Drugs, Health Technologies, Health Systems
Reimbursement Review
Tofacitinib
Requester: Public drug programs
Therapeutic area: Juvenile idiopathic arthritis, idiopathic arthritis
Summary
What Is Juvenile Idiopathic Arthritis?
Juvenile idiopathic arthritis (JIA) is a chronic inflammatory disorder primarily involving joints, with persistent joint swelling that can cause degradation of the articular cartilage and deformities of affected joints. JIA can be associated with chronic and acute uveitis. The existence of uveitis can be discordant with the activity of the joint disease. Uveitis can result in visual impairment if left untreated. Uveitis can be associated with a number of sequelae, even if treated, including glaucoma, band keratopathy, and cataracts. Patients living with JIA experience physical challenges that negatively affect their functioning, ability to participate in work, activities of daily living and recreation, and mental health.
For the fiscal year 2022–2023, it was estimated that 6,545 children aged 0 to 15 years were living with JIA in Canada (age-standardized prevalence rate of 0.10%) and 865 children in this age group were newly diagnosed with JIA (age-standardized incidence rate of 13 per 100,000 persons per year).
What Are the Treatment Goals and Current Treatment Options for Juvenile Idiopathic Arthritis?
Treatment options with improved forms of administration that can reduce disease symptoms, enable participation in activities of daily living and recreation, and reduce parent or caregiver burden were identified as important in patient group inputs. Goals of treatment identified through clinician inputs include controlling inflammation, reducing pain, preserving or improving physical function, preventing joint damage, achieving inactive disease, prolonging clinical remission, supporting physical growth and mental development, and restoring health-related quality of life.
Tumour necrosis factor (TNF) inhibitors, abatacept, and tocilizumab were considered relevant treatments to compare with tofacitinib.
What Is Tofacitinib and Why Did Canada’s Drug Agency Conduct This Review?
Tofacitinib is an oral medication that treats inflammation in patients with autoimmune diseases including inflammatory arthritis, such as JIA. Health Canada has approved tofacitinib for the treatment of active polyarticular juvenile idiopathic arthritis (pJIA; rheumatoid factor positive [RF-positive] or negative [RF-negative] polyarthritis, extended oligoarthritis, and systemic JIA [sJIA] without systemic manifestations), and juvenile psoriatic arthritis (JPsA) in children weighing ≥ 40 kg, who have responded inadequately or are intolerant to tumour necrosis factor (TNF) inhibitors or when use of those therapies is inadvisable [wording from original source].
At the request of the participating public drug programs, Canada’s Drug Agency (CDA-AMC) reviewed tofacitinib (with or without methotrexate) to inform a recommendation on whether it should be reimbursed for the treatment of active pJIA (RF-positive or RF-negative polyarthritis, extended oligoarthritis, and sJIA without systemic manifestations), JPsA, and enthesitis-related arthritis (ERA) in children who have responded inadequately or are intolerant to TNF inhibitors or when use of those therapies is inadvisable [wording from original source].
How Did CDA-AMC Evaluate Tofacitinib?
CDA-AMC reviewed the clinical evidence on the beneficial and harmful effects of tofacitinib and compared costs of tofacitinib (with or without methotrexate) versus other treatments used in Canada for active pJIA (RF-positive or RF-negative polyarthritis, extended oligoarthritis, and sJIA without systemic manifestations), JPsA, and ERA in children who have responded inadequately or are intolerant to TNF inhibitors or when use of those therapies is inadvisable [wording from original source]. TNF inhibitors, abatacept, and tocilizumab were considered relevant treatments to compare with tofacitinib.
The clinical evidence was identified through systematic searches of databases and grey literature for available studies.
The review was also informed by 2 patient group submissions and 1 clinician group submission in response to a CDA-AMC call for input and by input from the participating public drug programs around issues that may affect their ability to implement a recommendation.
Two pediatric rheumatologists with representation from the Prairies were consulted as part of the review process.
What Were the Findings?
Clinical Evidence
CDA-AMC reviewed the following clinical evidence:
one randomized withdrawal phase III trial comparing tofacitinib with placebo in 225 patients with polyarticular course JIA (pcJIA) (RF‑positive or RF-negative polyarthritis, extended oligoarthritis, and sJIA without systemic manifestations), JPsA, or ERA
one long-term extension study of the phase III trial
one network meta-analysis of tofacitinib and biologic disease-modifying antirheumatic drugs (e.g., adalimumab, etanercept, golimumab, infliximab, abatacept, tocilizumab) with or without methotrexate.
Based on evidence without a comparison:
Findings during the 18-week run-in period of the phase III trial (study part 1) were at risk of bias and had limited interpretability for efficacy and harms based on the open-label noncomparative design.
Findings from an additional 48 weeks of tofacitinib treatment in the long-term extension study were supportive of those in the phase III trial; however, the open-label study design limited the ability to distinguish effects of treatment from concomitant therapies, the natural history of the disease, or other factors.
For the comparison of tofacitinib versus placebo:
The results suggest a difference between treatments, with tofacitinib showing a greater improvement in JIA flare rate by week 44; greater improvements in rates of JIA–American College of Rheumatology (ACR) 30, 50, and 70 responses (criteria used to measure at least 30%, 50%, and 70% improvement from baseline, respectively, in at least 3 of 6 core set variables of JIA, with no more than 1 core set variable worsened by 30% or more) at week 44; and greater improvements in Childhood Health Assessment Questionnaire Disability Index scores from baseline in the double-blind withdrawal phase (study part 2) to week 44 for patients with pcJIA.
The results suggest a difference between treatments, with tofacitinib showing a greater improvement in the JIA-ACR physician’s global evaluation of overall disease activity at week 44; in the JIA-ACR assessment of number of joints with active arthritis from study part 2 baseline to week 44; in the JIA-ACR patient or parent assessment of overall well-being from study part 2 baseline to week 44; and in the 27-point Juvenile Arthritis Disease Activity Score (JADAS-27), C-reactive protein levels, and erythrocyte sedimentation rate scores at week 44 for patients with pcJIA.
The results do not suggest a difference between treatment groups in the JIA-ACR assessments of inactive disease rate and JIA-ACR clinical remission rate at week 44; in the JADAS-27 minimum disease activity rate and JADAS-27 inactive disease rate at week 44; in the JIA-ACR assessment of number of joints with limitation of motion from part 2 baseline to week 44; in patient or parent assessments of bodily pain from part 2 baseline to week 44 using the Child Health Questionnaire; or in patient or parent assessments of global health or of physical functioning from part 2 baseline to week 44 using the Child Health Questionnaire for patients with pcJIA.
The evidence was insufficient to demonstrate a difference between treatment groups for JIA flare rate or JADAS-27 scores at week 44 for patients with JPsA or ERA.
For the comparison of tofacitinib versus biologic disease-modifying antirheumatic drugs:
The results do not suggest differences between treatment groups in JIA-ACR 70 response rates at week 16 or in serious adverse event incidence rates for patients with all subtypes of nonsystemic JIA (excluding sJIA with active systemic features).
There was no evidence to inform how tofacitinib compares with other available treatments (e.g., TNF inhibitors, abatacept, or tocilizumab) on the occurrence of flare, occurrence of remission, pain, health-related quality of life, or parent or caregiver burden.
The safety profile of tofacitinib was as expected with no new safety signals in patients with JIA.
Economic Evidence
Reimbursing tofacitinib for active pJIA (RF-positive or RF-negative polyarthritis, extended oligoarthritis, and sJIA without systemic manifestations), JPsA, and ERA in children who have responded inadequately or are intolerant to TNF inhibitors or when use of those therapies is inadvisable [wording from original source] is expected to decrease costs to the public drug programs.
Abbreviations
ACE
Arthritis Consumer Experts
ACR
American College of Rheumatology
AE
adverse event
AESI
adverse event of special interest
bDMARD
biologic disease-modifying antirheumatic drug
CDA-AMC
Canada’s Drug Agency
CDEC
Canadian Drug Expert Committee
CHAQ
Childhood Health Assessment Questionnaire
CHAQ-DI
Childhood Health Assessment Questionnaire Disability Index
CHQ
Child Health Questionnaire
CI
confidence interval
CrI
credible interval
CRP
C-reactive protein
csDMARD
conventional synthetic disease-modifying antirheumatic drug
DMARD
disease-modifying antirheumatic drug
ERA
enthesitis-related arthritis
ESR
erythrocyte sedimentation rate
HRQoL
health-related quality of life
IL
interleukin
ILAR
International League of Associations for Rheumatology
IQR
interquartile range
ITC
indirect treatment comparison
JADAS
Juvenile Arthritis Disease Activity Score
JAK
Janus kinase
JIA
juvenile idiopathic arthritis
JPsA
juvenile psoriatic arthritis
LTE
long-term extension
MACE
major adverse cardiovascular event
NMA
network meta-analysis
NSAID
nonsteroidal anti-inflammatory drug
pcJIA
polyarticular course juvenile idiopathic arthritis
pediACR
Pediatric American College of Rheumatology
pJIA
polyarticular juvenile idiopathic arthritis
RA
rheumatoid arthritis
RCT
randomized controlled trial
RF
rheumatoid factor
SAE
serious adverse event
SC
subcutaneous
sJIA
systemic juvenile idiopathic arthritis
TEAE
treatment-emergent adverse event
TNF
tumour necrosis factor
Background
Introduction
The objective of the Clinical Review is to review and critically appraise the evidence on any beneficial and harmful effects of tofacitinib 5 mg oral tablets in the treatment of active polyarticular juvenile idiopathic arthritis (pJIA) (rheumatoid factor [RF]–positive or RF-negative polyarthritis, extended oligoarthritis, and systemic JIA [sJIA] without systemic manifestations), juvenile psoriatic arthritis (JPsA), and enthesitis-related arthritis (ERA) in children who have responded inadequately or are intolerant to tumour necrosis factor (TNF) inhibitors or when use of those therapies is inadvisable [wording from original source]. The focus will be placed on comparing tofacitinib to relevant comparators in clinical practice in Canada and identifying gaps in the current evidence. The Economic Review consists of a cost comparison of tofacitinib and relevant comparators for the same population. The comparators considered relevant to the reviews were biologic disease-modifying antirheumatic drugs (bDMARDs) (e.g., TNF inhibitors such as etanercept and adalimumab, interleukin [IL]-6 inhibitors such as tocilizumab, and T-cell co-stimulation blockers such as abatacept).
Table 1: Information on the Drug Under Review and on the CDA-AMC Review
Item | Description |
|---|---|
Information on the drug under review | |
Drug | Tofacitinib 5 mg and 10 mg oral tablets |
Relevant Health Canada indication | Tofacitinib is indicated for the treatment of active polyarticular JIA (pJIA; rheumatoid factor positive [RF+] or negative [RF-] polyarthritis, extended oligoarthritis, and systemic JIA without systemic manifestations), and juvenile psoriatic arthritis (JPsA) in children weighing ≥ 40 kg, who have responded inadequately or are intolerant to tumour necrosis factor (TNF) inhibitors or when use of those therapies is inadvisable. |
Mechanism of action | Targeted synthetic disease-modifying antirheumatic drug; potent selective inhibitor of the JAK family of kinases, particularly JAK1 and JAK3, thereby decreasing associated inflammation and symptoms |
Recommended dosage per Health Canada indication | Active JIA:
|
Data protection status | October 17, 2022 |
Status of generic drugs | Several generic formulations of tofacitinib are currently marketed in Canada. |
Information on the CDA-AMC review | |
Requester | Formulary Working Group |
Indication under consideration for reimbursement | For the treatment of active pJIA (RF-positive or RF-negative polyarthritis, extended oligoarthritis, and systemic JIA without active systemic manifestations), psoriatic arthritis, and enthesitis-related arthritis in children who have responded inadequately or are intolerant to TNF inhibitors or when use of those therapies is inadvisable |
CDA-AMC = Canada’s Drug Agency; JAK = Janus kinase; JIA = juvenile idiopathic arthritis; JPsA = juvenile psoriatic arthritis; pJIA = polyarticular juvenile idiopathic arthritis; RF = rheumatoid factor; TNF = tumour necrosis factor.
Context for the Review
The following drugs were reviewed by the Canadian Drug Expert Committee (CDEC) for the indicated population with JIA and received a final positive recommendation for reimbursement:
abatacept (Orencia): moderate to severe active pJIA in patients aged 6 years or older whose disease has responded inadequately to 1 or more disease-modifying antirheumatic drugs (DMARDs) such as methotrexate
adalimumab (Humira): pJIA, inadequate response to 1 or more DMARDs
canakinumab (Ilaris): active sJIA in patients aged 2 years or older whose disease has responded inadequately or who are intolerant to oral steroids or methotrexate; treatment to be discontinued if there is no improvement after day 15
etanercept (Erelzi): rheumatoid arthritis (RA), pJIA, and ankylosing spondylitis
tocilizumab (Actemra): pJIA, inadequate response to 1 or more DMARDs.
Anakinra, golimumab, infliximab, rilonacept, rituximab, and secukinumab (for active JPsA) have not been reviewed by CDEC for JIA. Anakinra and canakinumab are IL-1 blockers that are used specifically for patients with sJIA with active systemic features only; therefore, they are inappropriate for the requested reimbursement population. According to the clinical experts consulted, infliximab, golimumab, and secukinumab are used off-label for the treatment of JIA. Certolizumab pegol is not routinely used for JIA in Canada. Certolizumab pegol and secukinumab have been approved for the treatment of adult patients with inflammatory arthritis.
Submission History for the Drug Under Review
Canada’s Drug Agency (CDA-AMC) previously reviewed tofacitinib through the Reimbursement Review process in combination with methotrexate for reducing the signs and symptoms of RA in adult patients with moderately to severely active RA or as monotherapy in those who were intolerant to methotrexate. CDA‑AMC issued a recommendation of reimburse with clinical criterion and conditions in April 2015.
In addition, the public drug programs via the Formulary Working Group have requested a review of tofacitinib as per the indication under Table 1 for patients who have responded inadequately or are intolerant to TNF inhibitors or when use of those therapies is inadvisable [wording from original source]. However, the drug programs have also noted that the requested indication may require further discussion during deliberation, depending on the available efficacy and safety evidence for the review and other related considerations based on clinical practice and unmet needs.
Sources of Information
The contents of the Clinical Review are informed by studies identified through systematic literature searches and input received from interested parties.
Calls for input from patient, clinician, and industry groups are issued for each Non-Sponsored Reimbursement Review. The following submissions were received: 2 patient group submissions from Arthritis Consumer Experts (ACE) and a combined submission (from Arthritis Society Canada, the Canadian Arthritis Patient Alliance, the Canadian Spondyloarthritis Association, the Cassie and Friends Society, and Psoriasis Canada) and 1 clinician group submission from the Canadian Rheumatology Association. ACE conducted multiple anonymous surveys on private health insurance, health inequities, exercise, self-advocacy, and arthritis medications between May 2021 and July 2024 and summarized input from 7 respondents living with JIA. The combined patient group submission was based on an international survey that was conducted by the Canadian Arthritis Patient Alliance and the Take a Pain Check Foundation in 2022 of 56 youth and young adults living with rheumatic disease. The Canadian Rheumatology Association sourced information from its repository of guidelines, position papers, previous submissions to CDA-AMC, a search of the Trip Database (for additional guidelines, syntheses, and primary evidence), a MEDLINE search (for complementary information), and additional references from experts. The full submissions received are available on the project landing page in the consolidated input document.
Input from patient and clinician groups is considered throughout the review, including in the selection of outcomes to include in the Clinical Review and in the interpretation of the clinical evidence. Relevant patient and clinician group input and industry input is summarized in the Disease Background, Current Management, and Unmet Needs and Existing Challenges sections.
The drug programs provide input on each drug being reviewed through the Reimbursement Review process by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized and provided to the expert committee in a separate document.
Each review team includes at least 1 clinical expert with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process. Two pediatric rheumatologists with expertise in the diagnosis and management of patients with JIA participated as part of the review team, with representation from the Prairies (Manitoba and Saskatchewan).
Disease Background
JIA is a chronic inflammatory disorder primarily involving joints. Pathogenesis is unclear but likely involves interactions among underlying genetic predispositions, immune dysregulation, and environmental exposures (e.g., viral infections).1 The cardinal clinical manifestation of JIA is persistent joint swelling that results from hypertrophy and synovial fluid accumulation.1 Active inflammation from synovitis can result in articular cartilage damage from the release of enzymes (e.g., metalloproteinase) that remodel the cartilage and bone of the joint. Bone demineralization and erosions, also cardinal radiologic signs of inflammatory arthritis, will result.1 If the disease is not adequately controlled, the cartilage would be completely destroyed, resulting in joint space narrowing and ultimately a bone-on-bone appearance. This can result in obliteration of the joint space (ankylosis, or bony fusion), leading to deformities. The anatomic derangements from cartilage destruction will further result in pain and functional limitations.
JIA is defined by the onset of arthritis (joint swelling or pain with limitation in the range of motion) before 16 years of age and persisting for at least 6 weeks’ duration (except for sJIA) in the absence of other medical causes.2 The International League of Associations for Rheumatology (ILAR) has outlined criteria for at least 7 mutually exclusive subtypes of JIA: sJIA, oligoarthritis, pJIA (RF-positive or RF-negative), JPsA, ERA, and undifferentiated.2,3 They are defined as follows:
sJIA is defined as the presence of arthritis in 1 or more joints and fever of at least 2 weeks’ duration (with at least 3 days of a quotidian pattern) with no other etiology (e.g., infections, other autoimmune and autoinflammatory disorders, malignancy, and malaria)4 or fever plus at least 1 or more of the following: evanescent rash, generalized lymphadenopathy, hepatomegaly and/or splenomegaly, and serositis.2 sJIA is associated with life-threatening complications including macrophage activation syndrome, pericarditis, and interstitial lung disease.3,5 sJIA is termed “adult-onset Still disease” when it begins in patients older than 16 years.4
Oligoarthritis (oligoarticular JIA) is defined as arthritis affecting fewer than 5 joints within the first 6 months of disease. Patients with a persistent course continue to have fewer than 5 affected joints throughout their disease course, while those with an extended course develop more extensive arthritis affecting 5 or more joints more than 6 months after diagnosis.2
pJIA is defined as arthritis affecting 5 or more joints during the first 6 months of disease. pJIA is subcategorized as RF-positive or RF-negative, determined by the presence or absence of immunoglobulin M RF antibodies in blood tests on at least 2 occasions 12 weeks apart).2
JPsA is defined as the simultaneous presence of arthritis and psoriasis or arthritis plus at least 2 of the following: dactylitis, nail pitting or onycholysis, and psoriasis in a first-degree relative.2
ERA requires dual presentation of arthritis and enthesitis, or arthritis or enthesitis plus at least 2 of the following: presence of or history of sacroiliac joint tenderness and/or inflammatory lumbosacral pain, HLA-B27 positivity, onset of arthritis in a male aged older than 6 years, acute anterior uveitis, and positive family history of HLA-B27 associated diseases in a first-degree relative. HLA-B27 associated diseases include ankylosing spondylitis, sacroiliitis with inflammatory bowel disease, Reiter syndrome, and acute anterior uveitis.2
Patients who meet criteria for multiple categories or who do not meet criteria for any category are classified as having undifferentiated arthritis.2
Although newer classification systems are being developed to better reflect the complexity and overlap among JIA subtypes, these criteria still require validation and have not been adopted in routine clinical care. The revised ILAR criteria remain the most widely used framework for classification in clinical practice in Canada.
