Drugs, Health Technologies, Health Systems
Reimbursement Review
Antihemophilic Factor VIII (Recombinant, B-Domain Deleted), Fc-VWF-XTEN Fusion Protein (Altuviiio)
Sponsor: Sanofi-Aventis Canada Inc.
Therapeutic area: Congenital factor VIII deficiency
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
ABR
annualized bleeding rate
AE
adverse event
AHCDC
Association of Hemophilia Clinic Directors of Canada
AjBR
annualized joint bleeding rate
BU
Bethesda units
CANHC
Canadian Association of Nurses in Hemophilia Care
CDA-AMC
Canada’s Drug Agency
CHS
Canadian Hemophilia Society
CI
confidence interval
CPHC
Canadian Physiotherapists in Hemophilia Care
EHL
extended half-life
ESS
effective sample size
FAS
full analysis set
FVIII
factor VIII
GRADE
Grading of Recommendations, Assessment, Development and Evaluations
Haem-A-QoL
Haemophilia Quality of Life questionnaire for adults
Haemo-QoL
Haemophilia Quality of Life questionnaire for children
HJHS
Hemophilia Joint Health Score
HRQoL
health-related quality of life
IQR
interquartile range
IRR
incidence rate ratio
ISTH
International Society on Thrombosis and Haemostasis
ITC
indirect treatment comparison
LS
least squares
LTE
long-term extension
MAIC
matching-adjusted indirect comparison
MD
mean difference
MedDRA
Medical Dictionary for Regulatory Activities
MID
minimal important difference
MMRM
mixed-effects model with repeated measures
PK
pharmacokinetics
PPS
per-protocol set
PROMIS
Patient-Reported Outcomes Measurement Information System
PSM
propensity score matching
QoL
quality of life
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
SHL
standard half-life
SLR
systematic literature review
TEAE
treatment-emergent adverse event
TESAE
treatment-emergent serious adverse event
VWF
von Willebrand factor
WDAE
withdrawal due to adverse event
WFH
World Federation of Hemophilia
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Drug product: Altuviiio Strength: 250 IU, 500 IU, 1,000 IU, 2,000 IU, 3,000 IU, and 4,000 IU vials Formulation: lyophilized powder for reconstitution Route of administration: IV use after reconstitution |
Sponsor | Sanofi-Aventis Canada Inc. |
Indication | Altuviiio is indicated in adults, adolescents, and children with hemophilia A (congenital factor VIII [FVIII] deficiency) for:
|
Reimbursement request | As per proposed indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | March 26, 2025 |
Recommended dose | Prophylactic dosing: 50 IU/kg IV administered once weekly On-demand dosing: 50 IU/kg IV administered as a single dose Perioperative dosing: 50 IU/kg IV administered as a single dose prior to surgery |
FVIII = factor VIII; NOC = Notice of Compliance.
Introduction
Hemophilia is a bleeding disorder caused by deficiencies in coagulation factor VIII (FVIII) or factor IX.1-3 Hemophilia A is the most common form.4 It is a rare, congenital bleeding disorder caused by mutations in the gene that produces FVIII, a glycoprotein critical for hemostasis, which leads to excessive bleeding due to the inability to form blood clots.2,3,5 It predominantly affects male patients, although females who are heterozygous carriers can have factor levels in the hemophilia range.4 According to a 2023 Canadian Bleeding Disorders Registry report, there were 3,510 people in Canada living with hemophilia A, of whom 1,158 had severe disease.6 Disease severity is categorized as mild, moderate, or severe and is based on factor levels.7 For reference, normal FVIII activity is considered to be 40% or higher. Mild hemophilia A is defined by factor levels 5% to 40% of typical FVIII activity levels in the blood, moderate is defined by levels of 1% to 5%, and severe defined by levels less than 1% of typical FVIII activity levels.8 Patients with hemophilia A experience symptoms such as bleeding into joints, soft tissues and muscles, the mouth, and urine, as well as surface bleeding and easy bruising.9 Bleeding associated with hemophilia A can result in complications such as joint damage from repetitive bleeding, deep internal bleeding, and neurologic problems or death associated with bleeding in the brain.7 The challenges experienced by patients with hemophilia A can substantially impact patient quality of life (QoL) and physical, mental, social, and educational well-being.4,10 Hemophilia A is diagnosed based on a combination of clinical evaluation, family history, and laboratory tests (factor assay obtained through blood tests, such as the 1-stage clotting assay and chromogenic assay, which measure FVIII activity levels to demonstrate factor deficiency and FVIII genetic testing).11 Patients with a family history of hemophilia A are typically diagnosed at birth, while those without a family history are typically diagnosed after bleeding is observed, often during the first year of life for those with severe hemophilia.12
The International World Federation of Hemophilia (WFH) guidelines recommend primary prophylaxis as the standard of care for all patients with severe hemophilia A.8 Primary prophylaxis is defined as regular infusion of missing FVIII or administration of a factor mimetic to increase factor activity. The key for primary prophylaxis is to start a patient on treatment before a bleeding event. The goal of prophylactic therapy is to maintain factor levels greater than 3% to 5% (3 IU/dL to 5 IU/dL) to reduce the risk of spontaneous bleeding and for better preservation of joint function.13 Three options for primary prophylactic treatment exist in the current Canadian landscape: regular IV infusion of standard half-life (SHL) FVIII concentrate (Kovaltry, Xyntha, Zonovate), regular IV infusion of extended half-life (EHL) FVIII concentrate (Adynovate, Jivi, Eloctate, Esperoct), or regular subcutaneous injection of emicizumab. Apart from emicizumab, which provides a FVIII activity equivalence level of 10% to 15%, the trough levels of SHL and EHL FVIII concentrates are between 3% to 5% immediately before the next infusion. Based on the typical frequency of administration for currently available SHL and EHL products (2 to 3 times per week), the trough levels are often inadequate to provide bleeding protection. Patients with hemophilia A who participate in regular physical activities may time their prophylactic infusion to align with their physical activities or require additional doses on top of their prophylaxis just before certain physical activities to mitigate the risk of provoked bleeding.
Altuviiio is a recombinant FVIII analogue fusion protein that temporarily replaces the missing coagulation factor (FVIII) required for effective hemostasis. It is a new class of FVIII replacement therapy that functions independently of endogenous von Willebrand factor (VWF) to overcome the half-life limit imposed by FVIII–VWF interactions. FVIII half-life is significantly influenced by VWF plasma levels while circulating together as a noncovalent complex. Under typical physiological circumstances, the half-life of free FVIII is substantially reduced (approximately 2 hours) compared to that of VWF-bound FVIII (approximately 12 hours). By circulating independently of endogenous VWF, Altuviiio has demonstrated a 3-fold to 4-fold prolonged half-life compared to other SHL and EHL FVIII molecules. Altuviiio is approved by Health Canada for the treatment of hemophilia A (congenital FVIII deficiency) in adults and children for routine prophylaxis to reduce the frequency of bleeding episodes, on-demand treatment and control of bleeding episodes, and perioperative management of bleeding. The sponsor’s reimbursement request aligns with the Health Canada indication. The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of Altuviiio (lyophilized powder for IV infusion; 250 IU, 500 IU, 1,000 IU, 2,000 IU, 3,000 IU, and 4,000 IU per vial) for the treatment of adults, adolescents, and children with hemophilia A (congenital FVIII deficiency) for routine prophylaxis to prevent or reduce the frequency of bleeding episodes, treatment and control of bleeding episodes, or perioperative management of bleeding (surgical prophylaxis).
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to our call for input and from clinical expert(s) consulted by for the purpose of this review.
Patient Input
One patient group submission from the Canadian Hemophilia Society (CHS) was received for this review. The CHS is a national voluntary health charity that advocates for improvements in health and QoL for patients living with inherited bleeding disorders in Canada. Information provided for this submission was gathered through a national online survey distributed both in English and French between April 1, 2024, to June 1, 2024. A total of 104 responses were received. This included 57 patients with severe hemophilia A, 33 with mild hemophilia A, and 14 with moderate hemophilia A. Of these patients, 33 reported a history of FVIII inhibitors.
The patients highlighted joint pain and loss of function, pain from bleeding episodes, invasive medical procedures, surgical complications, restrictions on sport participation, difficulty performing everyday tasks, and long recovery times from bleeding episodes as significant symptoms and challenges associated with hemophilia A disease. Respondents also noted the detrimental effects of hemophilia A on their social and psychological well-being.
Overall, 11 patients were receiving FVIII prophylaxis; 6 were on FVIII on-demand therapy while most patients were receiving emicizumab for prophylaxis. Notably, 1 patient had undergone gene therapy. Overall, most respondents considered their current treatment regimen as “very effective” or “quite effective” in stopping or preventing bleeding. Although most respondents indicated that hemophilia A treatment has become simpler and less burdensome with emicizumab, the pain associated with the injection is a challenge and breakthrough bleeding still occurs. Patients reported that new therapies that can improve hemophilia A disease outcomes such as higher bleed protection, less pain management, reduced frequency (fewer doses with longer half-life) of treatment are needed to improve disease outcomes.
One patient with severe hemophilia A who had received Altuviiio through a special access program reported that since initiating treatment, this patient reported experiencing a sustained high FVIII level, with a factor trough of approximately 15%, which has reduced their risk of major and subclinical bleeding. Similar effects were reported by other patients, which appear to be maintained even if the injection is up to 2 days late. According to the patient, this has helped reduce the risk of bleeding, and travel has become easier with Altuviiio, due to the more flexible storage requirements compared to previous treatments. The patient reported no disadvantages or side effects of Altuviiio.
Clinician Input
Input From Clinical Experts Consulted for This Review
The clinical experts consulted for this review indicated that the most important treatment goals for patients with hemophilia A are to prevent bleeding, including spontaneous and traumatic bleeding events, reduce joint pain, improve health-related QoL (HRQoL), and achieve an unrestricted lifestyle comparable to the general population. The clinical experts noted that the current standard of care for patients with severe hemophilia A in Canada is primary prophylactic therapy. The goal of prophylactic treatment is to prevent bleeding, and as newer treatments are available, the overall goal is for patients to attain higher factor levels or near-normal factor levels. According to the clinical experts consulted for this review, there is no current therapy that can modify the underlying disease mechanism of hemophilia A outside of gene therapy which is currently unavailable in Canada. In addition, apart from emicizumab, which provides bleeding protection roughly equivalent to a steady-state trough FVIII level of 10% to 15%, the trough levels of available SHL and EHL FVIII concentrates are between 3% to 5% before the next infusion, with subsequent clearance dependent on the product half-life (but generally 14 to 18 hours) resulting in less bleed protection. Patients who participate in regular physical activities are at risk of bleeding with current prophylactic regimens when their FVIII levels are suboptimal. As a result, these patients need additional doses of factor concentrates on top of their regular prophylaxis just before certain physical activities to mitigate the risk of provoked bleeding.
According to the clinical experts, Altuviiio will change the treatment landscape for acute bleed and perioperative management, but they do not envision Altuviiio altering the underlying disease process of congenital hemophilia A. Both clinical experts indicated that Altuviiio will be the first agent in which a period of “normal hemostasis” (FVIII activity > 40% for the first 4 days of treatment) can be achieved without a trade-off in burden of treatment. Compared to available treatment options, the clinical experts suggested that Altuviiio would likely be used as a first-line therapy for patients who desire to use FVIII replacement rather than FVIII mimetic therapy or as an alternative or complementary therapy to emicizumab. The clinical experts noted that with the availability of Altuviiio, there would be no need for SHL FVIII products as the same FVIII levels could be achieved with fewer doses of Altuviiio.
The clinical experts noted that patients on emicizumab who have experienced a suboptimal response due to breakthrough bleeding events or adverse events (AEs) like injection pain, or more rarely, neutralizing antibodies to emicizumab, and those on SHL or EHL FVIII prophylaxis who still struggle with the frequent dosing interval would be best suited for Altuviiio. In addition, patients participating in high-level physical activities may benefit from the sustained high FVIII activity level with improved bleed protection as would patients requiring surgery regardless of their current FVIII treatment. Conversely, both experts indicated that Altuviiio will not be suitable for patients who have developed inhibitors to FVIII.
The clinical experts noted that outcomes used in clinical practice are largely aligned with those used in the pivotal trials, particularly regarding annualized bleeding rate (ABR), which is a common trial end point. Other clinical trial outcomes including joint health, QoL (Haemophilia Quality of Life questionnaire for adults [Haem-A-QoL]), and FVIII activity levels, are also closely monitored in clinical practice. Both clinical experts indicated that treatment with Altuviiio will be discontinued if there is evidence of the development of FVIII inhibitors, no evidence of improvement in bleeding episodes, occurrence of AEs with treatment administration (allergy or anaphylaxis), or loss of IV access.
According to the clinical experts, treatment with Altuviiio should be primarily managed within a hemophilia treatment centre, where specialized hematologists and multidisciplinary teams can monitor treatment including pharmacokinetics (PK) testing, manage complications, and provide perioperative or periprocedural guidance.
Clinician Group Input
Three clinician groups, the Association of Hemophilia Clinic Directors of Canada (AHCDC; 5 clinicians contributed to the input), Canadian Association of Nurses in Hemophilia Care (CANHC; 6 clinicians contributed), and Canadian Physiotherapists in Hemophilia Care (CPHC; 5 clinicians contributed), provided input for this review. AHCDC gathered input through national advisory boards, expert opinions, and clinical trial experience with Altuviiio. Information from CANHC was provided by members who responded to the call for input while the submission from CPHC was gathered via information from clinician experience, conferences attended, and in-services.
Clinician groups noted that the ultimate treatment goal for patients with hemophilia A is to minimize the number of bleeds while slowing hemophilic arthropathy progression. With currently available treatments, achieving this goal requires frequent administration of high treatment doses to overcome short treatment half-lives. This treatment burden is particularly notable in patients who require elevated trough levels due to recent surgical procedures, compromised joint health, or high physical activity levels, according to clinician group input. Consistent with expert input, the clinician groups agreed that current therapies demonstrate variable efficacy.
Aligning with expert input, clinician groups noted that Altuviiio could be used as first-line therapy for patients aged 2 years or older with hemophilia A or offered as an alternative treatment to those receiving other therapies. Patients best suited for treatment with Altuviiio, as identified by clinician groups, were consistent with that of the clinical expert input. Additional patient populations who clinicians noted may benefit from Altuviiio treatment included patients with hemophilic arthropathy or poor venous access. In addition, clinician groups noted that patients with mild hemophilia A receiving on-demand or episodic therapy and those undergoing surgery or procedures may benefit from Altuviiio. The clinician groups indicated that patients least likely to benefit from Altuviiio are those who are averse to IV infusions, have developed FVIII inhibitors, or have achieved 0 bleeds on prophylaxis and who feel that switching therapies would have a minimal positive impact on QoL.
The clinician groups agreed with the consulted experts that the outcomes used in the trials to assess response are realistic for clinical practice, adding that patients should be assessed every 6 months to 2 years, depending on disease severity. CANHC noted that a clinically meaningful response to Altuviiio treatment would involve favourable PK profile (half-life improved to near-normal levels), an absence of FVIII inhibitors, absence of bleeding events, improved stable joint health, improved QoL, and infrequent hospitalizations. The clinician groups’ suggested criteria for discontinuation aligned with expert input. AHCDC and CANHC also suggested discontinuation if the patient switches to a nonfactor replacement therapy, other experimental therapies, or if the treatment centre is unable to perform the required clotting assay. The input received from the clinician group regarding Altuviiio prescribing considerations, including the follow-up of patients by a hemophilia clinic director, was consistent with the clinical expert inputs received for this review.
Drug Program Input
Input was obtained from the drug programs that participate in our reimbursement review process. Please refer to Table 5 for further information. The following were identified as key factors that could potentially impact the implementation of Altuviiio:
considerations for initiation of therapy
considerations for continuation or renewal of therapy
considerations for prescribing of therapy
generalizability.
Clinical Evidence
Systematic Review
Description of Studies
Two pivotal, phase III, open-label, nonrandomized, multicentre studies (XTEND-1 and XTEND-Kids) were included in the systematic literature review (SLR) conducted by the sponsor.
A total of 159 patients who were at least 12 years of age with severe hemophilia A without inhibitors were enrolled in the XTEND-1 trial (including 8 patients living in Canada from 2 study sites) and divided into 2 treatment groups: arm A (n = 133) and arm B (n = 26). Patients on a current FVIII prophylactic treatment regimen and who participated in an observational prestudy (242HA201/OBS16221) for at least 6 months before baseline of the XTEND-1 trial were assigned to arm A. Those on an on-demand treatment regimen for hemophilia A were assigned to arm B. Patients in arm A received a dose of 50 IU/kg once-weekly Altuviiio as prophylactic treatment for 52 weeks and those in arm B received Altuviiio 50 IU/kg as on-demand treatment of bleeding episodes for the first 26 weeks and then switched to the 50 IU/kg weekly prophylactic treatment regimen with Altuviiio for another 26 weeks. The primary objective of the XTEND-1 trial was to evaluate the efficacy of Altuviiio as a prophylactic treatment based on the ABR in arm A (described in the following). The key secondary end point was to evaluate the efficacy of Altuviiio as a prophylactic treatment based on the intrapatient comparison of ABR during the trial compared to the historical prophylaxis ABR in the 78 patients in arm A who participated in the observation study.
The XTEND-Kids trial included a total of 74 previously treated patients with severe hemophilia A who were aged younger than 12 years old and comprised 2 age cohorts: children aged less than 6 years (n = 38) and children aged between 6 years to 12 years (n = 36) (including 9 patients living in Canada from 4 study sites). All 74 patients received once-weekly IV doses of 50 IU/kg Altuviiio prophylactic treatment for 52 weeks. The primary objective of the XTEND-Kids trial was to evaluate the safety of Altuviiio in previously treated pediatric patients with severe hemophilia A based on occurrence of inhibitors. The key secondary end point was to evaluate the efficacy of Altuviiio as prophylactic treatment based on ABR, annualized joint bleeding rate (AjBR), joint health, and QoL outcomes.
In both trials, patients with a history of a positive inhibitor test result at screening (defined as ≥ 0.6 Bethesda units [BU]/mL at screening), those with serious active bacterial or viral infection within 30 days of screening, history of hypersensitivity or anaphylaxis associated with any FVIII product, or who had received emicizumab within the 20 weeks before screening were excluded. Both trials evaluated the safety, efficacy, and PK of Altuviiio administered IV once weekly as prophylactic or on-demand treatment in previously treated patients with severe hemophilia A without inhibitors.
The objective of both trials was to assess the safety and efficacy of Altuviiio to maintain hemostasis in the following settings: routine prophylaxis, control and prevention of bleeding, and perioperative management, as measured by ABR and AjBR; intrapatient comparison of ABR (only XTEND-1; participants served as their own controls); and development of FVIII inhibitors (only XTEND-Kids) at week 52 following Altuviiio infusion. Other efficacy and safety end points in both trials included joint health (Hemophilia Joint Health Score [HJHS]); Haem-A-QoL and Haemophilia Quality of Life questionnaire for children (Haemo-QoL); withdrawal due to AEs (WDAEs); treatment-emergent AEs (TEAEs); treatment-emergent serious AEs (TESAEs); deaths; and notable harms. In the XTEND-1 trial, efficacy end points were tested hierarchically to maintain the overall type I error rate of 0.05 or less. All analyses in the XTEND-Kids trial were descriptive in nature and adjustments for multiplicity were not applied.
In the XTEND-1 trial, the mean age of patients at baseline was 35.4 years (standard deviation [SD] = 15.1 years), ranging from 12 years to 72 years, and most patients (78.6%) had no family history of FVIII inhibitors. In the 12 months before the study, the mean number of bleeding episodes reported was 3.2 (SD = 5.4) in arm A patients who all previously received a different prophylaxis regimen and 35.7 (SD = 22.2) in arm B patients who previously received on-demand treatment. In the XTEND-Kids trial, the mean age at baseline was 5.99 years (SD = 2.91 years; ages ranged from 1.4 to 11.0 years), the majority (77%) had no family history of FVIII inhibitor, and the mean bleeding episodes in patients on a prophylactic regimen before the study was 2.1 (SD = 4.2). The XTEND-1 study was completed on February 3, 2022, and the XTEND-Kids trial was completed on January 18, 2023. There were no reported important protocol deviations that could potentially influence the efficacy results in either the XTEND-1 or XTEND-Kids studies.
Efficacy Results
XTEND-1 Trial
Bleeding Outcomes
Annualized bleeding rate: The primary efficacy end point in the XTEND-1 trial was ABR in arm A (prophylaxis arm) assessed following 52 weeks of Altuviiio for prophylactic use. In the full analysis set (FAS), a total of 86 bleeding episodes were treated with Altuviiio in 133 patients in arm A during the efficacy period. The median ABR at week 52 was 0.00 (interquartile range [IQR], 0.00 to 1.04) and the mean ABR was 0.71 (95% confidence interval [CI], 0.52 to 0.97). In arm A, 131 (98.5%) patients had 5 or fewer bleeding episodes per year and 86 (64.7%) patients had no bleeding episodes during the study (Table 15). Sensitivity analyses were consistent with those of the primary analysis.
Annualized joint bleeding rate: Results for AjBR were consistent with the results for ABR. In arm A, 37 patients in the FAS had a total of 61 treated joint bleeds. The estimated mean AjBR at week 52 was 0.51 (95% CI, 0.36 to 0.72). Of the 133 patients in arm A, 131 (98.5%) patients had an AjBR of 5 or fewer episodes per year with 96 (72.2%) patients with no joint bleeds during the study.
In arm B, estimated mean AjBR at week 52 was 17.48 (95% CI, 14.88 to 20.54). The mean AjBR in arm B was similar to arm A after patients had switched to prophylactic treatment: mean exposure days were 0.62 (95% CI, 0.25 to 1.52). In an intrapatient comparison of AjBR in arm B, the joint bleeding rate ratio for prophylaxis versus on-demand treatment was 0.04 (95% CI, 0.01 to 0.08).
Intrapatient comparison of ABR between Altuviiio prophylaxis versus historical prophylaxis: Overall, the number of patients with ABR of 0 who had historical prophylaxis or received Altuviiio was 42.3% and 64.1%, respectively. In the FAS (N = 78), intrapatient comparison in arm A showed a mean ABR reduction of 77% (ABR ratio = 0.23; 95% CI, 0.13 to 0.42; P < 0.0001) in the efanesoctocog prophylaxis group compared to historical prophylaxis.
For the 26 patients in arm B, the bleeding rate ratio for prophylaxis versus on-demand treatment was 0.03 (95% CI, 0.02 to 0.07). With on-demand treatment, most patients (96.2%) had an ABR greater than 10, whereas most patients (76.9%) had no bleeds after switching to prophylactic treatment.
Physical Functioning and Pain (QoL)
Haem-A-QoL Physical Health score and Haemo-QoL score: In the XTEND-1 study, QoL data were collected in adult patients aged 17 years or older via the Haem-A-QoL Physical Health score and in adolescent patients aged 12 years to 16 years via the Haemo-QoL. In arm A, for patients aged 17 years or older (n = 98), the estimated mean change from baseline to week 52 in Haem-A-QoL Physical Health score was –6.74 (95% CI, −10.13 to –3.36; P = 0.0001). The Haemo-QoL results in the study’s adolescent population (all in arm A) mirrored those of the group aged 17 years or older, with improvements in Haemo-QoL Physical Health score (mean change from baseline to week 52 of −2.18 [SD = 22.05]) and total score (−3.45 [SD = 8.83]), in the group aged 13 years to 16 years (n = 18). In arm B, a mean change in Haem-A-QoL Physical Health score of –25.91 (SD = 22.29) by week 52 was reported. A sensitivity analysis performed for patients aged 17 years or older in arm A who had rolled over from the OBS16221 study (n = 66) also showed an improvement in Haem-A-QoL Physical Health score (least squares [LS] mean change from baseline to week 52 = −4.04; 95% CI, −8.06 to −0.03).
Patient-Reported Outcomes Measurement Information System (PROMIS) pain intensity and physical function: Item 3a of the PROMIS instrument assessed a patient’s worst pain in the last 7 days. This item was used to assess pain intensity in the XTEND trials. In arm A, in participants aged 12 years or older, the estimated mean change from baseline to week 52 in pain intensity was a difference in score of −0.21 (95% CI, −0.41 to −0.02; P = 0.0276). In arm B, the mean change from baseline to week 52 pain intensity was a difference in score of −0.77 (SD = 0.81).
The PROMIS instrument was also used to assess physical function in adult patients only (aged ≥ 18 years). In arm A, of 108 patients, 103 completed the PROMIS Short Form Physical Function questionnaire at baseline and 102 at week 52. The mean change in Physical Health score was 46.80 (SD = 8.82) at baseline to 47.35 (SD = 9.28) with a mean change from baseline to week 52 of 0.62 (SD = 4.77).
Joint Health
Hemophilia Joint Health Score: In arm A, the mean HJHS total score at baseline was 18.1 (SD = 18.4). The estimated mean change in the HJHS total score from baseline to week 52 was −1.54 (95% CI, −2.70 to −0.37; P = 0.0101). In arm B, the mean change from baseline to week 52 in HJHS total score was −4.1 (SD = 8.7). A sensitivity analysis performed using the data of patients in arm A who rolled over from the prospective observational OBS16221 study also showed an improvement in HJHS total score. The LS mean change from baseline to week 52 was −0.86 (95% CI, −2.38 to 0.66).
Perioperative Management Outcomes
Number of injections and dose to maintain hemostasis during major surgery: In the XTEND-1 study, 11 out of 12 major surgeries that occurred during the treatment regimen required a single injection of Altuviiio (i.e., the preoperative loading dose) to maintain hemostasis. The mean dose per injection was 41.65 IU/kg (SD = 15.21 IU/kg). For 1 surgery, conducted during routine prophylaxis, no preoperative loading dose was reported on the day before or the day of the surgery.
XTEND-Kids Trial
Inhibitor Development to FVIII
The primary end point of the XTEND-Kids study was the occurrence of inhibitor development against FVIII based on all patients who had reached at least 50 exposure days. Overall, 65 patients who had reached at least 50 exposure days were analyzed for inhibitors. The incidences of inhibitor development to FVIII were 0.0% (95% CI, 0.0 to 5.5) in patients with 50 exposure days or more to Altuviiio and 0.0% (95% CI, 0.0 to 4.9) in all treated patients.
Bleeding Outcomes
Annualized bleeding rate: The overall mean ABR at week 52 was 0.89 (95% CI, 0.56 to 1.42) and a median ABR was 0 (IQR, 0 to 1.02). Of the 74 patients, 47 (63.5%) had an ABR of 0, and 25 (33.8%) had an ABR of greater than 0 to 5 at 52 weeks. A total of 64 bleeding episodes were treated with Altuviiio in 27 of the 74 patients. Results of sensitivity analyses based on mean ABR at 52 weeks on the per-protocol set (PPS) or mean ABR on FAS including patients with data at week 26 were consistent with the primary analysis.
Annualized joint bleeding rate: The overall estimated mean AjBR was 0.59 (95% CI, 0.27 to 1.28), with 0.19 (95% CI, 0.06 to 0.62) in the cohort aged younger than 6 years, and 0.99 (95% CI, 0.38 to 2.60) in the cohort aged 6 years to younger than 12 years. Of the 74 patients who were included in the analysis, 61 (82.4%) patients reported no joint bleeds, while 12 (16.2%) patients reported 1 to 5 joint bleeds. One (1.4%) patient had 21 joint bleeds as per analysis, 18 of which were not confirmed by the investigator nor reported by the patient. A sensitivity analysis excluding the participant who did not receive the weekly prophylactic treatment for an extended period of time showed that the estimated mean AjBR in the cohort aged 6 years to younger than 12 years decreased to 0.41 (95% CI, 0.19 to 0.89) and the overall estimated mean AjBR to 0.30 (95% CI, 0.16 to 0.57).
Physical Functioning and Pain (QoL)
Haemo-QoLPhysical Health score and Haem-A-QoL score: For patients aged 4 years to 7 years, 8 years to younger than 12 years, and their respective caregivers, data were collected using 4 separated Haemo-QoL. For patients between the age of 4 years and 7 years, the mean change from baseline to week 52 was –5.31 (SD = 10.83) in the cohort aged younger than 6 years and 4.69 (SD = 5.41) in the cohort aged 6 years to younger than 12 years. In patients aged 4 years to 7 years, overall, the mean change from baseline to week 52 was −2.46 (SD = 10.49). Parents of children between the age of 4 years and 7 years were also asked to complete the Haemo-QoL for a parent-proxy assessment of this outcome. For the overall group, the mean change from baseline based on the parent proxy was –2.85 (SD = 11.82), which is aligned with the patient-reported results. For patients aged 8 years and older, the mean change from baseline to week 52 was –9.79 (SD = 12.18).
PROMIS Pain Intensity and Physical Function: Similar to the XTEND-1 trial, pain intensity was assessed in the XTEND-Kids study using item “a” of the PROMIS pediatric instrument as a change from baseline to week 52. For patients between the age of 5 years and 12 years, a parent or caregiver response was used as a proxy for the child. In the cohort of patients aged less than 6 years, the mean change from baseline was –0.44 (SD = 2.65) and for patients between the age of 6 years and 12 years, the mean change from baseline was −0.75 (SD = 2.53). Overall, the mean change in scores form baseline was –0.62 (SD = 2.52). Patients between the age of 8 years and 12 years responded to this outcome independently. For patients aged 8 years or older in the cohort aged 6 years to 12 years, the mean change from baseline was 0.00 (SD = 2.98).
In the cohort aged younger than 6 years, 8 parents of participants 5 years of age or older completed the PROMIS Short Form Physical Function questionnaire at baseline, and 8 parents at week 52. The mean change from baseline to week 52 in the cohort aged younger than 6 years was 3.96 (SD = 6.73; n = 7). In the cohort aged 6 years to younger than 12 years, 14 participants aged 8 years or older completed the PROMIS Short Form Physical Function questionnaire at baseline, and 16 participants at week 52. The mean change from baseline to week 52 was 0.78 (SD = 10.48; n = 10). In the cohort aged 6 years to younger than 12 years, 16 parents of participants aged younger than 12 years completed the questionnaire at baseline, and 16 parents at week 52. The mean change from baseline to week 52 was −1.36 (SD = 12.15; n = 10).
Joint Health
Hemophilia Joint Health Score: In the cohort aged younger than 6 years, 20 patients were aged 4 years or older and the mean change in HJHS total score from baseline to week 52 was 0.2 (SD = 8.3). In the cohort aged 6 years to younger than 12 years, the mean change in HJHS total score from baseline to week 52 was −1.1 (SD = 4.3) in 33 patients.
Perioperative Management Outcomes
Number of injections and dose to maintain hemostasis during major surgery: In the XTEND-Kids study, both major surgeries required a single injection of Altuviiio to maintain hemostasis. The mean dose per injection was 61.13 IU/kg (SD = 1.06 IU/kg).
Harms Results: AEs
XTEND-1: Of the 159 patients in the safety analysis set, 123 (77.4%) patients experienced at least 1 TEAE, resulting in a total of 394 TEAEs in the study. The most frequently reported TEAEs in more than 3% of patients were headache (20.1%); arthralgia (16.4%); fall (6.3%); back pain (5.7%); COVID-19 and fatigue (4.4% each); contusion, hemophilic arthropathy, and nasopharyngitis (3.8% each); and joint injury, pain in extremity, and toothache (3.1% each). Of the 159 patients, 77 (48.4%) patients had no TEAEs classified as moderate or severe but at least 1 TEAE that was classified as mild. In addition, 39 (24.5%) patients had no TEAEs classified as severe but at least 1 TEAE was classified as moderate, and 7 (4.4%) patients had at least 1 TEAE classified as severe.
XTEND-Kids: Of the 74 patients in the safety analysis set, 62 (83.8%) experienced at least 1 TEAE, resulting in a total of 255 TEAEs. The most frequently reported TEAEs (> 5% of patients overall) were SARS-CoV-2 test positive and upper respiratory tract infection (14.9% each); pyrexia (12.2%); asymptomatic COVID-19 (9.5%); gastroenteritis viral, head injury, and nasopharyngitis (8.1% each); arthralgia, pain in extremity, and vomiting (6.8% each); and contusion, diarrhea, viral infection, and viral upper respiratory tract infection (5.4% each). The majority of TEAEs were assessed by the investigator as mild in severity. Of the 74 patients, 43 (58.1%) had at least 1 TEAE of mild intensity and 13 (17.6%) patients had at least 1 TEAE of moderate intensity.
Harms Results: Serious AEs
XTEND-1: A total of 18 TESAEs were experienced in 15 (9.4%) patients, of which 16 TESAEs were reported in 13 patients in arm A and 2 TESAEs in 2 patients in arm B. Hemophilic arthropathy was the most commonly reported serious adverse event (SAE), which was reported in 2 (1.3%) patients in arm A. All other TESAEs were reported in 1 (0.6%) patient each. The majority of TESAEs were assessed by the investigator as mild to moderate in severity.
XTEND-Kids: A total of 10 TESAEs were experienced in 9 (12.2%) patients. The majority of TESAEs were assessed by the investigator as mild to moderate in severity. The 5 TESAEs assessed by the investigator as severe were TESAEs of circumcision and bacteremia, each in 1 patient aged less than 6 years and TESAEs of vascular device occlusion, head injury, and eosinophilic esophagitis, each in 1 patient aged 6 to less than 12 years.
Withdrawals Due to AEs
XTEND-1: Two TEAEs in 2 (1.3%) patients resulted in permanent treatment discontinuation. The reason for WDAE was due to a decrease in CD4 lymphocytes in 1 patient with a history of HIV infection (reported as a TESAE), and due to a combined tibia–fibula fracture in the other patient who withdrew due to an AE.
XTEND-Kids: No patients discontinued Altuviiio treatment due to a TEAE during the study.
Mortality
XTEND-1: Death was reported in 1 patient overall who was in arm B. The patient had a medical history of hepatitis C and died of metastatic pancreatic carcinoma, which was reported as a TESAE. The TESAE was assessed by the investigator as not related to Altuviiio treatment.
XTEND-Kids: There were no deaths reported during the study.
AEs of Special Interest
XTEND-1: There were no reports of inhibitor development to FVIII nor thromboembolic events during the study.
XTEND-Kids: An event of “hives around eyes, mouth, face, and chest” was reported in a 2-year-old patient after “eating chocolate.” This patient had no history of allergies at baseline. The event occurred approximately 3 months after the first dose of Altuviiio (weekly prophylaxis) and 3 days after the last injection. There were no reports of thromboembolic events during the study.
Critical Appraisal
The 2 pivotal trials (XTEND-1 and XTEND-Kids) included in the sponsor’s SLR were phase III, single-arm, open-label clinical trials. Although nonrandomized, open-label, single-arm design limits the interpretation of the efficacy results for both pivotal trials, the clinical experts consulted by Canada’s Drug Agency (CDA-AMC) for this review indicated that while traditional randomized controlled trials (RCTs) remain the gold standard for many conditions, these are not feasible in hemophilia A due to ethical constraints, challenges in patient recruitment, and the availability of effective treatments. According to the clinical experts, alternative designs like intrapatient comparisons and historical controls provide practical evaluation of new therapies such as Altuviiio. It was noted that participants in both trials were previously treated patients with severe hemophilia A without inhibitors and in particular, 92 patients (n = 82 in arm A and n = 10 in arm B) in the XTEND-1 trial were previously enrolled in an observational prestudy (242HA201/OBS16221). This was determined by CDA-AMC to be a potential selection bias. Additionally, although the sponsor provided data on the baseline characteristics of all participants in the prestudy observational study, the baseline clinical characteristics specific to the patients who continued into the XTEND-1 study from the observational study were not provided. CDA-AMC notes that this limits the ability to identify preexisting differences, potentially introducing bias and confounding, although the clinical experts indicated that the rolled over patients and those in the XTEND-1 study were likely similar and were not systematically different based on the baseline characteristics for the overall group.
In both trials, bleeding outcomes were measured using ABR and AjBR, both of which are widely accepted end points in hemophilia research that provides an objective assessment of bleeding outcomes. Joint health was measured using HJHS, which is a validated outcome measure but is subject to potential bias particularly due inter-rater variability. Additionally, although the study design was deemed appropriate for data collection across varied populations, the lack of blinding introduces potential bias, as knowledge of treatment assignment may influence reporting on subjective or patient-reported outcomes such as, HRQoL, physical function, and pain outcomes (outcomes related to the Haem-A-QoL and PROMIS instruments). As such, reliable assessments of these outcomes could not be made and there is potential for risk of bias that could lead to the overestimation of the treatment effect of Altuviiio.
Both trials appear to be adequately powered for assessing ABR and joint health outcomes; however, smaller subgroup analyses, such as surgery, perioperative management, or specific age groups, may not be fully powered to detect AEs or efficacy. Both trials included follow-up safety assessments for a few weeks after the last dose, but the duration of the XTEND-1 and XTEND-Kids trials was considered too short to sufficiently evaluate delayed adverse effects and assess the long-term safety of Altuviiio.
While the XTEND-1 study included a historical control through intrapatient comparison with patients’ prior prophylactic regimens in a previous study, this approach lacks randomization, is affected by temporal trend, and is prone to measurement bias, making causal inferences less robust compared to a concurrent randomized control group. Additionally, CDA-AMC notes that the reliance on historical data may introduce variability due to changes in patients' current conditions or other external factors unrelated to treatment efficacy such as carryover effects, making causal inferences less robust. Specifically, the XTEND-1 trial was conducted during the COVID-19 pandemic while the observational prestudy was conducted a few years before the pandemic (patients in these studies were enrolled between 2009 to 2017) (Table 23). Therefore, impact from possible change in physical activities related to social distancing which was required on many occasions during the pandemic (e.g., intensity, types of physical activities) on the risk of bleeding in patients with hemophilia is uncertain. In both trials, perioperative management outcomes were assessed descriptively based on the rating of hemostatic response on a 4-point ordinal scale performed 24 hours after the surgery by the surgeon or study investigator, as well as the number of injections and mean dose needed to maintain hemostasis per major surgery. CDA-AMC notes that while this subjective assessment is aligned with how this outcome would be assessed in clinical practice, it is likely subject to bias, especially given that the assessments were performed by those involved in the study.
CDA-AMC identified several considerations related to the generalizability of the XTEND trials in evaluating the efficacy and safety of Altuviiio. The trials enrolled patients from 6 study sites in Canada and included both adults and children with severe hemophilia A, which enhances the generalizability of the findings. In contrast, the results from the 2 trials may have limited generalizability as the study population was restricted to patients (without inhibitors) with severe hemophilia A. The clinical experts consulted for this review indicated that there is a subset of patients with mild or moderate hemophilia who may require prophylaxis. The design of the XTEND-1 and XTEND-Kids studies did not include these patients and therefore, the magnitude of the treatment effect in patients with mild and moderate hemophilia A is unclear. According to the clinical experts, the once-weekly dosing of 50 IU/kg used in the trials reflects what is expected in clinical practice. However, specific subgroups, such as patients with obesity or those participating in higher-risk physical activity may require adjusted dosing, which was not explored in the trials, and this limits the generalizability of the results to these populations. Although the clinical experts indicated the difficulty in the direct comparison of efanesoctocog with current standard of care, CDA-AMC notes that the lack of direct head-to-head comparison with the current standard of care, such EHL FVIII products or nonfactor therapies like emicizumab, limits external validity regarding the effectiveness and safety of Altuviiio compared to currently available therapies.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, Grading of Recommendations, Assessment, Development and Evaluations (GRADE) assessment was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.14,15
Although GRADE guidance is not available for noncomparative studies, the CDA-AMC review team assessed the 2 single-arm trials for study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias to present these important considerations. Because the lack of a comparator arm does not allow for a conclusion to be drawn on the effect of the intervention versus any comparator, the certainty of evidence for single-arm trials started at very low certainty with no opportunity for rating up.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
Results of GRADE Assessments
Table 2 and Table 3 present the GRADE summary of findings for Altuviiio from the XTEND-1 and XTEND-Kids trials for routine prophylaxis to reduce the frequency of bleeding episodes, on-demand treatment and control of bleeding episodes, and perioperative management of bleeding in adults and children with hemophilia A. The selection of outcomes for the GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
ABR
AjBR
intrapatient comparison of ABR
FVIII inhibitor formation
HJHS
physical functioning and pain outcome (Haem-A-QoL, Haemo-QoL)
perioperative management outcome (mean number of injections to maintain hemostasis during major surgery)
harms (TESAEs, TEAEs, mortality)
Long-Term Extension Studies
Description of Studies
One long-term extension (LTE) study was submitted for review, XTEND-ed (NCT04644575).18 The XTEND-ed LTE study is an ongoing phase III, open-label, multicentre study to assess long-term safety and efficacy of Altuviiio in previously treated patients with severe hemophilia A. The study began in February 2021 and is estimated for completion in 2027.18 At the time of this submission, the available evidence was limited to interim analyses based on conference presentations.19,20 The submitted interim analyses pertain only to patients rolled over from the XTEND-1 and XTEND-Kids studies into arm A of the XTEND-ed LTE study and reports on efficacy and safety-related outcomes over 2 additional years of treatment with Altuviiio.
Table 2: Summary of Findings for the Efficacy and Safety of Altuviiio for Adults With Hemophilia A (XTEND-1 Trial)
Outcome and follow-up | Patients, N (studies) | Effect | Certaintya | What happens |
|---|---|---|---|---|
Bleeding outcomes | ||||
ABR: treated bleeding episodes per year Follow-up: 121.2 total patient-years | 133 (1 single-arm trial with intrapatient comparison) | ABR (single-arm analysis):
ABR (intrapatient comparison):
| Lowb | Altuviiio may result in an improved ABR compared to historical prophylaxis (other marketed standard of care factor VIII prophylaxis) although the evidence is still uncertain. |
AjBR: treated joint bleeding episodes per year Follow-up: 121.2 total patient-years | 133 (1 single-arm trial) |
| Very lowc | The evidence is very uncertain about the effect of Altuviiio on AjBR compared to any comparator. |
Joint health | ||||
HJHS: change from baseline in total score Score: 0 (best) to 124 (worst) Follow-up: 52 weeks | 133 (1 single-arm trial) |
| Very lowd | The evidence is very uncertain about the effect of Altuviiio on HJHS compared to any comparator. |
Physical function and pain (QoL) | ||||
Haem-A-QoL: change from baseline in physical health score Total score: 0 (best) to 100 (worst) Follow-up: 52 weeks | 133 (1 single-arm trial) |
| Very lowd | The evidence is very uncertain about the effect of Altuviiio on Haem-A-QoL compared to any comparator. |
PROMIS Pain Intensity: change from baseline in worst pain intensity in the past 7 days Score: 0 (no pain) to 5 (very severe) Follow-up: 52 weeks | 133 (1 single-arm trial) |
| Very lowd | The evidence is very uncertain about the effect of Altuviiio on PROMIS Pain Intensity compared to any comparator. |
Perioperative management | ||||
Perioperative management: number of major surgeries with hemostatic response rated as excellent or good by investigator Follow-up: 52 weeks | 133 (1 single-arm trial) | Number of major surgeries with hemostatic response rated as excellent or good by investigator: 12 (100%) | Very lowe | The evidence is uncertain about the effect of Altuviiio for the perioperative management of bleeding in adults with hemophilia A compared to any comparator. |
Harms | ||||
TESAEs Follow-up: 52 weeks | 133 (1 single-arm trial) | Number of patients with ≥ 1 TESAE = 98 per 1,000 | Very lowf | The evidence is very uncertain about the effect of Altuviiio on the risk of TESAE compared to any comparator. |
ABR = annualized bleeding rate; AjBR = annualized joint bleeding rate; CI = confidence interval; Haem-A-QoL = Haemophilia Quality of Life questionnaire for adults; HJHS = Hemophilia Joint Health Score; PROMIS = Patient-Reported Outcomes Measurement Information System; QoL = quality of life; SD = standard deviation; TESAE = treatment-emergent serious adverse event.
Notes: All serious concerns with study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias are documented in the table footnotes.
aIn absence of a comparator arm, conclusions about efficacy relative to any comparator cannot be drawn and certainty of evidence started at very low for all end points. In addition, all outcomes were rated down 1 level for indirectness due to the exclusion of patients with mild or moderate hemophilia A.
bDespite the study limitations resulting in the certainty of evidence starting as “very low,” the CDA-AMC review team considered the strength of evidence sufficient to rate up 1 level to “low” based on the proportion of patients with an ABR of 0 (i.e., no bleeds) reported during the trial, a mean ABR < 1 that was considered as clinically meaningful by the clinical experts consulted for this review, and an intrapatient comparison suggestive of an improvement in ABR compared to prior historical prophylactic treatment.
cRated down 1 level for serious study limitations: risk of bias due to the nonrandomized study design.
dRated down 1 level for serious study limitations: risk of bias due to the nonrandomized, open-label study design.
eRated down 1 level for serious study limitations due to risk bias in measurement of the outcome because of the nonrandomized, open-label study design. Rated down 1 level due to imprecision due to insufficient sample size.
fRated down 1 level due to imprecision due to insufficient sample size.
Source: XTEND-1 Clinical Study Report.16 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 3: Summary of Findings for the Efficacy and Safety of Altuviiio for Children With Hemophilia A (XTEND-Kids Trial)
Outcome and follow-up | Patients, N (studies) | Effect | Certaintya | What happens |
|---|---|---|---|---|
Inhibitor formation | ||||
Factor VIII inhibitor formation: occurrence of neutralizing antibodies (inhibitor result of at least 0.6 BU/mL) Follow-up: 52 weeks | 74 (1 single-arm trial) |
| Very lowb | The evidence is uncertain about the effect of Altuviiio on the development of factor VIII inhibitors compared to any comparator. |
Bleeding outcomes | ||||
ABR: treated bleeding episodes per year Follow-up: 70.6 patient-years | 74 (1 single-arm trial) |
| Very lowc | The evidence is uncertain about the effect of Altuviiio on ABR compared to any comparator. |
AjBR: treated joint bleeding episodes per year Follow-up: 70.6 patient-years | 74 (1 single-arm trial) |
| Very lowc | The evidence is very uncertain about the effect of Altuviiio on AjBR compared to any comparator. |
Joint health | ||||
HJHS: change from baseline in total score Score: 0 (best) to 124 (worst) Follow-up: 52 weeks | 74 (1 single-arm trial) | Mean change from baseline to week 52 = –0.6 (SD = 6.0) | Very lowd | The evidence is very uncertain about the effect of Altuviiio on HJHS compared to any comparator. |
Physical function and pain (QoL) | ||||
Haemo-QoL: change from baseline in physical health score Total score: 0 (best) to 100 (worst) Follow-up: 52 weeks | 74 (1 single-arm trial) | Mean change from baseline to week 52:
| Very lowe | The evidence is very uncertain about the effect of Altuviiio on Haem-A-QoL compared to any comparator. |
PROMIS Pediatric Pain Intensity: change from baseline in worst pain intensity in the past 7 days Score: 0 (no pain) to 10 (worse pain) Follow-up: 52 weeks | 74 (1 single-arm trial) | Mean change from baseline to week 52:
| Very lowe | The evidence is very uncertain about the effect of Altuviiio on PROMIS Pediatric Pain Intensity compared to any comparator. |
Perioperative management | ||||
Perioperative management: number of major surgeries with hemostatic response rated as excellent or good by investigator Follow-up: 52 weeks | 74 (1 single-arm trial) | Number of major surgeries with hemostatic response rated as excellent or good by investigator = 2 (100%) | Very lowf | The evidence is uncertain about the effect of Altuviiio for the perioperative management of bleeding in children with hemophilia A compared to any comparator. |
Harms | ||||
TESAEs Follow-up: 52 weeks | 74 (1 single-arm trial) | Number of patients with ≥ 1 TESAE = 122 per 1,000 | Very lowg | The evidence is very uncertain about the effect of Altuviiio on the risk of TESAE outcomes compared to any comparator. |
ABR = annualized bleeding rate; AjBR = annualized joint bleeding rate; BU = Bethesda unit; CI = confidence interval; Haem-A-QoL = Haemophilia Quality of Life questionnaire for adults; HJHS = Hemophilia Joint Health Score; PROMIS = Patient-Reported Outcomes Measurement Information System; QoL = quality of life; SD = standard deviation; TESAE = treatment-emergent serious adverse event.
Note: All serious concerns with study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias are documented in the table footnotes.
aIn absence of a comparator arm, conclusions about efficacy relative to any comparator cannot be drawn and certainty of evidence started at very low for all end points. In addition, all outcomes were rated down 1 level for indirectness due to the exclusion of patients with mild or moderate hemophilia A.
bRated down 1 level for serious study limitations due to risk of bias due to missing data and nonrandomized study design. Of note, this outcome was the primary end point for XTEND-Kids and assessed as a safety end point. It was reported descriptively and not adjusted for multiple comparisons.
cRated down 1 level for serious study limitations: risk of bias due to the nonrandomized study design.
dRated down 1 level for serious study limitations: risk of bias due to the nonrandomized and missing data.
eRated down 1 level for serious study limitations: risk of bias due to the nonrandomized, open-label study design.
fRated down 1 level for serious study limitations due to risk bias in measurement of the outcome because of the nonrandomized, open-label study design. Rated down 1 level due to imprecision due to insufficient sample size.
gRated down 1 level due to imprecision due to insufficient sample size.
Source: XTEND-Kids Clinical Study Report.17 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Efficacy Results
In the first 2 years of the XTEND-ed study, the mean overall ABR was 0.72 (SD = 1.26) for patients in arm A of the XTEND-1 study (prophylaxis arm), 0.42 (SD = 0.89) for patients in arm B of the XTEND-1 study (on-demand switch to prophylaxis), and 0.70 (SD = 1.27) for patients from the XTEND-Kids study. Mean ABR in the XTEND-Kids study was also comparable across age groups, between patients aged younger than 6 years (ABR = 0.63; SD = 1.18) and aged 6 years to 12 years (ABR = 0.77; SD = 1.37).
Harms Results
As of the XTEND-ed study’s interim analysis cut-off date, 74% of patients from the XTEND-1 study group had at least 1 TEAE and 12% had at least 1 serious TEAE. The most common TEAEs (in > 5% of patients) included COVID-19 (22%), arthralgia (13%), headache (9%), nasopharyngitis (8%), and influenza (6%). Two patients discontinued therapy due to TEAEs.
In the XTEND-Kids study group, 61% of patients experienced at least 1 TEAE and 3% had at least 1 serious TEAE. The most common TEAEs (in > 5% of patients) included pyrexia (9%), arthralgia (7%), cough (7%), upper respiratory tract infection (6%), viral upper respiratory tract infection (6%), and oropharyngeal pain (6%). There were no treatment discontinuations in this group due to TEAEs.
As of the XTEND-ed study’s interim analysis cut-off date, there was no development of FVIII inhibitors in either group and no deaths were reported.
Critical Appraisal
The XTEND-ed LTE study was designed as an open-label extension to assess long-term efficacy and safety of Altuviiio for the treatment of patients with hemophilia A. This open-label design could bias the magnitude of treatment effect for subjective efficacy outcomes and reporting of safety parameters due to unblinded exposure to the study medication during the treatment period. Statistical hypothesis testing was not part of the design and there was no active comparator or placebo arm. The mean treatment duration in the XTEND-Kids study group was less than half of that of the XTEND-1 study group, 36.2 weeks and 82.5 weeks, respectively. Clinical experts noted that while 36 weeks is likely sufficient to assess treatment efficacy, more time is needed to evaluate long-term safety outcomes, such as inhibitor development.
The XTEND-ed arm A study population for this interim analysis consisted of patients who took part in the XTEND-1 and XTEND-Kids studies, and therefore it is reasonable to expect that the same strengths and limitations related to generalizability apply to the LTE. Given that patients needed to complete the XTEND-1 or XTEND-Kids studies before enrolling, the LTE population is inherently enriched and introduces some selection bias for responders.
Indirect Comparisons
Description of Studies
In the absence of head-to-head evidence comparing Altuviiio to other relevant therapies used to manage hemophilia A, the sponsor submitted 1 indirect treatment comparison (ITC) report21 comparing relative treatment effects of Altuviiio versus relevant comparator therapies as prophylactic treatment for adult patients with severe hemophilia A. The ITC report included 2 matching-adjusted indirect comparisons (MAICs) for comparing Altuviiio with a nonfactor replacement therapy agent (emicizumab) or an SHL product (octocog alfa), and 1 analysis using a propensity score matching (PSM) method for comparing Altuviiio with an EHL agent (efmoroctocog alfa). Outcome measures assessed in this ITC included ABRs for any bleeding, spontaneous bleeding, and joint bleeding.
Efficacy Results
Compared to Emicizumab
Emicizumab once weekly was assessed in 63 patients in arm D of the HAVEN 3 trial and 119 patients in arm A of the XTEND-1 trial. The estimated effective sample size (ESS) for arm A in the XTEND-1 study was reduced from 119 patients to 76 patients following matching, which corresponded to 63.8% of the original sample.
