associations for Rheumatology; KDIGO = Kidney Disease: Improving Global Outcomes; NMA = network meta-analysis; vs. = versus.
Note: Full details of the issues identified by CDA-AMC are provided in Appendix 11 in the Supplemental Material document.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions in consultation with clinical experts (e.g., refer to Table 18 in Appendix 11 in the Supplemental Material document). Detailed information about the CDA-AMC base case is provided in Appendix 11 in the Supplemental Material document.
Impact on Health Care Costs
Obinutuzumab plus standard therapy is predicted to be associated with additional health care costs compared to standard therapy alone (incremental costs = $40,437). This increase in health care spending results from drug acquisition costs associated with obinutuzumab (refer to Figure 1).
Figure 1: Impact of Obinutuzumab Plus Standard Therapy vs. Standard Therapy Alone on Health Care Costs
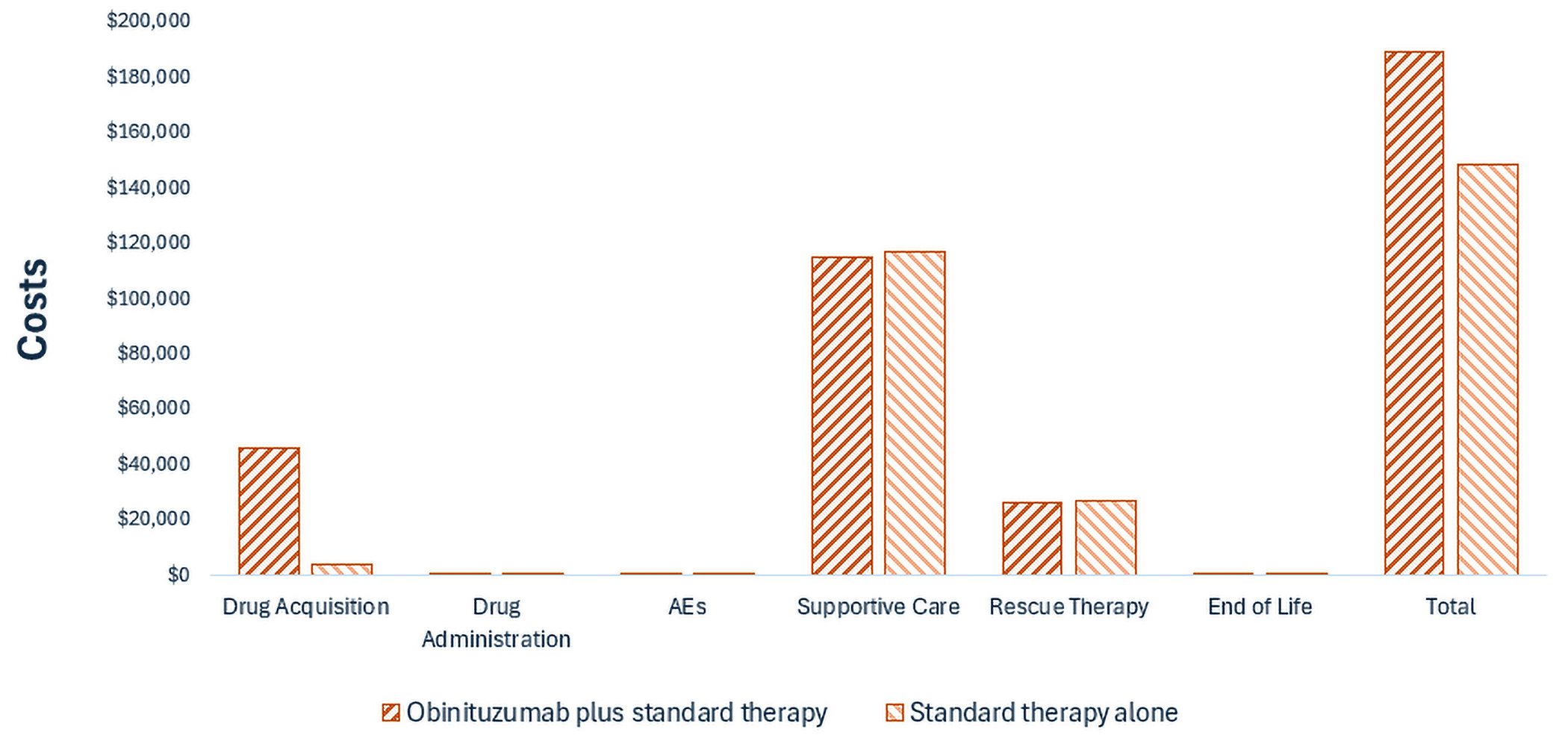
AE = adverse event; vs. = versus.