The lack of standard diagnostic criteria has complicated epidemiologic studies of JIA.1 Proportions of cases represented by JIA subtypes are estimated to be: 10% to 20% as sJIA,3,4 40% to 50% as oligoarthritis (75% persistent and 25% extended oligoarticular course),3,6 20% to 30% as pJIA7 (15% to 20% as RF-negative and 5% as RF-positive pJIA),3 0% to 11% as JPsA,3,8 and 9% to 19% as ERA (33% in East Asia and Southeast Asia).3 JIA occurs with a peak incidence at 1 to 3 years of age and a greater ratio of females to males (e.g., 2:1 to 3:1); variations exist depending on age at onset and type of disease.1 The incidence of JIA may differ by race and ethnicity.1 Other than ERA being more prevalent in people of East Asian and Southeast Asian heritage, RF-positive pJIA is more common among Indigenous populations in Canada, among whom 50% of patients with pJIA have the RF-positive subtype.
For the fiscal year 2022–2023, it was estimated that 6,545 children aged between 0 and 15 years were living with JIA in Canada (age-standardized prevalence rate of 0.10%).9 A total of 865 children in this age group were newly diagnosed with JIA (age-standardized incidence rate of 13 per 100,000 persons per year), with higher rates in females (age-standardized incidence rate of 16 per 100,000 persons per year) than in males (age-standardized incidence rate of 10 per 100,000 persons per year).9
While JIA is a predominantly joint-limited disorder, 10% to 20% of patients also have concomitant uveitis (inflammation of the eyes). This could be a potentially blinding disease if undetected and untreated. Antinuclear antibody positivity is associated with an increased risk of chronic asymptomatic uveitis (80% to 90%). The child will not be able to discern the involvement of their eyes. The condition is usually detected on eye exams. The frequency of eye exams is personalized according to the patient’s level of risk based on their JIA subtype and antinuclear antibody positivity status. A smaller proportion of patients have acute symptomatic uveitis (10% to 20%). These patients present with a red and painful eye. Acute uveitis occurs almost exclusively in those with ERA and in some patients with psoriatic arthritis.
Patient Group Input
Due to their disease, patients living with JIA experience physical challenges that negatively affect their functioning; ability to participate in work, activities of daily living, and recreation; and mental health. Patients also expressed challenges related to accessing health care (e.g., access to rheumatologists, time and distance required for travel to clinic) and the need to balance health care needs with other priorities.
Current Management
Treatment Goals
Patient Group Input
Patients living with JIA and other rheumatic diseases and their caregivers want access to additional treatment options, particularly those with improved forms of administration (i.e., oral medication). They expressed that the goals of treatment should align with the Outcome Measures in Rheumatology (OMERACT) core outcome domains for JIA (e.g., reductions in symptoms such as pain, joint swelling), with patient-reported outcomes extending beyond generic health-related quality of life (HRQoL) measures to include school participation, engagement in social activities, and parent or caregiver burden. Patients and their parents or caregivers identified several barriers and factors as important for ensuring access to high-quality care. These included reducing ageism, reducing barriers to participation at school and work (e.g., improved processes for accommodations and information on how to access accommodations), having information on both pharmacologic and nonpharmacologic treatments (e.g., acupuncture, physiotherapy, occupational therapy), improving access to insurance coverage for prescription medications (e.g., universal drug coverage), and having extended health benefits, including in adulthood (e.g., postgraduation), greater support from public and private drug plans in terms of coverage processes and administrative requirements (e.g., correct forms and guidance to complete them for reimbursement), reduced medication costs, and improved access to rheumatologists.
Clinician Input
The clinician group indicated that the goal of treatment is to achieve inactive disease with full, pain-free function by controlling joint inflammation and pain, thereby improving daily functional ability in patients with JIA.
The clinical experts identified goals of treatment as controlling both systemic and extra-articular inflammation; preventing joint damage or deformity; preserving age-appropriate physical function; and preventing visual morbidities from arthritis-related uveitis (e.g., reduced visual acuity, visual impairment, cataracts). Additional goals included restoring HRQoL; preserving, restoring, or improving physical function; achieving and maintaining clinical remission, ideally within the first 3 to 6 months of therapy; and supporting growth and development. Clinicians also highlighted the need to support growth in the mental and physical development of pediatric patients as they mature and the need to reduce disruptions to the lives of both patients and their families.
Current Treatment Options
Current pharmacologic treatments for JIA aim to alleviate symptoms and treat the disease by modifying the underlying immunopathogenic mechanisms of the disease. Commonly used pharmacologic therapies include nonsteroidal anti-inflammatory drugs (NSAIDs) (e.g., celecoxib, ibuprofen, naproxen), systemic glucocorticoids (e.g., methylprednisolone, prednisolone), topical glucocorticoids (e.g., prednisolone eye drops), intra-articular glucocorticoids (e.g., triamcinolone acetonide, triamcinolone hexacetonide), conventional synthetic disease-modifying antirheumatic drugs (csDMARDs) (e.g., leflunomide, methotrexate, sulfasalazine), and bDMARDs (e.g., TNF inhibitors [adalimumab, etanercept, certolizumab pegol, golimumab], IL-1 inhibitors [e.g., anakinra, canakimumab], IL-6 inhibitors [e.g., tocilizumab], IL-17 inhibitors [e.g., secukinumab], IL-12 and -23 inhibitors [e.g., ustekinumab], and T- or B-cell modulating agents [e.g., abatacept]).3
NSAIDs are typically used as first-line therapy for symptom relief. Glucocorticoids, prescribed either as intra-articular injections or short-term oral bridging therapy (generally less than 3 to 6 months), are used to achieve rapid control of inflammation, especially in patients with high levels of disease activity. For patients with persistent or moderate to severe disease activity, csDMARDs such as methotrexate are commonly initiated. If disease activity persists despite csDMARD use, bDMARDs are introduced. More recently, JAK inhibitors (e.g., tofacitinib) along with newer biologic drugs such as IL-17 inhibitors (e.g., secukinumab) have emerged as therapeutic options. The recommended sequence of pharmacologic treatments in patients with JIA is guided by disease severity and, in part, by the specific subtype of JIA.
Systemic JIA
Initial treatment of patients with sJIA without concomitant macrophage activation syndrome depends on the severity of symptoms. The general approach is to use NSAIDs for patients with mild disease and to add glucocorticoids for patients with severe cases, with the addition of IL-1 or IL-6 inhibitors for patients with moderate to severe disease. In the US, patients are able to receive anti-IL1 or anti-IL6 drugs without a previous unsuccessful trial of corticosteroids.10 Early use of an IL-1 inhibitor within 3 months of sJIA onset may change the course of the disease.10 Tocilizumab, an IL-6 inhibitor, is frequently used in the management of sJIA without macrophage activation syndrome.10 Patients are considered to have refractory chronic disease if after 6 months or more, they continue to have persistent, worsening, or recurrent systemic and/or arthritic symptoms despite separate trials of an IL-1 and an IL-6 inhibitor and/or if they have been unable to taper off systemic glucocorticoids.10 The overarching goals of therapy for patients with sJIA are the rapid control of symptoms to minimize complications related to uncontrolled disease while using immunosuppression judiciously to limit the adverse effects from therapy, particularly those associated with systemic glucocorticoid use.10
Oligoarthritis
Treatment of patients with oligoarticular JIA is guided by the number and type of joints involved and whether there are factors associated with a poorer prognosis (e.g., involvement of ankle, wrist, hip, or cervical spine; presence of erosive disease; delay in diagnosis; or elevated levels of inflammation markers and symmetric disease).11 For patients without identified risk factors, initial therapy consists of intra-articular glucocorticoids with a short-term course of an NSAID. For patients with 1 or more risk factors, a csDMARD is typically added at initiation.11 Patients who have persistent disease despite glucocorticoid joint injections and NSAIDs and those who progress to extended oligoarthritis are typically prescribed methotrexate.11 If a patient’s disease does not respond adequately to methotrexate, treatment is escalated by adding or switching to a bDMARD.11 A patient’s disease is considered refractory if they have active disease despite a 4-month trial of a bDMARD (with or without a csDMARD), intra-articular glucocorticoids, and/or NSAIDs.11
Polyarticular Juvenile Idiopathic Arthritis (RF-Positive, RF-Negative)
Initial recommended therapy for patients with pJIA is a DMARD, most commonly methotrexate at a dose of 10 mg/m2 to 15 mg/m2 of body surface area per week. In patients who have not previously been treated with a biologic drug and who have moderate to high levels of disease activity, biologic therapy may be initiated either alone or in combination with methotrexate. NSAIDs and intra-articular glucocorticoid injections may be used as adjuncts for symptom management but are not appropriate as monotherapy. The overall goal of treatment in patients with pJIA is to control synovitis and reduce associated inflammation.12
Juvenile Psoriatic Arthritis
The treatment of patients with JPsA depends on the type and severity of disease manifestations, including the presence of axial arthritis, uveitis, and psoriasis.12 NSAIDs are often used as initial therapy or as temporary monotherapy for those with mild disease.12 For patients with moderate to severe peripheral arthritis, a csDMARD (usually methotrexate) may be added to an NSAID.12 A bDMARD is added if the patient’s disease fails to respond substantially after 3 months.12 Patients are considered to have refractory disease if their disease does not respond to NSAIDs or intra-articular glucocorticoids within 6 weeks or to csDMARDs within 12 weeks.12 Subsequent treatment depends on the specific clinical manifestations.12
Enthesitis-Related Arthritis
Treatment of ERA focuses on controlling symptoms and preventing progression to structural damage.12 For patients with peripheral arthritis, NSAIDs are recommended as first-line therapy.12 If disease persists despite NSAIDs, csDMARDs are recommended followed by anti-TNF therapy if the response is inadequate.12 For enthesitis, NSAIDs are recommended as first-line therapy.12 If disease persists despite NSAIDs, a TNF inhibitor is conditionally recommended over methotrexate or sulfasalazine.12 Bridging with a short course of oral glucocorticoids may be considered in severe cases with articular involvement.12 For sacroiliitis, initial treatment with NSAIDs is also strongly recommended, but if the patient’s disease remains active, a TNF inhibitor should be introduced.12 Methotrexate monotherapy is not recommended, while sulfasalazine may be considered only when TNF inhibitors are contraindicated or have been ineffective.12 Short courses of oral or intra-articular glucocorticoids and physical therapy may serve as adjunctive therapies.12
The management of patients with JIA in Canada is centred on a multidisciplinary, evidence-based approach aimed at achieving optimal disease control and improving long-term outcomes. Management strategies are guided by international and national treatment guidelines11-13 for broad JIA clinical phenotypes that emphasize early diagnosis, prompt initiation of therapy, and regular monitoring using a treat-to-target approach. Treatment selections are chosen according to the patient’s disease severity and JIA subtype, modified by adverse prognostic factors as well as clinician and parent or caregiver preferences. Treatment is then adjusted according to clinical response.14
Unmet Needs and Existing Challenges
Patient Group Input
Arthritis Consumer Experts
Among the 7 patients living with JIA surveyed by ACE, sex was reported for 6 individuals, of whom 5 identified as female and 1 as male. One patient identified as being Indigenous and another patient identified as being a person of colour. Geographic information was available for 3 respondents (1 each in British Columbia, Manitoba, and Ontario). Reported time since JIA diagnosis varied, with 2 patients diagnosed 1 to 5 years ago, 1 patient diagnosed in the past 10 years, 1 patient diagnosed 11 to 15 years ago, and 3 patients diagnosed 15 years ago or longer. Patients described joint pain and stiffness as key challenges, leading to difficulties with participating in or maintaining exercise, requiring assistance from family and friends with daily activities and recreation, and relying on both social supports and their rheumatologist for emotional well-being. Some also reported the need for workplace accommodations, challenges with balancing time needed to receive care with other priorities, and barriers related to location and accessibility. Travel distances for appointments with a rheumatologist ranged from 1 km to 10 km (n = 1), to 26 km to 50 km (n = 1), to 101 km to 250 km (n = 1), with each patient attending more than 4 clinic visits a year.
While none of the respondents had experience with tofacitinib, patients shared that they had been treated with celecoxib (n = 1), celecoxib and methotrexate (n = 1), diclofenac potassium (n = 1), hydroxychloroquine (n = 1), and an unspecified originator biologic drug (n = 1). Several patients also had used nonpharmacologic treatments such as physiotherapy, occupational therapy, and exercise. ACE emphasized the importance of broadening treatment options for patients with JIA to allow for shorter time to remission and alternative options in the event of adverse effects or drug shortages. The patient group highlighted that a greater availability of treatment options would enable clinicians to individualize treatment based on each patient’s unique needs.
Arthritis Society Canada, the Canadian Arthritis Patient Alliance, the Canadian Spondyloarthritis Association, the Cassie and Friends Society, and Psoriasis Canada
The combined patient group submission included contact with families of patients with JIA and 2 co-authors who have lived experience of JIA, in addition to information from an international survey of 56 youth aged 12 to 30 years who have rheumatic disease (28 of whom were from Canada). Depending on the severity of disease (including frequency of flare-ups) and effects of treatment, children with JIA experience a range of symptoms (e.g., pain, fatigue, brain fog) that can change from day to day. The fluctuating nature of symptoms affects patients’ ability to undertake daily activities (e.g., getting out of bed, self-care) and participate in physical activities, school, and work. About 70% of youth with rheumatic disease said they felt they were treated differently at school or work because of their disease. The condition also affected patients’ confidence and sense of self, with 43% of respondents indicating they did not feel good about themselves and their bodies. Although 69% of respondents indicated receiving support from family, 2 in 5 individuals did not feel supported by their romantic partner and friends in managing their disease. In addition to adolescence being a challenging time, JIA also affects the emotional and psychological well-being of youth and young adults. Among youth living with rheumatic disease who were surveyed, 89% experienced a negative effect of the disease on their mental health, nearly 80% were worried about their mental health, and 79% were worried about their overall health.
Patients with JIA shared that they experienced several barriers due to their disease. One in 4 survey respondents with rheumatic disease said they were not heard by their treating physician, and they expressed the desire to be taken seriously by health care providers to have them understand their symptoms and needs. Two in 3 survey respondents found it difficult to access accommodations to allow them to participate in school and 64% expressed concerns about attending school.
While treatments for JIA have improved over time, the lack of an oral medication was emphasized as a substantial unmet need, with parents and caregivers highlighting regular infusions and injections as being associated with pain and anxiety. Current medications not only add to the burden of care but can also result in reluctance for or poor adherence to treatment, requiring further interventions (e.g., switch to hospital-based infusions, general anesthesia, or cognitive behavioural therapy). Additionally, responses to treatments were noted to vary from child to child and within the same patient (i.e., effective for a short time until the immune system adapts and is no longer responsive, requiring a switch to another medication).
Four families of patients with JIA had experience with tofacitinib. Improvements with tofacitinib treatment were observed in reduced clubbing of fingers and reduced swelling in the knees and wrists in 1 patient. Another family noted their child had improved sleep, anxiety, and overall well-being after starting treatment with tofacitinib. One family indicated that while their child did not continue treatment with tofacitinib, the switch to an oral medication had a profound impact on the patient’s quality of life and enabled a return to better function (including ease of travel and the ability for medication administration by another caregiver). One patient experienced elevated low-density lipoprotein levels with tofacitinib and required medications to lower their cholesterol level, but they otherwise expressed satisfaction with the treatment.
Whereas 1 family was able to receive tofacitinib through a compassionate care program, another family was unsuccessful in securing insurance coverage, resulting in out-of-pocket costs. More than half of respondents (55%) were worried about the cost of medications once they completed school, with approximately two-thirds of respondents reporting lack of knowledge on how to navigate drug insurance plans.
Finally, JIA has a substantial impact on parents and caregivers. Care-related activities such as attending medical appointments, administering medication, dealing with side effects, working with schools, and balancing parental commitments and work obligations take a toll on caregivers, including ongoing stress, fatigue, and anxiety. Parents and caregivers also experienced financial challenges due to the cost of medications, expenses related to travel and time away from work for treatment, and costs of extended health care. Concerns about current and future health status weighed on patients and their caregivers, including how to manage the transition from pediatric to adult care and how these concerns may influence decisions regarding employment and career aspirations.
Clinician Input
According to the clinical experts consulted, the introduction of biologic therapies in the past 2 decades has markedly improved outcomes in patients with JIA, enabling many patients to achieve sustained disease remission. However, despite treatment with 2 or more sequential bDMARDs, 45% to 52% of patients continue to experience active disease, according to the clinical group input, leaving them at risk for irreversible joint damage and long-term disability. The clinician group input also highlighted that 38% to 47% of patients do not achieve minimal disease activity, and 32% to 39% of patients do not reach a 50% improvement in symptoms. Some patients demonstrate primary nonresponse to biologic drugs, while secondary loss of efficacy, particularly with anti-TNF therapies, remains a common challenge that is often linked to the development of neutralizing antidrug antibodies and necessitates adjusting doses or switching agents.
Although advancements in immunopathology have led to the development of innovative therapies that target cytokines, these treatments largely remain approved only for adults with RA and are currently inaccessible to pediatric populations in Canada. Both the clinical experts and the clinician group noted that because nearly all DMARDs (with the exception of leflunomide and methotrexate) require parenteral administration via subcutaneous (SC) injection or IV infusion (which are often associated with pain and local injection site reactions), there remains a substantial unmet need for therapies that offer improved tolerance, acceptance, and adherence in the pediatric population. For example, children with needle phobia often experience substantial distress and trauma with frequent injections, which could be administered daily (e.g., anakinra, which is a painful injection), weekly (e.g., etanercept), or every 2 weeks (e.g., adalimumab). In addition, the clinical experts emphasized the need for hospital-based infusions, frequent laboratory testing (e.g., before each infusion every 3 to 4 months), and regular specialist visits (e.g., rheumatologist or ophthalmologist) is highly disruptive. These demands lead to time away from school for children and missed work for caregivers, contributing to reduced quality of life and, in some cases, poor academic performance. Finally, the experts emphasized that given tofacitinib’s availability in the US and EU for the management of refractory JIA, expanding access in Canada would help close the therapeutic gap and ensure equitable, evidence-based care for all children with JIA.
Considerations for Using the Drug Under Review
Contents within this section have been informed by input from the clinical experts consulted for the purpose of this review and from clinician groups. The following has been summarized by the review team.
Place in Therapy
Patient Population
The clinical experts emphasized tofacitinib’s different mechanism of action by targeting multiple intracellular pathways, unlike biologic drugs, which act extracellularly and on single cytokines. They further noted that its oral administration is an important option that is currently lacking in biologic drugs. This represents a meaningful advantage for pediatric patients and can help address challenges related to needle phobia, which could become a persistent stressor for the child and family throughout treatment and sometimes may result in avoidance of follow-ups and treatment; this sets the child up for permanent joint damage.