Compared to emicizumab once weekly, treatment with Altuviiio was associated with lower rate of any bleeding (treated and untreated) (incidence rate ratio [IRR] = 0.32; 95% CI, 0.19 to 0.56), treated spontaneous bleeding (IRR = 0.62; 95% CI, 0.25 to 1.50), and treated joint bleeding (IRR = 0.48; 95% CI, 0.24 to 0.95).
Compared to Octocog Alfa
Octocog alfa was assessed on 62 patients in arms A and B of the LEOPOLD I trial and 159 patients in pooled arms A and B of the XTEND-1 trial. Baseline characteristics of the XTEND-1 study pooled arms were adequately matched to aggregated data from the LEOPOLD I study arms A and B. The estimated ESS was reduced from 128 patients to 29 patients following matching, which corresponded to 22.7% of the original sample.
Compared to octocog alfa, treatment with Altuviiio was associated with lower rate of any bleeding (mean difference [MD] = –2.97; 95% CI, –4.28 to −1.67), spontaneous bleeding (MD = –2.23; 95% CI, –3.10 to –1.35), and joint bleeding (MD = –2.67; 95% CI, –3.85 to –1.49).
Compared to Efmoroctocog Alfa
Efmoroctocog alfa was assessed on 117 patients with individualized prophylaxis data in the A-LONG trial and 159 patients in the pooled arms A and B of the XTEND-1 trial. The estimated ESS for the XTEND-1 study was reduced from 145 patients to 87 patients following matching which corresponded to 60% of the original sample, and for the A-LONG study individual patient data were reduced from 116 patients to 30 patients following matching which corresponded to 26% of the original sample.
Compared to efmoroctocog alfa, treatment with Altuviiio was associated with lower frequency of any treated bleeding (IRR = 0.29; 95% CI, 0.17 to 0.51), spontaneous bleeding (IRR = 0.21; 95% CI, 0.09 to 0.49), and joint bleeding (IRR = 0.37; 95% CI, 0.20 to 0.71).
Harms Results
Harms outcomes were not assessed in these analyses.
Critical Appraisal
In this ITC, unanchored MAIC or a PSM method was used in balancing the baseline characteristics between the included trials. In the MAICs, these potential effect modifiers or prognostic factors were adjusted for if adequate data were reported in the comparator studies: age, body weight, race, prior treatment regimen, prior frequency of bleeding, presence of targeted joints, comorbidities, and baseline patient-reported outcome values. The clinical experts consulted for this review agreed that these are relevant effect modifiers and prognostic variables and also noted that physical activity level at baseline is an important factor in result interpretation. In addition, the use of historical control for intrapatient comparison of ABR in the XTEND-1 study may introduce variability due to changes in patients’ characteristics or external factors including temporal events unrelated to treatment efficacy. For example, the XTEND-1 trial coincided with the COVID-19 pandemic, where changes in the level of physical activity before or during the COVID-19 pandemic may have affected patients’ risk of bleeding due to changes in lifestyle and behaviour related to physical activity. As such, there is potential for risk of bias in the included studies due to potential confounding by the heterogeneity in physical activity level at baseline, and the time that patients were treated and evaluated (prepandemic versus during pandemic); however, the direction of bias is unclear, and the clinical experts consulted by CDA-AMC did not expect this to significantly impact the results. Furthermore, the clinical experts consulted for this review noted that clinical practice and management of patients with hemophilia A have evolved considerably in the past 10 years. For example, there has been an increase in the use of factor prophylaxis in patients of all ages and disease severities, and factor prophylaxis dosing and frequency are tailored based on patient’s own PK profile, bleeding profile, activity levels, and potential impact of a bleeding event. Further, the risk of severe bleeding in patients with factor levels indicative of mild to moderate disease range is recognized and such patients can still benefit from prophylactic treatment (either factor or nonfactor). In addition, many clinicians in Canada have adopted the WFH clinical practice guidelines. All these changes have not been considered in the ITC analyses. Therefore, the study results may be biased.
In the MAIC and PSM analyses, the reduction in ESS after the weighting process ranged from 36% to 77% of the original sample size in the included studies. A significant reduction in sample size can contribute to imprecision and increase uncertainty of the results. A notable reduction in ESS also suggests that the study results may be heavily influenced by a subset of the sample in the trials that may not be representative of the full sample. Harms outcomes, which are important to patients and clinicians, were not assessed in the analyses, representing a gap in evidence.
In the sponsor-submitted ITC report, indirect comparisons were conducted to compare Altuviiio (XTEND-1) against emicizumab (HAVEN 3), EHL products such as efmoroctocog alfa (A-LONG), and SHL products such as octocog alfa (LEOPOLD I). The clinical experts consulted for this review agreed with the sponsor’s assumption that all currently reimbursed drugs in EHL or SHL classes are expected to demonstrate equivalent efficacy within their own classes; therefore, 1 single drug in the EHL or SHL class can represent all currently available drugs in that particular drug class.
Studies Addressing Gaps in the Evidence From the Systematic Review
No relevant studies addressing gaps in the evidence from the systematic review were submitted by the sponsor.
Conclusions
Two pivotal phase III, nonrandomized, open-label clinical trials, XTEND-1 (N = 159) and XTEND-Kids (N = 74), were included in the sponsor’s systemic review to provide evidence on the efficacy and safety of Altuviiio in adult and pediatric patients with severe hemophilia A. Based on the inputs from patients, clinicians, and clinical experts, the most important treatment goals for patients with hemophilia A are to prevent bleeding, including spontaneous and traumatic bleeding events, reduce joint pain, improve HRQoL, and achieve unrestricted lifestyle comparable to the general population. In the XTEND-1 and XTEND-Kids studies, patients who received once-weekly Altuviiio administered as weekly prophylaxis for 52 weeks experienced an annual rate of treated bleeds that was considered clinically meaningful, with a mean ABR of 0.71 (95% CI, 0.52 to 0.97) and 0.89 (95% CI, 0.56 to 1.42) in the 2 trials, respectively. Further, the proportion of patients who did not report experiencing a treated bleed was 64.7% and 63.5% in the XTEND-1 and XTEND-Kids studies, respectively. Additionally, the XTEND-1 study included an intrapatient comparison that suggests that Altuviiio may result in an improved ABR compared to the patients’ historical prophylactic therapy, although the evidence is still uncertain. Patients enrolled in the pivotal trials also experienced an annual rate of joint bleeds that was considered clinically meaningful, and although the change from baseline to week 52 in HJHS indicated an improvement in joint health, the magnitude of this effect is unclear due to the small within-group differences and the lack of validated minimal important differences (MIDs) for this outcome. In adults (XTEND-1) the change from baseline in HRQoL based on the Haem-A-QoL Physical Health score indicated an improvement in QoL; however, the change was not considered clinically meaningful based on the within-group MID identified in the literature. Results for Haem-A-QoL total score in the XTEND-Kids study suggest an improvement for children aged 8 years to 12 years, but these data are highly uncertain due to a small sample size. While the results suggest an improvement in PROMIS Pain Intensity scores at week 52 for adults in the XTEND-1 study, it is unclear whether this change corresponds to a clinically meaningful difference due to the absence of validated MIDs for this outcome. Additionally, the evidence was insufficient to support an improvement in pain intensity among pediatric patients based on the results from the XTEND-Kids study. Perioperative management of bleeds was also assessed and although the evidence is limited by a small sample size, all surgeries were reported as having a good or excellent hemostatic response to perioperative use of Altuviiio. Regarding safety, Altuviiio was well tolerated and the reported TESAEs were generally consistent with what is anticipated in an adult, adolescent, and pediatric population with severe hemophilia A. The majority of TESAEs across both pivotal studies were assessed to be mild in severity. There was no development of inhibitors to FVIII detected in either study, and there were no reports of serious allergic reactions, anaphylaxis, or embolic or thrombotic events.
It should be noted that the certainty of evidence was assessed as very low for all outcomes identified as clinically relevant for this review, with the exception of ABR in adults, which was rated to be of low certainty due to the intrapatient comparison and proportion of patients reporting no bleeds. The review team acknowledges that there are challenges associated with conducting a traditional RCT for rare diseases such as hemophilia A, and that the study design used in the XTEND trials is consistent with other trials for this condition. Nonetheless, the study design limits the interpretation of the safety and efficacy of Altuviiio, particularly as it relates to any comparator. The sponsor submitted indirect evidence to address this gap in the evidence, which suggested that prophylactic treatment with Altuviiio was associated with improved bleeding outcomes compared with the other treatments currently available in practice, such as EHL agents, SHL agents, or emicizumab. However, the magnitude of the clinical benefit of Altuviiio versus these comparator therapies is uncertain and likely overestimated by the study findings due to the limitations of the available indirect evidence, such as a sizable reduction in the ESS from the original sample size after the propensity score weighting analyses, and inadequate or lack of adjustment for potential prognostic factors, which may introduce unmeasurable confounding in the relative treatment effect estimates. Overall, Altuviiio used prophylactically resulted in clinically meaningful rates of bleeding outcomes over 52 weeks of use. Additionally, due to the prolonged half-life relative to other SHL and EHL FVIII molecules that is noted in the product monograph, the evidence suggests that Altuviiio represents an additional option that supports a patient-centred approach to the management of hemophilia A.
In the perioperative setting, hemostatic response by Altuviiio was rated as excellent by the investigators or surgeons for all major surgeries. The clinical experts consulted for this review were in agreement that alternative designs like single-arm trials and intrapatient comparison provide practical evaluation of new therapies. Further concerns with generalizability of the results included no control for multiplicity in the XTEND-Kids study, and potential biases introduced by assumptions in the statistical models used to make the comparisons. In addition, the lack of control for physical activity and the potential temporal trends due to the pandemic limit the interpretation of the efficacy outcomes.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of Altuviiio 50 IU/kg IV in the treatment of hemophilia A (congenital FVIII deficiency) in adults and children.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
Hemophilia is a bleeding disorder caused by deficiencies in coagulation FVIII and factor IX .1-3 Hemophilia A is the most common form.4 It is a rare, congenital bleeding disorder caused by mutations in the gene that produces FVIII, a glycoprotein critical for hemostasis, which leads to excessive bleeding due to the inability to form blood clots.2,3,5 FVIII circulates as a noncovalent complex with VWF to regulate platelet aggregation and clot formation.22 VWF is required for normal survival of FVIII in circulation.22
Hemophilia A is an X-linked disorder predominantly affecting male patients, although females who are heterozygous carriers can have factor levels in the hemophilic range.4 Disease severity is categorized as mild, moderate, or severe, and is based on factor levels.7 For reference, normal FVIII activity is considered 40% or higher. Mild hemophilia A is defined by factor levels of 5% to 40% of FVIII activity levels in the blood, moderate is defined by levels of 1% to 5%, and severe is defined by levels less than 1% of normal FVIII activity levels.8 According to a 2023 Canadian Bleeding Disorders Registry report, there were 3,510 Canadians living with hemophilia A, of whom 1,158 had severe disease.23
Patients with hemophilia A experience symptoms such as bleeding into joints, soft tissues and muscles, the mouth, and urine, as well as surface bleeding and easy bruising.9 In some cases, bleeding is due to a known cause such as an injury; alternatively, some causes of bleeding may be unknown and are referred to as a spontaneous bleed.7 Spontaneous bleeding events are more common in patients with severe hemophilia A compared to mild or moderate disease.7,24 Bleeding associated with hemophilia A can result in complications such as joint damage from repetitive bleeding, deep internal bleeding, and neurologic problems or death associated with bleeding in the brain.7 Additionally, the challenges experienced by patients with hemophilia A can substantially impact patient QoL and physical, mental, social, and educational well-being.4,10 In patients with moderate to severe disease, reduced mobility due to joint bleeding is associated with increased reported pain, discomfort, and mental health challenges.1,25 In children and adolescents, restriction on certain sport-related activities to prevent trauma, frequent school absences due to repeated bleeding events, and accompanying pain associated with bleeding events, can impact physical, social, and intellectual development.26 In patients with severe hemophilia, joint bleeding is the most common clinical manifestation and can progress to hemophilic arthropathy, a disabling condition characterized by joint remodelling and chronic pain, which may necessitate future joint replacement.4,10
A definitive hemophilia A diagnosis is based on a factor assay, obtained through blood tests, such as the 1-stage clotting assay and chromogenic assay, which measure FVIII activity levels and can demonstrate factor deficiency.11 Patients with a family history of hemophilia A are typically diagnosed at birth, while those without a family history are typically diagnosed after bleeding is observed, often during the first year of life for individuals with severe hemophilia.12 In Canada, patients with hemophilia A are typically treated in a hemophilia treatment centre by a multidisciplinary care team that includes a physician with specific skills in treating bleeding disorders, a nurse, a physiotherapist, and a social worker.2 In patients with comorbidities, the care team may also include an orthopedic surgeon, internal medicine specialists, and infectious disease specialists.8
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
The international WFH guidelines for the management of hemophilia recommend primary prophylaxis as the standard of care for all patients with hemophilia A. This standard of treatment aligns with the recently released updated clinical practice guidelines for the treatment of hemophilia A by the International Society on Thrombosis and Haemostasis (ISTH).27 In patients with hemophilia A, primary prophylaxis is defined as regular infusion of missing FVIII to increase FVIII levels, or administration of a factor mimetic. The key for primary prophylaxis is to start patients on treatment before a bleeding event. According to the clinician group input and the 2 clinical experts consulted for this review, the main goal of primary prophylaxis is to prevent bleeding and enable patients with hemophilia A to live an active lifestyle and have an improved QoL, comparable to that of the general population. The goal of prophylactic therapy is to maintain factor levels greater than 3% to 5% (3 IU/dL to 5 IU/dL) to reduce the risk of spontaneous bleeding and for better preservation of joint function.8 Current WFH guidelines now recognize that patients remain at risk of bleeding at a trough level of 1%, and now recommend to target FVIII trough levels of 3% to 5% or higher,8 while higher FVIII levels around normal to near-normal levels (15% to 50%) may be needed achieve near-zero bleeds.28-33 However, normal FVIII activity levels have not been attainable with current treatments without trade-offs with burden of treatment.
Prophylactic treatment for hemophilia A includes FVIII replacement therapy, nonfactor therapy, and gene therapy. FVIII replacement therapies used in prophylactic, on-demand, and perioperative treatment settings for hemophilia A are categorized as SHL and EHL therapies.8 SHL therapies include virally inactivated products made from donated plasma or recombinant products manufactured using genetically engineered cells. The WFH do not express a preference for recombinant over plasma-derived clotting factor concentrates, and clinical experts consulted by the sponsor suggest the use of plasma-derived products is nearly nonexistent in the population of people with hemophilia A living in Canada.8,34 The half-life of SHL therapies is approximately 12 hours in adults (shorter in young children and increasing with age), requiring IV administration 3 to 4 times weekly .8 According to the clinical experts consulted for this review, available SHL therapies used in clinical practice in Canada are Kovaltry, Xyntha, and Zonovate. With EHL therapies (agents used in clinical practice are Adynovate, Jivi, Eloctate, and Esperoct), fusion technologies and PEGylation are used to extend product half-life by 1.4 times to 1.6 times that of SHL products, to approximately 19 hours (half-life is shorter in pediatric populations). The dosing of EHL therapies is thus reduced to twice weekly or every 3 days.8 If patients develop inhibitors to FVIII replacement therapy, immune tolerance induction therapy aimed at eradicating inhibitors using infusions of varying FVIII doses, or bypassing agents with mechanisms that bypass the need for FVIII replacement can be used.
Nonfactor replacement therapy provides hemostasis through a different mechanism than FVIII replacement. Currently the only available nonfactor therapy in Canada is emicizumab, a bispecific monoclonal antibody that mimics the function of missing FVIII by bridging activated factor IXa and factor X, to facilitate the coagulation cascade and achieve hemostasis. Based on the typical frequency of administration for currently available SHL and EHL products (2 to 3 times per week), the trough levels are often inadequate to provide bleed protection. Unlike SHL and EHL therapies, emicizumab is not approved for the treatment of bleeding episodes or for perioperative management.35 Additionally, Alhemo (concizumab injection) is another nonfactor therapy (antitissue factor pathway inhibitor antibody) that has been approved by Health Canada for the treatment of adolescent and adult patients with hemophilia A and B who have FVIII or factor IX inhibitors;36 Alhemo has not undergone review by CDA-AMC at the time of this review. Emerging adeno-associated virus gene therapy for hemophilia A offers promise of increased FVIII activity levels and protection against bleeds with a single dose and has been approved for use in Europe and in the US.37,38 There are several limitations associated with expected use of gene therapy including high proportions of individuals who carry adeno-associated virus neutralizing antibodies,39,40 substantial variability in factor trough levels achieved across individuals,41 and unknown long-term persistence of gene expression following gene therapy.42 Gene therapy for hemophilia A is not currently available in Canada.
The clinical experts consulted for this review confirmed that these approaches align with the Canadian clinical practice. According to the clinical experts, while the standard of care for severe hemophilia A is FVIII prophylaxis, some patients choose to remain on an episodic FVIII regimen.43 The experts also indicated that Hemlibra (emicizumab), Eloctate (efmoroctocog alfa), Kovaltry (octocog alfa), Xyntha (antihemophilic factor [recombinant]), Zonovate (antihemophilic factor [recombinant]), Adynovate (antihemophilic factor [recombinant], PEGylated), Jivi (antihemophilic factor [recombinant], PEGylated-aucl), and Esperoct (antihemophilic factor [recombinant], Fc fusion protein) are relevant comparators for the drug under review.
Drug Under Review
Key characteristics of Altuviiio are summarized in Table 4 with other treatments available for hemophilia A (congenital FVIII deficiency).
Altuviiio is a recombinant FVIII analogue fusion protein that temporarily replaces the missing coagulation factor (FVIII) required for effective hemostasis. Altuviiio is a new class of FVIII replacement therapy that functions independently of endogenous VWF to overcome the half-life limit imposed by FVIII–VWF interactions. FVIII half-life is significantly influenced by VWF plasma levels while circulating together as a noncovalent complex. Under typical physiological circumstances, the half-life of free FVIII is substantially reduced (approximately 2 hours) compared to that of VWF-bound FVIII (approximately 12 hours). By circulating independently of endogenous VWF, Altuviiio has demonstrated 3-fold to 4-fold prolonged half-life compared to other SHL and EHL FVIII molecules.
Altuviiio is approved by Health Canada for the treatment of hemophilia A (congenital FVIII deficiency) in adults, adolescents, and children with hemophilia A (congenital FVIII deficiency) for routine prophylaxis for prevention or reduction in the frequency of bleeding episodes, treatment and control of bleeding episodes, and perioperative management of bleeding (surgical prophylaxis). Altuviiio is administered intravenously as a single dose of 50 IU/kg once weekly for routine prophylaxis.44 The sponsor’s reimbursement request is as per the approved Health Canda indication. In the US, Altuviiio indication aligns with that of the requested Health Canada indication.45 In the European Union, Altuviiio is indicated for the treatment and prophylaxis of bleeding in patients with hemophilia A (congenital FVIII deficiency) in all age groups.46
Table 4: Key Characteristics of FVIII Replacements Available in Canada
Characteristic | Altuviiio44 | Kovaltry47 | Eloctate48 | Emicizumab49 |
|---|---|---|---|---|
Standard half-life, recombinant | Extended half-life, recombinant | |||
Mechanism |
| FVIII replacement | Bridges activated factor IX and factor X to restore the natural function of activated FVIII | |
Indicationa | For the treatment of hemophilia A (congenital FVIII deficiency) in adults and children for: routine prophylaxis to reduce the frequency of bleeding episodes, on-demand treatment and control of bleeding episodes, and perioperative management of bleeding | For the treatment of adults and children with hemophilia A for:
| For the treatment of patients with hemophilia A (congenital FVIII deficiency) with or without FVIII inhibitors as routine prophylaxis to prevent bleeding or reduce the frequency of bleeding episodes | |
Route of administration | IV | IV | IV | SC |
Recommended dose | Prophylaxis: Single dose of 50 IU/kg once weekly | Prophylaxis: 50 IU/kg every 3 to 5 days The dose may be adjusted based on patient response in the range of 25 IU/kg to 65 IU/kg; more frequent or higher doses up to 80 IU/kg may be required in pediatric patients aged < 12 years | Prophylaxis: Adults and adolescents (aged > 12 years): 20 IU/kg to 40 IU/kg of body weight, 2 or 3 times per week Children aged ≤ 12 years: 20 IU/kg to 50 IU/kg body weight, twice weekly, 3 times weekly, or every other day according to individual requirements |
|
Serious adverse effects or safety issues |
| Development of inhibitors to FVIII |
| |
FVIII = factor VIII; SC = subcutaneous.
Note: The generic name for Kovaltry is antihemophilic factor (recombinant). The generic name for Eloctate is antihemophilic factor (recombinant) B-domain deleted Fc fusion protein.
aHealth Canada indication.
Source: Altuviiio product monograph,44 Kovaltry product monograph,47 Eloctate product monograph,48 and emicizumab product monograph.49
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
One patient group input submission from the CHS was received for this review. The CHS is a national voluntary health charity that advocates for improvements in health and QoL for patients living with inherited bleeding disorders in Canada. The CHS is a member of the Network of Rare Blood Disorder Organization which follows safety, supply, and access issues for the broader category of blood and plasma-derived medicinal products in Canada. The CHS gathered input for this submission through a national online survey distributed both in English and French between April 1, 2024, to June 1, 2024. The survey received a total of 104 responses from patients in Canada with hemophilia A. This included 57 patients with severe hemophilia A, 33 with mild hemophilia A, and 14 with moderate hemophilia A. Of these patients, 33 reported a history of FVIII inhibitors. Among the 104 patients, 59 were receiving regular prophylactic therapy, 30 were receiving on-demand treatment, and 15 were not receiving treatment for hemophilia A.
When asked about their disease experience, survey respondents highlighted the following symptoms and challenges associated with hemophilia A: joint pain and loss of function, pain from bleeding episodes, invasive medical procedures, treatment side effects, surgery complications, and sleep difficulty. Respondents further indicated that hemophilia A-related restrictions impact their day-to-day functioning through limitations on physical activity and sport participation, loss of mobility, long recovery times from bleeding episodes, and restrictions on choice of jobs. Respondents also noted the detrimental effects on their social and psychological well-being, impacts on travelling due to concern over emergency hemophilia care, financial and educational constraints due to the risks of needing to stop working or days lost from work due to hospitalization, and the logistics of needing factor products with them at all times.
Of 104 survey respondents, 41% of patients were receiving emicizumab prophylaxis, 11% were receiving FVIII prophylaxis, and 6% were receiving FVIII on-demand therapy. Notably, 1 patient had undergone gene therapy. Most respondents indicated that they were “very satisfied” (49%) or “quite satisfied” (37%) with their current treatment. Only a few patients reported that they were “not very satisfied” (11%), or “not satisfied at all” (4%) with their current treatment. While most respondents (58%) noted no challenges in administering their current treatment, some indicated that geographical challenges were barriers to accessing treatment centres for bleeding episodes. Overall, most respondents considered their current treatment regimen “very effective” (48%) or “quite effective” (20%) in stopping or preventing bleeding. Most respondents indicated that treatment has become simpler and less burdensome with emicizumab as it can be self-administered at home, but injection pain is a challenge. Some respondents (6%) reported that emicizumab is effective in preventing bleeding episodes but not stopping active bleeding. Patients reported that new therapies that can improve hemophilia A disease outcomes such as higher bleed protection, improved pain management, and reduced frequency (fewer doses with longer half-life) of treatment are needed to improve their ability to engage in everyday activities.
One patient who had received Altuviiio through a special access program provided detailed input on their experience with the therapy. This patient has severe hemophilia, with a history of significant musculoskeletal bleeding and continued ankle joint pain that persisted even after total ankle replacement. In early 2024, this patient switched from emicizumab prophylaxis for breakthrough bleeds with an EHL FVIII agent (Jivi) to Altuviiio (5,000 IU) once per week. Since initiating treatment, this patient reported experiencing a sustained high FVIII level, with a factor trough of approximately 15%, that has reduced their risk of major and subclinical bleeding. Similar effects were reported by other patients, which appear to be maintained even if the injection is up to 2 days late. The patient indicated travel has become easier with Altuviiio, due to the more flexible storage requirements compared to previous treatments. The patient reported no disadvantages or side effects of Altuviiio. The patient indicated they have become self-sufficient, infuse themselves, and no longer require a caregiver, and highlighted substantial improvements in treatment burden, stress, and QoL.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of hemophilia A.
Unmet Needs
The 2 clinical experts consulted for this review indicated that the most important treatment goals for patients with hemophilia A are to prevent bleeding, including spontaneous and traumatic bleeding events, reduce joint pain, improve HRQoL, and achieve unrestricted lifestyle comparable to the general population. The clinical experts noted that the current standard of care for patients with severe hemophilia A in Canada is primary prophylactic therapy to increase patients’ FVIII levels to normal or near normal. There are 3 options for primary prophylactic treatment in the Canadian landscape: regular IV infusion of SHL FVIII concentrate (Kovaltry, Xyntha, Zonovate), regular IV infusion of EHL FVIII concentrate (Adynovate, Jivi, Eloctate, Esperoct), or regular emicizumab subcutaneous injection. Regarding unmet needs for patients with hemophilia A, the 2 clinical experts indicated that at this time, there is no therapy that can modify the underlying disease mechanism of hemophilia A outside of gene therapy which is currently unavailable in Canada. In addition, apart from emicizumab which provides bleeding protection roughly equivalent to the steady-state trough levels of 10% to 15%, the trough level of currently available SHL and EHL FVIII concentrates are between 3% to 5% before the next infusion, with subsequent clearance dependent on the product half-life (but generally 14 to 18 hours) resulting in less bleed protection. Patients with hemophilia A who participate in regular physical activities are at risk of bleeding with the present prophylactic regimen when their FVIII levels are suboptimal. As a result, patients need additional doses on top of their prophylaxis just before certain physical activities to mitigate the risk of provoked bleeding, even with emicizumab.
Place in Therapy
Overall, the experts indicated that Altuviiio will change the treatment landscape for acute bleed and perioperative management, but do not envision Altuviiio to alter the underlying disease process of congenital hemophilia A. Both clinical experts suggested that Altuviiio will be the first agent in which a period of “normal hemostasis” (FVIII activity > 40% for the first 4 days of treatment) can be achieved without a trade-off in burden of treatment and this is clinically relevant. Compared to available treatment options, the clinical experts suggested Altuviiio would likely be used as a first-line therapy for patients who desire to use FVIII replacement rather than FVIII mimetic therapy or as an alternative or complementary therapy to emicizumab. Individuals receiving emicizumab for prophylaxis may benefit from the EHL of Altuviiio for bleeding or perioperative management in place of SHL or EHL FVIII concentrates because of reduced infusion frequency. If approved as a first-line treatment, there would be no need for SHL FVIII products as Altuviiio will provide sustained FVIII levels with fewer doses, which SHL would require a higher frequency of infusions and doses to attain.
Patient Population
The 2 clinical experts indicated that all patients with congenital hemophilia A (without inhibitors) are expected to respond to Altuviiio. According to the experts, identifying patients best suited for Altuviiio should be based on individual patient circumstances, including bleeding phenotype, activity level, PK, and current treatment responses to tailor treatment options. The clinical experts noted that patients best suited for Altuviiio include patients receiving emicizumab who have experienced a suboptimal response due to breakthrough bleeding events or AEs like injection pain or neutralizing anti-emicizumab antibodies; patients on SHL or EHL FVIII prophylaxis who still struggle with the frequent dosing interval; patients participating in high-level physical activities who may benefit from the sustained high FVIII activity level with improved bleed protection; and patients requiring surgery regardless of their current treatment. The clinical experts noted that there would be no issues with diagnosis, misdiagnosis, or underdiagnosis of hemophilia A in routine clinical practice, given the presence of positive family history in some cases, and availability of clear diagnostic criteria and genetic testing. According to the clinical experts, several factors modify FVIII PK and influence dosing regimens in patients with hemophilia A as younger patients, particularly children, have faster FVIII clearance, leading to shorter half-lives and the need for more frequent dosing compared to adults. Patients living with elevated body mass index may experience altered FVIII distribution, requiring dose adjustments to achieve target levels. Both experts indicated that like other FVIII therapies, Altuviiio is not suitable for patients with inhibitors to FVIII.
Assessing the Response Treatment
The clinical experts noted that in general, outcomes used in clinical practice are largely aligned with those used in clinical trials, particularly regarding ABR, which is a common trial end point. Other clinical trial outcomes including joint health, QoL (Haem-A-QoL), and FVIII activity levels are also closely monitored in clinical practice. Patients and patient organizations also expressed a desire to experience a “hemophilia-free mind,” which was further described as freedom from bleeding, arthropathy, pain, and treatment burden.
Discontinuing Treatment
Both clinical experts indicated that treatment with Altuviiio will be discontinued if there is evidence of the development of FVIII inhibitors, no evidence of improvement in bleeding episodes, occurrence of AEs with treatment administration (allergy/anaphylaxis), or loss of IV access.
Prescribing Considerations
According to the clinical experts, treatment with Altuviiio should be primarily managed within a hemophilia treatment centre, where specialized hematologists and multidisciplinary teams can monitor treatment including PK testing, manage complications, and provide perioperative or periprocedural guidance. Additionally, 1 clinical expert noted that Altuviiio, like other FVIII therapies, is well-suited for home therapy, as many patients can self-administer IV treatment following training by hemophilia treatment centre staff; however, regular follow-up at the hemophilia treatment centre remains crucial to monitor treatment response, while benefiting from the convenience of home administration.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
Three clinician groups provided input for this review: AHCDC (5 clinicians contributed to the input), CANHC (6 clinicians contributed), and CPHC (5 clinicians contributed). AHCDC is a national, nonprofit organization of hemophilia clinic directors in Canada aiming to improve care for individuals with bleeding disorders through clinical services, research, and education. CANHC is a Canadian association of nurses who work in hemophilia treatment centres caring for individuals with bleeding disorders and offers resources for education, research, and professional development. CPHC comprises physiotherapists working in hemophilia treatment centres with experience managing hemophilia-related musculoskeletal complications and aims to assess and make recommendations around hemophilia management. AHCDC gathered input through national advisory boards, expert opinions, and clinical trial experience with Altuviiio. Information from CANHC was provided by members who responded to the call for input while the submission from CPHC was gathered via information from clinician experience, conferences attended, and in-services.
The clinician groups noted that the current standard of care for patients with hemophilia A consists of regular prophylactic replacement therapy with clotting factor concentrates or nonfactor subcutaneous therapy. Clinician groups noted that the ultimate treatment goal for patients with hemophilia A is to minimize the number of bleeds to 0 or near-zero while slowing hemophilic arthropathy progression, enabling patients to live healthier, active lives. According to clinician groups, achieving this goal currently requires frequent administration of high therapeutic doses to overcome short treatment half-lives, which may not be feasible in pediatric populations or individuals with poor venous access. This treatment burden is particularly notable in patients who require elevated trough levels due to recent surgical procedures, compromised joint health, or high physical activity levels, according to clinician group input. Consistent with expert input, the clinician groups agreed that current therapies demonstrate variable efficacy.
Aligning with expert input, clinician groups noted that Altuviiio could be used as first-line therapy for patients aged 2 years or older with hemophilia A or offered as an alternative treatment to those receiving other therapies. Patients best suited for treatment with Altuviiio, as identified by clinician groups, were consistent with that of the clinical expert input. Additional patient populations that clinician groups noted may benefit from Altuviiio treatment included patients with hemophilic arthropathy or poor venous access, patients with mild hemophilia A receiving on-demand or episodic therapy, and those undergoing surgery or procedures. The clinician groups indicated that patients least likely to benefit from Altuviiio are those who are averse to IV infusions, have developed FVIII inhibitors, or who have achieved 0 bleeds on prophylaxis and who feel that switching therapies would have a minimal positive impact on QoL.
The clinician groups agreed with consulted experts that the outcomes used in the trials to assess response are realistic for clinical practice, adding that patients should be assessed every 6 months to 2 years, depending on disease severity. CANHC noted that a clinically meaningful response to Altuviiio treatment would involve favourable PK profile (half-life improved to near-normal levels), an absence of FVIII inhibitors, absence of bleeding events, improved stable joint health, improved QoL, and infrequent hospitalizations. The clinician groups’ suggested criteria for discontinuation aligned with expert input. AHCDC and CANHC also suggested discontinuation if the patient switches to a nonfactor replacement therapy, other experimental therapies, or if the treatment centre is unable to perform the required clotting assay. The clinician group input regarding Altuviiio prescribing considerations, including the follow-up of patients receiving treatments in hemophilia treatment centres, was consistent with the expert input received for this review.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by for this review are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Considerations for initiation of therapy | |
Are there any special considerations for monitoring response to therapy (e.g., factor levels)? | According to the clinical experts, the special consideration for monitoring response to therapy is an assessment of FVIII levels. The clinical experts recommended a 1-stage assay50 as an ideal assessment of FVIII levels to monitor response to therapy as this is reliable compared to chromogenic assay. |
Is there any minimum age for treatment eligibility? | The clinical experts noted that there is no minimum age for treatment eligibility when considering initiation of therapy. |
If there is treatment failure, is it appropriate for a patient to switch back to comparator therapies, and how long should the interval (washout) be before doing so? | The clinical experts indicated that in the case of a treatment failure, decision-making regarding a switch to comparator therapies and the washout period before initiation of new therapies should be guided by a clinician who has experience treating patients with hemophilia. |
The sponsor claims that a significant advantage and safety feature of Altuviiio is its lack of association with the development of FVIII inhibitors. Only 3.9% of patients in arm A (5 patients) had a family history of FVIII inhibitors. All patients previously received factor therapies, so previously untreated patients were not included which are the population at highest risk of developing inhibitors. | According to the clinical experts, clinicians need to continuously assess for the development of inhibitors, especially if Altuviiio is to be used in a previously untreated patient with hemophilia A or initiated in patients with < 50 days of exposure to other FVIII concentrates. |
Do patients need to receive another therapy before starting this one, and what is the recommended timing between prior prophylactic therapy and the infusion of Altuviiio? | The clinical experts indicated that Altuviiio should not be restricted to patients with hemophilia A who have been on a prior therapy. Both clinical experts noted that if a patient is on another therapy, the timing between their prior prophylaxis and initiation of Altuviiio should be based on the half-life of the prior product, patient characteristics, and FVIII activity levels. |
Consider alignment with reimbursement criteria for SHL, EHL, and emicizumab products | This is a comment from the drug plans to inform expert committee deliberations. |
Considerations for continuation or renewal of therapy | |
What objective markers should be used to assess initial and ongoing response to treatment? | The clinical experts consulted for this review noted that the objective markers to consider for continuation or renewal of therapy includes initial and ongoing response to therapy such as assessing ABR, joint health status, frequency and severity of bleeds (including spontaneous, traumatic and target joints), and breakthrough bleeds. |
Consider alignment with renewal criteria for SHL, EHL, and emicizumab products | This is a comment from the drug plans to inform expert committee deliberations. |
Considerations for prescribing of therapy | |
Do you anticipate any tailoring therapy (personalizing medical treatment based on individual patient characteristics, such as level of activity, bleeding pattern [minor surgery], presence of inhibitors, and so forth)? | The 2 clinical experts noted they anticipate tailoring of Altuviiio based on pharmacokinetics and individual patient profile including bleeding pattern, level of physical activity, and joint health status. |
Altuviiio is reported to provide a mean FVIII activity of more than 40 IU/dL for most days of the week and 15 IU/dL on day 7. Do you envision any alternate dose or frequency in clinical practice? | According to the clinical experts consulted CDA-AMC, most patients on Altuviiio would receive the standard dosing of 50 IU/kg once weekly; however, tailoring could be done based on patient physical activity levels and surgeries and procedures, all under the guidance of an experienced hemophilia clinician. |
Consider alignment with prescribing criteria for SHL, EHL, emicizumab | This is a comment from the drug plans to inform expert committee deliberations. |
Generalizability | |
The pivotal trials only included patients who had previously received treatment with FVIII therapies; would previously untreated individuals (i.e., those who have a risk of inhibitor development) be eligible for Altuviiio? | The 2 clinical experts indicated that Altuviiio should not be restricted to persons with hemophilia A who have been on a prior therapy; however, if a previously untreated patient is started on Altuviiio, the clinician should closely monitor for inhibitor formation. |
EHL = extended half-life; FVIII = factor VIII; SHL = standard half-life.
Clinical Evidence
The objective of the CDA-AMC Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of Altuviiio (lyophilized powder for IV infusion, 250 IU, 500 IU, 1,000 IU, 2,000 IU, 3,000 IU, and 4,000 IU per vial) for the treatment of adults and children with hemophilia A for routine prophylaxis to reduce the frequency of bleeding episodes, on-demand treatment and control of bleeding episodes, or perioperative management of bleeding. The focus will be placed on comparing Altuviiio to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of Altuviiio is presented in 4 sections with the CDA-AMC critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The CDA-AMC assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes sponsor-submitted LTE study. The third section includes indirect evidence from the sponsor. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following are included in this review and appraised in this document:
two pivotal studies, the XTEND-1 study (NCT04161495) and the XTEND-Kids study (NCT04759131) identified in systematic review
one LTE study (XTEND-ed: ongoing)
one ITC using 3 different approaches.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
Description of Studies
Two studies (XTEND-1, XTEND-Kids) met the inclusion criteria of the sponsor-submitted systematic review.
The XTEND-1 study (NCT04161495) was a phase III, open-label, multicentre study to determine the safety, efficacy, and PK of Altuviiio administered IV once weekly as prophylaxis or on-demand treatment in previously treated patients aged at least 12 years with severe hemophilia A without inhibitors. The study consisted of a screening period of up to 8 weeks, a washout period (≥ 96 hours if patients previously received a SHL FVIII product and ≥ 120 hours for a EHL FVIII product), and an open-label treatment period up to 52 weeks. Safety was assessed throughout the study and for 2 weeks to 3 weeks after the last study treatment dose. The XTEND-1 study was completed on February 3, 2022. A total of 159 patients were enrolled in the XTEND-1 trial (including 8 patients living in Canada from 2 study sites) and divided into 2 treatment groups.
Arm A: Altuviiio prophylaxis (n = 133) — Patients who were on a current FVIII prophylactic treatment regimen and participated in an observational prestudy (242HA201/OBS16221) for at least 6 months before baseline of the XTEND-1 trial were assigned to arm A. This included a total of 78 patients who had at least 6 months of participation in the observational prestudy. The observational prestudy was a global, multicentre, prospective study of up to 12 months conducted in patients aged 12 years or older with severe hemophilia A who were receiving a marketed FVIII product. Patients in arm A received a once-weekly dose of 50 IU/kg Altuviiio as prophylactic treatment for 52 weeks.
Arm B: Altuviiio on-demand treatment followed by prophylaxis (n = 26) — Participants who were receiving an on-demand treatment regimen for hemophilia A before the baseline of the XTEND-1 trial were assigned to arm B. Patients in arm B received Altuviiio 50 IU/kg as on-demand treatment of bleeding episodes for the first 26 weeks and then switched to the 50 IU/kg weekly prophylactic treatment regimen with Altuviiio for another 26 weeks.
The primary objective of the XTEND-1 study was to evaluate the efficacy of Altuviiio as a prophylactic treatment based on the ABR in arm A (described in the following). The key secondary end point was to evaluate the efficacy of Altuviiio as a prophylactic treatment based on the intrapatient comparison of ABR during the trial compared to the historical prophylaxis ABR in the 78 patients in arm A who participated in the observation study.
All patients underwent PK assessments after the initial Altuviiio dose on day 1, with a subgroup of arm A (the sequential PK subgroup) receiving more extensive PK assessments at baseline and week 26. In addition, peak and trough FVIII sampling was performed in all patients throughout the study. Patients from either arm who underwent major surgery after the first dose of the study drug were included in the surgery subgroup to assess the control and prevention of bleeding during use of Altuviiio in the surgical setting.
Figure 1: Study Design of XTEND-1
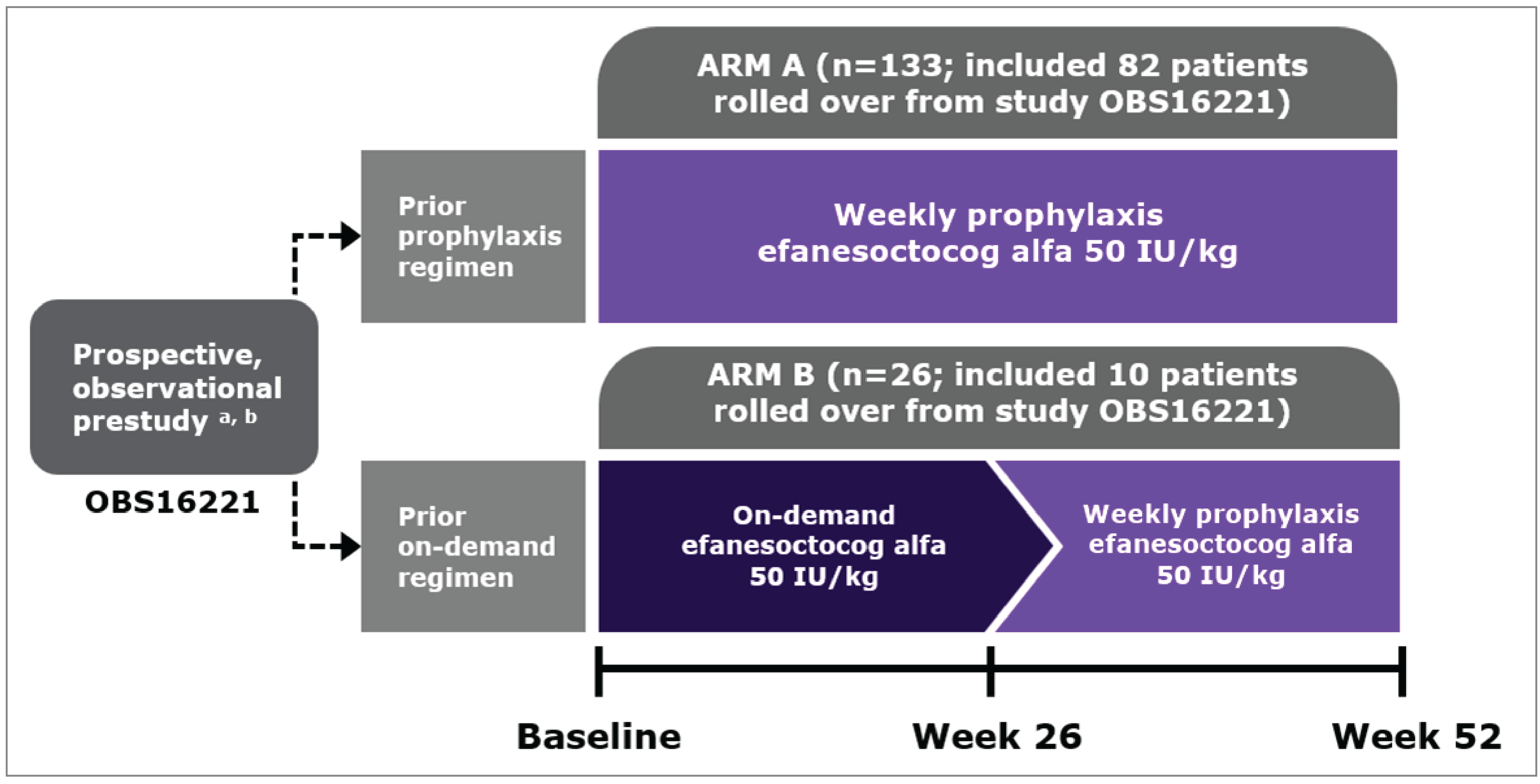
Note: “Efanesoctocog alfa” indicates Altuviiio.
aProspective prestudy is Study 242HA201/OBS16221.
bA total of 92 patients rolled over from the observational prestudy into the XTEND-1 study, including 82 patients into arm A and 10 into arm B.
Source: Sponsor’s clinical evidence summary.51
The XTEND-Kids study (NCT04759131) was a phase III, open-label, multicentre, nonrandomized study that evaluated the safety, efficacy, and PK of weekly Altuviiio prophylaxis in patients with previously treated severe hemophilia A who were aged younger than 12 years. The XTEND-Kids study also evaluated the efficacy of Altuviiio for treating bleeding episodes and for perioperative management. The trial comprised 2 age cohorts, children aged younger than 6 years (n = 38) and children aged 6 years to younger than 12 years (n = 36) (including 9 patients living in Canada from 4 study sites). There was a screening period up to 8 weeks and washout period of 96 hours or longer for children aged 6 years to younger than 12 years, and 72 hours or longer for patients aged younger than 6 years, followed by an open-label, 52-week treatment period. All 74 patients received once-weekly IV doses of 50 IU/kg Altuviiio for prophylactic treatment. Safety was assessed throughout the study and for 2 weeks to 3 weeks after the last Altuviiio dose. The XTEND-Kids study was completed on January 18, 2023. PK assessments were performed at baseline (day 1), and peak and trough activity levels were evaluated for all patients in the study. A total of 37 patients (19 patients aged < 6 years and 18 patients aged 6 to < 12 years) were included in the PK subgroup which had PK sampling conducted at baseline and up to 7 days (168 hours) after Altuviiio injection. Patients enrolled in the XTEND-Kids study who underwent major surgery during the study were included in a surgery subgroup.
The primary objective of the XTEND-Kids study was to evaluate the safety of Altuviiio in previously treated pediatric patients with severe hemophilia A based on occurrence of inhibitor development. The key secondary end point was to evaluate the efficacy of Altuviiio as prophylactic treatment based on ABR, AjBR, joint health, and QoL outcomes.
Characteristics of the included studies are summarized in Table 6.
Table 6: Details of Studies Included in the Systematic Review
Characteristic | XTEND-1 | XTEND-Kids |
|---|---|---|
Designs and populations | ||
Study design | Phase III, open-label, multicentre trial | Phase III, open-label, multicentre trial |
Locations | Patients were enrolled in 48 sites in 19 countries across Asia and Pacific Islands, Europe, North America, and South America Countries included Argentina, Australia, Belgium, Brazil, Bulgaria, Canada (2 study sites), France, Germany, Greece, Hungary, Italy, Japan, Mexico, the Netherlands, Spain, South Korea, Taiwan, UK, and US There were 2 Canadian sites that enrolled a total of 8 patients in Canada | Patients were enrolled in 40 sites in 15 countries across Asia and Pacific Islands, Europe, and North America Countries included: US, Canada (4 study sites), France, Germany, Hungary, Ireland, Italy, the Netherlands, Spain, Sweden, Switzerland, UK, Türkiye, Australia, and Taiwan There were 4 Canadian sites that enrolled a total of 9 patients in Canada |
Patient enrolment dates | Start date: December 4, 2019 End date: April 23, 2021 | Start date: March 22, 2021 End date: March 16, 2022 |
Enrolled (N) | 159 The study comprised 2 arms:
| 74 The study comprised 2 age cohorts receiving Altuviiio prophylaxis:
|
Inclusion criteria |
|
|
Exclusion criteria |
|
|
Drugs | ||
Intervention | Arm A: Altuviiio prophylaxis, 50 IU/kg administered intravenously once a week for 52 weeks. Arm B: Altuviiio on-demand treatment (50 IU/kg administered intravenously as needed) for 26 weeks followed by Altuviiio prophylaxis (50 IU/kg administered intravenously once a week) for another 26 weeks. | Altuviiio prophylaxis, 50 IU/kg administered intravenously once a week for 52 weeks. |
Comparator(s) | NA | NA |
Study duration | ||
Screening phase | Up to 8 weeks | Up to 8 weeks |
Washout period | ≥ 96 hours if patients previously received a SHL FVIII product and ≥ 120 hours if they had previously received an EHL FVIII product | ≥ 96 hours for children aged 6years to < 12 years, and ≥ 72 hours for patients aged < 6 years |
Treatment phase | Up to 52 weeks | Up to 52 weeks |
Follow-up phase | Safety was assessed throughout the study and for 2 weeks to 3 weeks after the last study treatment dose. Patients could also choose to continue in an open-label extension study (XTEND-ed). | Safety was assessed throughout the study and for 2 weeks to 3 weeks after the last study treatment dose. Patients could also choose to continue in an open-label extension study (XTEND-ed). |
Outcomes | ||
Primary end point | The mean ABR for patients receiving Altuviiio prophylactic treatment in arm A. | Occurrence of inhibitor development (neutralizing antibodies against FVIII as determined via the modified Nijmegen-Bethesda assay). |
Secondary and exploratory end points | End points of the XTEND-1 trial assessed the ability of Altuviiio to maintain hemostasis during routine prophylaxis, bleeding episodes, and in the perioperative setting. Secondary end points Routine prophylaxis:
Treatment of bleeds:
Consumption of Altuviiio:
Perioperative management:
Safety and tolerability:
Exploratory:
| End points of the XTEND-Kids trial assessed ability of Altuviiio to maintain hemostasis during routine prophylaxis, bleeding episodes, and in the perioperative setting. Secondary end points Routine prophylaxis:
Treatment of bleeds:
Consumption of Altuviiio:
Perioperative management:
Safety and tolerability:
Exploratory:
|
Publication status | ||
Publications | NCT04161495 von Drygalski A, Chowdary P, Kulkarni R, et al. efanesoctocog alfa Prophylaxis for Patients with Severe Hemophilia A. New England Journal of Medicine. 2023;388(4):310-318.52 | NCT04759131 Malec L, Peyvandi F, Chan AKC, et al. efanesoctocog alfa Prophylaxis for Children with Severe Hemophilia A. New England Journal of Medicine. 2024;391(3):235-246.53 |
ABR = annualized bleeding rate; AE = adverse event; AI = accumulation index; AjBR = annualized joint bleeding rate; ALT = alanine aminotransferase; ASA = acetylsalicylic acid; AST = aspartate aminotransferase; AUC = area under the activity-time curve; BU = Bethesda unit; CL = total clearance; CLss = total clearance at steady state; Cmax = maximum FVIII activity; Ctrough = trough activity; DNAUC = dose-normalized area under the activity-time curve; EHL = extended half-life; EQ-5D-5L = 5-Level EQ-5D; EQ-5D-Y = EQ-5D Youth; FVIII = factor VIII; Haem-A-QoL = Haemophilia Quality of Life questionnaire for adults; Haemo-QoL = Haemophilia Quality of Life questionnaire for children; HAL = Haemophilia Activities List; HCV = hepatitis C virus; HEAD-US = Hemophilia Early Arthropathy Detection with Ultrasound; HJHS = Hemophilia Joint Health Score; IR = incremental recovery; ISTH = International Society on Thrombosis and Haemostasis; JADE = Joint Activity and Damage Exam; MRT = mean residence time; NA = not applicable; NSAID = nonsteroidal anti-inflammatory drug; PGIC = Patient Global Impression of Change; PGIS = Patient Global Impression of Severity; PK = pharmacokinetics; PRO = patient-reported outcome; PROMIS = Patient-Reported Outcomes Measurement Information System; SAE = serious adverse event; SHL = standard half-life; t1/2 = elimination half-life; TSQM-9 = 9-item Treatment Satisfaction Questionnaire for Medication; ULN = upper limit of normal; Vss = volume of distribution at steady state.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Source: XTEND-1 Clinical Study Report54 and XTEND-Kids Clinical Study Report.17
Populations
Inclusion and Exclusion Criteria
Both the XTEND-1 and XTEND-Kids trials included patients with severe hemophilia A (< 1% endogenous FVIII activity), who had previously received treatment with any recombinant or plasma-derived FVIII replacement product or cryoprecipitate. The XTEND-1 study evaluated Altuviiio in adult and adolescent patients aged 12 years or older, while the XTEND-Kids study included children aged at least 12 years. Patients with a history of a positive inhibitor test or a positive inhibitor result at screening (defined as ≥ 0.6 BU/mL at screening), serious active bacterial or viral infection within 30 days of screening, history of hypersensitivity or anaphylaxis associated with any FVIII product, or abnormal renal function (defined as serum creatinine > 2.0 mg/dL) at screening and who had used emicizumab within the 20 weeks before screening were excluded from both trials. A summary of the inclusion and exclusion criteria for both trials are presented in Table 5. In the XTEND-1 trial, a key exclusion criterion was the presence of concurrent clinically significant liver disease that, in the opinion of the investigator, would make the patient unsuitable for enrolment. This may include, but was not limited to cirrhosis, portal hypertension, and acute hepatitis.
