Drugs, Health Technologies, Health Systems
Reimbursement Review
Seladelpar (Lyvdelzi)
Sponsor: Gilead Sciences Canada, Inc.
Therapeutic area: Primary biliary cholangitis
Summary
What Is Primary Biliary Cholangitis?
Primary biliary cholangitis (PBC) is a rare, progressive autoimmune disease that damages the bile ducts in the liver. Over time, if left untreated, PBC can lead to permanent scarring of liver tissue, which is called cirrhosis of the liver, and can cause liver failure and death.
In 2015, there were about 318 patients in Canada diagnosed with PBC.
What Are the Treatment Goals and Current Treatment Options for PBC?
Although many patients with PBC respond to first-line treatment with ursodeoxycholic acid (UDCA), approximately one-third of patients have PBC that has an inadequate response to UDCA or have UDCA intolerance.
There is a need for treatments that can cure the disease, normalize alkaline phosphatase (ALP) levels, reduce symptoms such as pruritus, and improve patients’ quality of life.
Current treatment options for PBC in Canada, beyond first-line treatment with UDCA, are obeticholic acid (OCA) and elafibranor used in combination with UDCA or as monotherapy in patients with UDCA intolerance.
What Is Seladelpar and Why Did Canada’s Drug Agency Conduct This Review?
Seladelpar activates the peroxisome proliferator-activated receptor delta, which plays a key role in the pathobiology of PBC in cell types including hepatocytes, cholangiocytes, Kupffer cells, and stellate cells.
The recommended dose for seladelpar is 10 mg orally once daily, with or without food. The Health Canada indication is for “the treatment of PBC in combination with UDCA in adults who have an inadequate response to UDCA alone, or as monotherapy in adults unable to tolerate UDCA.”
Canada’s Drug Agency (CDA-AMC) reviewed seladelpar to inform a recommendation to the participating public drug programs on whether it should be reimbursed for the indication under review by Health Canada.
How Did CDA-AMC Evaluate Seladelpar?
CDA-AMC reviewed the clinical evidence on the beneficial and harmful effects, as well as the economic evidence, of seladelpar versus other treatments used in Canada for the treatment of PBC, in combination with UDCA in adults whose PBC has an inadequate response to UDCA alone, or as monotherapy in adults with UDCA intolerance.
OCA and elafibranor were considered relevant comparators for the review of the clinical evidence.
CDA-AMC identified equity and ethical considerations relevant to seladelpar and PBC.
The review was informed by materials submitted by the sponsor, which included clinical and economic evidence.
The review was also informed by 1 patient group submission and 1 clinician group submission, in response to the call for input by CDA-AMC, and by input from the participating public drug programs on issues that may impact implementation of a recommendation.
Two clinical experts (gastroenterologists) with representation from Ontario and Alberta were consulted as part of the review process.
What Were the Findings?
Clinical Evidence
CDA-AMC reviewed the following clinical evidence:
1 phase III, randomized, double-blind controlled trial (RESPONSE) comparing seladelpar 10 mg taken orally once daily to placebo in 193 adults whose PBC had an inadequate response to UDCA or with UDCA intolerance. During the trial, seladelpar 10 mg was administered as add-on to UDCA therapy for patients with UDCA tolerance; for patients with UDCA intolerance, seladelpar was administered as monotherapy
1 ongoing, open-label long-term extension study (ASSURE) evaluating the safety of seladelpar 10 mg, taken orally once daily, in patients with PBC from the RESPONSE trial and 4 other trials
1 network meta-analysis (NMA) and 1 anchored matching-adjusted indirect comparison (MAIC) to assess the relative efficacy and safety of seladelpar with OCA and elafibranor in adults whose PBC had an inadequate response to UDCA or who had UDCA intolerance.
For the comparison of seladelpar versus placebo:
The RESPONSE trial showed high certainty of evidence that treatment with seladelpar in combination with UDCA in adults whose PBC had an inadequate response to UDCA, or as monotherapy in adults with UDCA intolerance, results in a clinically important increase in the proportion of patients with biochemical response and ALP normalization at 12 months.
The trial showed moderate and low certainty of evidence that treatment with seladelpar results in a clinically important decrease in pruritus at 6 months and 12 months, respectively.
There was low certainty of evidence of little to no difference in health-related quality of life (HRQoL) at 12 months.
Based on the evidence submitted to CDA-AMC, the effect of seladelpar on long-term outcomes such as progression to end-stage liver disease, transplant-free survival, or overall survival is unknown.
There was no new safety signals identified, and the safety of seladelpar in combination with UDCA was consistent with the known safety profiles of the individual drugs.
For the comparison of seladelpar versus OCA and elafibranor:
Based on the NMA, seladelpar may be associated with fewer occurrences of pruritus as an adverse event compared to OCA; however, due to limitations in both the NMA and MAIC, primarily attributed to serious imprecision, no conclusions can be drawn regarding the relative efficacy and safety of seladelpar compared with OCA and elafibranor.
Global HRQoL was not assessed in the NMA or MAIC.
Although no new safety signals were identified in the long-term extension study (the ASSURE study), there is uncertainty regarding the long-term safety of seladelpar due to the open-label design and lack of a comparator group.
The evidence submitted to CDA-AMC did not include head-to-head comparisons between seladelpar and relevant comparators (e.g., elafibranor and OCA) in the second-line setting, which represents a gap in the available direct evidence.
Economic Evidence
Seladelpar is available as a 10 mg oral capsule. At the submitted price of $235.33 per capsule, the annual cost of seladelpar is expected to be $85,955 per patient, based on the Health Canada–recommended dosage. When administered in combination with UDCA, the annual cost of seladelpar is estimated to range from $86,485 to $86,624 per patient, depending on dose of UDCA.
Key clinical efficacy inputs in the economic analysis (i.e., transition probabilities between PBC biomarker health states, specifically to states characterized by normal and mildly elevated ALP levels) for seladelpar, with or without UDCA, were derived from the RESPONSE trial. Evidence submitted by the sponsor indicates that seladelpar in combination with UDCA in adults whose PBC has an inadequate response to UDCA alone, or as monotherapy in adults with UDCA intolerance results in a clinically important increase in the proportion of patients with biochemical response and ALP normalization at 12 months compared to placebo in adults with PBC. The trial also showed moderate and low certainty of evidence that treatment with seladelpar results in a clinically important decrease in pruritus at 6 months and 12 months, respectively. The evidence for a difference in HRQoL at 12 months was of low certainty. The impact of seladelpar on long-term outcomes such as transplant-free survival is unknown.
Clinical efficacy comparisons of seladelpar (with or without UDCA) to elafibranor and OCA (each with or without UDCA) were informed by the sponsor-submitted NMA and anchored MAIC, respectively. Due to methodological limitations and imprecision associated with the sponsor-submitted indirect treatment comparisons, no definite conclusions could be drawn regarding the relative efficacy and safety of seladelpar compared to OCA and elafibranor (refer to the CDA-AMC Clinical Review section of this report). Based on the NMA, seladelpar may be associated with fewer occurrences of pruritus as an adverse event compared to OCA; however, this finding is highly uncertain. Overall, definite conclusions about the comparative harms and the impact on global HRQoL, which was not assessed in the sponsor-conducted indirect treatment comparisons, could not be made.
CDA-AMC estimates that the budget impact of reimbursing seladelpar, with or without UDCA, for use in the indicated population will be $770 million over the first 3 years compared to the amount currently spent on comparators. The expenditure on seladelpar, with or without UDCA, over this period is predicted to be $785 million. The estimated budget impact was driven by market share assumptions and the estimated population of eligible patients, both of which were highly uncertain. The economic feasibility must be addressed, given that the predicted incremental budget impact of reimbursing seladelpar, with or without UDCA, is predicted to be greater than $40 million in year 1, year 2, and year 3, and given the magnitude of uncertainty in the estimated budget impact.
Based on the sponsor’s submitted economic evaluation, seladelpar is expected to increase health care system costs if reimbursed by public drug plans. However, there is insufficient evidence to suggest that seladelpar provides greater health benefit to patients than OCA or elafibranor. If there are no differences in health outcomes between seladelpar and OCA or elafibranor, then the total cost of seladelpar to the health system should not exceed that of OCA or elafibranor for the treatment of adult patients with PBC, either in combination with UDCA or as monotherapy.
Abbreviations
AE
adverse event
AMA
antimitochondrial antibody
ANA
antinuclear antibody
BSC
best supportive care
CaNAL
Canadian Network for Autoimmune Liver Disease
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
Crl
credible interval
ESS
effective sample size
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
ITC
indirect treatment comparison
ITT
intention to treat
LS
least squares
LTE
long-term extension
MAIC
matching-adjusted indirect comparison
MID
minimal important difference
MMRM
mixed-effect model for repeated measures
MSPN
moderate to severe pruritus numerical rating scale
NMA
network meta-analysis
NRS
numerical rating scale
OCA
obeticholic acid
OR
odds ratio
PBC
primary biliary cholangitis
PBC-40
primary biliary cholangitis-40
RCT
randomized controlled trial
RR
risk ratio
SD
standard deviation
TB
total bilirubin
TEAE
treatment emergent adverse event
UDCA
ursodeoxycholic acid
ULN
upper limit of normal
Background
Introduction
The objectives of this report are as follows:
Review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of seladelpar 10 mg capsules, taken orally once daily, for the treatment of primary biliary cholangitis (PBC) in combination with ursodeoxycholic acid (UDCA) in adults whose PBC has an inadequate response to UDCA alone, or as monotherapy in adults with UDCA intolerance. The focus will be on comparing seladelpar to relevant comparators used in clinical practice in Canada and identifying gaps in the current evidence, as outlined in Table 1.
Review and critically appraise the economic information submitted by the sponsor, including a cost-effectiveness analysis and budget impact analysis. The focus of the economic review is aligned with the scope of the clinical review, unless otherwise stated. For most reviews, a Canada’s Drug Agency (CDA-AMC) base case is developed, informed by clinical expert input, the available clinical evidence, and the best interpretation of the economic evidence based on the information provided by the sponsor.
The application was submitted by the sponsor before receiving a Notice of Compliance from Health Canada. This report reflects the indication and recommended dosage for seladelpar during the initial CDA-AMC review period.
Table 1: Information on the Application Submitted for Review and on the CDA-AMC Review
Item | Description |
|---|---|
Information on the application submitted for review | |
Drug | Seladelpar (Lyvdelzi), 10 mg, capsule, oral |
Sponsor | Gilead Sciences Canada, Inc. |
Health Canada indication | For the treatment of PBC in combination with UDCA in adults who have an inadequate response to UDCA alone, or as monotherapy in adults unable to tolerate UDCA |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | October 16, 2025 |
Mechanism of action | Seladelpar activates the peroxisome proliferator-activated receptor delta that plays a key role in the pathobiology of PBC in cell types including hepatocytes, cholangiocytes, Kupffer cells, and stellate cells. |
Recommended dosage | 10 mg orally once daily, with or without food |
Submission type | Initial |
Sponsor’s reimbursement request | Per indication |
Submitted price | $235.33 per 10 mg capsule |
Information on the CDA-AMC review | |
Review type | Standard |
Clinical review focusa | Population: as defined in the Health Canada indication Intervention: per recommended dosage Comparators: UDCA monotherapy; obeticholic acid and elafibranor, as monotherapy or in combination with UDCA Outcomes:
|
CDA-AMC = Canada’s Drug Agency; NOC = Notice of Compliance; NRS = numerical rating scale; PBC = primary biliary cholangitis; PBC-40 = primary biliary cholangitis 40; QoL = quality of life; UDCA = ursodeoxycholic acid; ULN = upper limit of normal.
aThe economic review aligns with the scope of the clinical review, unless otherwise stated.
Submission History for the Drug Under Review
CDA-AMC has not previously reviewed seladelpar through the reimbursement review process.
Sources of Information
The contents of the Reimbursement Review report are informed by materials submitted by the sponsor, input received from interested parties (patient groups, clinician groups, and drug programs), and input from clinical experts consulted by CDA-AMC for this review.
Calls for patient group and clinician group input are issued for each reimbursement review. One patient group submission from Canadian PBC Society and 1 clinician group submission from the Canadian Network for Autoimmune Liver Disease (CaNAL) were received, in accordance with standard procedures. Patient input was gathered via a national survey of 380 patients, regional in-person meetings with more than 300 patients and caregivers, and a national conference in June 2025 with more than 100 participants. Clinician input was provided through expert consensus, steering committee discussions, and a June 2025 symposium. Both submissions were independently authored, with disclosed financial relationships. The full patient group submission is available in the consolidated input document.
Input from patient and clinician groups is considered throughout the review, including in the selection of outcomes for the clinical review and in the interpretation of the clinical and economic evidence. Relevant patient and clinician group input is summarized in the Disease Background, Current Management, and Unmet Needs and Existing Challenges sections.
The drug programs provide input on each drug being reviewed through the reimbursement review process by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in the Summary of Drug Program Input and Clinical Expert Responses table in Appendix 1 in the Supplemental Material document.
