CADTH Reimbursement Review
Pemigatinib (Pemazyre)
Sponsor: Incyte Biosciences Canada Corporation
Therapeutic area: Cholangiocarcinoma
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
ASC
active symptom control
BTC
biliary tract cancer
CCA
cholangiocarcinoma
CGOEN
Canadian Gastrointestinal Oncology Evidence Network
CI
confidence interval
CR
complete response
CYP3A4
cytochrome P3A4
DCR
disease control rate
DOR
duration of response
eCCA
extrahepatic cholangiocarcinoma
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EORTC
European Organisation for Research and Treatment of Cancer
EORTC QLQ-BIL21
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Cholangiocarcinomas and Gallbladder Cancer Module 21
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
FGF
fibroblast growth factor
FGFR
fibroblast growth factor receptor
FGFR2
fibroblast growth factor receptor 2
FOLFIRI
folinic acid, fluorouracil, and irinotecan hydrochloride
FOLFOX
folinic acid, fluorouracil, and oxaliplatin
HR
hazard ratio
HRQoL
health-related quality of life
ICC
intraclass correlation coefficient
iCCA
intrahepatic cholangiocarcinoma
IDH
isocitrate dehydrogenase
IRC
independent review committee
ITC
indirect treatment comparison
KM
Kaplan-Meier
KPS
Karnofsky performance status
MAIC
matching-adjusted indirect comparison
mFOLFOX
modified folinic acid, fluorouracil, and oxaliplatin
MID
minimally important difference
NDA
New Drug Application
ORR
objective response rate
OS
overall survival
PD
progressive disease
PFS
progression-free survival
PP
per protocol
PR
partial response
QoL
quality of life
RCT
randomized controlled trial
RECIST 1.1
Response Evaluation Criteria in Solid Tumours Version 1.1
SAE
serious adverse event
SD
standard deviation
TEAE
treatment-emergent adverse event
WDAE
withdrawal due to adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Pemigatinib (Pemazyre) tablets, 4.5 mg, 9 mg, and 13.5 mg, oral |
Indication | For the treatment of adults with previously treated, unresectable, locally advanced, or metastatic CCA with an FGFR2 fusion or other rearrangement |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | September 17, 2021 |
Sponsor | Incyte Biosciences Canada Corporation |
CCA = cholangiocarcinoma; FGFR2 = fibroblast growth factor receptor 2; NOC = Notice of Compliance.
Introduction
Gallbladder cancer and cholangiocarcinoma (CCA) are known as biliary tract cancers (BTCs) accounting for 10% to 15% of all primary liver cancer.1,2 CCAs are most commonly adenocarcinomas and comprise 2 main subtypes: intrahepatic (iCCA), initiating from the biliary tree within the liver, and extrahepatic (eCCA), initiating outside the liver parenchyma.2,3 In Canada and the US, respectively, there are approximately 400 and 5,000 new cases of CCA diagnosed each year.4 The median age at diagnosis is 65 years in Western industrialized nations.5 The 5-year relative survival rates for iCCA and eCCA, respectively, are 9% and 10%.6 Diagnosis of CCA is most commonly made in advanced stages (70% of patients are diagnosed with unresectable, locally advanced or metastatic disease)7 due to an absence of symptoms until later in the course of the disease.8 The rate of recurrence is high in the minority of patients who are able to undergo potentially curative surgery.9 Symptoms commonly appear when a bile duct is blocked and include jaundice, itching, light-coloured and greasy stools, dark urine, abdominal pain, loss of appetite or weight loss, fever, and nausea and vomiting.8
One of the most frequent genetic alterations in patients with iCCA involve the fibroblast growth factor receptor (FGFR) 2 (FGFR2).7 The FGFR2 fusions or rearrangements are found in 10% to 20%10 of patients with iCCA, while they rarely occur in eCCA. Alterations involving other members of the FGFR are rare, with an incidence below 0.5%.11 While there is strong genetic and functional evidence that FGFR genetic alterations can drive the formation of tumours,7 it is currently not known whether patients whose disease is positive for an FGFR2 alteration represent a distinct prognostic subgroup.11
For patients with advanced-stage or unresectable CCA and a good Eastern Cooperative Oncology Group Performance Status (ECOG PS) score of 0 or 1, standard-of-care first-line treatment is gemcitabine and cisplatin.9 If there are concerns about a patient’s renal function, oxaliplatin may be substituted for cisplatin.2 For patients with an ECOG PS of 2, gemcitabine monotherapy may be considered as first-line therapy.2 The median overall survival (OS), median progression-free survival (PFS), and objective response rate (ORR) in patients with BTCs treated with standard-care first-line palliative treatment with gemcitabine and cisplatin ranges from 11.2 to 11.7 months, 5.8 to 8.0 months, and 19.5% to 26.1%, respectively.12 The clinical experts consulted by CADTH noted there are currently no funded standard treatment options for patients in the second-line setting once the disease has progressed on first-line treatment. In the absence of proven treatment options in the second-line setting, participation in a clinical trial and best supportive care are recommended, including alleviating biliary obstruction and full access to palliative care and symptom management.2 According to the clinical experts consulted by CADTH, the second-line therapies used in Canadian clinical practice include folinic acid, fluorouracil, and oxaliplatin (FOLFOX); folinic acid, fluorouracil, and irinotecan hydrochloride (FOLFIRI); fluorouracil (alone or in combination with cisplatin or oxaliplatin); and capecitabine (alone or in combination with cisplatin or oxaliplatin). Second-line treatment with FOLFOX is currently the only drug based on phase III trial data in this setting.5 The ABC-06 trial13 evaluated the efficacy and safety of modified folinic acid, fluorouracil, and oxaliplatin (mFOLFOX) plus active symptom control (ASC) compared with ASC alone in patients with locally advanced or metastatic BTC (including CCA and gallbladder or ampullary carcinoma) whose disease had progressed on first-line cisplatin and gemcitabine therapy. At the median follow-up time of 21.7 months, median OS was 6.2 months in the FOLFOX group and 5.3 months in the control group (hazard ratio [HR] = 0.69; 95% confidence interval [CI], 0.50 to 0.97; P = 0.031); median PFS was 4 months in the FOLFOX group, and an objective response was observed in 5% of patients in the FOLFOX group. The clinical experts consulted by CADTH agreed there is an unmet need for effective therapies with an acceptable toxicity profile that achieve disease control, delay worsening of symptoms, maintain health-related quality of life (HRQoL), delay disease progression, and prolong survival.
Pemigatinib is a molecule kinase inhibitor with antitumour activity that inhibits FGFRs. FGFRs are receptor tyrosine kinases that activate signalling pathways in tumour cells.14 On September 17, 2021, pemigatinib was approved by Health Canada for the treatment of adults with previously treated, unresectable, locally advanced, or metastatic CCA with an FGFR2 fusion or other rearrangement. The sponsor’s requested reimbursement criteria for pemigatinib are per the Health Canada–approved indication. Pemigatinib underwent review by Health Canada through a standard review pathway. Pemigatinib has not previously been reviewed by CADTH. Oral pemigatinib is available as 4.5 mg, 9 mg, and 13.5 mg tablets. The recommended starting dose is 13.5 mg administered orally for 14 consecutive days followed by 7 days off therapy, in 21-day cycles. The product monograph states that treatment is to be continued until disease progression or unacceptable toxicity. Furthermore, it is recommended that a low-phosphate diet be initiated when the phosphate level is greater than 5.5 mg/dL, and that adding a phosphate-lowering therapy should be considered when the level is greater than 7 mg/dL. The dose of phosphate-lowering therapy is to be adjusted until the phosphate level returns to less than 7 mg/dL. It is recommended that discontinuation of phosphate-lowering therapy be considered during pemigatinib treatment breaks or if the phosphate level falls below normal.15 The objective of this CADTH review is to perform a systematic review of the beneficial and harmful effects of pemigatinib for the treatment of adult patients with previously treated, unresectable, locally advanced, or metastatic CCA with an FGFR2 fusion or other rearrangement.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from the clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Three patient groups, the Canadian Liver Foundation, the Canadian Organization for Rare Disorders, and the Cholangiocarcinoma Foundation, co-created 1 patient input for this review. This input was based on an online survey and a virtual focus group with a total of 27 respondents: 15 were patients diagnosed with CCA (4 of whom had CCA with FGFR2 fusions), and 2 were patients who had symptoms of CCA but did not have a diagnosis of CCA; 10 respondents were caregivers or family members of patients with CCA.
Respondents indicated a varying range of CCA symptoms affecting patients’ daily activities (including their social, work, and school lives and their relationships) causing detrimental effects on patients’ quality of life (QoL). Respondents highlighted problems with intimacy or sexual desire, fatigue, and anxiety. Other commonly experienced symptoms indicated by respondents included unintended weight loss, insomnia, gastrointestinal problems, abdominal pain, constipation, depression, and neuropathy. According to the 3 patient groups, delayed diagnosis, misdiagnosis, and a lack of specialists and treatment options available for this rare cancer significantly contribute to patients’ feelings of stress and anxiety and may delay or eliminate treatment options.
According to the patient input received, respondents reported they expect the following key outcomes to be improved from any new drug or treatment: QoL, tumour response, delay in disease progression, and additional treatment choice. Additionally, it was highlighted by the 3 patient groups that the identification of gene mutations and the development of targeted therapies was perceived by respondents to be very important and would spur hope for curable options. Four respondents indicated they had direct experience with taking pemigatinib. Respondents indicated little overall challenge dealing with the side effects from pemigatinib.
Clinician Input
Input From the Clinical Experts Consulted by CADTH
The clinical experts consulted by CADTH indicated there are currently no standard publicly funded second-line treatment options. Palliative therapy (e.g., FOLFOX, FOLFIRI, fluorouracil, and capecitabine) and best supportive care are recommended for patients in the present target setting. The clinical experts identified an unmet need for effective therapies with acceptable toxicity profiles that achieve disease control, delay worsening of symptoms, maintain HRQoL, delay disease progression, and prolong survival. The clinical experts consulted by CADTH stated that pemigatinib was to be used in adult patients with previously treated, unresectable, locally advanced, or metastatic CCA with an FGFR2 fusion or other rearrangement, as per the FIGHT-202 trial. Among patients enrolled in cohort A of the FIGHT-202 trial, the clinical experts did not identify any subgroups of patients who would potentially be best suited for, or benefit the least from, pemigatinib. The clinical experts consulted by CADTH felt it would be reasonable to generalize the results from cohort A to patients with FGFR2 alterations who are intolerant to first-line therapy.
The clinical experts agreed that patients would be identified as possible candidates for pemigatinib if they had the FGFR2 alteration. Clinical assessment to evaluate response to treatment with pemigatinib would include regular radiological imaging (i.e., CT or MRI) and a CA19 to 9 biomarker test every 2 to 3 months to determine whether a patient has experienced disease progression. In addition, patients would be seen by an oncologist every 3 to 4 weeks for clinical assessment (i.e., to assess disease symptoms and a patient’s ECOG PS). The clinical experts indicated that the most clinically meaningful responses to treatment include disease control (i.e., disease stability or response), improvement in disease-related symptoms, better pain control, weight gain, regaining a more active lifestyle, maintenance of HRQoL, and prolonged PFS and OS. Acceptable drug-related toxicity was also noted as a clinically meaningful outcome.
In the opinion of the clinical experts consulted by CADTH, treatment with pemigatinib should be discontinued if a patient experiences disease progression, has a worsening ECOG PS, is intolerant to or experiences unacceptable toxicity from pemigatinib (which cannot be improved with dose delays or reductions), or the patient is not interested in continuing treatment.
Clinician Group Input
Two clinician group inputs were provided, 1 from the Ontario Health (Cancer Care Ontario) Gastrointestinal Cancer Drug Advisory Committee and 1 from the Canadian Gastrointestinal Oncology Evidence Network (CGOEN) and other physicians who treat CCA. The views of the clinician groups overall were consistent with the clinical experts consulted by CADTH, indicating that the most important treatment goals are achieving disease control, delaying worsening of symptoms, maintaining HRQoL, delaying disease progression, and prolonging survival. It also important that the drug have an acceptable safety profile. The clinicians from the CGOEN also highlighted that the convenient oral route of administration of pemigatinib would contribute to improvements in QoL for patients, as fewer visits to a cancer centre and less chair time would be required compared with alternative treatment options. The clinicians from the CGOEN further suggested it would be reasonable to consider pemigatinib upfront for patients deemed unsuitable for standard first-line chemotherapy. This clinician group also noted that patients with compromised hepatic function or significant hyperbilirubinemia would be least suitable for treatment with pemigatinib. The clinicians from both inputs anticipated that pemigatinib would offer a clinically meaningful benefit and improved efficacy to patients with the potential for improved QoL.
Drug Program Input
The Provincial Advisory Group identified the following jurisdictional implementation issues: relevant comparators, consideration for initiation of therapy, consideration for discontinuation of therapy, generalizability, care provision, system issues, and economic considerations. The clinical experts consulted by CADTH weighed evidence from the FIGHT-202 trial and other clinical considerations to provide responses to the Provincial Advisory Group’s drug program implementation questions. Refer to Table 4 for more details.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
The FIGHT-202 trial is an ongoing, multi-centre, open-label, single-arm phase II trial evaluating the efficacy and safety of pemigatinib in patients with advanced, metastatic, or surgically unresectable CCA with FGFR2 alterations, other fibroblast growth factor (FGF) or FGFR alterations, or no FGF/FGFR alterations, whose disease did not respond to previous therapy. Patients were assigned to 3 cohorts, depending on the patient’s FGF/FGFR status (cohort A: FGFR2 fusions or rearrangements; cohort B: FGF/FGFR alterations other than FGFR2 fusions or rearrangements; cohort C: negative for FGF/FGFR alterations). This CADTH review focuses on cohort A, as cohorts B and C were not part of the requested reimbursement criteria submitted to CADTH and not approved in the Health Canada Notification of Compliance with conditions; therefore, they are beyond the scope of this review. Selected results for cohorts B and C have been included in Appendix 3. A total of 147 patients were enrolled to received oral pemigatinib (13.5 mg orally once daily on a schedule of 2 weeks on and 1 week off for each 21-day cycle). The primary outcome was ORR in cohort A, and secondary outcomes included ORR in cohorts B, A plus B, and C; PFS; duration of response (DOR); disease control rate (DCR); OS; and safety assessed in all 3 cohorts, respectively. Exploratory end points included HRQoL and symptom severity.