Interventions
XTEND-1 Trial
In the XTEND-1 trial, patients who were currently receiving a FVIII prophylactic treatment regimen who participated in an observational prestudy (242HA201/OBS16221) for at least 6 months before baseline were assigned to arm A. Patients who were receiving an on-demand treatment regimen for hemophilia A were assigned to arm B. All patients enrolled in the XTEND-1 trial received a 50 IU/kg dose of Altuviiio delivered via a slow push IV injection of 8 minutes (± 2 minutes) with the rate of administration determined by patient’s comfort level. Patients enrolled in arm A received Altuviiio prophylaxis once every week for 52 weeks. Individuals enrolled in arm B received on-demand treatment with Altuviiio as needed during the first 26 weeks, followed by Altuviiio prophylaxis once every week for the last 26 weeks. A single dose of 50 IU/kg was recommended for treating all bleeding episodes. Additional and adjusted doses were only given after consultation with the investigator. If a bleeding episode did not improve, additional doses of 30 IU/kg or 50 IU/kg every 2 days to 3 days were considered. For minor or moderate bleeding episodes occurring within 2 days to 3 days after a recent prophylactic dose, an initial 30 IU/kg dose may also be used. A single dose of Altuviiio before surgery was used to treat bleeding during a minor surgery whereas additional doses of 30 IU/kg or 50 IU/kg every 2 days to 3 days were administered to treat bleeds during a major surgery (allowed after 6 exposure days).
In the XTEND-1 trial, the concomitant mediations most frequently used by patients were paracetamol, tozinameran, and celecoxib. The intake of FVIII products other than Altuviiio was a major deviation to the protocol leading to treatment discontinuation unless it occurred in a life-threatening emergency or because of an accidental use and the sponsor agreed to retain the participant in the study.
XTEND-Kids Trial
In the XTEND-Kids trial, all patients received Altuviiio prophylaxis at a dose of 50 IU/kg once weekly during scheduled study visits via a slow push IV injection at a rate of administration determined by the patient’s comfort level, taking into account minimum injection duration per vial as per protocol. Treatment of bleeding episodes with Altuviiio during prophylaxis and dosing during minor or major surgeries aligns with dosing in the XTEND-1 trial, as summarized previously. In the XTEND-Kids trial, the most frequently used concomitant medications (< 10% of participants) were paracetamol, tranexamic acid, sodium chloride, amoxicillin, and tozinameran (COVID-19 vaccine). Outside of a clinical trial setting, Altuviiio was available for administration in hospital or at home in both the XTEND-1 and XTEND-Kids studies. The only criteria for stopping the administration of Altuviiio was the development of inhibitors against FVIII. All participants who completed the study in both trials were invited to enrol in an ongoing open-label extension study (XTEND-ed, NCT04644575), based on eligibility criteria.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 7, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review according to the clinical expert(s) consulted by CDA-AMC and input from patient and clinician groups and public drug plans. Using the same considerations, the CDA-AMC review team selected end points that were considered to be most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE.
Table 7: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | XTEND-1 | XTEND-Kids |
|---|---|---|---|
Bleeding outcomes | |||
ABRa,b | Throughout the study | Primaryc (arm A) | Key secondary |
AjBRd | Throughout the study | Secondary | Secondary |
Intrapatient comparison of ABR | |||
Intrapatient comparison of ABR between Altuviiio versus historical prophylaxisb | — | Secondary | — |
FVIII inhibitor formation | |||
Number (%) of patients who developed FVIII inhibitorsa | Baseline, weeks 4,13, 26, 39, and 52 | Safety end point | Primary |
Physical functioning and pain (QoL) | |||
Haemo-QoL Physical Healtha,b | At baseline, week 26, and week 52 | Secondaryc | Secondary |
PROMIS Pain Intensitya,b | At baseline, week 26, and week 52 | Secondaryc | Exploratory |
Joint health | |||
HJHSa,b | At baseline, week 26, and week 52 | Secondaryc | Secondary |
Response during perioperative management | |||
Mean (SD) number of injections to maintain hemostasis per major surgeryb | — | Secondary | Secondary |
Harms | |||
TESAEb | Baseline, weeks 4, 13, 26, 39, and 52 | Secondary | Secondary |
ABR = annualized bleeding rate; AjBR = annualized joint bleeding rate; FVIII = factor VIII; Haem-A-QoL = Haemophilia Quality of Life questionnaire for adults; Haemo-QoL = Haemophilia Quality of Life questionnaire for children; HJHS = Hemophilia Joint Health Score; PROMIS = Patient-Reported Outcomes Measurement Information System; QoL = quality of life; SD = standard deviation; TESAE = treatment-emergent serious adverse event.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
aIdentified as important to clinician groups.
bIdentified as important by consulting clinical experts.
cStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
dIdentified as important for the pharmacoeconomic analysis.
Source: XTEND-1 Clinical Study Report54 and XTEND-Kids Clinical Study Report.17
Bleeding Outcomes
Annualized Bleeding Rate
The primary efficacy end point in the XTEND-1 study was the ABR in the prophylaxis arm (arm A). The ABR for each individual patient was calculated using the following formula:
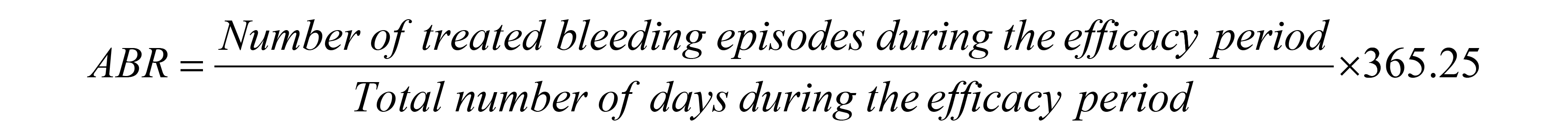
A bleeding episode was defined as an episode that starts from the first sign of bleeding and ends no more than 72 hours after the last injection to treat the bleeding episode. A bleeding episode was counted in the primary analysis if it was treated with Altuviiio. All types of bleeding episodes were included in determining the annualized number. An intrapatient comparison of ABR, based on the same definition, was also included in the studies.
Annualized Joint Bleeding Rate
AjBR was calculated using the number and location of all bleeds (spontaneous, traumatic, and type unknown) occurring in all joints during the efficacy period. In the XTEND-1 trial, an intrapatient comparison of AjBR in arm B between Altuviiio weekly prophylaxis and on-demand treatment was performed.
Development of Inhibitor Development Against FVIII
The primary end point in the XTEND-Kids study was the occurrence of inhibitor development against FVIII. This was defined as an inhibitor result of greater than 0.6 BU/mL, confirmed by a second test result from a separate sample, drawn 2 weeks to 4 weeks following the date when the original sample was drawn. Both tests were performed using the modified Nijmegen-Bethesda assay. The date of inhibitor development was the date of the sample with the first positive test result which was subsequently confirmed by the second sample. The overall incidence of positive inhibitor formation was calculated using the following formula:
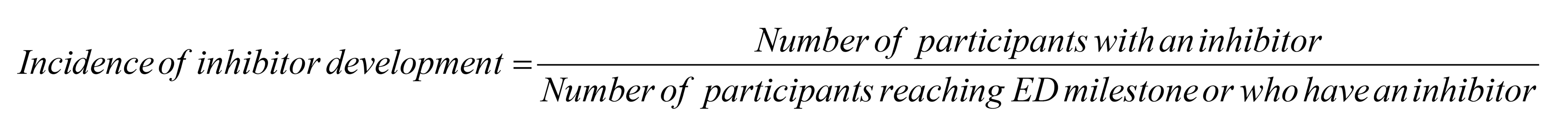
Haem-A-QoL
Across the XTEND trials, QoL data were collected in patients aged 17 years or older using the Haem-A-QoL, and via the Haemophilia Quality of Life Questionnaire for Children (Haem-A-QoL Kids short version) in patients aged 4 years to 16 years. Details of these questionnaires are in Table 8. In the XTEND-Kids study, parents also completed the Haem-A-QoL parent-proxy versions of the questionnaire. Questionnaires were summarized in terms of subscale scores and a total score. Scores were transformed to produce a Transformed Scale Score ranging from 0% to 100% with higher Transformed Scale Score values representing a worse QoL. Changes in total scores of Haem-A-QoL from baseline to week 52 were analyzed as secondary end points for the XTEND-1 study (arm A) and the XTEND-Kids study, respectively.
PROMIS Instruments
PROMIS instruments were used to evaluate QoL. In arm A of the XTEND-1 study, changes in PROMIS Pain Intensity and PROMIS Short Form Physical Function (in those aged ≥ 18 years) from baseline to week 52 were secondary end points. The assessment of pain in the XTEND-1 study was based on item 3a (intensity of pain at its worst score over the past 7 days). In the XTEND-Kids study, 3 PROMIS instruments (Pediatric Pain Intensity 1a, Pediatric Short Form Pain Interference, and Pediatric Short Form Physical Function) were used in exploratory end point analyses, completed by pediatric patients aged between 6 years to 12 years; if patients were aged younger than 6 years, parents completed the questionnaire as a proxy. The Pain Intensity instrument is a 3-item scale that evaluates pain severity using a 5-point Likert scale ranging from 0 (no pain) to 5 (very severe). The first 2 items assess pain utilizing a 7-day recall period, while the last item asks patients to rate their current level of pain. This tool is available for adults aged 18 years and older. The PROMIS Pediatric Pain Intensity form includes 1 item, rating pain intensity over the past 7 days, with a numeric rating scale ranging from 0 to 10, with 0 indicating no pain and 10 indicating worse pain. For each PROMIS instrument, the total raw score was converted to a standardized t score with a mean of 50 and a SD of 10; for negatively worded concepts like pain intensity, a t score of 60 was 1 SD worse than average, whereas for positively worded concepts like physical function, a t score of 60 was 1 SD better than average.
Hemophilia Joint Health Score
HJHS is a functional measure of joint health which was used to evaluate the effect of Altuviiio prophylaxis on joint health outcomes. HJHS was assessed in the XTEND-Kids study for patients aged 4 years and older and in arm A of the XTEND-1 study. The HJHS total score was calculated based on the sum of scores from 6 evaluated joints (ankles, elbows, and knees evaluated in terms of swelling, muscle atrophy, crepitus, flexion loss, extension loss, joint pain, and strength) and gait score (based on walking and climbing stairs). Total score ranges from 0 to 124, with higher scores indicating increased disease severity.
Response to Perioperative Management
Patients who underwent major surgery during the XTEND trials were included in the surgery subgroups to assess the efficacy of Altuviiio in the control and prevention of bleeding in the perioperative setting. Response to perioperative management was assessed as investigator’s or surgeon’s assessment of the patient’s hemostatic response to treatment and the number of injections and dose needed to maintain hemostasis during major surgery. Investigator or surgeon assessment of patient’s hemostatic response to Altuviiio treatment was collected 24 hours postsurgery based on the ISTH 4-point scale. Responses were given a numeric score (excellent = 1, good = 2, fair = 3, poor or none = 4), with a lower average score indicating better patient response to surgery with Altuviiio treatment. The number of injections, mean dose per injection (IU/kg), and total dose (IU/kg) required to maintain hemostasis during surgery was summarized for all major surgeries for patients in the surgery subgroup.
Additional Outcomes
Other outcomes of interest to the CDA-AMC review team including patient response to Altuviiio treatment of bleeding episodes and total annualized consumption of Altuviiio are shown in Appendix 1 (the XTEND-1 study) and Appendix 2 (the XTEND-Kids study).
Harms
All AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 24.1. The primary focus of AE reporting was on TEAEs. An AE with incomplete or missing date or time of onset (occurrence, worsening, or becoming serious) was classified as a TEAE unless there was definitive information to determine it was a pretreatment AE.
The occurrence of embolic and thrombotic events was also described. Embolic and thrombotic events were defined as arterial or venous thrombosis, confirmed by imaging. The analysis consisted of a search of TEAE data using the embolic and thrombotic events Standardized MedDRA Query.
Table 8: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
Haem-A-QoL Physical Health score | The Haem-A-QoL questionnaire assesses the quality of life of adults (aged ≥ 17 years) with hemophilia over the past 4 weeks. It contains 46 items in 10 domains: physical health (5 items), feelings (4 items), view of self (5 items), sports and leisure (5 items), work and school (4 items), dealing with hemophilia (3 items), treatment (8 items), future (5 items), family planning (4 items), and partnership and sexuality (3 items); the total score is also considered. Change in total score and the physical health domain from baseline to week 52 were assessed as secondary end points in arm A of XTEND-1. Each item was answered considering the last month on the 5-point Likert scale ranging from “never” to “all the time,” with several items having a “not applicable” option. Nonmissing scores were rescaled to express the score as a percentage from 0% to 100%, where a lower score represents a higher quality of life.55,56 | Validity: In a severe hemophilia (A or B) population (aged ≥ 12 years), several Haem-A-QoL domains and “total score” demonstrated known groups and convergent validity when compared with other trial measures, including the EQ-5D questionnaire (items and total scores) and joint impairment. Reliability: Internal consistency and reliability was previously reported to be sufficiently adequate (Cronbach alpha > 0.70) for 9 of the 10 Haem-A-QoL domains and for “total score” in a severe hemophilia (A or B) population (aged ≥ 12 years) at baseline. Responsiveness: Change in score correlations (baseline to 28 weeks) between the EQ-5D and the Haem-A-QoL total score, and physical health and feelings domains were moderate in magnitude (r ≥ 0.33) demonstrating sensitivity to change for these outcome measures in patients with hemophilia A.57 | In a severe hemophilia (A or B) population (aged ≥ 12 years), the most indicative, meaningful within-patient change was a reduction of 7.1 points for the total score and 10.0 points for Physical Health score over 6 months based on anchor- and distribution-based methods.55 In the same population, the Haem-A-QoL Physical Health domain threshold for meaningful change ranged from 8.0 points to 11.9 points using distribution-based methods.55 |
PROMIS Pain Intensity | PROMIS instruments are standardized tools used to evaluate different measures of physical and social well-being.58,59 These tools can be used in patients with chronic diseases across a variety of health domains to provide assessments of outcomes such as pain, fatigue, or emotional distress.59 The Pain Intensity instrument is a 3-item scale that evaluates pain severity using a 5-point Likert scale ranging from 0 (had no pain) to 5 (very severe).58,60 The first 2 items assess pain utilizing a 7-day recall period, while the last item asks patients to rate their current level of pain.58,60 This tool is available for adults aged ≥ 18 years of age.58,60 The pediatric version (Pediatric Pain Intensity 1a) was used in exploratory end point analyses for the XTEND-Kids study, completed by pediatric patients aged 6 years to 12 years; if patients were < 6 years of age, parents completed the questionnaire as a proxy. The PROMIS Pediatric Pain Intensity form includes 1 item, rating pain intensity over the past 7 days, with a numeric rating scale ranging from 0 to 10, with 0 indicating no pain and 10 indicating worse pain.61 For each PROMIS instrument, the total raw score is converted to a standardized t score with a mean of 50 and a SD of 10 to compare pain to the general population.60 For negatively worded concepts like pain intensity, a t score of 60 is 1 SD worse than average; whereas, for positively worded concepts like physical function, a t score of 60 was 1 SD better than average.60 | The PROMIS Short Form Pain Intensity questionnaire has not yet been validated among adult patients with hemophilia. However, 1 study found that the Pain Intensity questionnaire demonstrated acceptable construct validity, reliability, and responsiveness in patients receiving total hip arthroplasty.62 No studies were identified that investigated the PROMIS Pain Intensity 1a in pediatric patients. Validity: Scale-specific hypothesis testing was employed to assess construct validity using correlations with OHS total score and OHS pain and function subdomains at baseline and 12 months, and symptom-specific well-being at 12 months. Results showed high validity with PROMIS Pain Intensity.62 Reliability: PROMIS Pain Intensity demonstrated internal consistency was evaluated using Cronbach alpha, with values between 0.70 to 0.95. Additionally, it met the criteria for test-retest reliability (correlation coefficient ≥ 0.70). The PROMIS Pain Intensity instrument demonstrated the highest effect size, based on standard error of measurement calculated from the mean change score.62 Responsiveness: As hypothesized, negative change in score correlations were observed between OHS and PROMIS Pain Intensity, (r ≥ 0.5) demonstrating acceptable sensitivity to change.62 | No MID was identified for the PROMIS Pain Intensity questionnaire in adult or pediatric patients with hemophilia A. |
HJHS | The HJHS assesses specific features, or items, of the 6 index joints (elbows, knees, and ankles) as well as an assessment of global gait in pediatric and adult patients. For each of the 6 joints, the following items are scored:
The maximum score for an individual index joint is 20. Gait is scored 0 to 4 based on walking, stairs, running, and hopping on 1 leg. The total score is the sum of all joint and gait scores (range = 0 to 124), with a higher number indicating more severe joint damage.63 | Validity: In a multicentre international study containing patients with hemophilia and healthy adults, HJHS total scores were highly correlated with the WFH Gilbert scores (Spearman correlation, rs = 0.95), which is the original WFH Orthopedic Joint Score, demonstrating convergent construct validity.63 Discriminant (known groups) construct validity was evaluated by the Kruskal-Wallis nonparametric analysis of variance. The HJHS total score significantly differentiated between age groups (Kruskal-Wallis t = 35.02; P < 0.001) and disease severity in persons with hemophilia.63 Reliability: In a study consisting of male patients with hemophilia in the US, the Cronbach alpha value was 0.97 for the HJHS total score, exceeding the threshold of 0.70 established in previous studies, indicating sufficient internal consistency.64 All items on the HJHS had been reported to capture sufficient correlation with their respective joint total scores (r = 0.34 to 0.83, where r is the Pearson product moment correlation coefficient).64 In another study consisting of male patients with hemophilia in the US, the HJHS ankle domain reached a correlation of r > 0.5 for several domains and summary scores related to physical function, including scores specific to activity of the lower extremities. HJHS total scores also demonstrated similar correlations for similar domains and summary scores, except use of transportation. However, HJHS global gait did not reach a correlation of r > 0.5 with any patient-reported outcome instrument domain or summary scores.65 In a multicentre, international study of patients with hemophilia and healthy adults, the HJHS 2.1 items demonstrated adequate internal reliability (Cronbach alpha = 0.88).66 Item scores were correlated with total scores, with almost all HJHS items (muscle atrophy, crepitus, flexion and extension loss, joint pain, and strength) being highly correlated (alpha > 0.70), except for swelling and duration of swelling, which were only moderately correlated.63 Responsiveness: The HJHS is more sensitive to early joint changes than the Gilbert score.67 It can reportedly distinguish between different prophylactic strategies in young adults with severe hemophilia,68 between severe and nonsevere hemophilia in children,67,69 and is responsive to changes following physiotherapy treatment.70 However, due to high sensitivity, it also showed positive scores in 40% of unaffected young adults (total score ≤ 3 points).71,72 | No MID was identified for this population. |
Haem-A-QoL = Haemophilia Quality of Life questionnaire for adults; HJHS = Hemophilia Joint Health Score; MID = minimal important difference; OHS = Oxford Hip Score; PROMIS = Patient-Reported Outcomes Measurement Information System; SD = standard deviation; WFH = World Federation of Hemophilia.
Statistical Analysis
Sample Size and Power Calculation
XTEND-1 Trial
The sample size in the XTEND-1 study was based on risk of immunogenicity, the primary end point, and the key secondary end point.
Immunogenicity: To rule out a greater than acceptable risk of immunogenicity, assuming a dropout rate of 15% with a sample size of 124 patients in the prophylaxis arm, there would be 104 evaluable patients with 50 exposure days or more. If 2 patients or less out of 104 evaluable patients develop an inhibitor, then the upper bound of an exact 95% CI would exclude 6.8%, a threshold determined at the FDA FVIII Inhibitor Workshop that was held in 2003.
Primary end point: The primary efficacy objective of this study was to evaluate the efficacy of Altuviiio weekly prophylaxis as estimated by the mean ABR and 1-sided 97.5% CI in arm A. Based on currently marketed FVIII products, the mean ABR during clinical trials ranges from approximately 2 bleeds to 5 bleeds per year but can be as high as 6 bleeds per year.73-76 To show adequate control of bleeding consistent with currently marketed FVIII products, and to account for this variability, a clinically meaningful treatment effect may be claimed if the upper bound of the CI of the estimated ABR is 6 or less. In a phase III study of a recombinant FVIII Fc fusion protein, the mean ABR for an individualized prophylaxis arm was 2.9 and the dispersion factor was estimated at 2.3.54 Based on 2,000 simulations of a negative binomial regression model with mean ABR of 2.9 and dispersion factor of 2.3, a sample size of 124 patients will provide at least 90% power for the upper bound of the CI to exclude an ABR greater than 6, assuming a 15% dropout rate.
Key secondary end point: For the key secondary efficacy end point, an intrapatient comparison of ABR during the Altuviiio weekly prophylactic treatment period versus the historical prophylaxis ABR was performed using noninferiority testing for patients in arm A. The noninferiority margin was estimated based on the known treatment effect between on-demand and prophylactic treatment. A meta-analysis of phase III registrational studies for recombinant FVIII products that include both on-demand and prophylactic treatment arms estimated an average reduction of 31 bleeds per year between on-demand and prophylactic treatment regimens. The lower bound of this treatment effect was 27 bleeds per year. Using a fixed margin approach to maintain a substantial amount (85%) of the treatment effect results in a noninferiority margin of 4. For a noninferiority test, a sample size of 63 achieves 90% power to detect noninferiority using a 1-sided paired Wilcoxon signed rank test at a 0.025 significance level when the actual mean of paired differences is 0 and the noninferiority margin is 4. Without prior knowledge of the SD of the paired differences, a conservative estimate of 10 was assumed. To account for dropout and the use of the PPS, a total of at least 75 subjects who had completed 6 or more months of participation in the observational prestudy 242HA201/OBS166221 were to be enrolled in arm A.
XTEND-Kids Trial
In the XTEND-Kids study, the primary end point was FVIII inhibitor development and key secondary end points were bleeding outcomes including ABR and AjBR. The determination of the number of patients was based on clinical rather than statistical considerations. Taking into consideration the guideline from the Committee for Medicinal Products for Human Use,77 approximately 65 previously treated patients would need to be enrolled to obtain 50 or more patients with 50 or more exposure days at the end of the study. At least 12 patients in each age cohort needed to have completed adequate blood sample collection to assess key PK parameters.
Statistical Testing
XTEND-1 Trial
In the XTEND-1 trial, the primary end point, key secondary end point, and selected secondary end points of high clinical importance in hemophilia treatment were tested hierarchically to maintain the overall type I error rate of 0.05 or less. The end points included in the hierarchy were measured in arm A (the prophylaxis arm) in the following order.
Primary end point: The primary efficacy end point was ABR in arm A. The primary analysis of the primary end point estimated mean ABR and 1-sided 97.5% CI using a negative binomial model for the weekly prophylaxis arm (arm A) was based on the FAS. If the upper limit of the CI is less than or equal to 6, the weekly prophylactic treatment regimen was considered to have provided adequate bleeding control. Sensitivity analyses of the primary end point were performed using the PPS, and the FAS which included participants with an efficacy period of at least 26 weeks.
Key secondary end point: A key secondary end point was intrapatient comparison of ABR during Altuviiio weekly prophylaxis versus ABR during historical prophylaxis — noninferiority analysis. The intraparticipant comparison of ABR between Altuviiio weekly prophylaxis and historical prophylaxis was performed using a negative binomial regression model accounting for overdispersion with the dependent variable as “total bleeding episodes,” covariate as “treatment regimen,” repeated variable as “subject,” and log time as an offset variable. The mean paired difference and 95% CI were estimated using the PPS (as primary analysis) and FAS (as supportive analysis). Noninferiority was declared if the upper bound of the 1-sided 97.5% CI is less than 4. As a supportive analysis, the noninferiority analysis was also performed using the Wilcoxon signed rank test using the PPS. The null hypothesis that the median difference is 4 or more will be tested against the alternative hypothesis that the median difference is less than 4. The null hypothesis was rejected if P was less than 0.025, establishing noninferiority of Altuviiio weekly prophylactic treatment to historical prophylaxis. The 95% CI of the median of the ABR difference was calculated using Walsh averages and a Hodges-Lehmann estimate of the median difference.
Key secondary end point: Another key secondary end point was intrapatient comparison of ABR during Altuviiio weekly prophylaxis versus ABR during historical prophylaxis — superiority analysis. If noninferiority is achieved, then superiority was evaluated sequentially using a negative binomial regression model as specified previously. The paired ABR ratio and 95% CI were estimated using the FAS. The null hypothesis that the paired ABR ratio (Altuviiio: historical prophylaxis) is 1 or more was tested against the alternative hypothesis that the ABR ratio is less than 1. Superiority was declared if the upper bound of the 1-sided 97.5% CI is less than 1. A supportive analysis will also be performed for the superiority test when noninferiority is achieved. The mean paired ABR difference and 95% CI were estimated using the same negative binomial regression model as specified previously based on the FAS. The null hypothesis that the paired ABR difference is 0 or more was tested against the alternative hypothesis that the ABR difference is less 0. Superiority was declared if the upper bound of the 1-sided 97.5% CI is less than 0.
Selected secondary end points: These included Haem-A-QoL Physical Health domain, PROMIS Pain intensity 3a first item (i.e., PAINQU6), and HJHS total score. These outcomes were analyzed as a change from baseline to week 52. The change from baseline to week 52 was analyzed using a mixed-effects model with repeated measures (MMRM) analysis using observed data in arm A and excluding visits that are coincidental with a major surgical or rehabilitation period. The MMRM model included the baseline value of the end point as a covariate and visit as a fixed effect. The adjusted mean change in Haem-A-QoL Physical Health score from baseline to week 52, along with its 95% CI, were estimated by the MMRM model. A sensitivity analysis was performed for study participants who rolled over from the lead-in observational study using the same MMRM model.
XTEND-Kids Trial
All analyses in the XTEND-Kids trial were descriptive in nature and adjustments for multiplicity were not applied.
Sensitivity Analyses
A summary of the sensitivity analyses for select end points is provided in Table 9. Sensitivity analyses of the primary end point were performed using the PPS, and with FAS, including participants with an efficacy period of at least 26 weeks.
Subgroup Analyses
XTEND-1 Trial Primary End Point: ABR in Arm A
To assess the homogeneity of the treatment effect across various subgroups, analyses were performed on the primary end point across the following subgroups (categories with < 5 patients could be combined with other categories):
by age (12 to 17 years, 18 to 64 years, ≥ 65 years)
baseline bleeding phenotype (estimated bleeds in prior 12 months = 0, < 0 to 5, > 5 to 10, or > 10)
by number of target joints (none, less than or equal to median of number present, greater than median of number present)
by dosing and dosing interval compliance (< 80%, ≥ 80%).
The estimated mean ABR and the corresponding 95% CI were provided, using a negative binomial model for the weekly prophylaxis arm (arm A) based on the FAS for each subgroup, and using the same method as applied to the primary analysis. If the upper limit of the CI is less than or equal to 6, the weekly prophylactic treatment regimen was considered to provide adequate bleeding control.
XTEND-Kids Trial Secondary End Point: ABR
To assess the homogeneity of the treatment effect across various subgroups, analyses of the ABR were performed across the following subgroups (categories with < 5 patients could be combined with other categories):
baseline bleeding phenotype (estimated bleeds in prior 12 months = 0, < 0 to 5, > 5 to 10, or > 10)
by number of target joints (none, less than or equal to median of number present, greater than median of number present)
by dosing and dosing interval compliance (< 80%, ≥ 80%).
The mean and 95% CI of ABR were estimated using a negative binomial model. The model included the number of treated bleeding episodes during the efficacy period as the response variable and the log-transformed duration of efficacy period as the offset variable to account for variable duration. Individual ABR was calculated for each participant and summarized descriptively. The summaries were presented by age cohort and overall, for the FAS.
Table 9: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
XTEND-1 | ||||
ABR in arm A | Mean ABR and 1-sided 97.5% CI was estimated using a negative binomial model with the total number of treated bleeding episodes during the efficacy period as the response variable and log-transformed efficacy period duration (in years) as an offset variable. | None | NR | Sensitivity analyses of the primary end point were performed using the per-protocol set, as well as using the FAS including patients with an efficacy period of at least 26 weeks. |
Intrapatient ABR comparison: Altuviiio prophylaxis versus historical prophylaxis in arm A (noninferiority) | The comparison was performed using a negative binomial regression model with treatment (Altuviiio prophylaxis versus historical prophylaxis) as covariate. Noninferiority was declared if the upper bound of the 1-sided 97.5% CI was < 4. | None | NR | Not performed |
Intrapatient ABR comparison: Altuviiio prophylaxis versus historical prophylaxis in arm A (superiority) | If noninferiority is achieved, then superiority was evaluated sequentially using a negative binomial regression model. The paired ABR ratio and 95% CI was estimated using the FAS. Superiority was declared if the upper bound of the 1-sided 97.5% CI was < 1. | None | NR | Not performed |
Intrapatient ABR comparison: Altuviiio on-demand treatment versus prophylaxis in arm B | Estimated using a repeated negative binomial model with treatment (Altuviiio prophylaxis versus historical prophylaxis) as covariate. P value relates to paired rate ratio (Altuviiio prophylaxis or on demand); P ≥ 0.5. | None | NR | Not performed |
Percentage of patients maintaining FVIII activity exceeding prespecified levels | Descriptive statistics. Listing is provided for each patient’s baseline-corrected FVIII activity levels. | None | NR | Not performed |
AjBR | An intrapatient comparison of AjBR between weekly Altuviiio prophylaxis and on-demand treatment in arm B was performed using a repeated negative binomial regression model with treatment (Altuviiio prophylaxis versus on demand) as covariate. P value relates to paired rate ratio (Altuviiio prophylaxis/on demand); P ≥ 0.5. | None | NR | Not performed |
Change from baseline to week 52 in HJHS total score | The LS mean (SE) and 95% CI were estimated by MMRM with visit as fixed effect, and baseline HJHS total score as covariate in arm A. | None | The total joint score was set as missing if any 1 of the individual item scores at any joint was missing or if scores are evaluated within 2 weeks after a joint or muscle bleeding episode. The total joint score was re-derived if surgery was performed on a joint. The scores for that joint were replaced with the scores of the same joint at the last visit before the surgery using the LOCF technique. | In the XTEND-1 trial, a sensitivity analysis was performed for study patients who rollover from the lead-in observational study using the same MMRM model. |
Haem-A-QoL and Haemo-QoL questionnaires | LS mean (SE) and 95% CI were estimated by MMRM in arm A with visit as fixed effect, and baseline Haem-A-QoL Physical Health score as covariate. | Assessments during major surgical/ rehabilitation periods were excluded | When missing data are presented, a subscale score can only be calculated if at least 50% of questions for that subscale are answered (nonmissing and not “NA”). | A sensitivity analysis was performed for study patients who rollover from lead-in observational study using the same MMRM model as described. |
PROMIS Pain Intensity | LS mean (SE) and 95% CI were estimated by MMRM with visit as fixed effect, and baseline PROMIS Pain Intensity 3a first item (i.e., past 7 days intensity of pain at its worst score) as covariate in arm A. The adjusted mean change in PROMIS Pain Intensity 3a score from baseline to week 52, along with its 95% CI, was estimated by the MMRM model. | Assessments during major surgical/rehabilitation periods were excluded | NR | A sensitivity analysis was performed for study patients who rollover from the lead-in observational study using the same MMRM model. |
Number of injections and dose to maintain hemostasis for major surgery | Descriptive statistics | None | NR | Not performed |
Total Altuviiio consumption for major surgery | Descriptive statistics | None | NR | Not performed |
XTEND-Kids | ||||
FVIII inhibitor development | The 95% CI was calculated using Clopper-Pearson exact method. | None | NR | Not performed |
ABR | Estimated using a negative binomial model with the total number of treated bleeding episodes during the efficacy period as the response variable and log-transformed efficacy period duration (in years) as an offset variable. | None | NR | Sensitivity analysis of the ABR was performed using the per-protocol set, as well as using the FAS including patients with an efficacy period of at least 26 weeks. |
Percentage of patients maintaining FVIII activity exceeding prespecified levels | Descriptive statistics | None | NR | Not performed |
AjBR | Estimated using a negative binomial model with the total number of treated bleeding episodes during the efficacy period as the response variable and log-transformed efficacy period duration (in years) as an offset variable. | None | NR | An ad hoc sensitivity analysis was performed excluding the patient who did not receive the weekly prophylactic treatment for an extended period of time. |
Change from baseline to week 52 in HJHS total score and domain score | Descriptive statistics | None | The total joint score was set as missing if any 1 of the individual item score at any joint was missing or if scores are evaluated within 2 weeks after a joint or muscle bleeding episode. The total joint score was re-derived if surgery was performed on a joint. The scores for that joint were replaced with the scores of the same joint at the last visit before the surgery using the LOCF technique. | Not performed |
Haemo-QoL questionnaire | Descriptive statistics | None | When missing data were presented, a subscale or total score could only be calculated if at least 50% of questions for that subscale or questionnaire were answered (nonmissing and not “NA”). | Not performed |
Number of injections and dose to maintain hemostasis for major surgery | Descriptive statistics | None | NR | Not performed |
Total Altuviiio consumption for major surgery | Descriptive statistics | None | NR | Not performed |
ABR = annualized bleeding rate; AjBR = annualized joint bleeding rate; CI = confidence interval; FAS = full analysis set; FVIII = factor VIII; Haem-A-QoL = Haemophilia Quality of Life questionnaire for adults; Haemo-QoL = Haemophilia Quality of Life questionnaire for children; HJHS = Hemophilia Joint Health Score; LOCF = last observation carried forward; MMRM = mixed-effects model of repeated measures; NA = not applicable; NR = not reported; PROMIS = Patient-Reported Outcomes Measurement Information System; SE = standard error.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Source: XTEND-1 Clinical Study Report54 and XTEND-Kids Clinical Study Report.17
Analysis Populations
Analysis populations of the XTEND-1 and XTEND-Kids trials are presented in Table 10.
Table 10: Analysis Populations of XTEND-1 and XTEND-Kids Trials
Study | Population | Definition | Application |
|---|---|---|---|
XTEND-1 | All-enrolled analysis set | All patients who were enrolled in the study, regardless of whether they were dosed with study drug or not. Patients were considered enrolled when the investigator had verified that they were eligible according to the criteria in the protocol. | Patient disposition and enrolment summaries were based on the all-enrolled analysis set. |
FAS | All patients who received at least 1 dose of study drug. | All analyses of demographics, baseline characteristics, and efficacy were based on the FAS, unless otherwise specified. | |
PPS | A subset of the FAS including patients who did not have important protocol deviations potentially impacting efficacy. | The PPS was utilized for analysis of the key secondary efficacy end point, as well as sensitivity analysis of the primary end point. | |
Safety analysis set | The safety analysis set was the same as the FAS and included all patients who received at least 1 dose of study drug. | All analyses of safety were based on the safety analysis set, unless otherwise specified. | |
PKAS | All patients who had completed adequate blood sample collection to assess key PK parameters, as determined by the PK scientist. | All analyses of PK were based on the PKAS, unless otherwise specified. | |
Sequential PK subgroup | All patients who had evaluable PK profiles for both the baseline and repeat PK profiles, as determined by the PK scientist. Select patients from arm A participated in the sequential PK subgroup and underwent more extensive PK sampling at baseline and again at week 26. | Conducted to better characterize elimination half-life of Altuviiio. | |
Surgery subgroup | All patients who underwent major surgery after the first dose of study drug. | All analyses of surgical end points were based on the surgery subgroup. | |
XTEND-Kids | All-enrolled analysis set | All patients who were enrolled in the study, regardless of whether or not they were dosed with study drug. | Patients will be considered enrolled when the investigator has verified that they are eligible according to the criteria in the protocol. Patient disposition and enrolment summaries will be based on the all-enrolled analysis set. |
FAS | All patients who took at least 1 dose of study intervention. | All analyses of demographics, baseline characteristics, and efficacy will be based on the FAS, unless otherwise specified. | |
PPS | A subset of the FAS including patients who do not have important protocol deviations potentially impacting efficacy. | The PPS was utilized for sensitivity analysis of the efficacy end point of annualized bleeding rate. | |
Safety analysis set | The safety analysis is the same as the FAS and includes all patients who received at least 1 dose of study drug. | All analyses of safety were based on the safety analysis set, unless otherwise specified. | |
PKAS | All patients who have completed adequate blood sample collection to assess key PK parameters. | All analyses of PK were based on the PKAS. | |
Surgery subgroup | All patients who have undergone major surgery after the first dose of study drug. | All analyses of surgical end points were based on the surgery subgroup. |
FAS = full analysis set; PK = pharmacokinetics; PKAS = pharmacokinetic analysis set; PPS = per-protocol set.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Source: XTEND-1 Clinical Study Report54 and XTEND-Kids Clinical Study Report.17
Results
Patient Disposition
Overall, 159 patients were included in the XTEND-1 trial. This consisted of 133 participants in arm A and 26 participants in the arm B. Participants in the arm B were assigned to on-demand treatment followed by prophylaxis. The XTEND-Kids trial included a total of 74 patients, with 38 aged younger than 6 years and 36 between the age of 6 years and 12 years. Screening failure rates were similar in both the XTEND-1 trial (6.5%; 11 of 170) and the XTEND-Kids trial (6.3%; 5 of 79). The proportion of patients who discontinued from the study in the XTEND-1 trial was 6.8% (9 of 133) in arm A and 3.8% (1 of 26) in arm B. In the XTEND-Kids trial, 5.3% (2 of 38) of patients who were in the cohort of patients aged younger than 6 years discontinued from study.
The reasons for discontinuation in the XTEND-1 trial were mostly withdrawn consent or prohibited concomitant medication; other reasons were cited for discontinuation in the XTEND-Kids trial. There were no reported important protocol deviations that could potentially influence the efficacy results in either the XTEND-1 or XTEND-Kids trial. The summary of patient disposition is presented in Table 11.
Table 11: Summary of Patient Disposition in XTEND-1 and XTEND-Kids Trials
Patient disposition | XTEND-1 | XTEND-Kids | |||
|---|---|---|---|---|---|
Arm A Prophylaxis: Altuviiio, 50 IU/kg once weekly n = 133 | Arm B | Aged < 6 years n = 38 | Aged 6 to < 12 years n = 36 | ||
On demand: Altuviiio, 50 IU/kg n = 26 | Prophylaxis: Altuviiio, 50 IU/kg once weekly n = 26 | ||||
Screened, N | 170 | 79 | |||
Screen failure, n (%) | 11 (6.5) | 5 (6.3) | |||
Reason for screening failure, n (%) | |||||
Inclusion criteria | 4 (2.4) | 2 (2.5) | |||
Exclusion criteria | 2 (1.2) | 3 (3.8) | |||
Enrolled, N | 133 | 26 | 26 | 38 | 36 |
Discontinued from study, n (%) | 9 (6.8) | 0 | 1 (3.8) | 2 (5.3) | 0 |
Reason for discontinuation, n (%) | |||||
Adverse events | 1 (0.8) | 0 | 0 | 0 | 0 |
Lost to follow-up | 0 | 0 | 0 | 0 | 0 |
Protocol violation | 1 (0.8) | 0 | 0 | 0 | 0 |
Death | 0 | 0 | 1 (3.8) | 0 | 0 |
Consent withdrawn | 3 (2.3) | 0 | 0 | 0 | 0 |
Inability/unwillingness to comply with protocol | 0 | 0 | 0 | 0 | 0 |
Prohibited concomitant medications due to medical need as determined by investigator | 3 (2.3) | 0 | 0 | 0 | 0 |
Investigator decision | 0 | 0 | 0 | 0 | 0 |
Withdrawal criteria | 0 | 0 | 0 | 0 | 0 |
Other | 1 (0.8) | 0 | 0 | 2 (5.3) | 0 |
FAS, Na | 133 (100) | 26 (100) | 26 (100) | 38 (100) | 36 (100) |
Patients with an efficacy periodb | 133 (100) | 26 (100) | 26 (100) | 38 (100) | 36 (100) |
Safety analysis set, Nc | 133 (100) | 26 (100) | 26 (100) | 38 (100) | 36 (100) |
Per-protocol set, nd | 129 (97.0) | 25 (96.2) | 25 (96.2) | 38 (100) | 36 (100) |
PK analysis sete | 133 (100) | 26 (100) | 26 (100) | 19 (50.0) | 18 (50.0) |
Sequential PK subgroupf | 17 (12.8) | 0 | 0 | NA | NA |
Surgery subgroupg | 10 (7.5) | 0 | 1 (3.8) | 2 (5.3) | 0 |
Number of major surgeries | 11 | 0 | 1 | 2 | 0 |
Patients with at least 1 minor surgery | 12 (9.0) | 2 (7.7) | 1 (3.8) | 3 (7.9) | 5 (13.9) |
Number of minor surgeries | 15 | 2 | 1 | 3 | 6 |
COVID-19 nonimpacted populationh | 132 (99.2) | 26 (100) | 26 (100) | 38 (100) | 36 (100) |
FAS = full analysis set; NA = not available; PK = pharmacokinetics.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
aAll patients who received at least 1 dose of study drug.
bFAS population with at least 1 day of treatment for episodic regimen or at least 2 prophylactic injections for prophylactic regimen.
cAll patients who received at least 1 dose of study drug.
dA subset of FAS including patients who do not have important protocol deviations potentially impacting efficacy.
ePatients who have completed adequate blood sample collection to assess key PK parameters, as determined by the PK scientist.
fPatients who have evaluable PK profiles for both the baseline and repeat PK profiles, as determined by the PK scientist.
gPatients who have undergone major surgery after the first dose of study drug.
hPatients are without any major deviation related to COVID-19.
Source: XTEND-1 Clinical Study Report54 and XTEND-Kids Clinical Study Report.17
Baseline Characteristics
The baseline characteristics of the XTEND trials are summarized in Table 12. In the XTEND-1 study, the mean age of patients was 35.4 years (SD = 15.1 years), ranging from 12 years to 72 years; there were 25 (15.7%) adolescents patients aged 12 years to 17 years (all were in arm A), 129 (81.1%) adult patients aged 18 years to 64 years, and 5 (3.1%) patients aged greater than 65 years. One female patient was enrolled; all other patients were male. Additionally, most patients (78.6%) had no family history of FVIII inhibitors. In the 12 months before the study, the mean number of bleeding episodes reported was 3.2 (SD = 5.4) in arm A for patients who all previously received a different prophylactic regimen and 35.7 (SD = 22.2) in arm B patients who previously receiving on-demand treatment. When the analysis was restricted to a North American subset, the mean age of patients was 31.1 years (SD = 16.7 years), ranging from 12 years to 72 years. The majority of patients were male, excluding 1 female patient. Only 1 (3.9%) patient had a family history of inhibitors.
In the XTEND-Kids trial, all 74 pediatric patients were male with a mean age of 6.0 years (SD = 2.9 years); ages ranged from 1.4 years to 11.0 years. The majority (77%) had no family history of an inhibitor. In the 12 months before the study, the mean bleeding episodes in patients on a prophylactic regimen was 2.1 (SD = 4.2) in the overall study population.
Table 12: Summary of Baseline Characteristics From Studies Included in the Systematic Review
Characteristic | XTEND-1 | XTEND-Kids | ||||
|---|---|---|---|---|---|---|
Arm A Prophylaxis: Altuviiio, 50 IU/kg once weekly n = 133 | Arm B On demand: Altuviiio, 50 IU/kg and prophylactic Altuviiio, 50 IU/kg once weekly n = 26 | Overall N = 159 | Aged < 6 years n = 38 | Aged 6 years to < 12 years n = 36 | Overall N = 74 | |
Age (years) | ||||||
Mean (SD) | 33.9 (15.3) | 42.8 (11.7) | 35.4 (15.1) | 3.69 (1.21) | 8.42 (2.08) | 6.0 (2.9) |
Median (range) | 34.0 (12 to 72) | 39.0 (23 to 68) | 35.0 (12 to 72) | 4.0 (1.4 to 5.0) | 8.0 (6.0 to 11.0) | 5.0 (1.4 to 11.0) |
12 to 17, n (%) | 25 (18.8) | 0 | 25 (15.7) | — | — | — |
18 to 64, n (%) | 104 (78.2) | 25 (96.2) | 129 (81.1) | — | — | — |
≥ 65, n (%) | 4 (3.0) | 1 (3.8) | 5 (3.1) | — | — | — |
Sex, n (%) | ||||||
Female | 1 (0.8) | 0 (0) | 1 (0.6) | 0 (0) | 0 (0) | 0 (0) |
Male | 132 (99.2) | 26 (100) | 158 (99.4) | 38 (100) | 36 (100) | 74 (100) |
Race, n (%) | ||||||
Asian | 29 (21.8) | 0 | 29 (18.2) | 4 (10.5) | 4 (11.1) | 8 (10.8) |
Black or African American | 3 (2.3) | 0 | 3 (1.9) | 1 (2.6) | 2 (5.6) | 3 (4.1) |
White | 71 (53.4) | 26 (100) | 97 (61.0) | 30 (78.9) | 25 (69.4) | 55 (74.3) |
Not reported | 26 (19.5) | 0 | 26 (16.4) | 0 | 4 (11.1) | 4 (5.4) |
Other | 4 (3.0) | 0 | 4 (2.5) | 3 (7.9) | 1 (2.8) | 4 (5.4) |
Region, n (%) | ||||||
Asia and Pacific Islands | 33 (24.8) | 0 | 33 (20.8) | 11 (28.9) | 8 (22.2) | 19 (25.7) |
Europe | 67 (50.4) | 14 (53.8) | 81 (50.9) | 7 (18.4) | 20 (55.6) | 27 (36.5) |
North America | 26 (19.5) | 0 | 26 (16.4) | 20 (52.6) | 8 (22.2) | 28 (37.8) |
South America | 7 (5.3) | 12 (46.2) | 19 (11.9) | 0 | 0 | 0 |
Weight (kg) | ||||||
Mean (SD) | 78.00 (19.29) | 80.80 (18.04) | 78.46 (19.06) | 17.93 (3.53) | 35.79 (12.86) | 26.62 (12.90) |
Median (range) | 78.0 (33.9 to 132.8) | 77.90 (50.0 to 119.5) | 78.00 (33.9 to 132.8) | 18.00 (11.4 to 25.7) | 32.85 (17.2 to 66.5) | 22.05 (11.4 to 66.5) |
Body mass index (kg/m2) | ||||||
Mean (SD) | 25.59 (5.00) | 26.91 (5.56) | 25.80 (5.10) | 16.62 (1.73) | 18.83 (3.90) | 17.70 (3.17) |
Median (range) | 25.51 (15.0 to 39.8) | 26.35 (16.7 to 40.8) | 25.74 (15.0 to 40.8) | 16.41 (13.9 to 21.5) | 18.22 (13.2 to 31.0) | 16.89 (13.2 to 31.0) |
< 25 , n (%) | 57 (42.9) | 9 (36.0) | 66 (41.8) | — | — | — |
≥ 25 to < 30, n (%) | 52 (39.1) | 9 (36.0) | 61 (38.6) | — | — | — |
≥ 30, n (%) | 24 (18.0) | 7 (28.0) | 31 (19.6) | — | — | — |
Age at start of first prophylaxis (years), median (range) | 1.0 (0 to 35) | 3.0 (0 to 62) | 1.0 (0 to 62) | 1.0 (0 to 4) | 1.0 (0 to 5) | 1.0 (0 to 5) |
Family history of FVIII inhibitor, n (%) | ||||||
Yes | 5 (4) | 0 | 5 (3) | 7 (18.4) | 1 (2.8) | 8 (10.8) |
No | 100 (75) | 25 (96) | 125 (79) | 26 (68.4) | 31 (86.1) | 57 (77.0) |
Unknown | 28 (21) | 1 (4) | 29 (18) | 5 (13.2) | 4 (11.1) | 9 (12.2) |
Bleeding episodes in past 12 months, mean (SD) | 3.2 (5.4) | 35.7 (22.2) | 8.3 (15.5) | 2.0 (2.5) | 2.1 (5.4) | 2.1 (4.2) |
Bleeding episodes into joints in past 12 months, mean (SD) | 2.3 (4.5) | 27.4 (18.6) | 6.0 (12.1) | 0.8 (2.1) | 1.3 (5.1) | 1.1 (3.9) |
≥ 1 target joint, n (%)a | ||||||
Yes | 26 (20) | 23 (88) | 49 (31) | 1 (3) | 1 (3) | 2 (3) |
No | 107 (80) | 3 (12) | 110 (69) | 37 (97) | 35 (97) | 72 (97) |
Hemophilia Joint Health Scoreb | ||||||
Number of patients evaluated | 116 | 25 | — | — | — | — |
Mean score (SD) | 18.1 (18.4) | 26.3 (13.2) | — | — | — | — |
Mean endogenous VWF level (SD) | 115.9 (42.9) | 138.1 (102.5) | 119.5 (57.2) | — | — | — |
FVIII = factor VIII; SD = standard deviation; VWF = von Willebrand factor.
Notes: Percentages based on the number of patients with nonmissing data in the full analysis set.
Details included in the table are from the sponsor’s Summary of Clinical Evidence.
aA target joint was defined as a major joint with at least 3 spontaneous bleeding episodes in a consecutive 6-month period before study entry.
bValues for the Hemophilia Joint Health Score range from 0 to 124, with higher scores indicating worse joint health.
Source: XTEND-1 Clinical Study Report54 and XTEND-Kids Clinical Study Report.17
The mean bleeding episodes and bleeding into joint in the past 12 months were both higher in the XTEND-1 trial compared to the XTEND-Kids trial. The baseline characteristics outlined in Table 12 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
Exposure to Study Treatments
All 159 patients included in the XTEND-1 study received at least 1 dose of Altuviiio. A total of 152 (95.6%) patients were treated for at least 39 weeks and 98 (61.6%) patients for at least 52 weeks. The overall mean duration of Altuviiio dosing (calculated from the date and time of the first Altuviiio dose to the end date and time of the last treatment regimen in the study) was 49.64 weeks (SD = 8.34), 49.76 (SD = 8.74) in arm A and 26.13 (SD = 1.21) in arm B with on-demand treatment, and 22.89 (SD = 6.00) in arm B with prophylactic treatment. Of the 159 patients who received at least 1 dose of Altuviiio, 115 (72.3%) had 50 or more exposure days. The mean total number of exposure days was 48.4 (SD = 10.5) in the overall population, 50.9 (SD = 9.1) in arm A, 11.9 (SD = 4.3) in arm B with on-demand treatment, and 23.8 (SD = 6.1) in arm B with prophylactic treatment. The mean total numbers of injections per patient were similar to the respective mean number of exposure days in each arm.
In the XTEND-Kids trial, all the 74 pediatric patients received at least 1 dose of Altuviiio, 66 (89.2%) achieved 50 or more exposure days, with 31 (81.6%) patients in the cohort aged younger than 6 years and 35 (97.2%) patients in the cohort aged 6 years to younger than 12 years. There were 73 (98.6%) patients treated for at least 39 weeks and 56 (75.7%) patients treated for at least 52 weeks. The overall mean duration of Altuviiio dosing was 51.08 weeks (SD = 6.15 weeks), 49.80 weeks (SD = 8.40 weeks) in the cohort aged younger than 6 years and 52.43 weeks (SD = 0.85 weeks) in the cohort aged 6 years to younger than 12 years. The total number of exposure days was 52.5 (SD = 7.2) in the overall population, 51.2 (SD = 8.6) in the cohort aged younger than 6 years and 53.9 (SD = 4.9) in the cohort aged 6 years to younger than 12 years; the mean total number of injections per patient were similar to the respective mean number of exposure days in each age cohort.
Table 13: Summary of Patient Exposure From Studies Included in the Systematic Review
Exposure | XTEND-1 | XTEND-Kids | |||||
|---|---|---|---|---|---|---|---|
Arm A Prophylaxis: Altuviiio, 50 IU/kg once weekly n = 133 | Arm B | Overall N = 159 | Aged < 6 years n = 38 | Aged 6 to < 12 years n = 36 | Overall N = 74 | ||
On demand: Altuviiio, 50 IU/kg n = 26 | Prophylaxis: Altuviiio, 50 IU/kg once weekly n = 26 | ||||||
Total EDs | |||||||
< 5, n (%) | 2 (1.5) | 0 | 1 (3.8) | 2 (1.3) | 1 (2.6) | 0 | 1 (1.4) |
5 to < 10, n (%) | 0 | 7 (26.9) | 0 | 0 | 0 | 0 | 0 |
10 to < 25, n (%) | 3 (2.3) | 19 (73.1) | 8 (30.8) | 5 (3.1) | 0 | 0 | 0 |
25 to < 50, n (%) | 13 (9.8) | 0 | 17 (65.4) | 37 (23.3) | 6 (15.8) | 1 (2.8) | 7 (9.5) |
≥ 50, n (%) | 115 (86.5) | 0 | 0 | 115 (72.3) | 31 (81.6) | 35 (97.2) | 66 (89.2) |
≥ 5 EDs, n (%) | 131 (98.5) | 26 (100) | 25 (96.2) | 157 (98.7) | 37 (97.4) | 36 (100) | 73 (98.6) |
≥ 10 EDs, n (%) | 131 (98.5) | 19 (73.1) | 25 (96.2) | 157 (98.7) | 37 (97.4) | 36 (100) | 73 (98.6) |
≥ 25 EDs, n (%) | 128 (96.2) | 0 | 17 (65.4) | 152 (95.6) | 37 (97.4) | 36 (100) | 73 (98.6) |
Mean (SD) | 50.9 (9.1) | 11.9 (4.3) | 23.8 (6.1) | 48.4 (10.5) | 51.2 (8.6) | 53.9 (4.9) | 52.5 (7.2) |
Median (range) | 53.0 (2 to 63) | 12.0 (5 to 21) | 26.0 (2 to 28) | 53.0 (2 to 63) | 53.0 (3 to 57) | 54.0 (33 to 72) | 53.0 (3 to 72) |
Total injections per patient overall | |||||||
Mean (SD) | 51.1 (9.2) | 12.0 (4.2) | 23.9 (6.0) | 48.6 (10.6) | 51.3 (8.7) | 54.1 (5.2) | 52.6 (7.3) |
Median (range) | 53.0 (2 to 63) | 12.0 (5 to 21) | 26.0 (2 to 28) | 53.0 (2 to 63) | 53.0 (3 to 58) | 54.0 (33 to 74) | 53.0 (3 to 74) |
ED = exposure day; SD = standard deviation.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
aAn ED is a 24-hour period in which ≥ 1 Altuviiio injections are given. All injections over the study course are counted. For the XTEND-1 trial, the count from the overall column is the summation of EDs from arm A and arm B overall (i.e., the total EDs are based on the final treatment regimen that patients arrive at; thus, the overall count might not be equal to the summation of the count from the columns of treatment arm and regimen).