Each review team includes at least 1 clinical expert with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process. Two clinical experts (gastroenterologists) with expertise in the diagnosis and management of PBC participated as part of the review team.
Disease Background
PBC is a rare, progressive autoimmune liver disease characterized by immune-mediated destruction of cholangiocytes in the intrahepatic bile ducts, typically triggered by environmental factors in genetically predisposed individuals1. It predominantly affects middle-aged females, with a female-to-male ratio of 4:6:1 and a global prevalence of 1 in 1,000 females aged over 40 years.2,3 Incidence and prevalence range from 0.33 to 5.8 and 1.91 to 40.2 per 100,000 persons, respectively, with regional variability.4,5 In Canada (excluding Quebec), the best estimate of prevalence is 17,316 cases, with provincial variation.
Disease progression typically follows a path from cholestasis to hepatic inflammation, fibrosis, cirrhosis, and potentially liver failure or hepatocellular carcinoma.2,3,6 Up to 80% of patients experience pruritus and fatigue, with 20% reporting life-altering fatigue.7-10 Extrahepatic manifestations, including Sjögren syndrome, thyroid disorders, metabolic bone disease, and hypercholesterolemia, further increase clinical and humanistic burden.1,2,11-14
Diagnosis is confirmed when 2 of 3 criteria are met: elevated alkaline phosphatase (ALP), presence of antimitochondrial antibody (AMA) or PBC-specific antinuclear autoantibodies, and histologic evidence of bile duct destruction.15-18 AMA is positive in 90% to 95% of cases.3,18 Liver biopsy may be required in AMA-negative or unclear cases, and fibrosis staging is recommended early in diagnosis using imaging or elastography.1,16,18-20 Jurisdictions in Canada generally have access to all necessary diagnostics.18,21,22 Importantly, treatment with seladelpar requires no special pretreatment laboratory assessments, which may support equitable access to care across jurisdictions. If reimbursement is recommended, this feature may facilitate broad and timely availability of seladelpar, reducing barriers to treatment initiation, and enhance access for patients regardless of geographic location. Posttreatment care includes routine blood tests and liver monitoring.
Patient Group Input
Patients described PBC as a rare but profoundly disruptive autoimmune liver disease. Most commonly affecting females in midlife, the condition leads to unpredictable progression, persistent symptoms, and a reduced quality of life. Even when disease markers improve, the burden of fatigue, itch, and cognitive fog continues to interfere with employment, daily functioning, and social connection. Many patients live with multiple comorbidities and experience emotional distress related to both the disease and the limitations of current treatments.
Current Management
Treatment Goals
Patient Group Input
Patients expressed a need for treatment options that not only reduce the risk of disease progression and liver-related complications, but also address persistent symptoms such as fatigue, itch, and cognitive dysfunction. According to the Canadian PBC Society, fatigue, often described as debilitating and life-altering, was identified as the most impactful symptom, affecting independence and social functioning. Pruritus and brain fog were also highlighted as major burdens that interfere with daily functioning and quality of life. Patients emphasized that effective treatment should go beyond biochemical response to support energy, mental clarity, and overall well-being. Many patients expressed frustration that current therapies fail to relieve these symptoms and called for safe and accessible options that enable them to live more fully. The overarching treatment goal was not only survival but improved daily functioning, productivity, and autonomy. Survey data and patient testimonials revealed that PBC symptoms frequently result in loss of employment and reliance on disability benefits. Nearly one-third of respondents reported needing help with routine household tasks. Social isolation was common, with 34% reporting cancelled plans due to symptoms; some patients described losing friendships or family connections. In several cases, patients required caregiver support, often from spouses or other family members, to manage transportation, household duties, and emotional needs. This reliance introduced additional burdens on caregivers. Patients also reported feelings of guilt, depression, and diminished self-worth due to their dependence and inability to fulfill roles such as parenting or professional responsibilities. These impacts underscore the need for treatments that restore not only physical health but also patient dignity, autonomy, and social participation.
Clinician Input
The clinical experts and the clinician group indicated that, for patients whose PBC has an inadequate response to first-line UDCA, the most important treatment goals are to control disease activity by normalizing ALP and bilirubin levels, improve PBC-related symptoms (including pruritus), and prevent end-stage liver disease or transplant.
Current Treatment Options
UDCA is the first-line therapy for all patients with PBC.1,3 According to the clinical experts, up to 40% of patients will have a suboptimal response to UDCA and require second-line therapy to improve outcomes. Options for second-line therapy include obeticholic acid (OCA) in combination with UDCA (or as monotherapy in patients with UDCA intolerance) and elafibranor plus UDCA (or as monotherapy in patients with UDCA intolerance) for patients with an incomplete response (or intolerance) to UDCA. The experts noted that bezafibrate is an off-label option that has been demonstrated to improve biochemical response in patients with PBC with an incomplete response to UDCA. They also noted that treatment with OCA is associated with increased pruritus, elafibranor is associated with significant gastrointestinal upset, and bezafibrate is associated with hepatotoxicity, renal dysfunction, and myopathy. They noted that in general, treatments for pruritus are poorly tolerated in patients with PBC. According to the clinical experts, off-label treatment of pruritus is based on limited data, with bile acid resin (e.g., cholestyramine) as a first option, followed by rifampicin, sertraline, naltrexone, and gabapentin. However, these therapies are often poorly tolerated by patients, which limits their use.
Key characteristics of seladelpar, along with other treatments available for PBC, are summarized in the Key Characteristics table in Appendix 1 in the Supplemental Material document.
Unmet Needs and Existing Challenges
Patient Group Input
Many patients with PBC continue to experience persistent symptoms such as fatigue, pruritus, and cognitive difficulties despite available therapies. Some reported that OCA may worsen pruritus and noted that it is contraindicated in patients with advanced liver disease; others were uncertain about elafibranor due to limited experience with it in Canada. Patients expressed frustration with delayed diagnosis, inconsistent access to specialist care, and the lack of treatments targeting what matters most to them. Many advocated for equitable access to seladelpar, viewing it as a promising and better-tolerated second-line option that does not worsen pruritus. In several cases, patients reported needing caregiver support to manage daily responsibilities such as transportation, household tasks, and decision-making. This reliance introduced emotional and psychological strain for both patients and caregivers, particularly when independence was lost or symptoms were unpredictable. Financial stress was also noted, especially among those who had to leave the workforce or navigate disability benefits.
Clinician Group Input
According to the clinical experts, the unmet needs in PBC include new treatments that control disease activity (i.e., ALP and bilirubin normalization) and improve PBC-related symptoms, including pruritus. The experts noted that the availability of effective treatments for pruritus that have fewer side effects remains an important unmet need in PBC. Pruritus is debilitating and can be severe and have a substantial impact on quality of life, and currently available treatments are limited in efficacy, are poorly tolerated, and add to the polypharmacy burden for patients.
The CaNAL group identified persistent challenges in managing PBC, citing the limited effectiveness and tolerability of current treatment options. UDCA provides only partial biochemical control and does not address key symptoms such as pruritus and fatigue. While OCA is reimbursed as a second-line therapy, it offers modest biochemical benefit, often worsens pruritus, and is contraindicated in advanced liver disease. Elafibranor, though recently approved, demonstrated inconsistent effects on pruritus relief in clinical trials. Off-label fibrates are prescribed inconsistently due to tolerability and safety concerns and variability in specialist familiarity.
The CaNAL group emphasized that pruritus can interfere with sleep, concentration, and emotional well-being, and may prevent patients from engaging in work, social activities, or self-care. The symptom burden often leads to psychological distress and functional limitations, which in turn may necessitate caregiver support for transportation, medication management, or daily routines. The CaNAL group also highlighted the need for equitable access to effective therapies across diverse patient populations, including those in rural, remote, and equity-deserving communities. The group advocated for broader therapeutic options, such as seladelpar, to support personalized and timely care in routine clinical practice.
Considerations for Using the Drug Under Review
Contents within this section have been informed by input from the clinical experts consulted for the purpose of this review and, as well as the reimbursement conditions proposed by the sponsor (refer to the Initiation, Renewal, Discontinuation, and Prescribing Conditions Proposed by the Sponsor section in Appendix 1 in the Supplemental Material document). The implementation questions from the public drug programs and corresponding responses from the clinical experts consulted for this review are summarized in the Summary of Drug Program Input and Clinical Expert Responses table in Appendix 1 in the Supplemental Material document. The following has been summarized by the review team.
Place in Therapy
The clinical experts indicated that seladelpar could be used as a second-line treatment for patients whose PBC has an inadequate response to first-line UDCA or patients with UDCA intolerance. The experts noted that seladelpar could be used as add-on therapy with UDCA in patients whose PBC has an inadequate response to UDCA, or as monotherapy in patients with UDCA intolerance, including patients with severe pruritus. The experts noted that individualized decision-making regarding which second-line drug to use would depend on the individual patient’s symptom burden, comorbidities, and preferences.
Patient Population
The clinical experts noted that patients with PBC most in need of treatment include those whose PBC has an inadequate response to UDCA who are at risk of progressive disease and those with significant symptoms, including pruritus, given the lack of approved, available, effective, and well-tolerated treatment options for pruritus. The experts also noted that Indigenous Peoples and males have a higher prevalence of severe PBC and are at higher risk of poor outcomes. The experts noted that PBC is an uncommon disease and delays in diagnosis are common, particularly among patients with limited access to primary care (e.g., in rural areas).
The experts indicated that patients whose PBC has an inadequate response to UDCA, with or without pruritus, are best suited for therapy with seladelpar. The experts noted that there are limited efficacy data to support the use of seladelpar in patients with cirrhosis, with or without portal hypertension. The experts also noted that patients with early-stage disease may have a higher likelihood of response to treatment; however, those with advanced disease are in greater need of therapy.
Assessing the Response to Treatment
The clinical experts noted that, for patients with PBC, the response to treatments observed in clinical trials generally parallels those seen in clinical practice. Improvements in biochemical markers of cholestasis, such as decreases in ALP or the normalization of bilirubin, are important outcomes used to define beneficial treatment effects in the target patient population, as decline in ALP levels is generally considered an indicator of treatment success and may be associated with improved clinical outcomes. Specifically, the experts noted that a target ALP level of less than 1.67 times the upper limit of normal (ULN) at 1 year of treatment and/or a decrease of at least 15% from baseline are critical markers of effectiveness. They noted that biochemical responses are assessed at 6 months and 12 months after treatment initiation. They also noted that improvement in symptoms, particularly pruritus, is another clinically meaningful outcome in patients with PBC. The experts noted that, in clinical practice, liver stiffness is also measured to evaluate disease progression over time when transient elastography (FibroScan) is available. According to the clinical experts, clinical trials with longer duration (i.e., at least 2 years) would be required to observe a meaningful change in liver stiffness measurements.
Discontinuing Treatment
The clinical experts indicated that treatment with seladelpar should be discontinued if the disease does not respond to treatment (i.e., persistent ALP elevation > 1.67 × ULN or increasing conjugated bilirubin > ULN after 1 year of treatment, or worsening FibroScan readings over time) or if the treatment is not tolerated by the patient.
Prescribing Considerations
The clinical experts noted that a specialist with experience managing patients with liver disease (e.g., a gastroenterologist or hepatologist) is required to prescribe licensed second-line therapies for PBC, although in general, monitoring for patients with PBC can be provided within primary care ambulatory clinics, with specialist oversight.
Clinical Review
Methods
The review team considered studies in the sponsor’s systematic review (pivotal studies and randomized controlled trials [RCTs]), sponsor-submitted long-term extension (LTE) studies, indirect treatment comparisons (ITCs), and studies addressing gaps in the evidence. Eligible studies for the systematic review included published and unpublished pivotal studies and phase III and phase IV RCTs. Relevant patients and interventions were defined by the indication and the recommended dosage in the product monograph. Baseline ALP levels (ALP ≥ 350 U/L and ALP < 350 U/L) and cirrhosis at baseline were considered potentially important subgroups for interest for informing the reimbursement recommendation. Relevant comparators were drugs used in clinical practice in Canada to treat patients described in the indication under review. These included UDCA monotherapy, OCA, and elafibranor, as monotherapy or in combination with UDCA. LTE studies of included pivotal studies and RCTs were included, regardless of whether a comparison group was available. ITCs and gap-filling studies submitted by the sponsor were included when they addressed an identified gap in the systematic review evidence (e.g., missing comparator or longer follow-up time).
The review team selected outcomes and follow-up times for review based on the sponsor’s Summary of Clinical Evidence, clinical expert input, and patient and clinician group input. Included outcomes are those considered relevant to expert committee deliberations, and they were selected in consultation with committee members. Evidence from the systematic review for the most important outcomes was assessed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach. The following outcomes were assessed using GRADE because they address key treatment goals for PBC: biochemical response (defined by ALP < 1.67 × ULN, ≥ 15% decrease in ALP, and total bilirubin (TB) ≤ 1.0 × ULN), ALP normalization, and pruritus numerical rating scale (NRS) in patients with a baseline NRS of 4 or higher. The primary biliary cholangitis-40 (PBC-40) quality of life total score was also included, as quality of life and disease and treatment burden were identified as important outcomes in the patient group input.