Adults diagnosed with locally advanced, metastatic, or surgically unresectable CCA with FGFR2-positive disease who had documented disease progression after at least 1 line of prior systemic therapy were enrolled into cohort A of the Fight-202 trial. At baseline, 107 patients were identified as having FGFR2 fusions or rearrangements and were grouped into cohort A. Cohort B included 20 patients with other FGF/ FGFR alterations other than FGFR2, and cohort C included 18 patients with no identified FGF/FGFR alterations. One patient who was placed into an “undetermined” group was not assigned to any of the 3 cohorts, as their FGF and FGFR status results could not be confirmed by the central genomics laboratory. For patients in cohort A, the mean age was 55.3 years (standard deviation [SD] = 12.02). Most patients were female (60.7%) and enrolled in trial sites in North America (59.8%) or Europe (29.9%). Almost all patients (89% of patients overall and 98.1% of patients in cohort A) had iCCA. The majority of patients in cohort A had metastatic disease (82.2%) ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Median time since diagnosis was 1.28 years (range, 0.03 to 11.1 years) for patients in cohort A. The majority of patients in cohort A had an ECOG PS of 1 (53.3%), and all patients had received at least 1 line of prior systemic therapy for advanced or metastatic disease (60.7%, 27.1%, and 12.1% of patients received 1, 2, and ≥ 3 prior lines, respectively). Renal and hepatic impairment grades were normal or mild for most patients in cohort A (39.3% and 43.9% normal and mild renal impairment grades, respectively; 44.9% and 48.6% normal and mild hepatic grades, respectively).16
The futility analysis, which was performed on October 12, 2017,17 was pre-specified a priori in the statistical analysis plan. The timing of the subsequent analysis (March 22, 2019), at which point the predetermined threshold (i.e., lower limit of the 95% CI for an ORR > 15%) would be assessed, was not pre-specified a priori in the statistical analysis plan; however, the sponsor’s proposed timing was agreed upon by the FDA during its review process for pemigatinib. Two additional updated analyses occurred at the August 2019 and April 2020 data cut-off dates; the former was a 4-month safety update required for the FDA New Drug Application (NDA), the latter was performed to support the safety data summaries for another indication outside of Canada.17 The trial is still ongoing, with an estimated completion date in the first quarter of 2022.17
Efficacy Results
The key efficacy results from cohort A of the FIGHT-202 trial are summarized in Table 2. At the latest data cut-off date (April 7, 2020), the median duration of follow-up was 27.9 months in cohort A. Median OS was 17.48 (95% CI, 14.42 to 22.93) months. The survival probabilities of patients surviving for 6 and 12 months were ||||||||||||||||||||||||||||||||||||||| and 67.3% (95% CI, 57.4 to 75.4), respectively. Median PFS was 7.03 months (95% CI, 6.08 to 10.48). The PFS probabilities at 6 and 12 months were |||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||. The PFS results for the subgroup of interest, as pre-specified a priori in the protocol for this CADTH review, suggested that the treatment effects on PFS for the subgroups of patients with an ECOG PS of 0 and 1 plus 2 were generally consistent with the overall population in cohort A.
As of the April 7, 2020, data cut-off date, the proportion of patients who achieved an objective response was 37.0% (N = 40) (95% CI, 27.94 to 46.86), including 4 patients (3.7%) with a complete response (CR) and 36 patients (33.3%) with a partial response (PR). The ORR results for the subgroup of interest suggested that the treatment effects on ORR for the subgroups of patients with an ECOG PS of 0 and 1 plus 2 were generally consistent with the overall population in cohort A. Among the 40 patients who achieved an objective response, the median DOR was 8.08 months (95% CI, 5.65 to 13.14). The probabilities of maintaining a response for at least 6 and 12 months were ||||||||||||||||||||||||||||||||| and |||||||||||||||||||||||||||||||||||||, respectively.
The results for DCR were reported only for the March 22, 2019, data cut-off date. The proportion of patients with a best response of CR, PR, or stable disease was 82.2% (N = 88) (95% CI, 73.7 to 89.0), including 3 patients (2.8%) with a CR, 35 patients (32.7%) with a PR, and 50 patients (46.7%) with stable disease for 39 or more days since the first pemigatinib dose. The DCR results based on investigator assessment showed generally consistent results with those of an independent review committee (IRC); DCR was ||||||||||||||||||||||||||||||||||||||||||.
The descriptive summary statistics of observed scores for the European Organisation for Research and Treatment of Cancer (EORTC) Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) and the EORTC Quality of Life Questionnaire Cholangiocarcinomas and Gallbladder Cancer Module 21 (EORTC QLQ-BIL21) from baseline to ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||.16 A definition for what constituted a clinically meaningful change from baseline in the present target population was not provided. A post-hoc analysis assessed observed mean changes from baseline to week 16 by subgroups of patients (i.e., patients with a CR or PR, stable disease, or progressive disease [PD]). Results suggested that changes from baseline appeared directionally more favourable in patients with a CR or PR, or stable disease than in patients with PD.
Harms Results
Key harms reported in cohort A of the FIGHT-202 trial are summarized in Table 2. All patients in cohort A experienced at least 1 treatment-emergent adverse event (TEAE) (100.0%). The most commonly reported TEAEs were alopecia (|||||||), hyperphosphatemia (|||||||), diarrhea (|||||||), dysgeusia (|||||||), fatigue (|||||||), and nausea (|||||||). The percentage of patients experiencing serious TEAEs was ||||||| in cohort A. The most common serious TEAEs were |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Adverse events (AEs) led to discontinuation of study treatment in ||||||| of patients in cohort A. None of the patients withdrew from the FIGHT-202 study due to an AE as primary reason. TEAEs leading to treatment discontinuation included |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. TEAEs leading to death occurred relatively rarely in cohort A (N = 3; 2.8%) and included failure to thrive and bile duct obstruction. None of the TEAEs leading to death were considered treatment-related.16
The percentage of patients experiencing nail toxicity TEAEs was ||||||| in patients in cohort A. The most commonly reported nail toxicity included ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.16
The percentage of patients experiencing serous retinal detachment TEAEs in cohort A was |||||||. The most commonly reported serous retinal detachment was |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.16
The percentage of patients experiencing hyperphosphatemia TEAEs in cohort A was |||||||. The most commonly reported hyperphosphatemia events were ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.16
The percentage of patients experiencing hypophosphatemia TEAEs in cohort A was |||||||. The most commonly reported hypophosphatemia events were ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.16
Table 2: Summary of Key Results From Pivotal and Protocol Selected Study
Variable | Pemigatinib, cohort A outcome | |
|---|---|---|
N = 107 | N = 108a | |
Data cut-off date | March 22, 2019 | April 7, 2020 |
Median follow-up time,b months (range) | 15.44 (7.0 to 24.7) | 27.9 (||||||||||||) |
Secondary outcome: OS | ||
Median OS, months (95% CI)c | 21.06 (14.82 to NE) | 17.48 (14.42 to 22.93) |
Events (death), n (%) | ||||||||||||| | ||||||||||| |
Censored, n (%) | 67 (62.6) | ||||||||||||| |
KM estimates of OS at: | ||
3 months (95% CI) | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||||| |
6 months (95% CI) | 88.6 (80.8 to 93.4) | |||||||||||||||||||||||||||||||||||| |
9 months (95% CI) | ||||||||||||||||||||||||||||| | 76.1 (66.7 to 83.2) |
12 months (95% CI) | 67.5 (56.4 to 76.3) | 67.3 (57.4 to 75.4) |
Secondary outcome: PFS (IRC assessment) | ||
Median PFS, months (95% CI)c | 6.93 (6.18 to 9.59) | 7.03 (6.08 to 10.48) |
Events (disease progression or death), n (%) | 71 (66.4) | |||||||||||| |
Disease progression, n (%) | 63 (58.9) | |||||||||||| |
Death, n (%) | 8 (7.5) | |||||||||||| |
Censored, n (%) | 36 (33.6) | |||||||||||| |
KM estimates of PFS at: | ||
3 months (95% CI) | 78.9 (69.7 to 85.5) | ||||||||||||||||||||||||||||||| |
6 months (95% CI) | 61.7 (51.5 to 70.4) | ||||||||||||||||||||||||||||||| |
9 months (95% CI) | 45.3 (34.9 to 55.1) | ||||||||||||||||||||||||||||||| |
12 months (95% CI) | 29.2 (18.9 to 40.2) | ||||||||||||||||||||||||||||||| |
Primary outcome: ORR (IRC assessment) | ||
Objective response,d n (%) | 38 (35.5) | 40 (37.0) |
95% CIe | 26.50 to 45.35 | 27.94 to 46.86 |
Best overall response, n (%) | ||
Confirmed complete response | 3 (2.8) | 4 (3.7) |
Confirmed partial response | 35 (32.7) | 36 (33.3) |
Stable disease | 50 (46.7) | 49 (45.4) |
Progressive disease | 16 (15.0) | 16 (14.8) |
Not evaluablef | 3 (2.8) | 3 (2.8) |
Secondary outcome: DOR (IRC assessment)g | ||
Participants with confirmed objective responses, n (%) | 38 (35.5) | 40 (37.0) |
Participants with events, n (%) | 21 (55.3) | |||||||||||| |
Disease progression | 20 (52.6) | |||||||||||| |
Death | 1 (2.6) | |||||||||||| |
Participants censored, n (%) | 17 (44.7) | |||||||||||| |
Median DOR, months (95% CI)c | 7.49 (5.65 to 14.49) | 8.08 (5.65 to 13.14) |
KM estimates of DOR | ||
3 months (95% CI) | 100.0 (100.0 to 100.0) | ||||||||||||||||||||||||||||||| |
6 months (95% CI) | 68.5 (49.0 to 81.8) | ||||||||||||||||||||||||||||||| |
9 months (95% CI) | 47.4 (27.6 to 64.9) | ||||||||||||||||||||||||||||||| |
12 months (95% CI) | 37.4 (18.6 to 56.2) | ||||||||||||||||||||||||||||||| |
Secondary outcome: DCR (IRC assessment) | ||
Disease control,h n (%) | 88 (82.2) | Not availablei |
95% CIe | 73.7 to 89.0 | Not availablei |
Best response, n (%) | — | Not availablei |
Confirmed complete response | 3 (2.8) | Not availablei |
Confirmed partial response | 35 (32.7) | Not availablei |
Stable disease ≥ 39 days | 50 (46.7) | Not availablei |
Harms, n (%) (safety population) | N = 107 | N = 108 |
TEAEs | 107 (100.0) | |||||||||||| |
Hyperphosphatemia | ||||| (55.1) | |||||||||||| |
Alopecia | |||||| (58.9) | |||||||||||| |
Diarrhea | ||||||| (52.3) | |||||||||||| |
Fatigue | |||||| (44.9) | |||||||||||| |
Nausea | |||||| (40.2) | |||||||||||| |
Dysgeusia | |||||| (47.7) | |||||||||||| |
Serious TEAEs | |||||| (40.2) | |||||||||||| |
Pyrexia | ||| (4.7) | |||||||||||| |
Cholangitis | ||| (3.7) | |||||||||||| |
Abdominal pain | |||| (3.7) | |||||||||||| |
Infective cholangitis | ||| (2.8) | |||||||||||| |
Small intestinal obstruction | |||| (1.9) | |||||||||||| |
Discontinued treatment due to TEAEs | 5 (4.7) | |||||||||||| |
Deaths | 3 (|||||) | |||||||||||| |
Notable harms | ||
Nail toxicity (any-grade TEAEs), n (%) | 56 (52.3) | |||||||||||| |
Onychomadesis | 13 (12.1) | |||||||||||| |
Nail discoloration | 12 (11.2) | |||||||||||| |
Nail dystrophy | 10 (9.3) | |||||||||||| |
Onycholysis | 10 (9.3) | |||||||||||| |
Paronychia | 9 (8.4) | |||||||||||| |
Serous retinal detachment (any-grade TEAEs), n (%) | 4 (3.7) | |||||||||||| |
Retinal detachment | |||||||||||| | |||||||||||| |
Chorioretinal folds | |||||||||||| | |||||||||||| |
Detachment of retinal pigment epithelium | |||||||||||| | |||||||||||| |
Maculopathy | |||||||||||| | |||||||||||| |
Hyperphosphatemia (any-grade TEAEs), n (%) | |||||||||||| | |||||||||||| |
Hyperphosphatemia | |||| (55.1) | |||||||||||| |
Blood phosphorus increased | |||||||||||| | |||||||||||| |
Hypophosphatemia (any-grade TEAEs), n (%) | |||| (25.2) | |||||||||||| |
Hypophosphatemia | |||| (24.3) | |||||||||||| |
Blood phosphorus decreased | |||||||||||| | |||||||||||| |
CI = confidence interval; DCR = disease control rate; DOR = duration of response; IRC = independent review committee; KM = Kaplan–Meier; NA = not applicable; NE = not evaluable; NR = not reported; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; TEAE = treatment-emergent adverse event.