Source: XTEND-1 Clinical Study Report54 and XTEND-Kids Clinical Study Report.17
Concomitant Medications
Table 14 summarizes the concomitant medication used in the XTEND-1 trial. Most of the patients in the XTEND-1 trial (n = 147; 92.5%) received at least 1 concomitant medication during the study. The most frequently used concomitant medication was paracetamol in 65 (40.9%) patients, tozinameran in 57 (35.8%) patients, and celecoxib in 25 (15.7%) patients. Medication for hemophilia-related pain was taken by 115 (72.3%) patients. In total, 4 patients in arm A took a prohibited concomitant medication (FVIII products other than Altuviiio in 3 patients and Aspirin in 1 patient) that was reported as a major deviation to the protocol. This use of FVIII products other than Altuviiio was prohibited and led to permanent study treatment discontinuation unless it occurred in a life-threatening emergency or as a result of an accidental use.
In the XTEND-Kids trial, the use of concomitant medications or procedures were reported by 59 (79.7%) and 9 (12.2%) patients, respectively, in the safety analysis set. The most frequently used concomitant medications (in > 10% of patients) were paracetamol in 42 (56.8%) patients, tranexamic acid in 13 (17.6%) patients, sodium chloride in 9 (12.2%) patients, amoxicillin in 9 (12.2%) patients, and tozinameran in 8 (10.8%) patients. Medication for hemophilia-related pain was taken by 6 (8.1%) patients within 14 days before study visits.
Efficacy
XTEND-1 Trial
A summary of key efficacy outcomes in the XTEND-1 trial are presented in Table 15.
Bleeding Outcomes
Annualized bleeding rate: The primary efficacy end point in the XTEND-1 trial was ABR in arm A (prophylaxis arm) assessed at week 52. In the FAS, a total of 86 bleeding episodes were treated with Altuviiio in 133 patients in arm A during the efficacy period. The median ABR at week 52 was 0.00 (IQR, 0.00 to 1.04) and the mean ABR was 0.71 (95% CI, 0.52 to 0.97). In arm A, 131 (98.5%) patients had 5 or fewer bleeding episodes per year and 86 (64.7%) patients had no bleeding episodes during the study (Table 15). Two sensitivity analyses of the mean ABR at 52 weeks were performed and both results were consistent with those of the primary analysis.
Table 14: Concomitant Medication (≥ 10% of Patients in Either Treatment Arm) in XTEND-1 and XTEND-Kids Trials — Safety Analysis Set
Concomitant medicationa | XTEND-1 | XTEND-Kids | |||||
|---|---|---|---|---|---|---|---|
Arm A Prophylaxis: Altuviiio, 50 IU/kg once weekly n = 133 | Arm B | Overall N = 159 | Aged < 6 years n = 38 | Aged 6 to < 12 years n = 36 | Overall N = 74 | ||
On demand: Altuviiio, 50 IU/kg n = 26 | Prophylaxis: Altuviiio, 50 IU/kg once weekly n = 26 | ||||||
Patients with any concomitant medication, n (%) | 125 (94.0) | 22 (84.6) | 12 (46.2) | 147 (92.5) | 31 (81.6) | 28 (77.8) | 59 (79.7) |
Paracetamol | 59 (44.4) | 5 (19.2) | 2 (7.7) | 65 (40.9) | 23 (60.5) | 19 (52.8) | 42 (56.8) |
Tozinameran | 55 (41.4) | 1 (3.8) | 1 (3.8) | 57 (35.8) | 2 (5.3) | 6 (16.7) | 8 (10.8) |
Celecoxib | 24 (18.0) | 1 (3.8) | 0 | 25 (15.7) | — | — | — |
COVID-19 vaccine NRVV AD (CHADOX1 NCOV-19) | 15 (11.3) | 5 (19.2) | 6 (23.1) | 24 (15.1) | — | — | — |
Ibuprofen | 12 (9.0) | 3 (11.5) | 1 (3.8) | 16 (10.1) | — | — | — |
Etoricoxib | 10 (7.5) | 3 (11.5) | 1 (3.8) | 14 (8.8) | — | — | — |
Diclofenac | 5 (3.8) | 4 (15.4) | 1 (3.8) | 10 (6.3) | — | — | — |
COVID-19 vaccine NNRV AD26 (GAM-COVID-VAC) | 1 (0.8) | 4 (15.4) | 0 | 5 (3.1) | — | — | — |
Tranexamic acid | — | — | — | — | 6 (15.8) | 7 (19.4) | 13 (17.6) |
Amoxicillin | — | — | — | — | 6 (15.8) | 3 (8.3) | 9 (12.2) |
Sodium chloride | — | — | — | — | 6 (15.8) | 3 (8.3) | 9 (12.2) |
Colecalciferol | — | — | — | — | 1 (2.6) | 4 (11.1) | 5 (6.8) |
Heparin sodium | — | — | — | — | 4 (10.5) | 1 (2.8) | 5 (6.8) |
Fluticasone propionate | — | — | — | — | 4 (10.5) | 0 | 4 (5.4) |
Propofol | — | — | — | — | 4 (10.5) | 0 | 4 (5.4) |
aMedications are coded using the World Health Organization Drug Dictionary.
Source: XTEND-1 Clinical Study Report54 and XTEND-Kids Clinical Study Report.17 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Annualized joint bleeding rate: Results for AjBR were consistent with the results for ABR. In arm A, 37 patients in the FAS had a total of 61 treated joint bleeds. The estimated mean AjBR at week 52 was 0.51 (95% CI, 0.36 to 0.72). Of the 133 patients in arm A, 131 (98.5%) patients had an AjBR of 5 or fewer episodes per year with 96 (72.2%) patients having no joint bleeds during the study.
In arm B, the estimated mean AjBR at week 52 was 17.48 (95% CI, 14.88 to 20.54). The mean AjBR in arm B was similar to arm A after patients had switched to prophylactic treatment (mean = 0.62; 95% CI, 0.25 to 1.52). In an intrapatient comparison of AjBR in arm B, the joint bleeding rate ratio for prophylaxis versus on-demand treatment was 0.04 (95% CI, 0.01 to 0.08). Nine patients (34.6%) had an AjBR of more than 20 with on-demand treatment while no patients had an AjBR greater than 5 with prophylactic treatment.
Intrapatient Comparison of ABR
Intrapatient comparison of ABR between Altuviiio prophylaxis versus historical prophylaxis: Overall, the number of patients with an ABR of 0 who had historical prophylaxis or Altuviiio was 42.3% and 64.1%, respectively. In the FAS (n = 78), the intrapatient comparison in arm A reported a mean ABR reduction of 77% (ABR ratio = 0.23; 95% CI, 0.13 to 0.42; P < 0.0001) in the efanesoctocog prophylaxis group compared to historical prophylaxis. The assessment for noninferiority based on the PPS met the prespecified criteria. In the PPS (n = 77), intrapatient comparison between Altuviiio prophylaxis and historical prophylaxis led to a MD of −2.30 (95% CI, −3.49 to −1.11).
Intrapatient comparison of ABR between prophylaxis and on-demand use of Altuviiio: For the 26 patients in arm B, the efficacy of weekly prophylaxis in terms of ABR was compared to on-demand treatment with Altuviiio, as a secondary end point. The bleeding rate ratio for prophylaxis versus on-demand treatment was 0.03 (95% CI, 0.02 to 0.07). With on-demand treatment, most patients (96.2%) had an ABR of more than 10; whereas, after switching to prophylactic treatment, most patients (76.9%) had no bleeds.
Physical Functioning and Pain (QoL)
Haem-A-QoL Physical Health score and Haem-A-QoL score: In the XTEND-1 study, QoL data were collected in adult patients aged 17 years or older via the Haem-A-QoL Physical Health score and in adolescent patients aged 12 years to 16 years via the Haem-A-QoL. In arm A, for patients aged 17 years or older (n = 98), the estimated mean change from baseline to week 52 in Haem-A-QoL Physical Health score was –6.74 (95% CI, –10.13 to –3.36; P = 0.0001). The Haem-A-QoL results in the study’s adolescent population (all in arm A) mirrored those of the group aged 17 years or older, with improvements in Haem-A-QoL Physical Health score (mean change from baseline to week 52 = –2.18 [SD = 22.05]) and total score (mean change from baseline to week 52 = –3.45 [SD = 8.83]) in the group aged 13 years to 16 years (n = 18). In arm B, a mean change in Haem-A-QoL Physical Health score of –25.91 (SD = 22.29) by week 52 was reported. A sensitivity analysis performed for patients aged 17 years or older in arm A who had rolled over from the OBS16221 study (n = 66) also showed an improvement in Haem-A-QoL Physical Health score (LS mean change from baseline to week 52 = –4.04; 95% CI, –8.06 to –0.03).
PROMIS Pain Intensity and Physical Function: Item 3a of the PROMIS instrument assessed a patient’s worst pain in the last 7 days. This item was used to assess pain intensity in the XTEND trials. In arm A, in participants aged 12 years or older, the estimated mean change from baseline to week 52 in pain intensity was a difference in score of –0.21 (95% CI, –0.41 to –0.02). In arm B, the mean change from baseline to week 52 pain intensity was a difference in score of –0.77 (SD = 0.81).
The PROMIS instrument was also used to assess physical function in adult patients only (aged at least 18 years). In arm A, of 108 patients, 103 completed the PROMIS Short Form Physical Function questionnaire at baseline and 102 at week 52. The mean Physical Health score at baseline was 46.80 (SD = 8.82) and 47.35 (SD = 9.28) at week 52 and the mean change from baseline to week 52 of 0.62 (SD = 4.77).
Joint Health
HJHS: In arm A, the mean HJHS total score at baseline was 18.1 (SD = 18.4). The estimated mean change in the HJHS total score from baseline to week 52 was −1.54 (95% CI, −2.70 to −0.37; P = 0.0101). In arm B, the mean change from baseline to week 52 in HJHS total score was −4.1 (SD = 8.7). A sensitivity analysis performed using the data of patients in arm A who rolled over from the prospective observational OBS16221 study also showed an improvement in HJHS total score. The LS mean change from baseline to week 52 was −0.86 (95% CI, −2.38 to 0.66).
Table 15: Summary of Key Efficacy Results From the XTEND-1 Trial (Full Analysis Set)
Outcome | Arm A Prophylaxis: Altuviiio, 50 IU/kg once weekly n = 133 | Arm B | ||
|---|---|---|---|---|
On demand: Altuviiio, 50 IU/kg n = 26 | Prophylaxis: Altuviiio, 50 IU/kg once weekly n = 26 | |||
ABR | ||||
Total number of treated bleeding episodes, n | 86 | — | — | |
Total patient-years followed, n | 121.2 | 12.5 | 11.4 | |
ABR (model based), mean (95% CI) | 0.71 (0.52 to 0.97)a | 21.41 (18.81 to 24.36)b | 0.70 (0.33 to 1.48)b | |
ABR, median (IQR) | 0.00 (0.00 to 1.04) | 21.13 (15.12 to 27.13) | 0.00 (0.00 to 0.00) | |
Patients with ABR = 0, (%) | 86 (64.7) | 0 (0) | 20 (76.9) | |
AjBR | ||||
AjBR (model based),a mean (95% CI) | 0.51 (0.36 to 0.72) | 17.48 (14.88 to 20.54) | 0.62 (0.25 to 1.52) | |
AjBR, median (IQR) | 0.00 (0.00 to 1.02) | 18.42 (10.80 to 23.90) | 0.00 (0.00 to 0.00) | |
Patients with AjBR = 0, n (%) | 96 (72.2) | 0 (0) | 21 (80.8) | |
Intrapatient ABR comparisons | ||||
Intrapatient comparison of ABR between Altuviiio prophylaxis versus | ||||
Comparison groups | Historical prophylaxis n = 78 | Altuviiio n = 78 | — | |
ABR, mean (95% CI)b | 2.96 (2.00 to 4.37) | 0.69 (0.43 to 1.11) | — | |
Mean difference (95% CI)b | −2.27 (−3.44 to −1.10) | — | ||
Rate ratio (95% CI)b | 0.23 (0.13 to 0.42) | — | ||
P value (superiority)c | < 0.0001 | — | ||
Intrapatient comparison of ABR for prophylaxis use and on-demand use of Altuviiio | ||||
Rate ratio (95% CI) | NA | 0.03 (0.02 to 0.07) | ||
Number of patients with trough FVIII activity level achieved, n (%) | ||||
> 1% | 103 (100) | — | — | |
> 5% | 102 (99.0) | — | — | |
> 10% | 86 (83.5) | — | — | |
> 15% | 42 (40.8) | — | — | |
> 20% | 18 (17.5) | — | — | |
Joint health | ||||
HJHS total score | ||||
n | 133 | 26 | ||
Baseline, mean (SD) | 18.1 (18.4) | 26.3 (13.2) | ||
Change from baseline, mean (SD) | −1.5 (6.4) | −4.1 (8.7) | ||
95% CI | −2.70 to −0.37 | — | ||
P value | 0.0101 | — | ||
Physical functioning and pain (QoL) | ||||
Haem-A-QoL Physical Health score (aged ≥ 17 years) | ||||
n | 110 | 26 | ||
Baseline, mean (SD) | 37.02 (23.83) | 46.09 (21.79) | ||
Change between baseline and follow-up at week 52, mean (SD) | −6.79 (18.59) | −25.91 (22.29) | ||
95% CI | −10.13 to −3.36 | — | ||
P value | 0.0001 | — | ||
PROMIS Short Form Physical Function (aged ≥ 18 years) | ||||
n | 108 | 26 | ||
Baseline, mean (SD) | 46.80 (8.82) | — | ||
Change between baseline and follow-up at week 52, mean (SD) | 0.62 (4.77) | 4.08 (3.63) | ||
95% CI | — | — | ||
P value | — | — | ||
PROMIS Pain Intensity, worst pain intensity in past 7 days | ||||
n | 133 | 26 | ||
Baseline, mean (SD) | 2.47 (1.15) | 2.74 (1.05) | ||
Change between baseline and follow-up at week 52, mean (SD) | −0.21 (1.20) | −0.77 (0.81) | ||
95% CI | (−0.41 to −0.02) | — | ||
P value | 0.0276 | — | ||
ABR = annualized bleeding rate; AjBR = annualized joint bleeding rate; aPTT = activated partial thromboplastin time; CI = confidence interval; FAS = full analysis set; FVIII = factor VIII; Haem-A-QoL = Haemophilia Quality of Life questionnaire for adults; HJHS = Hemophilia Joint Health Score; IQR = interquartile range; NA = not available; PROMIS = Patient-Reported Outcomes Measurement Information System; QoL = quality of life; SD = standard deviation.
Note: All data are reported for FAS unless otherwise stated. Mean change from baseline is reported for mean change from baseline at week 52.
aEstimated using a negative binomial model with the total number of treated bleeding episodes during the efficacy period as the response variable and log-transformed efficacy period duration (in years) as an offset variable.
bEstimated using a repeated negative binomial model with treatment (Altuviiio prophylaxis versus historical prophylaxis) as covariate.
cAchieving trough FVIII activity levels > x% are based on average trough samples from each scheduled visit using the aPTT-based 1-stage clotting assay. Patients with trough samples outside 168 ± 5 hours from previous dose are excluded from this analysis.
Source: XTEND-1 Clinical Study Report.54
XTEND-Kids Trial
A summary of key efficacy outcomes in the XTEND-Kids trial is presented in Table 16.
FVIII Inhibitor Formation
Inhibitor development to FVIII: The primary end point of the XTEND-Kids study was the occurrence of inhibitor development against FVIII based on all patients who had reached at least 50 exposure days. Overall, 65 patients who had reached at least 50 exposure days were analyzed for inhibitors. The incidences of inhibitor development to FVIII were 0.0% (95% CI, 0.0 to 5.5) in patients with 50 or more days of exposure to Altuviiio and 0.0% (95% CI, 0.0 to 4.9) in all treated patients.
Bleeding Outcomes
Annualized bleeding rate: The overall mean ABR at week 52 was 0.89 (95% CI, 0.56 to 1.42) and a median ABR was 0 (IQR, 0 to 1.02). Of the 74 patients, 47 (63.5%) had an ABR of 0 and 25 (33.8%) had an ABR of greater than 0 to 5 at 52 weeks. Two patients in the cohort aged 6 years to younger than 12 years had a higher ABR with 7.2 in 1 patient and 21.4 in the other patient. A total of 64 bleeding episodes were treated with Altuviiio in 27 of the 74 patients (17 bleeding episodes in 14 patients in the cohort aged < 6 years and 47 bleeding episodes in 13 patients in the cohort aged < 12 years). Results of sensitivity analyses based on mean ABR at 52 weeks on the PPS or mean ABR on FAS including patients with data at week 26 were consistent with the primary analysis.
Annualized joint bleeding rate: In the FAS, there were a total of 42 treated joint bleeds throughout the study. The overall estimated mean AjBR was 0.59 (95% CI, 0.27 to 1.28), with 0.19 (95% CI, 0.06 to 0.62) in the cohort aged younger than 6 years and 0.99 (95% CI, 0.38 to 2.60) in the cohort aged 6 years to younger than 12 years. Of the 74 patients who had an efficacy period, 61 (82.4%) patients reported no joint bleeds, while 12 (16.2%) patients reported 1 to 5 joint bleeds. One (1.4%) patient had 21 joint bleeds as per analysis, 18 of which were not confirmed by the investigator nor reported by the patient. This patient received an intensive consolidation treatment regimen of Altuviiio 2 to 3 times per week for a total of approximately 4 months following the treatment of 2 traumatic hip bleeds. Consequently, the intensive consolidation therapy resulted in 18 derived “new” treated bleeds of unknown type as per statistical analysis A sensitivity analysis excluding the participant who did not receive the weekly prophylactic treatment for an extended period of time showed that the estimated mean AjBR in the cohort aged 6 years to younger than 12 years decreased to 0.41 (95% CI, 0.19 to 0.89) and the overall estimated mean AjBR to 0.30 (95% CI, 0.16 to 0.57)
Physical Functioning and Pain (QoL)
Haemo-QoL: QoL data were assessed using the Haem-A-QoL total score for patients between the age of 4 years and 7 years, and the Haemo-QoL Kids Short Version was used for patients of at least 8 years of age. For patients aged 4 years to 7 years, 8 years to younger than 12 years, and in respective caregivers, data were collected using 4 separated Haem-A-QoL. For patients between the age of 4 years and 7 years, the mean change from baseline to week 52 was −5.31 (SD = 10.83) in the cohort aged younger than 6 years and 4.69 (SD = 5.41) in the cohort aged 6 years to younger than 12 years. In patients aged 4 years to 7 years overall, the mean change from baseline to week 52 was −2.46 (SD = 10.49). Parents of children between the age of 4 years and 7 years were also asked to complete the Haem-A-QoL for a parent-proxy assessment of this outcome. For the overall group, the mean change from baseline based on the parent proxy was −2.85 (SD = 11.82), which is aligned with the patient-reported results. For patients aged 8 years and older, the mean change from baseline to week 52 was –9.79 (SD = 12.18).
PROMIS Pain Intensity and Physical Function: Similar to the XTEND-1 trial, pain intensity was assessed in the XTEND-Kids study using item “a” of the PROMIS Pediatric instrument as a change from baseline to week 52. For patients between the age of 5 years and 12 years, a parent or caregiver response was used as a proxy for the child. In the cohort of patients aged less than 6 years, the mean change from baseline was −0.44 (SD = 2.65) and for patients between the age of 6 years and 12 years, the mean change from baseline was −0.75 (SD = 2.53). Overall, the mean change from baseline was −0.62 (SD = 2.52). Patients between the age of 8 years and 12 years responded to this outcome independently. For patients aged 8 years or older in the cohort aged 6 years to 12 years, the mean change from baseline was 0.00 (SD = 2.98).
In the cohort aged younger than 6 years, 8 parents of participants aged 5 years or older completed the PROMIS Short Form Physical Function questionnaire at baseline, and 8 parents completed it at week 52. The mean change from baseline to week 52 was 3.96 (SD = 6.73; n = 7). In the cohort aged 6 years to younger than 12 years, 14 participants aged 8 years or older completed the PROMIS Short Form Physical Function questionnaire at baseline, and 16 participants at week 52. The mean change from baseline to week 52 was 0.78 (SD = 10.48; n = 10). In the cohort aged 6 years to younger than 12 years, 16 parents of participants aged younger than 12 years completed the questionnaire at baseline, and 16 parents at week 52. The mean change from baseline to week 52 was −1.36 (SD = 12.15; n = 10).
Joint Health
HJHS: In the XTEND-Kids study, the questionnaire primarily designed for children aged 4 years to 18 years was used. In the cohort aged younger than 6 years, 20 patients were aged 4 years or older and the mean change in HJHS total score from baseline to week 52 was 0.2 (SD = 8.3). In the cohort aged 6 years to younger than 12 years, the mean change in HJHS total score from baseline to week 52 was −1.1 (SD = 4.3) in 33 patients.
Response During Perioperative Management
Table 17 is a summary of the perioperative management outcomes of Altuviiio in the XTEND trials.
Number of injections and dose to maintain hemostasis during major surgery: A summary of the results available from both the XTEND-1 and XTEND-Kids studies for the use of Altuviiio for perioperative management of bleeding is available in Table 17.
Table 16: Summary of Key Efficacy Results From XTEND-Kids Trial (Full Analysis Set)
Detail | Age cohort | Overall N = 74 | |
|---|---|---|---|
Aged < 6 years n = 38 | Aged 6 to < 12 years n = 36 | ||
FVIII inhibitor development | |||
Patients with ≥ 50 exposure days to Altuviiio at 52 weeks | |||
Patients with an inhibitor test, n | 30 | 35 | 65 |
Patients with an inhibitor, n | 0 | 0 | 0 |
Incidence of inhibitor formation,% (95% CI)a | 0.0 (0.0 to 11.6) | 0.0 (0.0 to 10.0) | 0.0 (0.0 to 5.5) |
Patients with ≥ 25 exposure days to Altuviiio at 52 weeks | |||
Patients with an inhibitor test, n | 37 | 36 | 73 |
Patients with an inhibitor, n | 0 | 0 | 0 |
Incidence of inhibitor formation, % (95% CI) a | 0.0 (0.0 to 9.5) | 0.0 (0.0 to 9.7) | 0.0 (0.0 to 4.9) |
All treated patients at 52 weeks | |||
Patients with an inhibitor test, n | 38 | 36 | 74 |
Patients with an inhibitor, n | 0 | 0 | 0 |
Incidence of inhibitor formation,% (95% CI)a | 0.0 (0.0 to 9.3) | 0.0 (0.0 to 9.7) | 0.0 (0.0 to 4.9) |
ABR | |||
Total patient-years followed, n | 35.4 | 35.2 | 70.6 |
ABR (model based),a mean (95% CI) | 0.48 (0.30 to 0.77) | 1.33 (0.64 to 2.76) | 0.89 (0.56 to 1.42) |
ABR, median (IRQ) | 0.00 (0.00 to 1.00) | 0.00 (0.00 to 1.51) | 0.00 (0.0 to; 1.02) |
Patients with ABR = 0, n (%) | 24 (63.2) | 23 (63.9) | 47 (63.5) |
AjBR | |||
AjBR (model based),a mean (95% CI) | 0.19 (0.06 to 0.62) | 0.99 (0.38 to 2.60) | 0.59 (0.27 to 1.28) |
AjBR, median (Q1 to Q3) | 0.00 (0.00 to 0.00) | 0.00 (0.00 to 0.48) | 0.00 (0.00 to 0.00) |
All bleeds (treated and untreated) | |||
ABR (model based),a mean (95% CI) | 2.78 (1.39 to 5.58) | 2.85 (1.59 to 5.12) | 2.82 (1.79 to 4.42) |
ABR, median (Q1 to Q3) | 0.00 (0.00 to 1.97) | 1.00 (0.00 to 3.00) | 0.49 (0.00 to 2.05) |
Patients with ABR for all bleeds = 0, n (%) | 21 (55.3) | 16 (44.4) | 37 (50.0) |
Efficacy of Altuviiio in the treatment of bleeds | |||
Number of treated bleeds | 17 | 47 | 64 |
Number of injections to treat a bleed, mean (SD) | 1.12 (0.33) | 1.30 (0.69) | 1.25 (0.62) |
Bleeds treated with a single injection, n (%) | 15 (88.2) | 37 (78.7) | 52 (81.3) |
Total dose for resolution of a bleed (IU/kg) | |||
Mean (SD) | 52.29 (15.84) | 66.90 (37.01) | 63.02 (33.26) |
Median (Q1 to Q3) | 57.14 (50.00 to 60.98) | 52.58 (51.89 to 55.56) | 52.63 (51.64 to 60.98) |
Patients with trough FVIII activity levels,b n (%) | |||
Number of patients with all trough samples that are within 168 ± 5 hours from the previous dose | 32 | 29 | 61 |
> 1% | 32 (100) | 29 (100) | 61 (100) |
> 3% | 32 (100) | 29 (100) | 61 (100) |
> 5% | 24 (75.0) | 29 (100) | 53 (86.9) |
> 10% | 6 (18.8) | 15 (51.7) | 21 (34.4) |
> 15% | 3 (9.4) | 2 (6.9) | 5 (8.2) |
> 20% | 1 (3.1) | 2 (6.9) | 3 (4.9) |
Joint health | |||
HJHS total score | |||
n | 21 | 36 | 57 |
Baseline mean (SD) | 2.4 (7.1) | 2.1 (4.5) | 2.2 (5.5) |
Change from baseline to week 52, mean (SD) | 0.2 (8.3) | −1.1 (4.3) | −0.6 (6.0) |
Physical functioning and pain (QoL) | |||
Haem-A-QoL Short Form total score and Haemo-QoL Kids Short Form total score | |||
Aged 4 years to 7 years (Haem-A-QoL) | |||
n | 13 | 8 | 21 |
Baseline mean (SD) | 26.44 (14.69) | 19.53 (13.44) | 23.81 (14.30) |
Change from baseline to week 52, mean (SD) | −5.31 (10.83) | 4.69 (5.41) | −2.46 (10.49) |
≥ 8 years (Haem-A-QoL Kids Short Form) | |||
n | NA | 14 | — |
Baseline | NA | 22.09 (13.73) | — |
Change from baseline to week 52, mean (SD) | NA | −9.79 (12.18) | — |
PROMIS Short Form Physical Function | |||
Aged 5 to < 12 years, parent proxy | |||
n | 7 | 10 | — |
Baseline mean (SD) | 50.93 (3.47) | 50.74 (10.55) | — |
Change from baseline to week 52, mean (SD) | 3.96 (6.73) | −1.36 (12.15) | — |
Aged 8 years to < 12 years | |||
n | NA | 10 | — |
Baseline | NA | 51.71 (10.44) | — |
Change from baseline to week 52, mean (SD) | NA | 0.78 (10.48) | — |
PROMIS Pediatric Pain Intensity, worst pain intensity in past 7 days | |||
Aged 5 years to < 12 years, parent proxy | |||
n | 10 | 19 | 29 |
Baseline mean (SD) | 1.20 (2.25) | 1.05 (1.84) | 1.10 (1.95) |
Change from baseline to week 52, mean (SD) | −0.44 (2.65) | −0.75 (2.53) | −0.62 (2.52) |
Aged 8 years to < 12 years | |||
n | NA | 14 | — |
Baseline mean (SD) | NA | 1.71 (2.52) | — |
Change from baseline to week 52, mean (SD) | NA | 0.00 (2.98) | — |
ABR = annualized bleeding rate; AjBR = annualized joint bleeding rate; aPTT = activated partial thromboplastin time; CI = confidence interval; FVIII = factor VIII; Haem-A-QoL = Haemophilia Quality of Life questionnaire for adults; HJHS = Hemophilia Joint Health Score; IQR = interquartile range; NA = not available; PROMIS = Patient-Reported Outcomes Measurement Information System; QoL = quality of life; SD = standard deviation.
Notes: Mean change from baseline is reported for mean change from baseline at week 52.
Details included in the table are from the sponsor’s Summary of Clinical Evidence.
aEstimated using a negative binomial model with the total number of treated bleeding episodes during the efficacy period as the response variable and log-transformed efficacy period duration (in years) as an offset variable.
bAchieving trough FVIII activity levels > x% are based on average trough samples from each scheduled visit using the aPTT-based 1-stage clotting assay. Patients with trough samples outside 168 ± 5 hours from previous dose are excluded from this analysis.
Source: XTEND-Kids Clinical Study Report.17
In the XTEND-1 study, 11 of 12 major surgeries that occurred during the treatment regimen required a single injection of Altuviiio (i.e., the preoperative loading dose) to maintain hemostasis. The mean dose per injection was 41.65 IU/kg (SD = 15.21 IU/kg). For 1 surgery, conducted during routine prophylaxis, no preoperative loading dose was reported on the day before or the day of the surgery.
In the XTEND-Kids study, both major surgeries required a single injection of Altuviiio to maintain hemostasis. The mean dose per injection was 61.13 IU/kg (SD = 1.06 IU/kg).
Harms
XTEND-1 Trial
Harms data for the XTEND-1 trial are summarized in Table 18. The summary of harms is presented for arm A, arm B, and the overall population included in the XTEND-1 trial. CDA-AMC focused on reporting the harms results in the overall population rather than in different arms.
Table 17: Summary of Perioperative Management Outcomes (Surgery Subgroup) From XTEND-1 and XTEND-Kids Trials
Detail | XTEND-1 N = 185 | XTEND-Kids N = 74 |
|---|---|---|
na | 13 | 2 |
Number of major surgeries with hemostatic response rated as excellent or good by the investigator or surgeon, n (%) | 12 (100) | 2 (100) |
Number of injections to maintain hemostasis per major surgery,b mean (SD) | 1.0 (0.0) | 1.0 (0.0) |
Dose to maintain hemostasis during major surgery (IU/kg), mean (SD) | 41.65 (15.21) | 61.13 (1.06) |
SD = standard deviation.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
aAnalysis is based on the major surgeries conducted during the treatment regimen, excluding the surgeries conducted after the last Altuviiio dosing.
bThe number of injections to maintain hemostasis during surgery includes all injections, from loading dose (i.e., the preoperative injection, administered either on the day of surgery or one day before the surgery), to the end of surgery.
Source: XTEND-1 Clinical Study Report 54 and XTEND-Kids Clinical Study Report.17
Adverse Events
Of the 159 patients in the safety analysis set, 123 (77.4%) patients experienced at least 1 TEAE, resulting in a total of 391 TEAEs in the study. The most frequently reported TEAEs in greater than 3% of patients overall were headache (20.1%); arthralgia (16.4%); fall (6.3%); back pain (5.7%); COVID-19 and fatigue (4.4% each); contusion, hemophilic arthropathy, and nasopharyngitis (3.8% each); and joint injury, pain in extremity, and toothache (3.1% each).
The majority of TEAEs were also assessed to be mild in severity. Of the 159 patients, 77 (48.4%) patients had no TEAEs classified as moderate or severe but at least 1 TEAE was classified as mild. In addition, 39 (24.5%) patients had no TEAEs classified as severe but at least 1 TEAE was classified as moderate, and 7 (4.4%) patients had at least 1 TEAE classified as severe. TEAEs classified as severe were reported in 1 (0.6%) of each of the 7 patients and included angina pectoris, CD4 lymphocytes decreased, pancreatic carcinoma metastatic, mobility decreased, migraine, status epilepticus, and device breakage. Of the 8 TEAEs assessed as severe, 6 were also classified by the investigator as SAEs.
Serious Adverse Events
A total of 18 TESAEs were experienced in 15 (9.4%) patients, of which 16 TESAEs were reported in 13 patients in arm A and 2 TESAEs in 2 patients in arm B. Hemophilic arthropathy was the most commonly reported SAE, which was reported in 2 (1.3%) patients in arm A. All other TESAEs were reported in 1 (0.6%) patient each. The majority of TESAEs were assessed by the investigator as mild to moderate in severity.
Withdrawals Due to AEs
Two TEAEs in 2 (1.3%) patients resulted in permanent treatment discontinuation. The reason for WDAE was due to a decrease in CD4 lymphocytes in 1 patient with a history of HIV infection (reported as a TESAE), and due to a combined tibia–fibula fracture in the other patient who withdrew due to an AE.
Mortality
Death was reported in 1 patient overall, in arm B. The patient had a medical history of hepatitis C virus and died of metastatic pancreatic carcinoma, which was reported as a TESAE. The TESAE was assessed by the investigator as not related to Altuviiio treatment.
AEs of Special Interest
AEs of special interest were chosen for their relevance to hemophilia A or treatment and were prespecified in the study protocols. These AEs included the development of inhibitors, grade 3 or higher allergic reactions or anaphylactic reactions, and embolic or thrombotic events (except for injection site thrombophlebitis). There were no reports of inhibitor development to FVIII during the study. There were no reports of grade 3 or greater allergic reactions or anaphylaxis in association with Altuviiio administration during the study. There were no reports of embolic and thrombotic events during the study.
Table 18: Summary of Harms Results From XTEND-1 Trial (Safety Analysis Set)
Result | Arm A | Arm B | Overall N = 159 | |
|---|---|---|---|---|
Prophylaxis: Altuviiio, 50 IU/kg once weekly n = 133 | On demand: Altuviiio, 50 IU/kg n = 26 | Prophylaxis: Altuviiio, 50 IU/kg once weekly n = 26 | ||
Total number of TEAEs, n | 358 | 22 | 11 | 391 |
Number of patients with ≥ 1 TEAE, n (%) | 108 (81.2) | 12 (46.2) | 8 (30.8) | 123 (77.4) |
Most common (≥ 3%) TEAEs by MedDRA preferred term, n (%) | ||||
Headache | 26 (19.5) | 5 (19.2) | 1 (3.8) | 32 (20.1) |
Arthralgia | 25 (18.8) | 1 (3.8) | 0 | 26 (16.4) |
Fall | 10 (7.5) | 0 | 0 | 10 (6.3) |
Back pain | 8 (6.0) | 1 (3.8) | 0 | 9 (5.7) |
COVID-19 | 5 (3.8) | 1 (3.8) | 1 (3.8) | 7 (4.4) |
Fatigue | 7 (5.3) | 0 | 0 | 7 (4.4) |
Contusion | 6 (4.5) | 0 | 0 | 6 (3.8) |
Hemophilic arthropathy | 6 (4.5) | 0 | 0 | 6 (3.8) |
Nasopharyngitis | 6 (4.5) | 0 | 0 | 6 (3.8) |
Joint injury | 5 (3.8) | 0 | 0 | 5 (3.1) |
Pain in extremity | 5 (3.8) | 0 | 0 | 5 (3.1) |
Toothache | 4 (3.0) | 0 | 1 (3.8) | 5 (3.1) |
Gastroesophageal reflux disease | 4 (3.0) | 0 | 0 | 4 (2.5) |
Influenza like illness | 4 (3.0) | 0 | 0 | 4 (2.5) |
Limb injury | 4 (3.0) | 0 | 0 | 4 (2.5) |
SAEs reported in ≥ 1 patient by MedDRA preferred term, n (%) | ||||
Patients with ≥ 1 TESAE | 13 (9.8) | 2 (7.7) | 0 | 15 (9.4) |
Hemophilic arthropathy | 2 (1.5) | 0 | 0 | 2 (1.3) |
Basal cell carcinoma | 1 (0.8) | 0 | 0 | 1 (0.6) |
Cubital tunnel syndrome | 1 (0.8) | 0 | 0 | 1 (0.6) |
Status epilepticus | 1 (0.8) | 0 | 0 | 1 (0.6) |
Ulnar tunnel syndrome | 1 (0.8) | 0 | 0 | 1 (0.6) |
Angina pectoris | 1 (0.8) | 0 | 0 | 1 (0.6) |
Arthropathy | 1 (0.8) | 0 | 0 | 1 (0.6) |
Mobility decreased | 1 (0.8) | 0 | 0 | 1 (0.6) |
Blood glucose increased | 1 (0.8) | 0 | 0 | 1 (0.6) |
CD4 lymphocytes decreased | 1 (0.8) | 0 | 0 | 1 (0.6) |
Combined tibia–fibula fracture | 1 (0.8) | 0 | 0 | 1 (0.6) |
Traumatic hemorrhage | 1 (0.8) | 0 | 0 | 1 (0.6) |
Central venous catheter removal | 1 (0.8) | 0 | 0 | 1 (0.6) |
Device breakage | 1 (0.8) | 0 | 0 | 1 (0.6) |
COVID-19 pneumonia | 0 | 1 (3.8) | 0 | 1 (0.6) |
Pancreatic carcinoma metastatic | 0 | 1 (3.8) | 0 | 1 (0.6) |
Patients who stopped treatment due to adverse events (WDAE), n (%) | ||||
Patients with at least 1 WDAE | 2 (1.5) | 0 | 0 | 2 (1.3) |
CD4 lymphocytes decreased | 1 (0.8) | 0 | 0 | 1 (0.6) |
Combined tibia–fibula fracture | 1 (0.8) | 0 | 0 | 1 (0.6) |
Deaths, n (%) | ||||
TEAEs leading to death | 0 | 1 (3.8) | 0 | 1 (0.6) |
CD4 = clusters of differentiation 4; MedDRA = Medical Dictionary for Regulatory Activities; SAE = serious adverse event; TEAE = treatment-emergent adverse event; TESAE = treatment-emergent serious adverse event; WDAE = withdrawal due to adverse event.
Source: XTEND-1 Clinical Study Report.54
XTEND-Kids Trial
A summary of harms for the XTEND-Kids study is available in Table 19.
Adverse Events
Of the 74 patients in the safety analysis set, 62 (83.8%) experienced at least 1 TEAE, resulting in a total of 255 TEAEs. The most frequently reported TEAEs (in > 5% of patients overall) were SARS-CoV-2 test positive and upper respiratory tract infection (14.9% each); pyrexia (12.2%); asymptomatic COVID-19 (9.5%); gastroenteritis viral, head injury, and nasopharyngitis (8.1% each); arthralgia, pain in extremity, and vomiting (6.8% each); and contusion, diarrhea, viral infection, and viral upper respiratory tract infection (5.4% each). The majority of TEAEs were assessed by the investigator as mild in severity. Of the 74 patients, 43 (58.1%) patients had at least 1 TEAE of mild intensity and 13 (17.6%) patients had at least 1 TEAE of moderate intensity. There were 6 (8.1%) patients with at least 1 TEAE classified as severe. Of those with severe TEAEs, 3 patients aged younger than 6 years reported bacteremia, circumcision, or a fear of injection and 3 patients aged 6 to less than 12 years reported an eosinophilic esophagitis, vascular device occlusion, or chest or head injury. All patients with severe events recovered except 1 patient with extreme fear of blood draw who prematurely withdrew from the study 4 months later.
Table 19: Summary of Harms Results From XTEND-Kids Trial (Safety Analysis Set)
Results | Aged < 6 Years n = 38 | Aged 6 to < 12 years n = 36 | Overall N = 74 |
|---|---|---|---|
TEAEs | |||
Total number of TEAEs, n | 146 | 108 | 255 |
Number of patients with ≥ 1 TEAE, n (%) | 33 (86.8) | 29 (80.6) | 62 (83.8) |
Most common (> 5%) TEAEs by MedDRA preferred term, n (%) | |||
SARS-CoV-2 test positive | 7 (18.4) | 4 (11.1) | 11 (14.9) |
Upper respiratory tract infection | 6 (15.8) | 5 (13.9) | 11 (14.9) |
Pyrexia | 8 (21.1) | 1 (2.8) | 9 (12.2) |
Asymptomatic COVID-19 | 2 (5.3) | 5 (13.9) | 7 (9.5) |
Gastroenteritis viral | 5 (13.2) | 1 (2.8) | 6 (8.1) |
Head injury | 1 (2.6) | 5 (13.9) | 6 (8.1) |
Nasopharyngitis | 3 (7.9) | 3 (8.3) | 6 (8.1) |
Arthralgia | 0 | 5 (13.9) | 5 (6.8) |
Pain in extremity | 2 (5.3) | 3 (8.3) | 5 (6.8) |
Vomiting | 4 (10.5) | 1 (2.8) | 5 (6.8) |
Contusion | 1 (2.6) | 3 (8.3) | 4 (5.4) |
Diarrhea | 3 (7.9) | 1 (2.8) | 4 (5.4) |
Viral infection | 3 (7.9) | 1 (2.8) | 4 (5.4) |
Viral upper respiratory tract infection | 3 (7.9) | 1 (2.8) | 4 (5.4) |
SAEs reported in ≥ 1 patient by MedDRA preferred term, n (%) | |||
Number of patients with at least 1 TESAE | 5 (13.2) | 4 (11.1) | 9 (12.2) |
Vascular device infection | 2 (5.3) | 0 | 2 (2.7) |
Bacteremia | 1 (2.6) | 0 | 1 (1.4) |
Dehydration | 1 (2.6) | 0 | 1 (1.4) |
Asthma | 1 (2.6) | 0 | 1 (1.4) |
Eosinophilic esophagitis | 0 | 1 (2.8) | 1 (1.4) |
Vascular device occlusion | 0 | 1 (2.8) | 1 (1.4) |
Head injury | 0 | 1 (2.8) | 1 (1.4) |
Circumcision | 1 (2.6) | 0 | 1 (1.4) |
Device malfunction | 0 | 1 (2.8) | 1 (1.4) |
Patients who stopped treatment due to adverse events (WDAE), n (%) | |||
Patients with at least 1 WDAE | 0 | 0 | 0 |
Deaths, n (%) | |||
TEAEs leading to death | 0 | 0 | 0 |
MedDRA = Medical Dictionary for Regulatory Activities; SAE = serious adverse event; TEAE = treatment-emergent adverse event; TESAE = treatment-emergent serious adverse event; WDAE = withdrawal due to adverse event.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Source: XTEND-Kids Clinical Study Report.17
Serious Adverse Events
A total of 10 TESAEs were experienced in 9 (12.2%) patients, of which 5 TESAEs were reported in patients aged younger than 6 years and 5 TESAEs in patients aged 6 years to younger than 12 years. All TESAEs were reported in 1 (1.4%) patient each, except vascular device infection, which was reported in 2 (2.7%) patients. The majority of TESAEs were assessed by the investigator as mild to moderate in severity. The 5 TESAEs assessed by the investigator as severe were TESAEs of circumcision and bacteremia, each in 1 patient aged less than 6 years and TESAEs of vascular device occlusion, head injury, and eosinophilic esophagitis, each in 1 patient aged 6 years to less than 12 years.
Withdrawals Due to AEs
Although 1 patient withdrew from the study after 4 months due to extreme fear of blood draw, no patients discontinued Altuviiio treatment due to a TEAE during the study.
Mortality
There were no deaths reported during the study.
AEs of Special Interest
An event of “hives around eyes, mouth, face, and chest” was reported in one 2-year-old patient. The event occurred approximately 3 months after the first dose of Altuviiio (used for weekly prophylaxis) and 3 days after the last injection. There were no reports of embolic and thrombotic events during the study.
Critical Appraisal
Internal Validity
The 2 pivotal trials identified in the sponsor-conducted SLR were phase III, single-arm, open-label clinical trials (XTEND-1 and XTEND-Kids). Patients enrolled in both trials were previously treated with FVIII products for at least 6 months before baseline. In the XTEND-1 trial, 92 patients (n = 82 in arm A and n = 10 in arm B) were rolled over from an observational prestudy (242HA201/OBS16221) that assessed treatment patterns and outcomes in patients with severe hemophilia A previously treated with marketed FVIII replacement products. There were missing data related to efficacy outcomes such as FVIII inhibitor formation (patients with ≥ 50 exposure days), HJHS, and pain and physical function outcomes. Missing data on such critical outcomes are a potential risk of bias or imprecision regarding the efficacy of Altuviiio. Although the nonrandomized, open-label, single-arm design limits the interpretation of the efficacy results for both the XTEND-1 and XTEND-Kids studies, the clinical experts consulted by CDA-AMC for this review indicated that while traditional RCTs remain the gold standard for many conditions, it is not feasible in hemophilia A due to ethical constraints, challenges in patient recruitment, and the availability of effective treatments. According to the clinical experts, alternative designs like intrapatient comparisons and historical controls provide practical evaluation of new therapies such as Altuviiio.
The clinical experts indicated that the exclusion and inclusion criteria for the XTEND-1 and XTEND-Kids trials aligned with the patients they treat in clinical practice. It was noted that participants in both trials were previously treated patients with hemophilia A without inhibitors. Although the XTEND-1 trial started after the completion of the prestudy observational study, only 92 out of 156 participants were rolled over to the XTEND-1 study. All rolled over patients from the prestudy observational study were previously treated (≥ 150 exposure days) with any recombinant FVIII or plasma-derived FVIII product (prophylaxis or on demand). Although an appropriate washout period of 96 hours or longer for the rolled over patients who previously received a marketed FVIII product was observed, the criteria for enrolling patients from the prestudy observational study to the XTEND-1 study were not provided by the sponsor. This was determined by CDA-AMC as a potential selection bias. Additionally, although the sponsor provided data on the baseline characteristics of all patients in the prestudy observational study and those of the XTEND-1 study, the baseline clinical characteristics of the rolled over patients alone were not provided. CDA-AMC notes that this limits the ability to identify preexisting differences, potentially introducing bias and confounding; however, although this is speculative, the clinical experts indicated that the rolled over patients and those in the XTEND-1 study were likely similar and were not systematically different.
Patient compliance with FVIII prophylactic treatment during the prestudy observational study remained unclear, as the sponsor did not provide summary-level statistics on compliance. Although CDA-AMC recognizes that in the XTEND-1 study, a patient was considered compliant if their compliance rate was at least 80%, the reliability of the intrapatient comparison of ABR remains a concern. Although retention and compliance in the XTEND-Kids study was reported to be high, CDA-AMC notes that the administration of FVIII in the XTEND-Kids study is particularly challenging because it can be difficult to obtain venous access in children, the time commitment required for infusion may hinder compliance, and the cost associated with frequent administration may be burdensome. In both trials, efficacy was measured using ABR, a widely accepted end point in hemophilia research that provides an objective outcome.78 Joint health and QoL were measured using the HJHS and Haem-A-QoL, which are validated outcome measures although still subject to some patient-reported bias, especially in open-label trials. Both trials appear to be adequately powered for assessing ABR and joint health outcomes, with sample sizes of 159 in the XTEND-1 study and 74 in the XTEND-Kids study, which should provide a robust estimate of efficacy for this population. However, smaller subgroup analyses, such as surgery, perioperative management, or specific age groups, may not be fully powered to detect efficacy in subpopulations. Both trials included follow-up safety assessments for 2 weeks to 3 weeks after the last dose; however, a longer follow-up period is needed to sufficiently evaluate delayed effects, which is critical for assessing the long-term efficacy and safety of Altuviiio. Although an interim report on the LTE study concerning safety of Altuviiio is consistent with results from the XTEND trials, complete results are needed to evaluate long-term safety outcomes.
In the primary analyses, documentation of bleeding events in both trials via electronic patient diary or electronic case report form was reviewed and assessed by the investigator. CDA-AMC notes that investigator-reviewed documentation of bleeding events could overestimate the efficacy of Altuviiio, particularly on bleeding outcomes such as ABR, AjBR, and intrapatient comparison of ABR. This risk arises from potential reporting bias, subjective assessments, underreporting, and potentially inconsistent bleeding event verification. The absence of independent adjudication and differences in documentation methods is also of concern. While the XTEND-1 study included a historical control through intrapatient comparison of ABR based on patients’ prior prophylactic regimens in a previous study, this approach is inherently weaker compared to a concurrent randomized control group as the reliance on historical data may introduce variability due to changes in patients' characteristics or external factors unrelated to treatment efficacy. This may result in potential confounding, particularly due to temporal events. For example, the clinical experts consulted for the review noted that physical activity is an important indicator of hemophilia A disease outcome. Given that the XTEND-1 trial was conducted during the COVID-19 pandemic while the prestudy observational study was conducted some years before pandemic, impact from possible change in physical activities related to the social distancing that was required on many occasions during the pandemic (e.g., intensity, types of physical activities) on the risk of bleeding in patients with hemophilia is uncertain. However, the clinical experts consulted by CDA-AMC did not expect this to significantly impact the results. Additionally, although the study design was deemed appropriate for data collection across varied populations, the lack of blinding introduces bias, as both participants and investigators were aware of the treatment, which could influence patient-reported outcomes such as physical function and pain outcomes (Haem-A-QoL, Haemo-QoL, PROMIS Pain Intensity) and therefore reliable assessments of these outcomes could not be made.
In both trials, perioperative management outcomes were assessed based on the rating of response by the surgeon or study investigator, as well as the number of injections and mean dose needed to maintain hemostasis per major surgery. Specifically, the investigators or surgeons who completed the surgical procedures assessed the participant’s response to surgery with efanesoctocog treatment using a 4-point clinical scale: excellent, good, fair, and poor. This assessment was to be performed 24 hours after the surgery. The clinical experts consulted for this review noted that there is no available objective evaluation of perioperative outcomes. CDA-AMC notes that the reliance on the opinion of the surgeon seems subjective and likely a potential source of bias to inform decision-making especially given that several surgeons or investigators were involved in the study.
Concomitant medications were reported by 92.5% and 79.7% in the XTEND-1 and XTEND-Kids studies, respectively. In both trials, paracetamol was reported as the most frequently used concomitant medication followed by tozinameran and tranexamic acid. In the XTEND-1 trial, medication for hemophilia-related pain was taken by 115 (72.3%) patients and 4 patients took a prohibited concomitant medication (FVIII products other than Altuviiio in 3 patients and Aspirin in 1 patient), and this was reported as a major deviation to the protocol. In the XTEND-Kids trial, medication for hemophilia-related pain was taken by 6 (8.1%) patients within 14 days before study visits overall. According to the clinical experts consulted by CDA-AMC, there were no serious concerns with the use of these prohibited concomitant medications unless they were taken by accident or occurred in life-threatening emergency situations.
In the XTEND-1 trial, the primary end point, key secondary end point, and selected secondary end points of high clinical importance in hemophilia treatment were tested hierarchically to maintain the overall type I error rate of 0.05 or less. All analyses in the XTEND-Kids trial were descriptive and did not include adjustments for multiplicity. There were some concerns about the assumptions for the statistical model that were adopted to inform the relative efficacy of Altuviiio against historical prophylaxis. The use of a negative binomial regression model in the XTEND-1 trial to estimate intrapatient ABR, although considered appropriate by the clinical experts, presents specific challenges. While the approach reduces variability by using patients as their own controls, concerns include potential misalignment with model assumptions, such as overdispersion and handling of excess zeros, which could bias estimates. Additionally, unmeasured confounders, patient heterogeneity, and missing or incomplete prestudy data may limit the reliability of comparisons. The high proportion of zero ABRs during prophylaxis suggests that alternative models, like zero-inflated regression, might better capture the data distribution. Additionally, the assumption that a negative binomial model implies a constant bleed rate within each period of study, can make it challenging to interpret the magnitude of the effect estimates of Altuviiio compared to historical prophylaxis. This is of particular concern as the prestudy observational study and the trial were conducted before and during the pandemic, respectively, likely impacting the overall bleeding rate. Specifically, the reported estimates of the relative ABRs describe a weighted average of the rate of bleeding over time. CDA-AMC notes that this weighted average can be overly optimistic and fail to accurately capture waning efficacy over time.