Methods for data extraction, risk of bias appraisal, and certainty of evidence assessment are described in Appendix 2 in the Supplemental Material document.
Clinical Evidence
In this report, the following sources of evidence submitted by the sponsor are reviewed and appraised:
1 pivotal RCT study included in the systematic review (RESPONSE)
1 LTE study (ASSURE)
2 ITCs.
Systematic Review
Description of Studies
Study Characteristics
Characteristics of the included study are summarized in Table 2. Details pertaining to the eligibility criteria, interventions and comparators, and relevant outcome measures are in Appendix 3 in the Supplemental Material document.
The RESPONSE study was a pivotal phase III, multicentre, randomized, double-blind, placebo-controlled trial.23 The objective of the RESPONSE trial was to assess the efficacy and safety of seladelpar 10 mg, taken orally once daily, compared with placebo in adults whose PBC had an inadequate response to UDCA or adults with UDCA intolerance. During the trial, seladelpar 10 mg was administered as an add-on therapy to UDCA for patients who had UDCA tolerance; for patients who had UDCA intolerance, seladelpar was administered as monotherapy. Enrolled patients were required to have confirmed PBC, defined as meeting any 2 of the following 3 diagnostic criteria: a history of ALP greater than 1.0 times ULN for at least 6 months; positive AMA titre (> 1:40 on immunofluorescence or M2 positive by enzyme-linked immunosorbent assay) or positive PBC-specific antinuclear antibodies (ANAs) titre; and documented liver biopsy results consistent with PBC. The screening period was up to 3 weeks, the run-in period was up to 2 weeks, and the treatment period was up to 12 months. The study was conducted across 90 sites in 24 countries across North America (including 3 sites in Canada), Europe, Asia, and Latin America. Among the 360 subjects screened, 193 were randomly assigned in a 2:1 ratio to receive seladelpar 10 mg (N = 128) or placebo (N = 65). Randomization was performed using interactive response technology and was stratified by ALP level (< 350 U/L versus ≥ 350 U/L) and the presence of clinically important pruritus (NRS < 4 versus NRS ≥ 4). Treatment with antipruritic drugs was permitted if patients were on a stable dose within 1 month before screening. Treatment with OCA, fibrates, and elafibranor 6 weeks before screening were prohibited. Efficacy and safety assessments occurred during study visits at weeks 1, 3, 6, and 12 months. After completion of the treatment period, patients were invited to enrol into an open-label, LTE safety trial (the ASSURE study) in which all patients received seladelpar 10 mg, and patients previously randomized to placebo in the RESPONSE trial initiated seladelpar 10 mg. Data from the safety extension phase are presented in the Long-Term Extension Studies section of this report.
Table 2: Characteristics of the Study Included in the Systematic Review
Study name, design, and sample size | Key inclusion criteria | Key exclusion criteria | Intervention and comparator | Relevant end points |
|---|---|---|---|---|
RESPONSE study: phase III, double-blind, placebo-controlled RCT Total N = 193 |
|
|
| Primary:
Key secondary:
Other secondary:
|
AMA = antimitochondrial antibody; ANA = antinuclear antibody; CFB = change from baseline; ELISA = enzyme-linked immunosorbent assay; NRS = numerical rating scale; OCA = obeticholic acid; PBC = primary biliary cholangitis; PBC-40 = primary biliary cholangitis-40; QoL = quality of life; RCT = randomized controlled trial; UDCA = ursodeoxycholic acid; ULN = upper limit of normal.
aHistory of liver transplant, complications of portal hypertension, and cirrhosis with complications.
Sources: RESPONSE Clinical Study Report.23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Statistical Testing and Analysis Populations
For the sample size estimation, response rates for the primary outcome (biochemical response evaluated at 12 months) were estimated to be 20% the placebo group and 55% in the seladelpar group. Using a 2-sided test of equality of binomial proportions (Fisher’s exact test) at a 0.05 level of significance, a sample size of 180 randomized patients who received study drug provided greater than 90% power to detect a difference between the seladelpar and placebo groups. For the key secondary outcome of ALP normalization, response rates were estimated to be 2.5% in the placebo group and 25.5% in the seladelpar group. Under these assumptions, a sample size of 180 randomized patients who received study drug provided greater than 90% power to detect a difference between groups, based on a 2-sided Fisher’s exact test at a 0.05 level of significance. For the biochemical response and ALP normalization outcomes, any patient who did not have data reported at 12-month assessment was considered a nonresponder. The analysis of the key secondary outcome of change from baseline in weekly averaged pruritus NRS at month 6 sample size calculation was based on a 2-sample, 2-sided t test with a significance level of 0.05. The common standard deviation (SD) was estimated as 2 points. Under these assumptions, a total of 48 randomized patients who received study drug and had a baseline NRS greater than or equal to 4 and an NRS assessment at month 6 provided greater than 80% power to detect a treatment difference of greater than or equal to 2 NRS points between the seladelpar and placebo groups. For responder analyses, a dropout rate of approximately 10% was assumed. The assumptions for these power calculations were based on results from a prior seladelpar study (NCT03602560).
Control of the study-wide type I error rate was maintained at 5% using a hierarchical fixed-sequence methodology for the primary and key secondary efficacy analyses. If the primary efficacy analysis was statistically significant for seladelpar versus placebo at a 2-sided 0.05 significance level, then the 2 key secondary outcomes were analyzed hierarchically in the following order:
Normalization of ALP at month 12: If not statistically significant at a 2-sided 0.05 significance level, no further inferential testing was performed. If results were statistically significant, further testing proceeded.
Change from baseline to month 6 in pruritus NRS: This outcome was evaluated in the moderate to severe pruritus NRS (MSPN; baseline NRS ≥ 4) analysis set at a 2-sided 0.05 significance level.
For the composite biochemical response and ALP normalization outcomes, the incidence of response was analyzed using a Cochran-Mantel-Haenszel test adjusted for the randomization stratification variables. Statistical significance between groups was determined using a 2-sided P value threshold of 0.05 or less, and the risk difference and 95% confidence interval (CI) were calculated using the method of Miettinen and Nurminen. Continuous outcomes (pruritus NRS and PBC-40 quality of life total score) were analyzed using a mixed-effect model for repeated measures (MMRM), which estimated least squares (LS) mean changes from baseline by treatment (with associated standard errors), LS mean differences between treatment groups (with associated 2-sided 95% Cis), and 2-sided P values.
The intention-to-treat (ITT) analysis set was the primary analysis set for efficacy analyses, except of secondary outcomes evaluated in patients with MSPN. The MSPN analysis set was the primary analysis set for secondary outcomes based on NRS evaluations. All safety analyses were based on the safety analysis set, defined as any patient who received at least 1 dose of treatment.
Patient Disposition
Of the 360 patients screened for eligibility, 162 (45%) were screened out. The most common reasons for screen failure were not meeting laboratory parameter thresholds for ALP, ALT, AST, estimated glomerular filtration rate, or TB as measured by the central laboratory at screening. In total, 193 patients were randomized in a 2:1 ratio to receive seladelpar 10 mg (N = 128) or placebo (N = 65). Study discontinuation was reported in 8.6% of patients in the seladelpar group and 12.3% of patients in the placebo group. The most frequently reported reasons for discontinuation were adverse events (AEs) (2.3% versus 6.2% of patients) and patient withdrawal (3.9% versus 3.1% of patients). Protocol deviations were infrequent and similar in both groups.
Baseline Characteristics
A summary of key baseline patient characteristics for the ITT population is presented in Table 3. The baseline characteristics are limited to those most relevant to this review or considered likely to affect the outcomes or interpretation of the study results. Overall, key baseline characteristics were generally balanced between treatment groups. The trial population was mostly female (94.8%), white (88.1%), and non-Hispanic or Latino (69.9%), and the mean age was 56.7 years. In addition to patients with mild disease severity, the trial population included patients with moderate disease severity (e.g., 13.0% of patients had TB levels above the ULN at baseline, 14.0% of patients had moderate Rotterdam stage, and 14.0% of patients had cirrhosis at baseline). Mean ALP, TB levels, and pruritus NRS scores were balanced between treatment groups at baseline. Most patients (93.8%) received seladelpar 10 mg or placebo in addition to UDCA; 6.2% of patients had UDCA intolerance and received seladelpar 10 mg or placebo as monotherapy. Prior use of OCA or fibrates was reported in 15.6% of patients in the seladelpar group and 20.0% of patients in the placebo group.
Table 3: Summary of Baseline Characteristics for the ITT Population (RESPONSE Trial)
Characteristic | Seladelpar 10 mg (N = 128) | Placebo (N = 65) |
|---|---|---|
Age (years), mean (SD) | 56.6 (10.0) | 57.0 (9.2) |
Age at diagnosis (years), mean (SD) | 49.2 (9.9) | 49.3 (10.9) |
Female sex, n (%) | 123 (96.1) | 60 (92.3) |
Race, n (%) | ||
American Indian or Alaska Native | 3 (2.3) | 3 (4.6) |
Asian | 7 (5.5) | 4 (6.2) |
Black or African American | 2 (1.6) | 2 (3.1) |
White | 114 (89.1) | 56 (86.2) |
Patients with cirrhosis at baseline, n (%) | 18 (14.1) | 9 (13.8) |
Portal hypertension | 0 | 3 (4.6) |
Duration of disease (years), mean (SD) | 8.2 (6.7) | 8.6 (6.5) |
Positive for antimitochondrial antibodies, n (%) | 106 (82.8) | 55 (84.6) |
Ursodeoxycholic acid | ||
Intolerance, n (%) | 8 (6.2) | 4 (6.2) |
Daily dose (mg/kg), mean (SD) | 15.0 (3.1) | 14.9 (3.3) |
Prior use of OCA and/or fibrates, n (%) | 20 (15.6) | 13 (20.0) |
Pruritus NRS, mean (SD) | 3.0 (2.8) | 3.0 (3.0) |
< 4, n (%) | 79 (61.7) | 42 (64.6) |
≥ 4, n (%) | 49 (38.3) | 23 (35.4) |
≥ 4, mean score (SD) | 6.1 (1.4) | 6.6 (1.4) |
Alkaline phosphatase level (U/L), mean (SD) | 314.6 (123.0) | 313.8 (117.7) |
≥ 350, n (%) | 35 (27.3) | 18 (27.7) |
Total bilirubin level (mg/dL), mean (SD) | 0.769 (0.3) | 0.737 (0.3) |
≤ 1 x ULN, n (%) | 108 (84.4) | 60 (92.3) |
> 1 and ≤ 2 x ULN, n (%) | 20 (15.6) | 5 (7.7) |
ALT (U/L), mean (SD) | 47.4 (23.5) | 48.2 (22.8) |
AST (U/L), mean (SD) | 39.6 (16.1) | 41.7 (16.0) |
Gamma-glutamyltransferase (U/L), mean (SD) | 269.0 (240.0) | 287.5 (249.6) |
Albumin level (g/dL), mean (SD) | 4.2 (0.3) | 4.1 (0.2) |
Platelet count (x 103/mm3), mean (SD) | 241.7 (78.9) | 241.9 (84.5) |
History of pruritus, n (%) | 91 (71.1) | 48 (73.8) |
Liver stiffness (kPa), mean (SD) | 9.8 (6.2) | 8.7 (4.2) |
ITT = intention to treat; NRS = numerical rating scale; OCA = obeticholic acid; SD = standard deviation; ULN = upper limit of normal.
Note: Racial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
Sources: RESPONSE Clinical Study Report.23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Treatment Exposure and Concomitant Medications
Exposure to study treatments was similar between groups. The mean duration of exposure was 50.5 weeks (SD = 7.4 weeks) in the seladelpar group and 48.3 weeks (SD = 11.6 weeks) in the placebo group, and the mean average daily dose was ████ ███ █ █████ ██ in the seladelpar group and ████ ██ ███ █ █████ in the placebo group. The majority of patients (seladelpar ██████; placebo ██████) in both treatment groups received study drug for ██ █████ ██ ██████. Most patients in both groups (93.8% in the seladelpar group; 95.4% in the placebo group) received seladelpar or placebo in addition to UDCA; 6.2% of all patients had UDCA intolerance and received seladelpar as monotherapy. The mean duration of exposure to UDCA was ████ █████ ███ █ ████ in the seladelpar group and ████ █████ ███ █ █████ in the placebo group. The mean average daily dose of UDCA was 15.0 mg/kg/day (SD = 3.1 mg/kg/day) and 14.9 mg/kg/day (SD = 3.3 mg/kg/day) in the seladelpar and placebo groups, respectively. A total of ████ ███ ████ of patients in the seladelpar and placebo groups, respectively, had a dose interruption. The most frequent reason for dose interruption was AEs (6.3% in the seladelpar group; 6.2% in the placebo group), and most patients had only 1 dose interruption. Only 1 patient, in the seladelpar group, underwent a dose reduction from 10 mg to 5 mg, which was attributed to an AE, and subsequently uptitrated to 10 mg after approximately 2 months. Most patients in both groups (97.7% and 98.5% in the seladelpar and placebo groups, respectively) received concomitant medications. The most frequently used medications were paracetamol (21.1% and 33.8%), colecalciferol (19.5% and 23.1%), and omeprazole (20.3% and 16.9%). Colestyramine was used in 4.7% and 12.3% of patients in the seladelpar and placebo groups, respectively.