Impact on Health
Relative to standard therapy alone, obinutuzumab is expected to result in 0.51 additional QALYs per patient over the lifetime horizon (refer to Figure 2). This is because patients are expected to spend more time with complete response.
Figure 2: Impact of Obinutuzumab Plus Standard Therapy vs. Standard Therapy Alone on Patient Health
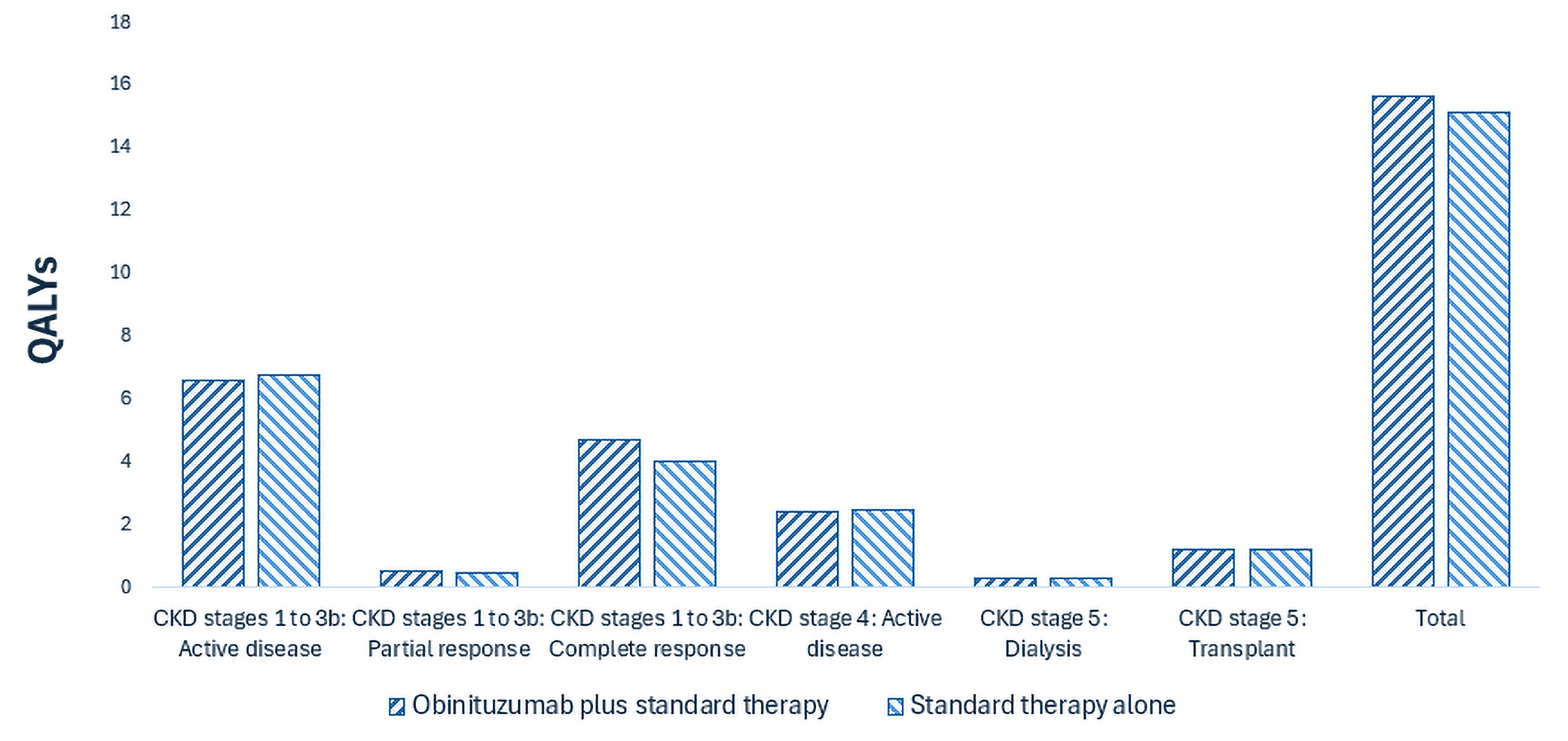
CKD = chronic kidney disease; QALY = quality-adjusted life-year; vs. = versus.