External Validity
CDA-AMC identified several considerations related to the generalizability of the XTEND-1 and XTEND-Kids studies in evaluating the efficacy and safety of Altuviiio. All patients included in the XTEND-1 and XTEND-Kids trials were patients with severe hemophilia A (< 1 IU/dL [< 1%] endogenous FVIII activity) who had been on previous prophylaxis or on-demand treatment with any recombinant and/or plasma-derived FVIII or cryoprecipitate for at least 150 exposure days. According to the clinical experts, this is reflective of current Canadian clinical practice where the standard of care for patients with severe hemophilia A is treatment with FVIII prophylaxis. The inclusion of both adults and children and patients from 6 study sites in Canada and from multiple countries across Asia, Europe, North America, and South America enhances the generalizability of the findings. The clinical experts consulted by CDA-AMC indicated that selection of eligible patients for treatment with Altuviiio should follow the inclusion and exclusion criteria used in both pivotal trials.
The results of the XTEND-1 and XTEND-Kids studies may have limited generalizability to the population of patients living in Canada as the study population was restricted to patients (without inhibitors) with severe hemophilia A. According to clinical experts consulted for this review, there is a subset of patients with mild or moderate hemophilia who require prophylaxis. The design of the XTEND-1 and XTEND-Kids studies did not include these patients. CDA-AMC notes that the magnitude of treatment effect in patients with mild and moderate hemophilia A is unclear. Additionally, CDA-AMC notes that the exclusion of patients with FVIII inhibitors limits the applicability of findings to new patients who have a higher risk of developing inhibitors such as those who have not received previous FVIII therapies. However, the clinical experts noted that patients are generally closely monitored for inhibitor development during the first few weeks of starting treatment. The impact of including patients with severe hemophilia A with a history of at least 12 bleeding episodes within 12 months, although acceptable, may exaggerate the efficacy of Altuviiio compared to what is seen in clinical practice as data on patients with mild or moderate hemophilia A are not available and generalizability to those patients remains uncertain. As a result, the magnitude of treatment effect in patients with less than 12 bleeding episodes within 12 months, especially for patients receiving on-demand therapy, compared to those included in the trials is unknown. There is limited evidence from an intrapatient comparison that supports efficacy of Altuviiio in patients previously treated with FVIII prophylaxis. Although the clinical experts indicated the difficulty in the direct comparison of efanesoctocog with other FVIII therapies, CDA-AMC notes that the lack of direct head-to-head comparison with the current standard of care, such as EHL FVIII products or nonfactor therapies like emicizumab, limits external validity regarding the real-world effectiveness and safety of Altuviiio compared to these alternative treatments. The lack of baseline data from the rolled over participants from the prestudy observational study also limits the generalizability and robustness of intrapatient comparisons, which rely on baseline data.
All patients included in the XTEND-1 and the XTEND-Kids trials were male except the inclusion of 1 female in the XTEND-1 trial. This was not considered to be an issue for the external validity of the study as hemophilia A predominantly affects males. Because hemophilia A can affect females, the clinical experts indicated that there is no biological reason that the safety and efficacy of Altuviiio would differ in females. According to the clinical experts, the once-weekly dosing of 50 IU/kg used in the trials aligns with the standard of treatment in hemophilia treatment centres, making it applicable to clinical settings. Specific subgroups, such as patients with obesity or those participating in higher physical activity, may require adjusted dosing, which was not thoroughly explored in the trials, limiting the generalizability of Altuviiio to these populations.
Results from the prestudy observational study included patients treated with EHL or SHL agents. However, information based on the type of FVIII concentrate (e.g., EHL versus SHL) that patients were previously treated with was not available as all patients were included in the analyses regardless of prior treatment with an EHL or SHL agent. According to the clinical experts consulted by CDA-AMC, the key difference between EHL and SHL FVIII agents in treating patients is the frequency of infusion. Due to its half-life, EHL FVIII may be associated with a lower ABR. However, this was not considered a serious generalizability issue by the clinical experts.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.14,15
High certainty: Very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: Moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. The word “likely” is used for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. The word “may” is used for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: Very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. Evidence of very low certainty is described as “very uncertain.”
Although GRADE guidance is not available for noncomparative studies, the CDA-AMC review team assessed pivotal single-arm trials for study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias to present these important considerations. Because the lack of a comparator arm does not allow for a conclusion to be drawn on the effect of the intervention versus any comparator, the certainty of evidence for single-arm trials started at very low certainty with no opportunity for rating up.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The target of the certainty of evidence assessment was the presence of a clinically important improvement in bleeding outcomes (ABR, AjBR), joint health (HJHS), physical function and pain (Haem-A-QoL, Haemo-QoL), and perioperative management, which were considered the most important outcomes to treatment by the clinical experts consulted for this review, and the clinician group and patient group inputs. According to the clinical experts, the target of the certainty of evidence assessment was the presence or absence of any (non-null) effect for the ABR and AjBR due to the lack of a formal estimate of a MID. According to the clinical experts, the target of the certainty of evidence assessment for perioperative management was the opinion of the surgeon’s or investigator’s rating of response due to the lack of a formal estimate of MIDs. The certainty of evidence was summarized narratively for the HJHS, Haem-A-QoL (Physical Health score and total score), and perioperative management as well as harms outcomes due to lack of comparator.
Results of GRADE Assessments
Table 2 and Table 3 present the GRADE summary of findings for Altuviiio for routine prophylaxis to reduce the frequency of bleeding episodes, and perioperative management of bleeding in adults and children with severe hemophilia A.
Long-Term Extension Studies
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
Description of Studies
One LTE study was submitted for review, the XTEND-ed study (NCT04644575).18 The XTEND-ed LTE study is an ongoing phase III, open-label, multicentre study to assess long-term safety and efficacy of Altuviiio in previously treated patients with severe hemophilia A. The study began in February 2021 and is estimated for completion in 2027.18 At the time of this submission, the available evidence was limited to interim analyses based on conference presentations.19,20 The reported data cut-off for the interim analysis was June 8, 2023. The submitted interim analyses pertain only to patients rolled over from the XTEND-1 and XTEND-Kids studies into arm A of the XTEND-ed LTE study and report on efficacy and safety-related outcomes over 2 additional years of treatment with Altuviiio.
The study comprises 3 treatment arms. Arm A included patients rolled over from the XTEND-1 and XTEND-Kids studies, patients from arm B or C of the XTEND-ed LTE study who agreed to roll over, or patients from other Altuviiio studies. Enrolment in arm B was limited to patients living in China; arm C enrolled patients globally with an upcoming planned major surgery and included patients living in Canada. Key exclusion criteria across all study arms included a positive inhibitor test. Patients in all study arms were administered weekly IV doses of 50 IU/kg Altuviiio prophylactic treatment. Arm A participants received the study drug for up to 48 months.
Interim outcomes included in the analysis were occurrence of inhibitor development (primary outcome), ABRs, treatment of bleeding episodes, safety and tolerability, and perioperative management. Statistical analyses were descriptive in nature. No formal comparisons were planned, and no hypotheses were formally tested.
Results
Patient Disposition
A total of 217 patients rolled over to the LTE from the 2 parent studies: from the XTEND-1 study, 98% (n = 121 of 124) of patients from arm A and 100% (n = 25 of 25) of patients from arm B; as well as 96% patients (n = 71 of 74) from the XTEND-Kids study. During the LTE, 9 patients discontinued therapy, 8 patients from the XTEND-1 study arm and 1 patient from the XTEND-Kids study arm. Discontinuations were due to the use of prohibited medications due to medical needs (44%), consent withdrawal (33%), AE occurrence (11%), and “other” reasons (11%).
Table 20: Patient Disposition in XTEND-ed Trial
Patient Disposition | XTEND-1 | XTEND-Kids | |
|---|---|---|---|
Arm A | Arm B | ||
Total number of patients in parent study, n | 124 | 25 | 74 |
Patients rolled over from parent study, n (%) | 121 (98) | 25 (100) | 71 (96) |
Patients remaining in study,a n (%) | 138 (95) | 70 (98.6) | |
Discontinued, n (%) | 8 (5.5) | 1 (1.4) | |
Adverse event | 1 (0.7) | — | |
Consent withdrawn | 2 (1.4) | 1 (1.4) | |
Use of prohibited medications due to medical needs | 4 (2.7) | — | |
Other | 1 (0.7) | — | |
aPatients remaining in study as of data cut-off date June 8, 2023.
Source: Sponsor’s clinical evidence.51
Exposure to Study Treatments
As of the XTEND-ed study interim analysis cut-off date, the mean treatment exposure days for patients enrolled from the XTEND-1 and XTEND-Kids studies were 83.1 days (SD = 14.8) and 35.6 days (SD = 14.7), respectively. The mean treatment duration was 82.5 weeks (SD = 14.3) for the XTEND-1 study group and 36.2 weeks (SD = 14.3) for the XTEND-Kids study group. The XTEND-1 and XTEND-Kids study groups were both reported to have high adherence to both treatment dose and treatment interval. Data on co-interventions and subsequent interventions for patients who participated in the XTEND-ed study were not provided by the sponsor.
Efficacy
Annualized Bleeding Rate
In the first 2 years of the XTEND-ed study, the mean overall ABR was 0.72 (SD = 1.26) for patients in arm A of the XTEND-1 study (prophylaxis arm), 0.42 (SD = 0.89) for patients in arm B of the XTEND-1 study (on-demand switch to prophylaxis), and 0.70 (SD = 1.27) for patients from the XTEND-Kids study. The mean ABR for patients from the XTEND-Kids study was also comparable across age groups: for patients aged younger 6 years, the mean ABR was 0.63 (SD = 1.18), and for patients aged 6 years to 12 years of age, the mean ABR was 0.77 (SD = 1.37).
Harms
Over the first 2 years of the XTEND-ed study, up to the interim analysis cut-off date, 74% of patients from the XTEND-1 study group had at least 1 TEAE and 12% had at least 1 serious TEAE. Two patients had 1 or more related TEAEs, which included facial paralysis and decreased coagulation FVIII level. The most common TEAEs (in > 5% of patients) included COVID-19 (22%), arthralgia (13%), headache (9%), nasopharyngitis (8%), and influenza (6%). Two patients discontinued therapy due to TEAEs.
In the XTEND-Kids study group, 61% of patients experienced at least 1 TEAE and 3% had at least 1 serious TEAE. The most common TEAEs (in > 5% of patients) included pyrexia (9%), arthralgia (7%), cough (7%), upper respiratory tract infection (6%), viral upper respiratory tract infection (6%), and oropharyngeal pain (6%). There were no treatment discontinuations due to TEAEs in this group.
As of the XTEND-ed study interim analysis cut-off date, there was no development of FVIII inhibitors in either group and no patient deaths.
Critical Appraisal
At the time of submission, the available evidence was limited to interim analyses based on conference presentations, which may impact the ability to sufficiently review and critically appraise the evidence, as well as the robustness of evidence and conclusions. Statistical hypothesis testing was not part of the study design and there was no active comparator or placebo arm. The XTEND-ed LTE study was designed as an open-label extension to assess long-term efficacy and safety of Altuviiio for the treatment of patients with hemophilia A. This open-label design could bias the magnitude of treatment effect for subjective efficacy outcomes and reporting of safety parameters due to unblinded exposure to the study medication during the treatment period. In addition, the mean treatment duration in the XTEND-Kids study group was less than half of that of the XTEND-1 study group, at 36.2 weeks and 82.5 weeks, respectively. Clinical experts noted that while 36 weeks is likely sufficient to assess treatment efficacy, more time is needed to evaluate long-term safety outcomes, such as inhibitor development.
The XTEND-ed arm A study population for this interim analysis consisted of patients who took part in the XTEND-1 and XTEND-Kids studies, and therefore it is reasonable to expect that the same strengths and limitations related to generalizability apply to the LTE. Given that patients needed to complete XTEND-1 or XTEND-Kids studies before enrolling, the LTE population is inherently enriched and introduces some selection bias for responders.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
Objectives for the Summary of Indirect Evidence
There were no studies that provided a direct comparison between Altuviiio, and relevant comparators were identified. In an effort to address this evidence gap, the sponsor conducted indirect comparison analyses (ITCs) that compared Altuviiio with currently available active treatments as prophylactic treatment for adult patients with hemophilia A. The objective of this section is to summarize and critically appraise the methods and findings of these studies.
Description of Indirect Comparisons
The sponsor submitted 1 ITC report21 comparing relative treatment effects of Altuviiio versus relevant comparator therapies as prophylactic treatment on patients with hemophilia A. Outcome measures assessed in this ITC included ABR, annualized spontaneous bleeding rate, and AjBR.
Study Selection Methods
To inform the ITC, a SLR was performed to identify phase III clinical trials of FVIII replacement therapies and nonfactor replacement therapies in patients with hemophilia A. The search was run in MEDLINE, Embase, and Cochrane clinical trials register through the OvidSP gate using a search strategy constructed based on the inclusion or exclusion criteria presented in the following. The last search was conducted February 10, 2021. The ITC was focused on selected comparators and outcomes in compliance with the inclusion or exclusion criteria specified in the population, intervention, comparator, and outcome (PICO) criteria for SLR. The feasibility analysis and the extractions were reviewed and critically assessed by an independent external reviewer. Additionally, a targeted literature review search was conducted aiming to identify further clinical evidence published up to the end of June 2022 (i.e., beyond the SLR timespan) from studies pivotal for registration of SLR-identified comparators. The targeted search included National Center for Biotechnology Information databases, the ClinicalTrials.gov registry, the drug registration documentation of the FDA and European Medicines Agency, and company websites containing information essential for product registration, not always found in the main publications.
According to the authors for this ITC report, all currently reimbursed drugs in the EHL and SHL classes are expected to have equivalent efficacy within their own classes. Therefore, ITCs were conducted to compare Altuviiio (from the XTEND-1 study) to emicizumab (from the HAVEN 3 study), EHL products such as efmoroctocog alfa (from the A-LONG study), and SHL products such as octocog alfa (from the LEOPOLD I study).
Details of selection criteria for these ITCs are provided in Table 21.
Table 21: Study Selection Criteria and Methods for ITCs Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Patients or patient subgroup with hemophilia A with or without inhibitors |
Intervention | Altuviiio: q.w. 50 IU per kg of prophylactic therapy (arm A of the XTEND-1 trial) |
Comparator | Prophylactic FVIII replacement therapies relevant to the Canadian treatment setting Final comparators to align with the Canadian pharmacoeconomic model, and accepted Pharmacoeconomic Deviation Request included:
|
Outcome | Annualized bleeding rate Annualized spontaneous bleeding rate Annualized joint bleeding rate |
Study designs | RCTs and non-RCTs (single-arm trials and open-label extension trials) |
Publication characteristics | Journal articles and abstracts (English language only) |
Exclusion criteria | The following were excluded:
|
Databases searched | MEDLINE, Embase, and Cochrane clinical trials register through the OvidSP gate |
Selection process | All studies were screened using predefined inclusion and exclusion criteria following the standard population, intervention, comparator, and outcome elements. The screening was conducted in 2 steps including level I title and abstract screening and level II full-text screening. Duplicates of studies (e.g., due to overlap in the coverage of databases) were deleted before screening. |
Data extraction process | Data were extracted from the relevant full-text publications identified in the current SLR by 1 reviewer and validated for accuracy by a second reviewer. Any discrepancies that arose between the 2 reviewers were reconciled by both reviewers and/or a third reviewer, if needed, to reach consensus. A data extraction template was developed to extract study design, baseline characteristics, and outcomes. Mean, median, standard deviation, standard error, and range were extracted for continuous variables where possible. For categorical variables, frequency and percentage were extracted. |
Quality assessment | Each RCT identified in the SLR underwent a comprehensive quality assessment using guidelines from NICE.79 |
EHL = extended half-life; FVIII = factor VIII: ITC = indirect treatment comparison; NICE = National Institute for Health and Care Excellence; q.w. = every week; RCT = randomized controlled trial; SHL = standard half-life; SLR = systematic literature review.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Source: Sponsor-submitted ITC report.21
ITC Analysis Methods
The XTEND-1 trial assessing Altuviiio is a 2-arm, parallel-design, nonrandomized trial. Patients who had been previously receiving FVIII prophylaxis were allocated to Altuviiio prophylaxis for 52 weeks (arm A). Those receiving on-demand treatment were allocated to arm B, in which they received on-demand Altuviiio for 26 weeks followed by 26 weeks of prophylactic Altuviiio. Because the design of the XTEND-1 trial does not allow the creation of connected networks with any of the comparator trials, the anchored comparisons (such as Bucher indirect comparison or network metanalysis) are not feasible for the comparison between Altuviiio and alternative treatments.80 Therefore, unanchored indirect comparison is considered when networks are disconnected and only single arms from respective trials can be used.80
In this ITC report, 2 methods were used with an intent of reducing or minimizing the risk of bias through balancing of the between-treatment differences in baseline characteristics of the included studies.
Unanchored MAIC, when only aggregated data regarding baseline characteristic and outcomes were available:81,82 to compare the mean estimates between outcomes of treatments A and B using standard statistical methods; between-trial imbalances in patient characteristics should be adjusted for, including all effect modifiers and prognostic factors.
An observational comparison using a PSM approach when patient-level data for both compared studies are available: the propensity score can be interpreted as the conditional probability of being assigned to the treatment, given the baseline characteristics. Propensity scores are generated using logistic regression with the treatment use as the outcome and the observed background characteristics as predictors. These propensity scores can then be applied to minimize the bias of the comparison by balancing baseline characteristics between groups.81,82
The unanchored MAIC was conducted for the comparison between Altuviiio and emicizumab or octocog alfa to minimize the risk of bias associated with unanchored comparisons, while the PSM method was used in the comparison between Altuviiio (XTEND-1) and efmoroctocog alfa (A-LONG).
In all analyses using the MAIC method, the XTEND-1 study patient-level data were adjusted for every baseline characteristic provided that adequate data were reported in the comparator studies. The following variables were considered to be adjusted for:
age (mean, median, SD)
body weight or body mass index (mean, median, SD)
race (proportion of Asian patients and white patients)
prior treatment regimen (proportion of patients receiving prophylaxis before entry)
prior frequency of bleeding (mean, median, SD)
presence of target joints (mean, median, SD, proportion of patients with 0, 1, or ≥ 2)
comorbidities (proportion of patients with HIV and or hepatitis C virus infections)
baseline patient-reported outcome values (mean, SD).
In the MAIC analyses, the model used for bleeding rate estimation from the XTEND-1 study individual patient data depended on the method of estimation used in the comparator study (e.g., negative binomial, crude mean), so that the same measure was used for treatment effect comparison. In particular, the method of bleeding rate estimation also determined the method of outcome comparison. The rates for comparators estimated using a count model (e.g., negative binomial) are directly reported from the model as the log of the rates. The between-treatment comparison expressed on the log scale and exponentiated results in the estimate of IRR. On the other hand, the absolute difference in rates calculated from 2 mean values results in the comparison following normally distributed MD in the incidence rate. Refer to Table 22 for a comparison of definitions for bleeding outcomes in the sponsor’s ITC report.
The PSM method allowed for estimation of both IRR and MD for incidence rate comparison between Altuviiio and efmoroctocog alfa, due to availability of individual patient data from both studies. In the comparison between the XTEND-1 and A-LONG trials, all patients from the XTEND-1 study arms A and B and A-LONG study individualized prophylaxis arm were included, assessing treatment effect comparison after matching patients for baseline characteristics using a PSM full matching method. The following baseline characteristics in the 2 trials were adjusted for in the model:
age (mean and SD)
body weight (mean and SD)
proportion of patients treated prophylactically before enrolment
presence of target joints, including:
proportion of patients with 0 target joint
number of target joint per patient (mean and SD)
prior bleeds (mean and SD)
proportion of patients who are HIV positive
proportion of patients who are hepatitis C positive
baseline Haem-A-QoL scores (mean and SD), including:
total score for change from baseline in Haem-A-QoL total score
physical score for change from baseline in Haem-A-QoL Physical Health score
The sponsor noted that in the comparison between Altuviiio and efmoroctocog alfa, during matching an additional caliper was imposed on inferential units that discard the patients if the units are more than a set distance (in propensity score units) away from predefined radius of their closest match. Given the lack of clarity about the optimal caliper width, the caliper ranges were manually chosen for selected variables to maximize the covariates adjustment and minimize information loss.
Definition for Bleeding Outcomes
Two commonly used methods to compare incidence rates are the IRR and the MD in the incidence rates. Both methods produce qualitatively similar estimates and statistically consistent inference but differ in the interpretation of results. IRR gives clinically interpretable results on the relative scale (treatment results in percent change in risk relative to comparator), while MD gives clinically interpretable results on the absolute scale (mean change in risk).83
Table 22: Comparison of Definitions for Bleeding Outcomes
Comparator | Trial | ABR (IRR) | ABR (MD) |
|---|---|---|---|
Efmoroctocog | A-LONG |
| NA |
Octocog alfa | LEOPOLD I | NA |
|
Emicizumab | HAVEN 3 |
| NA |
ABR = annualized bleeding rate; IRR = incidence rate ratio; MD = mean difference; NA = not available.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Source: Sponsor-submitted indirect treatment comparison report.21
Results
Summary of Included Studies
The XTEND-1 trial and another 12 publications of potential comparators contributed to the ITC analyses and 8 drugs were involved: Altuviiio, 1 nonfactor replacement therapy (emicizumab), 4 EHL therapies (efmoroctocog alfa, damoctocog alfa pegol, rurioctocog alfa pegol, and turoctocog alfa pegol), 2 SHL therapies (octocog alfa [2 different drugs]), and a gene therapy (valoctocogene roxaparvovec). These drugs were used as prophylactic therapy, on-demand therapy, or a mix of prophylactic and on-demand therapy. Among these studies, 3 evaluated the effect of prior prophylaxis regimen on bleeding outcomes, and 9 evaluated the effect of prior prophylaxis or on-demand regimen on bleeding outcomes and HRQoL. Outcomes of interest assessed in this ITC report included ABRs (all, treated, joint, and spontaneous), HRQoL (measured by Haem-A-QoL), and joint health scores (as measured by HJHS). ABR outcomes were evaluated in all 13 included trials, while HRQoL and joint health outcomes were less commonly reported. HRQoL was evaluated in 4 trials of Altuviiio, efmoroctocog alfa, turoctocog alfa, and emicizumab used as prophylaxis or for an on-demand regimen. Joint health outcome was evaluated in 2 trials of Altuviiio and emicizumab (every 4 weeks). Therefore, the current report primarily focused on the bleeding outcomes. Because gene therapy for hemophilia A is not currently available in Canada, the currently report summarizes the results for the comparisons between Altuviiio and EHL therapies, SHL therapies, and nonfactor replacement therapy.
As per the sponsor, all currently reimbursed drugs in the EHL and SHL classes are expected to demonstrate similar efficacy within their own classes. The sponsor also noted that to align with the Canadian pharmacoeconomic model as well as the submitted pharmacoeconomic deviation request which was accepted for this review, the comparators for Altuviiio in this ITC report included efmoroctocog alfa (to represent the EHL group) and octocog alfa (to represent the SHL group). Trial characteristics of the included studies are shown in Table 23. The primary outcome of the XTEND-1 trial was assessed at 52 weeks, which was similar to the LEOPOLD I and A-LONG studies. In the HAVEN 3 study, bleeding rates were assessed at 24 weeks. In the XTEND-1, LEOPOLD I, and A-LONG studies, the study drugs were used as prophylactically or on demand. In the HAVEN 3 study, emicizumab was used prophylactically only.
Baseline patient characteristics in the XTEND-1 trial before and after matching with the HAVEN 3 study (arm D) and LEOPOLD I study are presented in Appendix 3. The baseline patient characteristics before and after matching for the XTEND-1 and A-LONG studies are presented in Table 27.
All patients in the included studies were aged 12 years or older and had a diagnosis of severe hemophilia A. In all studies, severe hemophilia A was defined as less than 1 IU/dL (< 1%) endogenous FVIII activity. Before matching, there were some imbalances between the studies in proportion of patients treated prophylactically before enrolment, presence of target joints, history of prior bleeding, and proportion of HIV or hepatitis C virus infections.
Table 23: Assessment of Homogeneity
Authors | Study | Study type | Population | Intervention (class) | Arms |
|---|---|---|---|---|---|
von Drygalski et al. | XTEND-1 | Open-label, phase III study Patients were enrolled between 2019 and 2021 | Previously treated patients aged ≥ 12 years; severe hemophilia A without inhibitors | Altuviiio (recombinant antihemophilic factor) | Group A: 50 IU/kg of Altuviiio prophylaxis for 52 weeks (n = 133). Group B: 26 weeks of on-demand use of Altuviiio (50 IU/kg) followed by 26 weeks of 50 IU/kg of Altuviiio prophylaxis (n = 26). |
Mahlangu et al. (2018) | HAVEN 3 | Phase III, open-label, RCT Patients were enrolled between 2015 and 2017 | Aged ≥ 12 years with severe congenital hemophilia A without inhibitors | Emicizumab (nonfactor replacement) | Emicizumab prophylactic regimens included 4 initial loading doses of 3.0 mg/kg q.w., followed by a dose of either 1.5 mg/kg q.w. (n = 36) or 3.0 mg/kg q.2.w. (n = 35), or no prophylaxis (n = 18). |
Mahlangu et al. (2014) | A-LONG | Phase III open-label, RCT Patients were enrolled between 2010 and 2012 | Aged ≥ 12 years with severe congenital hemophilia A without inhibitors | Efmoroctocog alfa (EHL) | Arm 1: individualized prophylaxis (twice-weekly dosing; 25 IU/kg on day 1 and 50 IU/kg on day 4 to start, followed by 25 IU/kg to 65 IU/kg every 3 days to 5 days [n = 118]). Arm 2: weekly prophylaxis (65 IU/kg [n = 24]). Arm 3: episodic (on-demand) treatment as needed for bleeding episodes (10 IU/kg to 50 IU/kg, depending on bleeding severity [n = 23]). |
Saxena et al. | LEOPOLD I | A 4-part, open-label, crossover RCT, phase I/III study Patients were enrolled between 2009 and 2012 | Aged 12 years to 65 years with severe congenital hemophilia A without inhibitors | Octocog alfa (SHL) | Part A (Pharmacokinetics study): Patients received a single 50 IU/kg dose of IV octocog alfa and rFVIII-FS separated by at least a 3-day washout period in a crossover design. Part B: Patients received prophylactic 20 IU/kg to 50 IU/kg (randomized to high or low potency) octocog alfa administered 2 to 3 times per week for 6 months; patients then crossed over to the alternative potency. Patients had the option to remain on therapy for an additional year extension. Part C: Patients not enrolled in part A or B received octocog alfa as part of major surgery. Dosing for all parts was determined by chromogenic substrate assay. |
EHL = extended half-life; FVIII = factor VIII; q.w. = once a week; q.2.w. = every 2 weeks; RCT = randomized controlled trial; rFVIII-FS = sucrose-formulated recombinant factor VIII; SHL = standard half-life.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Source: Sponsor-submitted indirect treatment comparison report,21 von Drygalski et al. (the XTEND-1 study),84 Mahlangu et al. (2018) (the HAVEN 3 study),85 Mahlangu et al. (2014) (the A-LONG study),76 XTEND-1 Clinical Study Report, and Saxena et al. (the LEOPOLD I study).86
Results
Efficacy
MAIC for Altuviiio compared to emicizumab once weekly (for patients with prior prophylactic therapy): Emicizumab once weekly was assessed in arm D of the HAVEN 3 trial in 63 patients, with severe hemophilia A, who had been receiving prophylactic treatment before enrolment. Based on the available publication, the age of patients ranged from 13 years to 68 years, while the body weight ranged from 52.8 kg to 139 kg. Arm A of the XTEND-1 trial was compared with arm D of the HAVEN 3 study, because both cohorts recruited patients receiving prophylactic treatment before enrolment. Fourteen of 133 patients in the XTEND-1 study arm A with age and/or body weight outside the ranges reported in the corresponding HAVEN 3 study cohort were excluded from the analysis, so that 119 patients receiving Altuviiio were finally included in the MAIC (Table 24).
Table 24: Preselection of XTEND-1 Patients With Comparable Baseline Characteristics in HAVEN 3 Trial
Results | Arm D of the HAVEN 3 trial (prior prophylaxis) | XTEND-1 arm A (prophylactic use) individual patient data |
|---|---|---|
Age at baseline (years), range | 13 to 68 | 12 to 72 |
Body weight at baseline (kg), range | 52.8 to 139 | 33.9 to 132.8 |
Patients remaining after restrictions, n | 119 | |
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Source: Sponsor-submitted indirect treatment comparison report.21
For the comparison between prophylactic regimens of Altuviiio and emicizumab once weekly, the baseline variables that were adjusted for in the matching process included: age, body weight, presence of target joints (proportion of patients without target joints, with 1 target joint, or with at least 2 target joints), and most abundant racial group (proportion of white patients, proportion of Asian patients). The estimated ESS was reduced from 119 to 76 patients following matching, which corresponds to 63.8% of the initial sample.
All ABRs in the HAVEN 3 study were calculated using a negative binomial model with stratification for the history of previous bleeds (< 9 or ≥ 9 bleeding events in the previous 24 weeks). ABRs for the XTEND-1 study were estimated using the same regression model, but without stratification factor due to lack of data regarding history of bleeds within 24 weeks before enrolment. A summary of the results for the MAIC of Altuviiio to emicizumab is available in Figure 2. Based on estimated effects from the model, Altuviiio compared to emicizumab once weekly (prior prophylactic use) was associated with lower rate of any bleeding (treated and untreated) (IRR = 0.32; 95% CI, 0.19 to 0.56) and joint treated bleeding (IRR = 0.48; 95% CI, 0.24 to 0.95). There was no evidence for significant differences regarding frequency of treated spontaneous bleeding (IRR = 0.62; 95% CI, 0.25 to 1.50).
Figure 2: Incidence Rate Ratio for ABR of Altuviiio vs. Emicizumab
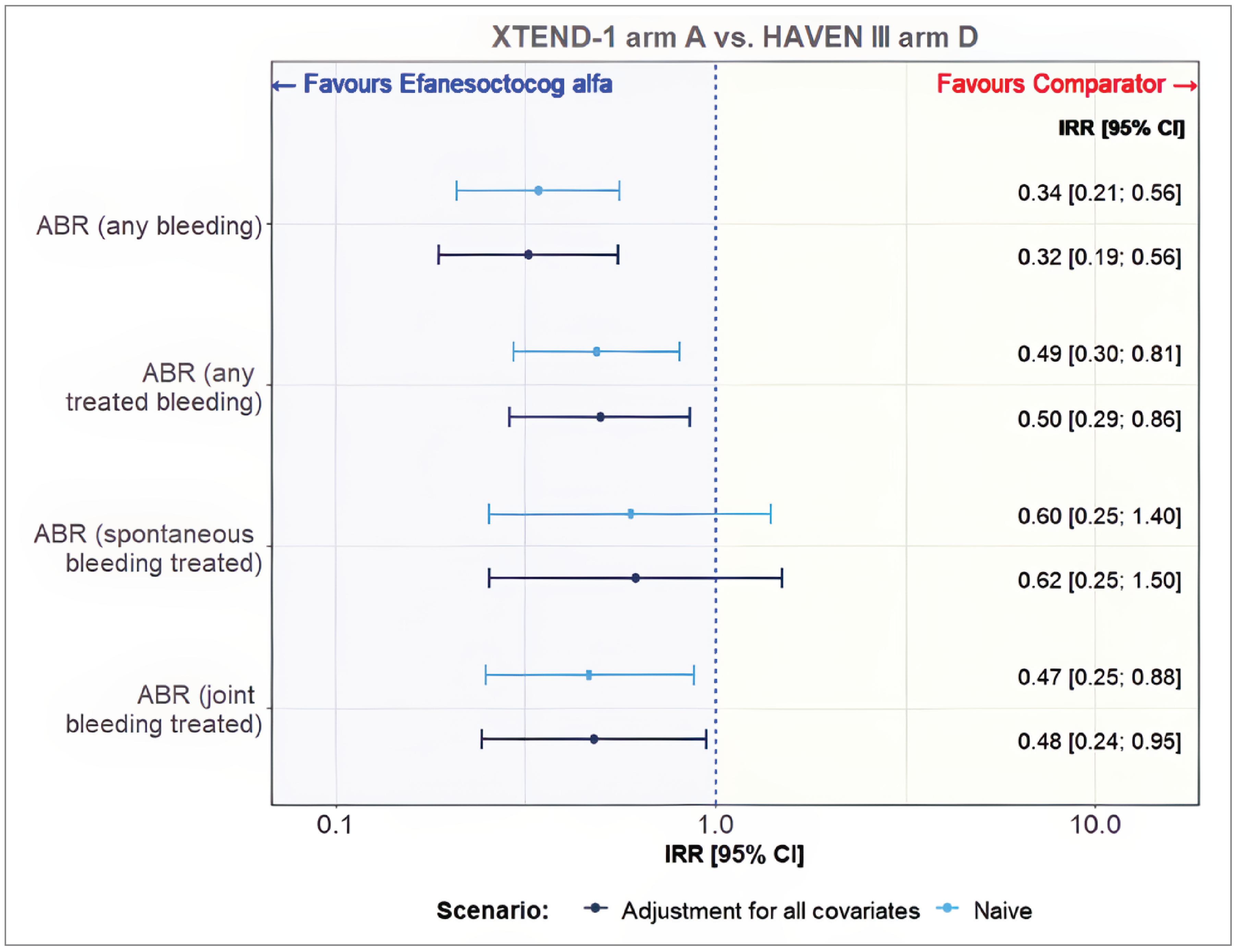
ABR = annualized bleeding rate; CI = confidence interval; IRR = incidence rate ratio; MAIC = matching-adjusted indirect comparison; vs. = versus.
Source: Sponsor-submitted indirect treatment comparison report.21
MAIC for Altuviiio compared to octocog alfa (SHL class) (for patients with prior prophylactic and on-demand therapy): Octocog alfa (Kovaltry) was assessed in arms A and B of the LEOPOLD I trial in 62 patients with severe hemophilia A, who had been receiving prophylactic and on-demand treatment before enrolment. Based on the available publication, the age of patients ranged from 12 years to 61 years, while the body weight ranged from 39 kg to 121 kg. Pooled arms A and B of the XTEND-1 trial were compared with the LEOPOLD I study, because both cohorts recruited patients receiving prophylactic and on-demand treatment before enrolment. Seventeen of 159 patients in the XTEND-1 study with age and/or body weight outside the ranges reported in the corresponding LEOPOLD I study cohort were excluded from the analysis, so that 142 patients receiving Altuviiio were finally included in the MAIC (Table 25). Data from the LEOPOLD II study were not included in this ITC as these patients exclusively received on-demand therapy and not had previously received prophylactic therapy.
Table 25: Preselection of XTEND-1 Patients With Comparable Baseline Characteristics in LEOPOLD I Trial
Results | LEOPOLD I (prior prophylaxis and on-demand therapy use) pooled arms N = 159 | XTEND-1 arm A (prophylactic use) individual patient data |
|---|---|---|
Age at baseline (years), range | 12 to 61 | 12 to 72 |
Body weight at baseline (kg), range | 39 to 121 | 33.9 to 132.8 |
Patients remaining after restrictions, n | 142 | |
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Source: Sponsor-submitted indirect treatment comparison report.21
The comparison between interventions was adjusted for the following baseline variables in the MAIC model: age, body weight, proportion of patients with 1 or more target joints, proportion of white patients, proportion of patients treated prophylactically before enrolment, and prior bleeds. After matching, the estimated ESS was reduced from 128 to 29 patients following matching, which corresponds to 22.7% of the initial sample.
All ABRs in the LEOPOLD I study were reported as mean and SD. ABRs for the XTEND-1 study were estimated analogously. A summary of the results for the MAIC of Altuviiio to octocog alfa is available in Figure 3. Altuviiio compared to octocog alfa was associated with lower frequency of any bleeding (MD = −2.97; 95% CI, –4.28 to −1.67), spontaneous bleeding (MD = −2.23; 95% CI, –3.10 to –1.35), and joint bleeding (MD = −2.67; 95% CI, –3.85 to –1.49).
PSM for Altuviiio compared to efmoroctocog alfa (EHL class) (individualized prophylaxis): Efmoroctocog alfa was assessed in the A-LONG trial in 165 patients with severe hemophilia recruited to individualized prophylaxis (n = 118), weekly prophylaxis (n = 24), or on-demand treatment (n = 23); only 117 patients with assessed individualized prophylaxis were included in the comparison with Altuviiio, as this treatment regimen corresponds to dosing specified in regulatory labels. Pooled arms A and B of the XTEND-1 trial were compared with the A-LONG study, because both cohorts recruited patients receiving prophylactic and on-demand treatment before enrolment. The age of A-LONG study patients ranged from 12 years to 65 years and the body weight ranged from 42 kg to 127 kg; thus, the population from the XTEND-1 study had a slightly wider range of age and body weight values compared to the A-LONG study. In the analysis, all patients from the XTEND-1 study arm A and B and the A-LONG study individualized prophylaxis arm were included, assessing treatment effect comparison after matching patients for baseline characteristics using the PSM full matching method.
Figure 3: Mean Difference of ABRs Between Altuviiio and Octocog Alfa
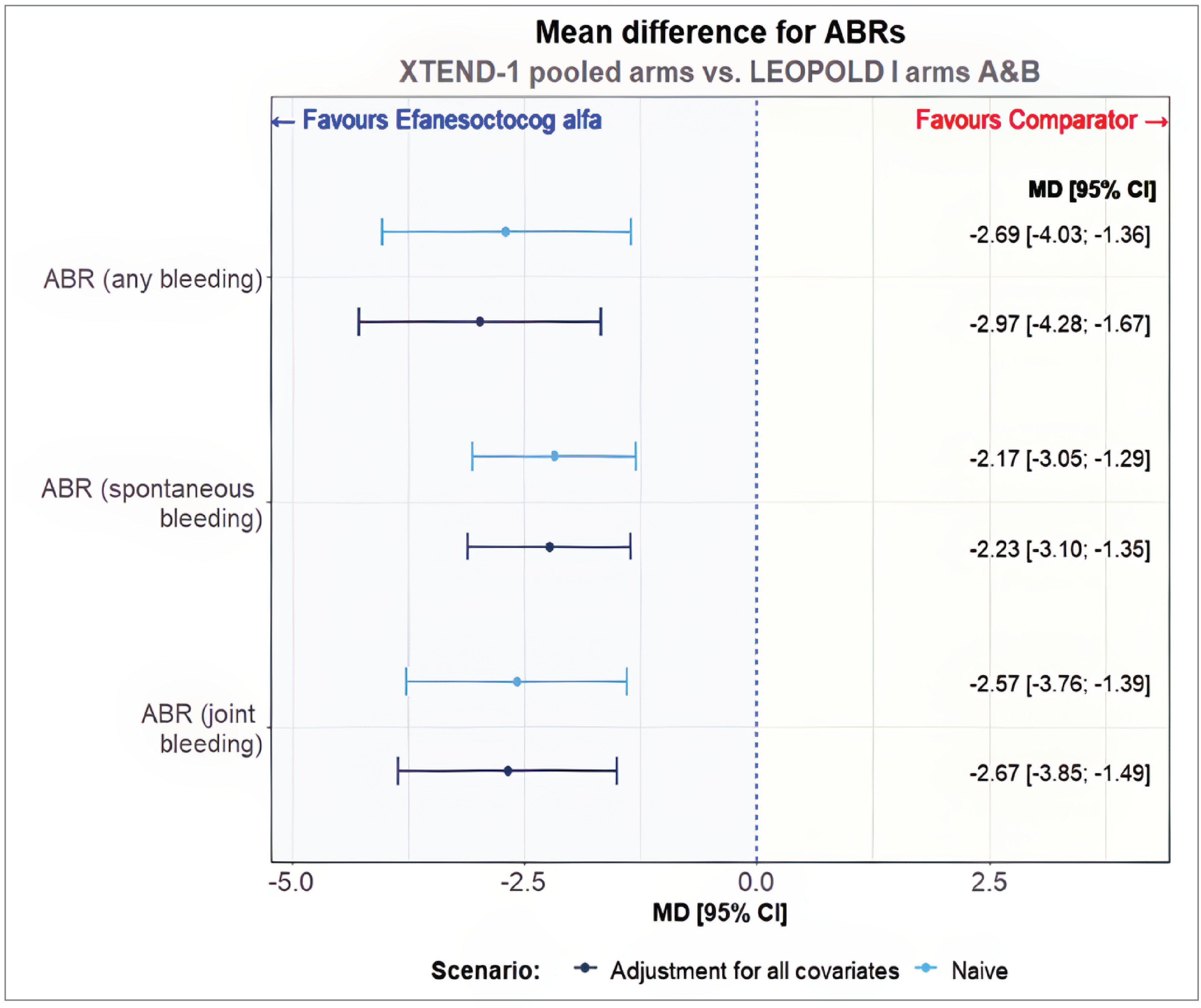
ABR = annualized bleeding rate; CI = confidence interval; MAIC = matching-adjusted indirect comparison; MD = mean difference; vs. = versus.
Source: Sponsor-submitted indirect treatment comparison report.21
Table 26: Preselection of XTEND-1 Patients With Comparable Baseline Characteristics in the A-LONG Trial
Results | A-LONG individualized prophylaxis N = 117 | XTEND-1 individual patient data (pooled arms) N = 159 |
|---|---|---|
Age at baseline (years), range | 12 to 65 | 12 to 72 |
Body weight at baseline (kg), range | 42 to 127 | 33.9 to 132.8 |
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Source: Sponsor-submitted indirect treatment comparison report.21
The following baseline variables were adjusted for in the comparison between Altuviiio and efmoroctocog alfa: age, body weight, proportion of patients treated prophylactically before enrolment, presence of target joints (proportion of patients with 0 target joint, number of target joints per patient), prior bleeds, proportion of patients living with HIV or hepatitis C virus infections, and baseline Haem-A-QoL scores (total score and physical score).
The estimated ESS in models for bleeding for the XTEND-1 study individual patient data was reduced from 145 to 87 patients following matching which corresponds to 60% of the initial sample, and for the A-LONG study individual patient data was reduced from 116 to 30 patients following matching which corresponds to 26% of the initial sample (Table 27). The number of patients analyzed may be less than the number of patients recruited due to removed patients with missing information on baseline characteristics or outcomes.
The comparison between prophylactic regimens of Altuviiio and the efmoroctocog alfa individualized prophylaxis arm in patients receiving prior prophylactic or prior on demand therapy was feasible for FVIII consumption, the ABRs for any treated bleeding episodes, spontaneous treated bleeding episodes, and joint treated bleeding episodes. Treatment effect estimate for ABRs between the XTEND-1 and A-LONG studies was estimated with a negative binomial model with treatment duration as offset. A summary of the results for the PSM analysis of Altuviiio to efmoroctocog alfa is available in Figure 4. Altuviiio compared to efmoroctocog alfa individualized prophylaxis was associated with favourable (less frequent) estimates for treated bleeding rate (IRR = 0.29; 95% CI, 0.17 to 0.51), spontaneous bleeding rate (IRR = 0.21; 95% CI, 0.09 to 0.49), and joint bleeding rate (IRR = 0.37; 95% CI, 0.20 to 0.71]). The estimated IRRs were statistically significant.
Additional results: compared to other comparator therapies: Altuviiio was also compared to damoctocog alfa pegol, rurioctocog alfa pegol, and turoctocog alfa pegol, and the results on ABR favoured Altuviiio (Table 28). The direction of the changes in these results are comparable to those with emicizumab, octocog alfa, and efmoroctocog alfa.
Table 27: Matching of Baseline Characteristics Between XTEND-1 Pooled Arms and A-LONG for Models Assessing Bleeding Outcomes and Factor VIII
Variables | Before matching | After matching | P value for difference | ||||
|---|---|---|---|---|---|---|---|
XTEND-1 (arm A and B) N = 145 | A-LONG (individual prophylaxis) N = 116 | P value for difference | XTEND-1 (arm A and B) ESS = 87 (60%) | A-LONG (individual prophylaxis) ESS = 30 (26%) | |||
Estimate (SD) | Estimate (SD) | Estimate (SD) | Estimate (SD) | Balance improvement (%) | |||
Mean age (years) | 35.36 (15.61) | 33.05 (12.79) | 0.190 | 29.52 (12.39) | 30.73 (11.75) | 48 | 0.632 |
Mean weight (kg) | 77.42 (19.01) | 73.35 (15.15) | 0.056 | 73.53 (13.97) | 73.65 (12.47) | 97 | 0.964 |
Prior prophylaxis (%) | 84.1 (NR) | 73.3 (NR) | 0.031a | 89.7 (NR) | 95.4 (NR) | 47 | 0.338 |
Mean number of TJs | 0.938 (1.741) | 1.672 (2.072) | 0.003a | 0.609 (1.417) | 0.730 (1.489) | 84 | 0.698 |
Patients with 0 TJs (%) | 70.3 (NR) | 37.9 (NR) | < 0.001a | 78.2 (NR) | 75.7 (NR) | 92 | 0.777 |
Mean number of prior bleeds | 8.34 (15.55) | 18.31 (22.37) | < 0.001a | 4.40 (8.39) | 5.04 (7.89) | 94 | 0.710 |
HIV rate (%) | 13.8 (NR) | 21.6 (21.6) | 0.099 | 9.2 (NR) | 6.9 (NR) | 70 | 0.698 |
HCV rate (%) | 34.5 (NR) | 47.4 (47.4) | 0.034a | 33.3 (NR) | 37.7 (NR) | 66 | 0.660 |
ESS = effective sample size; HCV = hepatitis C virus; NR = not reported; PSM = propensity score matching; SD = standard deviation; TJ = target joint.
Notes: The statistical tests used were the 2-sample t test for continuous variables and the 2-sample test for equality of proportions for binary variables. PSM model = age + weight + prior regimen + TJ + prior bleeds + HIV + HCV (caliper: SD = 0.1, age = 20, weight = 20, TJ = 5, prior bleeds = 5).
Details included in the table are from the sponsor’s Summary of Clinical Evidence.
aStatistically significant difference in baseline characteristic between studies.
Source: Sponsor-submitted indirect treatment comparison report.21
Figure 4: Incidence Rate Ratio for ABRS of Altuviiio vs. Efmoroctocog Alfa
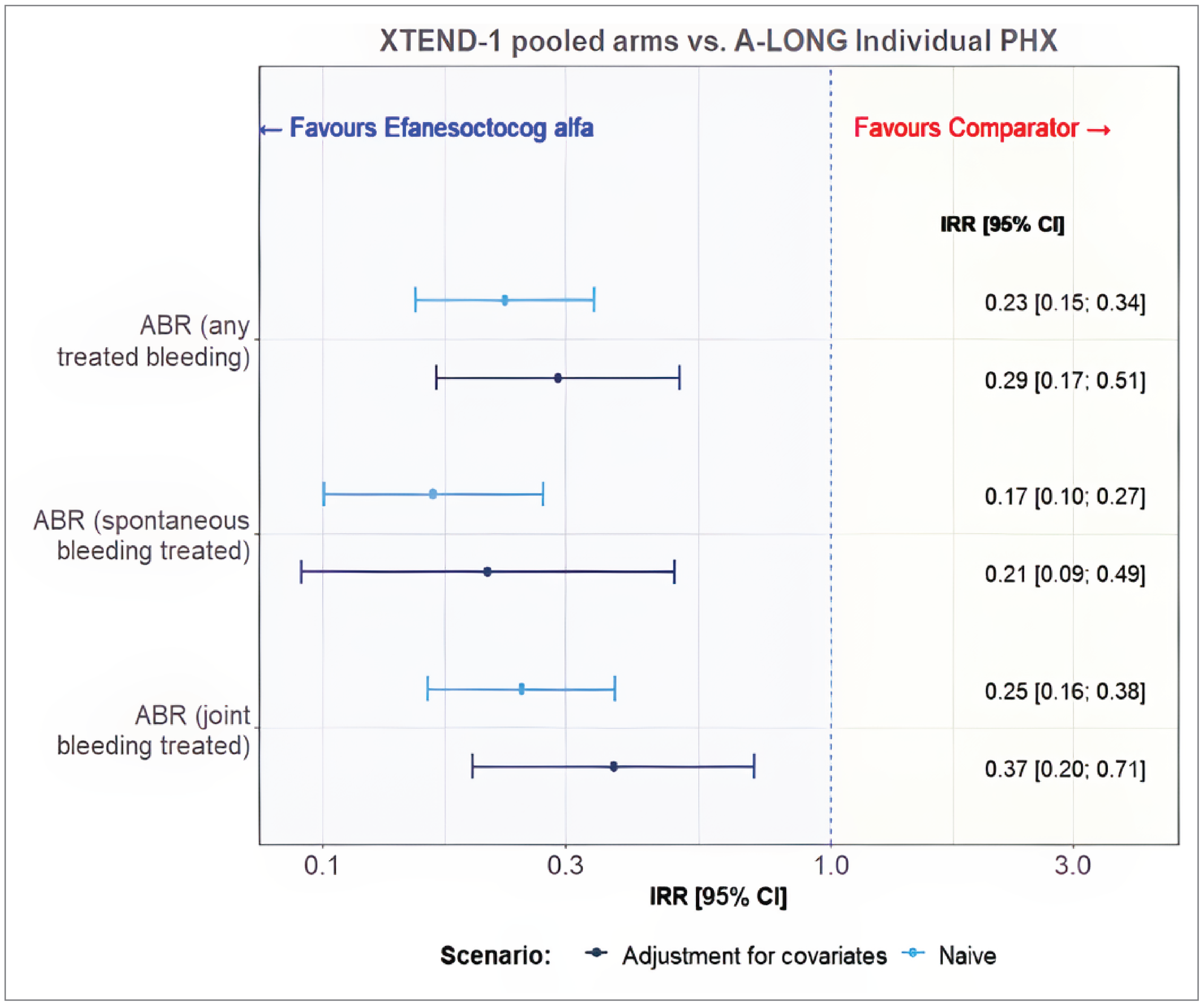
ABR = annualized bleeding rate; CI = confidence interval; Haem-A-QoL = Haemophilia Quality of Life questionnaire for adults; IRR = incidence rate ratio; PHX = prophylaxis; vs. = versus.
Note: Change from baseline in Haem-A-QoL total score or physical score in the comparisons between Altuviiio vs. efmoroctocog alfa as prophylactic therapy was −2.43 (95% CI, −8.48 to 3.62) and −7.01 (95% CI, −14.69 to 0.67), respectively.
Source: Sponsor-submitted indirect treatment comparison report.21
Table 28: Summary of Additional Efficacy Outcome Measures for Other EHL Therapies in the Sponsor-Submitted ITC (MAIC and PSM)
Outcomes | Damoctocog alfa pegol (pooled PHX and O-D)a | Rurioctocog alfa pegolb | Turoctocog alfac |
|---|---|---|---|
ABR (any bleeding), IRR (95% CI) | NA | 0.20 (0.12 to 0.31) | 0.24 (0.15 to 0.37) |
ABR (any bleeding), mean difference (95% CI) | −3.79 (−5.22 to −2.36) | NA | −2.50 (−3.20 to −1.80) |
ABR (spontaneous bleeding), IRR (95% CI) | NA | NA | NA |
ABR (spontaneous bleeding), mean difference (95% CI) | −3.05 (−4.32 to −1.79) | NA | −1.46 (−1.99 to −0.93) |
ABR (joint bleeding), IRR (95% CI) | NA | NA | NA |
ABR (joint bleeding), mean difference (95% CI) | −3.18 (−4.39 to −1.97) | NA | −1.90 (−2.51 to −1.29) |
Haem-A-QoL total score change from baseline, mean difference (95% CI) | NA | NA | −5.18 (−7.02 to −3.34) |
Haem-A-QoL Physical Health score change from baseline (MD) | NA | NA | −3.01 (−6.16 to 0.15) |
ABR = annualized bleeding rate; CI = confidence interval; EHL = extended half-life; Haem-A-QoL = Haemophilia Quality of Life questionnaire for adults; IRR = incidence rate ratio; ITC = indirect treatment comparison; MAIC = matching-adjusted indirect treatment comparison; MD = mean difference; NA = not available; O-D = on demand; PHX = prophylactic; PSM = propensity score matching; vs. = versus.
Note: IRR < 1.0 or MD < 0 indicates favouring Altuviiio; IRR > 1.0 or MD > 0 indicates favouring comparators.
aBased on the PROTECT VIII trial.
bBased on the PROPEL and PROLONG-ATE studies.
cBased on the PATHFINDER 2 study.
Source: Sponsor-submitted indirect treatment comparison report.21
Harms
Harms data were not collected as part of the SLR and thus not included as part of this analysis of indirect evidence.
Critical Appraisal of Sponsor-Submitted Indirect Evidence
In this ITC report including MAICs and propensity score analysis, indirect evidence was identified by searching multiple databases based on prespecified inclusion and exclusion criteria. It was unknown if the studies were selected by 2 independent reviewers. Appropriate methods and tools were used to reduce the risk of bias and error in data extraction and quality assessment of the included studies. However, it was unknown if the risk of bias of the included studies was assessed by 2 independent reviewers and the results of quality assessment were not provided. In addition, risk of bias might have been assessed at the level of the trial, rather than at the level of the reported results (i.e., per outcome), which ignores that risk of bias can vary by reported result within a trial. It is worth noting that hemophilia A is a rare disease, therefore the number of clinical trials of hemophilia A and the number of patients recruited in the available trials could be limited. In total, 4 studies were included in this ITC report. They are all small open-label trials, randomized or nonrandomized; therefore, the evidence base for indirect comparisons between Altuviiio and comparator therapies is small.