Note: Outcomes are presented in order of priority as identified in the CADTH review protocol.
aThe April 7, 2020, data cut-off date included 1 additional patient in cohort A who had been enrolled after the August 30, 2019, data cut-off date.17
bFollow-up time for all patients in cohort A in the efficacy evaluable population.
cThe 95% CI was calculated using the Brookmeyer and Crowley method (1982).
dParticipants who had a best overall response of complete response or partial response.
eThe CI was calculated based on the exact method for binomial distribution.
fPost-baseline tumour assessment was either not performed due to study discontinuation (2 participants) or was performed before the minimum interval of 39 days for an assessment of stable disease (1 participant).
gComplete and partial responses were confirmed.
hParticipants who had a best overall response of complete response, partial response, or stable disease with measurements that met the stable disease criteria after the date of the first dose at a minimum interval of 39 days.
iThe DCR outcome was not generated for the April 7, 2020, data cut-off date since the primary focus of the analyses at that date was for the integrated safety summary for a new regulatory submission.17
Critical Appraisal
The primary objective of phase II (randomized or non-randomized) trials is to document the safety outcomes and investigate whether the estimate of effect for a new drug is large enough to use it in confirmatory phase III trials. Phase II trials may not accurately predict the harm and/or effectiveness of treatments. The clinical experts consulted by CADTH noted that, despite the high unmet need, conducting a randomized controlled trial (RCT) in this setting with a targeted therapy, such as pemigatinib, compared with currently available therapies in the second line in Canadian clinical practice would likely not be feasible. The FIGHT-202 trial included no formal statistical significance and hypotheses testing and point estimates with 95% CIs were reported to estimate the magnitude of treatment effect. A greater than 95% probability of having a 95% CI for ORR in cohort A with a lower limit larger than 15% was the basis for the sample size determination and was regarded as the threshold for a positive study outcome. The subgroup analyses were non-inferential; wide CIs reflected uncertainty in the effect estimates, and small sample sizes limited the generalizability to a broader population. Interpretation of time‐to‐event end points such as OS and PFS is limited in single‐arm studies; since all patients in cohort A received the same treatment, the extent to which the observed survival is due to the natural history of the tumour or the intervention remains unclear. While there is strong genetic and functional evidence that FGFR genetic alterations can drive the formation of tumours,7 it is currently not known whether patients with an FGFR2 alteration represent a distinct prognostic subgroup.11 The clinical experts agreed that progression on prior systematic therapy is a major prognostic factor in the present target population, and they did not anticipate that patients would derive any substantial benefit from their underlying disease biology at the time they were enrolled into the FIGHT-202 trial. The results for patient-reported outcomes were inconclusive, given the non-comparative, open-label design of the trial, the lack of a pre-specified analysis of the patient-reported outcomes data, the substantial decline in the number of patients available to provide assessments over time, and the lack of a definition for what constituted a clinically meaningful change from baseline in the target population.
In the absence of a direct comparison of pemigatinib with relevant treatment options, the sponsor submitted an indirect treatment comparison (ITC) in the form of a matching-adjusted indirect comparison (MAIC) comparing the efficacy of pemigatinib (cohort A of the FIGHT-202 trial) with each of the 2 treatment groups in the ABC-06 study. The results of the ITC favoured pemigatinib for PFS and OS in comparison with FOLFOX plus ASC as well as compared with ASC alone. The clinical experts agreed with the CADTH clinical review team that, given the absence of robust comparative data on PFS and OS, the ability to interpret the relative treatment effects observed between pemigatinib and FOLFOX plus ASC and ASC alone was limited, and no firm conclusions could be drawn on how pemigatinib compared with other relevant treatment options. The clinical experts consulted by CADTH anticipated, however, that based on the FIGHT-202 results and on the poor results with existing treatment options in clinical practice, pemigatinib appeared to offer at least similar or improved clinical benefits compared with current therapies, with better tolerability.
Indirect Comparisons
Description of Studies
Two studies, the FIGHT-202 trial and the ABC-06 study, were included in the sponsor’s ITC. The sponsor submitted an ITC in the form of an MAIC between cohort A of the FIGHT-202 study and each of the 2 treatment groups in the ABC-06 study. The ABC-06 study compared mFOLFOX plus ASC versus ASC alone in patients with locally advanced or metastatic BTC. Cohort A of the FIGHT trial included only patients with unresectable, locally advanced, or metastatic CCA who had the FGFR2 mutation.
Efficacy Results
OS: Pemigatinib Versus mFOLFOX Plus ASC
The results of the ITC favoured pemigatinib for PFS and OS in comparison with mFOLFOX plus ASC as well as with ASC alone. Median OS was |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| for the pemigatinib group versus ||||||||||||||||||||||||||||||||||||||||||||||||||||||| for the mFOLFOX plus ASC group, based on the March 22, 2019, data cut-off for the FIGHT-202 study. The corresponding HR was |||||||||||||||||||||||||||||||||||||||| and the HR using the results from the April 7, 2020, data cut-off was |||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||.
OS: Pemigatinib Versus ASC Alone
Median OS was |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| for the pemigatinib group versus ||||||||||||||||||||||||||||||||||||||| months for the ASC alone group, based on the March 22, 2019, data cut-off for the FIGHT-202 study. The corresponding HR was |||||||||||||||||||||||||||| ||||||||||||||||||||||||||| and the HR using the results from the April 7, 2020, data cut-off was ||||||||||||||||||||||||||||||||||||||||||||||.
PFS: Pemigatinib Versus mFOLFOX Plus ASC
Median PFS was ||||||||||||||||||||||||||||||||||||||| months versus ||||||||||||||||||||||||||||||||||||||||| months for the pemigatinib versus mFOLFOX plus ASC groups, based on the March 22, 2019, data cut-off for the FIGHT-202 study. The corresponding HR was |||||||||||||||||||||||||| ||||||||||||||||||||||||||||| and the HR using the results from the April 7, 2020, data cut-off was |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
PFS for pemigatinib versus ASC alone was not assessed.
Harms Results
No comparisons for harms or safety were incorporated in the sponsor’s ITC.
Critical Appraisal
There were potentially important underlying differences between the FIGHT-202 and ABC-06 studies. In particular, the FGFR2 alterations were not reported in the ABC-06 trial. Given that FGFR2 alterations occur almost exclusively in iCCA and that the prevalence of FGFR2 alterations is less than 20%8 in patients with iCCA, there is likely a large disparity in FGFR2 mutation status between the study populations. While the FIGHT-202 study included only patients with CCA, the ABC-06 study included patients with BTC, which encompasses gallbladder cancer and ampullary cancer in addition to CCA. Ninety-eight percent of patients in cohort A of the FIGHT-202 study had iCCA compared with 42% and 47% in the mFOLFOX plus ASC and ASC alone groups, respectively. Since disease type and FGFR2 status were more restricted in the FIGHT-202 study, these differences could not be addressed through the weighting of patients in the pemigatinib group.
The covariates chosen for adjustment were based on age, sex, ECOG PS, and serum albumin. The following baseline characteristics were also available for both studies and did not appear to be considered: disease stage, percentage of patients with prior surgery for cancer, and number of lines of prior systemic therapy for advanced or metastatic cancer. The clinical experts consulted by CADTH for this review were of the opinion that the number of lines of previous therapy was of key importance in terms of prognosis. The clinical experts were not aware of any additional prognostic factors and/or effect modifiers that were not reported in both studies and should have been considered.
While there are retrospective studies suggesting that the presence of FGFR2 mutations in CCA may be associated with a better prognosis,18,19 the clinical experts consulted by CADTH were of the opinion that FGFR2 mutation status was not an important prognostic factor in the indicated patient population. The clinical experts considered the fact that patients in both the FIGHT-202 and ABC-06 trials had progressed on prior systemic therapy to be of greater importance in terms of prognosis. The clinical experts expected patients in the FIGHT-202 study to have more advanced disease than patients in the ABC-06 study because the FIGHT-202 study population was more heavily pre-treated overall. It is unclear whether the pemigatinib group was more or less similar to the ASC-06 groups in this respect following weighting, as the weighting process did not take the number of prior lines of systemic therapy into account.
The effective sample size of the pemigatinib group was reduced by approximately 50% after weighting to the mFOLFOX plus ASC and ASC alone groups, and it is unclear how representative the post-weighting pemigatinib groups are of cohort A of the FIGHT-202 study.
Comparisons of pemigatinib with other relevant comparators (FOLFIRI, fluorouracil alone or in combination with cisplatin or oxaliplatin, and capecitabine alone or in combination with cisplatin or oxaliplatin) were not available. Given that mFOLFOX plus ASC is the only therapy beyond the first-line setting with RCT evidence of an OS benefit, the clinical experts consulted by CADTH expected that mFOLFOX plus ASC would have the greatest efficacy out of all the relevant comparators.
In summary, for the unanchored MAIC to produce unbiased treatment effect estimates, both the effect modifiers and prognostic variables need to be adjusted for in the analysis. Residual confounding remains the major limitation of the MAIC despite adjusting for age, sex, ECOG PS, and serum albumin in the comparisons of pemigatinib with mFOLFOX plus ASC and ASC alone. While any bias introduced by the differences between the FIGHT-202 and ASC-06 studies in the number of prior lines of systemic therapy may have been against pemigatinib, the substantial differences in FGFR2 mutation status and tumour site between trials introduce a high degree of uncertainty in the OS and PFS results. Furthermore, MAICs cannot account for unknown cross-trial differences; thus, the MAIC estimates are susceptible to bias from unknown confounding. An evaluation of potential bias from residual confounding was not reported; therefore, the magnitude of this bias in the relative treatment effect estimates is unclear. Overall, it remains uncertain whether pemigatinib provides additional OS or PFS benefit versus mFOLFOX plus ASC or ASC alone.
Other Relevant Evidence
One additional relevant report was summarized that was included in the sponsor’s submission to CADTH. FIGHT-101 is an ongoing, open-label phase I/II dose-escalation and expansion study of pemigatinib among participants with previously treated advanced malignancies with and without FGF/FGFR alterations. As of February 2019, FIGHT-101 had enrolled 160 participants from 14 study sites in the US and Denmark. Of these, 116 received at least 1 dose of pemigatinib monotherapy. Sixteen participants who were treated with pemigatinib monotherapy had CCA; 6 of these patients had FGFR2 rearrangements or fusions and received pemigatinib 13.5 mg orally once daily on a schedule of 2 weeks on, 1 week off for each 21-day cycle. All 6 of these patients had a reduction in tumour volume during the study. The best overall response of PR was observed in 1 of the 6 aforementioned patients with a DOR of 101 days, and the remaining 5 participants had a best overall response of stable disease. In terms of safety outcomes, 1 TEAE of non-serous grade 2 retinal edema occurred in 1 participant with CCA who was receiving 13.5 mg of pemigatinib monotherapy on an intermittent schedule. However, due to the open-label design and the limited data on the efficacy of pemigatinib on CCA within the report, the ability to interpret these results is considerably limited.
Conclusions
One phase II, singe-arm, open-label trial (FIGHT-202) provided evidence regarding the efficacy and safety of pemigatinib in patients with advanced, metastatic, or surgically unresectable CCA with FGFR2 alterations (cohort A) whose disease did not respond to previous therapy. The FIGHT-202 trial achieved the predetermined threshold for a positive outcome (a lower limit of the 95% CI for ORR > 15%) in cohort A. The clinical experts consulted by CADTH felt that the achieved ORR of 37% (April 7, 2020, data cut-off date) was clinically meaningful for the target population and durable (median of 8.08 months; range, 5.65 to 13.14). In the opinion of the clinical experts, the observed responses appeared higher than what is seen with the therapies currently used in the second line in this setting. There was uncertainty around the magnitude of the clinical benefit, given the limitations in the evidence from the non-comparative phase II clinical trial. The clinical experts consulted by CADTH noted that, despite the high unmet need, conducting an RCT with a targeted therapy such as pemigatinib, compared with the therapies that are currently available in the second line in Canadian clinical practice, would likely not be feasible. While the secondary efficacy outcomes, OS and PFS, appeared supportive of the observed ORR achievements, the non-randomized design of the FIGHT-202 trial made interpreting PFS and OS events attributable to pemigatinib challenging. In the absence of a direct comparison of pemigatinib with relevant treatment options, the sponsor submitted an ITC. However, the CADTH critical assessment identified limitations with the sponsor’s submitted unanchored MAIC (including heterogeneity across study designs and populations and the inability to adjust for all potential confounders and prognostic variables), which limited the ability to interpret the relative treatments effects observed between pemigatinib and other treatments. The results for the HRQoL and symptom severity exploratory outcomes remained inconclusive due to a number of important limitations. The toxicity profile of pemigatinib was considered manageable by the clinical experts consulted by CADTH and appeared favourable compared with currently available chemotherapy options. However, the non-comparative design of the FIGHT-202 trial made interpreting the safety events attributable to pemigatinib challenging, since all patients in cohort A received the same treatment.