One clinical expert suggested that tofacitinib could be used as a first-line targeted DMARD following methotrexate, without requiring prior failure of biologic therapy. This view was based not only on its distinct mechanism and oral administration but also on evidence from the PROPEL trial,15 which demonstrated benefit for patients treated with tofacitinib in the first-line setting after methotrexate. In contrast, another expert emphasized that patients should generally trial other targeted therapies before initiating tofacitinib, citing the established effectiveness and safety of bDMARDs such as TNF inhibitors, IL-6 monoclonal antibody, and T-cell co-stimulation modulators. They also noted that the PROPEL trial showed benefit among patients who had previously received TNF inhibitors.15
Current clinical guidelines support a stepwise treatment strategy, beginning with csDMARDs (e.g., methotrexate) and progressing to biologic drugs for patients with inadequate response. Within this framework, the experts agreed that tofacitinib represents a valuable option for patients who experience secondary loss of efficacy with biologic therapies, often due to the development of antidrug antibodies or for those with complex, treatment-refractory disease. In such cases, where other disease-modifying agents are ineffective, unsafe, or poorly tolerated, tofacitinib may address a critical unmet clinical need. Overall, the experts concluded that its role should be primarily reserved for patients with refractory disease or those who are intolerant to standard therapies.
According to the clinical experts, tofacitinib would be best suited for patients with moderate to severe JIA, including pJIA, JPsA, sJIA without systemic features, and ERA. Individuals most in need of an intervention include patients with persistent joint disease despite appropriate treatment. In particular, individuals whose disease responds inadequately to, or who are intolerant of, currently approved treatments, including csDMARDs (e.g., methotrexate) and bDMARDs (e.g., TNF inhibitors, IL-6 inhibitors, abatacept), were identified as most likely to benefit from tofacitinib.
The clinical experts indicated that eligible patients can be identified through a combination of clinical assessment (e.g., swollen or tender joints, morning stiffness, functional limitations), laboratory testing, and advanced diagnostic imaging (e.g., musculoskeletal ultrasound or MRI to detect radiographically persistent disease such as subclinical synovitis, bone marrow edema, or joint effusion).
While JIA remains a primarily clinical diagnosis, established classification systems (such as ILAR criteria) provide important support for diagnostic accuracy and therapeutic planning. Nevertheless, diagnostic challenges do exist, particularly in the early stages or in patients with atypical presentations. In some cases, symptoms may be mistaken for mechanical injuries or infections, leading to delayed referral and postponement of effective treatment. Additionally, overlapping features such as psoriasis, uveitis, or axial involvement can complicate precise subtype classification, potentially affecting therapeutic planning and access to certain treatments. These diagnostic complexities highlight the need for early evaluation by pediatric rheumatology specialists and underscore the importance of increasing awareness among primary care providers to ensure timely identification and management of JIA.
Assessing the Response to Treatment
According to the clinical experts, treatment response is assessed in clinical practice using tender joint count, swollen joint count, and physician global assessments of disease activity. Composite measures like the Pediatric American College of Rheumatology (pediACR) core set criteria are not routinely used in clinical practice. They are more commonly used in trial settings. The ACR core set criteria includes clinical assessment (i.e., swollen joint count, joints with limited range of movements), global assessment (i.e., physician and parent global assessments of disease on a 10 cm visual analogue scale), functional status (e.g., Childhood Health Assessment Questionnaire [CHAQ] to evaluate physical function and pain), and laboratory test markers (i.e., erythrocyte sedimentation rate [ESR] or C-reactive protein [CRP] levels to monitor systemic inflammation). Other composite indices such as the 10-point Juvenile Arthritis Disease Activity Score (JADAS-10) (assessed in 10 joints) or JADAS-27 (assessed in 27 joints) are also used to provide an overall disease activity score (by disease subtype) with an external set standard for improvement. Expert consensus indicated that a minimum of 50% improvement from baseline in pediACR criteria would be considered clinically meaningful.16 One clinical expert emphasized that definitions of meaningful response may vary among physicians; for example, a partial response in a patient with severe pJIA might be considered acceptable by some but regarded as treatment failure requiring escalation by others, highlighting the usefulness of composite indices in standardizing assessments and reducing variability. Another clinical expert noted that having a patient achieve inactive disease within 3 to 6 months of treatment would be an ideal therapeutic goal.
The experts also indicated that certain assessments are clinic dependent, as time constraints can limit the routine use of formal tools such as the CHAQ or composite indices. While clinical trials generally employ structured, multidomain assessments to calculate composite outcome measures, routine practice tends to focus more heavily on active joint counts, tolerance to treatment, and overall HRQoL.
Discontinuing Treatment
According to the experts, tofacitinib should be discontinued under the following circumstances:
Inadequate response: No meaningful clinical improvement after an adequate treatment trial of 3 to 4 months or worsening of disease within the first 3 months of therapy.
Adverse events (AEs): including serious AEs (SAEs) (e.g., requiring additional intervention, IV antimicrobials, or hospitalization), notable harms (e.g., thromboembolic events, major adverse cardiovascular events [MACE]), and new or recurrent AEs (e.g., infections, cytopenia, or thromboembolic events).
Disease remission: Achievement of complete disease remission for a minimum of 6 months while receiving therapy. There is no consensus or high-quality evidence to guide the optimal approach to tapering or discontinuation of therapy (e.g., gradual dose reduction vs. extended dosing intervals). In practice, decisions are individualized based on patient factors, preferences, and clinician judgment.
Safety concerns: Development of a new serious medical condition (e.g., malignancy), pregnancy or plans for pregnancy, or contraindications due to newly initiated medications.
Prescribing Considerations
The clinical experts consulted for this review emphasized that tofacitinib should be prescribed and monitored by clinicians with expertise in the management of patients with JIA (e.g., pediatric rheumatologist) in a tertiary care hospital, academic setting, or community setting.
The clinician group aligned with the clinical experts regarding tofacitinib’s anticipated role in therapy, the patient population most likely to benefit from tofacitinib, considerations for treatment discontinuation, and prescribing requirements. There was broad agreement on how to assess treatment response, with both the clinician group and clinical experts highlighting the importance of core outcomes endorsed by the Outcome Measures in Rheumatology organization including joint pain, signs of joint inflammation, limitations in activity or physical function, patient-reported perception of disease activity (overall well-being), and AEs. Inflammation associated with extra-articular features was also noted as relevant for specific JIA categories.17 Additionally, the clinician group reported that the JADAS-27 is the most commonly used tool in Canada, with a reduction of at least 5.5 points (out of a total possible score of 57, with higher scores indicating greater levels of disease activity) considered a clinically meaningful improvement.18
Clinical Review
Methods
The review team conducted a systematic review to identify evidence for tofacitinib for the treatment of active pJIA (RF-positive or RF-negative polyarthritis, extended oligoarthritis, and sJIA without systemic manifestations), JPsA, and ERA in children who have responded inadequately or are intolerant to TNF inhibitors or when use of those therapies is inadvisable [wording from original source].
An information specialist conducted a literature search of key bibliographic databases, trial registries, and grey literature sources using a peer-reviewed search strategy. The initial search was completed on June 13, 2025, with alerts maintained until the Formulary Management Expert Committee meeting on September 18, 2025. Search strategies are detailed in Appendix 1 in the Supplemental Material.
Studies were selected according to the eligibility criteria listed in Table 2. Long-term extension (LTE) studies of included randomized controlled trials (RCTs), indirect treatment comparisons (ITCs) that met the eligibility criteria except for the study design criteria, and studies that did not meet the eligibility criteria but were considered to address important gaps in the systematic review evidence were included. Given that direct evidence comparing tofacitinib with relevant comparators (TNF inhibitors, abatacept, and tocilizumab) was lacking, a search for ITCs was also conducted.
Relevant comparators included treatments used in clinical practice in Canada in the patient population under review. Clinical expert input, patient group input, and clinician group input were considered when selecting outcomes (and follow-up times) for review. Selected outcomes are those considered relevant to expert committee deliberations. Detailed methods for study selection, data extraction, and risk of bias appraisal are provided in Appendix 1 in the Supplemental Material.
Table 2: Systematic Review Eligibility Criteria
Criteria | Description |
|---|---|
Population | Active pJIA (RF-positive or RF-negative polyarthritis, extended oligoarthritis, and sJIA without systemic manifestations), JPsA, and ERA in children who have responded inadequately or are intolerant of TNF inhibitors or when use of those therapies is inadvisable [wording from original source] |
Intervention | Tofacitinib with or without methotrexate |
Comparator | Any of the following in addition to background treatments (e.g., NSAIDs, glucocorticoids, csDMARDs):
|
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study design | Published phase III and IV RCTs |
AE = adverse event; AESI = adverse event of special interest; csDMARD = conventional synthetic disease-modifying antirheumatic drug; ERA = enthesitis-related arthritis; HRQoL = health-related quality of life; JPsA = juvenile psoriatic arthritis; MACE = major adverse cardiovascular event; NSAID = nonsteroidal anti-inflammatory drug; pJIA = polyarticular juvenile idiopathic arthritis; RCT = randomized controlled trial; RF = rheumatoid factor; SAE = serious adverse event; sJIA = systemic juvenile idiopathic arthritis; TNF = tumour necrosis factor.
Clinical Evidence
From the search for primary studies, the review team identified 181 unique records via the searches of databases and registers, of which 172 were excluded based on the title and abstract. The review team screened the full text of 9 records and included 2 reports15,19 of 1 study (the PROPEL study). One report of an LTE of the PROPEL study was also included.20 No potentially relevant records were identified from other sources.
From the search for ITCs, the review team identified 33 unique records via the searches of databases, of which 31 were excluded based on the title and abstract. The review team screened the full text of 2 records and included 1 report of 1 study.21
A list of excluded studies, including reasons for their exclusion, is provided in Appendix 1 of the Supplemental Material.
Systematic Review
Description of Studies
Study Characteristics
The PROPEL (Ruperto et al. [2021]) study15 (A3921104; NCT02592434)22 was a multicentre (64 sites across 14 countries: Argentina, Australia, Belgium, Brazil, Canada (3 sites), Israel, Mexico, Poland, Russia, Spain, Türkiye, Ukraine, UK, and US), phase III, placebo-controlled, 2-part withdrawal RCT that enrolled 225 patients between June 10, 2016, and May 16, 2019. Sources of funding for the trial included the manufacturer (Pfizer). Eligible patients were children aged 2 years to younger than 18 years with RF-negative polyarthritis (n = 104), RF-positive polyarthritis (n = 39), extended oligoarthritis (n = 28), sJIA without systemic features (n = 13), JPsA (n = 20), or ERA (n = 21).
The rationale reported for a randomized withdrawal study design was to limit patients’ exposure to placebo given the availability of effective treatments for JIA. Study part 1 was an open-label run-in phase during which patients received tofacitinib from week 0 to week 18 or until disease flare. Only patients who achieved a JIA-ACR 30 response or better by the end of study part 1 were eligible for randomization in the placebo-controlled withdrawal phase (study part 2); those who did not achieve a JIA-ACR 30 response by week 18 of study part 1 were discontinued from the study. Study part 2 was a double-blind phase in which patients were randomized in a 1:1 ratio to tofacitinib or placebo from week 18 to week 44 or until disease flare. Randomization was stratified by JIA category (extended oligoarthritis, RF-positive polyarthritis, RF-negative polyarthritis, sJIA without systemic features, JPsA, or ERA) and by baseline CRP levels (normal [reference range of 0 mg/dL to 0.287 mg/dL] vs. elevated [> 0.287 mg/dL] levels) in patients with polyarticular course JIA (pcJIA).
The primary end point was the occurrence of disease flare by week 44 (or end of study; week 26 of the double-blind phase) according to Pediatric Rheumatology Collaborative Study Group and Paediatric Rheumatology International Trials Organisation disease flare criteria. Key secondary end points in patients with pcJIA were JIA-ACR 30, 50, and 70 response rates at week 44 and mean change in CHAQ-DI scores from baseline of study part 2 (week 18) to week 44. Additional (non-key) secondary end points at scheduled visits over the study duration (study part 1 and part 2) included:
JIA-ACR 30, 50, and 70 responses
rates of JIA-ACR inactive disease
JADAS-27 CRP and JADAS-27 ESR scores
rates of JADAS-27 minimum disease activity and JADAS-27 inactive disease
achievement of JIA-ACR clinical remission at least once
change from baseline in each of 6 JIA-ACR core set variables (physician’s global assessment of overall disease activity, patient or parent assessment of overall well-being, number of joints with active arthritis, number of joints with limitation of motion, CHAQ-DI scores, and ESR)
Child Health Questionnaire (CHQ) scores (global health, physical functioning, and bodily pain)
safety (with adverse events of special interest [AESIs] including serious infections, cytopenias, malignancies, and cardiovascular diseases).
The primary and secondary end points were assessed in patients with pcJIA. In patients with JPsA and ERA, exploratory analyses were prespecified for the end points of JIA flare rate by week 44 and mean JADAS-27 score during study part 1 and part 2. Post hoc analyses included JADAS-10 (score, minimal disease activity, and clinically inactive disease); ACR-defined clinically inactive disease and clinical remission; JADAS-10 clinically inactive disease or remission plus normal physical function; and ACR clinically inactive disease plus normal physical function during study parts 1 and 2.19
Patients with extended oligoarthritis, RF-positive or RF-negative polyarthritis, or sJIA without systemic features were collectively referred to as pcJIA; they were required to have active disease, defined as 5 or more active joints at enrolment, with an inadequate response to 1 or more DMARDs (including methotrexate or bDMARDs). Patients with JPsA or ERA were required to have active disease, defined as 3 or more active joints at enrolment, and an inadequate response to NSAIDs. The trial excluded patients with: sJIA with active systemic features other than active joints and elevated acute-phase reactants within 6 months of enrolment; persistent oligoarthritis; undifferentiated JIA; and active uveitis within 3 months of enrolment.
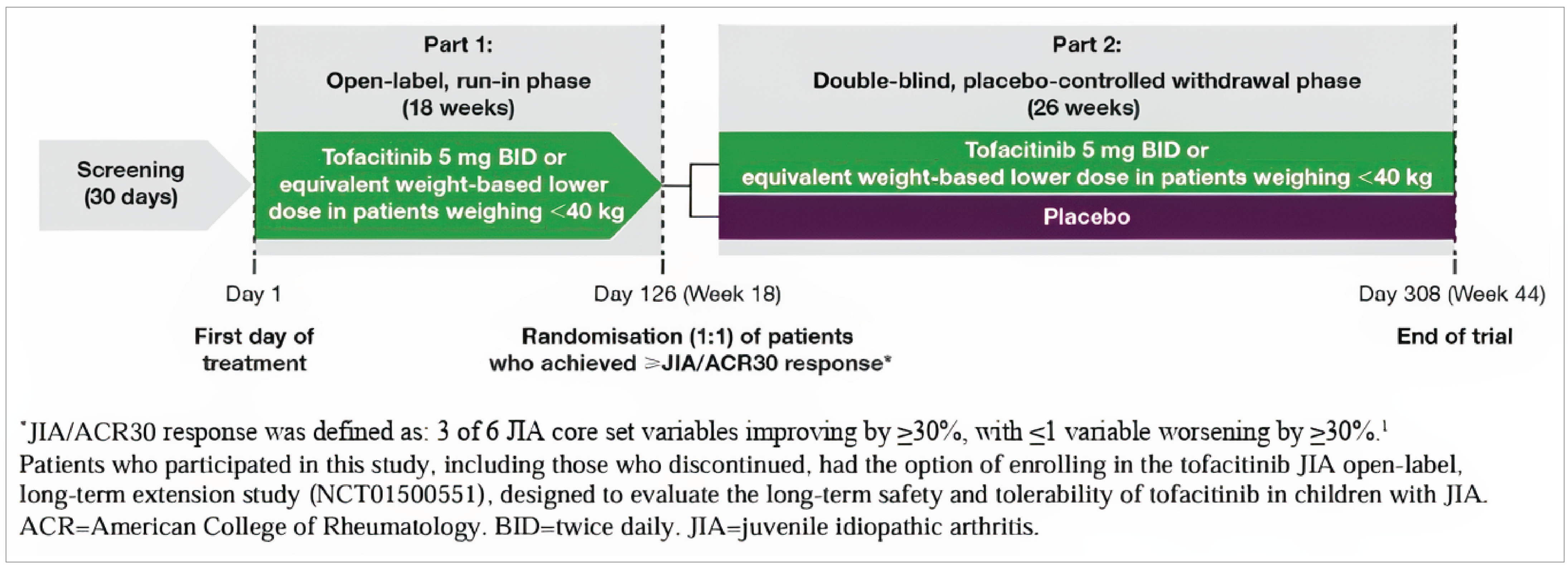
Source: Reprinted from The Lancet, Vol. 398, Ruperto N, Brunner HI, Synoverska O, et al., Tofacitinib in juvenile idiopathic arthritis: a double-blind, placebo-controlled, withdrawal phase 3 randomized trial, Pages No. 1984-1996. Copyright (2021), with permission from Elsevier.
Tofacitinib was available in 2 formulations with matching placebo for oral administration: 5 mg oral tablet and 1 mg/mL grape-flavoured oral solution. Patients weighing 40 kg or more received tofacitinib (5 mg) tablets taken twice daily or a 5 mL oral solution if they were unable to swallow tablets. Patients weighing less than 40 kg received tofacitinib (1 mg/mL) oral solution with doses selected to match the predicted steady-state average during plasma concentrations in patients weighing 40 kg or more after receiving 5 mg twice daily: 2 mg twice daily (for 5 kg to < 7 kg body weight), 2.5 mg twice daily (for 7 kg to < 10 kg body weight), 3 mg twice daily (for 10 kg to < 15 kg body weight), 3.5 mg twice daily (for 15 kg to < 25 kg body weight), and 4 mg twice daily (for 25 kg to < 40 kg body weight).
Concomitant medications that study participants were allowed to use included NSAIDs, a stable dose of an oral glucocorticoid (maximum dose of prednisone 0.2 mg equivalent per kg per day or 10 mg per day for ≥ 2 weeks before baseline, whichever is lower), and a stable dose of methotrexate (maximum 25 mg/week or 20 mg/m2/week orally or parenterally, whichever is lower; methotrexate must have been used for at least 3 months’ duration and at stable dose for at least 6 weeks before baseline). Intra-articular corticosteroids were to be administered in a total dosage of up to 2 mg/kg (up to 80 mg) of methylprednisolone equivalent every 6 months; no more than 2 joints were to be injected in any given 6-month period and individual joints were not to be injected any more frequently than once in a 6-month period. Intra-articular corticosteroids were to be avoided for 6 weeks before any study visit. Patients with JPsA were also allowed concomitant topical treatments for psoriasis (e.g., topical steroids). Biologic and nonbiologic DMARDs (other than methotrexate) were not allowed at any time during the study.
Details regarding relevant outcome measures are provided in Appendix 2 in the Supplemental Material.