The relative efficacy of Altuviiio (as prophylactic treatment) compared to the currently available active treatments in the target population was examined in this ITC. One of the major concerns for ITCs is that the included trials could be highly heterogeneous in terms of study design and patient characteristics at baseline. The studies included in the ITC were published between 2014 to 2023. The selection criteria in these 4 studies were similar, where patients who were aged 12 years or older with severe hemophilia A were enrolled. With the goal of reducing the risk of bias in result interpretation, unanchored MAIC or a PSM method was used in balancing the baseline characteristics between the included trials. The selection of conducting a MAIC or PSM analysis was appropriately justified in the sponsor’s ITC report. In the MAICs, the sponsor noted that the following potential effect modifier or prognostic factors were adjusted for if adequate data were reported in the comparator studies: age, body weight or body mass index, race, prior treatment regimen, prior frequency of bleeding, presence of targeted joints, comorbidities, and baseline patient-reported outcome values. There was no information provided on how these variables were identified, if based on literature review or being identified through a deliberating process by the clinicians. In general, the clinical experts consulted for this review agreed that these are relevant effect modifiers and prognostic variables. In addition, the XTEND-1 trial coincided with the COVID-19 pandemic, where changes in the level of physical activity before or during the COVID-19 pandemic may have affected patients’ risk of bleeding due to changes in lifestyle and behaviour related to physical activity. As such, there is potential for risk of bias in the included studies due to potential confounding by the heterogeneity in physical activity level at baseline, and the time that patients were treated and evaluated (prepandemic versus during pandemic). However, the direction of bias is unclear, and the clinical experts consulted by CDA-AMC did not expect this to significantly impact the results.
According to the clinical expert consulted for this review, clinical practice and management of patients with hemophilia A have evolved considerably in the past 10 years. For example, because there has been an increase in the use of factor prophylaxis in patients of all ages and disease severities, factor prophylaxis dosing and frequency are tailored based on patient’s own PK profile, bleeding profile, activity levels, and potential impact of a bleeding event. Further, the risk of severe bleeding in patients with factor levels indicative of mild to moderate disease range is recognized and such patients can still benefit from prophylactic treatment (either factor or nonfactor). In addition, many clinicians in Canada have adopted the WFH clinical practice guidelines. All these improvements have not been considered in the analyses, and as a result, the reported study results may be confounded and biased. After weighting in the PSM analysis, most of the patient characteristics were balanced between the XTEND-1 and A-LONG studies, although remaining imbalance were still observed. According to the clinical experts consulted on this review, the remaining differences observed between the 2 studies were small and unlikely to have substantial impact on study results.
In this ITC report, the sponsor noted that all currently reimbursed drugs in the EHL and SHL classes are expected to have equivalent efficacy within their own classes, based on the key opinion leaders’ input. Therefore, ITCs were conducted to compare Altuviiio to emicizumab, efmoroctocog alfa (representing EHL products), and octocog alfa (representing SHL products). The clinical experts consulted for this review agreed that this assumption was reasonable. However, a rationale of why certain drugs were selected in the ITC analyses instead of others in a same drug class was not provided by the sponsor. The analysis was primarily focused on patients who had been receiving prophylactic regimens before study entry, which was consistent with the inclusion criteria for arm A of the XTEND-1 trial for Altuviiio. However, comparisons with patients receiving prior on-demand treatment and the mixed populations (e.g., prophylactic and on-demand treatments) were also attempted.
The estimated ESS was reduced to 63.8% of the initial sample of the XTEND-1 study in the comparison with emicizumab and to 22.7% of the initial sample of the XTEND-1 study in the comparison with octocog alfa. In the comparison with efmoroctocog alfa, the ESS was reduced to 60% of the initial XTEND-1 study sample and 26% of the initial A-LONG study sample. A significant reduction in sample size can contribute to imprecision and increase uncertainty of the results. A notable reduction in ESS also suggests that the study results may be heavily influenced by a subset of the sample in the trials which may not be representative of the full sample, thus limiting generalizability to the full population presented by the XTEND-1 trial. The ABR was the main outcome of interest in this ITC report. HRQoL (measured with Haem-A-QoL) was also assessed in patients receiving prophylactic treatment. The results were briefly reported for the comparisons between Altuviiio and 2 EHL therapies (efmoroctocog alfa and turoctocog alfa) without sufficient details, and the study findings were inconsistent for these 2 EHL therapies. Other important clinical outcomes of interest to patients and clinicians, such as harms or inhibitor development, were not assessed in this ITC report. Therefore, the relative treatment effect of Altuviiio versus relevant comparators on these outcomes remains unknown. Moreover, the comparative efficacy and safety of Altuviiio versus comparators in the pediatric population, or its use as on-demand treatment or perioperative management was unclear due to the lack of clinical evidence.
Studies Addressing Gaps in the Systematic Review Evidence
There were no relevant studies addressing the gaps in the systematic review evidence submitted for this review.
Discussion
Summary of Available Evidence
Two pivotal, nonrandomized, open-label, multicentre, phase III trials (XTEND-1, N = 159; XTEND-Kids, N = 74) were included in the SLR conducted by the sponsor. The XTEND-1 and XTEND-Kids studies evaluated the safety and efficacy of weekly Altuviiio for prophylactic use for 52 weeks in an adolescent and adult (aged ≥ 12 years) population and pediatric (patients aged < 12 years) population, respectively, with severe congenital hemophilia A (defined as < 1 IU/dL [< 1%] endogenous FVIII activity) without FVIII inhibitors. The XTEND-1 study also assessed the use of efanesoctocog as on-demand treatment compared to prophylactic use in terms of the ABR. Patients in the XTEND-1 trial were divided into 2 treatment groups based on the treatment regimen before entering the study: Altuviiio prophylaxis group (arm A) and Altuviiio on-demand treatment followed by prophylaxis (arm B). Arm A included participants who were on a current FVIII prophylactic treatment regimen and participated in an observational prestudy (242HA201/OBS16221) for at least 6 months before baseline of the XTEND-1 study. The XTEND-Kids study comprised 2 age cohorts, children aged less than 6 years (n = 38) and children aged 6 years to less than 12 years (n = 36), and all patients received once-weekly IV doses of 50 IU/kg Altuviiio prophylactic treatment. Lastly, Altuviiio for perioperative management of bleeds was also assessed in any patient from the XTEND-1 and XTEND-Kids studies who underwent major surgery after the first dose of the study drug.
In the XTEND-1 study, the primary efficacy outcome was ABR and in the XTEND-Kids study, the primary outcome was formation of FVIII inhibitors. Other efficacy outcomes in both trials were AjBR, physical function and pain (Haemo-QoL, Haem-A-QoL), HJHS, response to perioperative management (number of injections to maintain hemostasis before major surgery), and harms outcomes. In the XTEND-1 study, the mean age of patients was 35.4 years (SD = 15.1 years), ranging from 12 years to 72 years, and 78.6% of patients had no family history of FVIII inhibitors. Further, the mean number of bleeding episodes 12 months before the study was 3.2 (SD = 5.4) in arm A and 35.7 (SD = 22.2) in arm B. In the XTEND-Kids trial, the mean age was 5.99 years (SD = 2.91 years), 77% of patients had no family history of an inhibitor, and the mean number of bleeding episodes during the 12 months before the study was 2.1 (SD = 4.2).
The key limitations of the body of evidence were related to the open-label study design, absence of randomized, direct comparative evidence between Altuviiio and other FVIII prophylaxis, and the exclusion of patients with FVIII inhibitors and patients who have not received prior FVIII therapies.
The sponsor submitted indirect evidence comparing efanesoctocog to relevant comparator therapies as prophylactic treatment for adult patients with severe hemophilia A. This included 2 MAICs that compared Altuviiio with a nonfactor replacement therapy agent (emicizumab) and an octocog alfa agent (as a proxy for SHL products), and 1 observational study using a PSM method to compare Altuviiio with efmoroctocog alfa (as a proxy for EHL products). Outcome measures assessed in this ITC included ABRs for any bleeding, spontaneous bleeding, and joint bleeding.
Evidence for long-term efficacy and safety of Altuviiio for prophylactic use was included in the submission based on the ongoing phase III, open-label, multicentre study, XTEND-ed (N = 217) that enrolled patients who were previously enrolled in the Altuviiio trials. This submitted interim analyses pertains only to patients rolled over from the XTEND-1 and XTEND-Kids studies into arm A of the XTEND-ed study and reports on efficacy and safety-related outcomes over up to an additional 2 years of treatment with Altuviiio.
Interpretation of Results
Efficacy
Input from patients’ groups, clinician groups, Canadian Blood Services, and the clinical experts consulted for this review indicated that the most important treatment goals for patients with hemophilia A are to prevent bleeding, including spontaneous and traumatic bleeding events, reduce joint pain, improve HRQoL, and achieve unrestricted lifestyle comparable to the general population. With currently available treatments, achieving this goal requires frequent administration of high treatment doses to overcome short treatment half-lives. According to the clinical expert and clinician group input, treatment burden is particularly notable in patients who require elevated trough levels due to recent surgical procedures, have compromised joint health, or have high physical activity levels. Consistent with clinical expert input, the clinician group and patient group agreed that current existing therapies demonstrate variable efficacy.
The efficacy of Altuviiio as routine prophylaxis to reduce the frequency of bleeding episodes, and perioperative management of bleeding in adults and children with hemophilia A was assessed using 2 pivotal trials, the XTEND-1 and XTEND-Kids studies. The results of the XTEND-1 and XTEND-Kids trials suggest that Altuviiio (50 IU/kg) prophylaxis for up to 52 weeks reduces bleeding associated with hemophilia A based on the number of bleeds per year and the number of joint bleeds per year, assessed with ABR and AjBR, respectively. Although all analyses in the XTEND-Kids study were descriptive, and no formal hypothesis testing was performed, bleeding rates following 52 weeks of Altuviiio for prophylactic use in the XTEND-Kids study were consistent with those observed in the XTEND-1 study and considered clinically relevant by the clinical experts consulted by CDA-AMC. Additionally, once-weekly prophylaxis with Altuviiio for 52 weeks may result in an improved ABR compared to a prestudy standard of care FVIII prophylaxis based on the intrapatient analysis, although the evidence is uncertain as the magnitude of effect may be overestimated based on limitations of external and internal validity as the prior observational study and the XTEND-1 trial were conducted before and during the pandemic, respectively. However, the differences in magnitude of efficacy results are not expected to invalidate the clinical significance of the bleeding outcomes in the XTEND-1 study, which are expected to address a gap in therapy experienced by patients currently treated with standard-care FVIII. The clinical experts were in agreement that the reduction in bleeding outcomes was clinically meaningful, albeit there are several limitations associated with the open-label, nonrandomized design, which are reflected by the GRADE assessment as low level of certainty for ABR and very low level of certainty for AjBR. In rare disease conditions like hemophilia A, challenges such as patient recruitment, and availability of effective treatment make it practically impossible to use traditional RCTs in evaluating treatment efficacy. The clinical experts consulted for this review indicated that although RCTs remain the gold standard, alternative designs like single-arm trials and intrapatient comparison are accepted in hemophilia A as being able to provide a practical evaluation of new therapies. Overall, the data for bleeding outcomes in the XTEND-1 study were considered robust by the clinical experts consulted for this review given the consistency of the primary results with various sensitivity analyses, as well as the proportion of patients who were reported as experiencing no bleeds requiring treatment during the trials. Specific subgroups, such as patients with obesity or those participating in higher-intensity physical activity who may require adjusted dosing, were considered relevant to the assessment of Altuviiio; however, these were not studied in the trials.
The current standard of care for patients with hemophilia A in Canada is primary prophylactic therapy. According to the clinical experts consulted for this review, the goal of prophylaxis is to increase a patient’s FVIII levels to normal or near-normal levels to prevent spontaneous bleeding, prevent arthropathy, and to attain an active lifestyle comparable to individuals without hemophilia A. However, current products do not provide these normal or near-normal FVIII levels. Apart from emicizumab, which provides a FVIII activity equivalence level of 10% to 15%, the trough levels of available SHL and EHL FVIII concentrates are between 3% to 5% immediately after infusion with subsequent clearance dependent on the product half-life (but generally 14 to 18 hours), resulting in less bleeding protection. For patients who participate in regular physical activities, infusions may be planned to coincide with physical activity, otherwise additional doses on top of their regular prophylaxis are needed before certain physical activities to mitigate the risk of provoked bleeding. According to the patient group input received for this review, patients reported that new therapies that can improve hemophilia A disease outcomes such as improved bleeding protection with a reduced administration frequency (fewer doses with longer half-life) are needed to improve disease outcomes. As described, in both the XTEND-1 and XTEND-Kids studies, once-weekly Altuviiio for prophylactic use in patients with severe hemophilia A resulted in an ABR that was considered clinically relevant. Further, FVIII activity was descriptively reported as being greater than 40% for the first 4 days of treatment, and greater than 10% for nearly 7 days. As noted by the clinical experts consulted by CDA-AMC, clinically, this is reflective of effective bleeding control without a trade-off in burden of treatment. This is consistent with the reported half-life for Altuviiio87 as well as input received from a patient group for this submission, where 1 patient who had been treated with Altuviiio via a special access program indicated that Altuviiio has helped reduced her risk of bleeding due to the sustained high FVIII level even after several days post infusion.
Outcomes that assessed joint health (HJHS), physical function and pain outcomes (PROMIS instruments), and HRQoL (Haem-A-QoL) were also identified as outcomes of importance to patients based on the patient and clinician group input received for this review. The input from the patient and clinician groups noted that up to 46% of patients with hemophilia report living with chronic pain which negatively affect physical and psychological health. Regarding joint health, the evidence did not suggest an improvement in joint health based on the HJHS, but it also did not suggest a worsening of joint health. The clinical experts indicated that while this is an outcome of importance for patients and an unmet need, an improvement in joint health is not expected based on the mechanism of action for Altuviiio. In adults (the XTEND-1 study), the change from baseline to week 52 in HRQoL based on the Haem-A-QoL Physical Health score indicated an improvement in QoL; however, this within-group change was not considered clinically meaningful based on the within-group MID identified in the literature.55 In the XTEND-Kids study, the results for Haem-A-QoL total score did suggest an improvement for children aged 8 years to 12 years, but these data are highly uncertain due to a small sample size. While the results suggest an improvement in PROMIS Pain Intensity scores at week 52 for adults in the XTEND-1 study, it is unclear whether this change corresponds to a clinically meaningful difference due to the absence of validated MIDs for this outcome. Additionally, the evidence was insufficient to support an improvement in pain intensity among pediatric patients based on the results from the XTEND-Kids study. Assessments of pain and physical function did not correspond to an improvement in either trial. In consultation with the clinical experts consulted for this review, assessments at week 52 may not be a sufficient amount of time to evaluate an improvement in these outcomes and long-term results are likely needed to for an adequate assessment of these outcomes.
In view of this, the sponsor submitted an interim analysis of the ongoing LTE study, XTEND-ed. Outcomes included in the interim analysis include: the occurrence of inhibitor development (primary outcome), ABR, treatment of bleeding episodes, safety and tolerability, and perioperative management. Evidence for ABR was included in this review report, and the results over 2 additional years of therapy were consistent with what was observed in the pivotal trials. However, the available evidence was only limited to analyses based on conference presentations, which likely impacts the robustness of evidence and conclusions. The XTEND-ed arm A study population for this interim analysis consisted of patients who took part in the XTEND-1 and XTEND-Kids studies, and therefore it is reasonable to expect that the same strengths and limitations related to generalizability apply to the LTE.
In the XTEND-Kids trial, the primary end point was a safety outcome, that is, the incidence of inhibitor development to FVIII which was 0.0% in all treated patients and 0.0% in patients with 50 or more exposure days to Altuviiio. The inclusion of previously treated patients with hemophilia A and inclusion of patients with no family history of FVIII inhibitors (the majority of the study population) limits the generalizability of these findings, which was supported by input from the clinical experts. CDA-AMC notes that a larger sample of previously treated patients followed over a longer period and assessments of previously untreated patients would be useful in understanding the immune response and the potential immunogenicity of Altuviiio. Although the clinical experts consulted for this review agreed that the inclusion of patients with no history of FVIII treatment is a limitation, they indicated that this was not a major concern, because in clinical practice, patients are closely monitored for inhibitor development during the first few weeks of starting treatment consistent with standards of care in the Canadian landscape.
The patient group input received for this review highlighted surgery complications as one of the challenges associated with hemophilia A. Based on the patient group input, surgery is normally required to break the cycle of chronic synovitis in patients with hemophilia A; however, patients who present for such invasive procedures are at increased risk of bleeding during surgery. As such, perioperative management of hemophilia A is an important need. In both trials, a single injection of 50 IU/kg Altuviiio was effective in terms of achieving a hemostatic response in the perioperative setting, in which the hemostatic response was rated as excellent by the investigators or surgeons for all 14 major surgeries that occurred during the XTEND-1 and XTEND-Kids studies. While both trials appear adequately powered for assessing bleeding and joint health outcomes, outcomes concerning perioperative management may not be fully powered to detect rare AEs or efficacy in subpopulations, limiting the interpretation of this outcome.
No studies were identified in the sponsor’s systematic review that provided a direct comparison between Altuviiio and relevant comparators. As such, indirect evidence that compared Altuviiio with efmoroctocog alfa, octocog alfa, and emicizumab as prophylactic treatment for adult patients with hemophilia A was submitted. An anchored comparison was not feasible; therefore, 2 unanchored MAICs and a PSM analysis were conducted. Results from these indirect comparisons suggested that prophylactic treatment with Altuviiio was associated with improved bleeding outcomes (any bleeding, spontaneous bleeding, and joint bleeding) compared with comparator therapies currently available in clinical practice for adult patients with severe hemophilia A, such as EHL agents, SHL agents, or emicizumab. However, a definite conclusion on the magnitude of the clinical benefit of Altuviiio cannot be drawn due to limitations of the available indirect evidence, such as a sizable reduction in ESS, and inadequate or lack of adjustment for potential prognostic factors (e.g., physical activity level at baseline and the impact of the COVID-19 pandemic) that may introduce unmeasurable confounding in the relative treatment effect estimates. The effect of Altuviiio relative to other active therapies on patients’ HRQoL is also unknown.
Harms
In both pivotal trials, safety analyses were conducted for all patients who received at least 1 dose of Altuviiio. In both studies, AEs and SAEs were reported by the patient (or, when appropriate, by a caregiver, surrogate, or the patient's legally authorized representative). Overall, Altuviiio was well tolerated and reported TEAEs were generally consistent with what is anticipated in an adult and adolescent population with severe hemophilia A. There were no reports of serious allergic reaction, anaphylaxis, or vascular thrombotic events. In addition, no clinically meaningful patterns or trends were identified in the laboratory or vital sign parameters.
In the XTEND-1 trial, the most frequently reported TEAEs in greater than 3% of patients were headache (20.1%); arthralgia (16.4%); fall (6.3%); back pain (5.7%); COVID-19 and fatigue (74.4% each); contusion, hemophilic arthropathy, and nasopharyngitis (3.8% each); and joint injury, pain in extremity, and toothache (3.1% each). A total of 18 TESAEs were experienced by 15 (9.4%) patients. Hemophilic arthropathy was reported in 2 (1.3%) patients. The majority of TESAEs were assessed by the investigator as mild to moderate in severity. Two TEAEs in 2 (1.3%) patients resulted in permanent treatment discontinuation. Death was reported in 1 patient, in arm B. This patient had a medical history of hepatitis C virus and died of pancreatic carcinoma metastatic, which was reported as a TESAE. The TESAE was assessed by the investigator as not related to Altuviiio treatment. There were no reports of inhibitor development to FVIII, or embolic or thrombotic events during the study.
In the XTEND-Kids trial, the most frequently reported TEAEs (in < 5% of patients overall) were SARS-CoV-2 test positive and upper respiratory tract infection (14.9% each); pyrexia (12.2%); asymptomatic COVID-19 (9.5%); gastroenteritis viral, head injury, and nasopharyngitis (8.1% each); arthralgia, pain in extremity, and vomiting (6.8% each); and contusion, diarrhea, viral infection, and viral upper respiratory tract infection (5.4% each). The majority of TEAEs were assessed by the investigator as mild in severity. A total of 10 TESAEs were experienced in 9 (12.2%) patients. All TESAEs were reported in 1 (1.4%) patient each, except vascular device infection which was reported in 2 (2.7%) patients. The majority of TESAEs were assessed by the investigator as mild to moderate in severity. The 5 TESAEs assessed by the investigator as severe were TESAEs of circumcision and bacteremia. No patients discontinued Altuviiio treatment due to a TEAE during the study and no deaths were reported.
The 52-week assessment period of the XTEND-1 and XTEND-Kids studies were determined to be sufficient duration according to input provided by the clinical experts consulted in this review. Both trials included follow-up safety assessments for a few weeks after the last dose, but a longer follow-up period is needed to evaluate delayed adverse effects, which is critical for assessing the long-term safety of Altuviiio. Overall, the safety profile of Altuviiio compared to FVIII prophylaxis remains unknown based on the absence of direct, comparative evidence. An assessment of harms data was not conducted in the ITCs submitted by the sponsor.
Conclusion
Two pivotal phase III, nonrandomized, open-label clinical trials, the XTEND-1 (N = 159) and XTEND-Kids (N = 74) studies, were included in the sponsor’s systemic review to provide evidence on the efficacy and safety of Altuviiio in adult and pediatric patients with severe hemophilia A. Based on the inputs from patients, clinicians, and clinical experts, the most important treatment goals for patients with hemophilia A are to prevent bleeding, including spontaneous and traumatic bleeding events, reduce joint pain, improve HRQoL, and to achieve an unrestricted lifestyle comparable to the general population. In the XTEND-1 and XTEND-Kids studies, patients who received once-weekly Altuviiio administered as weekly prophylaxis for 52 weeks experienced an annual rate of treated bleeds that was considered clinically meaningful, with a mean ABR of 0.71 (95% CI, 0.52 to 0.97) and 0.89 (95% CI, 0.56 to 1.42) in the 2 trials, respectively. Further, the proportion of patients who did not report experiencing a treated bleed was 64.7% and 63.5% in the XTEND-1 and XTEND-Kids studies, respectively. Additionally, the XTEND-1 study included an intrapatient comparison that suggests that Altuviiio may result in an improved ABR compared to the patients’ historical prophylactic therapy, although the evidence is still uncertain. Patients enrolled in the pivotal trials also experienced an annual rate of joint bleeds that was considered clinically meaningful, and although the change from baseline to week 52 in HJHS indicated an improvement in joint health, the magnitude of this effect is unclear due to the small within-group differences and the lack of validated MIDs for this outcome. In adults (the XTEND-1 study) the change from baseline in HRQoL based on the Haem-A-QoL Physical Health score indicated an improvement in QoL; however, the change was not considered clinically meaningful based on the within-group MID identified in the literature. Results for Haem-A-QoL total score in the XTEND-Kids study suggest an improvement for children aged 8 years to 12 years, but these data are highly uncertain due to a small sample size. Although we observed improvement in PROMIS Pain Intensity scores at week 52 in the XTEND-1 study, the magnitude of this effect is unclear given the small within-group differences and the lack of MIDs for this outcome. Perioperative management of bleeds was also assessed and although the evidence is limited by a small sample size, all surgeries were reported as having a good or excellent hemostatic response to perioperative use of Altuviiio. Regarding safety, Altuviiio was well tolerated and the reported TESAEs were generally consistent with what is anticipated in an adult, adolescent, and pediatric population with severe hemophilia A. The majority of TESAEs across both pivotal studies were assessed to be mild in severity. There was no development of inhibitors to FVIII detected in either study, and there were no reports of serious allergic reactions, anaphylaxis, or embolic or thrombotic events.
It should be noted that the certainty of evidence was assessed as very low for all outcomes identified as clinically relevant for this review, with the exception of ABR in adults, which was considered to be of low certainty due to the intrapatient comparison and proportion of patients reporting no bleeds. The review team acknowledges that there are challenges associated with conducting a traditional RCT for rare diseases such as hemophilia A, and that the study design used in the XTEND trials is consistent with other trials for this condition. Nonetheless, the study design limits the interpretation of the safety and efficacy of Altuviiio, particularly as it relates to any comparator. The sponsor submitted indirect evidence to address this gap in the evidence, which suggested that prophylactic treatment with Altuviiio was associated with improved bleeding outcomes compared with the other treatments currently available in practice, such as EHL agents, SHL agents, or emicizumab. However, the magnitude of the clinical benefit of Altuviiio versus these comparator therapies is uncertain and likely overestimated by the study findings due to the limitations of the available indirect evidence, such as a sizable reduction in the ESS from the original sample size after the propensity score weighting analyses, and inadequate or lack of adjustment for potential prognostic factors, which may introduce unmeasurable confounding in the relative treatment effect estimates. Overall, Altuviiio used prophylactically resulted in clinically meaningful rates of bleeding outcomes over 52 weeks of use. Additionally, due to the prolonged half-life relative to other standard and EHL FVIII molecules that is noted in the product monograph, the evidence suggests that Altuviiio represents an additional option that supports a patient-centred approach to the management of hemophilia A.
In the perioperative setting, hemostatic response by Altuviiio was rated as excellent by the investigators and surgeons for all major surgeries. The clinical experts consulted for this review were in agreement that alternative designs like single-arm trials and intrapatient comparison provide practical evaluation of new therapies. Further, concerns with generalizability of the results included no control for multiplicity in the XTEND-Kids study and potential biases introduced by assumptions in the statistical models used to make the comparisons. In addition, the lack of control for physical activity and the potential temporal trends due to the pandemic limits the interpretation of the efficacy outcomes.
References
1.O'Hara J, Walsh S, Camp C, et al. The impact of severe haemophilia and the presence of target joints on health-related quality-of-life. Health Qual Life Outcomes. 2018;16(1):84. PubMed
2.Foundation NBD. Hemophilia A. n.d.: https://www.bleeding.org/bleeding-disorders-a-z/types/hemophilia-a. Accessed 2024 Oct 10.
3.Castaman G, Matino D. Hemophilia A and B: molecular and clinical similarities and differences. Haematologica. 2019;104(9):1702-1709. PubMed
4.Berntorp E, Fischer K, Hart DP, et al. Haemophilia. Nat Rev Dis Primers. 2021;7(1):45. PubMed
5.Society CH. Hemophilia A and B. 2018: https://www.hemophilia.ca/hemophilia-a-and-b/#:%7E:text=Hemophilia%20A%20affects%20fewer%20than,people%2C%20or%20about%20600%20Canadians. Accessed 2024 Oct 10.
6.Canadian Blood Disorders Registry. Factor FVIII Deficiency. Available from: https://www.ahcdc.ca/storage/files/20240911110116-hemophilia-a-tables.pdf.
7.Hoots WK, Malex L. Clinical manifestations and diagnosis of hemophilia. In: Shapiro A, ed. UpToDate. Waltham (MA)2024. Accessed 2024 Oct 10.
8.Srivastava A, Santagostino E, Dougall A, et al. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia. 2020.
9.Ahmed AM. Bleeding Disorders: Insights into Aetiology, Pathogenesis, Diagnosis and Management. Int J Hematol Disord. 2014;1(1):22-26.
10.Gualtierotti R, Solimeno LP, Peyvandi F. Hemophilic arthropathy: Current knowledge and future perspectives. J Thromb Haemost. 2021;19(9):2112-2121. PubMed
11.Bowyer AE, Gosselin RC. Factor VIII and Factor IX Activity Measurements for Hemophilia Diagnosis and Related Treatments. Semin Thromb Hemost. 2023;49(6):609-620. PubMed
12.[US] CfDCaP. Diagnosing Hemophilia. 2024: https://www.cdc.gov/hemophilia/testing/index.html#:~:text=Those%20with%20severe%20hemophilia%20can,diagnosed%20until%20later%20in%20life. Accessed Oct 16 2024.
13.Srivastava A, Santagostino E, Dougall A, et al. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia. 2020;26(S6):1-158.
14.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
15.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
16.Clinical Study Report: EFC16293 (XTEND-1) A Phase 3 Open-Label, Multicenter Study of the Safety, Efficacy, and Pharmacokinetics of Intravenous Recombinant Coagulation Factor VIII Fc-von Willebrand Factor-XTEN Fusion Protein (rFVIIIFc-VWF-XTEN; BIVV001) in Previously Treated Patients ≥12 Years of Age With Severe Hemophilia A. Bioverativ Therapeutics Inc. (a Sanofi Company). 225 Second Avenue Waltham, MA 02451 United States. June 2022.
17.Clinical Study Report: EFC16295 (XTEND-Kids). A Phase 3 open-label, multicenter study of the safety, efficacy, and pharmacokinetics of intravenous recombinant coagulation Factor VIII Fc-von Willebrand Factor-XTEN fusion protein (rFVIIIFc-VWF-XTEN; BIVV001) in previously treated pediatric patients <12 years of age with severe hemophilia A. May 2023. Bioverativ Therapeutics Inc. (a Sanofi Company). 225 Second Avenue Waltham, MA 02451 United States.
18.Sanofi. NCT04644575: Long-term Safety and Efficacy of Efanesoctocog Alfa (BIVV001) in Previously Treated Patients With Hemophilia A (XTEND-ed). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2024: https://clinicaltrials.gov/study/NCT04644575?intr=Efanesoctocog%20Alfa%20BIVV001&rank=1. Accessed 2024 Oct 30.
19.Drygalski A, Konigs C, Konkle BA, et al. Interim Analysis of Joint Outcomes in Adult and Adolescent Patients With Severe Hemophilia A Receiving Efanesoctocog Alfa During the Phase 3 XTEND-ed Long-Term Extension Study ISTH 2024 Congress, June 22, 2024 [Oral Presentation]. Bankok, TH2024. Accessed 2024 Oct 30.
20.Chan A, Susen S, Khoo L, et al. Perioperative Management with Efanesoctocog Alfa in Adults, Adolescents, and Children with Severe Hemophilia A in the Phase 3 XTEND Clinical Program. ISTH 2024 Congress, June 23, 2024 [Oral Presentation]. Bangkok, TH2024. Accessed 2024 Oct 30.
21.Indirect treatment comparison (ITC) between efanesoctocog alfa and selected comparators in patients with hemophilia A without inhibitors - technical report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: efanesoctocog alfa, lyophilized powder, 250, 500, 1000, 2000, 3000, or 4000 IU/vial, for solution for Intravenous infusion. City (PROV): Sanofi-Aventis Canada Inc.; 2023 Jan 24. 2023.
22.Pipe SW, Montgomery RR, Pratt KP, Lenting PJ, Lillicrap D. Life in the shadow of a dominant partner: the FVIII-VWF association and its clinical implications for hemophilia A. Blood. 2016;128(16):2007-2016. PubMed
23.Canadian Blood Disorders Registry. Factor FVIII Deficiency. Available from: https://ahcdc.ca/storage/pdf/21/FVIII-2021.pdf.
24.Franchini M, Favaloro EJ, Lippi G. Mild hemophilia A. J Thromb Haemost. 2010;8(3):421-432. PubMed
25.Måseide RJ, Berntorp E, Astermark J, et al. Health-related quality of life and physical activity in Nordic patients with moderate haemophilia A and B (the MoHem study). Haemophilia. 2024;30(1):98-105. PubMed
26.Azeredo-da-Silva AF, Zanotto BS, Kuwabara YS, Mata VE. Quality of life in children and adolescents with hemophilia A: A systematic review and meta-analysis. Res Pract Thromb Haemost. 2023;7(1):100008. PubMed
27.Rezende SM, Neumann I, Angchaisuksiri P, et al. International Society on Thrombosis and Haemostasis clinical practice guideline for treatment of congenital hemophilia A and B based on the Grading of Recommendations Assessment, Development, and Evaluation methodology. J Thromb Haemost.
28.Malec L, Matino D. Targeting higher factor VIII levels for prophylaxis in haemophilia A: a narrative review. Haemophilia. 2023;29(6):1419-1429. PubMed
29.den Uijl IE, Fischer K, Van Der Bom JG, Grobbee DE, Rosendaal FR, Plug I. Analysis of low frequency bleeding data: the association of joint bleeds according to baseline FVIII activity levels. Haemophilia. 2011;17(1):41-44. PubMed
30.Soucie JM, Monahan PE, Kulkarni R, Konkle BA, Mazepa MA. The frequency of joint hemorrhages and procedures in nonsevere hemophilia A vs B. Blood Adv. 2018;2(16):2136-2144. PubMed
31.Chowdary P, Fischer K, Collins PW, et al. Modeling to Predict Factor VIII Levels Associated with Zero Bleeds in Patients with Severe Hemophilia A Initiated on Tertiary Prophylaxis. Thromb Haemost. 2020;120(5):728-736. PubMed
32.Tiede A, Abdul Karim F, Jiménez-Yuste V, et al. Factor VIII activity and bleeding risk during prophylaxis for severe hemophilia A: a population pharmacokinetic model. Haematologica. 2021;106(7):1902-1909. PubMed
33.Valentino LA, Pipe SW, Collins PW, et al. Association of peak factor VIII levels and area under the curve with bleeding in patients with haemophilia A on every third day pharmacokinetic-guided prophylaxis. Haemophilia. 2016;22(4):514-520. PubMed
34.Data on File. Sanofi-aventis Canada Inc. February 2024.
35.Gringeri A, Ewenstein B, Reininger A. The burden of bleeding in haemophilia: is one bleed too many? Haemophilia. 2014;20(4):459-463. PubMed
36.ALHEMO (concizumab injection). Product Monograph, 2023. Accessed at: https://pdf.hres.ca/dpd_pm/00071786.PDF.
37.Duncan N, Shapiro A, Ye X, Epstein J, Luo MP. Treatment patterns, health-related quality of life and adherence to prophylaxis among haemophilia A patients in the United States. Haemophilia. 2012;18(5):760-765. PubMed
38.Sun J, Zhou X, Hu N. Factor VIII replacement prophylaxis in patients with hemophilia A transitioning to adults: a systematic literature review. Orphanet J Rare Dis. 2021;16(1):287. PubMed
39.Kruzik A, Fetahagic D, Hartlieb B, et al. Prevalence of Anti-Adeno-Associated Virus Immune Responses in International Cohorts of Healthy Donors. Mol Ther Methods Clin Dev. 2019;14:126-133. PubMed
40.Greenberg B, Butler J, Felker GM, et al. Prevalence of AAV1 neutralizing antibodies and consequences for a clinical trial of gene transfer for advanced heart failure. Gene Ther. 2016;23(3):313-319. PubMed
41.Mahlangu JN, Blanchette V, Klamroth R. Redefining prophylaxis in the modern era. Haemophilia. 2021;27(S3):21-27. PubMed
42.Mannucci PM. Hemophilia therapy: the future has begun. Haematologica. 2020;105(3):545-553. PubMed
43.CADTH Clinical Review Report - Emicizumab (HEMLIBRA), 2021. Accessed at: https://www.cadth.ca/sites/default/files/cdr/clinical/st0651-hemlibra-clinical-review-report.pdf.
44.ALTUVIIIO (efanesoctocog alfa): Lyophilized powder, 250, 500, 1000, 2000, 3000, or 4000 IU/vial, for solution for Intravenous infusion [Product Monograph]. Toronto (ON): Sanofi-Aventis Canada Inc.; TBD.
45.Bioverativ Therapeutics Inc. ALTUVIIIO (Antihemophilic Factor [Recombinant], Fc-Von Willebrand Factor-XTEN Fusion Protein) US Prescribing Information. February 2023.
46.ALTUVOCT (efanesoctocog alfa): Powder and solvent for solution for injection [Product Information]. Stockholm, SE: Swedish Orphan Biovitrum AB: https://www.ema.europa.eu/en/documents/product-information/altuvoct-epar-product-information_en.pdf.
47.Bayer Inc. KOVALTRY (antihemophilic factor [recombinant]), Product Monograph. March 2023. https://pdf.hres.ca/dpd_pm/00069834.PDF.
48.Sanofi-Aventis Canada Inc. ELOCTATE (antihemophilic factor [recombinant BDD], Fc fusion protein), Product Monograph. Nov 2021. https://pdf.hres.ca/dpd_pm/00063552.PDF.
49.Hoffmann-La Roche Limited. HEMLIBRA (emicizumab injection), Product Monograph. Dec 23, 2022. https://pdf.hres.ca/dpd_pm/00068868.PDF.
50.Pipe S S-KA, Konkle BA, Kitchen S, Negrier C, Liu M, Santagostino E, Willemze A, Abad-Franch L, Knobe K, Seth Chhabra E. . A global comparative field study to evaluate the factor VIII activity of efanesoctocog alfa by one-stage clotting and chromogenic substrate assays at clinical haemostasis laboratories. Haemophilia. 2024;30(1). PubMed
51.CDA-AMC Sponsory Summary of Clinical Evidence Template: Efanesoctocog alfa for the treatment of adults and children with hemophilia A (congenital factor VIII deficiency) for: Routine prophylaxis to reduce the frequency of bleeding episodes On-demand treatment and control of bleeding episodes Perioperative management of bleeding [internal sponsor report]. Sanofi Canada; 18 Sept 2024. Accessed 24 Oct 2024.
52.von Drygalski A, Chowdary P, Kulkarni R, et al. Efanesoctocog Alfa Prophylaxis for Patients with Severe Hemophilia A. N Engl J Med. 2023;388(4):310-318. PubMed
53.Malec L, Peyvandi F, Chan AKC, et al. Efanesoctocog Alfa Prophylaxis for Children with Severe Hemophilia A. N Engl J Med. 2024;391(3):235-246. PubMed
54.Bioverativ Therapeutics Inc. (a Sanofi Company). XTEND-1 Clinical Study Report, June 2022.
55.Wyrwich KW, Krishnan S, Poon JL, et al. Interpreting important health-related quality of life change using the Haem-A-QoL. Haemophilia. 2015;21(5):578-584. PubMed
56.von Mackensen S, Catalani O, Asikanius E, Paz-Priel I, Lehle M, Trask P. Determining meaningful health-related quality-of-life improvement in persons with haemophilia A using the Haemophilia Quality of Life Questionnaire for Adults (Haem-A-QoL). Haemophilia. 2020;26(6):1019-1030. PubMed
57.von Mackensen S, Eldar-Lissai A, Auguste P, et al. Measurement properties of the Haem-A-QoL in haemophilia clinical trials. Haemophilia. 2017;23(3):383-391. PubMed
58.Health NIo. Instrument: PROMIS Pain Intensity - Short Form 3a v1.0. National Institute on Drug Abuse (NIDA) Clinical Trials Network (CTN)-recommended Common Data Elements (CDEs) of Substance Use Disordersn.d.: https://cde.nida.nih.gov/instrument/0a481bfb-a5e6-3c84-e050-bb89ad43314d. Accessed 2024 Oct 30.
59.Revicki D, Cook K. PROMIS Pain-Related Measures: An Overview. Practical Pain Management. 2015;15(3).
60.HealthMeasures. PAIN INTENSITY: A brief guide to the PROMIS® Pain Intensity instruments. 2020: https://www.healthmeasures.net/images/promis/manuals/PROMIS_Pain_Intensity_Scoring_Manual.pdf. Accessed 2024 Oct 30.
61.Scale PPNR. Pain Intensity - 1a. healthmeasures.net2017: https://www.healthmeasures.net/administrator/components/com_instruments/uploads/PROMIS%20Pediatric%20Numeric%20Rating%20Scale%20v1.0%20-%20Pain%20Intensity%201a%2010-5-2017.pdf. Accessed Dec 23, 2024.
62.Stephan A, Stadelmann VA, Leunig M, Impellizzeri FM. Measurement properties of PROMIS short forms for pain and function in total hip arthroplasty patients. J Patient Rep Outcomes. 2021;5(1):41. PubMed
63.St-Louis J, Abad A, Funk S, et al. The Hemophilia Joint Health Score version 2.1 Validation in Adult Patients Study: A multicenter international study. Res Pract Thromb Haemost. 2022;6(2):e12690. PubMed
64.Wang M, Batt K, Kessler C, et al. Internal consistency and item-total correlation of patient-reported outcome instruments and hemophilia joint health score v2.1 in US adult people with hemophilia: results from the Pain, Functional Impairment, and Quality of life (P-FiQ) study. Patient Prefer Adherence. 2017;11:1831-1839. PubMed
65.Batt K, Recht M, Cooper DL, Iyer NN, Kempton CL. Construct validity of patient-reported outcome instruments in US adults with hemophilia: results from the Pain, Functional Impairment, and Quality of life (P-FiQ) study. Patient Prefer Adherence. 2017;11:1369-1380. PubMed
66.Nunnally JC. Psychometric Theory (2nd. ed.). New York (NY): McGraw-Hill; 1978.
67.Feldman BM, Funk SM, Bergstrom BM, et al. Validation of a new pediatric joint scoring system from the International Hemophilia Prophylaxis Study Group: validity of the hemophilia joint health score. Arthritis Care Res (Hoboken). 2011;63(2):223-230. PubMed
68.Fischer K, Steen Carlsson K, Petrini P, et al. Intermediate-dose versus high-dose prophylaxis for severe hemophilia: comparing outcome and costs since the 1970s. Blood. 2013;122(7):1129-1136. PubMed
69.Bladen M, Main E, Hubert N, Koutoumanou E, Liesner R, Khair K. Factors affecting the Haemophilia Joint Health Score in children with severe haemophilia. Haemophilia. 2013;19(4):626-631. PubMed
70.Groen W, van der Net J, Lacatusu AM, Serban M, Helders PJ, Fischer K. Functional limitations in Romanian children with haemophilia: further testing of psychometric properties of the Paediatric Haemophilia Activities List. Haemophilia. 2013;19(3):e116-125. PubMed
71.Fischer K, Poonnoose P, Dunn AL, et al. Choosing outcome assessment tools in haemophilia care and research: a multidisciplinary perspective. Haemophilia. 2017;23(1):11-24. PubMed
72.Sluiter D, Foppen W, de Kleijn P, Fischer K. Haemophilia Joint Health Score in healthy adults playing sports. Haemophilia. 2014;20(2):282-286. PubMed
73.Kavakli K, Yang R, Rusen L, Beckmann H, Tseneklidou-Stoeter D, Maas Enriquez M. Prophylaxis vs. on-demand treatment with BAY 81-8973, a full-length plasma protein-free recombinant factor VIII product: results from a randomized trial (LEOPOLD II). J Thromb Haemost. 2015;13(3):360-369. PubMed
74.Konkle BA, Stasyshyn O, Chowdary P, et al. Pegylated, full-length, recombinant factor VIII for prophylactic and on-demand treatment of severe hemophilia A. Blood. 2015;126(9):1078-1085. PubMed
75.Mahlangu J, Kuliczkowski K, Karim FA, et al. Efficacy and safety of rVIII-SingleChain: results of a phase 1/3 multicenter clinical trial in severe hemophilia A. Blood. 2016;128(5):630-637. PubMed
76.Mahlangu J, Powell JS, Ragni MV, et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood. 2014;123(3):317-325. PubMed
77.Guideline on the clinical investigation of recombinant and human plasma-derived factor VIII products. European Medicines Agency, July 2018. Accessed at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-investigation-recombinant-and-human-plasma-derived-factor-viii-products-revision-2_en.pdf.
78.Keipert C M-OM, Gauly F, Arras-Reiter C, Hilger A. Annual Bleeding Rates: Pitfalls of Clinical Trial Outcomes in Hemophilia Patients. Clin Transl Sci 2020;13(6):1127-1136. PubMed
79.National Institute for Health and Care Excellence. Developing NICE guidelines: the manual, Appendix H: Appraisal checklists, evidence tables, GRADE and economic profiles, 2023. Accessed at: https://www.nice.org.uk/process/pmg20/resources/appendix-h-appraisal-checklists-evidence-tables-grade-and-economic-profiles-pdf-8779777885.
80.Phillippo, D., Ades, T., Dias, S., Palmer, S., Abrams, K. R., & Welton, N. (2016). NICE DSU Technical Support Document 18: Methods for population-adjusted indirect comparisons in submissions to NICE. (Technical Support Documents). NICE Decision Support Unit. Accessed at: https://research-information.bris.ac.uk/ws/portalfiles/portal/94868463/Population_adjustment_TSD_FINAL.pdf.
81.Faria, R., Hernandez Alva, M., Manca, A., Wailoo, AJ. (2015). NICE DSU Technical Support Document 17: The Use of Observational Data to Inform Estimates of Treatment Effectiveness in Technology Appraisal: Methods for Comparative Individual Patient Data. Accessed at: https://www.sheffield.ac.uk/media/34204/download?attachment.
82.Rosenbaum PR, Rubin DB. The central role of the propensity score in observational studies for causal effects. Biometrika. 1983;70(1):41-55.
83.Guevara JP, Berlin JA, Wolf FM. Meta-analytic methods for pooling rates when follow-up duration varies: a case study. BMC Med Res Methodol. 2004;4(1):17. PubMed
84.Bioverativ Therapeutics Inc. (a Sanofi Company). XTEND-1 Clinical Trial Protocol. August 2021.
85.Mahlangu J, Oldenburg J, Paz-Priel I, et al. Emicizumab Prophylaxis in Patients Who Have Hemophilia A without Inhibitors. N Engl J Med. 2018;379(9):811-822. PubMed
86.Saxena K, Lalezari S, Oldenburg J, et al. Efficacy and safety of BAY 81-8973, a full-length recombinant factor VIII: results from the LEOPOLD I trial. Haemophilia. 2016;22(5):706-712. PubMed
87.Lissitchkov T, Willemze A, Jan C, Zillberstein M, Katragadda S. A. Pharmacokinetics of recombinant factor VIII in adults with severe hemophilia A: fixed-sequence single-dose study of octocog alfa, rurioctocog alfa pegol, and efanesoctocog alfa. Res Pract Thromb Haemost. May 2023; 7(6). PubMed
Appendix 1: Summary of Other Outcomes in XTEND-1
Please note that this appendix has not been copy-edited.
Table 29: Summary of Other Outcomes in XTEND-1
Outcomes | Arm A Prophylaxis (N = 133) | Arm B | |
|---|---|---|---|
On Demand (N = 26) | Prophylaxis (N = 26) | ||
Efficacy of Altuviiio as Prophylaxis Treatment | |||
Spontaneous Bleeds | |||
Mean AsBR, model baseda (95% CI) | 0.27 (0.18; 0.41) | 15.83 (12.27; 20.43) | 0.44 (0.16; 1.20) |
Median AsBR (Q1:Q3) | 0.00 (0.00; 0.00) | 16.69 (8.64; 23.76) | 0.00 (0.00; 0.00) |
Number (%) of patients with AsBR = 0 | 107 (80.5) | 1 (3.8) | 22 (84.6) |
All Bleeds (treated and untreated) | |||
Mean ABR, model baseda (95% CI) | 1.11 (0.83; 1.48) | 22.21 (19.41; 25.42) | 0.88 (0.42; 1.84) |
Median ABR (Q1:Q3) | 0.00 (0.00; 1.15) | 21.13 (16.80; 27.13) | 0.00 (0.00; 1.93) |
Number (%) of patients with ABR = 0 | 75 (56.4) | 0 (0) | 19 (73.1) |
Efficacy of Altuviiio in Treatment of Bleeds | |||
Treatment of Bleeds | |||
Number of treated bleeds | 86 | 268 | 8 |
Mean (SD) number of injections to treat a bleed | 1.1 (0.3) | 1.0 (0.2) | 1.0 (0.0) |
Number (%) of bleeds treated with a single injection | 81 (94.2) | 261 (97.4) | 8 (100) |
Mean (SD) total dose for resolution of a bleed (IU/kg) | 49.95 (16.63) | 51.74 (6.56) | 45.74 (9.52) |
Patient Assessment of Response to Treatment of Bleeds | |||
Number of injections with an evaluation | 73 | 255 | 6 |
Number (%) of injections with response rated as excellent or good | 60 (82.2) | 251 (98.4) | 6 (100) |
Physician’s Global Assessment of Patient Response to Treatment | |||
Number of total responses across all visits | 622 | 77 | 44 |
Number (%) of injections with response rated as excellent or good | 595 (95.7) | 74 (96.1) | 41 (93.2) |
Altuviiio Consumption | |||
Annualized Altuviiio Consumption per Patient (IU/kg) | |||
Mean (SD) | 3,131.75 (4,113.19) | 1,135.32 (404.94) | 2,751.54 (88.26) |
Median (Q1; Q3) | 2,756.99 (2,705.66; 2,805.81) | 1,212.27 (769.93; 1,382.15) | 2,737.53 (2,718.69; 2,818.43) |
ABR = annualized bleeding rate; aPTT = activated partial thromboplastin time; AsBR: annualized spontaneous bleeding rate; AjBR: annualized joint bleeding rate; CI = confidence interval; FAS = full analysis set; FVIII = factor VIII; SD = standard deviation
Note: All data are reported for FAS unless otherwise stated. Mean change from baseline is reported for mean change from baseline at week 52.
aEstimated using a negative binomial model with the total number of treated bleeding episodes during the efficacy period as the response variable and log-transformed efficacy period duration (in years) as an offset variable.
Source: XTEND-1 Clinical Study Report,54
Appendix 2: Summary of Other Outcomes in XTEND-Kids
Please note that this appendix has not been copy-edited.
Table 30: Summary of Other Outcomes in XTEND-Kids
Outcomes | Age Cohort | Overall (N = 74) | |
|---|---|---|---|
< 6 years (N = 38) | 6 to < 12 years (N = 36) | ||
Efficacy of Altuviiio as Prophylaxis Treatment | |||
Spontaneous Bleeds | |||
Mean AsBR, model baseda (95% CI) | 0.17 (0.08; 0.38) | 0.14 (0.04; 0.53) | 0.16 (0.08; 0.30) |
Median AsBR (Q1; Q3) | 0.00 (0.00; 0.00) | 0.00 (0.00; 0.00) | 0.00 (0.00; 0.00) |
Number (%) of patients with AsBR = 0 | 32 (84.2) | 33 (91.7) | 65 (87.8) |
All bleeds (treated and untreated) | |||
Mean ABR, model baseda (95% CI) | 2.78 (1.39; 5.58) | 2.85 (1.59; 5.12) | 2.82 (1.79; 4.42) |
Median ABR (Q1; Q3) | 0.00 (0.00; 1.97) | 1.00 (0.00; 3.00) | 0.49 (0.00; 2.05) |
Number (%) of patients with ABR for all bleeds = 0 | 21 (55.3) | 16 (44.4) | 37 (50.0) |
Physician’s Global Assessment of Patients’ Response to Treatment | |||
Number of total responses across week 13 and week 52 EOS/ET visits | 74 | 73 | 147 |
Number (%) of assessments rated as excellent | 71 (95.9) | 71 (97.3) | 142 (96.6) |
Efficacy of Altuviiio in the Treatment of Bleeds | |||
Number of treated bleeds | 17 | 47 | 64 |
Mean (SD) number of injections to treat a bleed | 1.12 (0.33) | 1.30 (0.69) | 1.25 (0.62) |
Number (%) of bleeds treated with a single injection | 15 (88.2) | 37 (78.7) | 52 (81.3) |
Total dose for resolution of a bleed (IU/kg) | |||
Mean (SD) | 52.29 (15.84) | 66.90 (37.01) | 63.02 (33.26) |
Median (Q1; Q3) | 57.14 (50.00; 60.98) | 52.58 (51.89; 55.56) | 52.63 (51.64; 60.98) |
Patient’s Assessment of Response to Treatment of Bleeds | |||
Number of first injections with an evaluation | 16 | 24 | 40 |
Number (%) of injections with response rated as excellent or good | 15 (93.8) | 24 (100.0) | 39 (97.5) |
> 20% | 1 (3.1) | 2 (6.9) | 3 (4.9) |
Annualized Altuviiio Consumption per Patient (IU/kg) | |||
Mean (SD) IU/kg | 3,115.57 (488.64) | 2,884.67 (207.88) | 3,003.24 (394.01) |
Median (Q1; Q3) | 2,986.26 (2,888.27; 3,101.37) | 2,837.09 (2,779.56; 2,957.02) | 2,912.69 (2,822.93; 3,026.40) |
ABR = annualized bleeding rate; aPTT = activated partial thromboplastin time; AjBR = annualized joint bleeding rate; AsBR = annualized spontaneous bleeding rate; CI = confidence interval; EOS = end of study; ET = early termination; FVIII = factor VIII; SD = standard deviation
Note: Mean change from baseline is reported for mean change from baseline at week 52
aEstimated using a negative binomial model with the total number of treated bleeding episodes during the efficacy period as the response variable and log-transformed efficacy period duration (in years) as an offset variable.
Source: XTEND-Kids Clinical Study Report17
Appendix 3: Summary of Baseline Characteristics of Included Studies in Sponsor-Submitted ITCs
Please note that this appendix has not been copy-edited.