Critical Appraisal
Internal Validity
The RESPONSE trial was a randomized, double-blind, placebo-controlled, phase III trial. Randomization and allocation concealment procedures, including stratification by ALP level and the presence of clinically important pruritus, were appropriate and were conducted using interactive response technology. Overall, key baseline demographic and disease characteristics were reasonably balanced between treatment groups.
Sample size and power calculations were based on the primary (composite biochemical response) and key secondary outcomes (ALP normalization and pruritus NRS), and the trial was powered to detect significant differences for these outcomes. The primary and key secondary outcomes were appropriately controlled for multiple comparisons. All other analyses were descriptive, (i.e., not controlled for type I error), including the HRQoL outcome. The sample size for the prespecified subgroup analyses of interest for the primary and key secondary outcomes were small; therefore, the trial may not have been powered to detect subgroup differences.
Rates of study discontinuation were generally low and similar between groups, and were primarily due to AEs and patient withdrawal. Nonresponder imputation was used for missing data in the primary analysis for the primary and key secondary binary outcomes, and prespecified sensitivity analyses were conducted under different assumptions. Nonresponder imputation assumes that missing outcomes occur at random and are unrelated to observed or unobserved variables; however, this assumption may be unrealistic, and violations may introduce bias, particularly when between-group differences are pronounced. In the RESPONSE trial, between-group differences in missing data did not appear pronounced, and appropriate sensitivity analyses (e.g., tipping point and multiple imputation analyses) supported the robustness of results. For the pruritus NRS outcome at 12 months in patients with a baseline NRS of 4 or higher, results were subject to bias due to missing data (seladelpar = 20%; placebo = 30%), with a higher proportion of missing data in the placebo group; however, the direction and magnitude of any potential bias are unclear. For this outcome, no analyses other than MMRM were reported to evaluate the impact of missing data (e.g., alternative imputation methods or sensitivity analyses).
The investigators, patients, and study personnel were blinded to the treatment, which reduces the risk of bias due to deviations from the intended intervention. While it is possible for patients and personnel to become unblinded due to knowns AEs or clear efficacy of the study drug, there was not clear evidence that this occurred. The proportion of patients who prematurely stopped study treatment was similar across groups, primarily due to AEs and withdrawal by patients. The use of concomitant therapies and prior UDCA therapy was also generally balanced between the treatment groups. Similarly, the blinding of the trial supported a low potential for bias in outcome assessment.
PBC-related biomarkers such as changes in ALP and TB were used as surrogate end points in the RESPONSE trial to predict treatment benefit in the absence of direct outcomes on disease progression (e.g., cirrhosis) and survival, including liver transplant-free survival, which are considered important outcomes for patients and clinicians. The primary outcome, biochemical response at 12 months (i.e., ALP < 1.67 × ULN, TB ≤ ULN, and an ALP decrease ≥ 15%), although nonvalidated, has been adopted in several clinical trials involving patients with PBC. The clinical experts consulted for this review agreed that this is a reasonable clinical trial outcome in the study population. They indicated that biochemical measures have prognostic value within individual patients and are used in clinical practice to monitor disease progression. There is evidence to suggest that reductions in ALP and TB levels are associated with longer overall survival and transplant-free survival in patients with PBC,24-26 which was confirmed by the clinical experts consulted for this review. However, direct evidence of clinically important treatment effect on slowing disease progression and improving survival outcomes remains unavailable.
HRQoL was assessed by the PBC-40 quality of life total score, which has been validated in patients with PBC, although no minimally important differences were identified in the literature. The result of this outcome was subject to potential bias due to missing outcome data (> 20% in each group), although the direction and magnitude of the bias is unclear given no other alternative imputation methods (in addition to MMRM) or sensitivity analyses were done to assess the impact of missing data.
External Validity
The population requested for reimbursement aligns with the Health Canada indication and overall trial population. The dosing and administration of seladelpar were consistent with the product monograph. In the RESPONSE trial, the PBC of most patients had an inadequate response to UDCA and received seladelpar in combination with UDCA, whereas a small proportion of patients had UDCA intolerance and received seladelpar monotherapy. According to the clinical experts consulted by CDA-AMC, the eligibility criteria and baseline characteristics of the RESPONSE trial are generalizable to adults with PBC in the second-line setting whose PBC has an inadequate response to UDCA or adults with UDCA intolerance in the setting in Canada.
The clinical experts also noted that PBC is more common among First Nations populations in Canada, in whom treatment response is impaired and the need for transplant is higher; however, it is unclear whether this population was represented in the RESPONSE trial.
The evidence submitted to CDA-AMC did not include head-to-head comparisons between seladelpar and relevant comparators such as elafibranor and OCA, which represents a gap in the available direct evidence.
The follow-up duration in the RESPONSE trial was insufficient to assess clinically important outcomes that the clinical experts considered important, such as survival or disease progression to fibrosis, cirrhosis, or liver transplant.
Results
The key efficacy results and findings from the GRADE assessment are presented in this section. Detailed efficacy and harms results can be found in Appendix 4 in the Supplemental Material document.
Efficacy
Summary of findings and certainty of the evidence: For all outcomes, the clinical experts were unable to suggest a specific threshold for a clinically important effect; therefore, certainty was rated for the presence of a non-null effect. Table 4 presents the GRADE summary of findings for seladelpar versus placebo.
Subgroup analysis: The biochemical response results were consistent across the subgroup analyses of interest by baseline ALP level (ALP ≥ 350 U/L and ALP < 350 U/L) and by cirrhosis status at baseline, favouring seladelpar (Figure 1). The key secondary outcomes were also consistent across the subgroup analyses of interest, favouring seladelpar (data not shown).
Figure 1: Forest Plot of the Biochemical Response Rate at 12 Months by Subgroup (ITT Population, RESPONSE Trial)
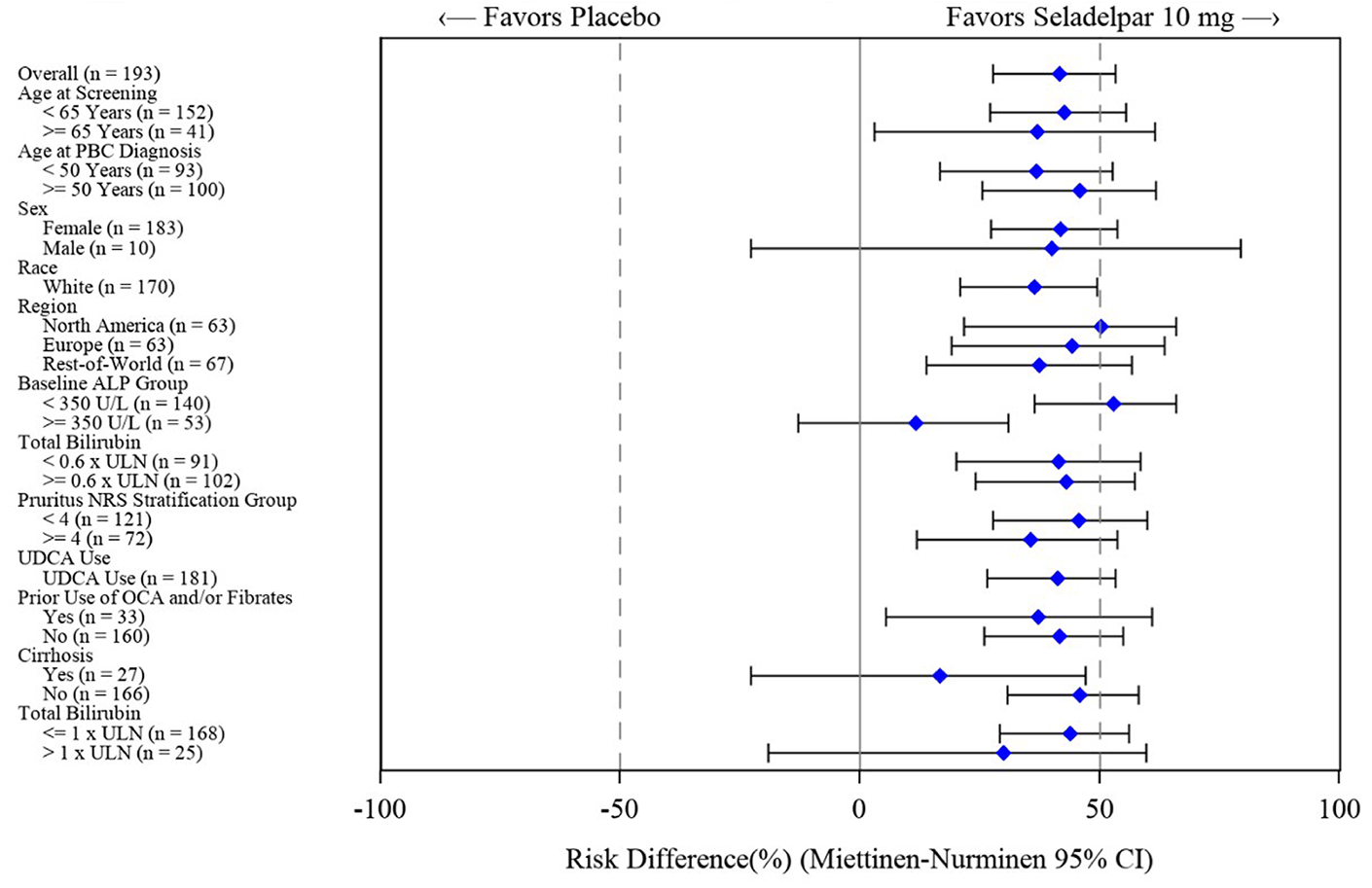
CI = confidence interval; ITT = intention to treat; NRS = numerical rating scale; OCA = obeticholic acid; PBC= primary biliary cholangitis; UDCA = ursodeoxycholic acid; ULN = upper limit of normal.
Source: RESPONSE Clinical Study Report.23
Harms
Key harms results include the following:
Most patients in both groups reported at least 1 treatment emergent adverse event (TEAE) (86.7% in the seladelpar group versus 84.6% in the placebo group). The most frequently reported TEAEs (≥ 5% in either group) in the seladelpar group were COVID-19 (18.0% versus 15.4%), headache (7.8% versus 3.1%), and abdominal pain (7.0% versus 1.5%), with a higher proportion of patients in the seladelpar group reporting these events than the placebo group. The most frequently reported TEAEs in the placebo group were COVID-19 (15.4% versus 18.0%), pruritus (15.4% versus 4.7%), and upper respiratory tract infection (9.2% versus 0.8%), with pruritus and abdominal pain being reported more frequently in the placebo group. Grade 3 or higher TEAEs were similar between groups (seladelpar group: 10.9%; placebo group: 7.7%).
The incidence of serious TEAEs was similar between the seladelpar group (7.0%) and the placebo group (6.2%), with COVID-19 being the most frequently reported event in both groups (0.8% versus 1.5%).
The incidence of TEAEs leading to study treatment discontinuation was infrequent and similar between the seladelpar group (3.1%) and the placebo group (4.6%).
The incidence of notable harms in both treatment groups was infrequent and comparable between groups. Liver-related toxicity occurred in 6.3% and 9.2% of patients, muscle-related toxicity occurred in 6.3% and 7.7% of patients, and pancreatic-related toxicity occurred in 1.6% and 1.5% of patients in the seladelpar and placebo groups, respectively. There were no renal-related toxicities in either group.
No deaths were reported in either group.