Introduction
Disease Background
Hepatobiliary cancers are highly lethal cancers and refer to cancers arising in the liver (hepatocellular carcinoma), gall bladder, and bile ducts (iCCA and eCCA). Gallbladder cancer and CCA are known as BTC.3 The most common type of liver cancer is hepatocellular carcinoma, followed by BTC, accounting for 70% to 85% and 10% to 15% of all primary liver cancers, respectively.1,2 Gallbladder cancer is the most common type of BTC.3
CCAs are most commonly adenocarcinomas3 and comprise 2 main subtypes: iCCA, initiating from the biliary tree within the liver, and eCCA, initiating outside the liver parenchyma; eCCA is subdivided into perihilar CCA or Klatskin tumour and distal CCA.2 With eCCA accounting for 80% to 90% of all CCA, iCCA is the least frequently reported subtype.2 The incidence of CCA is generally low (0.3 to 3.5 per 100,000) in Europe, the US, and Australia, but is higher in other parts of the world where certain parasite infections are common (e.g., Thailand, China, and Korea); northeast Thailand has the highest incidence (90 per 100,000) of CCA in the world.2 In Canada and the US, respectively, there are approximately 400 and 5,000 new cases of CCA diagnosed each year.4 The median age at diagnosis is 65 years in Western industrialized nations.5 The 5-year relative survival rates for iCCA and eCCA, respectively, are 9% and 10%. The 5-year relative survival rates broken down by stages of disease are 25%, 8%, and 2%, respectively, for localized, regional, and distant iCCA; and 15%, 16%, and 2% for localized, regional, and distant eCCA, respectively.6
While most CCAs arise spontaneously, without any known risk factors,7 established risk factors for CCAs include primary sclerosing cholangitis, chronic ulcerative colitis, cysts in the bile ducts, and infection with a Chinese liver fluke parasite.20 Additionally, in Western countries, hepatitis C and liver cirrhosis have been identified as risk factors for iCCA, while obesity, diabetes mellitus, metabolic disease, and certain substances (alcohol, tobacco, oral contraceptive pills, dioxin, and asbestos) have also been suggested as risk factor for CCA.7
Diagnosis of CCA is most commonly made in advanced stages (70% of patients are diagnosed with unresectable, locally advanced or metastatic disease) due to an absence of symptoms until later in the course of the disease.7 The rate of recurrence is high among the minority of patients who are able to undergo potentially curative surgery.9 Symptoms commonly appear when a bile duct is blocked and include jaundice, itching, light-coloured and greasy stools, dark urine, abdominal pain, loss of appetite or weight loss, fever, and nausea and vomiting.8
Different genetic alterations in BTC with oncogenic properties have been identified in recent years. Nearly 40% of patients harbour genetic alterations (e.g., isocitrate dehydrogenase 1 [IDH1] or isocitrate dehydrogenase 2 [IDH2], FGFR2, BRAF, and HE2/neu)9; however, evaluation of targeted treatment options is hampered by the low overall patient numbers.11 One of the most frequent genetic alterations in patients with iCCA involves FGFR2.7 FGFR2 fusions or rearrangements are found in 10% to 20%10 of patients with iCCA, while they rarely occur in eCCA.11 Alterations involving other members of the FGFR are rare, with an incidence below 0.5%.11 While there is strong genetic and functional evidence that FGFR genetic alterations can drive the formation of tumours,7 it is currently not known whether FGFR2 alteration–positive patients represent a distinct prognostic subgroup11 and/or respond differently to chemotherapy compared with patients with unselected CCA.12 Retrospective studies19,21-23 in the first-line setting in patients with CCA suggest that patients with FGFR alterations appear to have a better prognosis compared with patients with unselected CCA, and that FGFR alterations occur more frequently in young women.9 A number of phase II studies have been published that report the results of FGFR2-directed therapies. Notably, 2 non-comparative phase II trials have reported very similar results for patients with iCCA that is positive for FGFR2 fusions or rearrangements: objective responses were 35.5% (38 out of 107), with an estimated median PFS of 6.9 months for pemigatinib, and 31% (22 out of 71) with a median PFS of 5.8 months in patients treated with infigratinib.11 Pemigatinib was approved by the FDA in April 2020 for the treatment of adults with previously treated, unresectable, locally advanced, or metastatic CCA with an FGFR2 fusion or other rearrangement as detected by an FDA-approved test.24 Since March 2021, pemigatinib has been authorized in the European Union25 as monotherapy and is indicated for the treatment of adults with locally advanced or metastatic CCA with an FGFR2 fusion or rearrangement whose cancer has progressed after at least 1 prior line of systemic therapy.26 The FDA has granted accelerated approval to infigratinib for the treatment of adult patients with previously treated, unresectable, locally advanced, or metastatic CCA with an FGFR2 fusion or other rearrangement as detected by an FDA-approved test.27 Currently, 3 phase III RCTs are recruiting patients with CCA that is positive for FGFR2 fusions or rearrangements that compare first-line standard of care (gemcitabine plus cisplatin) with the following treatments:
pemigatinib (FIGHT-302 [NCT03656536], which has an estimated completion date of June 28, 2026),28 which has a frequency of administration that is different than that in FIGHT-202 (where it is administered continuously)
infigratinib (PROOF [NCT03773302],29 which has an estimated completion date of September 30, 2024)
futibatinib (FOENIX-CCA3 [NCT04093362],30 which has an estimated completion date of February 2026).
All 3 trials have PFS as the primary end point.
Standards of Therapy
Most patients with CCA have advanced-stage disease at the time of diagnosis and although surgery is the preferred treatment option, only 35% of patients are eligible for surgical resection with curative intent.5 For patients with advanced-stage or unresectable CCA and a good ECOG PS (0 or 1), standard-of-care first-line treatment is gemcitabine and cisplatin.9 If there are concerns about a patient’s renal function, oxaliplatin may be substituted for cisplatin.2 For patients with an ECOG PS of 2, gemcitabine monotherapy may be considered as first-line therapy. The median OS, median PFS, and ORR in patients with BTCs treated with standard-care, first-line palliative treatment with gemcitabine and cisplatin range from 11.2 to 11.7 months, 5.8 to 8.0 months, and 19.5% to 26.1%, respectively.12 Patients with molecularly unselected iCCA treated with standard first-line therapy were shown to have median PFS and median OS of 8.4 and 15.4 months, respectively.12 There are currently no funded standard treatment options for patients in the second line once the disease has progressed on first-line treatment.9 In the absence of proven treatment options in the second-line setting for patients with CCA, participation in a clinical trial and best supportive care are recommended, including alleviating biliary obstruction and full access to palliative care and symptom management.2 According to the clinical experts consulted by CADTH, the second-line therapies used in Canadian clinical practice include FOLFOX, FOLFIRI, fluorouracil (alone or in combination with cisplatin or oxaliplatin), and capecitabine (alone or in combination with cisplatin or oxaliplatin). A systematic review,31 including 761 patients participating in case reports, retrospective analyses, or phase II trials of second-line therapies for advanced BTC, reported a mean OS of 7.2 months (95% CI, 6.2 to 8.2), a mean PFS of 3.2 months (95% CI, 2.7 to 3.7), and a response rate of 7.7% (95% CI, 4.6% to 10.9%).31
Second-line treatment with FOLFOX is currently the only drug based on phase III trial data in this setting.5 The ABC-06 trial13 evaluated the efficacy and safety of FOLFOX plus ASC compared with ASC alone in patients with locally advanced or metastatic BTC (including CCA and gallbladder or ampullary carcinoma) whose disease had progressed on first-line cisplatin and gemcitabine therapy. At the median follow-up time of 21.7 months, median OS was 6.2 months in the FOLFOX group and 5.3 months in the control group (HR = 0.69; 95% CI, 0.50 to 0.97; P = 0.031); in the FOLFOX group, median PFS was 4 months and an objective response was observed in 5% of patients.13
In Canada, there are currently no funded standard targeted treatment options for patients with CCA whose disease harbours generic alterations that have been identified for targeted therapeutics. Another common genetic alteration in iCCA are IDH1 and IDH2 mutations, which are found in 10% to 23% of iCCA, and a targeted treatment, ivosidentib,32 has received priority review by the FDA for the treatment of patients with previously treated, IDH1-mutant CCA.33
There was consensus among the clinicians that there is an unmet need for effective therapies with an acceptable toxicity profile that achieve disease control, delay worsening of symptoms, maintain HRQoL, delay disease progression, and prolong survival. It was also mentioned by the clinical experts that there are currently no biomarker-directed regimens specifically for patients with CCA that is FGFR2-positive. The experts anticipated more promising benefit with a targeted therapy option in later lines than with chemotherapy with a disease that is steadily growing more resistant.
Drug
Pemigatinib is a molecule kinase inhibitor with antitumour activity that inhibits FGFRs. FGFRs are receptor tyrosine kinases that activate signalling pathways in tumour cells.14
On September 17, 2021, pemigatinib was issued market authorization with conditions by Health Canada for the treatment of adults with previously treated, unresectable, locally advanced, or metastatic CCA with an FGFR2 fusion or other rearrangement. The sponsor’s requested reimbursement criteria for pemigatinib are as per the Health Canada–approved indication. In addition, the Health Canada indication states that the “clinical effectiveness of pemigatinib is based on ORR and DOR from a single-arm phase II trial in patients with specific FGFR2 rearrangements. Treatment with pemigatinib should be initiated following confirmation of a susceptible genetic alteration using a validated test.”15 Pemigatinib underwent review by Health Canada through a standard review pathway. Pemigatinib has no other Health Canada–approved indication and has not previously been reviewed by CADTH.
After being granted priority review and breakthrough therapy and orphan drug designations, pemigatinib received an accelerated approval by the FDA in April 2020 for the treatment of adults with previously treated, unresectable, locally advanced, or metastatic CCA with an FGFR2 fusion or other rearrangement, as detected by an FDA-approved test.24 Since March 2021, pemigatinib has been authorized in the European Union25 as monotherapy and is indicated for the treatment of adults with locally advanced or metastatic CCA with an FGFR2 fusion or rearrangement whose cancer has progressed after at least 1 prior line of systemic therapy.26 Pemigatinib has also been approved in Japan.34
Oral pemigatinib is available as 4.5 mg, 9 mg, and 13.5 mg tablets. The recommended starting dose is 13.5 mg administered orally for 14 consecutive days followed by 7 days off therapy, in 21-day cycles. The product monograph states that treatment is to be continued until disease progression or unacceptable toxicity. Furthermore, it is recommended that a low-phosphate diet be initiated when the phosphate level is greater than 5.5 mg/dL, and that consideration be given to adding a phosphate-lowering therapy when the level is greater than 7 mg/dL. The dose of phosphate-lowering therapy is to be adjusted until the phosphate level returns to less than 7 mg/dL. It is recommended that consideration be given to discontinuing phosphate-lowering therapy during pemigatinib treatment breaks or if the phosphate level falls below normal.15
Table 3: Key Characteristics of Pemigatinib
Characteristic | Description |
|---|---|
Mechanism of action | Inhibits FGFRs (1 to 3) by blocking signalling of FGFRs and reducing cell capabilities of cancerous cell lines that lead to constitutive activation of FGFR signalling pathways.35 |
Indicationa | For the treatment of adults with previously treated, unresectable, locally advanced, or metastatic cholangiocarcinoma with an FGFR2 fusion or other rearrangement. |
Route of administration | Oral. |
Recommended dose | 13.5 mg orally once daily for 14 consecutive days followed by 7 days off treatment every 21-day cycle. |
Serious adverse effects or safety issues | According to the product monograph, hyperphosphatemia was reported in 59% of all patients who received pemigatinib; therefore, recommendations include dietary phosphate restriction, administration of phosphate-lowering therapy, and dose modification when required. Serous retinal detachment occurred in 7.5% of all patients treated with pemigatinib; therefore, recommendations include performing ophthalmological examination before initiation of therapy, every 2 months for the first 6 months of treatment and every 3 months afterwards, and urgently at any time for visual symptoms. Pemigatinib treatment may cause fetal harm and may impair fertility in females. |
FGFR = fibroblast growth factor receptor.
aHealth Canada–approved indication. Full indication states:
• Pemazyre (pemigatinib) is indicated for the treatment of adults with previously treated, unresectable, locally advanced or metastatic cholangiocarcinoma with a fibroblast growth factor 2 (FGFR2) fusion or other rearrangement.
• Clinical effectiveness of Pemazyre is based on objective response rate and duration of response from a single-arm phase II trial in patients with specific FGFR2 rearrangements (see CLINICAL TRIALS).
• Treatment with Pemazyre should be initiated following confirmation of a susceptible genetic alteration using a validated test (see CLINICAL TRIALS) (page 4).15
Stakeholder Perspectives
Patient Group Input
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input for the purpose of this review. The full patient input received is included in Appendix 5.
Three patient groups, the Canadian Liver Foundation, the Canadian Organization for Rare Disorders, and the Cholangiocarcinoma Foundation, co-created 1 patient input for this review. The input was based on an online survey and a virtual focus group. A total of 27 respondents were included in the patient input. Of the respondents, 15 were patients who had been diagnosed with CCA (4 patients had CCA with FGFR2 fusions), 2 were patients who had symptoms of CCA but no diagnosis, and 10 were caregivers or family members of patients with CCA.
Respondents indicated a varying range of CCA symptoms affecting patients’ daily activities (including their social, work, and school lives and their relationships) and causing detrimental effects on patients’ QoL. Respondents highlighted problems with intimacy or sexual desire, fatigue, and anxiety. Other commonly experienced symptoms indicated by respondents included unintended weight loss, insomnia, gastrointestinal problems, abdominal pain, constipation, depression, and neuropathy. According to the 3 patient groups, delayed diagnosis, misdiagnosis, and a lack of specialists and treatment options available for this rare cancer significantly contribute to patients’ feelings of stress and anxiety and may delay or eliminate treatment options.
According to the patient input received, respondents reported they expect the following key outcomes to be improved from any new drug or treatment: QoL, tumour response, delay in disease progression, and additional treatment choice. Additionally, it was highlighted by the 3 patient groups that the identification of gene mutations and the development of targeted therapies was perceived to be very important by respondents and would spur the hope for curable options. Four respondents indicated they had direct experience with taking pemigatinib. Respondents indicated little overall challenge dealing with the side effects from pemigatinib.
Clinician Input
Input From the Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of CCA.