Table 31: Matching of Baseline Characteristics Between XTEND-1 Arm A and HAVEN 3 Arm D
Variables | XTEND-1 arm A baseline (N = 119) | HAVEN 3 arm D baseline | XTEND-1 arm A after matching | |||||
|---|---|---|---|---|---|---|---|---|
Estimate | SD | Estimate | SD | Estimate | SD | ESS | % | |
Mean age | 34.91 | 14.23 | 36.4 | 14.4 | 36.4 | 14.4 | 76 | 63.8% |
Mean weight | 81.26 | 16.74 | 79.0 | 15.4 | 79.0 | 15.4 | ||
% pts w/ 0 TJ | 78.2% | N/A | 58.7% | N/A | 58.7% | N/A | ||
% pts w/ 1 TJ | 5.9% | 12.7% | N/A | 12.7% | ||||
% pts w/ 2+ TJ | 16.0% | 28.6% | N/A | 28.6% | ||||
% White | 54.6% | 74.6% | N/A | 74.6% | ||||
% Asian | 21.0% | 19.0% | N/A | 19.0% | ||||
ESS = Effective sample size; N/A = not available; SD = Standard deviation; pts = patients; TJ = Target joint.
Source: Sponsor-Submitted Indirect Evidence.21
Table 32: Matching of Baseline Characteristics Between XTEND-1 Pooled Arms and LEOPOLD I Arms A and B
Variables | XTEND-1 pooled arms baseline (N = 128) | LEOPOLD I A and B baseline | XTEND-1 arm A after matching | |||||
|---|---|---|---|---|---|---|---|---|
Estimate | SD | Estimate | SD | Estimate | SD | ESS | % | |
Mean age | 33.88 | 13.14 | 31.5 | 12.7 | 31.5 | 12.7 | 29 | 22.7% |
Mean weight | 78.61 | 17.09 | 77.0 | 17.1 | 77.0 | 17.1 | ||
% pts w/ 1+ TJ | 31.0 | N/A | 71.0 | N/A | 71.0 | N/A | ||
% White | 59.9 | N/A | 88.7 | N/A | 88.7 | N/A | ||
% Prior prophylaxis | 83.8 | N/A | 80.6 | N/A | 80.6 | N/A | ||
Mean number of prior bleeds | 8.03 | 15.43 | 11.5 | 15.1 | 11.5 | 15.1 | ||
ESS = Effective sample size; N/A = not available; SD = Standard deviation; pts = patients; TJ = Target joint.
Source: Sponsor-Submitted Indirect Evidence.21
Pharmacoeconomic Review
Abbreviations
ABR
annualized bleeding rate
AE
adverse event
AjBR
annualized joint bleeding rate
BIA
budget impact analysis
CBDR
Canadian Bleeding Disorders Registry
CDA-AMC
Canada's Drug Agency
EHL
extended half-life
FVIII
factor VIII
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
MAIC
matching-adjusted indirect comparison
PMPRB
Patented Medicine Prices Review Board
PS
Pettersson score
QALY
quality-adjusted life-year
SHL
standard half-life
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Antihemophilic factor VIII (recombinant, B-domain deleted), Fc-VWF-XTEN fusion protein (Altuviiio) lyophilized powder for reconstitution 250 IU, 500 IU, 1,000 IU, 2,000 IU, 3,000 IU, and 4,000 IU vials |
Indication | Indicated in adults, adolescents, and children with hemophilia A (congenital factor VIII [FVIII] deficiency) for:
|
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | March 26, 2025 |
Reimbursement request | Same as indication |
Sponsor | Sanofi-Aventis Canada Inc. |
Submission history | No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov cohort model |
Target population | Adults with hemophilia A (congenital factor VIII deficiency) without inhibitors for routine prophylaxis to reduce the frequency of bleeding episodes Children were included in a scenario analysis only |
Treatment | Altuviiio lyophilized powder for reconstitution 250 IU, 500 IU, 1,000 IU, 2,000 IU, 3,000 IU, and 4,000 IU vials |
Dose regimen | 50 IU/kg IV administered once weekly |
Submitted price | Altuviiio 250 IU/vial: $827.50 Altuviiio 500 IU/vial: $1,655 Altuviiio 1,000 IU/vial: $3,310 Altuviiio 2,000 IU/vial: $6,620 Altuviiio 3,000 IU/vial: $9,930 Altuviiio 4,000 IU/vial: $13,240 $3.31 per IU for all vials |
Submitted treatment cost | $739,164 per year |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | 65 years (lifetime) |
Key data sources |
|
Submitted results |
|
Key limitations |
|
CDA-AMC reanalysis results |
|
ABR = annualized bleeding rate; AjBR = annualized joint bleeding rate; CDA-AMC = Canada's Drug Agency; EHL = extended half-life; ICER = incremental cost-effectiveness ratio; ITC = indirect treatment comparison; LY = life-year; MAIC = matching-adjusted indirect comparison; PS = Pettersson score; QALY = quality-adjusted life-year; SHL = standard half-life.
Conclusions
Evidence from the XTEND-1 and XTEND-Kids trials showed that 52 weeks of prophylactic treatment with Altuviiio resulted in a clinically relevant rate of bleeding outcomes and the potential to provide adequate perioperative management of bleeding in patients with severe hemophilia A. Based on the clinical team appraisal of both trials, there is a low to very low level of certainty for bleeding outcomes and very low level of certainty for the other efficacy outcomes. Given the nonrandomized open-label design and the lack of control for physical activity, there may be a potential risk of bias in the risk of bleeding events. In the absence of direct comparative evidence to extended half-life (EHL) agents, standard half-life (SHL) agents, and emicizumab, the sponsor’s submitted indirect treatment comparisons (ITCs) included unanchored matching-adjusted indirect comparisons (MAICs) and propensity score analysis to inform the comparative efficacy on annualized bleeding rate (ABR). The indirect evidence suggests that prophylaxis with Altuviiio was associated with improved bleeding outcomes compared to EHL agents, SHL agents, or emicizumab in adult patients with severe hemophilia A. The clinical review by Canada’s Drug Agency (CDA-AMC) concluded that the sponsor’s ITC had several limitations (i.e., sizable reduction in the effective sample size after propensity score weighting analyses, and inadequate or lack of adjustment for potential prognostic factors) which introduces uncertainty to the true magnitude of the comparative clinical benefits of Altuviiio but likely resulted in an overestimation.
CDA-AMC undertook reanalysis adjusting the on-demand dosage of SHL agents and including the costs of breakthrough bleeds associated with Altuviiio. Given limitations with the sponsor’s probabilistic analysis, CDA-AMC reports only the deterministic results. In adult patients with severe hemophilia A without inhibitors who require routine prophylaxis, the incremental cost-effectiveness ratio (ICER) of Altuviiio compared to SHL agents was $4,432,402 per quality-adjusted life-year (QALY) gained (incremental costs = $5,502,419; incremental QALYs = 1.25). While the CDA-AMC reanalysis results were largely consistent with the sponsor’s, the lower ICER value estimated by CDA-AMC compared to the sponsor’s submission was driven by the adjustment of on-demand dosage of SHL agents which increased the expected costs in the strategy for SHL agents, consequently reducing the incremental cost difference between Altuviiio and SHL therapies. Altuviiio dominated emicizumab and EHL agents (associated with less total costs and more QALYs). At the publicly available prices, a price reduction of at least 22% (from $3.31 to $2.58 per IU) is required for Altuviiio to be considered cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per QALY gained compared to SHL agents. A scenario analysis was conducted to align with the patient characteristics of the XTEND-Kids trial and applying the ABR reported in the XTEND-Kids trial. The scenario analysis resulted in similar results compared to the base case (i.e., adult population), with an ICER of $4,150,946 per QALY gained when comparing Altuviiio to SHL agents. Emicizumab and EHL therapies remained dominated by Altuviiio.
CDA-AMC notes that these results are highly uncertain given the available clinical evidence. The cost-effectiveness results are driven by the clinical outcomes that predict fewer joint bleeds associated with Altuviiio compared to relevant comparators. Estimates of comparative clinical benefits were derived from the sponsor’s ITC which, as noted previously, are uncertain and likely to be overestimated. The validity of the predicted QALY gains is further dependent on the validity of the surrogate relationship between ABR and joint health (i.e., Pettersson score [PS]) which is unsubstantiated. Within the submitted model, treatment acquisition costs may not reflect clinical practice given that they were calculated based on the exact dose required (per mg or per IU) rather than having the dose rounded to the nearest whole vial to minimize wastage. The price of the comparators is also a key driver in the cost-effectiveness of Altuviiio. As the analyses are based on publicly available list prices for comparators, the cost-effectiveness of Altuviiio is highly dependent on the actual prices. In light of all of these limitations, the cost-effectiveness of Altuviiio and the required price reduction estimates are highly uncertain. Several scenario analyses were conducted to highlight the sensitivity of the model to these inputs. When assuming a smaller clinical benefit in ABRs, the ICER of Altuviiio increased to $5,922,936 per QALY compared to SHL agents. When assuming lower prices for emicizumab (22% and 90%), emicizumab was no longer dominated by Altuviiio and the ICER for Altuviiio ranged from $10,668,293 to $43,301,657 per QALY gained compared to emicizumab. In light of these findings, further price reductions may be warranted to address these uncertainties.
Lastly, it is important to highlight that the sponsor’s submitted model is limited in scope as it does not address the full Health Canada indication. The cost-effectiveness of Altuviiio for routine prophylaxis in patients with mild or moderate hemophilia, or for on-demand treatment and perioperative management is unknown.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from the Canadian Hemophilia Society which gathered input from an online survey between April 1, 2024, to June 1, 2024, and received a total of 104 responses from patients living in Canada with hemophilia A. Of the 104 responses, 57 patients were diagnosed with severe hemophilia A, 14 with moderate, and 33 with mild hemophilia A. Of the total responses, 41% of patients were receiving emicizumab prophylaxis, 11% were receiving factor VIII (FVIII) prophylaxis, and 6% were receiving FVIII on-demand therapy. Most respondents (85%) expressed satisfaction with their current treatment, citing its effectiveness in preventing and stopping bleeding, though a minority faced geographical challenges accessing treatment centres. Emicizumab was noted to simplify treatment with at-home self-administration though injection pain was noted as a drawback. A small percentage (6%) indicated it was less effective for stopping active bleeding, and many emphasized the need for therapies offering better bleed protection, less frequent dosing, and improved quality of life. Only 1 patient was included who had experience using Altuviiio. This patient reported significant benefits, including sustained high FVIII levels, reduced bleeding risk, greater flexibility for travel, and improved independence and quality of life, with no reported side effects or disadvantages.
Clinicians from 3 clinician groups, the Association of Hemophilia Clinic Directors of Canada (5 clinicians), Canadian Association of Nurses in Hemophilia Care (6 clinicians), and Canadian Physiotherapists in Hemophilia Care (5 clinicians), provided input for the review of Altuviiio. Clinician groups stated that patients with hemophilia A currently receive regular prophylactic therapy with clotting factor concentrates or nonfactor subcutaneous treatments. Gene therapy is another treatment option for hemophilia A; however, it is not yet available in Canada. Clinicians noted that the main treatment goal is to reduce bleeds to zero and slow joint damage to improve patients' quality of life. Altuviiio could be a first-line or alternative treatment for patients aged 2 years and older, particularly those requiring higher factor levels with less frequent dosing or those with joint issues. It could also benefit patients undergoing surgery, with mild hemophilia needing on-demand treatment, or those with recurrent breakthrough bleeds despite optimized prophylaxis. Patients resistant to emicizumab may also benefit. The Association of Hemophilia Clinic Directors of Canada noted that those with zero bleeds on current prophylaxis might have limited benefit from switching therapies. Treatment response should be assessed every 6 months to 2 years, monitoring bleed rates, pharmacokinetics, joint health, and inhibitor development. Altuviiio should be discontinued if uncontrolled bleeding, inhibitor development, or adverse reactions occur, or if the patient switches to other therapies.
Drug plans and Canadian Blood Services provided input for the review of Altuviiio. Drug plans inquired if treatment with Altuviiio should be limited to patients who have previously undergone other therapies, or if patients who have not previously received treatment should be considered. Regarding treatment dosage and frequency, drug plans inquired about the possibility of tailoring therapy, wherein patients would receive personalized medical treatment based on a variety of factors including but not limited to bleeding pattern. Additionally, feedback from Canadian Blood Services noted that in practice, doses are rounded to the nearest vial. As the therapy is reported to provide a mean FVIII activity of more than 40 IU/dL for most days of the week and 15 IU/dL on day 7, drug plans questioned if alternative dose or frequency would be required in clinical practice. Additionally, plans inquired if there is a minimum age for treatment eligibility, and if there is a washout period should treatment fail and a patient switch to comparator therapies.
Several of these concerns were addressed in the sponsor’s model:
breakthrough bleeds were included in the prophylactic treatment group for both the pharmacoeconomic and budget impact analysis (BIA).
on-demand treatment was included in the budget impact submission.
In addition, CDA-AMC addressed some of these concerns as follows:
alternative dosage for FVIII therapies was explored in the BIA.
CDA-AMC was unable to address the following concerns raised from input:
the sponsor only included prophylactic treatment in their pharmacoeconomic analysis.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
Altuviiio is indicated in adults, adolescents, and children for routine prophylaxis to prevent or reduce frequency of bleeding episodes, for treatment and control of bleeding episodes, and for perioperative management of bleeding (surgical prophylaxis).1 CDA-AMC accepted a deviation request from the sponsor to focus the economic model on the prophylactic use of Altuviiio.2 The sponsor submitted a cost-utility analysis comparing costs and outcomes of Altuviiio for routine prophylaxis in patients with hemophilia A (congenital FVIII) compared with EHL agents, SHL agents, and emicizumab.3 The sponsor assumed that all currently reimbursed drugs in the EHL and SHL classes are expected to demonstrate equivalent efficacy and costs within their own classes. The modelled population reflected those recruited with the XTEND-1 trial population and consisted of adult patients with hemophilia A without inhibitors using routine prophylaxis to reduce the frequency of bleeding episodes.4 As per the CDA-AMC partially accepted deviation request,2 EHL agents and SHL agents were represented by a single drug within their class, efmoroctocog alfa (Eloctate) and octocog alfa (Kovaltry), respectively. The pediatric hemophilia A population was explored in a scenario analysis.
Altuviiio is available as lyophilized powder for solution for IV infusion. The treatment is available in 250 IU, 500 IU, 1,000 IU, 2,000 IU, 3,000 IU, or 4,000 IU per vial. The recommended dosage of Altuviiio is 50 IU/kg administered once weekly for routine prophylaxis.1 At the submitted price of $828 per 250 IU vial ($3.31 per IU across all vial sizes), the annual cost for an adult patient was estimated by the sponsor to be $739,164. The sponsor did not factor drug wastage in for Altuviiio nor the FVIII comparators and emicizumab, assuming treatment would be dispensed to the exact IU or nearest milligram.
The sponsor’s model predicts clinical outcomes in terms of QALYs and life-years. The economic evaluation was conducted over a 65-year time horizon from the perspective of a Canadian public health care payer. A 1.5% discount rate was applied to costs, QALYs, and life-years.3
Model Structure
The sponsor’s submitted model was a multistate Markov model with a 6-month cycle length.3 Two main health states were defined in the model: alive and dead. However, patients in the alive health state can transition through 29 substates based on their joint status, quantified using a PS of 0 to 28. At each cycle, all patients experience a probability of joint bleed and nonjoint bleed events based on ABR, regardless of their PS. The number of joint bleeds determines the probability of maintaining the same PS or transitioning to a higher PS substate. If a PS of 28 is reached, patients incur joint surgery and return to a PS of 15. The sponsor assumed that joint surgery would lead to a maximum reduction in patients’ PS score from 28 to ██ ██ ███████ █████████ ██ ██ ██████ for each joint).3 A figure illustrating the sponsor’s submitted model is presented in Appendix 3.
Model Inputs
In the sponsor’s base-case analysis, model population baseline characteristics (age = 35 years, weight = 85.9 kg, and PS = 14) were obtained from the XTEND-1 trial,4 assumptions, and the hemophilia A report completed by the Institute for Clinical and Economic Review in 2020.5 Body weight changes in the model as patients age within the simulation. A scenario analysis on the pediatric hemophilia A population was explored in which patients entered the model at 6 years of age and a PS of 0, aligning with the XTEND-Kids trial and key opinion leader feedback, respectively.
The main clinical inputs for all comparators are overall ABR (and its breakdown into nonjoint ABR and joint ABR [AjBR]) and the relationship between joint bleeds and joint health (i.e., PS). The ABR data (absolute rates) for Altuviiio were sourced from the XTEND-1 trial and were assumed to be the same over time. Relative measures for overall ABR and AjBR were derived from a sponsor-submitted MAIC (e.g., incidence rate ratios compared to emicizumab and EHL agents and mean absolute difference compared to SHL agents) and applied to the absolute rates of Altuviiio to determine the absolute ABR and AjBR for the comparators. Nonjoint ABRs for the comparators were estimated by subtracting their absolute AjBR from the overall ABR.
The transition probabilities across the PS health states were based on the relationship between joint bleeds and arthropathy established by the study by Coppola et al.6 At each cycle, patients experience a probability of joint bleed and nonjoint bleed events, and 6.52 joint bleeds in adults and 36.52 in younger patients (aged < 25 years) are associated with an increase in the PS of 1 point. As joint bleed events occur, the level of arthropathy increases (i.e., PS increases) until joint replacement surgery is required. The default assumption is that when a patient reaches a PS of 28, joint surgery is required.5,7,8
Utility values for each health substate (PS score) were based on the O’Hara et al. (2018) study that focused on EQ-5D utility values split by age and level of arthropathy.9,10 Specific disutility values informed individual bleeding episodes and joint replacement surgery. For patients who receive joint surgery, a utility value of 0.32 is applied for 1 month, based on a time trade-off utility found in a general hip replacement presurgery patient group reported in the literature.5,9-11 For a joint bleed and a nonjoint bleed, disutility values of 0.28 and 0.16 were applied for a duration of 2 days, respectively.9,12 No disutilities associated with adverse events (AEs) were applied in the model.
Cost in the model included those related to drug acquisition, resource utilization, hospitalizations, and surgery costs. Drug acquisition costs were calculated by the sponsor, using the submitted price per IU for Altuviiio and the per mg or IU prices from a previous CDA-AMC review for emicizumab16 to derive pricing for all comparators. Product monographs were sourced for recommended weight-based dosages. Resource use costs included those associated with outpatient physician, nurse, and X-ray visits and CT, MRI, and magnetic ultrasonography rates. Costs for resource use were obtained from the Ontario Schedule of Benefits.13 Nondrug costs associated with bleeds were informed by the Canadian Institute for Health Information patient cost estimator.14 Surgery costs were informed by Canadian Institute for Health Information’s Canadian Joint Replacement Registry annual report.15 No costs associated with AEs were applied in the model.
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (500 iterations), and the probabilistic results were similar to deterministic results. The probabilistic findings are presented in the following. The analyses were based on the sponsor-submitted pricing for Altuviiio. Comparator pricing was derived from the previous CDA-AMC review for emicizumab.16
Base-Case Results
In the sponsor’s base case, SHL agents and Altuviiio remained on the cost-effectiveness efficiency frontier. Among adults, Altuviiio was associated with higher QALYs (incremental QALY = 0.98) and higher costs (incremental cost = $5,246,289) compared to SHL agents, resulting in an ICER of $5,347,086 per QALY gained. Based on a WTP threshold of $50,000, there is a 3% probability of Altuviiio being cost-effective compared to SHL agents. Altuviiio dominated emicizumab and EHL agents (i.e., less costly and more effective). The majority of the incremental QALY gain of Altuviiio (99%) accrued beyond the XTEND-1 trial duration (52 weeks) and is based on extrapolations.
Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3. The disaggregated results showed that the additional QALYs associated with Altuviiio were mainly attributed to fewer joint bleeds compared to relevant comparators (derived from the sponsor-submitted MAIC) and the surrogate relationship between ABR and PS (derived from the study by Coppola et al.).6 Drug costs are also drivers of the results and the prophylactic drug costs are offset by savings with bleeding costs (except versus SHL agents).
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) | ||
|---|---|---|---|---|---|
Sponsor’s base case (probabilistic) | |||||
SHL agents | 19,650,743 | 19.76 | Reference | ||
Altuviiio | 24,879,032 | 20.74 | 5,347,086 | ||
Dominated treatments | |||||
Emicizumab | 25,236,613 | 20.39 | Dominated by Altuviiio | ||
EHL agents | 27,526,937 | 20.27 | Dominated by emicizumab and Altuviiio | ||
Sponsor base case (deterministic) | |||||
SHL agents | 18,422,637 | 19.37 | Reference | ||
Altuviiio | 24,752,439 | 20.62 | 5,098,890 | ||
Dominated treatments | |||||
Emicizumab | 25,098,797 | 20.14 | Dominated by Altuviiio | ||
EHL agents | 27,054,327 | 19.97 | Dominated by emicizumab and Altuviiio | ||
EHL = extended half-life; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SHL = standard half-life.
Source: Sponsor’s pharmacoeconomic submission.17
Sensitivity and Scenario Analysis Results
The sponsor provided deterministic scenario analyses exploring the impact of alternative model assumptions, including exploring uncertainty in discount rates, the impact of reimbursing Altuviiio in the pediatric population, variations in the time horizon, reducing adherence rate, and halving bleed rates for each comparator. The scenarios with the greatest impact on the ICER were the pediatric population and changes in the bleed rates. In the pediatric population, patients entered the model at age of 6 years, aligning with the XTEND-Kids trial mean population and the Health Canada indication. The results of the scenario analysis highlight that Altuviiio dominated both emicizumab and EHL agents; however, the ICER compared to SHL agents was 2,175,622. Similarly, when bleed rates were halved for each comparator, Altuviiio continued to dominate emicizumab and EHL agents, but the ICER compared to SHL agents was increased to 8,822,143.
The sponsor additionally conducted a scenario analysis from a societal perspective, capturing indirect costs incurred from lost workplace productivity and lost wages due to bleeds, hospitalizations, and medical appointments. The results of this analysis were similar to the sponsor’s base case.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The target population of the model does not reflect the Health Canada indication nor the sponsor’s reimbursement population: The Health Canada indication is for adults, adolescents, and children with hemophilia A (congenital FVIII deficiency) for routine prophylaxis to prevent or reduce frequency of bleeding episodes, treatment and control of bleeding episodes, and perioperative management of bleeding (surgical prophylaxis).1 While the sponsor reimbursement request aligns with the Health Canada indication, a deviation request was submitted by the sponsor to only examine the use of Altuviiio in adults with hemophilia A for routine prophylaxis to reduce the frequency of bleeding episodes.2 The sponsor noted that the exclusion of the on-demand and perioperative population was due to limited data availability and anticipating that only an extremely small proportion of patients would be expected to receive Altuviiio in both settings. This misaligns with the assumptions made in the sponsor’s submitted BIA, where the sponsor assumed that approximately ████% and ██% of adult and pediatric patients, respectively, with hemophilia A would require on-demand treatment.18 Clinical expert feedback obtained by CDA-AMC further confirmed that a significant proportion of patients with mild and severe hemophilia A are still receiving on-demand treatment and may decide to switch to prophylaxis with the availability of more convenient options. As no evidence was found on the comparison between on-demand treatment and prophylaxis, the CDA-AMC Clinical Review Report could not assess on-demand use of Altuviiio.
Key model parameters, including the efficacy of Altuviiio, were based on the XTEND-1 trial which only recruited patients with severe hemophilia A.3 The XTEND-1 study further excluded patients aged younger than 12 years.4 According to clinical experts consulted by CDA-AMC, there is no minimum age for treatment eligibility when considering initiation of therapy with Altuviiio. The pediatric population was examined by the sponsor in a scenario analysis by assuming identical relative efficacy as for adults.3 It is unclear whether this assumption on relative efficacy from the adult population is transferable to the pediatric population. In summary, the Health Canada indication and reimbursement request is broader as it includes the use of Altuviiio for prophylaxis, on-demand treatment, or perioperative management of bleeding, without any age or severity restriction.
As on-demand treatment, perioperative management, and prophylaxis for patients with mild and moderate hemophilia A were not modelled in the sponsor submission, CDA-AMC was unable to address this limitation. Therefore, the cost-effectiveness of Altuviiio for on-demand treatment and perioperative management and for prophylaxis in patients with mild and moderate hemophilia remains unknown.
To address limitations surrounding the pediatric population, CDA-AMC included several scenario analyses to explore the following: patients entering the model at the age of 6 years, patients entering the model at the age of 12 years, and patients entering the model at the age of 6 years and assuming the XTEND-Kids study ABR.
Comparative clinical efficacy of prophylaxis with Altuviiio is uncertain: In the absence of direct comparative evidence, the sponsor submitted an ITC, including unanchored MAICs and a propensity score analysis, to appraise the effect of prophylactic use of Altuviiio in adults on ABR, in comparison to emicizumab, EHL agents, and SHL agents. The pediatric population and other important clinical outcomes, including AEs, were not assessed in the ITCs. While the results from the ITCs suggest that prophylaxis with Altuviiio was associated with improved bleeding outcomes for adult patients with severe hemophilia compared to EHL agents, SHL agents, and emicizumab, the magnitude of clinical benefit is uncertain and likely overestimated. Limitations with the available indirect evidence includes a sizable reduction in the effective sample size from the original sample size after the propensity score weighting analyses, and inadequate or lack of adjustment for potential prognostic factors, which may introduce unmeasurable confounding in the relative treatment effect estimates.
Additionally, ABRs were assumed to remain stable over the patient’s lifetime and the sponsor-submitted model improperly characterized uncertainty around the relative effect on ABRs derived from the ITC, which was not included among the probabilistic parameters and contained programming errors. For example, the mean difference and incidence rate ratios relative to comparators were used deterministically to generate the absolute ABRs for the comparators and a distribution was assigned to the transformed data instead; the same standard error from emicizumab was used in the probabilistic sampling for EHL agents and SHL agents; and costs of breakthrough bleeds were included for the comparators but not for Altuviiio. This underestimates the uncertainty around the comparative efficacy of Altuviiio. Lastly, the sponsor’s submitted model included numerous IFERROR statements, which lead to situations in which the parameter value is overwritten with an alternative value without alerting the user to the automatized overwriting. The use of IFERROR statements makes thorough auditing of the sponsor’s model impractical. It remains unclear whether the model is running inappropriately by overriding errors.
CDA-AMC was unable to address the limitations with the comparative clinical evidence or the model programming for the appropriate characterization of uncertainty around ABRs. Owing to the lack of transparency implicit in the calculation of the probabilistic ICER, the CDA-AMC base-case and scenario analyses are presented deterministically, and the costs of breakthrough bleads were added to the Altuviiio treatment arm.
CDA-AMC included a scenario analysis deflating the estimated benefit on ABRs derived from the ITC by 25% to explore uncertainty around the overestimation of benefit. CDA-AMC also performed exploratory analysis assuming similar efficacy in joint bleeds for emicizumab and Altuviiio.
The strength of the association of number of joint bleeds needed to increase PS is uncertain: The sponsor assumed that, on average, the PS increases by 1 point for every 36.52 joint bleeds (if aged ≤ 24 years) or for every 6.52 joint bleeds (if aged > 25 years) based on published literature.6,19 This relationship between joint bleeds and arthropathy is uncertain because the methods by which Coppola et al. arrived at the values through the POTTER study19 were unclear. Additionally, the results reported in the POTTER study19 do not provide sufficient information to establish a surrogate relationship between AjBRs and PS. This adds uncertainty to the economic analysis because only the direct evidence from the pivotal trials incorporated into the economic model informs the number of annual bleeds (i.e., ABRs and AjBRs). The number of bleeds then indirectly estimates the patients’ PS, which in in turn determines their quality of life and number of joint replacements needed. Health-related quality of life across comparators was briefly reported for the comparisons between Altuviiio and 2 EHL therapies (efmoroctocog alfa and turoctocog alfa) without sufficient details, and the results were inconsistent for the 2 EHL agents. The pivotal trial used the Haemophilia Quality of Life questionnaire for adults to measure physical functioning and pain. In the trial, although there was a significant reduction in the Haemophilia Quality of Life questionnaire for adults Physical Health score at week 52, the magnitude of effect of Altuviiio on this outcome remains unclear. PS was not evaluated in the pivotal trial nor in the submitted ITCs. Therefore, the relationship between quality of life and number of annual bleeds is very uncertain. As the sponsor does not suggest a survival benefit with Altuviiio, the predicted clinical benefits are dependent on this surrogate relationship between ABRs and joint health.
Additionally, in the sponsor-submitted model, uncertainty around the association between joint bleeds and PS was not explored in the probabilistic analysis (i.e., deterministic parameter only). Given the insufficient evidence to support the strength of this relationship, the magnitude and effect on the cost-effectiveness estimates of variations in the number of bleeds needed to increase PS, and in turn, quality of life, is unclear.
CDA-AMC included 2 scenario analyses to explore the uncertainty of this association by assuming a 25% increase or decrease in the number of bleeds needed to increase PS by 1 point.
Dispensing of treatment is not reflective of clinical practice: In the sponsor’s submitted economic model, treatment was dispensed according to its respective product monographs with treatment acquisition costs calculated based on the exact dose (per mg or IU) required. The product monograph for Altuviiio states that Altuviiio is for single-use only and is available in preset concentrations. According to the CDA-AMC clinical experts and Canadian Blood Services, patients receiving Altuviiio, emicizumab, and other FVIII comparators would typically have their dose rounded to the nearest whole vial with drug dispensed accordingly to minimize wastage. How treatments are dispensed needs to be accounted for in the cost of treatment and, in the sponsor’s approach, treatment acquisition cost for both Altuviiio and the other comparators were underestimated.
Due to limitations in the sponsor-submitted model structure, assuming doses are rounded to the nearest whole vial to minimize wastage was not possible. Therefore, CDA-AMC was unable to conduct a reanalysis to address this limitation.
Underestimation of the costs of breakthrough bleed: In the sponsor-submitted model, the sponsor assumed, based on key opinion leader feedback, that in the case of a bleed event, patients treated with prophylactic SHL agents are assumed to be treated with 3 doses of 20 IU/kg on demand. Clinical expert feedback obtained by CDA-AMC noted that, in Canadian clinical practice, the breakthrough bleed dosage for SHL and EHL agents would be similar (i.e., 3 doses of 45 IU/kg).
While the clinical expert feedback agreed with the dose and frequency of administration for Altuviiio for breakthrough bleeds, it was evident that the model did not appropriately program bleed costs in the Markov trace to include breakthrough bleeds with Altuviiio (i.e., used an empty cell to determine the bleed costs in both the joint bleed and nonjoint bleed columns), underestimating total costs for Altuviiio.
Additionally, clinical expert feedback noted that certain patient groups, typically more active individuals, preemptively take additional prophylactic doses of EHL or SHL agents before physical activity to prevent bleeds. The clinical experts highlighted that the same could be expected for Altuviiio, should it be reimbursed, where patients would take an additional prophylactic dose before physical activity. This was not captured in the sponsor submission.
CDA-AMC conducted a base-case reanalysis including the accurate breakthrough bleed costs for both joint and nonjoint bleeds for Altuviiio and adjusting breakthrough bleed dosage for SHL agents to 3 doses of 45 IU/kg (same as the EHL dosage) to align with clinical expert opinion. CDA-AMC was unable to include additional doses before physical activities.
Comparator pricing based on publicly available prices: The sponsor’s analysis estimated the cost of comparators using a previous CDA-AMC review on emicizumab,16 which had estimated prices based on the published list prices of comparators in the Patented Medicine Prices Review Board (PMPRB) basket. The price of comparators does not reflect any confidential pricing that may have been negotiated by Canadian Blood Services. As such, the estimated drug acquisition costs are uncertain.
Due to confidentiality surrounding the negotiated price of comparators in Canada, CDA-AMC was unable to address this limitation in the base case.
To explore the uncertainty of the cost of comparators and the impact on the cost-utility analysis, CDA-AMC performed 2 scenario analyses with price reductions for emicizumab: 22% (similar to what was estimated for Altuviiio price reduction) and 90% (as per Hemlibra price reduction recommendation16).
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Recombinant FVIII concentrates (EHLs and SHLs agents) were pooled together, assuming similar efficacy, when defining model comparators. | Uncertain. According to the clinical experts consulted by CDA-AMC, the efficacy and potential harms among recombinant FVIII concentrates are likely similar. However, the costs of these products may differ, and thus, the cost-effectiveness of Altuviiio relative to individual recombinant FVIII concentrate products is unknown. |
Cost and impact of a treated bleed event were assumed similar (i.e., no difference by severity and type of bleeding event). | Inappropriate. According to clinical experts, the amount of prophylaxis needed to treat a bleed is dependent on the location and severity of the bleed, and the patient’s intrinsic factor levels. |
Perfect adherence was assumed. | This is an acceptable and conservative assumption. Lower adherence was explored in scenario analysis. |
Patients receiving prophylaxis with emicizumab were assumed to receive EHL treatment if a bleed occurred while on prophylaxis. | Uncertain. According to the clinical experts consulted by CDA-AMC, while this assumption is relatively reasonable, patients who were previously on SHL treatments before emicizumab would feel more comfortable using SHL agents on demand, should a bleed occur. Therefore, while most patients would receive EHL agents on demand if on emicizumab for prophylaxis, a proportion of patients may receive SHL on demand. As SHL agents are less costly than EHL agents, this assumption may have overestimated total costs in the emicizumab treatment arm. However, the impact is likely not substantial as the proportion of patients that would receive SHL agents for breakthrough bleeds may be relatively small. |
In the absence of data indicating a higher mortality risk for patients with hemophilia A without inhibitors than the general population, the model assumed that life expectancy followed the mortality rates of the Canadian general population. | Uncertain. Evidence on mortality rate for patients with hemophilia A varies, with some countries reporting the same mortality rate as the general population,20 and others reporting a higher rate. Due to uncertainty around the evidence, an exploratory scenario analysis was performed using the mortality rate of 1.4 from the study by Hassan et al.21 |
Adverse events were not included in the pharmacoeconomic model due to limited data and no serious adverse events reported in the XTEND-1 trial. | Reasonable. |
CDA-AMC = Canada's Drug Agency; EHL = extended half-life; FVIII = factor VIII; SHL = standard half-life.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
Several limitations within the sponsor’s submission could not be addressed, including the patient population, ABRs, missing comparators, wastage, exploring the relationship between PS and ABR, comparative efficacy, and other limitations. CDA-AMC undertook a reanalysis with 2 changes to align the dosage of SHL agents for breakthrough bleeds with clinical practice and to correctly program breakthrough bleeds for Altuviiio in the Markov trace. The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. On-demand dosage of SHL agents | 3 doses of 20 IU/kg | 3 doses of 45 IU/kg |
2. Breakthrough bleeds for Altuviiio | Empty cell used as input to calculate costs of joint and nonjoint bleeds | Joint bleed: $165.5 per kg Nonjoint bleed: $165.5 per kg |
CDA-AMC base case | ― | Reanalysis of 1 + 2 |
CDA-AMC = Canada's Drug Agency; SHL = standard half-life.
In the CDA-AMC base case, total costs associated with Altuviiio were $25,086,872, with 20.62 total QALYs. As the only changes made to the CDA-AMC base case were to increase the dosage of SHL agents for breakthrough bleeds and include the costs of breakthrough bleeds for Altuviiio, the CDA-AMC base case was broadly similar to the sponsor’s base case. Treatment with Altuviiio was more costly (incremental costs = $5,502,419) and more effective (incremental QALYs = 1.25) than SHL agents, with an ICER of $4,432,402 per QALY gained. Similar to the sponsor’s base case, Altuviiio dominated both emicizumab and EHL agents (e.g., was less costly and more effective). CDA-AMC notes that these results are highly uncertain as they are driven by fewer joint bleeds compared to relevant comparators (which rely on bleed rates for which the magnitude of benefit is highly uncertain, likely overestimated, and assumed to be stable over the patient’s lifetime) and the relationship between ABR and PS (with insufficient information to quantify this surrogate relationship).
Table 6: Summary of the CDA-AMC Reanalysis Results
Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
CDA-AMC base case (deterministic) | |||
SHL agents | 19,584,453 | 19.37 | Reference |
Altuviiio | 25,086,872 | 20.62 | 4,432,402 |
Dominated treatments | |||
Emicizumab | 25,098,797 | 20.14 | Dominated by Altuviiio |
EHL agents | 27,054,327 | 19.97 | Dominated by Altuviiio and emicizumab |
CDA-AMC = Canada's Drug Agency; EHL = extended half-life; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-years; SHL = standard half-life.
Note: The CDA-AMC base case is presented deterministically owing to the limitations with the sponsor’s probabilistic analysis.
Scenario Analysis Results
CDA-AMC undertook price reduction analysis based on the sponsor’s deterministic base case and the CDA-AMC deterministic base case (Table 7). The results suggest that an approximately 22% to 26% price reduction (in the CDA-AMC and sponsor’s base case, respectively) is required for Altuviiio to be considered cost-effective at a WTP threshold of $50,000 per QALY gained, compared to SHL agents.
Table 7: CDA-AMC Price Reduction Analyses
Analysis | Unit drug costa | ICERs for Altuviiio vs. SHL agents ($/QALY) | |
|---|---|---|---|
Price reduction | ($) | Sponsor base case | CDA-AMC reanalysis |
No price reduction | 3.31 | 5,098,890 | 4,432,402 |
10% | 2.98 | 3,126,001 | 2,459,513 |
20% | 2.65 | 1,153,113 | 486,625 |
30% | 2.32 | Dominant | Dominant |
CDA-AMC = Canada's Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SHL = standard half-life; vs. = versus.
aUnit drug cost per IU.
CDA-AMC conducted a series of scenario analyses to explore the impact of the following: patients entering the model at the age of 6 years; patients entering the model at the age of 12 years; assuming the ABR for Altuviiio prophylaxis from the XTEND-Kids study ABR for prophylaxis rather than from the XTEND-1 study (adults) for patients entering the model at the age of 6 years; increasing mortality rate for hemophilia A to align with the study by Hassan and colleagues;21 increasing by 25% the number of bleeds required to change PS by 1 point; decreasing by 25% the number of bleeds required to change PS by 1 point; reducing the estimated benefit derived from the ITC by 25%; and reducing the price of emicizumab by 90% to align with the Hemlibra CADTH review.16 An exploratory analysis was added assuming similar efficacy in joint bleeds for emicizumab and Altuviiio.
Results of these scenarios are presented in Appendix 4 (Table 14).
The results were mostly similar to the sponsor’s base case and the CDA-AMC base case (i.e., Altuviiio continued to dominate emicizumab and EHL agents and held similar ICERs compared to SHL agents), except when assuming reduced benefits derived from the ITC and reduced prices for emicizumab. When assuming a 25% reduction on the benefits derived from the ITCs, the ICER of Altuviiio increased to $5,922,936 per QALY compared to SHL agents. When assuming reduced prices for emicizumab, Altuviiio no longer dominated emicizumab. In these scenarios, emicizumab became either the next best treatment alternative after SHL agents (if confidential negotiations achieved a 22% price reduction for emicizumab) or the new reference scenario (if confidential negotiations achieved a 90% price reduction for emicizumab, as per CDA-AMC recommendation). In these scenarios with reduced emicizumab prices, Altuviiio ICERs ranged from $10,668,293 to $43,301,657 per QALY compared to emicizumab. These demonstrates that, in the submitted model, cost-effectiveness of Altuviiio is most sensitive to the negotiated prices of the comparators, which are confidential.
Issues for Consideration
CDA-AMC was unable to adequately assess the impact of potentially lower prices of the comparators on the cost-effectiveness of Altuviiio. Reduced price of comparators, arising from the tendering process by Canadian Blood Services22 as opposed to PMPRB’s maximum average potential price, may lead to different conclusions than the current analysis, potentially resulting in an even higher ICER for Altuviiio to which greater price reductions may be warranted.
Overall Conclusions
Evidence from the XTEND-1 and XTEND-Kids trials showed that 52 weeks of prophylactic therapy with Altuviiio resulted in a clinically relevant rate of bleeding outcomes (i.e., ABR, AjBR) and the potential to provide adequate perioperative management of bleeding, in patients with severe hemophilia A. Based on the clinical team appraisal of both trials, there is a low to very low level of certainty for bleeding outcomes and very low level of certainty for the other efficacy outcomes. Given the nonrandomized open-label design and the lack of control for physical activity, there may be a potential risk of bias in the risk of bleeding events. In the absence of direct comparative evidence to EHL agents, SHL agents, and emicizumab, the sponsor’s submitted ITCs included unanchored MAICs and propensity score analysis to inform the comparative efficacy on ABR. The indirect evidence suggests that prophylaxis with Altuviiio was associated with improved bleeding outcomes compared to EHL agents, SHL agents, or emicizumab in adult patients with severe hemophilia A. The CDA-AMC clinical review concluded that the sponsor’s ITC had several limitations (i.e., sizable reduction in the effective sample size after propensity score weighting analyses, and inadequate or lack of adjustment for potential prognostic factors) which introduces uncertainty to the true magnitude of the comparative clinical benefits of Altuviiio but likely resulted in an overestimation.
CDA-AMC undertook reanalysis adjusting the on-demand dosage of SHL agents and including the costs of breakthrough bleeds associated with Altuviiio. Given the limitations with the sponsor’s probabilistic analysis, CDA-AMC reports only the deterministic results. In adult patients with severe hemophilia A without inhibitors who require routine prophylaxis, the ICER of Altuviiio compared to SHL agents was $4,432,402 per QALY gained (incremental costs = $5,502,419; incremental QALYs = 1.25). While the CDA-AMC reanalysis results were largely consistent with the sponsor’s, the lower ICER value estimated by CDA-AMC compared to the sponsor’s submission were driven by the adjustment of on-demand dosage of SHL agents which increased the expected costs in the SHL strategy, consequently reducing the incremental cost difference between Altuviiio and SHL agents. Altuviiio dominated emicizumab and EHL agents (associated with less total costs and more QALYs). At the publicly available prices, a price reduction of at least 22% (from $3.31 to $2.58 per IU) is required for Altuviiio to be considered cost-effective at a WTP threshold of $50,000 per QALY gained compared to SHL agents. A scenario analysis was conducted to align with the patient characteristics of the XTEND-Kids trial and applied the ABR reported in the XTEND-Kids trials. The scenario analysis resulted in similar results compared to the base case (i.e., adult populations), with an ICER of $4,150,946 per QALY gained when comparing Altuviiio to SHL agents. Emicizumab and EHL agents remained dominated by Altuviiio.
CDA-AMC notes that these results are highly uncertain given the available clinical evidence. The cost-effectiveness results are driven by the clinical outcomes that predict fewer joint bleeds associated with Altuviiio compared to relevant comparators. Estimates of comparative clinical benefits were derived from the sponsor’s ITC which, as noted previously, are uncertain and likely to be overestimated. The validity of the predicted QALY gains is further dependent on the validity of the surrogate relationship between ABR and joint health (i.e., PS) which is unsubstantiated. Within the submitted model, treatment acquisition costs may not reflect clinical practice given that they were calculated based on the exact dose required (per mg or per IU) rather than rounding to the nearest whole vial to minimize wastage. The price of the comparators is also a key driver in the cost-effectiveness of Altuviiio. As the analyses are based on publicly available list prices for comparators, the cost-effectiveness of Altuviiio is highly dependent on the actual prices. In light of all these limitations, the cost-effectiveness of Altuviiio and the required price reduction estimates are highly uncertain. Several scenario analyses were conducted to highlight the sensitivity of the model to these inputs. When assuming a smaller clinical benefit in ABRs, the ICER of Altuviiio increased to $5,922,936 per QALY compared to SHL agents. When assuming lower prices for emicizumab (22% and 90%), emicizumab was no longer dominated by Altuviiio and the ICER for Altuviiio ranged from $609,402 to $43,301,657 per QALY gained compared to emicizumab. In light of these findings, further price reductions may be warranted to address these uncertainties.
Lastly, it is important to highlight that the sponsor’s submitted model is limited in scope as it does not address the full Health Canada indication. The cost-effectiveness of Altuviiio for routine prophylaxis in patients with mild and moderate hemophilia, or for on-demand treatment and perioperative management is unknown.
References
1.Sanofi-Aventis Canada Inc. Altuviiio (efanesoctocog alfa for injection): lyophilized powder, intravenous, 250, 500, 1000, 2000, 3000, or 4000 IU/vial [product monograph]. March 26, 2025.
2.Canada's Drug Agency. CDA pharmacoeconomic deviation request (approved) [sponsor supplied reference]. 2024.
3.Sanofi-Aventis Canada Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Altuviiio (efanesoctocog alfa), lyophilized powder, 250, 500, 1000, 2000, 3000, or 4000 IU/vial, for solution for intravenous infusion. September 18, 2024.
4.von Drygalski A, Chowdary P, Kulkarni R, et al. Efanesoctocog alfa prophylaxis for patients with severe hemophilia A (XTEND-1). N Engl J Med. 2023;388(4):310-318. doi:10.1056/NEJMoa2209226 PubMed
5.Institute for Clinical and Economic Review. Valoctocogene roxaparvovec and emicizumab for hemophilia A without inhibitors: effectiveness and value. Final report. November 20, 2020.
6.Coppola A, D'Ausilio A, Aiello A, et al. Cost-effectiveness analysis of late prophylaxis vs. on-demand treatment for severe haemophilia A in Italy. Haemophilia. 2017;23(3):422-429. doi:10.1111/hae.13185 PubMed
7.Fischer K, Collins P, Björkman S, et al. Trends in bleeding patterns during prophylaxis for severe haemophilia: observations from a series of prospective clinical trials. Haemophilia. 2011;17(3):433-8. doi:10.1111/j.1365-2516.2010.02450.x PubMed
8.Earnshaw SR, Graham CN, McDade CL, et al. Factor VIII alloantibody inhibitors: cost analysis of immune tolerance induction vs. prophylaxis and on-demand with bypass treatment. Haemophilia. 2015;21(3):310-319. doi:10.1111/hae.12621 PubMed
9.O'Hara J, Walsh S, Camp C, et al. The impact of severe haemophilia and the presence of target joints on health-related quality-of-life. Health Qual Life Outcomes. 2018;16(1):84. doi:10.1186/s12955-018-0908-9 PubMed
10.O'Hara J, Walsh S, Camp C, et al. The relationship between target joints and direct resource use in severe haemophilia. Health Econ Rev. 2018;8(1):1. doi:10.1186/s13561-018-0185-7 PubMed
11.Laupacis A, Bourne R, Rorabeck C, et al. The effect of elective total hip replacement on health-related quality of life. J Bone Joint Surg Am. 1993;75(11):1619-1626. doi:10.2106/00004623-199311000-00006 PubMed
12.Neufeld EJ, Recht M, Sabio H, et al. Effect of acute bleeding on daily quality of life assessments in patients with congenital hemophilia with inhibitors and their families: observations from the dosing observational study in hemophilia. Value Health. 2012;15(6):916-25. doi:10.1016/j.jval.2012.05.005 PubMed
13.OHIP. Ontario schedule of benefits and fees. 2024. Accessed December 1, 2024. https://www.ontario.ca/page/ohip-schedule-benefits-and-fees
14.Canadian Institute for Health Information. Patient cost estimator. 2022. Accessed December 1, 2024. https://www.cihi.ca/en/patient-cost-estimator
15.Canadian Institute for Health Information. Canadian joint replacement registry annual report: hip and knee replacements in Canada. 2022.
16.CADTH. Drug Reimbursement Review Pharmacoeconomic report report: Emicizumab (Hemlibra). 2021. Accessed December 1, 2024. https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/st0651-hemlibra-pharmacoeconomic-review-report.pdf
17.Sanofi-Aventis Canada Inc. Drug Reimbursement Review sponsor submission: Altuviiio (efanesoctocog alfa), lyophilized powder, 250, 500, 1000, 2000, 3000, or 4000 IU/vial, for solution for intravenous infusion [internal sponsor's package]. September 18, 2024.
18.Sanofi-Aventis Canada Inc. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Altuviiio (efanesoctocog alfa), lyophilized powder, 250, 500, 1000, 2000, 3000, or 4000 IU/vial, for solution for intravenous infusion. September 18, 2024.