Table 4: Summary of Findings for Seladelpar vs. Placebo for Patients With PBC
Outcome and follow-up | Patients (study), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Seladelpar | Difference | |||||
Cholestasis response (ITT analysis set) | |||||||
Biochemical response, defined as ALP < 1.67 × ULN, ALP decrease of ≥ 15%, and total bilirubin ≤ 1.0 × ULN Follow-up: 12 months | 193 (1 RCT) | NA | 200 per 1,000 | 617 per 1,000 (533 to 701) | 417 more per 1,000 (277 more to 534 more) | Higha | Seladelpar results in a clinically important increase in the proportion of patients with biochemical response at month 12 when compared with placebo. |
ALP normalization Follow-up: 12 months | 193 (1 RCT) | NA | 0 per 1,000 | 250 per 1,000 (175 to 325) | 250 more per 1,000 (183 more to 332 more) | Higha | Seladelpar results in a clinically important increase in the proportion of patients with ALP normalization at month 12 when compared with placebo. |
Pruritus WI-NRS (MSPN analysis set) | |||||||
Pruritus NRS scores in MSPN analysis set, LS mean change from baseline; scores range from 0 to 10, with higher scores indicating worse itch Follow-up: 6 months | 65 (1 RCT) | NA | −1.7 | −3.2 (SE: 0.28) | −1.5 (−2.5 to −0.5) | Moderateb | Seladelpar likely results in a clinically important decrease in pruritus at 6 months when compared with placebo. |
Pruritus NRS scores in MSPN analysis set, LS mean change from baseline; scores range from 0 to 10, with higher scores indicating worse itch Follow-up: 12 months | 55 (1 RCT) | NA | −1.5 | −3.3 (SE: 0.33) | −1.8 (−3.0 to −0.6) | Lowc | Seladelpar may result in a clinically important decrease in pruritus at 12 months when compared with placebo. |
PBC-40 QoL total score (ITT analysis set) | |||||||
LS mean change from baseline; total scores range from 40 to 200, with higher scores indicating worse QoL Follow-up: 12 months | 145 (1 RCT) | NA | −6.19 | −5.85 (SE: 1.64) | 0.33 (−4.98 to 5.64) | Lowd | Seladelpar may result in little to no difference in HRQoL at 12 months when compared with placebo. |
CI = confidence interval; HRQoL = health-related quality of life; ITT = intention to treat; LS = least square; MSPN = moderate to severe pruritus; NA = not applicable; NRS = numeric rating scale; PBC = primary biliary cholangitis; PBC-40 = primary biliary cholangitis-40; QoL = quality of life; RCT = randomized controlled trial; SE = standard error; ULN = upper limit of normal; WI = worst itch; vs. = versus.
Notes: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aFor biochemical response and ALP normalization, the clinical experts could not determine a minimal important difference, but they considered that the effect estimate and entire confidence interval were consistent with an important benefit in favour of seladelpar.
bRated down 1 level for serious imprecision. The sample size is small, suggesting some concern for overestimation of the true effect. The clinical experts could not determine a minimal important difference for pruritus, but they considered that the effect estimate and entire confidence interval were consistent with an important benefit in favour of seladelpar.
cRated down 1 level for serious risk of bias due to missing outcome data at 12 months, and 1 level for serious imprecision due to a small sample size, suggesting some concern for overestimation of the true effect. For pruritus, the clinical experts could not determine a minimal important difference, but they considered that the effect estimate and entire confidence interval were consistent with an important benefit in favour of seladelpar.
dRated down 2 levels for very serious imprecision due to the 95% CI for the between-group mean LS mean difference including the possibility of benefit and harm. There was no known minimal important difference, so the target of the certainty appraisal was any effect.
Sources: RESPONSE Clinical Study Report.23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Long-Term Extension Studies
Description of Studies
The ASSURE study (CB8025-31731-RE) is an ongoing, multicentre, open-label, LTE trial designed to evaluate the safety and tolerability of seladelpar in patients with PBC who previously participated in seladelpar clinical trials. Participants were enrolled from 5 parent studies, including 3 legacy trials (CB8025-21629, CB8025-31731, and CB8025-31735 [ENHANCE]), as well as the RESPONSE trial (CB8025-32048), and a hepatic impairment study (CB8025-21838). While eligibility criteria were generally aligned with those of the parent trials, participants had varying exposure histories; some resumed treatment after more than 1 year due to prior development holds, while others transitioned directly from active trials.
All participants received seladelpar 10 mg orally once daily, with the option to start at 5 mg for tolerability concerns and later uptitrate the dose. The study is expected to continue for approximately 5 years or until the first marketing approval is obtained. The primary objective is to evaluate long-term safety and tolerability through TEAEs, laboratory findings, and other clinical safety parameters. Secondary outcomes include long-term efficacy assessments based on biochemical markers (e.g., ALP and bilirubin) and symptom control (notably pruritus). The study also assesses clinical events of disease progression such as liver transplant, model for end-stage liver disease scores of 15 or higher, and ascites. Exploratory objectives include changes in liver histology and fibrosis, quality of life, biomarker profiles, and pharmacokinetics.
Patient Disposition
As of the June 29, 2023, interim data cut-off, 300 participants were screened, 286 were enrolled, and 280 received at least 1 dose of seladelpar and were included in the safety analysis set. Among these, 279 patients started on the 10 mg dose, while 1 initiated at 5 mg due to tolerability concerns. TEAEs led to treatment discontinuation in 9 participants (3.2%) and study withdrawal in 7 (2.5%). The study permitted continued follow-up assessments in patients who discontinued treatment but remained in the trial. No major subgroup differences in reasons for discontinuation were reported; although, stratification by parent study or treatment modality was not provided. No formal ITT, per-protocol, or MSPN-defined subgroup analyses were reported in the interim analysis.
Baseline Characteristics
Baseline demographic and disease characteristics were largely consistent with those observed in the parent trials. The population was predominantly female (94.3%) and middle-aged (mean age = 58.6 years), consistent with the known epidemiology of PBC. Approximately 16.4% of participants had cirrhosis, and over 83% had mild disease severity based on Rotterdam criteria. While values for age, liver biochemistry, and prior UDCA use were reported, the interim report did not present subgroup analyses stratified by prior trial, duration of prior seladelpar exposure, or treatment modality (e.g., monotherapy versus combination with UDCA).
Exposure to Study Treatments
All 280 participants received seladelpar as part of the ASSURE study: 279 initiated treatment at 10 mg once daily and 1 at 5 mg. Dose adjustments between 5 mg and 10 mg were permitted at the investigator's discretion based on individual tolerability. Seladelpar was administered as an adjunct therapy to UDCA in patients with UDCA tolerance, or as monotherapy for patients with UDCA intolerance. Exposure duration in the ASSURE study ranged from 0.1 to 124 weeks, reflecting varied timelines of enrolment and reinitiation.
There were no reported patterns of noncompliance, and reinitiation after dose interruptions was allowed when appropriate. Co-interventions were minimal; UDCA was the only concurrent therapy explicitly reported. No rescue or subsequent PBC-specific therapies was documented. Time-to-event data stratified by treatment group or co-intervention were not provided in this interim report.
The overall study discontinuation rate was 10%, primarily due to AEs or voluntary withdrawal. No major subgroup differences were reported; although, stratification by prior study was not provided.
Critical Appraisal
Internal Validity
The design of the ASSURE study provides evidence for the long-term safety of seladelpar but limits causal inference regarding efficacy due to its open-label design and the absence of a control group. Only patients who completed prior seladelpar trials were eligible, introducing potential selection bias, particularly for drug-tolerant individuals who had previously demonstrated treatment adherence and tolerability, and/or were more likely to have had a positive response to seladelpar. The interim analysis relied solely on descriptive statistics, with no formal hypothesis testing or inferential comparisons. Although the statistical analysis plan outlines the analytic framework, the full study protocol was not provided, limiting transparency regarding prespecified outcomes and procedures. The lack of blinding increases the risk of outcome reporting and performance bias, particularly for subjective end points such as pruritus and quality of life.
External Validity
The enrolled population reflects the typical demographic profile of individuals with PBC, which supports generalizability to this population in clinical practice. However, external validity is limited by the selective inclusion of trial completers from earlier seladelpar studies, many of whom had prior drug exposure to seladelpar. These participants may represent a more responsive, adherent, or drug-tolerant population. In addition, the lack of a real-world comparator group and the absence of subgroup analyses by geographic region, disease severity, or comorbidities limit the applicability of the findings to broader patient populations. Furthermore, the interim report did not include efficacy data beyond 1 year of treatment.
Results
Efficacy
No formal efficacy results were included in the interim Clinical Study Report. While efficacy assessments are part of the study protocol, the report explicitly states that only safety data were evaluated and presented at the interim June 29, 2023, data cut-off. Therefore, outcomes such as ALP normalization, TB reduction, or other adjudicated end points (e.g., liver transplant, model for end-stage liver disease score ≥ 15, and ascites) were prespecified in the study protocol but were not analyzed or reported at the interim cut-off.
Harms
Detailed results for harms are presented in Appendix 5 in the Supplemental Material document. Key results include the following.
Seladelpar was generally well-tolerated, and no new safety signals were identified as of the June 29, 2023, data cut-off. Of 14 hepatic safety events reviewed by the Clinical Events Review Committee, 13 were deemed “very unlikely” and 1 was deemed “unlikely” to be related to seladelpar. One case was considered indeterminate due to concurrent gastrointestinal bleeding. Clinically significant physical examination findings were reported in 15 participants, although no consistent pattern emerged. A single case of atrioventricular block was the only notable electrocardiogram abnormality and did not lead to treatment interruption. Muscle-, renal-, or pancreatic-related safety concerns were not reported in the interim analysis.
Indirect Evidence
Direct comparative evidence exists between seladelpar plus UDCA versus placebo plus UDCA for the majority of participants in the RESPONSE trial; seladelpar monotherapy versus placebo was assessed in a small number of participants. However, there is a gap in evidence comparing seladelpar with relevant comparators (OCA and elafibranor). ITCs were required to address this gap and to inform the pharmacoeconomic model.
Description of ITCs
One network meta-analysis (NMA) and 1 anchored matching-adjusted indirect comparison (MAIC) was submitted by the sponsor comparing the relative efficacy and safety of seladelpar with OCA and elafibranor, respectively, in adults whose PBC had an inadequate response to UDCA or who had UDCA intolerance.27
Study Selection and Review Methods
The ITCs were informed by a systematic literature review of evidence (as described in Section 2) in adults aged 18 years or older whose PBC had an inadequate response to UDCA or who had UDCA intolerance. RCTs evaluating OCA or elafibranor as monotherapy or in combination with UDCA were eligible. Relevant outcomes for the literature review included any efficacy, harms, or patient-reported end points.
Database searches in MEDLINE, Embase, the Cochrane Central Register of Controlled Trials, and the Cochrane Database of Systematic Reviews were conducted on August 5, 2024, with no date limits. Conference proceedings, clinical trial registries, and bibliographic reviews were also searched. Title and abstract screening, full-text screening, data extraction, and quality assessment of included studies were each performed by 2 reviewers independently. Discrepancies were resolved by a third reviewer. RCTs were assessed for quality using the Cochrane Risk of Bias 2.0 tool.
Search Results and Feasibility Assessment
The literature review identified 5 unique RCTs that were included in the feasibility assessment. Three trials assessed OCA versus placebo (POISE, NCT03633227, and COBALT), and 1 trial each assessed seladelpar (the RESPONSE trial) and elafibranor (the ELATIVE trial) versus placebo. The feasibility assessment assessed the extent of heterogeneity across the included trials and whether ITCs would be appropriate. The following potential sources of heterogeneity were explored: study design, patient eligibility criteria, baseline patient characteristics, outcome characteristics, and efficacy and safety results. The sponsor identified 5 key effect modifiers for PBC: age at diagnosis, ALP levels at baseline, bilirubin levels at baseline, the proportion of patients with cirrhosis at baseline, and ANA-positive status; although, the methods used to identify these effect modifiers were not reported. Based on the feasibility assessment, the sponsor determined that there was significant heterogeneity in patient and outcome characteristics in the COBALT and NCT03633227 trials versus the POISE and RESPONSE trials, and therefore excluded the COBALT and NCT03633227 trials from the base case. For the base case, an NMA was considered appropriate for comparing the RESPONSE and POISE trials, and an anchored MAIC was used for the RESPONSE and ELEVATE trials. The sponsor’s rationale for using an anchored MAIC was significant heterogeneity in 2 key effect modifiers: baseline bilirubin levels and the proportion of patients with cirrhosis at baseline. The ITCs were conducted for the following relevant outcomes of interest for this CDA-AMC review: composite biochemical response (ALP < 1.67 × ULN, ≥ 15% ALP decrease from baseline, and TB ≤ 1.0 × ULN), ALP normalization (ALP ≤ 1.0 × ULN), TEAEs, pruritus, and all-cause study discontinuations at 12 months.
NMA and MAIC Analysis Methods
For additional information on the analysis methods for the ITCs, refer to Appendix 6 in the Supplemental Material document.
Network meta-analysis: For the comparison between the RESPONSE and POISE trials, a random-effects Bayesian NMA using a generalized linear framework was conducted for biochemical response, ALP normalization, TEAEs, pruritus, and all-cause study discontinuations at 12 months. Efficacy outcomes were reported as risk ratios (RRs) with 95% credible intervals (CrIs), and safety outcomes were reported as odds ratios (ORs) with 95% CrIs.
Matching-adjusted indirect comparison: An anchored MAIC was performed by matching and adjusting patient characteristics in the RESPONSE trial to the patients in the ELEVATE trial. The MAIC was adjusted for 4 key effect modifiers: age at screening, ALP levels at baseline, bilirubin levels at baseline, and proportion of patients with cirrhosis at baseline (Table 5). Efficacy outcomes were reported as RRs with 95% CIs using binomial regression, and safety outcomes were reported as ORs with 95% CIs calculated using logistic regression. The effective sample size (ESS) was calculated to reflect the sample size of the weighted population.
Summary of Included Studies
For additional information on patient characteristics of the included trials, refer to Appendix 6 in the Supplemental Material document.