Assessments of efficacy and safety were conducted at regular intervals from day 1 (baseline) to week 44 (end of study or early discontinuation visit). Efficacy assessments (JIA-ACR core set variables [by certified joint assessors]; JIA-ACR 30, 50, 70, 90, 100 responses; JIA flare; JIA-ACR inactive disease and clinical remission; and JADAS-27) were conducted in real time according to validated criteria by independent evaluators at the centralized coordinating centres. Patients who experienced an episode of disease flare at any time (study part 1 or 2) were discontinued from the study. The study was planned to be conducted over approximately 3 years. At the end of the trial patients could enter the LTE study (A3921145; NCT01500551).20,23
Statistical Testing and Analysis Populations
Sample size calculations were based on the following assumptions to achieve approximately 90% power or higher at a 0.05 alpha level: the rate of JIA flares between tofacitinib and placebo in the double-blind phase (study part 2) were assumed to follow a normal approximation approach for binomial populations with a JIA-ACR 30 response rate of 54% to 65% (in study part 1) and a corresponding true difference of 31% in flare rates. Approximately 210 patients were targeted for enrolment in the open-label run-in phase. A sample size of 170 patients with pcJIA (at minimum 24 with extended oligoarthritis, 20 with RF-positive polyarthritis, and 62 with RF-negative arthritis [no minimum was required for the number of individuals with sJIA without active systemic features]) was calculated to be sufficient to achieve the desired power; sample sizes for the pcJIA categories were determined from prevalence data and precedents in the literature. Among those with pcJIA, stratification targeted enrolment of at least 50% with baseline CRP levels greater than the upper limit of normal. A minimum of 20 patients for each of JPsA, ERA, and age categories (2 years to < 6 years, 6 years to < 12 years, and 12 years to < 18 years) were targeted for study enrolment.
To control for type I error, the primary and key secondary end points (JIA flare rate; JIA-ACR 30, 50, and 70 response rates; and mean change in CHAQ-DI score from baseline at week 44) were tested using a gatekeeping sequential approach in which statistical significance was claimed for the second end point only if the first end point in the sequence met the requirements for significance. The analysis of the individual components of the composite end points (for JIA flare and response rates on JIA-ACR) did not specify whether the analysis approach accounted for multiple testing with an appropriate control of the type I error rate. Results for end points not included in the hierarchical testing were reported along with P values and associated 95% confidence intervals (CIs) but were not interpreted for statistical significance.
Binary end points, as with the primary end point, were analyzed using the normal approximation approach for binomial populations for the double-blind pcJIA analysis set, with associated 2-sided 95% CIs. JIA flare rate was measured relative to the previous visit in study part 1 and relative to the randomization visit in study part 2. Patients who discontinued study treatment for any reason were considered to have disease flare; those who discontinued after maintaining JIA-ACR inactive disease for at least 24 weeks in the double-blind phase were considered as having a non-disease flare. JIA-ACR response rates were calculated relative to study part 1 baseline measures. Forest plots for the primary and key secondary end points were provided. Continuous end points (changes from baseline in JADAS-27 CRP scores, JADAS-27 ESR scores, CHQ scores, CHAQ scores, JIA-ACR core set variables) were analyzed using a mixed-effect model with repeated measures without imputation for missing data; the estimated treatment difference and associated 95% CI were presented. Efficacy end points for patients with JPsA and ERA were assessed using summary and descriptive statistics by treatment group at each time point in the double-blind phase. Efficacy end points in the open-label run-in phase were presented using summary or descriptive statistics at each visit.
Prespecified subgroup analyses were conducted for the primary end point (JIA flare rate by week 44 in study part 2) and JIA-ACR 30 response rate at week 44 for the following subgroups: JIA category (extended oligoarthritis, RF-positive polyarthritis, RF-negative polyarthritis, or sJIA without active systemic features), baseline CRP level in study part 1 (normal or elevated), age, baseline body weight in study part 1 (< 40 kg or ≥ 40 kg), and geographic region: North America (Canada and US), South and Central America (Argentina, Brazil, and Mexico [classification used in original source]), Europe (Belgium, Poland, Spain, and UK), or all other (Australia, Israel, Russia, Türkiye, and Ukraine). No subgroups were identified as relevant to the review outside of the JIA patient categories already included in the reimbursement population of interest. Inferential statistics and a forest plot for treatment comparison by the subgroups were performed at week 26 of the double-blind phase for the efficacy end points.
Counts of AEs were based on the start date of the AE and were not double counted across phases. If the start date of an AE fell in the open-label phase, the end date of the AE fell in the double-blind phase, and the severity of the AE remained the same or was lower in the double-blind phase, then the AE was counted in the open-label phase and not in the double-blind phase. However, if the same AE started in the open-label phase and worsened in severity in the double-blind phase, the AE would then be counted in both phases. For AEs, SAEs, permanent discontinuations due to AEs, and AESIs, incidence rates (patients with events per 100 patient-years) were calculated. Analyses of AEs were restricted to assessments that occurred no more than 28 calendar days after the last dose of study drug.
All randomized patients who received at least 1 dose of study drug were included in the intention-to-treat population for the analysis of efficacy end points. Patients with pcJIA were included in the analysis of primary and secondary end points in study part 2. Patients with JPsA or ERA were included in exploratory efficacy analyses. Patients who received at least 1 dose of study drug were included in the safety analysis and grouped according to their assigned treatment. No interim analysis for efficacy was prespecified. An interim analysis was prespecified for the review of safety data by the data safety monitoring board. A data cut-off date for the primary efficacy analyses was not available.
Patient Disposition
Of 286 patients who were screened, a total of 61 individuals were ineligible due to not meeting inclusion criteria (n = 55), withdrawal by parent or guardian (n = 4), and “other reason” (unspecified; n = 2). A total of 225 patients were enrolled in study part 1: 184 patients with pcJIA, 20 patients with JPsA, and 21 patients with ERA. Fifty-two of 225 patients (23%) discontinued study part 1 due to JIA flare (14%), AEs (5%), protocol deviations (2%), and “other” (unspecified; 1%). A total of 185 patients (82%) completed study part 1. At week 18, a total of 12 patients (5%) (9 with pcJIA, 3 with JPsA or ERA) discontinued the study due to insufficient clinical response (11 patients: 8 with pcJIA, 3 with JPsA or ERA) or protocol deviation (1 patient with pcJIA).
A total of 173 patients (142 with pcJIA, 15 with JPsA, and 16 with ERA) were enrolled in study part 2. Patients were randomized to the tofacitinib group (n = 88) or the placebo group (n = 85). Seventy-four of 173 patients (43%) discontinued the study during part 2. Reasons for study discontinuations in the tofacitinib group and placebo groups were insufficient clinical response (25% and 52%, respectively), AEs (2% and 2%, respectively), medication error without associated AE (1% and 0, respectively), withdrawal by parent or guardian (1% and 0, respectively), protocol deviation (0% and 1%, respectively), and “other” (unspecified; 1% and 0, respectively). A total of 99 patients (57%) completed study part 2 (69% of patients in the tofacitinib group and 45% of patients in the placebo group).
Baseline Characteristics
Demographic and disease characteristics of patients with pcJIA, JPsA, or ERA in study parts 1 and 2 are summarized in Table 3.
Health Canada has approved tofacitinib for the treatment of active polyarticular JIA (rheumatoid factor–positive or rheumatoid factor–negative polyarthritis, extended oligoarthritis, and sJIA without systemic manifestations), and juvenile psoriatic arthritis in children weighing 40 kg or more, who have responded inadequately or are intolerant to TNF inhibitors or when use of those therapies is inadvisable [wording from original source].
In addition to the approved indication, the study population also included patients with ERA and those weighing less than 40 kg; the requested reimbursement indication aligns with the study population.
Table 3: Summary of Baseline Characteristics in the Included Study
Characteristic | PROPEL part 1 | PROPEL part 2 | |
|---|---|---|---|
Tofacitinib (N = 225) | Tofacitinib (N = 88) | Placebo (N = 85) | |
Sex, n (%) | |||
Female | 169 (75) | 66 (75) | 64 (75) |
Male | 56 (25) | 22 (25) | 21 (25) |
Age (years), n (%) | |||
2 to < 6 | 22 (10) | 11 (13) | 9 (11) |
6 to < 12 | 64 (28) | 22 (25) | 23 (27) |
12 to < 18 | 139 (62) | 55 (63) | 53 (62) |
Age (years), median (Q1 to Q3) | 13.0 (9.0 to 15.0) | 13.0 (9.0 to 15.0) | 13.0 (9.0 to 15.0) |
Age at diagnosis (years), median (Q1 to Q3) | 8.0 (4.0 to 12.3) | 8.4 (3.9 to 12.5) | 8.0 (4.4 to 12.2) |
Disease duration (years), median (Q1 to Q3) | 2.5 (1.0 to 5.6) | 2.5 (1.0 to 5.7) | 2.0 (1.0 to 5.1) |
Body weight, n (%) | |||
< 40 kg | 84 (37) | NR | NR |
≥ 40 kg | 141 (63) | 52 (59) | 54 (64) |
Race,a n (%) | |||
Black or African American | 5 (2) | NR | NR |
White | 196 (87) | 76 (86) | 74 (87) |
Other | 24 (11) | NR | NR |
ILAR JIA category, n (%) | |||
Extended oligoarthritis | 28 (12) | 8 (9) | 10 (12) |
Rheumatoid factor–positive | 39 (17) | 14 (16) | 14 (16) |
Rheumatoid factor–negative | 104 (46) | 45 (51) | 42 (49) |
Systemic JIA without active systemic features | 13 (6) | 5 (6) | 4 (5) |
Psoriatic arthritis | 20 (9) | 7 (8) | 8 (9) |
Enthesitis-related arthritis | 21 (9) | 9 (10) | 7 (8) |
Prior DMARD use, n (%) | |||
Prior conventional synthetic DMARD use, n (%) | 206 (92) | 80 (91) | 80 (94) |
Number of prior biologic DMARDs, n (%) | |||
0 | NR | 57 (65) | 58 (68) |
1 | NR | 18 (20) | 17 (20) |
≥ 2 | NR | 13 (15) | 10 (12) |
Prior biologic DMARD use, n (%) | 85 (38) | 31 (35) | 27 (32) |
Abatacept | NR | 7 (8) | 2 (2) |
Adalimumab | NR | 13 (15) | 18 (21) |
Etanercept | NR | 18 (20) | 13 (15) |
Infliximab | NR | 2 (2) | 1 (1) |
Tocilizumab | NR | 8 (9) | 5 (6) |
Disease activity measures | |||
Physician’s global assessment of overall disease activity, median (Q1 to Q3) | 6.0 (4.5 to 7.5) | 6.0 (4.5 to 7.5) | 6.0 (4.5 to 7.5) |
Number of joints with active arthritis, median (Q1 to Q3) | 10.0 (6.0 to 15.0) | 10.0 (7.0 to 16.0) | 9.0 (6.0 to 14.0) |
Number of joints with limitation of motion, median (Q1 to Q3) | 6.0 (3.0 to 10.0) | 6.0 (3.0 to 12.0) | 5.0 (3.0 to 8.0) |
CHAQ-DI score, median (Q1 to Q3) | 0.9 (0.3 to 1.5) | 0.8 (0.4 to 1.4) | 0.9 (0.3 to 1.5) |
Patient or parent assessment of overall well-being, median (Q1 to Q3) | 5.0 (3.0 to 7.0) | 5.0 (2.5 to 7.0) | 5.0 (3.0 to 7.0) |
Duration of morning stiffness, minutes, median (Q1 to Q3) | 30.0 (15.0 to 60.0) | 30.0 (15.0 to 60.0) | 30.0 (20.0 to 60.0) |
JADAS-27, median (Q1 to Q3) | 20.1 (16.2 to 26.6) | 19.7 (16.2 to 27.4) | 20.1 (14.7 to 25.4) |
Laboratory test parameters | |||
CRP, mg/dL, median (Q1 to Q3) | 0.3 (0.1 to 10.0) | 0.3 (0.1 to 1.3) | 0.2 (0.1 to 0.9) |
ESR, mm/h, median (Q1 to Q3) | 17.0 (10.0 to 32.0) | 19.0 (10.0 to 31.5) | 17.0 (9.0 to 35.0) |
CHAQ-DI = Childhood Health Assessment Questionnaire Disability Index; CRP = C-reactive protein; DMARD = disease-modifying antirheumatic drug; ESR = erythrocyte sedimentation rate; ILAR = International League of Associations for Rheumatology; JADAS-27 = Juvenile Arthritis Disease Activity Score in 27 joints, based on CRP; JIA = juvenile idiopathic arthritis; NR = not reported; Q1 = first quartile; Q3 = third quartile.
aThe category “Other” reflects wording from the original source.
Source: Ruperto et al. (2021)15
Treatment Exposure and Concomitant Medications
Duration of treatment exposure was not reported in study part 1 or part 2.
In study part 1 (N = 225), the number of patients (with pcJIA, JPsA, or ERA) with concomitant medications included 147 (65%) receiving methotrexate and 73 (32%) receiving oral glucocorticoids.
In study part 2, the proportions of patients (with pcJIA, JPsA, or ERA) with concomitant medications (on the date of the first dose of study drug) included 65% of those in the tofacitinib group and 70% in the placebo group receiving methotrexate, and 39% of those in the tofacitinib group and 26% in the placebo group were receiving glucocorticoids. The median dose of methotrexate was 15.0 mg per week (interquartile range [IQR] = 7.5 mg per week) in the tofacitinib group and 15.0 mg per week (IQR =10.0 mg per week) in the placebo group. The median prednisone equivalent dose was 5.0 mg per day (IQR = 4.0 mg per day) in the tofacitinib group and 5.5 mg per day (IQR = 3.0 mg per day) in the placebo group.
Critical Appraisal
Internal Validity
There was a potential risk of bias in the open-label design of the run-in phase due to patients and investigators being aware of the treatment received. This awareness has the potential to affect efficacy outcomes assessed by investigators or by patients or parents (e.g., physician’s global assessment of disease activity, patient or parent assessment of well-being). Given that disease activity measures (JIA-ACR response, JADAS-27, and JADAS-10) include subjective components in the composite end points, the open-label design increases the possibility for bias toward reporting favourable outcomes during the first 18 weeks of treatment.
While the 2-phase design (open-label run-in followed by randomized withdrawal) is commonly used in trials of new interventions in patients with pcJIA and is accepted by regulatory agencies, it introduces potential risks of bias. First, as described, the open-label nature of the run-in phase may have led to misclassifying patients as responders, inflating the number eligible for the randomized phase and increasing the risk that an enriched and biased sample—composed of those perceived to be more likely to benefit or tolerate treatment—were randomized. Second, any expectation-related effects during the open-label phase could artificially inflate response rates, leading to a greater apparent loss of response in the control arm upon withdrawal, thereby exaggerating treatment effects in the randomized phase. The randomized, double-blind design of the second phase mitigates some risks of bias by balancing baseline covariates and reducing the influence of expectations or observer bias between arms. However, it does not address the selection bias introduced by the run-in phase, particularly the exclusion of patients with early disease flare or AEs. In this trial, approximately 23% of participants discontinued during the run-in phase, including 14% due to JIA flare and 5% due to AEs, increasing the possibility that an enriched population of treatment-tolerant responders was selected for randomization. Although baseline characteristics were generally similar between the full run-in cohort and the randomized cohort, this does not rule out meaningful differences in unmeasured factors such as disease trajectory or treatment responsiveness. Consequently, the trial is at risk of bias due to enrichment and selection effects introduced during the open-label run-in phase, which limit the internal validity of treatment effect estimates in the randomized phase (and reduce generalizability of the accuracy of the treatment effect to the patient population with pcJIA).
There was low risk of bias in the randomization process used in the withdrawal phase. Patients were allocated to treatment groups in study part 2 using an interactive response technology system; patients, investigators, and the sponsor were masked to the assigned treatment. Moreover, baseline characteristics were balanced overall between treatment groups, supporting low concern for bias arising from the randomization process. However, there was a risk for unblinding in patients who experienced early disease flare or differential AEs. These events may have revealed treatment allocation, especially if initial classification of a participant as a responder during the open-label run-in phase was inaccurate. While this does not undermine the integrity of the randomization process, it may have introduced bias in outcome assessment during the withdrawal phase.
Carry-over effects of treatment from the open-label run-in to the randomized phase are an important consideration. However, given the twice-daily dosing and relatively short elimination half-life (on the order of hours to days), any pharmacologic carry-over is unlikely to have had a substantial effect on outcomes over the 44-week follow-up duration of the randomized phase.
As described, 23% of patients discontinued the study during the 18-week run-in phase. Subsequently, in study part 2, the proportions of patients who discontinued the study were 31% of those treated with tofacitinib and 55% in the placebo group, commonly due to insufficient clinical response, although the proportion of discontinuations was lower in the tofacitinib group (25%) compared with the placebo group (52%). This differential attrition raises concerns about bias due to missing outcome data, particularly if the reasons for dropout are related to both treatment group and likelihood of response. Although such patterns are expected in treatment withdrawal designs, the high and imbalanced rates of discontinuation may lead to overestimation of any treatment effect if patients in the placebo group whose disease did not respond to treatment discontinued earlier or were less likely to complete outcome assessments.
Overall, the risk of bias due to missing outcome data appears to be moderate to high, given the differential dropout by treatment group and the potential for informative censoring that could influence comparative estimates of sustained efficacy.
No imputation strategies were used to account for secondary end points with missing data. Data at week 44 were available for a subset of randomized individuals representing approximately 58% of patients with pcJIA (for secondary end points of JIA-ACR core set variables, CHAQ-DI scores, JADAS-27 scores, and CHQ scores), as well as 47% of patients with JPsA and 50% of patients with ERA for JADAS-27 scores. Given that only 57% of patients completed study part 2, outcome data may have been subject to attrition bias. If patients who remained in the study differed systematically from those who discontinued, the results may not accurately represent the treatment effect in the targeted population of patients with JIA.
Concomitant medications included NSAIDs, a stable dose of an oral glucocorticoid, a stable dose of methotrexate, and criteria for permitted intra-articular corticosteroids (maximum dosage and administration was to be avoided for 6 weeks before a study visit). While the proportions of patients who received methotrexate were similar between treatment groups, more patients received glucocorticoids in the tofacitinib group (39%) than in the placebo group (26%). It is unclear how this may affect treatment effects, including timing. Data on the number of patients who received NSAIDs or intra-articular corticosteroids were not reported, so the review team could not assess the proportions of patients involved or the potential impact of these concomitant medications on study treatment.
For the primary end point of JIA disease flare by week 44, flare was defined as a worsening of 30% or more in at least 3 of 6 JIA-ACR core set variables with up to 1 variable improving by 30% or more. Although the primary and key secondary end points were included in the hierarchical testing strategy, there was no reported adjustment for multiple comparisons of components of the JIA-ACR core set variables. This raises concerns about inflated type I error risk for interpreting results of individual components, particularly as stand-alone findings. While subgroup analyses were prespecified for JIA flare by week 44 and JIA-ACR 30 response at week 44, there was no prespecified hypothesis of differences in efficacy according to subgroup of patients or accordingly, test for interaction effects between subgroups. Therefore, findings from subgroup analyses should be considered exploratory. No adjustment of multiple comparisons was conducted for other (non-key) secondary end points, some of which were clinically relevant as stand-alone measures in JIA (e.g., number of joints with active arthritis, patient or parent assessment of well-being). Data for outcomes assessed during study part 1 lacked a comparator group, precluding robust causal inferences regarding efficacy and safety from that phase alone. Together, these issues suggest some concerns regarding risk of bias in the selection and reporting of results, especially for secondary and exploratory analyses.