19.Tagliaferri A, Matichecchia A, Rivolta GF, et al. Optimising prophylaxis outcomes and costs in haemophilia patients switching to recombinant FVIII-Fc: a single-centre real-world experience. Blood Transfus. 2020;18(5):374-385. doi:10.2450/2019.0220-19 PubMed
20.Walsh CE, Soucie JM, Miller CH, et al. Impact of inhibitors on hemophilia a mortality in the United States. Am J Hematol. 2015;90(5):400-405. doi:10.1002/ajh.23957 PubMed
21.Hassan S, Monahan RC, Mauser‐Bunschoten EP, et al. Mortality, life expectancy, and causes of death of persons with hemophilia in the Netherlands 2001–2018. J Thromb Haemost. 2021;19(3):645-653. doi:10.1111/jth.15182 PubMed
22.Canadian Hemophilia Society. The tender process (RFP) for clotting factor concentrates in Canada. Accessed December 18, 2024. https://www.hemophilia.ca/files/What%20is%20an%20RFP.pdf
23.Saskatchewan Drug Plan: search formulary. 2024. Accessed December 1, 2024. https://formulary.drugplan.ehealthsask.ca/SearchFormulary
24.CADTH. Drug Reimbursement Expert Review Committee final recommendation: Emicizumab (Hemlibra - Hoffmann-La Roche Ltd.). December, 2020. Accessed December 1, 2024. https://www.cadth.ca/sites/default/files/cdr/complete/ST0651%20Hemlibra%20-%20CPEC%20Final%20Recommendation%20December%2023%2C%202020_for%20posting.pdf
25.Association of Hemophilia Clinic Directors of Canada. Canadian blood disorders registry. Factor FVIII deficiency. 2021. Accessed December 1, 2024. https://ahcdc.ca/storage/pdf/21/FVIII-2021.pdf
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison Table for Prophylaxis in Patients With Hemophilia A Without FVIII
Treatment | Dosage form/ strength | Form | Price per unit ($) | Recommended dosage | Daily cost ($) | Course or annual cost ($) |
|---|---|---|---|---|---|---|
Antihemophilic factor VIII (recombinant, B-domain deleted), Fc-VWF-XTEN fusion protein (Altuviiio) | 250 IU vial 500 IU vial 1,000 IU vial 2,000 IU vial 3,000 IU vial 4,000 IU vial | Powder for IV injection | 827.5000a 1,665.0000 3,310.0000 6,620.0000 9,930.0000 13,240.0000 (3.31 per IU in all vials) | Prophylaxis 50 IU/kg once weekly. | 40 kg patient: 945.71 80 kg patient: 1,891.43 | 40 kg patient: 345,186 80 kg patient: 690,371 |
Factor VIII treatments, available through Canadian Blood Services | ||||||
Emicizumab (Hemlibra) | 30 mg/1 mL 60 mg/0.4 mL 105 mg/0.7mL 150 mg/1 mL | Solution for SC injection | 3,661.5180b 7,323.0360 12,815.3130 18,307.5900 (122.05 per mg in all vials) | Loading dose: 3 mg/kg once weekly for 4 weeks Maintenance dose: 1.5mg/kg once weekly or 3 mg/kg every 2 weeks or 6 mg/kg every 4 weeks, starting week 5 for patients ≥ 12 years of age who weigh ≥ 40 kg. Patients under 12 years or those who weigh < 40kg should receive 1.5 mg/kg every week or 3 mg/kg every 2 weeks starting week 5. | 40 kg patient: Year 1: 1,126.40 Thereafter: 1,046.15 80 kg patient: Year 1: 2,252.80 Thereafter: 2,092.30 | 40 kg patient: Year 1: 411,136 Thereafter: 381,841 80 kg patient: Year 1: 822,272 Thereafter: 763,688 |
Antihemophilic Factor Recombinant, PEGylated (Adynovate) | 250 IU 500 IU 750 IU 1,000 IU 1,500 IU 2,000 IU 3,000 IU | Powder for IV injection | 471.5500 943.1000 1,414.6500 1,886.2000 2,829.3000 3,772.4000 5,658.6000 1.8862 per IUb in all vials) | Patients < 12 years: 40 to 60 IU/kg twice weekly Patients ≥ 12 years: 40 to 50 IU/kg twice weekly | 40 kg patient: 943.10 to 1,347.29 80 kg patient: 1,751.47 to 2,155.66 | 40 kg patient: 344,232 to 491,759 80 kg patient: 639,287 to 786,815 |
Antihemophilic Factor Recombinant BDD, Fc Fusion Protein (Eloctate) | 250 IU 500 IU 750 IU 1,000 IU 1,500 IU 2,000 IU 3,000 IU | Powder for IV injection | 471.5500 943.1000 1,414.6500 1,886.2000 2,829.3000 3,772.4000 5,658.6000c (1.8862 per IUb in all vials) | 50 IU/kg every 3 to 5 days. | 40 kg patient: 754.48 to 1,257.47 80 kg patient: 1,508.96 to 2,514.93 | 40 kg patient 275,385 to 458,975 80 kg patient 550,770 to 917,951 |
Antihemophilic Factor Recombinant, BDD, PEGylated (Jivi) | 250 IU 500 IU 1,000 IU 2,000 IU 3,000 IU | Powder for IV injection | 471.5500 943.1000 1,886.2000 3,772.4000 5,658.6000 (1.8862 per IUb in all vials) | Patients ≥ 12 years: 30 to 40 IU/kg twice weekly | 40 kg patient: 673.64 to 808.37 80 kg patient: 1,347.29 to 1,751.47 | 40 kg patient: 245,880 to 295,055 80 kg patient: 491,759 to 639,287 |
Antihemophilic Factor Recombinant, (Kovaltry) | 250 IU 500 IU 1,000 IU 2,000 IU 3,000 IU | Powder for IV injection | 364.8000 729.6000 1,459.2000 2,918.4000 4,377.6000 (1.4592b per IU in all vials) | Patients ≤ 12 years: 20 to 50 IU/kg 2 to 3 times weekly, or every other day Patients > 12 years: 20 to 40 IU/kg 2 to 3 times weekly | 40 kg patient (≤ 12): 312.69 to 1,459.20 80 kg patient: 625.37 to 2,032.46 | 40 kg patient (≤ 12): 114,130 to 532,608 80 kg patient: 228,260 to 741,847 |
Antihemophilic Factor Recombinant, BDDrFVIII, (Xyntha) | 250 IU 500 IU 1,000 IU 2,000 IU | Powder in vial or prefilled syringe | 279.5500 559.1000 1,118.2000 2,236.4000 1.1182b per IU | Adults and adolescents: 30 ± 5 IU/kg 3 times weekly | 40 kg patient: 479.23 to 599.03 80 kg patient: 958.45 to 1,198.68 | 40 kg patient: 174,918 to 218,647 80 kg patient: 349,835 to 437,294 |
BDD = B-domain deleted; CDA-AMC = Canada's Drug Agency; FVIII = factor VIII; IU = international unit; rFVIII = recombinant factor VIII; SC = subcutaneous
Daily and annual costs (assuming 365 days per year) are rounded to the nearest available vial size; patients typically administer the full amount in the vial for factor VIII products, according to the clinical experts consulted by CDA-AMC. PMPRB pricing represents the maximum average potential price in Canada; it is likely that due to the Canadian Blood Services tendering system, actual costs per IU paid are substantially lower.
aSponsor’s submitted price.
bBased on priced used for the Hemlibra submission.16 For those prices unavailable from PMBRP. Eloctate price from PMBRP was used as the lowest available long-acting proxy. Australian prices were converted to CA$ based on exchange rate of 0.9508 from Bank of Canada Currency Converter, average August 13 through 19, 2020.
cSaskatchewan formulary (accessed November 2024).23
Table 9: CDA-AMC Cost Comparison Table for On-Demand Management of Bleeding in Patients With Hemophilia A Without Factor VIII
Treatment | Dosage form/ strength | Form | Price per unit ($) | Recommended dosage | Cost per bleed ($)d |
|---|---|---|---|---|---|
Antihemophilic factor VIII (recombinant, B-domain deleted), Fc-VWF-XTEN fusion protein (Altuviiio) | 250 IU vial 500 IU vial 1,000 IU vial 2,000 IU vial 3,000 IU vial 4,000 IU vial | Powder for IV infusion | 827.5000a 1,665.0000 3,310.0000 6,620.0000 9,930.0000 13,240.0000 (3.31 per IU in all vials) | 50 IU/kg once weekly. | 40 kg patient: 6,620 80 kg patient: 13,240 |
Factor VIII treatments, available through Canadian Blood Services | |||||
Antihemophilic Factor Recombinant, PEGylated (Adynovate) | 250 IU 500 IU 750 IU 1,000 IU 1,500 IU 2,000 IU 3,000 IU | Powder for IV injection | 471.5500 943.1000 1,414.6500 1,886.2000 2,829.3000 3,772.4000 5,658.6000 (1.8862 per IUb in all vials) | Minor bleed: 10 to 20 IU every 12 to 24 hours Moderate bleed: 15 to 30 IU every 12 to 24 hours Major bleed: 30 to 50 IU every 8 to 24 hours | 40 kg patient: 1,886 to 79,220 80 kg patient: 2,829 to 158,441 |
Antihemophilic Factor Recombinant BDD, Fc Fusion Protein (Eloctate) | 250 IU 500 IU 750 IU 1,000 IU 1,500 IU 2,000 IU 3,000 IU | Powder for IV injection | 471.5500 943.1000 1,414.6500 1,886.2000 2,829.3000 3,772.4000 5,658.6000 (1.8862 per IUb in all vials) | Patients ≥ 12 years: Minor and moderate bleed: 20 to 30 IU/kg every 24 to 48 hours Major bleed: 40 to 50 IU/kg every 12 to 24 hoursc Patients < 12 years: Minor and moderate bleed: 20 to 30 IU/kg every 12 to 24 hours Major bleed: 40 to 50 IU/kg every 8 to 24 hours | 40 kg patient: 2,829 to 79,220 80 kg patient: 2,829 to 105,627 |
Antihemophilic Factor Recombinant, BDD, PEGylated (Jivi) | 250 IU 500 IU 1,000 IU 2,000 IU 3,000 IU | Vial | 471.5500 943.1000 1,886.2000 3,772.4000 5,658.6000 (1.8862 per IUb in all vials) | Minor bleed: 10 to 20 IU every 12 to 24 hours Moderate bleed: 15 to 30 IU every 12 to 24 hours Major bleed: 30 to 50 IU every 8 to 24 hours | 40 kg patient: 1,886 to 79,220 80 kg patient: 2,829 to 158,441 |
Antihemophilic Factor Recombinant, (Kovaltry) | 250 IU 500 IU 1,000 IU 2,000 IU 3,000 IU | Vial | 364.8000 729.6000 1,459.2000 2,918.4000 4,377.6000 (1.4592 per IUb in all vials) | 10 to 30 IU/kgc Minor bleeds: every 12 to 24 hours (at least 1 day) Moderate bleeds: every 12 to 24 hours (3 to 4 days) Major bleeds: every 8 to 24 hours until resolved | 40 kg patient: 1,459 to 38,304 80 kg patient: 2,189 to 76,608 |
Antihemophilic Factor Recombinant, BDDrFVIII, (Xyntha) | 250 IU 500 IU 1,000 IU 2,000 IU | Powder in vial or prefilled syringe | 279.5500 559.1000 1,118.2000 2,236.4000 (1.1182 per IUb in all vials) | Minor bleeds: 20 to 40 IU/kg every 12 to 24 hours Moderate bleeds: 30 to 60 IU/kg every 12 to 24 hours Major bleeds: 60 to 100 IU/kg every 8 to 24 hours | 40 kg patient: 1,677 to 93,928 80 kg patient: 3,355 to 187,857 |
BDD = B-domain deleted; CDA-AMC = Canada's Drug Agency; FVIII = factor VIII; IU = international unit; IV = IV; rFVIII = recombinant factor VIII; SC = subcutaneous,
Costs per infusion are rounded to the nearest available vial size; patients typically administer the full amount in the vial for factor VIII products, according to the clinical experts consulted by CDA-AMC. PMPRB pricing represents the maximum average potential price in Canada; it is likely that due to the Canadian Blood Services tendering system, actual costs per IU paid are substantially lower.
Antihemophilic Factor/ VWF Complex (Humate-P) Human VWF and human Coagulation FVIII (Wilate) are used in a very selective numbers of patients and were deemed not relevant for this submission according to the clinical experts consulted by CDA-AMC and the Canadian Blood Services.
aSponsor’s submitted price.
bBased on priced used for the Hemlibra submission.16
dOn-demand treatment costs per bleed were calculated for administration of the different products ranging from one day to one week (minor to major bleeds).
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to key limitation that the target population in the model does not reflect the Health Canada indication nor the sponsor’s reimbursement population, and that relevant comparators are missing. |
Model has been adequately programmed and has sufficient face validity | No | Refer to key limitation on improper programming of breakthrough bleeds. Also, the sponsor’s submitted model included numerous IFERROR statements. |
Model structure is adequate for decision problem | No | Refer to key limitations on magnitude of benefit derived from the MAIC, and the strength of association between joint bleeds and PS. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Lack of transparency implicit in the calculation of the probabilistic ICER. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Refer to key limitations on dispensing of treatments, strength of association between joint bleeds and PS, and comparative clinical efficacy uncertainty. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
PS = Pettersson Score
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of the Sponsor’s Base Case
Table 11: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Altuviiio | Emicizumab | EHL | SHL |
|---|---|---|---|---|
Discounted LYs | ||||
Total | 33.33 | 33.33 | 33.33 | 33.33 |
Average number of joint bleeds | 16.84 | 35.11 | 45.79 | 105.98 |
Average number of nonjoint bleeds | 6.86 | 38.38 | 35.90 | 16.84 |
Number of joint surgeries | 0.00 | 0.01 | 0.02 | 0.24 |
Discounted QALYs | ||||
Total | 20.74 | 20.39 | 20.27 | 19.76 |
No bleed | 20.88 | 20.78 | 20.72 | 20.44 |
Joint bleed | −0.12 | −0.24 | −0.32 | −0.73 |
Nonjoint bleeds | −0.03 | −0.15 | −0.14 | −0.07 |
Joint surgery | 0.0000 | 0.0029 | 0.0117 | 0.1151 |
Discounted costs | ||||
Total | $24,879,032 | $25,236,613 | $27,526,937 | $19,650,743 |
Prophylactic drug costs | $24,618,330 | $22,394,466 | $24,371,036 | $16,078,358 |
Joint bleeds costs | $172,539 | $1,349,051 | $1,759,753 | $3,065,219 |
Nonjoint bleeds costs | $70,270 | $1,475,121 | $1,377,931 | $486,156 |
Joint surgery costs | $0.55 | $85 | $336 | $3,257 |
Adverse event costs | $0.00 | $0.00 | $0.00 | $0.00 |
Resource use costs | $17,893 | $17,890 | $17,880 | $17,754 |
EHL = extended half-life; LY = life-year; QALY = quality-adjusted life-year; SHL = standard half-life.
Note: adding row values may not equal total numbers as some disaggregate results were omitted from the table.
Source: Sponsor’s pharmacoeconomic submission17
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CDA-AMC Base Case
Table 12: Summary of the Stepped Analysis of the CDA-AMC Base-Case Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|---|
Sponsor base case (deterministic) | SHL | 18,422,637 | 19.37 | Reference |
Altuviiio | 24,752,439 | 20.62 | 5,098,890 | |
Dominated treatments | ||||
Emicizumab | 25,098,797 | 20.14 | Dominated by Altuviiio | |
EHL | 27,054,327 | 19.97 | Dominated by emicizumab and Altuviiio | |
1. CDA-AMC reanalysis 1: Increase SHL breakthrough bleed dosage | SHL | 19,584,453 | 19.37 | Reference |
Altuviiio | 24,752,439 | 20.62 | 4,163,004 | |
Dominated treatments | ||||
Emicizumab | 25,098,797 | 20.14 | Dominated by Altuviiio | |
EHL | 27,054,327 | 19.97 | Dominated by emicizumab and Altuviiio | |
2. CDA-AMC reanalysis 2: Program breakthrough bleeds for Altuviiio | SHL | 18,422,637 | 19.37 | Reference |
Altuviiio | 25,086,872 | 20.62 | 5,368,288 | |
Dominated treatments | ||||
Emicizumab | 25,098,797 | 20.14 | Dominated by Altuviiio | |
EHL | 27,054,327 | 19.97 | Dominated by emicizumab and Altuviiio | |
CDA-AMC base case (deterministic) (Reanalysis 1 + 2) | SHL | 19,584,453 | 19.37 | Reference |
Altuviiio | 25,086,872 | 20.62 | 4,432,402 | |
Dominated treatments | ||||
Emicizumab | 25,098,797 | 20.14 | Dominated by Altuviiio | |
EHL | 27,054,327 | 19.97 | Dominated by emicizumab and Altuviiio | |
CDA-AMC = Canada's Drug Agency; EHL = extended half-life; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SHL = standard half-life.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. The results of all steps are presented deterministically unless otherwise indicated. The cumulative CDA-AMC base case is presented deterministically owing to the limitations with the sponsor’s probabilistic analysis.
Table 13: Disaggregated Summary of the CDA-AMC Economic Evaluation Results
Parameter | Altuviiio | Emicizumab | EHL | SHL |
|---|---|---|---|---|
Discounted LYs | ||||
Total | 33.33 | 33.33 | 33.33 | 33.33 |
Average number of joint bleeds | 17.00 | 35.42 | 45.95 | 106.00 |
Average number of nonjoint bleeds | 6.67 | 38.54 | 35.66 | 18.33 |
Number of joint surgeries | 0.00 | 0.00 | 0.02 | 0.24 |
Discounted QALYs | ||||
Total | 20.62 | 20.14 | 19.97 | 19.37 |
No bleed | 20.76 | 20.54 | 20.42 | 20.06 |
Joint bleed | −0.12 | −0.24 | −0.32 | −0.73 |
Nonjoint bleeds | −0.03 | −0.15 | −0.14 | −0.07 |
Joint surgery | 0.0000 | 0.0022 | 0.0107 | 0.1150 |
Discounted costs | ||||
Total | $25,086,872 | $25,098,797 | $27,054,327 | $19,584,453 |
Prophylactic drug costs | $24,491,593 | $22,713,501 | $24,423,890 | $16,195,528 |
Joint bleeds costs | $414,785 | $1,133,686 | $1,470,727 | $2,871,329 |
Nonjoint bleeds costs | $162,661 | $1,233,717 | $1,141,579 | $496,614 |
Joint surgery costs | $0 | $64 | $311 | $3,288 |
Adverse event costs | $0 | $0 | $0 | $0 |
Resource use costs | $17,833 | $17,830 | $17,821 | $17,694 |
CDA-AMC = Canada's Drug Agency; EHL = extended half-life; LY = life-year; NA = not applicable; QALY = quality-adjusted life-year; SHL = standard half-life.
Note: adding row values may not equal total numbers as some disaggregate results were omitted from the table.
Scenario Analyses
Table 14: Summary of the Scenario Analyses
Scenario analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (deterministic) | SHL | 18,422,637 | 19.37 | Reference |
Altuviiio | 24,752,438 | 20.62 | 5,098,890 | |
Dominated treatments | ||||
Emicizumab | 25,098,797 | 20.14 | Dominated by Altuviiio | |
EHL | 27,054,327 | 19.97 | Dominated by emicizumab and Altuviiio | |
CDA-AMC base case (deterministic) | SHL | 19,584,453 | 19.37 | Reference |
Altuviiio | 25,086,872 | 20.62 | 4,432,402 | |
Dominated treatments | ||||
Emicizumab | 25,098,797 | 20.14 | Dominated by Altuviiio | |
EHL | 27,054,327 | 19.97 | Dominated by emicizumab and Altuviiio | |
1. CDA-AMC Scenario 1: Starting age 6 | SHL | 22,953,344 | 30.67 | Reference |
Altuviiio | 29,001,528 | 32.13 | 4,150,946 | |
Dominated treatments | ||||
Emicizumab | 28,916,992 | 31.58 | Ext. dominated by Altuviiio and SHL | |
EHL | 31,306,294 | 31.40 | Dominated by emicizumab and Altuviiio | |
2. CDA-AMC Scenario 2: Starting age 12 | SHL | 23,546,687 | 28.60 | Reference |
Altuviiio | 30,179,683 | 30.01 | 4,708,164 | |
Dominated treatments | ||||
Emicizumab | 30,084,569 | 29.48 | Ext. dominated by Altuviiio and SHL | |
EHL | 32,539,704 | 29.30 | Dominated by emicizumab and Altuviiio | |
3. CDA-AMC Scenario 3: XTEND-Kids ABR for prophylaxis aged 6 and up | SHL | 23,038,302 | 30.64 | Reference |
Altuviiio | 29,173,944 | 32.06 | 4,328,781 | |
Dominated treatments | ||||
Emicizumab | 29,620,778 | 31.41 | Dominated by Altuviiio | |
EHL | 32,083,049 | 31.21 | Dominated by emicizumab and Altuviiio | |
4. CDA-AMC Scenario 4: 1.4 mortality rate for hemophilia A | SHL | 18,643,794 | 18.53 | Reference |
Altuviiio | 23,889,269 | 19.71 | 4,474,279 | |
Dominated treatments | ||||
Emicizumab | 23,904,409 | 19.27 | Dominated by Altuviiio | |
EHL | 25,759,725 | 19.11 | Dominated by emicizumab and Altuviiio | |
5. CDA-AMC Scenario 5: + 25% in number of bleeds to change PS | SHL | 19,583,710 | 19.41 | Reference |
Altuviiio | 25,086,872 | 20.65 | 4,441,606 | |
Dominated treatments | ||||
Emicizumab | 25,098,748 | 20.22 | Dominated by Altuviiio | |
EHL | 27,054,113 | 20.06 | Dominated by emicizumab and Altuviiio | |
6. CDA-AMC Scenario 6: −25% in number of bleeds to change PS | SHL | 19,585,106 | 19.31 | Reference |
Altuviiio | 25,086,872 | 20.55 | 4,408,174 | |
Dominated treatments | ||||
Emicizumab | 25,099,055 | 20.04 | Dominated by Altuviiio | |
EHL | 27,055,012 | 19.88 | Dominated by emicizumab and Altuviiio | |
7. CDA-AMC Scenario 7: 25% reduction in benefit derived from MAIC | SHL | 18,980,892 | 19.58 | Reference |
Altuviiio | 25,086,872 | 20.62 | 5,922,936 | |
Dominated treatments | ||||
Emicizumab | 24,625,267 | 20.30 | Ext. dominated by Altuviiio and SHL | |
EHL | 26,531,646 | 20.16 | Dominated by emicizumab and Altuviiio | |
8. CDA-AMC Scenario 8: 22.2% price reduction of emicizumab | SHL | 19,584,453 | 19.37 | Reference |
Emicizumab | 20,053,447 | 20.14 | 609,402 | |
Altuviiio | 25,086,872 | 20.62 | 10,668,293 | |
Dominated treatments | ||||
EHL | 27,054,327 | 19.97 | Dominated by emicizumab and Altuviiio | |
9. CDA-AMC Scenario 9: 90% price reduction of emicizumab | Emicizumab | 4,656,646 | 20.14 | Reference |
Altuviiio | 25,086,872 | 20.62 | 43,301,657 | |
Dominated treatments | ||||
SHL | 19,584,453 | 19.37 | Dominated by emicizumab | |
EHL | 27,054,327 | 19.97 | Dominated by emicizumab | |
10. CDA-AMC Scenario 10: emicizumab ABR equal to Altuviiio ABR for joint bleeds | SHL | 19,584,453 | 19.37 | Reference |
Emicizumab | 24,509,219 | 20.49 | 4,413,898 | |
Altuviiio | 25,086,872 | 20.62 | 4,596,692 | |
Dominated treatments | ||||
EHL | 27,054,327 | 19.97 | Dominated by emicizumab and Altuviiio | |
ABR = annualized bleeding rate; CDA-AMC = Canada's Drug Agency; EHL = Extended half-life; ICER = incremental cost-effectiveness ratio; MAIC = matching-adjusted indirect comparison; PS = Pettersson score; QALY = quality-adjusted life-year; SHL = standard half-life.
aReference product is least costly alternative.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 15: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a BIA assessing the expected budgetary impact of reimbursing Altuviiio for the treatment of adults, adolescents and children with hemophilia A (congenital factor VIII deficiency) without inhibitors for: routine prophylaxis to prevent or reduce the frequency of bleeding episodes; treatment and control of bleeding episodes; and perioperative management of bleeding (surgical prophylaxis).18 The BIA was conducted using an epidemiological approach and was undertaken from the perspective of Canadian Blood Services over a 3-year time horizon (2025 to 2027). The costs of treating breakthrough bleeds for patients on prophylaxis and wastage (for all patients) was not considered in the base case. Drug costs calculations assumed an average weight of 85.9 kg for adults (86% of patients) and 35kg for pediatric patients (14% of patients). Key inputs to the BIA are documented in Table 16.
Table 16: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Proportion of hemophilia A (congenital FVIII deficiency) patients across Canada (0.0081)25 Without Inhibitors (97%)16 Adult (86%)16 Pediatric (14%)16 | 2,512 / 2,553 / 2,595 2,437 / 2,477 / 2,517 2,092 / 2,122 / 2,153 341 / 345 / 350 |
Number of patients eligible for Altuviiio (excluding Quebec) On demand Prophylaxis | 1,938 / 1,970 / 2,002 1,536 / 1,561 / 1,587 402 / 409 / 415 |
Annual bleeds: prophylaxis4 | |
Altuviiio SHLs EHLs Emicizumab | 0.71 3.73 2.46 2.22 |
Annual bleeds: on demand | |
Mild Moderate Severe Adult weighted average Pediatric weighted average | ████ 10 40 ███████████ ███████████ |
Market uptake (3 years)a | |
Uptake (reference scenario) SHLs EHLs Emicizumab | 15% / 12% / 10% 22% / 22% / 22% 63% / 66% / 68% |
Uptake (new drug scenario) Altuviiio SHLs EHLs Emicizumab | 10% / 15% 20% 11.5% / 7.5% / 5% 16.5% / 13% / 9% 62% / 64.5% / 66% |
Cost of treatment per patient, per annum (prophylaxis treatment / on-demand treatment)b | |
Altuviiio SHLs EHLs Emicizumab (initial year) Emicizumab (subsequent years) | $680,088 / $203,134 $457,644 / $138,294 $703,509 / $430,491 $810,025 / $430,491 $752,313 / $430,491 |
EHL = extended half-life ; SHL = standard half-life.
aFor the on-demand market, given that emicizumab is not indicated for on-demand therapy, it was assumed all of emicizumab’s market share would incur EHLs cost.
bWeighted average between pediatric and adult yearly costs.
Table 17: Sponsor’s Estimations and Assumptions on Population Size, Disease Severity, Treatment Regimen (Reference Scenario)
Condition | Patient Flow | Patient Numbers | |||
|---|---|---|---|---|---|
Age | Severity | Approach to Therapy | Proportion Treated | Base Year | |
Hemophilia A (without FVIII Inhibitors) | Pediatric (< 12) 14% | Severe 60% | Prophylaxis ██% | 100% | 131 |
On demand ██% | 100% | 70 | |||
Moderate 10% | Prophylaxis ██% | 80% | 17 | ||
On demand ██% | 100% | 12 | |||
Mild 30% | Prophylaxis ██% | 0% | 0 | ||
On demand ██% | 100% | 35 | |||
Adult (≥ 12) 86% | Severe 29% | Prophylaxis ████% | 100% | 194 | |
On demand ████% | 100% | 404 | |||
Moderate 10% | Prophylaxis ████% | 80% | 53 | ||
On demand ████% | 100% | 139 | |||
Mild 61% | Prophylaxis ████% | 0% | 0 | ||
On demand ████% | 100% | 850 | |||
Total Number of Patients Eligible for Altuviiio, Per Year | 1,907 | ||||
FVIII = factor VIII.
Note: figures may not add up due to rounding.
Source: Sponsor’s submitted BIA model.18
Summary of the Sponsor’s BIA Results
The sponsor estimated the 3-year budget impact of reimbursing Altuviiio for the treatment of adults, adolescents and children with hemophilia A (congenital factor VIII deficiency) without inhibitors for: routine prophylaxis to prevent or reduce the frequency of bleeding episodes; treatment and control of bleeding episodes; and perioperative management of bleeding (surgical prophylaxis) to result in cost savings of $93,521,203 over the first 3 years (Year 1: $17,124,878; Year 2: $30,051,621; and Year 3: $46,344,704).
The estimated 3-year budget impact for prophylaxis alone resulted in an incremental budget impact of $7,383,381 over the first 3 years (Year 1: $2,089,714, Year 2: $2,664,518 and Year 3: $2,629,149).
The estimated 3-year budget impact for on-demand treatment resulted in cost savings of $100,904,584 over 3 years (Year 1: $19,214,529, Year 2: $32,716,139 and Year 3: $48,973,853).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Uncertainty in the cost of comparators: The price of FVIII comparators was derived from the previous CDA-AMC Hemlibra review.16 As the previous submission noted as a limitation, estimated prices were based on the published list prices of comparators in the PMPRB basket. The price of these comparators does not reflect any confidential pricing that may have been negotiated by the Canadian Blood Services. As such, the estimated drug acquisition costs are uncertain.
CDA-AMC was unable to fully address this limitation. To explore the uncertainty surrounding comparator costs, 2 scenario analyses were conducted to reduce the cost of emicizumab by 22.2% and 90%.
Uncertainty in ABRs and drug costs associated with bleeds: In the sponsor-submitted model, for the on-demand treatment, ABR was assumed to be ████ bleeds for mild disease, 10 bleeds for moderate, and 40 bleeds for severe disease. The average of these bleeds was then multiplied by the proportion of severity for both the adult and pediatric groups. This resulted in a weighted ABR of approximately █████ for adults and █████ for pediatrics. However, the XTEND-1 trial, which included an on-demand treatment group, noted that on-demand ABR was 21.41.4 Notably, only severe hemophilia A patients without inhibitors were included in the XTEND-1 trial. Therefore, because an ABR of 40 bleeds was assumed for severe patients, the overall average of █████ is overestimated. It is reasonable to assume that in turn, the ABR for pediatrics and less severe patients are also overestimated.
Additionally, the drug costs to treat breakthrough bleeds for patients on prophylaxis were not included in the sponsors estimates. The XTEND-1 and XTEND-Kids trial data, which was used to estimate ABR and patient characteristics for patients receiving prophylaxis, only included severe hemophilia A patients without inhibitors. As the study population in XTEND-1 had greater uncontrolled bleeding than would be expected in Canadian practice across all severities, this may overstate the clinical efficacy of Altuviiio compared to what would be expected in the clinical setting, for patients on prophylaxis.
CDA-AMC conducted a base-case reanalysis which included the drug costs of treating breakthrough bleeds for patients on prophylaxis. CDA-AMC conducted scenario analyses exploring the use of Altuviiio limited to severe patients and reducing the on-demand ABR by half, reflecting the values reported in severe patients in the XTEND-1 trial.4
Uncertainty in the number of patients allocated across current treatment paradigms: The sponsor estimated the proportions of patients, by age group and severity, who would be currently receiving prophylaxis versus on-demand therapy based on a weighted average from the previous CDA-AMC Hemlibra review. In the review, it was noted that the proportions were based on clinical expert opinion and were uncertain, as the distribution of treatment is not publicly available. Notably, the prevalence of hemophilia A was captured from the Canadian Bleeding Disorders Registry (CBDR) that noted patients living with hemophilia A as of 2021. A more recent report from 2023 is available showing a greater number of existing patients than estimated by the sponsor and a different severity distribution among pediatric patients.25 Feedback from the Canadian Blood Services noted that the eligible population is likely higher than what was estimated by the sponsor and that the proportion of patients treated with moderate disease for both the adult and pediatric population is uncertain.
All severities of disease were modelled in the submitted BIA. However, when calculating the total number of patients eligible through the patient flow, the sponsor-submitted BIA removed 20% of adult patients with moderate disease assigned to prophylaxis (i.e., assuming they would not be actively managed but not included among the number of patients incurring on-demand costs). Additionally, CDA-AMC clinical experts noted that preference for prophylaxis or on-demand therapy varies across disease severity and age groups.
CDA-AMC conducted a base-case reanalysis re-estimating the number of eligible patients by adjusting the hemophilia A prevalence and severity distribution (aligned with the CBDR 2023 report); and assumptions to the % of patients opting for prophylaxis or on-demand treatment according to their disease severity (based on clinical expert input). Several scenario analyses were conducted to explore the uncertainties surrounding treatment options by disease severity and target population size including: a 25% increase in the target population, alternative proportion of patients treated on demand or prophylactically suggested by the Canadian Blood Services, and limited reimbursement to severe patients only.
Wastage was not included in the base case and was inappropriately modelled in scenario analysis: In the sponsor’s submitted BIA base case, treatment was dispensed according to its respective product monographs with treatment acquisition costs calculated based on the exact dose (per mg or IU) required. Therefore, the sponsor’s base case underestimated treatment acquisition cost for both Altuviiio and comparators. How treatments are dispensed needs to be accounted for in the cost of treatment. While a model functionality to include wastage is available, this option does not account for the lowest priced vial combination. When wastage was considered in the BIA model, the sponsor rounded the total dose according by the number of the lowest priced vials. This is inaccurate as it implies that patients will only receive a combination of the smallest vial size, which overestimates wastage across all comparators and therefore the relative cost savings. According to the CDA-AMC clinical experts, patients on both Altuviiio and other FVIII comparators would typically have their dose rounded to the nearest whole vial (up or down) with drug dispensed accordingly to minimize wastage.
Due to limitations in the sponsor-submitted model structure, assuming accurate wastage was not possible. Therefore, CDA-AMC was unable to conduct a reanalysis to assess this limitation.
Market shares in the reference scenario did not align with clinical expectations: The sponsor assumed that, to account for emicizumab not being indicated for on-demand use, the market shares allocated for emicizumab would incur EHLs costs in the on-demand subgroup. Clinical expert feedback obtained by CDA-AMC noted that this market capture is uncertain and likely overestimates the proportion of on-demand patients that would receive treatment with EHL. Clinical expert feedback obtained by CDA-AMC suggested that the sponsors market shares for the reference scenario are reasonable to represent the prophylactic use, however, for on-demand treatment, the experts noted that approximately 25% of patients remain on SHLs and the remaining 75% on EHLs.
CDA-AMC conducted a base-case reanalysis with distinct market shares for the reference scenario for on-demand treatment to reflect clinical expert opinion.
Uncertainty regarding uptake of Altuviiio among eligible patients across current treatment paradigms: In the sponsor-submitted BIA base case, the uptake of Altuviiio was not differentiated between prophylactic and on-demand use. The uptake of Altuviiio was assumed to be 10%, 15%, and 20% in years 1, 2, and 3, respectively, for both the prophylaxis and on-demand use. Feedback obtained from clinical experts consulted by CDA-AMC noted that Altuviiio is expected to capture a different proportion of market depending on the treatment type. For the prophylaxis group, the clinical experts agreed with a 10% and 15% market capture in years 1 and 2, however the experts predicted that the single dose per week will entice patients that are on other treatments to switch onto Altuviiio, increasing the market capture to 30% by year 3. Additionally, the experts expect that the introduction of Altuviiio will virtually eliminate the use of SHL over 3 years, prophylactic or on demand. The experts noted that they expect the uptake of Altuviiio among those patients opting for on-demand treatment to be significantly higher, with 40%, 60%, and 80% of on-demand patients using Altuviiio in years 1, 2, and 3, respectively.
In the sponsor submission, patients do not move between treatment type (e.g., on-demand patients cannot become prophylaxis patients). Clinical expert opinion suggested that a proportion of patients currently choosing on-demand treatment would opt to switch to prophylaxis if Altuviiio were reimbursed. Therefore, the sponsor submission may overestimate the proportion of patients that would continue to be treated on demand over time and consequently overestimating the cost savings from on-demand use.
CDA-AMC conducted a base-case reanalysis, which considered distinct market shares and uptake of Altuviiio in the new drug scenario between prophylaxis and on-demand use to reflect clinical expert opinion; and scenario analysis with alternative market uptake from input from the Canadian Blood Services.
Due to limitations in the sponsor-submitted model structure, estimating the budget impact of a proportion of patients assigned to on-demand treatment transitioning to prophylaxis was not possible. Therefore, CDA-AMC was unable to conduct a reanalysis to assess this limitation.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submission by including breakthrough bleeds on prophylaxis, aligning the number of the hemophilia A patients with the 2023 CBDR data, updating market shares in the reference scenario to differ between prophylaxis and on-demand use, and increasing the market uptake of Altuviiio in both the prophylactic and on-demand treatment groups.
Table 18: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Cost of breakthrough bleeds for patients on prophylaxis | Not included | Included |
2. Derivation of n. eligible patients to treatment type and by severity | Proportion of patients with hemophilia A: 0.0081 Pediatric severity distribution mild: 30% severe: 60% Proportion of moderate adult patients in prophylaxis assumed to be treated: 80% Proportion of patients opting for prophylaxis (same across all severities): Adults: 32.4% Pediatrics: 65% Proportion of patients opting for on-demand therapy (same across all severities): Adults: ████% Pediatrics: ██% | Proportion of patients with hemophilia A: 0.0115 Pediatric severity distribution mild: 37% severe: 53% Proportion of moderate adult patients in prophylaxis assumed to be treated: 100% Proportion of patients opting for prophylaxis by severity (mild, moderate severe) Adult: 5%, 80%, 100% Pediatrics: 5%, 50%, 95% Proportion of patients opting for on-demand treatment by severity (mild, moderate severe) Adult: 95%, 20%, 0% Pediatrics: 95%, 50%, 5% |
3. Market shares on reference scenario | Prophylaxis and On demand Year 1 / Year 2 / Year 3 EHLs: 22% / 22% / 22% SHLs: 15% / 12% / 10% Emicizumab: 63% / 66% / 68%a | Distinct market shares by prophylaxis and on-demand treatment. SHL market increased for on-demand treatment to align with clinical expert feedback. Prophylaxis: Year 1 / Year 2 / Year 3 EHLs: 22% / 22% / 22% SHLs: 15% / 12% / 10% Emicizumab: 63% / 66% / 68% (remain the same as assumed by sponsor) On demand: Year 1 / Year 2 / Year 3 EHLs: 75% / 75% / 75% SHLs: 25% / 25% / 25% Emicizumab: 0% / 0% / 0% |
4. Market shares in the new drug scenario | Prophylaxis and On demand: Year 1 / Year 2 / Year 3 Altuviiio: 10% / 15% / 20% SHL: 11.5% / 7.5% / 5% EHL: 16.5% / 13% / 9% Emicizumab: 62% / 64.5% / 66%a | Distinct market shares by prophylaxis and on-demand treatment Prophylaxis: Year 1 / Year 2 / Year 3 Altuviiio: 10% / 15% / 30% SHL: 10% / 7% / 0% EHL: 20% / 19% /15% Emicizumab: 60% / 59% / 55% On demand: Year 1 / Year 2 / Year 3 Altuviiio: 40% / 60% / 80% EHL: 50% / 35% / 20% SHL: 10% / 5% / 0% Emicizumab: 0% / 0% / 0% |
CDA-AMC base case | reanalysis 1 + 2 + 3 + 4 | |
CBDR = Canadian Bleeding Disorders Registry; CDA-AMC = Canada's Drug Agency; EHL = extended half-life; SHL = standard half-life.
aPatients assigned to on-demand treatment with emicizumab incurred EHL costs in the sponsor’s base case
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 19, and a more detailed breakdown is presented in Table 20. Applying these changes increase the 3-year cost savings to $471,472,317 ($87,462,805 in year 1, $158,553,584 in year 2, and $225,455,930 in year 3) and becomes cost savings in both prophylaxis ($6,549,544) and on-demand setting ($464,922,774).
Table 19: Summary of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total ($) | On demand | Prophylaxis |
|---|---|---|---|
Submitted base case | −93,521,203 | −100,904,584 | 7,383,381 |
CDA-AMC reanalysis 1 | −102,033,200 | −100,904,584 | −1,128,616 |
CDA-AMC reanalysis 2 | −102,813,124 | −127,959,526 | 25,146,401 |
CDA-AMC reanalysis 3 | 80,220,570 | 72,837,189 | 7,383,381 |
CDA-AMC reanalysis 4 | −359,676,002 | −366,821,906 | 7,145,904 |
CDA-AMC base case | −471,472,317 | −464,922,774 | −6,549,544 |
BIA = budget impact analysis; CDA-AMC = Canada's Drug Agency.
CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 20):
Assuming no breakthrough bleeds for prophylactic patients.
Assuming only severe patients would be eligible for reimbursement, to align with the XTEND-1 and XTEND-Kids trials.
Assuming a 25% increase in the proportion of patients eligible for treatment.
Assuming a 50% decrease in on-demand ABR, to align with the XTEND-1 trial.
Assuming a price reduction of 22.2% for emicizumab (similar price reduction for Altuviiio) from the cost-utility analysis based on public available prices includes all severities.
Assuming alternative proportion of patients opting for prophylaxis and on-demand treatment by severity (mild, moderate, severe) from input from the Canadian Blood Services:
Prophylaxis adult: 5%, 50%, 95%
On demand adult: 95%, 50%, 5%
Prophylaxis pediatrics: 5%, 65%, 95%
On demand pediatrics: 95%, 35%, 5%
Assuming alternative market shares for the new drug scenario (Year 1 / Year 2 / Year 3) from input from the Canadian Blood Services:
Prophylaxis:
Altuviiio: 10% / 20% / 30%,
SHL: 5% / 3% / 2%,
EHL: 20% / 15% / 8%,
emicizumab: 65% / 62% / 60%.
On demand:
Altuviiio: 15% / 25% / 35%,
SHL:30% / 25% / 20%,
EHL:55% / 50% / 45%.
Assuming a price reduction of 90% for emicizumab (exploratory price reduction suggested from the Hemlibra CDA-AMC pharmacoeconomic review) – includes all severities
Assuming reimbursement is limited to severe patients, a 50% decrease in on-demand ABR, and a price reduction of 90% for emicizumab (SA 2 + SA 4 + SA8 combined)
CDA-AMC base-case and scenario analyses indicate that the BIA results are highly uncertain and sensitive to the inclusion of breakthrough bleed costs among the prophylaxis costs (i.e., making prophylaxis cost savings), the reimbursement of Altuviiio to mild and moderate patients, and the price of comparators. As a higher proportion of mild and moderate patients are likely to be opting for on-demand treatment, the majority of the costs savings are estimated to come from the use of Altuviiio in the on-demand settings. These cost savings may be overestimated. CDA-AMC noted that the ABRs assumed in the model for patients on-demand treatment seemed to be overestimated when compared to the ABRs observed in on-demand arm of the XTEND-1 trial (severe patients). Additionally, the cost savings estimated from breakthrough bleeds avoided with prophylaxis were assumed the same across severities and based on the ABRs derived from severe patients, which were also likely overestimated as per the CDA-AMC clinical review. Finally, the BIA results are sensitive to the price of the comparators. In scenarios assuming reduced price of emicizumab (22% and 90%, including all severities), the reimbursement of Altuviiio was estimated to have an incremental budget impact to the drug plans ranging from $50 to $221 million over 3 years for prophylactic use but estimated to be offset by savings from on-demand use across all severities. CDA-AMC notes that the cost savings predicted by the model may be overestimated, as they rely on prophylaxis bleed rates from severe patients for which the magnitude of benefit is highly uncertain as is the assumption that these rates remain stable over the patient’s lifetime; and on-demand bleed rates seem overestimated compared to available evidence from severe patients (on-demand arm of XTEND-1 trial). Therefore, it may overstate the clinical efficacy of Altuviiio across all severities and treatment type, but especially for mild and moderate patients for which evidence is lacking.
In a multivariate scenario analysis (scenario 9) in which reimbursement of Altuviiio is aligned with the trial (i.e., only considered for severe patients and with bleed rates decreased by 50% for those treated on demand) and assuming lower confidential prices for emicizumab (by 90% as per Hemlibra submission24) the costs savings from on-demand use no longer offset the incremental costs with prophylaxis. Altuviiio is estimated to result in a 3-year budget impact of approximately $177 million if only reimbursed for patients with severe hemophilia A for prophylaxis and on-demand treatment.
Table 20: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Reanalysis | Scenario | Treatment type | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|---|
Submitted base case | Reference | Combined | 843,446,630 | 875,221,222 | 906,886,894 | 933,255,586 | 2,715,363,702 |
Prophylaxis | 272,337,669 | 281,279,354 | 289,516,727 | 296,485,159 | 867,281,239 | ||
On demand | 571,108,961 | 593,941,869 | 617,370,167 | 636,770,427 | 1,848,082,463 | ||
New drug | Combined | 843,446,630 | 858,096,345 | 876,835,273 | 886,910,882 | 2,621,842,500 | |
Prophylaxis | 272,337,669 | 283,369,067 | 292,181,245 | 299,114,308 | 874,664,620 | ||
On demand | 571,108,961 | 574,727,277 | 584,654,028 | 587,796,574 | 1,747,177,879 | ||
Budget impact | Combined | 0 | −17,124,878 | −30,051,621 | −46,344,704 | −93,521,203 | |
Prophylaxis | 0 | 2,089,714 | 2,664,518 | 2,629,149 | 7,383,381 | ||
On demand | 0 | −19,214,592 | −32,716,139 | −48,973,853 | −100,904,584 | ||
CDA-AMC base case | Reference | Combined | 1,689,367,580 | 1,733,234,696 | 1,774,938,122 | 1,812,243,116 | 5,320,415,934 |
Prophylaxis | 999,568,510 | 1,032,120,961 | 1,062,324,130 | 1,087,940,230 | 3,182,385,322 | ||
On demand | 689,799,070 | 701,113,735 | 712,613,992 | 724,302,886 | 2,138,030,613 | ||
New Drug | Combined | 1,689,367,580 | 1,645,771,890 | 1,616,384,540 | 1,586,787,186 | 4,848,943,617 | |
Prophylaxis | 999,568,510 | 1,036,325,420 | 1,058,068,150 | 1,081,442,208 | 3,175,835,778 | ||
On demand | 689,799,070 | 609,446,470 | 558,316,390 | 505,344,979 | 1,673,107,839 | ||
Budget impact | Combined | 0 | −87,462,805 | −158,553,582 | −225,455,930 | −471,472,317 | |
Prophylaxis | 0 | 4,204,459 | −4,255,980 | −6,498,023 | −6,549,544 | ||
On demand | 0 | −91,667,264 | −154,297,602 | −218,957,907 | −464,922,774 | ||
CDA-AMC scenario analysis 1 – no breakthrough bleeds | Reference | Combined | 1,617,329,698 | 1,659,098,051 | 1,698,653,244 | 1,734,075,301 | 5,091,826,596 |
Prophylaxis | 927,530,628 | 957,984,316 | 986,039,252 | 1,009,772,415 | 2,953,795,983 | ||
On demand | 689,799,070 | 701,113,735 | 712,613,992 | 724,302,886 | 2,138,030,613 | ||
New Drug | Combined | 1,617,329,698 | 1,576,680,735 | 1,548,568,854 | 1,525,991,833 | 4,651,241,421 | |
Prophylaxis | 927,530,628 | 967,234,264 | 990,252,464 | 1,020,646,854 | 2,978,133,582 | ||
On demand | 689,799,070 | 609,446,470 | 558,316,390 | 505,344,979 | 1,673,107,839 | ||
Budget impact | Combined | 0 | −82,417,316 | −150,084,390 | −208,083,469 | −440,585,174 | |
Prophylaxis | 0 | 9,249,948 | 4,213,212 | 10,874,439 | 24,337,599 | ||
On demand | 0 | −91,667,264 | −154,297,602 | −218,957,907 | −464,922,774 | ||
CDA-AMC scenario analysis 2 – no mild or moderate patients eligible | Reference | Combined | 811,629,882 | 838,004,564 | 862,480,956 | 883,249,965 | 2,583,735,486 |
Prophylaxis | 808,087,065 | 834,403,635 | 858,820,961 | 879,529,936 | 2,572,754,532 | ||
On demand | 3,542,817 | 3,600,930 | 3,659,995 | 3,720,029 | 10,980,954 | ||
New Drug | Combined | 811,629,882 | 840,927,397 | 858,239,403 | 876,860,679 | 2,576,027,479 | |
Prophylaxis | 808,087,065 | 837,802,670 | 855,380,274 | 874,276,701 | 2,567,459,645 | ||
On demand | 3,542,817 | 3,124,727 | 2,859,129 | 2,583,978 | 8,567,833 | ||
Budget impact | Combined | 0 | 2,992,833 | −4,241,553 | −6,289,286 | −7,708,007 | |
Prophylaxis | 0 | 3,399,036 | −3,440,687 | −5,253,235 | −5,294,886 | ||
On demand | 0 | −476,203 | −800,866 | −1,136,052 | −2,413,121 | ||
CDA-AMC scenario analysis 3 – 25% higher prevalence (all severities included) | Reference | Combined | 2,111,709,475 | 2,166,543,370 | 2,218,672,653 | 2,265,303,895 | 6,650,519,918 |
Prophylaxis | 1,249,460,637 | 1,290,151,201 | 1,327,905,163 | 1,359,925,288 | 3,977,981,652 | ||
On demand | 862,248,838 | 876,392,168 | 890,767,490 | 905,378,607 | 2,672,538,266 | ||
New Drug | Combined | 2,111,709,475 | 2,057,214,863 | 2,020,480,675 | 1,983,483,983 | 6,061,179,521 | |
Prophylaxis | 1,249,460,637 | 1,295,406,775 | 1,322,585,188 | 1,351,802,759 | 3,969,794,723 | ||
On demand | 862,248,838 | 761,808,088 | 697,895,487 | 631,681,223 | 2,091,384,799 | ||
Budget impact | Combined | 0 | −109,328,507 | −198,191,978 | −281,819,912 | −589,340,397 | |
Prophylaxis | 0 | 5,255,574 | −5,319,975 | −8,122,528 | −8,186,930 | ||
On demand | 0 | −114,584,080 | −192,872,002 | −273,697,384 | −581,153,467 | ||
CDA-AMC scenario analysis 4 – 50% decrease in on-demand ABR | Reference | Combined | 1,344,468,045 | 1,382,677,828 | 1,418,631,126 | 1,450,091,673 | 4,251,400,628 |
Prophylaxis | 999,568,510 | 1,032,120,961 | 1,062,324,130 | 1,087,940,230 | 3,182,385,322 | ||
On demand | 344,899,535 | 350,556,867 | 356,306,996 | 362,151,443 | 1,069,015,306 | ||
New Drug | Combined | 1,344,468,045 | 1,341,048,655 | 1,337,226,345 | 1,334,114,697 | 4,012,389,698 | |
Prophylaxis | 999,568,510 | 1,036,325,420 | 1,058,068,150 | 1,081,442,208 | 3,175,835,778 | ||
On demand | 344,899,535 | 304,723,235 | 279,158,195 | 252,672,489 | 836,553,920 | ||
Budget impact | Combined | 0 | −41,629,173 | −81,404,781 | −115,976,976 | −239,010,931 | |
Prophylaxis | 0 | 4,204,459 | −4,255,980 | −6,498,023 | −6,549,544 | ||
On demand | 0 | −45,833,632 | −77,148,801 | −109,478,954 | −232,461,387 | ||
CDA-AMC scenario analysis 5 – 22.2% price reduction for emicizumab | Reference | Combined | 1,554,376,665 | 1,588,473,163 | 1,620,820,672 | 1,651,045,379 | 4,860,339,214 |
Prophylaxis | 864,577,595 | 887,359,429 | 908,206,680 | 926,742,494 | 2,722,308,602 | ||
On demand | 689,799,070 | 701,113,735 | 712,613,992 | 724,302,886 | 2,138,030,613 | ||
New Drug | Combined | 1,554,376,665 | 1,508,396,882 | 1,479,258,752 | 1,457,408,288 | 4,445,063,922 | |
Prophylaxis | 864,577,595 | 898,950,411 | 920,942,362 | 952,063,309 | 2,771,956,083 | ||
On demand | 689,799,070 | 609,446,470 | 558,316,390 | 505,344,979 | 1,673,107,839 | ||
Budget impact | Combined | 0 | −80,076,282 | −141,561,919 | −193,637,092 | −415,275,293 | |
Prophylaxis | 0 | 11,590,983 | 12,735,682 | 25,320,815 | 49,647,481 | ||
On demand | 0 | −91,667,264 | −154,297,602 | −218,957,907 | −464,922,774 | ||
CDA-AMC scenario analysis 6 –alternative proportion of eligible patients by severity – from CBS | Reference | Combined | 1,641,351,495 | 1,682,954,129 | 1,722,619,486 | 1,758,317,570 | 5,163,891,185 |
Prophylaxis | 908,198,010 | 937,774,844 | 965,217,143 | 988,491,677 | 2,891,483,664 | ||
On demand | 733,153,485 | 745,179,285 | 757,402,343 | 769,825,893 | 2,272,407,521 | ||
New Drug | Combined | 1,641,351,495 | 1,589,345,635 | 1,554,757,217 | 1,519,693,947 | 4,663,796,798 | |
Prophylaxis | 908,198,010 | 941,594,973 | 961,350,202 | 982,587,638 | 2,885,532,813 | ||
On demand | 733,153,485 | 647,750,661 | 593,407,015 | 537,106,309 | 1,778,263,985 | ||
Budget impact | Combined | 0 | −93,608,495 | −167,862,269 | −238,623,623 | −500,094,387 | |
Prophylaxis | 0 | 3,820,130 | −3,866,941 | −5,904,039 | −5,950,850 | ||
On demand | 0 | −97,428,624 | −163,995,328 | −232,719,584 | −494,143,536 | ||
CDA-AMC scenario analysis 7 –market shares for new drug scenario from CBS | Reference | Combined | 1,689,367,580 | 1,733,234,696 | 1,774,938,122 | 1,812,243,116 | 5,320,415,934 |
Prophylaxis | 999,568,510 | 1,032,120,961 | 1,062,324,130 | 1,087,940,230 | 3,182,385,322 | ||
On demand | 689,799,070 | 701,113,735 | 712,613,992 | 724,302,886 | 2,138,030,613 | ||
New Drug | Combined | 1,689,367,580 | 1,667,913,345 | 1,667,914,090 | 1,671,291,914 | 5,007,119,348 | |
Prophylaxis | 999,568,510 | 1,061,978,015 | 1,068,061,766 | 1,077,885,182 | 3,207,924,963 | ||
On demand | 689,799,070 | 605,935,330 | 599,852,324 | 593,406,732 | 1,799,194,386 | ||
Budget impact | Combined | 0 | −65,321,351 | −107,024,033 | −140,951,202 | −313,296,586 | |
Prophylaxis | 0 | 29,857,054 | 5,737,635 | −10,055,048 | 25,539,641 | ||
On demand | 0 | −95,178,405 | −112,761,668 | −130,896,154 | −338,836,227 | ||
CDA-AMC scenario analysis 8 – 90% lower emicizumab price | Reference | Combined | 1,142,107,113 | 1,146,363,618 | 1,150,137,647 | 1,158,738,778 | 3,455,240,043 |
Prophylaxis | 452,308,043 | 445,249,883 | 437,523,655 | 434,435,892 | 1,317,209,430 | ||
On demand | 689,799,070 | 701,113,735 | 712,613,992 | 724,302,886 | 2,138,030,613 | ||
New Drug | Combined | 1,142,107,113 | 1,088,846,179 | 1,060,469,183 | 1,062,278,138 | 3,211,593,501 | |
Prophylaxis | 452,308,043 | 479,399,709 | 502,152,794 | 556,933,159 | 1,538,485,662 | ||
On demand | 689,799,070 | 609,446,470 | 558,316,390 | 505,344,979 | 1,673,107,839 | ||
Budget impact | Combined | 0 | −57,517,439 | −89,668,464 | −96,460,640 | −243,646,542 | |
Prophylaxis | 0 | 34,149,826 | 64,629,138 | 122,497,267 | 221,276,231 | ||
On demand | 0 | −91,667,264 | −154,297,602 | −218,957,907 | −464,922,774 | ||
CDA-AMC scenario analysis 9 – severe patients only, assuming a 50% decrease in on-demand ABR and 90% lower emicizumab price (SA2 + SA 4 + SA8 combined) | Reference | Combined | 367,433,467 | 361,756,453 | 355,539,826 | 353,073,585 | 1,070,369,864 |
Prophylaxis | 365,662,058 | 359,955,988 | 353,709,829 | 351,213,570 | 1,064,879,387 | ||
On demand | 1,771,409 | 1,800,465 | 1,829,998 | 1,860,015 | 5,490,477 | ||
New Drug | Combined | 367,433,467 | 389,126,297 | 407,387,909 | 451,563,747 | 1,248,050,952 | |
Prophylaxis | 365,662,058 | 387,563,933 | 405,958,344 | 450,244,758 | 1,243,767,035 | ||
On demand | 1,771,409 | 1,562,363 | 1,429,565 | 1,291,989 | 4,283,917 | ||
Budget impact | Combined | 0 | 27,369,844 | 51,848,082 | 98,463,162 | 177,681,088 | |
Prophylaxis | 0 | 27,607,945 | 52,248,515 | 99,031,188 | 178,887,649 | ||
On demand | 0 | −238,102 | −400,433 | −568,026 | −1,206,560 |
BIA = budget impact analysis; CBS = Canadian Blood Services; CDA-AMC = Canada's Drug Agency; SA = scenario analysis
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@cda-amc.ca.