The RESPONSE and POISE trials reported generally similar patient and trial characteristics, including the sponsor-identified effect modifiers. The mean age at PBC diagnosis was reported for the RESPONSE and POISE trials and was approximately 48 years; this was not reported in the ELATIVE trial. Across all trials, the proportion of females was higher than the proportion of males, ranging from 90.6% (the POISE trial) to 96% (the ELATIVE trial), and the mean age was similar (approximately 57 years). All trials reported a mean ALP at baseline ranging from 314.3 U/L to 323.0 U/L, and mean TB levels at baseline ranging from 0.56 mg/dL (the ELATIVE trial) to 0.76 mg/dL (the RESPONSE trial). The proportion of patients with TB above the ULN at baseline ranged from 3.7% (the ELATIVE trial) to 13.0% (the RESPONSE trial). The proportion of patients with baseline cirrhosis ranged from 9.9% (the ELATIVE trial) to 16.7% (the POISE trial). In particular, the RESPONSE and ELATIVE trials differed with respect to baseline bilirubin levels and the proportion of patients with cirrhosis at baseline. The RESPONSE and POISE trials were similar for 4 of the 5 effect modifiers; ANA status was not reported for all trials. All patients across the trials had prior use of UDCA (100%), most patients were receiving background UDCA therapy (approximately 94%), and the proportion of patients with prior OCA use ranged from 0% to 17.1%.
The composite biochemical response outcome in the ELATIVE trial had an extremely low placebo response rate compared to the other trials (3.8% versus 9.6% in the POISE trial and 20% in the RESPONSE trial).The RESPONSE and ELATIVE trials also used different ALP and bilirubin ULN cut-offs. The RESPONSE trial used uniform thresholds (ALP: 116 IU/L; bilirubin: 18.8 µmol/L), while the ELATIVE trial applied sex-specific cut-offs (ALP: 104 IU/L for females and 129 IU/L for males; bilirubin: 20.5 µmol/L). The RESPONSE and POISE trials also used different ULN cut-offs (ALP: 118 IU/L for females and 124 IU/L for males; bilirubin: 19.32 µmol/L for females and 25.48 µmol/L for males). For both the NMA and MAIC, ULN cut-offs for ALP and bilirubin in the RESPONSE trial were recalculated to align with the comparator trials for the biochemical response and ALP normalization outcomes.
Table 5: MAIC Summary of Baseline Characteristics Before and After Weighting (Base Case)
Characteristic | ELATIVE | RESPONSE | |
|---|---|---|---|
Raw | Adjusted | ||
N | 161 | 193 | 193 |
Age (years), mean (SD) | 57.1 (8.7) | 56.7 (9.8) | 57.1 (8.7) |
Cirrhosis (%) | 10 | 14 | 10 |
Bilirubin (µmol/L), mean (SD) | 9.6 (5.1) | 0.75 (0.30) 12.8 (5.13) | 9.6 (5.11) |
ALP (U/L), mean (SD) | 321.9 (150.9) | 314.3 (121.9) | 321.9 (151.3) |
MAIC = matching-adjusted indirect comparison; SD = standard deviation.
Sources: ITC Technical Report.27 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal of ITCs
The methods used to conduct the systematic literature review to inform the NMA and MAIC included appropriate criteria to search databases, select studies, extract data, and assess risk of bias of the included studies. Selection bias is expected to be low given the comprehensiveness of the searches and study selection methods; however, an a priori protocol was not reported. The NMA and MAIC included relevant efficacy and harm outcomes identified by the CDA-AMC team, but did not include a global HRQoL outcome. The risk of bias assessment at the outcome level and its potential impact on the NMA effect estimates were not explicitly assessed or discussed; although, the sponsor noted that, in general, trials were considered to be at low risk of bias.
For the NMA and MAIC, the following patient characteristics were identified by the sponsor as potential key effect modifiers: age at diagnosis, ALP levels at baseline, bilirubin levels at baseline, the proportion of patients with cirrhosis at baseline, and ANA-positive status. It was unclear how these effect modifiers were identified. Austin (2014)28 suggested that the identification of potentially prognostically important covariates be based on clinical expertise or a systematic literature review of available evidence. ANA status was not reported for any of the trials, and the clinical experts did not consider ANA status to be an effect modifier. Aside from ANA status, the clinical experts agreed that these were all relevant treatment effect modifiers for patients with PBC. In addition, the experts suggested that prior treatment experience, male sex, more severe pruritus at baseline, and the Indigenous patient populations in Canada are potential effect modifiers, as these patients may have suboptimal response to the treatment and poorer prognosis.
In the NMA, the RESPONSE and POISE trials reported similar patient and trial characteristics, including 4 of the sponsor-identified effect modifiers; however, other potential effect modifiers suggested by the clinical experts (previously mentioned in the Critical Appraisal of ITCs section) were not considered. Because the RESPONSE and POISE trials used different ULN cut-off values for ALP and bilirubin, the sponsor recalculated the values for biochemical response and ALP normalization from the RESPONSE trial to align with those used in the POISE trial, which was deemed appropriate. According to the sponsor, pairwise comparisons between trials were not performed; therefore, the assessment of statistical heterogeneity was not feasible. However, heterogeneity could have been evaluated and should have been considered to better understand potential differences across studies. The evidence network was informed by only 2 studies, and with no closed loops (aside from that formed by different doses in POISE), so it was not possible to assess the consistency between direct and indirect sources of evidence. It is unclear whether the combination of different doses in the POISE trial was appropriate, and it remains uncertain how this approach may have introduced heterogeneity or bias into the study estimates. All comparisons were therefore informed only by indirect evidence, which increases the level of uncertainty in the NMA. Sensitivity analyses using different assumptions and prior distributions for between-study heterogeneity were explored, and the results were similar to the base-case analysis.
In the MAIC, the RESPONSE and ELATIVE trials differed significantly in ALP and bilirubin ULN cut-off values, with the RESPONSE trial using uniform thresholds (ALP: 116 IU/L; bilirubin: 18.8 µmol/L) and the ELATIVE trial applying sex-specific thresholds (ALP: 104 IU/L for females and 129 IU/L for males; bilirubin: 20.5 µmol/L). To address these differences, the sponsor recalculated the composite biochemical response and ALP normalization outcomes from the RESPONSE trial to align with the sex-specific cut-off values from the ELATIVE trial. Additionally, the trials differed with respect to 2 key effect modifiers (baseline bilirubin levels and the proportion of patients with baseline cirrhosis), which were adjusted for in the analysis. However, other potential effect modifiers suggested by the clinical experts (previously mentioned in the Critical Appraisal of ITCs section) were not considered. After weighting to adjust for 4 effect modifiers, the ESS for the base-case analysis declined by more than 50% from the original sample size. Such reductions can contribute to imprecision, increasing uncertainty of the results. A notable reduction in ESS also suggests that results may be driven by a subset of the sample in the trials, which may not be representative of the full sample. Given that the population of these analyses were weighted to the ELATIVE trial, the results may be driven by a subset of patients in the seladelpar group, limiting generalizability to the full population presented by the RESPONSE trial.
In the NMA and MAIC, the 95% CrIs and CIs for the point estimates were wide for most outcomes and crossed the null when comparing seladelpar with OCA or elafibranor; therefore, confidence in the effect estimates for efficacy and harms of the study drugs was limited and precluded definite conclusions about which treatments may be favoured. In particular, the 95% CI for ALP normalization in both the NMA and MAIC was very wide, and the estimates were considered unstable.
Efficacy and Harms Results of ITCs
Key results of the NMA and MAIC are presented in Table 6 and Table 7, respectively.
Table 6: Summary of NMA Results for Seladelpar vs. OCA at 12 Months (Base Case)
Random-effects Bayesian NMA based on informative priors | Result |
|---|---|
Composite biochemical response | RR (95% CrI) |
Seladelpar vs. OCA 5 mg to 10 mg | ████ █████ ██ █████ |
Seladelpar vs. OCA 10 mg | ████ █████ ██ █████ |
ALP normalization (ALP ≤ 1.0 × ULN) | RR (95% CrI) |
Seladelpar vs. OCA 5 mg to 10 mg | █████ █████ ██ █████ |
Seladelpar vs. OCA 10 mg | ████ █████ ██ ██████ |
Any TEAE | OR (95% CrI) |
Seladelpar vs. OCA 5 mg to 10 mg | ████ █████ ██ █████ |
Seladelpar vs. OCA 10 mg | ███ █████ ██ █████ |
Pruritus | OR (95% CrI) |
Seladelpar vs. OCA 5 mg to 10 mg | ████ █████ ██ █████ |
Seladelpar vs. OCA 10 mg | ████ █████ ██ █████ |
All-cause discontinuations | OR (95% CrI) |
Seladelpar vs. OCA 5 mg to 10 mg | ████ █████ ██ █████ |
Seladelpar vs. OCA 10 mg | ████ █████ ██ █████ |
CrI = credible interval; NMA = network meta-analysis; OCA = obeticholic acid; OR = odds ratio; RR = risk ratio; TEAE = treatment emergent adverse event; ULN = upper limit of normal; vs. = versus.
Sources: ITC Technical Report.27 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 7: Summary of MAIC Results for Seladelpar vs. Elafibranor at 12 Months (Base Case)
Anchored MAIC (adjusted for 4 effect modifiersa) | Result |
|---|---|
Composite biochemical response | RR (95% CI) |
Seladelpar vs. elafibranor (ESS = 70) | ████ █████ ██ █████ |
ALP normalization | RR (95% CI) |
Seladelpar vs. elafibranor 80 mg (ESS = 70) | ████ █████ ██ ██████ |
Any TEAE | OR (95% CI) |
Seladelpar vs. elafibranor 80 mg (ESS = 70) | ████ █████ ██ █████ |
Pruritus | OR (95% CI) |
Seladelpar vs. elafibranor 80 mg (ESS = 70) | ████ █████ ██ █████ |
All-cause discontinuations | OR (95% CI) |
Seladelpar vs. elafibranor 80 mg (ESS = 70) | ████ █████ ██ █████ |
CI = confidence interval; ESS = effective sample size; MAIC = matching-adjusted indirect comparison; OR = odds ratio; RR = risk ratio; TEAE = treatment emergent adverse event; vs. = versus.
aAdjusted for age, ALP and bilirubin levels at baseline, and the proportion of patients with cirrhosis at baseline.
Sources: ITC Technical Report.27 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Discussion
One pivotal phase III, randomized, double-blind controlled trial, 1 LTE study, and 2 ITCs submitted by the sponsor were summarized in this report.
The RESPONSE pivotal trial (N = 193) met the inclusion criteria for the systematic review conducted by the sponsor. The objective of the RESPONSE trial was to assess the efficacy and safety of seladelpar 10 mg, taken orally once daily, compared to placebo in adults whose PBC had an inadequate response to UDCA or who had UDCA intolerance. During the trial, seladelpar 10 mg was administered as an add-on therapy to UDCA for patients with UDCA tolerance; for patients with UDCA intolerance, seladelpar was administered as monotherapy. Enrolled patients were required to have confirmed PBC, defined as meeting any 2 of the following 3 diagnostic criteria: 1) a history of ALP above 1.0 × ULN for at least 6 months; 2) positive AMA titre (> 1:40 on immunofluorescence or M2 positive by enzyme-linked immunosorbent assay) or positive PBC-specific ANA titre; and 3) documented liver biopsy results consistent with PBC. Treatment with antipruritic drugs was permitted if patients were receiving a stable dose for at least 1 month before screening. Treatment with OCA, fibrates, and elafibranor within 6 weeks before screening was prohibited. The Health Canada indication and reimbursement request aligned with the study population. The outcomes relevant to this review included the primary outcome of biochemical response; key secondary outcomes of ALP normalization and pruritus NRS in patients with a baseline NRS of 4 or higher at 6 months; and secondary outcomes of pruritus NRS in patients with a baseline NRS of 4 or higher at 12 months, HRQoL measured using the PBC-40 total score, and safety. Overall, key baseline characteristics were generally balanced between treatment groups. The trial population was mostly female (94.8%), white (88.1%), and non-Hispanic or Latino (69.9%), and the mean age was 56.7 years. In addition to patients with mild disease severity, the trial population included patients with moderate disease severity (e.g., 13.0% of patients had TB levels above ULN at baseline, 14.0% of patients had moderate Rotterdam stage, and 14.0% of patients had cirrhosis at baseline). Mean ALP, TB levels, and pruritus NRS were balanced between treatment groups at baseline. Most patients (93.8%) received seladelpar 10 mg or placebo in addition to UDCA; 6.2% of patients had UDCA intolerance and received seladelpar 10 mg or placebo as monotherapy. Prior use of OCA or fibrates was reported in 15.6% of patients in the seladelpar group and 20.0% of patients in the placebo groups.
After completion of the treatment period, patients were invited to enrol in an open-label, LTE safety trial (ASSURE), in which all participants received seladelpar 10 mg; patients previously randomized to the placebo group in the RESPONSE trial initiated seladelpar. No efficacy results were reported.