Although there are advantages of composite end points (e.g., capturing the effect of treatment on multiple outcomes) and as they apply to JIA (multifaceted measurement of disease activity and impact on patients), each component outcome is distinct and varies in their clinical importance, responsiveness to treatment, and relevance to patients. When components with differing clinical weight or frequency are combined, the overall composite may be disproportionately influenced by changes in less important or more common components, which may be misleading in terms of the overall result or interpretation of findings. This concern is important in pediatric populations, among whom the relative value of outcomes may differ between patients and caregivers. The challenge of using and interpreting results of composite end points in JIA may be attenuated by measures that are widely used and accepted, such as those of pediACR and JADAS that were included in the study. These have been designed to reflect meaningful and clinically relevant changes in disease activity, and their use helps standardize assessment and interpretation, improving internal validity of the results from these instruments.
Treatment exposure was reported as the number and proportion of patients who had exposure to tofacitinib during the entire study for each AE. Placebo exposure was reported as “might have contributed a maximum of 28 days to the risk period.” There was no information reported on actual duration of treatment exposure, either overall or by treatment group, for either study part 1 or 2. No information was collected or reported on adherence with tofacitinib, which is an important consideration in pediatric populations, among whom adherence with medication can vary. In the context of the open-label phase, this information may have been beneficial to assess the proportion of patients who were not able to adhere to treatment among those not meeting the response threshold.
Finally, the sponsor had direct involvement in the study beyond funding, including study design, management of the trial, data collection, data analysis, and manuscript draft and revisions. In the absence of information on how such study involvement may have affected the analysis and reporting, it is unclear as to the direction and magnitude of any potentially related bias.
External Validity
According to the clinical experts consulted, the trial population was generally aligned with the target population they would treat. However, in addition to individuals with pcJIA (extended oligoarthritis, RF‑positive or RF-negative polyarthritis, sJIA without systemic features), JPsA, and ERA who were eligible for enrolment in the pivotal trial, patients with JIA subtypes associated with a poor prognosis (i.e., persistent oligoarthritis and undifferentiated JIA), who were excluded from the trial, should also be eligible for tofacitinib treatment if prior treatments have failed. Although the study used criteria for the number of joints with active disease (≥ 5 active joints for pcJIA and ≥ 3 active joints for JPsA and ERA), the clinical experts expressed that eligibility criteria should not be restricted to set joint counts because patients with fewer active joints would benefit from treatment with tofacitinib.
The patient population in the trial was overall reflective of patients in clinical practice in Canada, according to the clinical experts, including the distribution of JIA subtypes and prior DMARD use. The clinical experts acknowledged that the most common JIA subtype in Canada is oligoarthritis, although most patients with this subtype do not require treatment with biologic drugs or other DMARDs. There might be varying needs in different provinces due to different clustering of ethnicities in the provinces (i.e., those associated with having distinct subtypes of JIA). A relatively high proportion of patients with ERA and RF-positive pJIA have been observed among Indigenous populations in Manitoba, Saskatchewan, and Alberta (i.e., Edmonton). RF-negative pJIA is more common in Ontario.
While the proportions of patients with prior bDMARD and DMARD use were reflective of clinical practice, the experts noted that anti-TNF drugs are commonly used whereas abatacept is less commonly used in Canada.
In the real world, given that there are several classes of biologic drugs available to patients with JIA, rheumatologists would not withdraw therapy and leave the patient without a biologic drug or other kinds of DMARD for long, unless it were to wait for some transient adverse effects to pass. This contributes to the difficulty of trying to obtain measurement outcomes in patients who withdraw from trials due to inefficacy of the placebo, as their physicians would have started them on another class of medication, and any measurement done while taking those medications would not be reflective of their treatment exposure.
Although more patients in the tofacitinib group received concomitant glucocorticoids compared with those in the placebo group, the clinical experts remarked that the trial specified criteria for the permitted glucocorticoid dose, the median dose was relatively low (5.0 mg/day [IQR = 4.0]), and the median dose was comparable between the treatment groups.
Whereas the study used a minimum response of JIA-ACR 30 (or better) at 18 weeks as a criterion for enrolment into the double-blind withdrawal phase, the clinical experts shared that JIA-ACR 30 measures minimal response and, therefore, is not clinically meaningful. The clinical experts highlighted JIA-ACR 50 or JIA-ACR 70 would be preferred, although ACR criteria are more applicable in trial settings and are cumbersome to conduct in practice. The experts added that a clinically relevant time point for assessing treatment response is at approximately 12 to 16 weeks. Evidence from trials evaluating biologic drugs for patients with pJIA showed that lack of response (JIA-ACR 50 or better) at 12 weeks was associated with low probability of treatment response at 14 to 16 weeks. Similarly, trials evaluating tofacitinib for adults with RA demonstrated that lack of treatment response by about 3 months was associated with low probability of low disease activity by 6 months.
The clinical experts outlined that in practice settings, disease activity is assessed using active joint counts, morning stiffness (frequency and duration), clinical JADAS, ESR or CRP levels for patients with some subtypes (JPsA, ERA), and enthesitis. Composite instruments such as those used in the trial (JIA-ACR) are less applicable (and practical) in clinical practice settings. Tools such as the clinical JADAS are continually revised to adopt new thresholds (e.g., applicable to patients with specific JIA subtypes) for determining disease activity, making it difficult to compare true response over time without recalculating all prior measures.
Results
Efficacy
Results for outcomes important to this review are presented in Table 4 for patients with pcJIA and Table 5 for patients with JPsA or ERA. Median duration of follow-up for the overall population was not reported. Key results include the following:
Disease Activity and Physical Function – JIA Flare Rate
For patients with pcJIA, the results suggest a difference between treatments, with those receiving tofacitinib showing a greater improvement in JIA flare rate than those receiving placebo by week 44.
For patients with pcJIA, between-group treatment effect differences from subgroup analyses of JIA flare rate by week 44 were overall consistent with the primary analysis; however, 95% CIs for most subgroups included the possibility of no benefit.
For patients with JPsA, 29% of patients (2 of 7) in the tofacitinib group and 75% of patients (6 of 8) in the placebo group experienced a flare by week 44. For patients with ERA, 55% of patients (4 of 9) in the tofacitinib group and 57% of patients (4 of 7) in the placebo group had JIA flares by week 44.
Disease Activity and Physical Function – JIA-ACR
For patients with pcJIA, the results suggest a difference between treatments, with those receiving tofacitinib showing greater improvements in JIA-ACR 30, JIA-ACR 50, and JIA-ACR 70 response rates at week 44.
For patients with pcJIA, between-group treatment effect differences from subgroup analyses of JIA-ACR 30 response rates at week 44 were overall consistent with the primary analysis; however, 95% CIs for most subgroups included the possibility of no benefit.
For patients with pcJIA, the results do not suggest a difference between treatment groups in JIA-ACR inactive disease rate and JIA-ACR clinical remission rate at week 44.
For patients with pcJIA, the results suggest a difference between treatments, with those receiving tofacitinib showing a greater improvement (lower score) in physician’s global assessment of overall disease activity at week 44.
Disease Activity and Physical Function – JADAS-27
For patients with pcJIA, the results do not suggest a difference between treatment groups in JADAS-27 minimum disease activity rate or JADAS-27 inactive disease rate at week 44.
For patients with JPsA, 31% of patients (5 of 16) had a mean score of 3.1 (variance not reported) in the tofacitinib group and 13% of patients (2 of 15) had a mean score of 4.3 (variance not reported) in the placebo group on the JADAS-27 at week 44. For patients with ERA, 31% of patients (5 of 16) had a mean score of 6.9 (variance not reported) in the tofacitinib group and 20% of patients (3 of 15) had a mean score of 1.3 (variance not reported) in the placebo group on the JADAS-27 at week 44.
For patients with pcJIA, the results suggest a difference between treatments, with those receiving tofacitinib showing a greater improvement (lower score) in JADAS-27 CRP score and JADAS-27 ESR score at week 44.
Table 4: Summary of Key Efficacy Results — pcJIA
Variable | PROPEL part 2 | |
|---|---|---|
Tofacitinib N = 72a | Placebo N = 70a | |
Disease activity and physical function | ||
JIA flareb rate | ||
Patients with data at part 1 week 12, n (%) | 166 | — |
Patients with JIA flare at part 1 week 12, n (%) | 12 (7.23) | — |
Patients with data at part 1 week 18, n (%) | 154 | — |
Patients with JIA flare at part 1 week 18, n (%) | 13 (8.44) | — |
Patients with data at week 44, n (%) | 72 (100) | 70 (100) |
Patients with JIA flare by week 44, n (%) | 21 (29.17) | 37 (52.86) |
Difference in JIA flare rate by week 44 (%) (95% CI)c | –23.7 (–39.4 to –8.0) | Reference |
P value | 0.0031 | Reference |
JIA-ACR | ||
JIA-ACR 30 responsed rate | ||
Patients with data at part 1 week 12, n (%) | 167 | — |
Patients achieving JIA-ACR 30 response at part 1 week 12, n (%) | 143 (85.63) | — |
Patients with data at part 1 week 18, n (%) | 154 | — |
Patients achieving JIA-ACR 30 response at part 1 week 18, n (%) | 142 (92.21) | — |
Patients with data at week 44, n (%) | 72 (100) | 70 (100) |
Patients achieving JIA-ACR 30 response at week 44, n (%) | 51 (70.83) | 33 (47.14) |
Difference in JIA-ACR 30 response rate at week 44, % (95% CI)e | 23.7 (8.0 to 39.4) | Reference |
P valuee | 0.0031 | Reference |
JIA-ACR 50 responsed rate | ||
Patients with data at part 1 week 12, n (%) | 167 | — |
Patients achieving JIA-ACR 50 response at part 1 week 12, n (%) | 120 (71.86) | — |
Patients with data at part 1 week 18, n (%) | 154 | — |
Patients achieving JIA-ACR 50 response at part 1 week 18, n (%) | 129 (83.77) | — |
Patients with data at week 44, n (%) | 72 (100) | 70 (100) |
Patients achieving JIA-AC R50 response at week 44, n (%) | 48 (66.67) | 33 (47.14) |
Difference in JIA-ACR 50 response rate, % (95% CI)e | 19.5 (3.6 to 35.5) | Reference |
P valuee | 0.0166 | Reference |
JIA-ACR 70 responsed rate | ||
Patients with data at part 1 week 12, n (%) | 167 | — |
Patients achieving JIA-ACR 70 response at part 1 week 12, n (%) | 78 (46.71) | — |
Patients with data at part 1 week 18, n (%) | 154 | — |
Patients achieving JIA-ACR 70 response at part 1 week 18, n (%) | 94 (61.04) | — |
Patients with data at week 44, n (%) | 72 (100) | 70 (100) |
Patients achieving JIA-ACR 70 response at week 44, n (%) | 39 (54.17) | 26 (37.14) |
Difference in JIA-ACR 70 response rate (95% CI)e | 17.0 (0.9 to 33.2) | Reference |
P valuee | 0.0387 | Reference |
JIA-ACR inactive diseasef rate | ||
Patients with data at part 1 week 12, n (%) | NR | — |
Patients achieving JIA-ACR inactive disease at part 1 week 12, n (%) | NR | — |
Patients with data at part 1 week 18, n (%) | 184 | — |
Patients achieving JIA-ACR inactive disease at part 1 week 18, n (%) | 26 (14) | — |
Patients achieving JIA-ACR inactive disease by week 44, n (%) | 19 (26.39) | 12 (17.14) |
Difference, % patients achieving JIA-ACR inactive disease by week 44 (95% CI)g | 9.25 (–4.23 to 22.72) | Reference |
P value | 0.1787 | Reference |
JIA-ACR clinical remissionh rate | ||
Patients with data at part 1 week 12, n (%) | NR | — |
Patients achieving JIA-ACR clinical remission at part 1 week 12, n (%) | NR | — |
Patients with data at part 1 week 18, n (%) | NR | — |
Patients achieving JIA-ACR clinical remission at part 1 week 18, n (%) | NR | — |
Patients achieving JIA-ACR clinical remission by week 44, n (%) | 3 (4.17) | 3 (4.29) |
Difference, % patients achieving JIA-ACR clinical remission by week 44 (95% CI)g | –0.12 (–6.74 to 6.50) | Reference |
P value | 0.9719 | Reference |
JIA-ACR core set variable: physician’s global assessment of overall disease activity using VAS (0 [no activity] to 10 [maximum disease activity]) | ||
Patients with data at part 1 week 12, n | 166 | — |
Change in physician global assessment of disease activity from part 1 baseline to week 12, mean (SD) | –4.04 (1.88) | — |
Patients with data at part 1 week 18, n | 154 | — |
Change in physician global assessment of disease activity from part 1 baseline to week 18, mean (SD) | –4.54 (1.92) | — |
Patients with data at week 44, n (%) | 49 (68) | 33 (47) |
Change in physician global assessment of disease activity from part 2 baseline to week 44, VAS, LS mean (SE) | –0.16 (0.29) | 1.42 (0.34) |
LS mean difference (95% CI)i | –1.58 (–2.44 to –0.71) | Reference |
P value | 0.0007 | Reference |
JADAS-27j (0 to 57, higher = more disease activity) | ||
JADAS-27 CRP scorek | ||
Patients with data at part 1 week 12, n (%) | 163 | — |
JADAS-27 CRP at part 1 week 12, score, mean (SD) | –14.33 (6.96) | — |
Patients with data at part 1 week 18, n (%) | 153 | — |
JADAS-27 CRP at part 1 week 18, score, mean (SD) | –15.80 (7.12) | — |
Patients with data at week 44, n (%) | 49 (68) | 32 (46) |
Change in JADAS-27 CRP from part 2 baseline to week 44, score, LS mean (SE) | 0.03 (0.91) | 4.39 (1.00) |
LS mean difference (95% CI)i | –4.36 (–7.02 to –1.71) | Reference |
P value | 0.0027 | Reference |
JADAS-27 ESR scorek | ||
Patients with data at part 1 week 12, n | 165 | — |
JADAS-27 ESR at part 1 week 12, score, mean (SD) | –14.54 (6.90) | — |
Patients with data at part 1 week 18, n | 154 | — |
JADAS-27 ESR at part 1 week 18, score, mean (SD) | –15.94 (7.17) | — |
Patients with data at week 44, n | 49 (68) | 33 (47) |
Change in JADAS-27 ESR from part 2 baseline to week 44, score, LS mean (SE) | 0.09 (0.91) | 4.50 (0.97) |
LS mean difference (95% CI)i | –4.41 (–6.99 to –1.82) | Reference |
P value | 0.0018 | Reference |
JADAS-27 minimum disease activityl rate | ||
Patients with data at part 1 week 12, n | 165 | — |
Patients achieving JADAS-27 minimum disease activity at part 1 week 12, n (%) | 48 (29.09) | — |
Patients with data at part 1 week 18, n | 154 | — |
Patients achieving JADAS-27 minimum disease activity at part 1 week 18, n (%) | 68 (44.16) | — |
Patients achieving JADAS-27 minimum disease activity at week 44, n (%) | 34 (47.22) | 23 (32.86) |
Difference, % patients achieving JADAS-27 minimum disease activity at week 44 (95% CI)g | 14.37 (–1.57 to 30.30) | Reference |
P value | 0.0773 | Reference |
JADAS-27 inactive diseasem rate | ||
Patients with data at part 1 week 12, n | 165 | — |
Patients achieving JADAS-27 inactive disease at part 1 week 12, n (%) | 6 (3.64) | — |
Patients with data at part 1 week 18, n | 154 | — |
Patients achieving JADAS-27 inactive disease at part 1 week 18, n (%) | 12 (7.79) | — |
Patients achieving JADAS-27 inactive disease by week 44, n (%) | 13 (18.06) | 7 (10.00) |
Difference, % patients achieving JADAS-27 inactive disease at part 2 baseline (95% CI)g | 8.06 (–3.27 to 19.38) | Reference |
P value | 0.1634 | Reference |
Physical function or activity limitation | ||
JIA-ACR core set variable: CHAQ-DI score (0 [no or minimal physical dysfunction] to 3 [very severe physical dysfunction]) | ||
Patients with data at part 1 week 12, n | 165 | — |
CHAQ-DI at part 1 week 12, score, mean (SD) | –0.41 (0.53) | — |
Patients with data at part 1 week 18, n | 154 | — |
CHAQ-DI at part 1 week 18, score, mean (SD) | –0.49 (0.57) | — |
Patients with data at week 44, n (%) | 49 (68) | 33 (47) |
Change in CHAQ-DI from part 2 baseline to week 44, score, LS mean (SE) | –0.9 (0.04) | 0.03 (0.04) |
LS mean difference (95% CI)e | –0.12 (–0.22 to –0.01) | Reference |
P valuee | 0.0292 | Reference |
JIA-ACR core set variable: number of joints with active arthritisn | ||
Patients with data at part 1 week 12, n | 166 | — |
Change in number of joints with active arthritis from part 1 baseline to week 12, mean (SD) | –9.76 (6.76) | — |
Patients with data at part 1 week 18, n | 154 | — |
Change in number of joints with active arthritis from part 1 baseline to week 18, mean (SD) | –10.29 (6.79) | — |
Patients with data at week 44, n (%) | 49 (68) | 33 (47) |
Change in number of joints with active arthritis from part 2 baseline to week 44, LS mean (SE) | 0.55 (0.74) | 2.79 (0.77) |
LS mean difference (95% CI)i | –2.24 (–4.36 to –0.13) | Reference |
P value | 0.0384 | Reference |
JIA-ACR core set variable: number of joints with limitation of motiono | ||
Patients with data at part 1 week 12, n | 166 | — |
Change in number of joints with limited range of motion from part 1 baseline to week 12, mean (SD) | –5.09 (5.79) | — |
Patients with data at part 1 week 18, n | 154 | — |
Change in number of joints with limited range of motion from part 1 baseline to week 18, mean (SD) | –5.77 (5.82) | — |
Patients with data at week 44, n (%) | 49 (68) | 33 (47) |
Change in number of joints with limited range of motion from part 2 baseline to week 44, mean (SE) | 0.38 (0.29) | 1.20 (0.34) |
LS mean difference (95% CI)i | –0.82 (–1.66 to 0.02) | Reference |
P value | 0.0549 | Reference |
Pain | ||
CHQ score: parent or caregiver assessment of child’s bodily pain and relative burden of disease on the parent or caregiver (0 [poor health status] to 100 [positive health status]) | ||
Patients with data at part 1 week 4, n | 171 | — |
Change in parent or caregiver assessment of bodily pain from part 1 baseline to week 4, score, mean (SD) | 19.42 (21.14) | — |
Patients with data at part 1 week 18, n | 149 | — |
Change in parent or caregiver assessment of bodily pain from part 1 baseline to week 18, score, mean (SD) | 30.60 (22.79) | — |
Patients with data at week 44, n (%) | 49 (68) | 31 (44) |
Change in patient or parent assessment of bodily pain from part 2 baseline to week 44, VAS, LS mean (SE) | 6.34 (3.13) | –1.91 (3.91) |
LS mean difference (95% CI)i | 8.26 (–0.43 to 16.94) | Reference |
P value | 0.0620 | Reference |
Health-related quality of life | ||
JIA-ACR core set variable: patient or parent assessment of overall well-being (0 [very good well-being] to 10 [very poor well-being]) | ||
Patients with data at part 1 week 12, n | 165 | — |
Change in patient or parent assessment of overall well-being from part 1 baseline to week 12, VAS, mean (SD) | –2.30 (2.15) | — |
Patients with data at part 1 week 18, n | — | — |
Change in patient or parent assessment of overall well-being from part 1 baseline to week 18, VAS, mean (SD) | –2.68 (2.33) | — |
Patients with data at week 44, n (%) | 49 (68) | 33 (47) |
Change in patient or parent assessment of overall well-being from part 2 baseline to week 44, VAS, LS mean (SE) | –0.49 (0.22) | 0.24 (0.24) |
LS mean difference (95% CI)i | –0.73 (–1.31 to –0.15) | Reference |
P value | 0.0154 | Reference |
CHQ score: parent or caregiver assessment of child’s well-being and relative burden of disease on the parent or caregiver (0 [poor health status] to 100 [positive health status]) | ||
Global health | ||
Patients with data at part 1 week 4, n | 171 | — |
Change in parent or caregiver assessment of global health from part 1 baseline to week 4, score, mean (SD) | 13.86 (22.40) | — |
Patients with data at part 1 week 18, n | 148 | — |
Change in parent or caregiver assessment of global health from part 1 baseline to week 18, score, mean (SD) | 21.28 (22.79) | — |
Patients with data at week 44, n (%) | 49 (68) | 30 (43) |
Change in patient or parent assessment of global health from part 2 baseline to week 44, VAS, LS mean (SE) | 5.46 (2.83) | 1.66 (3.72) |
LS mean difference (95% CI)i | 3.79 (–3.72 to 11.31) | Reference |
P value | 0.3179 | Reference |
Physical functioning | ||
Patients with data at part 1 week 4, n | 171 | — |
Change in parent or caregiver assessment of physical functioning from part 1 baseline to week 4, score, mean (SD) | 11.83 (24.47) | — |
Patients with data at part 1 week 18, n | 149 | — |
Change in parent or caregiver assessment of physical functioning from part 1 baseline to week 18, score, mean (SD) | 21.44 (26.78) | — |
Patients with data at week 44, n (%) | 49 (68) | 31 (44) |
Change in patient or parent assessment of physical functioning from part 2 baseline to week 44, VAS, LS mean (SE) | 1.45 (3.16) | –1.82 (3.92) |
LS mean difference (95% CI)i | 3.28 (–5.23 to 11.78) | Reference |
P value | 0.4452 | Reference |
ACR = American College of Rheumatology; CHAQ-DI = Childhood Health Assessment Questionnaire Disability Index; CHQ = Child Health Questionnaire; CI = confidence interval; CRP = C-reactive protein; ESR = erythrocyte sedimentation rate; JADAS = Juvenile Arthritis Disease Activity Score; JIA = juvenile idiopathic arthritis; JIA-ACR = juvenile idiopathic arthritis–American College of Rheumatology; LS = least squares; pcJIA = polyarticular course juvenile idiopathic arthritis; SD = standard deviation; SE = standard error; VAS = visual analogue scale.