Overall Results
The results of the CDA-AMC base case suggest an ICER of $79,492 per QALY gained for obinutuzumab plus standard therapy compared to standard therapy alone (refer to Table 6).
Table 6: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Standard therapy | 148,957 | 15.15 | Reference |
Obinutuzumab plus standard therapy | 189,394 | 15.65 | 79,492 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Publicly available list prices were used for all comparators. Full results are reported in Table 19 in Appendix 11 in the Supplemental Material document.
Uncertainty and Sensitivity
Uncertainty was explored in the scenario analyses outlined in Table 5. Uncertainty around treatment duration and treatment effect after discontinuation had the largest impact on cost-effectiveness (e.g., refer to Table 22 in Appendix 11 in the Supplemental Material document).
Summary of the Budget Impact
The sponsor submitted a budget impact analysis to estimate the 3-year (2026 to 2028) budget impact of reimbursing obinutuzumab for the treatment of adult patients with active lupus nephritis who are receiving standard therapy. The sponsor assumed the payer would be CDA-AMC–participating public drug plans and derived the size of the population of patients who would be eligible for treatment using an epidemiologic approach. The price of obinutuzumab plus standard therapy was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in Appendix 12 in the Supplemental Material document.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (e.g., refer to Appendix 12 in the Supplemental Material document). CDA-AMC estimated that by year 3 of reimbursement, 1,916 patients would be eligible for obinutuzumab plus standard therapy; of these, 433 patients are expected to receive obinutuzumab plus standard therapy. The estimated incremental budget impact of reimbursing obinutuzumab is predicted to be approximately $6 million over the first 3 years, with an expected expenditure of $14 million on obinutuzumab. The actual budget impact of reimbursing obinutuzumab will depend on its uptake and the displacement rate of standard therapy.
Conclusion
Based on the CDA-AMC base case, obinutuzumab plus standard therapy would be considered cost-effective at the submitted price if the public health care system were willing to pay at least $79,492 for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details of the impact of price reductions on cost-effectiveness are presented in Table 21 in Appendix 11 in the Supplemental Material document). The estimated cost-effectiveness of obinutuzumab plus standard therapy compared to belimumab plus standard therapy is uncertain because of the unknown comparative efficacy between the 2 treatments. As such, there is insufficient evidence to determine whether obinutuzumab plus standard therapy provides a greater health benefit than belimumab plus standard therapy. If there are no differences in health outcomes between obinutuzumab plus standard therapy and belimumab plus standard therapy, then the total cost of obinutuzumab to the health system should not exceed that of belimumab.
The budget impact of reimbursing obinutuzumab plus standard therapy to the public drug plans in the first 3 years is estimated to be approximately 6 million. The 3-year expenditure on obinutuzumab (i.e., not accounting for current expenditure on comparators) is estimated to be $14 million.