To inform the pharmacoeconomic model and address gaps in the comparative evidence for other treatments of interest for adults with PBC, the sponsor conducted 1 NMA and 1 anchored MAIC. The objective of the ITCs was to assess the relative efficacy and safety of seladelpar with OCA (POISE trial) and elafibranor (ELEVATE trial) in adults whose PBC had an inadequate response to UDCA or who had UDCA intolerance. Outcomes of interest included biochemical response, ALP normalization, TEAEs, pruritus, and all-cause study discontinuations at 12 months. The sponsor identified the following 5 key effect modifiers for PBC: age at diagnosis, ALP levels at baseline, bilirubin levels at baseline, the proportion of patients with cirrhosis at baseline, and ANA-positive status; although, the methods used to identify these effect modifiers were not reported. For the comparison between the RESPONSE and POISE trials, a random-effects Bayesian NMA using a generalized linear framework was conducted for the outcomes of interest and reported as RRs with 95% CrIs; safety outcomes were reported as ORs with 95% CrIs. For the comparison between seladelpar and elafibranor, an anchored MAIC was performed by matching and adjusting patient characteristics of the RESPONSE trial to the patients in the ELEVATE trial. The MAIC was adjusted for 4 key effect modifiers: age at screening, ALP levels at baseline, bilirubin levels at baseline, and the proportion of patients with cirrhosis at baseline. Efficacy outcomes were reported as RRs with 95% CIs using binomial regression, and safety outcomes were reported as ORs with 95% CIs using logistic regression.
Efficacy
The evidence from the pivotal trial, RESPONSE, addressed treatment outcomes noted to be important by both patients and clinicians. The patient group input indicated a need for effective and safe treatment options that can reduce the risk of disease progression, alleviate symptoms such as pruritus, improve HRQoL, and reduce treatment side effects. Similarly, the clinical experts consulted by the review team indicated that the unmet needs in PBC include treatments that control disease activity (e.g., ALP and bilirubin normalization), improve PBC-related symptoms (including pruritus), and prevent end-stage liver disease or transplant. However,, long-term survival outcomes such as end-stage liver disease or transplant-free survival were not reported in the RESPONSE trial.
The RESPONSE trial showed clinically meaningful improvements with seladelpar compared with placebo for biochemical response, ALP normalization, and pruritus in adults whose PBC had an inadequate response to UDCA or who had UDCA intolerance. For biochemical response and ALP normalization at 12 months, the estimated between-group differences were 41.7% (95% CI, 27.7 to 53.4) and 25.0% (95% CI, 18.3 to 33.2), respectively. For the GRADE assessment, the clinical experts consulted by CDA-AMC could not determine a minimal important difference (MID) for these outcomes, but they considered the effect estimates and confidence intervals clinically meaningful, with high certainty of evidence, favouring seladelpar. For pruritus NRS in patients with a baseline NRS of 4 or higher at 6 and 12 months, the between-group LS mean differences in change from baseline were −1.5 (95% CI, −2.5 to −0.5) and −1.8 (95% CI, −3.0 to −0.6), respectively. For the GRADE assessment, the clinical experts could not determine an MID for pruritus, but they considered the effect estimates and confidence intervals clinically meaningful, favouring seladelpar. The certainty of evidence was moderate at 6 months, due to small sample size. The certainty of evidence was low at 12 months, due to the risk of bias from missing outcome data and small sample size. The results for these outcomes listed were consistent across the prespecified subgroups of interest by baseline ALP (ALP ≥ 350 U/L and ALP < 350 U/L) and by cirrhosis status at baseline, favouring seladelpar. For the PBC-40 total score at 12 months, there was low certainty of evidence of little to no difference in HRQoL. Because no MID was identified for this outcome, the target of the certainty appraisal was any effect. The low certainty was attributed to very serious imprecision because the 95% CI for the between-group LS mean difference included the possibility of benefit and harm.
The evidence submitted to CDA-AMC did not include head-to-head comparisons between seladelpar and relevant comparators such as elafibranor and OCA, which represents a gap in the available direct evidence.
The ASSURE study did not report efficacy results in the submitted interim analysis, despite the protocol including relevant end points such as ALP normalization, bilirubin reduction, and pruritus improvement. Consequently, the study does not yet contribute new data on the durability of seladelpar’s biochemical or symptomatic efficacy beyond the duration of the RESPONSE trial, which represents a key evidence gap.
In the NMA and MAIC, the 95% CrIs and CIs for the point estimates were wide for all efficacy outcomes and crossed the null when comparing seladelpar with OCA or elafibranor; therefore, confidence in the effect estimates for efficacy and harms of the study drugs was limited and precludes definite conclusions as to which treatment may be favoured. For the NMA, the evidence network was informed by only 2 studies and had no closed loops aside from that formed by different doses in the POISE trial; therefore, it was not possible to assess the consistency between direct and indirect sources of evidence. All comparisons were informed only by indirect evidence, which increased the level of uncertainty in the NMA. For the MAIC, the ESS for the base-case analysis declined by more than 50% from the original sample size, which likely contributed to imprecision and increased uncertainty. A notable reduction in ESS also suggests that results may be heavily influenced by a subset of participants in the trials who may not be representative of the full sample. Given that the population of these analyses were weighted to the RESPONSE trial, the results may be driven by a subset of patients in the seladelpar group, limiting generalizability to the full population in the RESPONSE trial.
Harms
Most patients in both groups reported at least 1 TEAE. The most frequently reported TEAEs in the seladelpar group were COVID-19, headache, and abdominal pain, with a higher proportion of patients in the seladelpar group reporting these events than in the placebo group. The most frequently reported TEAEs in the placebo group were COVID-19, pruritus, and upper respiratory tract infection, with pruritus and abdominal pain being reported more frequently in the placebo group. Grade 3 or higher TEAEs were similar between groups. The incidence of serious TEAEs was similar between groups, with COVID-19 being the most frequently reported event. The incidence of TEAEs leading to study treatment discontinuation and notable harms were infrequent and similar between groups. There were no deaths reported in both groups.
Interim safety findings from the open-label LTE study ASSURE suggest that seladelpar continues to demonstrate an acceptable safety profile over the longer term. Most participants initiated treatment at 10 mg once daily, with limited need for dose modification. Serious TEAEs occurred in 6.4% of participants, and 3.2% discontinued treatment permanently due to TEAEs. There were no deaths reported, and notable harms were infrequent. Overall, no new safety signals were identified. However, several limitations constrain interpretation. The open-label design and absence of a control arm introduce potential performance and reporting bias, especially for subjective outcomes such as pruritus. The selective enrolment of trial completers introduces potential selection bias favouring individuals who are more adherent, tolerant, and/or responsive to seladelpar. The interim report did not include subgroup analyses (e.g., by geographic region, parent trial, disease severity, or comorbidities), limiting generalizability to broader patient populations.
In the NMA and MAIC, the 95% CrIs and CIs for the point estimates were wide for most safety outcomes and crossed the null when comparing seladelpar with OCA or elafibranor. Pruritus was the only outcome for which seladelpar appeared to be favoured compared to OCA, which seladelpar was favoured. Overall, confidence in the effect estimates for the harm outcomes precludes definite conclusions as to which treatment may be favoured.
Ethics and Equity Considerations
The lived experience of patients with PBC raises important ethical and equity considerations. Patients with PBC frequently report debilitating fatigue, pruritus, and cognitive dysfunction that impair their ability to work, maintain independence, and participate in social and family life. Nearly one-third of surveyed patients required help with daily tasks, and many described leaving the workforce, relying on disability benefits, and losing meaningful relationships due to symptom burden. These limitations contribute to feelings of guilt, depression, and diminished self-worth, particularly among those unable to fulfill parenting or professional roles. Social isolation and psychological distress were commonly reported, with some patients describing suicidal ideation linked to unrelenting symptoms such as itch. In several cases, patients required caregiver support, introducing additional emotional, psychological, and financial strain on family members. Caregivers were often spouses or close relatives, and their involvement was described as essential but taxing, especially when patients lost independence or required assistance with transportation and decision-making. One clinician group highlighted that pruritus can severely disrupt sleep and concentration, further compounding emotional distress and limiting patients’ ability to engage in daily activities. The need for multiple symptom-targeting medications adds to treatment complexity and burden, particularly for those managing care in rural or underserved settings. These burdens underscore the importance of access to therapies that not only slow disease progression but also alleviate symptoms and restore autonomy, dignity, and quality of life. Ensuring equitable access to such treatments across jurisdictions, including for patients in rural or underserved communities, is essential to reducing disparities and improving outcomes for both patients and caregivers. Additionally, the trial population lacked substantive sex and ethnic diversity, particularly representation of Indigenous Peoples, and therefore the findings may not be generalizable to diverse populations. The clinical experts also noted that Indigenous people and males have a higher prevalence of severe PBC and are at a higher risk of poor outcomes.
Conclusion
Evidence from 1 phase III, randomized, double-blind controlled trial (the RESPONSE study) reported outcomes that were important to both patients and clinicians. The trial showed high certainty of evidence that treatment with seladelpar, in combination with UDCA in patients whose PBC had an inadequate response to UDCA or as monotherapy in patients with UDCA intolerance, results in a clinically important increase in the proportion of patients with biochemical response and ALP normalization at 12 months compared to placebo in adults with PBC. The trial showed moderate and low certainty of evidence that treatment with seladelpar results in a clinically important decrease in pruritus at 6 months and 12 months, respectively. There was low certainty of evidence of little to no difference in HRQoL at 12 months. Based on the evidence submitted to CDA-AMC, the effect of seladelpar on long-term outcomes such as progression to end-stage liver disease or transplant-free survival is unknown. There were no new safety signals identified, and the safety of seladelpar in combination with UDCA was consistent with the known safety profiles of the individual drugs. Although no new safety signals were identified in the LTE study (ASSURE), efficacy outcomes were not reported, uncertainty remains regarding the long-term safety of seladelpar due to the open-label design and lack of a comparator group. The evidence submitted to CDA-AMC did not include head-to-head comparisons between seladelpar and relevant comparators such as elafibranor and OCA in the second-line setting, which represents a gap in the available direct evidence. Based on the NMA, seladelpar may be associated with fewer occurrences of pruritus as an AE compared to OCA; however, due to limitations in both the NMA and MAIC, mostly attributed to serious imprecision, no conclusions can be drawn on the relative efficacy and safety of seladelpar compared to OCA and elafibranor.
Economic Review
Methods
The review team appraised the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of seladelpar for the treatment of PBC, used in combination with UDCA in adults whose PBC has an inadequate response to UDCA alone, or as monotherapy in adults with UDCA intolerance. This was conducted by assessing the evidence in 2 subgroups: patients whose PBC had an inadequate response to UDCA (referred to as the UDCA-tolerant subgroup), and patients with UDCA intolerance (referred to as the UDCA-intolerant subgroup).
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of seladelpar, with or without UDCA, from the perspective of a public health care payer in Canada over a 50-year time horizon. The modelled population comprised adult patients requiring second-line treatment for PBC, either in combination with UDCA for those whose PBC had an inadequate response to UDCA alone, or as monotherapy in those with UDCA intolerance. The modelled population is aligned with the Health Canada indication and was based on the participants in the RESPONSE trial. In the UDCA-tolerant subgroup, comparators included elafibranor with UDCA, OCA with UDCA, and UDCA monotherapy. In the UDCA-intolerant subgroup, comparators were elafibranor, OCA, and best supportive care (BSC). The sponsor’s base-case analysis included costs related to drug acquisition and administration, AEs, pruritus management, health care resource use, and end-of-life care. Additional information about the sponsor’s submission is provided in Appendix 3 in the Supplemental Material document.
In the sponsor’s base case, seladelpar was more costly and effective than all comparators in both the UDCA-tolerant and UDCA-intolerant subgroups. Across comparisons, more than 95% of predicted incremental benefit accrued beyond the treatment duration of the RESPONSE trial (maximum follow-up period = 1 year).
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 8; full details are provided in Appendix 11 in the Supplemental Material document).