Note: Study part 1 (week 0 to week 18) and study part 2 (week 18 to week 44); In part 1, 184 patients with polyarticular JIA were enrolled.
aIncluded patients with pcJIA (extended oligoarthritis, RF-positive polyarthritis, RF-negative polyarthritis, and systemic JIA without active systemic features at enrolment).
bA flare was defined by the Pediatric Rheumatology Collaborative Study Group and the Paediatric Rheumatology International Trials Organisation as worsening of 30% or more in 3 or more of 6 JIA-ACR core set variables, with no or 1 variable improving by 30% or more.
cBased on normal approximation to the binomial.
dJIA-ACR 30, JIA-ACR 50, and JIA-ACR 70 response criteria were defined as 3 of 6 JIA core set variables improved by a minimum of 30%, 50%, and 70%, respectively, with no more than 1 JIA core set variable worsened by 30% or more. Response rates were calculated relative to part 1 baseline. Patients who discontinued for any reason, except while in clinical remission, were counted as their disease not responding to treatment at the discontinuation visit and at all subsequent visits with each study phase (part 1 or 2).
eBased on normal approximation to the binomial. JIA-ACR 30, JIA-ACR 50, and JIA-ACR 70 response rates at week 44 and CHAQ-DI scores at week 44 were adjusted for multiple testing.
fCriteria for achievement of JIA-ACR inactive disease were: no joints with active arthritis; no fever, rash, serositis, splenomegaly, hepatomegaly, or generalized lymphadenopathy attributable to systemic JIA; no active uveitis; normal ESR or, if elevated, not attributable to JIA; physician’s global evaluation of overall disease activity score of “best possible”; and, morning stiffness duration of maximum 15 minutes. Patients who discontinued for any reason, except while in clinical remission, were counted as having active disease at the discontinuation visit and at all subsequent visits with each study phase (part 1 or 2).
gBased on normal approximation to the binomial. The alpha has not been adjusted for multiple testing and there is an increased risk of type I error.
hAchievement of JIA-ACR clinical remission was defined as inactive disease for 6 continuous months.
iBased on a mixed model for repeated measures with fixed effects of treatment, visit, JIA category, open-label baseline CRP, treatment-by-visit interaction, and the double-blind baseline value. The alpha has not been adjusted for multiple testing and there is an increased risk of type I error.
jJADAS-27 score includes 4 components: physician global assessment of disease activity (assessed on a VAS of 0 [no activity] to 10 [maximum activity]); parent, legal guardian, or patient global assessment of overall well-being (assessed on a VAS of 0 [very well] to 10 [very poor]); number of joints with active disease (maximum of 27 and defined as joint with swelling or, in absence of swelling, limited range of motion accompanied by either pain on motion or tenderness); and CRP levels or ESR, as applicable.
kCRP levels were measured in mg/L and ESR was measured in mm/h. CRP and ESR values were normalized to a 0 to 10 scale.
lJADAS-27 minimum disease activity was defined as a JADAS-27 score of 3.8 or lower for participants with polyarthritis, and 2 or lower for participants with oligoarthritis (< 4 active joints).
mJADAS-27 inactive disease is defined as a JADAS-27 score of 1 or lower for both polyarthritis and oligoarthritis.
nNumber of joints with active arthritis was assessed on 71 joints.
oNumber of joints with limitation of motion was assessed on 67 joints.
Source: Ruperto et al. (2021)15 and NCT0259243422
Physical Function or Activity Limitation
For patients with pcJIA, the results suggest a difference between treatments, with those receiving tofacitinib showing a greater improvement (lower score) in CHAQ-DI scores between part 2 baseline and week 44.
For patients with pcJIA, the results suggest a difference between treatments, with those receiving tofacitinib showing a greater improvement in the change in number of joints with active arthritis on the JIA-ACR core set variables between part 2 baseline and week 44.
For patients with pcJIA, the results do not suggest a difference between treatment groups for change in the number of joints with limitation of motion on the JIA-ACR core set variables between part 2 baseline and week 44.
Pain
For patients with pcJIA, the results do not suggest a difference between treatment groups for change in patient or parent assessment of bodily pain on the CHQ between part 2 baseline and week 44.
HRQoL
For patients with pcJIA, the results suggest a difference between treatments, with those receiving tofacitinib showing a greater improvement (lower score) in JIA-ACR patient or parent assessment of overall well-being between part 2 baseline and week 44.
For patients with pcJIA, the results do not suggest a difference between treatment groups for change in patient or parent assessment of global health or of physical functioning on the CHQ between part 2 baseline and week 44.
Table 5: Summary of Key Efficacy Results — JPsA and ERA
Variable | PROPEL part 2 | |
|---|---|---|
Tofacitinib N = 16 | Placebo N = 15 | |
Disease activity and physical function | ||
JIA flare rate | ||
JPsA | ||
Patients with data at week 44, n | 7 | 8 |
Patients with JIA flare by week 44, n (%) | 2 (29) | 6 (75) |
ERA | ||
Patients with data at week 44, n | 9 | 7 |
Patients with JIA flare by week 44, n (%) | 4 (44) | 4 (57) |
JADAS-27a (0 to 57, higher = more disease activity) | ||
JPsA | ||
Patients with data at part 1 week 18, n (%) | 16 | — |
JADAS-27 at week 18, score, mean (SD) | 4.0 (NR) | — |
Patients with data at week 44, n | 5 | 2 |
JADAS-27 at week 44, score, mean (SD) | 3.1 (NR) | 4.3 (NR) |
ERA | ||
Patients with data at part 1 week 18, n (%) | 20 | — |
JADAS-27 at part 1 week 18, score, mean (SD) | 6.6 (NR) | — |
Patients with data at week 44, n | 5 | 3 |
JADAS-27 at week 44, score, mean (SD) | 6.9 (NR) | 1.3 (NR) |
CRP = C-reactive protein; ERA = enthesitis-related arthritis; ESR = erythrocyte sedimentation rate; JADAS = Juvenile Arthritis Disease Activity Score; JIA = juvenile idiopathic arthritis; JPsA = juvenile psoriatic arthritis; NR = not reported; SD = standard deviation; VAS = visual analogue scale.
aThe JADAS-27 score includes 4 components: physician global assessment of disease activity (assessed on a VAS of 0 [no activity] to 10 [maximum activity]); parent, legal guardian, or patient global assessment of overall well-being (assessed on a VAS of 0 [very well] to 10 [very poor]); number of joints with active disease (maximum of 27 and defined as joint with swelling or, in absence of swelling, limited range of motion accompanied by either pain on motion or tenderness); and CRP levels or ESR, as applicable.
Source: Ruperto et al. (2021)15 and NCT0259243422
Harms
Detailed results for harms for the included study are provided in the PROPEL study.15 Harms were reported by system organ class with select preferred terms (specific harms) reported.
Key results include the following:
In study part 1, a total of 153 of 225 patients (68%) experienced an AE:
Among treatment-emergent adverse events (TEAEs) specified, common TEAEs (excluding SAEs) occurring in at least 5% of patients were upper respiratory tract infection (11%), headache (7%), nausea (6%), vomiting (6%), and pyrexia (5%).
SAEs occurred in 7 patients (3%); these included Crohn disease (1 patient; 0.4%), diarrhea (0.4%), vomiting (0.4%), appendicitis (0.4%), epidural empyema (0.4%), pneumonia (0.4%), sinusitis (0.4%), subperiosteal abscess (0.4%), Still disease (0.4%), and condition aggravated (unspecified; 0.4%).
Permanent discontinuations due to AEs occurred in 26 patients (12%), with disease progression and JIA the most common reasons for the study discontinuation (numerical data not reported).
The total number of patients with trial-specified AESIs were not reported. AESIs included hepatic events (3 patients; 1%), herpes zoster (2 patients; 1%), and serious infection (3 patients; 1%).
In study part 2, TEAEs occurred in 68 of 88 patients (77%) in the tofacitinib group and 63 of 85 patients (74%) in the placebo group:
Permanent discontinuations due to AEs occurred in 16 patients (18%) in the tofacitinib group and 29 patients (34%) in the placebo group. Reasons for study discontinuations were not reported.
The total number of patients with trial-specified AESIs were not reported. One patient with a serious infection of pilonidal cyst repair in the tofacitinib group was adjudicated and deemed as not meeting opportunistic infection criteria. One patient experienced appendicitis in the placebo group.
No deaths occurred during study part 1 or 2.
AESIs also identified as relevant for this review of gastrointestinal perforation, interstitial lung disease, MACE, macrophage activation syndrome, malignancies, opportunistic infection, thrombotic events, and tuberculosis did not occur in any patient during study part 1 or 2.
LTE Studies
Description of Studies
One ongoing, multicentre (22 sites), phase III, open-label, single-arm extension study by Brunner et al. (2024)20 (NCT01500551)23 evaluated the long-term safety, tolerability, and efficacy of tofacitinib in patients aged between 2 and younger than 18 years (including those who turned 18 during the LTE) with JIA. Eligible patients had completed or discontinued 1 of 3 qualifying parent trials: a phase I pharmacokinetic study (NCT01513902)24 or 1 of 2 phase III placebo-controlled trials (the Ruperto et al. [2021] study15 and an ongoing NCT03000439 trial evaluating tofacitinib in patients with sJIA without active systemic features). Patients could enter the LTE study within 14 days of their last visit in the Ruperto et al. (2021) study15 (with the last parent study visit serving as the LTE baseline); otherwise, a new baseline was established on day 1 of enrolment in the LTE study. Key eligibility for enrolment in the LTE study was the same as in the pivotal study. Patients received open-label tofacitinib administered at a dose of 5 mg twice daily or a weight-based equivalent lower dose. Permitted background therapies remained the same as those of the pivotal study. The primary objective of the LTE study was to evaluate the long-term safety and tolerability of tofacitinib; efficacy was a secondary objective.
Safety outcomes were the same as in the pivotal study. All AEs were coded using the Medical Dictionary for Regulatory Activities, version 25.0, and were reported using preferred terms. Independent external adjudication committees and an internal sponsor committee conducted masked assessments of all safety events. Patients who received at least 1 dose of tofacitinib were included in the safety analyses. Safety outcomes were summarized descriptively and incidence rates were calculated as the number of patients experiencing a first event per 100 patient-years of follow-up. Safety was evaluated from the LTE baseline through the data cut-off date of July 11, 2022.
Efficacy assessments included those as evaluated and defined in the pivotal study: JIA core set variables; JIA-ACR 30, 50, 70, 90, and 100 response rates; JADAS-27; and CHAQ-DI scores. Additionally, parental assessment of their child’s pain was conducted at each visit using a visual analogue scale ranging from 0 to 10, with 0 indicating no pain. Pain levels were interpreted as follows: 1 to 4 = mild, 5 to 6 = moderate, and 7 to 10 = severe. Efficacy was assessed separately in patients with pcJIA, JPsA, and ERA. Data for patients with sJIA without active systemic features were collected but not analyzed separately due to small numbers. Study visits occurred at baseline, month 1, month 3, and every 3 months thereafter, with annual assessments for uveitis. Efficacy analyses were based on observed data without imputation and included patients who received at least 1 dose of tofacitinib. Baseline for efficacy assessments was taken from the qualifying or index study or from the LTE baseline data if more than 14 days had passed before LTE study entry. JIA-ACR response rates and disease activity scores were reported through to month 48. To determine a patient’s inactive disease status, the uveitis result from the last assessment was carried forward until the next available assessment. Patient-reported outcomes, including CHAQ-DI scores, pain, and overall well-being scores, were analyzed descriptively over time.
At the time of publication, the LTE study20 is ongoing; efficacy and safety data collection was planned for up to 10 years. Results for efficacy and safety are presented for 105 months of follow-up at the interim analysis (data cut-off of July 11, 2022).20
Patient Disposition
Of the 225 patients enrolled in the LTE study, 26 patients were from a phase I pharmacokinetic study24 and 199 patients were from the Ruperto et al. (2021) study,15 comprising 185 patients with pcJIA (extended oligoarthritis [n = 27], RF-positive polyarthritis [n = 36], RF-negative polyarthritis [n = 111], and sJIA without active systemic features [n = 11]), 19 patients with JPsA, and 21 patients with ERA. At the time of the interim analysis, 102 of 225 patients (45.3%) had discontinued the LTE study.20 Reasons for study discontinuation were insufficient clinical response (13.3%), AEs (12.0%), withdrawal of consent by the parent or guardian (5.8%), withdrawal of consent by the patient (4.9%), other unspecified reasons (4.4%), loss to follow-up (1.8%), and pregnancy (0.9%). Forty-nine patients (21.8%) completed the LTE and 74 (32.9%) remained in the study. All 225 enrolled patients were included in the safety analysis set.
Baseline Characteristics
Baseline characteristics of patients in the LTE study20 are summarized in Appendix 4 in the Supplemental Material.
Compared to patients in the Ruperto et al. (2021) study,15 patients in the LTE study20 were overall similar in demographic and disease characteristics. However, there were differences between the Ruperto et al. (2021) study15 and the LTE study20 in the number of patients with RF-negative polyarthritis (46% vs. 56%, respectively) and number of patients with prior bDMARDs (38% vs. 31%, respectively).
Exposure to Study Treatments and Concomitant Medications
The duration of tofacitinib treatment before entry into the LTE study20 ranged from 5 days to 44 weeks. During the LTE study,20 patients received tofacitinib for a median of 41.6 months (range, 1 to 103 months), with a total of 700 patient-years of follow-up and a median follow-up time of 42.3 months (range, 1 to 104 months).
Concomitant medications used during the LTE study20 included NSAIDs (76.9%), methotrexate (65.8%), corticosteroids (44.9%), and hormonal contraceptives (5.8%).
Critical Appraisal
Internal Validity
The LTE study was an open-label, noncomparative extension study without a control arm. This design inherently limits the ability to attribute observed long-term outcomes directly to tofacitinib, as there was no concurrent comparator. Additionally, patients with prior tofacitinib or placebo treatment from the Ruperto et al. study15 were eligible for enrolment, and results were not stratified by prior treatment exposure. This introduces heterogeneity and may limit the interpretation of whether long-term efficacy or safety findings are attributable to continuous tofacitinib use or differences in the timing of treatment initiation.
Only patients who completed prior phase I or III trials or who discontinued for reasons unrelated to treatment-related SAEs were eligible, introducing a risk of selection bias toward participants who tolerated and responded well to tofacitinib. The use of validated outcome measures, including JIA-ACR response criteria, JADAS scores, and CHAQ-DI scores, strengthens the clinical reliability of the efficacy assessments. However, the analyses were based on observed data only, without imputation for missing values. This increases the potential for bias if data are not missing at random, particularly at later time points when the sample size becomes smaller.
The open-label nature of the study increases the risk for reporting and assessment bias, particularly for subjective outcomes such as pain and overall well-being. Furthermore, the study did not perform formal hypothesis testing and results were presented descriptively, further limiting the strength of conclusions.
Although background medications (e.g., methotrexate, corticosteroids) were permitted and could be adjusted during the study, details regarding these changes were limited. This restricts the ability to attribute observed outcomes solely to tofacitinib, particularly with respect to long-term safety and efficacy signals.
External Validity
The study enrolled a relatively broad population of patients with pcJIA, JPsA, or ERA, reflecting common clinical subtypes for which tofacitinib may be used. However, the exclusion of patients whose disease falls into certain JIA categories (e.g., refractory persistent oligoarthritis and undifferentiated JIA) may limit generalizability to the entire target population. The clinical experts also noted that eligible patients were those who completed prior phase I or III trials (or discontinued for reasons unrelated to AEs) whereas in the real world, patients for whom 1 therapy failed would have been switched to another therapy. Therefore, it would not be possible to monitor patients who discontinued due to lack of efficacy, as they will have received another biologic drug, making it challenging to disentangle observed long-term effects of tofacitinib from those of another biologic drug. The study therefore reflects the safety and effectiveness profile of patients who continue to be treated with tofacitinib, consistent with real-world experience.
Baseline characteristics of patients were heterogeneous, as those who entered the LTE study more than 14 days after their last study visit had a new baseline determined at point of entry into the LTE study, whereas for the remainder of patients, the baseline was taken from the baseline of the previous trial. This issue also applies to concomitant treatments.
Treatment regimens and follow-up durations in the LTE study generally reflect clinical use of tofacitinib in a long-term, real-world setting, supporting external relevance. The oral solution formulation used in the trial for patients weighing less than 40 kg is not available in Canada and must be compounded, which may limit local applicability for younger or lower-weight patients.