Unmet Needs
The clinical experts consulted by CADTH noted there are currently no funded standard second-line treatment options for patients with unresectable, locally advanced, or metastatic CCA with FGFR2 alterations. Until recently, no high-quality evidence was available in this setting. The first randomized phase III RCT that included patients with CCA in the second line evaluated the chemotherapy option mFOLFOX plus ASC compared with ASC alone in patients with BTC after standard first-line chemotherapy. mFOLFOX was associated with high toxicity, low activity (the ORR achieved with the FOLFOX regimen was 5%; CR = 1%; PR = 4%; stable disease = 28%), and low efficacy (median OS of 6.2 months with the mFOLFOX regimen versus 5.3 months with ASC alone). The clinical expert noted that, currently, mFOLFOX is the most commonly used therapy in the target population in Canadian clinical practice. It was emphasized by the clinical experts that patients with disease that has progressed on first-line chemotherapy often have a rapidly declining ECOG PS, and only a small proportion of patients may be suitable for further systemic treatment. There was consensus among the clinicians that there is an unmet need for effective therapies with acceptable toxicity profiles that achieve disease control, delay worsening of symptoms, maintain HRQoL, delay disease progression, and prolong survival. It was also mentioned by the clinical experts that there are currently no biomarker-directed regimens specifically for patients with CCA whose disease is FGFR2-positive. The experts anticipated more promising benefit with a targeted therapy option in later lines than with another line of chemotherapy with a disease that is steadily growing more resistant.
Place in Therapy
The clinical experts consulted by CADTH stated that pemigatinib was to be used in adult patients with previously treated, unresectable, locally advanced, or metastatic CCA with an FGFR2 fusion or other rearrangement as per the FIGHT-202 trial. It was agreed that oral pemigatinib would likely shift the current treatment paradigm. It was also noted that in the absence of robust comparative data, no firm conclusions could be drawn on how pemigatinib compared with other relevant treatment options; however, the clinical experts anticipated that, based on the FIGHT-202 results and on poor results with existing treatment options in clinical practice, pemigatinib appeared to offer at least similar or improved clinical benefits compared with current therapies, with better tolerability. The FIGHT-202 trial excluded patients who would be intolerant to standard first-line therapy without experiencing PD. The clinical experts consulted by CADTH felt it would be reasonable to generalize the results from cohort A to patients with FGFR2 alterations who are intolerant to first-line therapy, given the favourable safety profile of oral pemigatinib. Furthermore, the clinical experts anticipated seeing the benefit of treatment with pemigatinib regardless of the number of previous lines of systemic therapy, as long as the patients’ cancer has the FGFR2 alteration. However, the clinical experts agreed that patients should not have been previously treated with an FGFR2-targeted therapy.
Patient Population
Overall, the clinical experts consulted by CADTH agreed that patients, as selected per the inclusion and exclusion criteria for cohort A of the FIGHT-202 trial, should be eligible for pemigatinib therapy. Among patients enrolled in cohort A of the FIGHT-202 trial, the clinical experts did not identify any subgroups of patients who would potentially be either best suited for or benefit the least from oral pemigatinib. It was emphasized that patients with CCA that does not have the FGFR2 alteration would not be expected to derive any benefit from pemigatinib. The clinical experts noted that patients with disease that has progressed on first-line chemotherapy often have a rapidly declining ECOG PS and only a small proportion of patients may be suitable for further systemic chemotherapy treatment; for example, platinum-based therapies can lead to neuropathy (after approximately 6 months), and patients who receive standard first-line platinum-based chemotherapy may not be able to be treated with second-line FOLFOX due to this toxicity.
The clinical experts considered that the determination of the presence of FGFR2 fusions or other rearrangements is required before the initiation of treatment with pemigatinib. The clinical experts noted it would be ideal to have molecular FGFR2 testing results available after commencing first-line therapy and before the patients’ disease progresses. A valid test would involve next-generation sequencing, which was used in the FIGHT-202 trial to enrol patients into cohort A. FGFR2 testing is currently not routinely available nor funded in jurisdictions in Canada.
Assessing Response to Treatment
In the opinion of the clinical experts consulted by CADTH, assessments to evaluate the response to treatment include regular radiological imaging (i.e., CT or MRI) and a CA19 to 9 biomarker test every 2 to 3 months to determine whether a patient’s disease has progressed. In addition, patients would be seen by an oncologist every 3 to 4 weeks for clinical assessment (i.e., to assess disease symptoms and the patient’s ECOG PS).
The clinical experts indicated that the most clinically meaningful responses to treatment include disease control (i.e., disease stability or response), improvement in disease-related symptoms, better pain control, weight gain, regaining a more active lifestyle, maintenance of HRQoL, and prolonged PFS and OS. Acceptable drug-related toxicities was also noted as a clinically meaningful outcome.
Discontinuing Treatment
In the opinion of the clinical experts consulted by CADTH, treatment with pemigatinib should be discontinued if a patient experiences disease progression, has a worsening ECOG PS, is intolerant to or experiences unacceptable toxicity from pemigatinib (which cannot be improved with dose delays or reductions), or the patient is not interested in continuing treatment.
Prescribing Conditions
The clinical experts consulted by CADTH noted that because pemigatinib is an oral drug that is self-administered at home, patients should have regular access to outpatient oncology clinics to confirm treatment tolerance and that the disease has not progressed.
Clinician Group Input
The information in this section is a summary of 2 inputs provided by the registered clinician groups that responded to CADTH’s call for clinician input for the purpose of this review.
Two clinician group inputs were provided, 1 from the Ontario Health (Cancer Care Ontario) Gastrointestinal Cancer Drug Advisory Committee and 1 from the CGOEN and other physicians who treat CCA. The views of the clinician groups overall were consistent with the clinical experts consulted by CADTH, indicating that the most important treatment goals are achieving disease control, delaying worsening of symptoms, maintaining HRQoL, delaying disease progression, and prolonging survival. It is also important that the drug have an acceptable safety profile. The clinicians from the CGOEN also highlighted that the convenient oral route of administration of pemigatinib would contribute to improvements in QoL for patients, as fewer visits to a cancer centre and less chair time would be required compared with alternative treatment options. The clinicians from CGOEN further suggested it would be reasonable to consider pemigatinib upfront for patients deemed unsuitable for standard first-line chemotherapy. This clinician group also noted that patients with compromised hepatic function or significant hyperbilirubinemia would be least suitable for treatment with pemigatinib. The clinicians from both inputs anticipated that pemigatinib would offer clinically meaningful benefit and improved efficacy to patients, with the potential for improved QoL.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.36
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Standard first-line treatment of advanced CCA in Canada is typically cisplatin and gemcitabine. In the FIGHT 202 study, patients were eligible as long as disease progression occurred with at least 1 line of chemotherapy. Should patients who have experienced disease progression on cisplatin and gemcitabine be eligible for pemigatinib? | The eligibility criteria of the FIGHT-202 trial did not restrict the number of previous lines of systemic therapy. The majority of patients in cohort A (60.7%) had 1 prior therapy for advanced or metastatic disease followed by patients who had 2 and 3 or more lines of prior therapy (27.1% and 12.1% of patients, respectively). The most commonly received anti-cancer therapy was cisplatin plus gemcitabine chemotherapy. The clinical experts felt it would be reasonable to generalize the results observed in cohort A to patients with FGFR2 alterations regardless of line of therapy, given the molecular targeted mechanism of action of pemigatinib, which is different from chemotherapy. However, the clinical experts agreed that patients should not have previously been treated with an FGFR2-targeted therapy. |
Patients with an ECOG PS of 0 to 2 were eligible for the FIGHT-202 trial. Should patients with an ECOG PS of > 2 also be eligible for treatment with pemigatinib? | Because patients with an ECOG PS greater than 2 were excluded from the FIGHT-202 trial, there is currently insufficient data to support the generalizability of treatment benefit with pemigatinib to patients with an ECOG PS greater than 2. |
For patients currently on treatment (e.g., second-line FOLFOX), should patients switch to pemigatinib or reserve pemigatinib for a subsequent line? | The clinical experts noted that patients who are currently on second-line therapy (e.g., second-line FOLFOX) and whose disease has not progressed should not be switched to pemigatinib, unless patients are experiencing intolerable toxicity from their second-line therapy. |
The sponsor estimates that 19% of patients would have the FGFR2 mutation. FGFR2 testing is not routinely available nor funded in jurisdictions. When is the best time for testing to determine FGFR2 mutation status? | The clinical experts considered that the determination of the presence of FGFR2 fusions or other rearrangements is required before the initiation of treatment with pemigatinib. The clinical experts noted it would be ideal to have molecular FGFR2 testing results available after commencing first-line therapy and before patients’ disease progresses. |
CCA = cholangiocarcinoma; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FGFR2 = fibroblast growth factor receptor 2; FOLFOX = folinic acid, fluorouracil, and oxaliplatin.
Clinical Evidence
The clinical evidence included in the review of pemigatinib is presented in 3 sections. The first section, the systematic review, includes the pivotal study or studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor. The third section includes an additional relevant study that was considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of pemigatinib tablets (13.5 mg orally once daily on a schedule of 2 weeks on and 1 week off for each 21-day cycle) for the treatment of adult patients with previously treated, unresectable, locally advanced, or metastatic CCA with an FGFR2 fusion or other rearrangement.
Methods
Studies selected for inclusion in the systematic review included the pivotal study provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Of note, the systematic review protocol presented subsequently was established before the granting of a Notice of Compliance by Health Canada.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adult patients with previously treated, unresectable, locally advanced, or metastatic CCA with an FGFR2 fusion or other rearrangement Subgroups: ECOG PS |
Intervention | Pemigatinib 13.5 mg orally once daily on a schedule of 2 weeks on and 1 week off for each 21-day cycle |
Comparator | mFOLFOX FOLFIRI 5-FU alone or in combination with cisplatin or oxaliplatin Capecitabine alone or in combination with cisplatin or oxaliplatin Best supportive care |
Outcomes | Efficacy outcomes:
Harms outcomes:
Notable harms and harms of special interest:
|
Study designs | Published and unpublished phase II, III, and IV RCTs |
5-FU = fluorouracil; AE = adverse event; CCA = cholangiocarcinoma; DCR = disease control rate; DOR = duration of response; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FGFR2 = fibroblast growth factor receptor 2; FOLFIRI = folinic acid, fluorouracil, and irinotecan hydrochloride; HRQoL = health-related quality of life; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; RCT = randomized controlled trial; TTP = time to progression; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
aThese outcomes were identified as being of particular importance to patients in the input received by CADTH from patient groups.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.37
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) through Ovid and Embase (1974–) through Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was pemigatinib. Clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. See Appendix 1 for the detailed search strategies.
The initial search was completed on July 19, 2021. Regular alerts updated the search until the meeting of the CADTH pan-Canadian Oncology Drug Review Expert Committee (pERC) on November 10, 2021.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.38 Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. See Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers and through contacts with appropriate experts. In addition, the manufacturer of the drug was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
A focused literature search for network meta-analyses dealing with pemigatinib or CCA was run in MEDLINE All (1946–) through Ovid and Embase (1974–) through Ovid on July 16, 2021. No limits were applied, and conference abstracts were excluded from the search results.
Findings From the Literature
A total of 4 reports presenting data from 1 unique study were identified from the literature for inclusion in the systematic review (Figure 1). The included study is summarized in Table 6. A list of excluded studies is presented in Appendix 2.
Table 6: Details of the FIGHT-202 Study
Characteristic | Description |
|---|---|
Designs and populations | |
Study design | Phase II, multi-centre, open-label, single-arm, multi-cohort trial |
Locations | Patients enrolled in 67 sites across 12 countries:
|
Patient enrolment dates | January 17, 2017, to March 22, 2019 |
Data cut-off dates |
|
Enrolled (N) | 146 patients:a
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Pemigatinib: 13.5 mg orally once daily on a schedule of 2 weeks on, 1 week off for each 21-day cycle. Treatment should continue until radiological disease progression, unacceptable toxicity, withdrawal of consent, or physician choice. |
Comparator(s) | NAd |
Outcomes | |
Primary end point | ORR (cohort A) |
Secondary and exploratory end points | Secondary:
Safety (cohort A, cohort B, and cohort C):
Exploratory:
|
Notes | |
Publicationsh | |
AE = adverse event; ALT = alanine transaminase; AST = aspartate transaminase; CCA = cholangiocarcinoma; CNS = central nervous system; CYP3A4 = cytochrome P3A4; DCR = disease control rate; DOR = duration of response; ECG = electrocardiogram; ECOG = Eastern Cooperative Oncology Group; EMA = European Medicines Agency; EORTC = European Organisation for Research and Treatment of Cancer; FGF = fibroblast growth factor; FGFR2 = fibroblast growth factor receptor 2; NA = not applicable; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PK = pharmacokinetic; QLQ-BIL21 = Quality of Life Questionnaire Cholangiocarcinoma and Gallbladder Cancer Module 21; QLQ-C30 = Quality of Life Questionnaire Core 30; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; ULN = upper limit of normal.
aOne patient was not assigned to any cohort because of an inadequate tissue sample. This patient was not included in the efficacy analyses.39
bTo be enrolled and assigned to their initial cohort, patients had to have results from a certified local laboratory; however, the final cohort assignment for statistical analyses was based on next-generation sequencing results (using the Foundation Medicine Clinical Trial Assay) from the central genomics laboratory.12
cAbnormal laboratory parameters: Total bilirubin ≥ 1.5 × ULN or ≥ 2.5 × ULN in the presence of Gilbert syndrome or disease involving liver; AST and ALT > 2.5 × ULN or > 5 × ULN in the presence of liver metastases; creatinine clearance ≤ 30 mL/min based on Cockroft-Gault; serum phosphate > institutional ULN; serum calcium outside of the institutional normal range or serum albumin—corrected calcium outside of the institutional normal range when serum albumin is outside of the institutional normal range; for potassium levels < institutional lower limit of normal, supplementation can be used to correct potassium level during the screening.
dFIGHT-202 was a non-comparative single-arm phase II trial.
eAs specified in the protocol of the FIGHT-202 trial, only patients from the US were allowed to enrol in cohort C.
fSpecific PK analyses were not specified a priori in the statistical analysis plan.12
gThe EORTC QLQ-BIL21 questionnaire is only translated and validated in the primary languages of those countries.12
hOne additional report was included: the Clinical Study Report, which had a data cut-off date of March 22, 2019 (addendum 1 had a data cut-off date of August 30, 2019, and addendum 2 had a data cut-off date of April 7, 2020). (The Clinical Study Report was received as part of the submission to CADTH.)