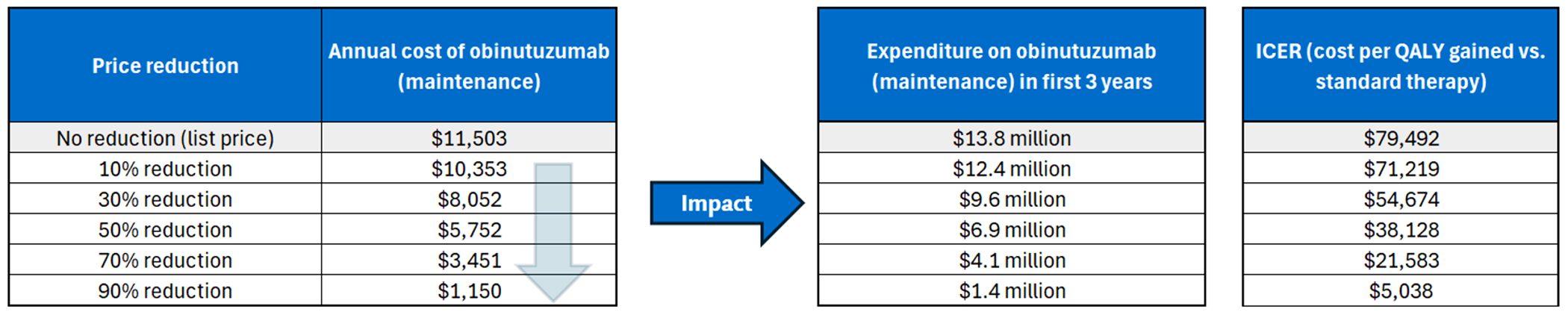
References
1.Asif S, Bargman J, Auguste B. A review of the AURORA and BLISS trials: will it revolutionize the treatment of lupus nephritis? Curr Opin Nephrol Hypertens. 2022;31(3):278-282. doi:10.1097/MNH.0000000000000792 PubMed
2.Parikh SV, Almaani S, Brodsky S, Rovin BH. Update on Lupus Nephritis: Core Curriculum 2020. Am J Kidney Dis. 2020;76(2):265-281. doi:10.1053/j.ajkd.2019.10.017 PubMed
3.Bernatsky S, Joseph L, Pineau CA, Tamblyn R, Feldman DE, Clarke AE. A population-based assessment of systemic lupus erythematosus incidence and prevalence—results and implications of using administrative data for epidemiological studies. Rheumatology. 2007;46(12):1814-1818. doi:10.1093/rheumatology/kem233 PubMed
4.Carter EE, Barr SG, Clarke AE. The global burden of SLE: prevalence, health disparities and socioeconomic impact. Nat Rev Rheumatol. 2016;12(10):605-20. doi:10.1038/nrrheum.2016.137 PubMed
5.Schur PH, Hahn BH. Pisetsky DS, Curtis MR, eds. Epidemiology and pathogenesis of systemic lupuserythematosus. UpToDate. 2020.
6.Anders H-J, Saxena R, Zhao M-h, Parodis I, Salmon JE, Mohan C. Lupus nephritis. Nat Rev Dis Prim. 2020;6(1):7. PubMed
7.Ferucci ED. Understanding the disproportionate burden of rheumatic diseases in Indigenous North American populations. Rheum Dis Clin North Am. 2020;46(4):651-660. PubMed
8.Hurd K, Barnabe C. Systematic review of rheumatic disease phenotypes and outcomes in the Indigenous populations of Canada, the USA, Australia and New Zealand. Rheumatol Int. 2017;37(4):503-521. PubMed
9.Fanouriakis A, Kostopoulou M, Anders H-J, et al. EULAR recommendations for the management of systemic lupus erythematosus with kidney involvement: 2025 update. Ann Rheum Dis. 2026;85(1):75-90. PubMed
10.Sammaritano LR, Askanase A, Bermas BL, et al. 2024 American College of Rheumatology (ACR) Guideline for the Screening, Treatment, and Management of Lupus Nephritis. Arthritis Care Res (Hoboken). 2025;77(9):1045-1065. doi:10.1002/acr.25528 PubMed
11.Hoffmann-La Roche Ltd. Gazyva (obinutuzumab): 25 mg/mL injection [draft product monograph]. March 4, 2025.
12.Kasitanon N, Boripatkosol P, Louthrenoo W. Response to combination of mycophenolate mofetil, cyclosporin A and corticosteroid treatment in lupus nephritis patients with persistent proteinuria. Int J Rheum Dis. 2018;21(1):200-207. PubMed
13.Chen W, Liu Q, Chen W, et al. Outcomes of maintenance therapy with tacrolimus versus azathioprine for active lupus nephritis: a multicenter randomized clinical trial. Lupus. 2012;21(9):944-952. PubMed
14.Hoffmann-La Roche Ltd. Clinical Study Report: Study CA41705 (REGENCY). A Phase III, Randomized, Double-Blind, Placebo-Controlled, Multicenter Study to Evaluate the Efficacy and Safety of Obinutuzumab in Patients with ISN/RPS 2003 Class III or IV Lupus Nephritis [internal sponsor's report]. December 2024.
15.Hoffmann-La Roche Ltd. Clinical Evidence Template (CET) for GAZYVA® (obinutuzumab) for the treatment of adult patients with active lupus nephritis who are receiving standard therapy [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Gazyva (obinutuzumab) 25 mg/mL injection. September 16, 2025.