The use of internationally accepted efficacy measures, including JIA-ACR response criteria and JADAS scores, enhances the relevance of findings to clinical practice. Nonetheless, HRQoL outcomes were not formally evaluated, despite being a priority from patients’ and caregivers’ perspectives. The available 48-month follow-up data provide substantial insights into long-term treatment sustainability, though longer-term risks beyond this period require further study (planned for 10 years).
Results
Efficacy
Results relevant to this review are presented in Appendix 4 in the Supplemental Material.
Key results in the LTE study include the following:
For patients with pcJIA, the flare rate was 1.7% (1 out of 60 patients) at month 48. For patients with JPsA (n = 7) or ERA (n = 9), the flare rate was 0%.
For patients with pcJIA, JIA-ACR 30, JIA-ACR 50, and JIA-ACR 70 response rates were 96.7%, 93.4%, and 85.2%, respectively, at month 48 (61 patients). For patients with JPsA, JIA-ACR 30, JIA-ACR 50, and JIA-ACR 70 response rates were 100.0% (among 7 patients), 100.0% (among 6 patients), and 100.0% (among 6 patients), respectively. For patients with ERA, JIA-ACR 30, JIA-ACR 50, and JIA-ACR 70 response rates were 100.0% (among 9 patients), 100.0% (among 9 patients), and 75.0% (6 of 8 patients), respectively.
For patients with pcJIA, the JIA-ACR inactive disease rate was 40.6% (26 of 64 patients) at month 48.
For patients with pcJIA, the JIA-ACR clinical remission rate was 1.6% (1 of 64 patients) at month 48.
For patients with pcJIA, the mean JADAS-27 score based on CRP levels was 2.8 (standard error = 0.5) at month 48.
For patients with pcJIA, the proportion who achieved JADAS inactive disease was 46.8% (29 of 62 patients) at month 48.
For patients with pcJIA, the proportion who achieved JADAS minimal disease activity was 74.2% (46 of 62 patients) at month 48. For patients with JPsA, the proportion who achieved JADAS minimal disease activity was 57.1% (4 of 7 patients) at month 48. For patients with ERA, the proportion who achieved JADAS minimal disease activity was 37.5% (3 of 8 patients) at month 48.
For patients with pcJIA, the median CHAQ-DI score was 0.0 (Q1 = 0.0, Q3 = 0.6) at month 48.
For patients with pcJIA, the median patient or parent–reported pain score was 0.5 (Q1 = 0.0, Q3 = 1.0) month 48.
For patients with pcJIA, the median patient or parent–reported overall well-being score was 0.5 (Q1 = 0.0, Q3 = 1.5) at month 48.
Harms
Detailed harms data in the LTE study are available in Appendix 4 in the Supplemental Material.
A total of 201 of 225 patients (89.3%) experienced at least 1 AE over 700 patient-years of follow-up. The most frequently reported AEs (occurring in ≥ 5% of patients) were upper respiratory tract infection (21.3%), JIA exacerbation (12.4%), nasopharyngitis (12.0%), arthralgia (9.8%), viral infection (9.8%), urinary tract infection (9.3%), headache (8.9%), fever (8.9%), cough (8.4%), vomiting (8.4%), abdominal pain (8.4%), sinusitis (7.6%), influenza (7.6%), severe acute respiratory syndrome coronavirus 2 positive test (7.1%), COVID-19 (6.7%), oropharyngeal pain (6.7%), arthritis (6.2%), nausea (6.2%), disease progression (5.8%), pharyngitis (5.8%), bronchitis (5.3%), and ear infection (5.3%).
SAEs were reported in 34 of 225 patients (15.1%). Serious infections occurred in 4.4% of patients (10 of 225), including herpes zoster (n = 3) and limb abscess, COVID-19, Escherichia coli pyelonephritis, infectious mononucleosis, influenza, molluscum contagiosum, rhinovirus infection, and urinary tract infection (n = 1 each). A total of 4 herpes zoster cases were reported, 3 of which met criteria for serious opportunistic infections.
No deaths were reported during the study.
AEs led to permanent treatment discontinuation in 12.9% of patients (29 of 225). The most common reasons for discontinuation (in 2 patients each) included interferon gamma release assay positivity, arthritis, spontaneous abortion, aggravated condition, disease risk factor, herpes zoster, JIA exacerbation, pregnancy, and suicide attempt.
AESIs included serious infections in 10 patients (4.4%) and uveitis in 2 patients (0.9%). Both cases of uveitis were mild in severity and resolved without dose adjustment.
There were no reported cases of MACE, thromboembolic events (including deep vein thrombosis), malignancies (including nonmelanoma skin cancer), gastrointestinal perforation, interstitial lung disease, macrophage activation syndrome, or tuberculosis.
Indirect Evidence
Because the phase III randomized withdrawal study was placebo-controlled, there is a gap in the evidence regarding the efficacy and safety of tofacitinib relative to the comparators used to treat patients with pJIA (RF-positive or RF-negative polyarthritis, extended oligoarthritis, or sJIA without systemic manifestations), JPsA, or ERA whose disease has responded inadequately or who are intolerant to TNF inhibitors, or in whom the use of those therapies is inadvisable. We identified 1 ITC that included relevant comparators for tofacitinib (TNF inhibitors [e.g., etanercept, adalimumab, infliximab, golimumab], abatacept, and tocilizumab) and aligned with the population, intervention, comparison, outcomes, and study design of interest for the Reimbursement Review.
Description of Indirect Comparison
The ITC by Li et al. (2025)21 evaluated the efficacy and safety of JAK inhibitors and bDMARDs when used with or without methotrexate for the treatment of patients with nonsystemic forms of JIA. The ITC was based on a systematic literature review and employed Bayesian network meta-analysis (NMA) methods to estimate the comparative efficacy for JIA-ACR 70 response rate by week 16 (± 4 weeks) and SAEs. Eligible treatments in the NMA included bDMARDs (abatacept, adalimumab, anakinra, canakinumab, certolizumab, etanercept, golimumab, infliximab, rilonacept, rituximab, secukinumab, and tocilizumab) and JAK inhibitors (tofacitinib and baricitinib).
Study Selection and Review Methods
The literature search for the ITC included English-language published RCTs (randomized parallel and randomized withdrawal designs) in 3 databases and 1 clinical trial registry for records from the inception of the databases to December 2023. Eligible studies needed to include pediatric patients who were diagnosed with nonsystemic JIA (including all JIA subtypes except for sJIA with active systemic features) that investigated 1 of the bDMARDs, JAK inhibitors, or other small molecule drugs as previously identified. Outcomes of interest included JIA-ACR 70 response within 16 weeks (standard deviation = 4 weeks) of the initiation of treatment and SAEs. Studies that were conducted exclusively in children diagnosed with active sJIA, focused on uveitis in patients with JIA, reported secondary analysis of RCTs, or were published as conference abstracts were excluded. Information was reported for characteristics of the study (registration number, years of study, sample size, study design, and treatment groups) and for patient demographic characteristics (age, sex, race). Information at baseline on disease characteristics (duration of disease, JIA categories), the proportion of patients receiving specified background therapies (methotrexate, sulfasalazine, hydroxychloroquine, NSAIDs, and glucocorticoids), or whether background therapies were mandated by the study or permitted were not available. Whether the study eligibility criteria required patients to have demonstrated inadequate responses to csDMARDs or NSAIDs was also not available. Screening, data extraction, and an assessment of the methodological quality of trials (using version 2 of the Cochrane risk of bias tool for randomized trials) were conducted independently by 2 reviewers. Data extraction for each study arm for parallel randomized trials and for the open-label lead-in phase only for randomized withdrawal trials was conducted by 2 reviewers.
ITC Analysis Methods
The NMA was conducted using arm-based Markov chain Monte Carlo Bayesian methods with noninformative prior distributions for the model parameters. A hierarchical modelling approach was used to pool information across 11 different drugs within 5 drug classes. Random effects models were used for estimates. Rather than a contrast-based approach, an arm-based NMA was used to handle the 2 different study designs and to account for between-trial and within-trial correlation by partial pooling based on a hierarchical data structure. Under the transitivity assumption, the arm-based sample should be homogeneous. However, due to the exclusion of patients with specific JIA subtypes in some studies, the distribution of patients with JIA subtypes varied by study. This was addressed by including the percentages of patients with JIA subtypes as arm-level covariates in the model. Sensitivity analyses were performed to exclude trials that focused exclusively on patients with JPsA and/or ERA and to exclude trials at high risk of bias. Geometry of the network for the analysis was summarized in a network diagram based on study type, number of patients, and trials. Convergence was evaluated using trace plots, autocorrelation functions, and potential scale reduction factor, with codes specified. The node-split method was used to evaluate the inconsistency between direct and indirect evidence. No information was provided on the selection of the primary end point follow-up time point.
Summary of Included Studies
The NMA included 16 trials (7 parallel and 9 randomized withdrawal) evaluating 9 bDMARDs (e.g., adalimumab, etanercept, golimumab, secukinumab; 3 were combined with methotrexate) and the 2 JAK inhibitors with a total of 1,821 patients. All the randomized parallel trials examined a TNF inhibitor. There were 2 trials (N = 404) of a JAK inhibitor; 5 trials (N = 400) of a TNF inhibitor; 7 trials (N = 284) of a TNF inhibitor plus methotrexate; single trials of each of T-cell inhibitor (abatacept; N = 190), IL-6 inhibitor (tocilizumab; N = 188), and IL-17a inhibitor (secukinumab; N = 86); and 269 controls. Risk of bias was assessed as low in 12 trials, with some concerns regarding selective outcome reporting in 3 trials (etanercept plus methotrexate vs. controls [2 trials]; etanercept), and it was assessed as high due to protocol deviations, outcome measurement, and missing outcome data in 1 trial (infliximab plus methotrexate). Across the trials, there was heterogeneity in the study start year (range, 1997 to 2018), years of study duration (range, 1 to 4 years), sample sizes of treatment groups (range, 12 to 220), proportion of females (range, 24% to 88%), age (range, 8.3 to 14 years), race distribution, years of JIA duration (range, 0.25 to 5.9 years), JIA subtype distribution, and background therapies. Baseline disease activity was also heterogeneous across the trials in mean active joint count (range, 5.4 to 25.5 joints), mean joint count with loss of range of motion (range, 2 to 18.4 joints), and mean score on the physician global assessment of disease activity (range, 4.73 to 8), mean score on the patient or parent assessment of overall well-being (range, 1.8 to 8 score), and mean CHAQ score (range, 0.51 to 1.5 score). No information was provided in the publication on dose, duration, or frequency of treatment(s) or follow-up duration.
Direct pairwise comparison was available for patients treated with adalimumab versus those treated with adalimumab plus methotrexate, with indirect evidence for the remaining bDMARD comparisons. There were no closed loops in the network and the study authors did not report on consistency.
Critical Appraisal of ITC
The systematic literature search and study selection process did not raise major concerns. Study inclusion criteria were defined, and screening, data extraction, and risk of bias assessments were performed in duplicate by independent reviewers. However, important trial-level information was not reported in the evidence base, which limited the interpretability of the results and generalizability to the population of interest for this review. Specifically, no information on dose, timing, or duration of treatment (aside from drug exposure by person-years) was reported per trial; this is critical information for determining whether the drugs were adequately similar for pooling within each drug class. This limitation is notable given that input from the clinical experts indicated that JAK inhibitors have an observed faster onset of effect compared with some bDMARDs; it is unclear whether the pooled trials and comparisons were similar in this respect. Harms outcomes were missing key safety information (specific AEs, discontinuations due to AEs, deaths, and AESIs); the safety outcome of SAEs did not specify types of SAEs or system organ class, which precluded meaningful interpretation. However, it is acknowledged that lower event frequencies for individual AEs would necessitate using the aggregate counts. Follow-up duration in the trials was also not stated, which may affect harms data and interpretability; follow-up likely varied across the trials based on the reported study periods.
The NMA was conducted using an arm-based Bayesian hierarchical model with partial pooling by drug class, integrating data from both parallel-group and treatment withdrawal RCTs. A common within-trial correlation statistic (ρ) was assumed for all multiarm trials. While the arm-based approach was justified to accommodate differences in study design and to account for within- and between-trial correlations, the validity of the key transitivity assumption is uncertain. In this analysis, transitivity implies that all arms within the same drug class—regardless of whether they came from parallel-group or withdrawal trials—share similar treatment effects. Given the heterogeneity across trials in key demographic characteristics, disease characteristics, baseline disease indicators, and unreported factors such as prior biologic drug use and disease duration, this assumption may not hold. Adjustment for JIA subtype distribution via arm-level covariates partially addresses 1 potential effect modifier, but it is unclear whether this fully mitigates bias. Other potentially important effect modifiers were not included.
Reporting of the statistical methods for the Bayesian NMA lacked detail on the prior probability distribution used for treatment effects and was missing information on the numbers of iterations and chains. Without these details, it is not possible to assess the stability of the posterior estimates or the influence of priors, which can be substantial in sparse networks. The network geometry was sparse, with no closed loops, and results were imprecise, with wide 95% credible intervals. Only local (node-split) inconsistency was reported; no global inconsistency assessment was described.
Rationale for the selected time point of 16 weeks (standard deviation = 4 weeks) was also not provided. Nevertheless, this appeared to be in line with the time point of 12 to 16 weeks for assessing treatment response, according to the clinical experts. Although the authors prespecified sensitivity analyses that excluded studies of patients with JPsA or ERA and trials with high risk of bias, it is unclear if other sensitivity analyses were important to address potential differential treatment effects (e.g., studies of patients with vs. without inadequate disease response to DMARDs at baseline).
Efficacy and Harms Results of the ITC
Key results of the ITC are presented in Table 6. Key results include the following:
Aggregate summary statistics for JIA-ACR 70 response rates ranged from 11.29% to 89.47% and SAE incidence rates ranged from 0 to 0.32 per person-year. Across all trials, treatment exposure ranged from 3.46 to 58.46 person-years.
For patients with all subtypes of nonsystemic JIA (excluding sJIA with active systemic features), the evidence was insufficient to demonstrate a difference in JIA-ACR 70 response rates at week 16 or in SAE incidence rates between treatment groups.
Results of sensitivity analyses were consistent with the base case.
Study authors reported there was good model convergence, and when the models were evaluated for consistency of direct to indirect evidence, the node-split method did not identify statistically significant evidence of inconsistency (P = 0.709).
Table 6: Summary of Efficacy Results in the NMA
Comparator | Tofacitinib vs. comparator | |
|---|---|---|
JIA-ACR 70a at 16 weeks, OR (95% CrI) | SAEs, IRR (95% CrI) | |
Control | 3.02 (0.98 to 9.52) | 1.28 (0.46 to 3.7) |
Etanercept | 1.04 (0.3 to 3.68) | 1.06 (0.34 to 3.73) |
Adalimumab | 1.39 (0.4 to 4.79) | 0.8 (0.25 to 2.37) |
Golimumab | 0.8 (0.19 o 3.02) | 0.92 (0.31 to 2.64) |
Abatacept | 1.68 (0.41 to 8.31) | 1.18 (0.37 to 3.86) |
Tocilizumab | 0.98 (0.23 to 3.9) | 0.76 (0.21 to 2.62) |
Etanercept plus methotrexate | 0.87 (0.3 to 2.67) | 1.33 (0.46 to 5.1) |
Adalimumab plus methotrexate | 0.76 (0.2 to 2.47) | 0.92 (0.28 to 3.08) |
Infliximab plus methotrexate | 1.02 (0.32 to 3.43) | 0.79 (0.29 to 2.23) |
CrI = credible interval; IRR = incidence rate ratio; JIA-ACR = juvenile idiopathic arthritis–American College of Rheumatology; OR = odds ratio; NMA = network meta-analysis; SAE = serious adverse event; VAS = visual analogue scale.
Note: Estimates were based on a random-effects model. The number of studies contributing to each effect estimate was not available.
aJIA-ACR 70 response criteria were defined as 3 of 6 JIA core set variables improved by a minimum of 70%, with no more than 1 JIA core set variable worsened by 30% or more.
Source: Li et al. (2025)21
Discussion
Efficacy
Patients living with JIA and their parents or caregivers reported challenges related to their disease (e.g., joint pain, swelling, and stiffness limited their ability to participate in activities of daily living and recreation, requiring accommodations at school and work). They also reported challenges related to accessing health care (e.g., access to rheumatologists, time and distance required for travel to clinic), receiving information on pharmacologic and nonpharmacologic treatments, accessing insurance coverage for medications and extended health benefits, balancing health care needs with other priorities, and experiencing negative effects on their mental health. Clinicians identified treatment goals for patients with JIA are to control disease inflammation, achieve inactive disease, prevent and minimize joint damage and deformity, preserve physical function, prevent visual morbidities, achieve and prolong disease remission, support growth and development, and restore HRQoL. Clinicians expressed that tofacitinib is a JAK inhibitor that offers an intracellular mechanism of action that is distinct from bDMARDs or csDMARDs and is the first orally administered advanced therapy for pediatric patients with persistent disease or intolerance to parenteral treatments.
The randomized withdrawal phase III trial demonstrated superiority of tofacitinib over placebo on several measures of disease activity and physical function.15 Tofacitinib reduced JIA flare rate by week 44; improved rates of JIA-ACR 30, 50, and 70 responses by week 44; and improved CHAQ-DI scores between the study part 2 baseline and week 44 for patients with pcJIA. Tofacitinib was also associated with greater improvement over placebo in most other disease end points, including JADAS-27 scores, but the lack of adjustment for type I error for these outcomes precludes drawing firm conclusions of superiority with tofacitinib compared with placebo. However, there is uncertainty about the clinical significance of the results, especially without an active comparator. No minimal important difference has been established for the JIA-ACR core set variable of number of joints with active arthritis, patient or parent JIA-ACR assessment of overall well-being, or JADAS-27 scores or scores for individual indices. No between-group thresholds were identified by the clinical experts and only within-group cut-offs for disease activity scores were found in the literature. On balance, the clinical experts consulted considered the efficacy results of the trial to be clinically important in favour of tofacitinib.
To address the lack of head-to-head trials comparing tofacitinib with currently available treatments for JIA, ITC evidence was needed. A literature search identified 1 published ITC based on NMA methods that aligned with the population, intervention, comparison, outcomes, and study designs of interest for this review. In the NMA, tofacitinib showed odds ratios ranging from 0.76 (favouring adalimumab plus methotrexate) to 1.68 (favouring tofacitinib over abatacept) when compared with other treatments for achieving JIA-ACR70 at week 16. All 95% credible intervals were wide and crossed the null value, indicating a high degree of statistical imprecision. This level of uncertainty means that the available data do not reliably establish whether tofacitinib provides a clinically important advantage or disadvantage compared with any included comparator. The statistical imprecision is compounded by methodological limitations in the NMA: absence of clear reporting on homogeneity and consistency assumptions, lack of details on prior distributions, and the need to assume a uniform within-trial arm-to-arm correlation for all multiarm studies. These factors limit the robustness of the relative effect estimates, particularly in a network in which several comparisons are informed by sparse data or indirect evidence only. Given the credible intervals include superiority, similarity, and inferiority, the current evidence base does not allow confident differentiation of tofacitinib’s efficacy from that of other active treatments at 16 weeks.