Source: Abou-Alfa et al. (2020),39 Clinical Study Report,16 sponsor’s response.17
Description of Studies
FIGHT-202 is an ongoing, multi-centre, open-label, single-arm phase II trial evaluating the efficacy and safety of pemigatinib in patients with advanced, metastatic, or surgically unresectable CCA with FGFR2 alterations, alterations in other FGFs or FGFRs, or no FGF or FGFR alterations, whose disease did not respond to previous therapy. Patients were assigned to 3 cohorts depending on their FGF/FGFR status (cohort A: FGFR2 fusions or rearrangements; cohort B: FGF/FGFR alterations other than FGFR2 fusions or rearrangements; or cohort C: negative for FGF/FGFR alterations). This CADTH review focuses on cohort A, since cohorts B and C were not part of the requested reimbursement criteria submitted to CADTH and not approved in the Health Canada Notice of Compliance with conditions and are therefore beyond the scope of this review. Selected results for cohorts B and C have been included in Appendix 3. The primary objective of the trial was to assess the efficacy of pemigatinib in patients with advanced, metastatic, or surgically unresectable CCA with FGFR2 alterations (patients in cohort A) that did not respond to at least 1 previous treatment. Patients in this international trial were enrolled at 67 sites across 12 countries, which are listed in Table 6. The majority of sites were in the US followed by Europe, with no sites in Canada. Enrolment started on January 12, 2017 and ended on March 22, 2019.16
A total of 146 patients were enrolled to received oral pemigatinib (13.5 mg orally once daily on a schedule of 2 weeks on, 1 week off for each 21-day cycle). Best supportive care was administered as needed and included but was not limited to palliative radiotherapy for bone lesions, stent placement, or replacement of blocked bile ducts.17 Pemigatinib was administered until documented disease progression or unacceptable toxicity.12 The study design is depicted in Figure 2.
To be enrolled and for the initial cohort assignment, patients had to have results from a certified local laboratory; however, final cohort assignment for the statistical analyses was based on the results of next-generation sequencing (using the Foundation Medicine Clinical Trial Assay) from the central genomics laboratory.12 The study consisted of 3 phases, the screening phase (lasting up to 28 days), the treatment phase, and the follow-up phase. During the follow-up phase, patients were followed for safety (final follow-up visit 30 to 35 days after end of treatment), for disease status every 9 weeks (for patients who discontinued pemigatinib for reasons other than disease progression), and for OS at least every 12 weeks.12 Response assessment, according to Response Evaluation Criteria in Solid Tumours Version 1.1 (RECIST 1.1) (Eisenhauer et al.)40 was based on radiologic imaging and performed by an IRC and occurred every 2 cycles (every 6 weeks) for the first 4 cycles and every 3 cycles (every 9 weeks) thereafter, and at the treatment discontinuation visit.12 Tumour response was also evaluated by the investigator.12 Safety and tolerability were evaluated by monitoring the frequency, duration, and severity of AEs.12
The predetermined threshold for a positive study outcome (lower limit of the 95% CI for ORR > 15%) was achieved as of the March 22, 2019, data cut-off date.16 While the timing of this data cut-off date was not pre-specified a priori in the statistical analysis plan, the sponsor’s proposed timing was agreed upon by the FDA during its review process for pemigatinib.5 Two additional updated analyses occurred at the August 2019 and April 2020 data cut-off dates: the former was a 4-month safety update required for the FDA NDA, the latter was performed to support the safety data summaries for another indication outside of Canada.17 The trial is still ongoing, with an estimated completion date in the first quarter of 2022.17 The FIGHT-202 trial was sponsored by Incyte Corporation.39
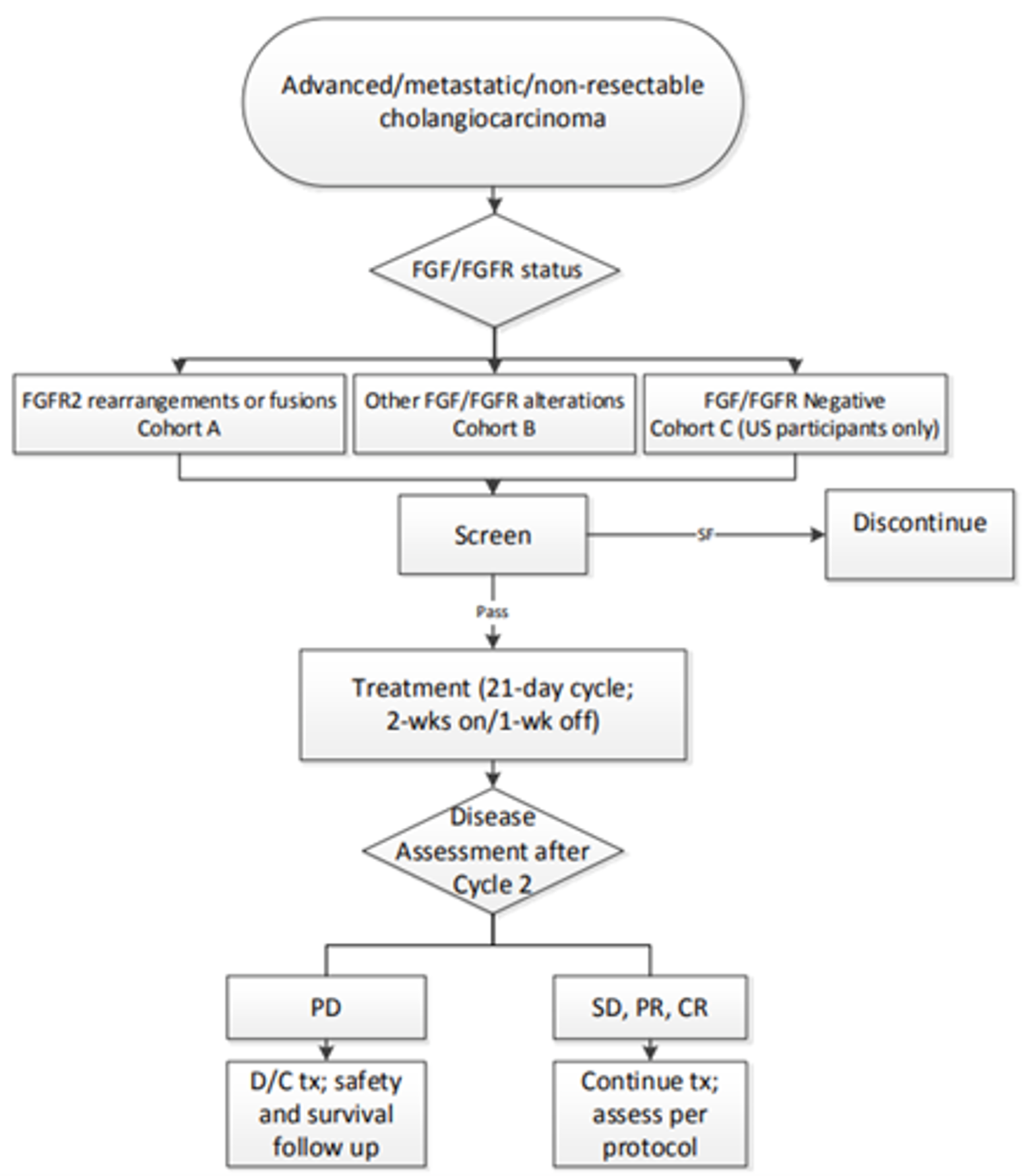
CR = complete response; D/C = discontinue; FGF = fibroblast growth factor; FGFR = fibroblast growth factor receptor; PD = progressive disease; PR = partial response; SD = stable disease; SF = screen fail; tx = therapy.
Source: Clinical Study Report.16
Populations
Inclusion and Exclusion Criteria
The key inclusion and exclusion criteria used in the FIGHT-202 trial are described in Table 6. Briefly, the trial enrolled adults, aged 18 years or older, diagnosed with advanced, metastatic, or surgically unresectable CCA, whose disease progressed after at least 1 line of prior systemic therapy (prior therapy with selective FGFR inhibitors was not permitted). Patients had to have documented FGF/FGFR gene alteration status and radiologically measurable disease according to RECIST 1.1. At screening, patients had to have an ECOG PS of 0 to 2, a life expectancy of at least 12 weeks, adequate hepatic and renal function, and no clinically significant corneal or retinal disorder, confirmed by ophthalmologic examination.16
Baseline Characteristics
The baseline characteristics of patients who comprised the safety population of the FIGHT-202 trial are summarized in Table 7. At baseline, 107 patients were identified as having FGFR2 fusions or rearrangements and were grouped into cohort A. Cohort B included 20 patients with other FGF/ FGFR alterations other than FGFR2, and cohort C included 18 patients with no identified FGF/FGFR alterations. One patient who was placed into an “undetermined” group was not assigned to any of the 3 cohorts, as the local FGF/FGFR status results could not be confirmed by the central genomics laboratory. For patients in cohort A, the mean age was 55.3 years (SD = 12.02); most patients were female (60.7%), and most were enrolled in trial sites in North America (59.8%) or Europe (29.9%). Almost all patients (89% of patients overall and 98.1% of patients in cohort A) had iCCA. The majority of patients in cohort A had metastatic disease (82.2%), with the ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Median time since diagnosis was 1.28 years (range, 0.03 to 11.1 years) in patients in cohort A. The majority of patients in cohort A had an ECOG PS of 1 (53.3%) or 0 (42.1%) and all patients had received at least 1 line of prior systemic therapy for advanced or metastatic disease (60.7%, 27.1%, and 12.1% of patients received 1, 2, and ≥ 3 prior lines, respectively). Renal and hepatic impairment grades were normal or mild for most patients in cohort A (39.3% and 43.9% had normal and mild renal impairment grades, respectively; 44.9% and 48.6% had normal and mild hepatic grades, respectively).16
Table 7: Summary of Baseline Characteristics, Safety Population for FIGHT-202
Characteristic | Cohort A FGFR2 fusions or rearrangements N = 107 | Cohort B other FGF/ FGFR alterations N = 20 | Cohort C no FGF/ FGFR alterations N = 18 | All patients N = 146a |
|---|---|---|---|---|
Age (years) | ||||
Mean (SD) | 55.3 (12.02) | 61.9 (10.99) | 63.7 (10.68) | 57.2 (12.08) |
Median (range) | 56.0 (26 to 77) | 63.0 (45 to 78) | 65.0 (31 to 78) | 59.0 (26 to 78) |
Age category, n (%) | ||||
< 65 years | 82 (76.6) | 10 (50.0) | 7 (38.9) | 100 (68.5) |
65 to < 75 years | 20 (18.7) | 7 (35.0) | 8 (44.4) | 35 (24.0) |
≥ 75 years | 5 (4.7) | 3 (15.0) | 3 (16.7) | 11 (7.5) |
Sex, n (%) | ||||
Male | 42 (39.3) | 9 (45.0) | 10 (55.6) | 62 (42.5) |
Female | 65 (60.7) | 11 (55.0) | 8 (44.4) | 84 (57.5) |
Geographical region, n (%) | ||||
North America | 64 (59.8) | 6 (30.0) | 18 (100.0) | 89 (61.0) |
Western Europe | 32 (29.9) | 3 (15.0) | 0 (0) | 35 (24.0) |
Rest of the worldb | 11 (10.3) | 11 (55.0) | 0 (0) | 22 (15.1) |
Race, n (%) | ||||
White | 79 (73.8) | 9 (45.0) | 15 (83.3) | 104 (71.2) |
Asian | 11 (10.3) | 11 (55.0) | 0 (0) | 22 (15.1) |
Black or African American | 7 (6.5) | 0 (0) | 1 (5.6) | 8 (5.5) |
American Indian or Alaska Native | 0 (0) | 0 (0) | 1 (5.6) | 1 (0.7) |
Otherc | 4 (3.7) | 0 (0) | 1 (5.6) | 5 (3.4) |
Missing | 6 (5.6) | 0 (0) | 0 (0) | 6 (4.1) |
ECOG Performance Status, n (%) | ||||
Grade 0 | 45 (42.1) | 7 (35.0) | 7 (38.9) | 59 (40.4) |
Grade 1 | 57 (53.3) | 10 (50.0) | 8 (44.4) | 76 (52.1) |
Grade 2 | 5 (4.7) | 3 (15.0) | 3 (16.7) | 11 (7.5) |
Grade ≥ 3 | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Renal impairment grade at baseline, n (%)d | ||||
Normal | 42 (39.3) | 6 (30.0) | 7 (38.9) | 55 (37.7) |
Mild | 47 (43.9) | 13 (65.0) | 7 (38.9) | 68 (46.6) |
Moderate | 18 (16.8) | 1 (5.0) | 3 (16.7) | 22 (15.1) |
Severe | 0 (0) | 0 (0) | 1 (5.6) | 1 (0.7) |
Hepatic impairment grade at baseline, n (%)e | ||||
Normal | 48 (44.9) | 13 (65.0) | 13 (72.2) | 75 (51.4) |
Mild | 52 (48.6) | 7 (35.0) | 4 (22.2) | 63 (43.2) |
Moderate | 7 (6.5) | 0 | 1 (5.6) | 8 (5.5) |
Cholangiocarcinoma location, n (%) | ||||
Intrahepatic | 105 (98.1) | 13 (65.0) | 11 (61.1) | 130 (89.0) |
Extrahepatic | 1 (0.9) | 4 (20.0) | 7 (38.9) | 12 (8.2) |
Other | 0 | 3 (15.0)f | 0 | 3 (2.1) |
Missing | 1 (0.9)g | 0 | 0 | 1 (0.7) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Time since diagnosis (years) | ||||
Mean (SD) | 1.57 (1.619) | 1.01 (0.676) | 1.52 (1.240) | 1.49 (1.481) |
Median | 1.28 | 0.73 | 0.98 | 1.10 |
Minimum, maximum | 0.03,h 11.1 | 0.2, 2.5 | 0.3, 4.3 | 0.03, 11.1 |
Metastatic disease, n (%) | ||||
Yes | 88 (82.2) | 20 (100.0) | 16 (88.9) | 125 (85.6) |
No | 16 (15.0) | 0 (0) | 2 (11.1) | 18 (12.3) |
Not evaluable | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Missing | 2 (1.9) | 0 (0) | 0 (0) | 2 (1.4) |
Number of previous systematic therapies for advanced or metastatic disease, n (%)i | ||||
1 | 65 (60.7) | 12 (60) | 12 (66.7) | 89 (61.0) |
2 | 29 (27.1) | 7 (35.0) | 2 (11.1) | 38 (26.0) |
≥ 3 | 13 (12.1) | 1 (5.0) | 4 (22.2) | 19 (13.0) |
Previous cancer surgery, n (%) | ||||
Yes | 38 (35.5) | 6 (30.0) | 4 (22.2) | 48 (32.9) |
No | 69 (64.5) | 14 (70.0) | 14 (77.8) | 98 (67.1) |
Previous radiotherapy, n (%) | ||||
Yes | 28 (26.2) | 3 (15.0) | 5 (27.8) | 36 (24.7) |
No | 79 (73.8) | 17 (85.0) | 13 (72.2) | 110 (75.3) |
History of hepatitis, n (%) | ||||
Hepatitis B | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Hepatitis C | 1 (0.9) | 1 (5.0) | 0 (0) | 2 (1.4) |
Current sites of disease, n (%) j | ||||
Liver | 101 (94.4) | 17 (85.0) | 18 (100.0) | |||||||||||| |
Lymph nodes | 57 (53.3) | 11 (55.0) | 10 (55.6) | |||||||||||| |
Lung | 58 (54.2) | 9 (45.0) | 10 (55.6) | 77 (52.7) |
Bone | 21 (19.6) | 4 (20.0) | 2 (11.1) | 27 (18.5) |
Ascites | 8 (7.5) | 5 (25.0) | 2 (11.1) | 15 (10.3) |
Pancreas | 7 (6.5) | 1 (5.0) | 2 (11.1) | |||||||||||| |
Pleural effusion | 4 (3.7) | 2 (10.0) | 0 (0) | 6 (4.1) |
Skin or subcutaneous tissue | 2 (1.9) | 0 (0) | 0 (0) | 2 (1.4) |
Bladder | 0 (0) | 1 (5.0) | 0 (0.0) | 1 (0.7) |
Colon | 1 (0.9) | 0 (0) | 0 (0) | 1 (0.7) |
Other | 31 (29.0) | 7 (35.0) | 12 (66.7) | 51 (34.9) |
ECOG = Eastern Cooperative Oncology Group; eGFR = estimated glomerular filtration rate; FGF = fibroblast growth factor; FGFR2 = fibroblast growth factor receptor 2; MDRD = Modification of Diet in Renal Disease; SD = standard deviation.