16.CADTH. Drug Reimbursement Expert Review Committee final recommendation: Obinutuzumab (Gazyva - Hoffmann-La Roche Limited). January 27, 2015. Accessed November 18, 2025. https://www.cda-amc.ca/sites/default/files/pcodr/pcodr-gazyva-cll-fn-rec.pdf
17.CADTH. Drug Reimbursement Expert Review Committee final recommendation: Obinutuzumab (Gazyva - Hoffmann-La Roche Limited). June 2, 2017. Accessed November 18, 2025. https://www.cda-amc.ca/sites/default/files/pcodr/pcodr_obinutuzumab_gazyva_fl_fn_rec.pdf
18.CADTH. Drug Reimbursement Expert Review Committee final recommendation: Obinutuzumab (Gazyva - Hoffmann-La Roche Limited). November 1, 2018. Accessed November 18, 2025. https://www.cda-amc.ca/sites/default/files/pcodr/pcodr_obinutuzumab_gazyva_fl_preun_fn_rec.pdf
19.Anders HJ, Saxena R, Zhao MH, Parodis I, Salmon JE, Mohan C. Lupus nephritis. Nat Rev Dis Primers. 2020;6(1):7. doi:10.1038/s41572-019-0141-9 PubMed
20.Markowitz GS, D'Agati VD. The ISN/RPS 2003 classification of lupus nephritis: an assessment at 3 years. Kidney Int. 2007;71(6):491-5. doi:10.1038/sj.ki.5002118 PubMed
21.KDIGO. 2024 Lupus Nephritis Guideline. 2024;105(1S). https://kdigo.org/wp-content/uploads/2024/01/KDIGO_2024_Lupus_Nephritis_Guideline.pdf [sponsor supplied reference]. 2024.
22.American College of Rheumatology. ACR 2024 Guideline for the Screening, Treatment, and Management of Lupus Nephritis. Published online 2024. Accessed April 16, 2025. https://assets.contentstack.io/v3/assets/bltee37abb6b278ab2c/blt4db6d0b451e88caf/lupus-nephritis-guideline-summary-2024.pdf [sponsor supplied reference]. 2024.
23.Furie R, Rovin BH, Houssiau F, et al. Two-Year, Randomized, Controlled Trial of Belimumab in Lupus Nephritis. N Engl J Med. 2020;383(12):1117-1128. doi:10.1056/NEJMoa2001180 PubMed
24.Rovin BH, Teng YKO, Ginzler EM, et al. Efficacy and safety of voclosporin versus placebo for lupus nephritis (AURORA 1): a double-blind, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet. 2021;397(10289):2070-2080. doi:10.1016/S0140-6736(21)00578-X PubMed
25.Rovin BH, Ayoub IM, Chan TM, Liu Z-H, Mejía-Vilet JM, Floege J. KDIGO 2024 Clinical Practice Guideline for the Management of Lupus Nephritis. Kidney Int. 2024;105(1S):S1-S69. PubMed
26.Hoffmann-La Roche Ltd. Statistical Analysis Plan [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Gazyva (obinutuzumab) 25 mg/mL injection. September 16, 2025.
27.NICE. Single technology appraisal and highly specialised technologies evaluation: User guide for company evidence submission template [sponsor supplied reference]. 2015:15-17.
28.Urowitz M, Georgiou ME, Touma Z, et al. Renal response status to predict long-term renal survival in patients with lupus nephritis: results from the Toronto Lupus Cohort. Lupus Sci Med. 2024;11(2):e001264. doi:10.1136/lupus-2024-001264 PubMed
29.Dall'Era M, Cisternas MG, Smilek DE, et al. Predictors of long-term renal outcome in lupus nephritis trials: lessons learned from the Euro‐Lupus Nephritis cohort. Arthritis Rheumatol. 2015;67(5):1305-1313. doi:10.1002/art.39026 PubMed
30.Tamirou F, Lauwerys BR, Dall'Era M, et al. A proteinuria cut-off level of 0.7 g/day after 12 months of treatment best predicts long-term renal outcome in lupus nephritis: data from the MAINTAIN Nephritis Trial. Lupus Sci Med. 2015;2(1). doi:10.1136/lupus-2015-000123 PubMed
31.Inker LA, Collier W, Greene T, et al. A meta-analysis of GFR slope as a surrogate endpoint for kidney failure. Nat Med. 2023;29(7):1867-1876. doi:10.1038/s41591-023-02418-0 PubMed
32.De Vriese AS, Sethi S, Fervenza FC. Lupus nephritis: redefining the treatment goals. Kidney Int. 2025;107(2):198-211. doi:10.1016/j.kint.2024.10.018 PubMed
33.Fanouriakis A. EULAR recommendations for lupus nephritis [abstract]. Lupus Sci Med. 2025;12(Suppl 1):A2-A3. doi:10.1136/lupus-2025-la.2PubMed
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.