The PROPEL trial15 involved a broader patient population than the one specified in the approved indication. Relevant subgroups in the reimbursement request include patients with JPsA or ERA. The findings for patients with JPsA or ERA were limited in interpretation due to very small sample sizes and the absence of between-group differences (likely precluded by the small sample size). Post hoc analyses of data from patients with pcJIA, JPsA, or ERA were consistent with the primary analyses of data from patients with pcJIA, suggesting greater improvements with tofacitinib over placebo in achievement of minimal disease activity, clinically inactive disease, and clinical remission based on JADAS-10, as well as greater improvements in rates of patients with clinically inactive disease and clinical remission based on JIA-ACR criteria and in rates of clinically inactive disease or clinical remission combined with normal physical function (CHAQ score of 0) at week 44. Clinical experts emphasized that despite the limited efficacy data for tofacitinib in patients with JPsA or ERA, these patients are included in their clinical population.
The indication specifies patients whose disease has responded inadequately or who are intolerant to TNF inhibitors, or in whom use of those therapies is inadvisable, yet the product monograph does not explicitly state why Health Canada included these criteria for use. In the PROPEL trial, about two-thirds of patients had not previously used bDMARDs while the remaining one-third of patients had used 1 or more bDMARDs. However, the PROPEL trial did not undertake analyses to assess the effect of previous TNF inhibitor use on treatment effects. The clinical experts agreed that it would be appropriate to position tofacitinib treatment after TNF inhibitors given the established efficacy and harms profiles of these drugs in pediatric and adult populations. However, 1 of the clinical experts suggested that tofacitinib could also be considered as first-line therapy following treatment with methotrexate (or other csDMARD) therapy, per the PROPEL trial. Of note, the health technology assessment of tofacitinib by the UK’s National Institute for Health Care Excellence recommended its use in patients whose disease has responded inadequately or who are intolerant to TNF inhibitors, or in whom the use of those therapies is inadvisable, citing similar efficacy as adalimumab and etanercept but higher costs, warranting a second-line placement.25
While the approved indication for tofacitinib is for patients weighing 40 kg or more, the reimbursement request does not specify a minimum body weight. The PROPEL trial included dosages less than 5 mg, which is the minimum strength available in Canada. Subgroups analyses of patients with JIA flare rate and JIA-ACR 30 response at week 44 were prespecified for body weight but were considered exploratory, with small sample sizes and often wide CIs that neared or crossed the null. A liquid formulation for children weighing less than 40 kg is available in other jurisdictions, but not in Canada. The clinical experts highlighted this as an access issue and a limitation to prescribing tofacitinib. They noted that the chemical instability of tofacitinib means that effective and stable compounding of strengths less than 5 mg is limited to 14 days, requiring a new bottle of medication to be picked up every 14 days. The clinical experts consulted for this review emphasized that tofacitinib as an oral drug would fulfill an unmet need for many pediatric patients, especially for whom an orally administered treatment may be more acceptable over SC injections or infusion treatments.
No rationale was provided by the PROPEL study15 for its exclusion of patients with refractory persistent oligoarthritis or undifferentiated JIA, so it is unclear whether the findings may be extrapolated to these subtypes with their associated poor disease prognosis and potential to benefit from tofacitinib, according to the clinical experts. In line with the PROPEL trial,15 the clinician group input identified these patients among those with JIA subtypes who would be least suitable for treatment with tofacitinib.
The pivotal trial15 specified that the rationale for the randomized withdrawal study design was to limit participants’ exposure to placebo treatment due to the availability of effective treatments for JIA. Theoretically, such a trial design may increase the probability of detecting a treatment effect, given that only patients demonstrating response were eligible for participation in the placebo-controlled withdrawal phase. A high rate of study discontinuations due to disease flare in both study parts 1 and 2 precluded a meaningful assessment of the impact of the run-in phase on the validity of the trial results. Moreover, interpretation of efficacy at the 12 to 16 week time point is hampered by the open-label and noncomparative design; it’s unclear how findings during study part 1 translate into the withdrawal phase or real-world practice.
Core outcomes in patients with JIA include joint pain, joint inflammatory signs, inflammation related to extra-articular features, physical function and activity limitations, AEs, perception of disease activity, and overall patient perception. The outcomes used in the trial are widely adopted in clinical trials, including the JIA-ACR response criteria, JADAS-27, and JADAS-10. Input from the clinician group indicated JADAS-27 is used most commonly in Canada. However, according to the clinical experts consulted, stand-alone assessments (e.g., select JIA-ACR core set variables) are more commonly undertaken in clinical practice over composite indices (e.g., JADAS-10) due to time constraints. They expressed that standardized response criteria such as JIA-ACR 30, 50, 70, 90, and 100 are often used in clinical trials but are cumbersome to assess in clinical practice. With evolving thresholds for interpretation of composite end points (e.g., based on JIA subtypes) and their limited applicability in clinical practice for consistent adoption, it is unclear whether findings from clinical trials — primarily the magnitude of treatment effect — are generalizable to patients in the real world.
Efficacy end points used in the LTE study were aligned with those of the PROPEL study, with findings supporting sustained benefit of tofacitinib after 48 weeks of treatment. However, the open-label design and descriptive analyses limit the interpretability of findings. Thus, the longer-term efficacy of tofacitinib is unclear.
Harms
While AEs in the tofacitinib and placebo groups were similar for patients with at least 1 AE and SAEs including serious infections, there were scant details on specific AEs experienced by patients in the trial. The frequency of study discontinuations due to AEs was lower for patients treated with tofacitinib and no deaths were observed over the course of the study.
A higher proportion of patients in the LTE study versus the PROPEL study experienced a TEAE (89% vs. 77%, respectively) including upper respiratory tract infection (21% vs. 15%), nasopharyngitis (12% vs. 8%), JIA exacerbation (12% vs. 3%), headache (9% vs. 2%), and vomiting (8% vs. 0%). A smaller proportion of patients experienced disease progression with tofacitinib during the LTE study (6%) relative to the PROPEL study (9%). A higher proportion of patients had SAEs during the LTE study (15%) than the PROPEL study (1%).
According to the clinical experts, the safety profile of tofacitinib was very comparable to other biologic drugs and possibly better than some approved biologic drugs that are commonly used for patients with JIA. The experts shared that thrombotic events have been highlighted due to cardiovascular-related AEs among adults; however, compared to adults who often have multiple secondary comorbidities (e.g., hypertension, diabetes, lipid abnormalities), documented thrombotic events have been rare among pediatric populations. Although the frequency of SAEs appeared to be high during the LTE study relative to the PROPEL study, the experts noted that issues of adherence with oral tablets should be considered in interpreting the data. Importantly, the clinical experts indicated that no deaths due to AEs were reported during the PROPEL study (44 weeks of treatment) or with prolonged tofacitinib exposure in the LTE study (48 weeks of treatment).
Harms information in the ITC only included SAEs. As with the efficacy analyses, there was not clear a distinction in the comparative safety between tofacitinib and other active treatments. The limitations that apply to the efficacy analyses also apply to the SAE analyses. Therefore, no firm conclusion about the comparative harms can be made.
Conclusion
Patients with JIA and their caregivers seek additional treatment options with improved forms of administration that can reduce disease symptoms, enable participation in activities of daily living and recreation, and reduce parent or caregiver burden. Clinicians advocate for patients with pJIA, JPsA, or ERA to have more efficacious treatment options to control disease inflammation, achieve inactive disease, prevent and minimize joint damage and deformity, preserve physical function, prevent visual morbidities, achieve and prolong disease remission, support growth and development, and restore HRQoL. Evidence from a randomized withdrawal phase III trial (PROPEL) that included 225 children aged 2 years to younger than 18 years with active pcJIA (RF-positive or RF-negative polyarthritis, extended oligoarthritis, or sJIA without systemic manifestations) demonstrated tofacitinib provides superior improvement versus placebo with reduced JIA flare rate by week 44; improved JIA-ACR 30, 50 and 70 response rates at week 44; and improved CHAQ-DI scores by week 44, and it addresses key treatment goals for patients with pJIA. While the clinical significance of the results is uncertain, clinical experts consulted by CDA-AMC considered the totality of the effects to represent an important benefit with tofacitinib for patients whose disease has responded inadequately to methotrexate and who have or have not previously used bDMARDs, including patients whose disease has responded inadequately or who are intolerant to a TNF inhibitor, or when use of those therapies is inadvisable. Results for patients with JPsA and ERA suggested greater improvement with tofacitinib compared with placebo for disease flare and appeared to be aligned with those for the pcJIA population; however, there is greater uncertainty considering the very small number of patients in the trial. An NMA did not identify clear differences in comparative efficacy or harms with tofacitinib and relevant comparators, but the analyses lacked precision and were limited by methodological limitations. The harms profile of tofacitinib was as expected with no new safety signals in patients with JIA. The LTE study of the phase III trial demonstrated findings supportive of the PROPEL study; however, the open-label study design limits the ability to distinguish effects of treatment from concomitant therapies, the natural history of the disease, or other factors.
Economic Review
Methods
CDA-AMC Assessment of Costs
The Economic Review consisted of a cost comparison for tofacitinib, with or without methotrexate, versus TNF inhibitors (etanercept, adalimumab, golimumab), abatacept, or tocilizumab for the treatment of active pJIA (RF-positive or RF-negative polyarthritis, extended oligoarthritis, and sJIA without systemic manifestations), JPsA, and ERA in children who have responded inadequately or are intolerant to TNF inhibitors or when use of those therapies is inadvisable [wording from original source].
Based on public list prices, tofacitinib with methotrexate is expected to have a per patient cost of $5,393 per patient per year (refer to Appendix 5 in the Supplemental Material). Etanercept, adalimumab, golimumab, abatacept, and tocilizumab (both SC and IV) are expected to have per patient costs between $6,321 (tocilizumab SC) and $20,837 (golimumab) per year. Therefore, the difference in the cost of tofacitinib compared to bDMARDs varies between –$928 (tocilizumab SC) and –$15,444 (golimumab) per patient per year. As such, the reimbursement of tofacitinib for the indication of this review is expected to decrease overall drug acquisition costs. Certain doses and comparators are different for patients with sJIA or ERA compared to those with other pJIA subtypes, and tofacitinib is expected to have lower costs for patients with these subtypes, as well. Additional items for consideration are provided in the following bullets:
According to the Clinical Review, evidence from the published literature was insufficient to demonstrate a difference in clinical outcomes and SAEs between bDMARDs for patients with all subtypes of nonsystemic JIA.
Several generic formulations of tofacitinib are currently marketed in Canada. According to the pan-Canadian Pharmaceutical Alliance Tiered Pricing Framework,26 oral solid generic drugs sourced by 3 or more manufacturers are priced at 25% of the brand reference price.
No health care resource use outcomes were reported in the studies included in the Clinical Review.
According to the clinical experts consulted for this review, hospitalizations due to the disease are uncommon in the population covered by this review and tofacitinib is not expected to lead to SAEs. Tofacitinib is expected to have similar monitoring costs relative to other comparators. Because tofacitinib is a tablet administered orally, its adoption is expected to decrease the utilization of drug administration–related health care resources when compared to drugs administered through IV infusion (by a health care professional in a specialized facility) or SC injection (requiring injection-related supplies, administration training, and cold chain logistics for transportation or storage). Moreover, tofacitinib may result in increased adherence to treatment given its oral administration, which in turn may reduce complications and use of associated health care resources. However, there is no evidence of differences in utilization of other health care resources due to the use of tofacitinib relative to its comparators.
Tofacitinib was not previously reviewed by CDA-AMC for this indication. As of August 2025, there are no drugs under review for this indication.
No cost-effectiveness studies conducted in Canada were identified based on a literature search conducted on June 13, 2025, with alerts maintained until the Formulary Management Expert Committee meeting on September 18, 2025.
Conclusion
The reimbursement of tofacitinib for the indication of this review is expected to decrease overall drug acquisition costs. Based on the Clinical Review conclusions, results from published studies do not suggest a difference in JIA-ACR 70 response rates at week 16 or in SAE incidence rates among patients treated with tofacitinib compared with those treated with bDMARDs. The comparative efficacy of tofacitinib versus bDMARDs on other clinical outcomes is unknown, as it has not yet been assessed in the published literature.
Given that tofacitinib is associated with lower drug acquisition costs and uncertain clinical benefit versus other active comparators, reimbursement of tofacitinib may result in reduced up front costs to the public health care system with uncertain benefit.
References
1.Grom AA. Post TW, ed. Juvenile idiopathic arthritis: epidemiology and immunopathogenesis. UpToDate; May 2025. Accessed August 5, 2025. http://www.uptodate.com
2.Nolan B. Post TW, ed. Classification of juvenile idiopathic arthritis. UpToDate; May 17, 2024. Accessed August 5, 2025. http://www.uptodate.com
3.Sandborg CI, Schulert GS, Kimura Y. Juvenile Idiopathic Arthritis. N Engl J Med. 2025;393(2):162-174. doi: 10.1056/NEJMra2402073 PubMed
4.Kimura Y, Schulert G. Post TW, ed. Systemic juvenile idiopathic arthritis: clinical manifestations and diagnosis. UpToDate; January 1, 2024. Accessed August 5, 2025. http://www.uptodate.com
5.Kimura Y, Schulert G. Post TW, ed. Systemic juvenile idiopathic arthritis: complications. UpToDate; December 9, 2024. Accessed August 5, 2025. http://www.uptodate.com
6.Weiss PF. Post TW, ed. Oligoarticular juvenile idiopathic arthritis. UpToDate; April 9, 2025. Accessed August 7, 2025. http://www.uptodate.com
7.Weiss PF. Post TW, ed. Polyarticular juvenile idiopathic arthritis: clinical manifestations, diagnosis, and complications. UpToDate; December 12, 2023. Accessed August 5, 2025. http://www.uptodate.com
8.Nigrovic PA. Post TW, ed. Psoriatic juvenile idiopathic arthritis: epidemiology, clinical manifestations, and diagnosis. UpToDate; December 9, 2023. Accessed August 5, 2025. http://www.uptodate.com
9.Public Health Agency of Canada. Canadian Chronic Disease Surveillance System (CCDSS) [Data Tool]. July 2024. Accessed August 5, 2025. https://health-infobase.canada.ca/ccdss/data-tool/
10.Kimura Y, Schulert G. Post TW, ed. Systemic juvenile idiopathic arthritis: treatment and prognosis. UpToDate; December 9, 2024. Accessed August 7, 2025. http://www.uptodate.com
11.Onel KB, Horton DB, Lovell DJ, et al. 2021 American College of Rheumatology Guideline for the Treatment of Juvenile Idiopathic Arthritis: Therapeutic Approaches for Oligoarthritis, Temporomandibular Joint Arthritis, and Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2022;74(4):553-569. doi: https://doi.org/10.1002/art.42037 PubMed
12.Ringold S, Angeles-Han ST, Beukelman T, et al. 2019 American College of Rheumatology/Arthritis Foundation Guideline for the Treatment of Juvenile Idiopathic Arthritis: Therapeutic Approaches for Non-Systemic Polyarthritis, Sacroiliitis, and Enthesitis. Arthritis Care Res (Hoboken). 2019;71(6):717-734. doi: 10.1002/acr.23870 PubMed
13.Fautrel B, Mitrovic S, De Matteis A, et al. EULAR/PReS recommendations for the diagnosis and management of Still's disease, comprising systemic juvenile idiopathic arthritis and adult-onset Still's disease. Ann Rheum Dis. 2024;83(12):1614-1627. doi: 10.1136/ard-2024-225851 PubMed
14.Weiss PF. Post TW, ed. Polyarticular juvenile idiopathic arthritis: treatment and prognosis. UpToDate; September 25, 2024. Accessed August 7, 2025. http://www.uptodate.com
15.Ruperto N, Brunner HI, Synoverska O, et al. Tofacitinib in juvenile idiopathic arthritis: a double-blind, placebo-controlled, withdrawal phase 3 randomised trial. Lancet. 2021;398(10315):1984-1996. doi: 10.1016/S0140-6736(21)01255-1 PubMed
16.Lim LSH, Shobhan S, Lokku A, Ringold S, Pullenayegum E. Latent classes of early response trajectories to biologics initiation in juvenile idiopathic arthritis: an analysis of four trials. Pediatr Rheumatol Online J. 2022;20(1):57. doi: 10.1186/s12969-022-00719-1 PubMed
17.Morgan EM, Munro JE, Horonjeff J, et al. Establishing an Updated Core Domain Set for Studies in Juvenile Idiopathic Arthritis: A Report from the OMERACT 2018 JIA Workshop. J Rheumatol. 2019;46(8):1006-1013. doi: 10.3899/jrheum.181088 PubMed
18.Bulatovic Calasan M, de Vries LD, Vastert SJ, Heijstek MW, Wulffraat NM. Interpretation of the Juvenile Arthritis Disease Activity Score: responsiveness, clinically important differences and levels of disease activity in prospective cohorts of patients with juvenile idiopathic arthritis. Rheumatology (Oxford). 2014;53(2):307-12. doi: 10.1093/rheumatology/ket310 PubMed
19.Consolaro A, Ruperto N, Lovell DJ, et al. Clinically Inactive Disease and Remission in Patients With Juvenile Idiopathic Arthritis Receiving Tofacitinib: Post Hoc Analysis of a Phase III Trial. J Rheumatol. 2025. doi: 10.3899/jrheum.2024-0536 PubMed
20.Brunner HI, Akikusa JD, Al-Abadi E, et al. Safety and efficacy of tofacitinib for the treatment of patients with juvenile idiopathic arthritis: preliminary results of an open-label, long-term extension study. Ann Rheum Dis. 2024;83(11):1561-1571. doi: 10.1136/ard-2023-225094 PubMed
21.Li Y, Huang B, Andorf S, Yue X, Lovell DJ, Brunner HI. Comparative Effectiveness and Safety of the JAK Inhibitors and Biologic Disease-Modifying Antirheumatic Drugs in Treating Children With Nonsystemic Juvenile Idiopathic Arthritis: A Bayesian Meta-Analysis of Randomized Controlled Trials. ACR Open Rheumatol. 2025;7(2):e11788. doi: 10.1002/acr2.11788 PubMed
22.Pfizer. Efficacy, Safety and Tolerability of Tofacitinib for Treatment of Polyarticular Course Juvenile Idiopathic Arthritis (Jia) in Children and Adolescent Subjects. ClinicalTrials.gov. Updated 2020/04/13/. https://clinicaltrials.gov/study/NCT02592434
23.Pfizer. A Long-Term, Open-Label Follow-up Study of Tofacitinib for Treatment of Juvenile Idiopathic Arthritis (Jia). ClinicalTrials.gov. Updated 2025/05/06/. https://clinicaltrials.gov/study/NCT01500551
24.Ruperto N, Brunner HI, Zuber Z, et al. Pharmacokinetic and safety profile of tofacitinib in children with polyarticular course juvenile idiopathic arthritis: results of a phase 1, open-label, multicenter study. Pediatr Rheumatol Online J. 2017;15(1):86. doi: 10.1186/s12969-017-0212-y PubMed
25.National Institute for Health and Care Excellence. Technology appraisal guidance. TA735: Tofacitinib for treating juvenile idiopathic arthritis. October 20, 2021. Accessed August 27, 2025. https://www.nice.org.uk/guidance/ta735
26.pan-Canadian Pharmaceutical Alliance. Generic drugs. Accessed July 29, 2025. https://www.pcpacanada.ca/generic-drug-framework
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@cda-amc.ca.