aOne participant from the safety population was assigned to a group labelled “undetermined” and excluded from the efficacy evaluable population because the local laboratory FGF and FGFR results could not be confirmed centrally due to technical issues with the tissue sample.
bRest of world consists of Israel, South Korea, Taiwan, Thailand, and Japan.
cIncludes Hispanic, Latino, or Spanish (n = 1) or not reported (n = 4).
dBaseline renal impairment grade (normal, mild, moderate, or severe) based on eGFR (calculated using the MDRD equation): normal renal function = eGFR ≥ 90 mL/min/1.73 m2, mild renal impairment = eGFR ≥ 60 and < 90 mL/min/1.73 m2, moderate renal impairment = eGFR ≥ 30 to < 60 mL/min/1.73 m,2, and severe renal impairment = eGFR < 30 mL/min/1.73 m2.
eDegree of hepatic impairment based on National Cancer Institute Hepatic Working Group Criteria.
fIncludes gallbladder (n = 2) and ampulla of Vater (n = 1).
gAt baseline, this participant had stage 4 cholangiocarcinoma (T3 N0 M1), presumed to be intrahepatic, with current sites of disease at the liver, omentum, and peritoneum.
hThe participant's date of diagnosis was entered incorrectly by trial site personnel. The time since diagnosis is 22.11 months, based on the correct date of diagnosis.
iThere was a maximum number of 5 therapies for patients with FGFR2 fusions or rearrangements and a maximum of 3 therapies for patients in the other cohorts.
jA patient could have multiple current disease sites.
Source: Clinical Study Report.16
All patients in cohort A had received at least 1 prior systemic cancer therapy (Table 8). The majority of patients had received platinum-based chemotherapy regimens before study enrolment, most commonly gemcitabine, reported as gemcitabine (85.0%) or gemcitabine hydrochloride (7.5%) and cisplatin (75.7%). The second and third most commonly received pyrimidine analogues were fluorouracil (29.0%) and ||||||||||||||||||||||||||||||||; the second most frequently administered platinum compound was oxaliplatin (38.3%).16 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.39
Table 8: Summary of Prior Systemic Cancer Therapy, Safety Population
Prior treatment, n (%) WHO drug class/ WHO drug term | Pemigatinib cohort A outcome (N = 107) |
|---|---|
Pyrimidine analogues | 106 (99.1) |
Gemcitabine | 91 (85.0) |
Fluorouracil | 31 (29.0) |
Capecitabine | |||||||||||| |
Gemcitabine hydrochloride | 8 (7.5) |
Tegafur | |||||||||||| |
Floxuridine | |||||||||||| |
Platinum compounds | 101 (94.4) |
Cisplatin | 81 (75.7) |
Oxaliplatin | 41 (38.3) |
Carboplatin | |||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
||||||||||||||||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||| | |||||||||||| |
Source: Clinical Study Report.16
All patients in cohort A (N = 107) had centrally confirmed FGFR2 fusions or rearrangements with 56 unique rearrangement or fusion partners identified (Table 9).16 The most frequently identified FGFR2 partner was BICC1 (31 patients; 29%). There were 5 patients with FGFR rearrangements referred to as FGFR2-N/A, as no specific partner could be identified.39
Table 9: FGF/FGFR Fusion Partners or Rearrangements Identified by Central Genomics Laboratory in 2 or More Patients, Cohort A
FGF/FGFR alterations, n | Pemigatinib cohort A outcome (N = 107) |
|---|---|
FGFR2-BICC1 | 31 |
FGFR2-N/A | 5 |
FGFR2-KIAA1217 | 4 |
FGFR2-AHCYL1 | 3 |
FGFR2-ARHGAP24 | 2 |
FGFR2-AFF4 | 2 |
FGFR2-CCDC6 | 2 |
FGFR2-MACF1 | 2 |
FGFR2-NOL4 | 2 |
FGFR2-NRAP | 2 |
FGFR2-PAWR | 2 |
FGFR2-SLMAP | 2 |
FGF = fibroblast growth factor; FGFR2 = fibroblast growth factor receptor 2.
Source: Clinical Study Report.16
Interventions
Patients enrolled in the FIGHT-202 trial self-administered oral pemigatinib once daily on a schedule of 2 weeks on, 1 week off. Treatment and dose are described in Table 10.
Table 10: Treatment Regimen in the FIGHT-202 Trial
Detail | Description |
|---|---|
Dose | Pemigatinib: 13.5 mg administered orally (tablets) once daily for 2 weeks continuously (14 days) followed by a 1-week (7 days) pause for each 21-day treatment cycle |
Treatment discontinuation | Patients were withdrawn from the study treatment when the following criteria were met:
Patients could be withdrawn from the study treatment when the following criteria were met:
|
AE = adverse event; ECG = electrocardiogram; FGF = fibroblast growth factor; FGFR = fibroblast growth factor receptor; IEC = independent ethics committee; IRB = institutional review board; QT = QT interval; QTc = corrected QT interval.
Source: Sponsor’s submission.12
Dose Modifications
Dose interruptions, delays, and modifications were permitted and were guided by the occurrence of toxicities (related or unrelated to the study drug). A maximum of 2 dose reductions (from a daily dose of 13.5 mg to 9 mg, and from 9 mg to 6 mg) was recommended; patients could not receive a dose below 6 mg daily. No modification of the treatment schedule was allowed. At the occurrence of any grade 3 toxicity not manageable by supportive care, or a level of aspartate transaminase and/or alanine transaminase greater than 5.0 times the upper limit of normal, treatment could be interrupted for up to 14 days until toxicity resolved to grade 1 or less. Once a dose interruption occurred, pemigatinib was either restarted at the same dose that was being taken before the dose interruption occurred, or at the next-lower dose and monitored as clinically indicated. Situations in which treatment was delayed for more than 14 days before restarting treatment had to be discussed on a case-by-case basis with the sponsor. In cases of recurrent grade 3 toxicity after 2 dose reductions or any other grade 4 toxicity, pemigatinib administration had to be discontinued; exceptions to that required the sponsor’s approval.16
Concomitant Medication
Concomitant medications were allowed to treat comorbidities or AEs as long as they did not include potent cytochrome P3A4 (CYP3A4) inhibitors and inducers or moderate CYP3A4 inducers (there was no restriction on topical ketoconazole), another selective FGFR inhibitor, an investigational study drug for any indication, or any anti-cancer medications other than the study drug.12
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trial included in this review is provided in Table 11. These end points are further summarized subsequently. A detailed discussion and critical appraisal of the HRQoL and symptom severity outcomes is provided in Appendix 4.
Table 11: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measurea | Outcome |
|---|---|
OS | Secondary (cohort A, cohort B, and cohort C)b |
PFS | Secondary (cohort A, cohort B, and cohort C)b |
ORR | Primary (cohort A) |
Secondary (cohort B)b | |
Secondary (cohort A plus cohort B)b | |
Secondary (cohort C)b | |
DOR | Secondary (cohort A, cohort B, and cohort C)b |
DCR | Secondary (cohort A, cohort B, and cohort C)b |
TTP | Not measured in FIGHT-202 |
HRQoL | |
EORTC QLQ-C30 | Exploratory (cohort A, cohort B, and cohort C)b |
Symptom severity | |
EORTC QLQ-BIL21 | Exploratory (cohort A, cohort B, and cohort C)b |
Safety | |
Frequency, duration, and severity of AEs | Secondary (cohort A, cohort B, and cohort C)b |
AE = adverse event; DCR = disease control rate; DOR = duration of response; EORTC = European Organisation for Research and Treatment of Cancer; HRQoL = health-related quality of life; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; QLQ-BIL21 = Quality of Life Questionnaire Cholangiocarcinoma and Gallbladder Cancer Module 21; QLQ-C30 = Quality of Life Questionnaire Core 30; TTP = time to progression.
aOutcomes are presented in order of priority, as identified in the CADTH review protocol.
bThis CADTH review presents results for cohort A, as cohorts B and C were not part of the requested reimbursement criteria and not approved in the Health Canada Notification of Compliance with conditions; therefore, they are beyond the scope of this review. For the interested reader, selected results for cohorts B and C have been included in Appendix 3.
Source: Statistical analysis plan.12
Overall Survival
OS was a secondary outcome of the FIGHT-202 trial and was defined as the time from the date of first dose until the date of death due to any cause.12 The OS analyses were performed for patients in cohorts A, B, and C, respectively.12
Progression-Free Survival
PFS was a secondary outcome of the FIGHT-202 trial and was measured as the time from the date of the first dose of the study drug until the date that PD was first recorded or death, whichever occurred first. The date of PD was the time point at which progression was first documented. Disease progression for the analysis of PFS was determined according to RECIST 1.1 criteria as assessed by an independent centralized radiological review committee (see Table 29 in Appendix 3 for definitions for the evaluation of the target lesions). PFS was also analyzed based on investigator assessment. The PFS analyses were performed for patients in cohorts A, B, and C, respectively.12
Objective Response Rate
The ORR assessed in cohort A was the primary outcome of the FIGHT-202 trial. The ORR evaluated in cohort B, cohort A plus B, and cohort C were secondary outcomes. ORR was defined as the proportion of patients who achieved a best overall response of CR (disappearance of all target lesions) or a PR (≥ 30% decrease in the sum of the longest diameters of target lesions) at any post-baseline visit before the first instance of PD. Best overall response was defined as the best response documented post baseline before and including the first instance of PD, in the order of CR, PR, stable disease, PD, and not evaluable. Clinical response for the analysis of ORR was determined based on RECIST 1.1, assessed by an independent radiological review committee using central genomics laboratory results, and required confirmation of a CR and PR at least 4 weeks after the initial assessment (see Table 29 in Appendix 3 for definitions for the evaluation of the target lesions). The only clinical data transmitted by the site to the IRC were radiation history, prior surgeries, and investigator-documented benign radiographic abnormalities. For stable disease, the stable disease criteria had to be met at least once after the date of the first dose at a minimum interval of 39 days; in case these criteria were not met, patients were recorded as having an overall response of PD if the next available assessment indicated PD, or not evaluable if there was no additional assessment available. ORR was also analyzed based on investigator assessment; however, confirmation of a CR and PR was not required.12
Duration of Response
DOR was a secondary outcome of the FIGHT-202 trial and was defined as the interval from the date of a CR or PR (i.e., an overall response contributing to an objective response) to the date of death or first overall response of PD, whichever occurred first.12 Clinical response for the analysis of DOR was determined based on RECIST 1.1, assessed by an independent radiological review committee. The DOR analyses were performed for patients in cohorts A, B, and C, respectively. DOR was also analyzed based on investigator assessment.12
Disease Control Rate
DCR was a secondary outcome of the FIGHT-202 trial and was defined as the proportion of patients with a best response of CR, PR, or stable disease. Clinical response for the analyses of DCR was determined based on RECIST 1.1, assessed by an independent radiological review committee using central genomics laboratory results, and required confirmation of a CR and PR at least 4 weeks after the initial assessment.12 The DCR analyses were performed for patients in cohorts A, B, and C, respectively. DCR was also analyzed based on investigator assessment.12
Time to Progression
This outcome was not assessed in the FIGHT-202 trial.