Table 8: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The efficacy and safety of seladelpar compared to OCA and elafibranor are uncertain. | The sponsor relied on a network meta-analysis (NMA) and matching-adjusted indirect comparison (MAIC) to assess efficacy and safety. Instead of using the relative measures of effect from these ITCs, the sponsor calibrated transition probabilities in the pharmacoeconomic model to align with prespecified end points. The uncertainty and methodological limitations in the ITCs, combined with the calibration approach, undermined the credibility of the model results. | CDA-AMC could not address this issue in the base case due to the lack of direct or robust indirect comparative evidence. | No scenario analysis was conducted. |
The long-term effectiveness and safety of seladelpar, with or without UDCA, compared to UDCA monotherapy (the UDCA-tolerant subgroup) and BSC (the UDCA-intolerant subgroup) are uncertain. | In the pharmacoeconomic model, nearly all of the estimated survival benefit and QALYs associated with seladelpar were accrued beyond the RESPONSE trial duration of 12 months. The open-label long-term extension ASSURE study lacked a comparator group and did not report efficacy outcomes, limiting the ability to assess long-term effectiveness and safety of seladelpar with or without UDCA. | CDA-AMC could not address this issue in the base case due to the lack of long-term comparative evidence. | No scenario analysis was conducted. |
The impact of seladelpar on long-term outcomes, such as transplant-free survival and overall survival, is uncertain. | The majority of life-years and QALY gains in the pharmacoeconomic model were due to increased time spent in health states characterized by normal or mildly elevated ALP levels. This reliance on biochemical markers to estimate downstream benefits in survival outcomes make the model results highly uncertain. | CDA-AMC could not address this issue in the base case due to the lack of direct evidence on the effect of seladelpar on long-term outcomes such as transplant-free and overall survival. | No scenario analysis was conducted. |
The modelling approach may misrepresent the relative treatment benefits (seladelpar, OCA, and elafibranor) with respect to pruritus management. | The relative effects of treatments (elafibranor and OCA) on pruritus as an AE were captured using odds ratios obtained from ITCs. However, the modelling approach assumed that the treatment effect was identical across all pruritus severity levels, which may bias the estimation of pruritus-related disutility. | CDA-AMC could not address this issue in the base case due to lack of evidence of treatment effects by pruritus severity. | No scenario analysis was conducted. |
The use of unadjusted discontinuation rates from respective trials potentially bias the model outcomes in favour of seladelpar. | Although the sponsor-conducted ITCs showed no statistically significant differences in all-cause discontinuation rates, the sponsor modelled a lower discontinuation rate for seladelpar compared to elafibranor and OCA, which likely overestimated seladelpar’s relative health benefits. | CDA-AMC could not address this issue in the base case due to model structure limitations. | No scenario analysis was conducted. |
AE = adverse event; BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; ITC = indirect treatment comparison; MAIC = matching-adjusted indirect comparison; NMA = network meta-analysis; OCA = obeticholic acid; QALY = quality-adjusted life-year; UDCA = ursodeoxycholic acid.
Note: Full details of the issues identified by CDA-AMC are provided in Appendix 3 in the Supplemental Material document.
CDA-AMC Assessment of Cost-Effectiveness
Based on the CDA-AMC clinical review of the RESPONSE trial, treatment with seladelpar, with or without UDCA, results in a clinically important increase in the proportion of patients with biochemical response and ALP normalization at 12 months compared to placebo in adults with PBC. The trial also showed moderate and low certainty of evidence that treatment with seladelpar results in a clinically important reduction in pruritus at 6 months and 12 months, respectively. The evidence for a difference in HRQoL at 12 months was of low certainty. However, due to the lack of long-term comparative clinical evidence to support assumptions on duration of treatment effect and the lack of evidence on the effect of seladelpar on long-term outcomes such as transplant-free and overall survival, the incremental costs and quality-adjusted life-years predicted by the submitted model are highly uncertain. More than 95% of incremental quality-adjusted life-years were estimated beyond the observation period of the trial. Based on feedback from clinical experts and data presented by the sponsor, the use of UDCA monotherapy in the UDCA-tolerant subgroup and BSC in the UDCA-intolerant subgroup for this indication is anticipated to be limited and would not be affected if seladelpar becomes available.
The evidence submitted to CDA-AMC did not include head-to-head comparisons between seladelpar and relevant comparators such as elafibranor and OCA in the second-line setting, which represents a gap in the available direct evidence. Based on the CDA-AMC clinical review of the sponsor-submitted NMA and MAIC comparing seladelpar, with or without UDCA, to elafibranor, with or without UDCA, and to OCA, with or without UDCA, results for biochemical response and ALP normalization were inconclusive, owing to methodological limitations and imprecision. Confidence in point estimates for most safety outcomes precluded definitive conclusions as to which treatment may be favoured. Global HRQoL was not assessed in the sponsor-conducted ITCs; therefore, whether seladelpar, with or without UDCA, improves HRQoL is unknown. Seladelpar may be associated with fewer occurrences of pruritus as an AE compared to OCA; however, this finding is highly uncertain due to serious imprecision in the submitted ITCs. Due to these limitations, no definitive conclusions could be drawn on the comparative efficacy and safety when comparing seladelpar with or without UDCA for the treatment of adult patients with PBC. As such, no reanalyses were performed. Seladelpar was associated with higher annual drug acquisition costs compared to all other comparators (refer to Table 11 in Appendix 8 in the Supplemental Material document).
Summary of the Budget Impact
The sponsor submitted a budget impact analysis to estimate the 3-year (2026 to 2028) budget impact of reimbursing seladelpar, with or without UDCA, for use in the Health Canada–indicated population. The sponsor assumed the payer would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The price of seladelpar was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in Appendix 5 in the Supplemental Material document.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (refer to Appendix 5 in the Supplemental Material document). CDA-AMC estimated that 8,066 patients would be eligible for seladelpar, with or without UDCA, over a 3-year period (year 1 = 7,271; year 2 = 7,665; year 3 = 8,066), of whom 4,033 were expected to receive seladelpar, with or without UDCA (year 1 = 1,985; year 2 = 3,066; year 3 = 4,033). The estimated incremental budget impact of reimbursing seladelpar, with or without UDCA is predicted to be approximately $770 million over the first 3 years, with an expected expenditure of $785 million on seladelpar, with or without UDCA. The magnitude of uncertainty in the budget impact must be addressed to ensure the feasibility of adoption.
Conclusion
Based on the CDA-AMC clinical review of the RESPONSE trial, seladelpar, with or without UDCA, likely results in a clinically important increase in the proportion of patients with biochemical response and ALP normalization at 12 months compared with placebo in adults with PBC. There was evidence of moderate to low certainty that seladelpar, with or without UDCA, may reduce pruritus at 6 months and 12 months; however, the evidence regarding a difference in HRQoL at 12 months was of low certainty. The effect of seladelpar on long-term outcomes such as progression to end-stage liver disease, transplant-free survival, and overall survival is unknown. The cost-effectiveness of seladelpar, with or without UDCA, compared to UDCA monotherapy in the UDCA-tolerant subgroup and BSC in the UDCA-intolerant subgroup is highly uncertain. However, based on feedback from clinical experts and data presented by the sponsor, the use of UDCA monotherapy or BSC for this indication is anticipated to be limited and would not be impacted if seladelpar becomes available.
Evidence from the sponsor-submitted NMA and MAIC was insufficient to show differences in efficacy for seladelpar, with or without UDCA, compared to elafibranor, with or without UDCA, or OCA, with or without UDCA. Overall, definite conclusions regarding comparative harms and impact on global HRQoL could not be drawn; global HRQoL was not assessed in the sponsor-conducted ITCs. Given the uncertainty in the comparative clinical evidence, there is insufficient evidence to determine whether seladelpar provides greater health benefit than elafibranor or OCA. Seladelpar was associated with higher annual drug acquisition costs than all other comparators (refer to the Cost Comparison table in Appendix 8 in the Supplemental Material document). If there are no differences in health outcomes between seladelpar and OCA or elafibranor, then the total cost of seladelpar to the health system should not exceed that of OCA or elafibranor for the treatment of adult patients with PBC, either in combination with UDCA or as monotherapy.
The budget impact of reimbursing seladelpar, with or without UDCA, to the public drug plans in the first 3 years is estimated to be approximately $770 million. The 3-year expenditure on seladelpar, with or without UDCA (i.e., not accounting for current expenditure on comparators), is estimated to be $785 million. The estimated budget impact is highly uncertain due to the uncertainty in the market share of seladelpar, with or without UDCA, the reimbursement status of elafibranor, and the prevalence of PBC.
References
1.Younossi, Z.M., et al., Diagnosis and Management of Primary Biliary Cholangitis. Am J Gastroenterol, 2019. 114(1): p. 48-63. PubMed
2.European Association for the Study of the Liver, EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J Hepatol, 2017. 67(1): p. 145-172. PubMed
3.Trivella, J., B.V. John, and C. Levy, Primary biliary cholangitis: Epidemiology, prognosis, and treatment. Hepatol Commun, 2023. 7(6). PubMed
4.Trivedi, P.J. and G.M. Hirschfield, Recent advances in clinical practice: epidemiology of autoimmune liver diseases. Gut, 2021. 70(10): p. 1989-2003. PubMed
5.Boonstra, K., U. Beuers, and C.Y. Ponsioen, Epidemiology of primary sclerosing cholangitis and primary biliary cirrhosis: a systematic review. J Hepatol, 2012. 56(5): p. 1181-1188. PubMed
6.Schnabl, B., PPAR Agonists in Primary Biliary Cholangitis. N Engl J Med, 2024. 390(9): p. 855-858. PubMed
7.Gungabissoon, U., et al., Disease burden of primary biliary cholangitis and associated pruritus based on a cross-sectional US claims analysis. BMJ Open Gastroenterol, 2022. 9(1). PubMed
8.Martin, M.L., et al., Development and adaptation of patient-reported outcome measures for patients who experience itch associated with primary biliary cholangitis. J Patient Rep Outcomes, 2019. 3(1): p. 2. PubMed
9.Mayo, M.J., et al., Impact of Pruritus on Quality of Life and Current Treatment Patterns in Patients with Primary Biliary Cholangitis. Dig Dis Sci, 2023. 68(3): p. 995-1005. PubMed
10.Onofrio, F.Q., G.M. Hirschfield, and A.F. Gulamhusein, A Practical Review of Primary Biliary Cholangitis for the Gastroenterologist. Gastroenterol Hepatol (N Y), 2019. 15(3): p. 145-154. PubMed
11.Chalifoux, S.L., et al., Extrahepatic Manifestations of Primary Biliary Cholangitis. Gut Liver, 2017. 11(6): p. 771-780. PubMed
12.Ebadi, M., et al., Vitamin D Is Associated with Clinical Outcomes in Patients with Primary Biliary Cholangitis. Nutrients, 2022. 14(4). PubMed
13.Hammitt, K.M., et al., Patient burden of Sjogren's: a comprehensive literature review revealing the range and heterogeneity of measures used in assessments of severity. RMD Open, 2017. 3(2): p. e000443. PubMed
14.Wah-Suarez, M.I., et al., Hyperlipidaemia in primary biliary cholangitis: treatment, safety and efficacy. Frontline Gastroenterol, 2019. 10(4): p. 401-408. PubMed
15.Canadian Liver Foundation. Primary Biliary Cholangitis [sponsor submitted reference]. [cited 2024 November 22, 2024]; Available from: https://www.liver.ca/wp-content/uploads/2022/09/Primary-Biliary-Cholangitis.pdf.
16.Hirschfield, G.M., et al., A consensus integrated care pathway for patients with primary biliary cholangitis: a guideline-based approach to clinical care of patients. Expert Rev Gastroenterol Hepatol, 2021. 15(8): p. 929-939. PubMed
17.Lindor, K.D., et al., Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology, 2019. 69(1): p. 394-419. PubMed
18.The P. B. C. Network. Primary biliary cholangitis (PBC): Living with your diagnosis [sponsor submitted reference]. [cited 2024 November 22, 2024]; Available from: https://www.liver.ca/wp-content/uploads/2018/08/EASL_PBC_Patient-Guidelines_ENG.pdf.
19.Bowlus, C.L. and M.E. Gershwin, The diagnosis of primary biliary cirrhosis. Autoimmun Rev, 2014. 13(4-5): p. 441-4. PubMed
20.Kowdley, K.V., et al., Application of the Latest Advances in Evidence-Based Medicine in Primary Biliary Cholangitis. Am J Gastroenterol, 2023. 118(2): p. 232-242. PubMed
21.Canadian P. B. C. Society. What you should know about PBC [sponsor submitted reference]. 2021 [cited 2025 February 20, 2025]; Available from: https://pbc-society.ca/what-is/.
22.Liver Canada. Primary Biliary Cholangitis [sponsor submitted reference]. 2025 [cited 2025 February 1, 2025]; Available from: https://liver.ca/primary-biliary-cholangitis-2/.
23.CymaBay Therapeutics Inc, Clinical Study Report: CB8025-32048. A placebo-controlled, randomized, phase 3 study to evaluate the efficacy and safety of seladelpar in patients with Primary Biliary Cholangitis (PBC) and an inadequate response to or an intolerance to ursodeoxycholic acid (UDCA) [internal sponsor's report]. November 30, 2023.
24.Beuers, U. and K.D. Lindor, A major step towards effective treatment evaluation in primary biliary cirrhosis. J Hepatol, 2011. 55(6): p. 1178-80. PubMed
25.Carbone, M., et al., Sex and age are determinants of the clinical phenotype of primary biliary cirrhosis and response to ursodeoxycholic acid. Gastroenterology, 2013. 144(3): p. 560-569.e7; quiz e13-4. PubMed
26.Lammers, W.J., et al., Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: an international follow-up study. Gastroenterology, 2014. 147(6): p. 1338-49.e5; quiz e15. PubMed
27.Gilead Sciences Canada Inc, Clinical efficacy, safety, and tolerability of approved medications for the treatment of primary biliary cholangitis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: seladelpar, 10 mg seladelpar (as seladelpar lysine dihydrate), oral. April 2, 2025.
28.Austin, P.C., The use of propensity score methods with survival or time-to-event outcomes: reporting measures of effect similar to those used in randomized experiments. Stat Med, 2014. 33(7): p. 1242-58. PubMed
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.