Health-Related Quality of Life
The HRQoL outcomes measured in the trial included the EORTC QLQ-C30. No analysis plan, objective, or minimally important difference (MID) for the EORTC QLQ-C30 instrument were specified a priori in the statistical analysis plan; it was noted, however, that the scores for each scale were to be calculated. According to the study’s protocol scores for each scale, changes from baseline to each visit were to be measured and summarized descriptively.12 The EORTC QLQ-C30 questionnaire was assessed at baseline and then every 3 cycles starting with cycle 3, until discontinuation of the study treatment and at the end-of-treatment visit.12 A detailed discussion and critical appraisal of the HRQoL measures is provided in Appendix 4.
The EORTC QLQ-C30 is 1 of the most commonly used patient-reported outcome measures in oncology clinical trials. It is a multi-dimensional, cancer-specific, evaluative measure of HRQoL. It was designed specifically for the purpose of assessing changes in participants’ HRQoL in clinical trials in response to treatment. The EORTC QLQ-C30 questionnaire consists of 30 questions that are scored to create 5 multi-item functional scales, 3 multi-item symptom scales, 6 single-item symptom scales, and a 2-item QoL scale. Most questions have 4 response options (not at all, a little, quite a bit, very much), with scores on these items ranging from 1 to 4. For the 2 items that form the global QoL scale, however, the response format is a 7-point Likert-type scale, with anchors between 1 (very poor) and 7 (excellent). Version 3.0 of the questionnaire, which was used in the included trials in this report, is the most current version. Each raw scale score is converted to a standardized score that ranges from 0 to 100 using a linear transformation, with a higher score reflecting better function on the function scales, higher symptoms on the symptom scales, and better QoL (i.e., higher scores simply reflect higher levels of response on that scale). Thus, a decline in score on the symptom scale would reflect an improvement, whereas an increase in score on the function and QoL scales would reflect an improvement. It is available in 118 different languages on the EORTC Quality of Life Group website and is intended for use in adult populations only.
The reliability of the EORTC QLQ-C30 instrument was evaluated in an international study in patients with BTC.41 Internal consistency was assessed and was acceptable for all scales except for the physical function, cognitive function, and nausea/ vomiting scales, which had mixed results. Results for test–retest reliability were also mixed, with the intraclass correlation coefficient (ICC) for the scales ranging from 0.52 to 0.92. Construct validity and responsiveness were not assessed for the EORTC QLQ-C30.41 Estimates for MIDs in the literature were not found for the EORTC QLQ-C30 in patients with CCA or BTC.
Symptom Severity
Symptom severity was assessed in the trial using the EORTC QLQ-BIL21. The EORTC QLQ-BIL21 was an exploratory outcome in the FIGHT-202 trial. No analysis plan, objective, or MID for the EORTC QLQ-BIL21 instrument were specified a priori in the statistical analysis plan; it was noted, however, that the scores for each scale were to be calculated. According to the study’s protocol scores for each scale, changes from baseline to each visit were to be measured and summarized descriptively.12 The EORTC QLQ-BIL21 questionnaire was assessed at baseline and then every 3 cycles starting with cycle 3, until discontinuation of the study treatment and at the end-of-treatment visit.12 A detailed discussion and critical appraisal of the HRQoL measures is provided in Appendix 4.
The EORTC QLQ-BIL21 is a disease-specific module to be used in addition to the EORTC QLQ-C30 to assess HRQoL in patients with CCA and gallbladder cancer.42 It consists of 21 questions, with 18 of the items grouped into 5 scales: eating symptoms (4 items), jaundice symptoms (3 items), tiredness (3 items), pain symptoms (4 items), and anxiety symptoms (4 items).42 The remaining 3 items are single-item assessments of treatment side effects, difficulties with drainage bags or tubes, and concerns about weight loss.42 Patients complete the questionnaire based on a 1-week recall period by rating each item on a 4-point Likert scale (1 = not at all, 2 = a little, 3 = quite a bit, 4 = very much).42 The scores are then transformed linearly to a 0 to 100 scale to yield scale scores using EORTC guidelines, with higher scores indicating more severe symptoms.41,42 The questions have been translated according to QoL group guidelines into Mandarin Chinese, Italian, German, Dutch, Spanish, and Hindi.41,42
An international study was conducted to validate the EORTC QLQ-BIL21 in patients with BTC.41 The study included 172 adult patients with CCA and 91 patients with gallbladder cancer who had an expected minimum survival of 3 months and were undergoing treatment.41 Internal consistency was assessed and was acceptable for all multi-item scales of the EORT QLQ-BIL21 instrument. Test–retest reliability was assessed using the ICC, which showed good reproducibility. Known group validity was assessed and shown to distinguish groups based on Karnofsky performance status (KPS). There was some evidence for responsiveness to the change in the eating, jaundice, tiredness, pain, treatment side effects, and anxiety scales. The single-item assessment of difficulties with drainage bags and tubes was considered irrelevant by 29 patients.41 The study authors noted that not all patients experience drains during their treatment and that perhaps there should be a “not applicable” option for responding to that item.41 Estimates for MIDs in the literature were not found for the EORTC QLQ-BIL30 in patients with CCA or BTC.
Safety
Safety was designated as a secondary outcome in the FIGHT-202 trial and was defined as any untoward medical occurrence associated with the use of a drug in humans, whether or not the occurrence was considered drug-related, that occurred after a patient provided informed consent.12 While the data listings included all AEs, the analysis of AEs was limited to TEAEs.12 A TEAE included any AE reported for the first time or a worsening of a pre-existing event after the first dose of pemigatinib.12 Abnormal laboratory values or test results observed in patients constituted AEs only if they were associated with clinical signs or symptoms, were considered clinically meaningful, required therapy (e.g., hematologic abnormality requiring transfusion), or required changes in the investigation study drug.12 Disease progression was recorded as an AE only if there were no other identifiable AEs or serious adverse events (SAEs) associated with the disease progression at the time of reporting.12
AEs were organized based on the Medical Dictionary for Regularity Activities (MedDRA) preferred term and system organ class. Severity of AEs was defined according to the US National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03 (CTCAE 4.03). If a toxicity was not included in the CTCAE 4.03 criteria, it was graded on a scale of 1 to 4 (where 1 = mild, 2 = moderate, 3 = severe, and 4 = life-threatening).12
All AEs were documented by the investigator from the date a patient signed the informed consent form to at least 30 to 35 days after the end-of-treatment visit (or the last dose of the study drug if the end-of-treatment visit was not performed).12
The FIGHT-202 trial included the following parameters for the analysis of AEs:
number of patients reporting TEAEs, SAEs, grade 3 or 4 TEAEs, fatal TEAEs, and temporarily interrupting pemigatinib or permanently discontinuing pemigatinib due to TEAEs
summary of TEAEs and grade 3 or 4 TEAEs by system organ class, preferred term, decreasing order of frequency, and maximum severity
summary of TEAEs leading to death, treatment-emergent SAEs, TEAEs leading to dose modifications (reductions, interruptions), and TEAEs leading to discontinuation of pemigatinib by system organ class and preferred term.12
The FIGHT-202 trial monitored the following parameters: patients’ physical examination, changes in vital signs, electrocardiogram findings, and changes in clinical laboratory blood and urine sample evaluations.12
Statistical Analysis
Sample Size Determination
For the final analysis of the primary end point (ORR in cohort A), a sample size of approximately 100 patients with FGFR2 translocation documented by the central genomics laboratory was planned.12 A sample size of 100 patients was selected to guarantee an adequate population for robust response data and safety assessments.39 With the assumption that 33% of patients treated with pemigatinib would achieve an objective response, a sample size of 100 patients (assuming 10% of patients would be lost to follow-up) was estimated to provide greater than 95% probability of having a 95% CI with a lower limit of greater than 15%.39 If the lower limit of the 95% CI for ORR exceeded 15%, it was predetermined that the trial results would be considered positive.16 The minimum clinically meaningful proportion of patients with an objective response was considered to be 15%, based on ORR results reported by previous studies43-45 of patients with CCA.39 For the analyses of cohort B and cohort C, a sample size of up to 20 patients was planned for each cohort, allowing a greater than 80% chance of observing at least 4 objective responders per cohort, if the underlying ORR was 30%.39
Interim Analysis
An interim analysis for futility for cohort A was planned after approximately 25 patients had enrolled and had at least 1 post-baseline tumour assessment or had permanently discontinued treatment.12 Enrolment into cohort A could have been terminated for futility if 2 or fewer of the 25 patients in cohort A had achieved a response. Based on a sample size of 60 patients in cohort A, there was a less than 10% probability of the proportion of patients with an objective response being greater than 15% at the final analysis.12 Initially, the trial was designed to enrol 60 patients into cohort A; however, protocol amendment 5 (October 3, 2017) was later approved to increase the sample size of cohort A to approximately 100 patients.12 The futility analysis was conducted on October 3, 2017. Since the futility boundary was not crossed, the study proceeded as planned.17
The timing of the final analysis, at which point the predetermined threshold (i.e., a lower limit of the 95% CI for ORR > 15%) would be assessed, was not pre-specified a priori in the statistical analysis plan. According to the FDA report, the sponsor proposed a data cut-off date of March 22, 2019, during the pre-NDA meeting on August 8, 2019, during which the FDA acknowledged that the suggested data cut-off date would provide a minimum of 7 months of follow-up for all patients in the efficacy set and a minimum of 6 months of follow-up, from time of initial response, for 92% of responders. The sponsor further agreed to provide additional follow-up data for DOR (DOR only; no other efficacy outcome was analyzed at that time). This corresponded to an August 30, 2019, data cut-off date, based on data with at least 6 months of follow-up from the time of initial response for all responders, and a minimum of 12 months of follow-up for all patients in the efficacy set. The data cut-off date of August 30, 2019, also aligned with the 4-month safety update required for the NDA submitted to the FDA.17 According to the FDA, the proposed data analyses were sufficient to support the filing of an NDA under the provision of accelerated approval.5
An updated data analysis occurred at the April 7, 2020, data cut-off date to support safety data summaries for another indication outside of Canada.17 Since the April 7, 2020, data cut-off date included 1 additional patient in cohort A who had been enrolled after the August 30, 2019, data cut-off date, some efficacy analyses (i.e., survival and response outcomes) were performed, in addition to safety analyses, and were provided to relevant regulatory authorities.17
For the purpose of regulatory requirements outside of Canada, a data cut-off date of July 8, 2021, was set. Data for the July 8, 2021, data cut-off date has not been submitted to CADTH. Two patients were still receiving treatment at that time and, therefore, a subsequent data cut-off date will be set once the database is closed, approximately in the first quarter of 2022.17
Primary Outcome
The primary outcome in the FIGHT-202 trial was ORR in patients enrolled in cohort A. A brief overview of statistical methods used for the primary outcome is provided in Table 12.
No statistical comparisons were planned between cohorts and no formal hypothesis testing or inferential analyses were performed.39 The 95% CI for ORR was estimated using the exact method for binomial distribution.12 Patients with insufficient baseline or on-study response assessment data were considered nonresponders and were included in the denominators in the calculation of ORR.12 One sensitivity analysis for ORR was planned in the per-protocol (PP) population.12
Secondary Outcomes
A brief overview of the statical methods used for the secondary outcomes is provided in Table 12.
Three secondary outcomes involved ORR in the FIGHT-202 trial: ORR in patients in cohort B, ORR in patients in cohort A plus cohort B, and ORR in patients in cohort C. The 3 secondary ORR outcomes were planned to be analyzed in the same manner as the primary ORR outcome, and the 95% CI for ORR was estimated using the exact method for binomial distribution. No sensitivity analyses were planned for the 3 secondary ORR outcomes.12
PFS was a secondary outcome in the FIGHT-202 trial and was assessed for cohorts A, B, and C, respectively; no statistical comparisons were planned between cohorts and no formal hypothesis testing or inferential analyses were performed. The number of patients who progressed, died, and were censored were summarized. A Kaplan–Meier (KM) plot of PFS was presented with its 95% CI; 95% CI was estimated using the Brookmeyer and Crowley method.46,12 Censoring was based on the FDA Guidance for Industry: Clinical Trial End points for the Approval of Cancer Drugs and Biologics (FDA, 201547; FDA, 201848).12 Reasons for censoring included no baseline tumour assessment, no adequate post-baseline response assessment, no progression, study discontinuation for undocumented progression or for toxicity or other reason, starting of new anti-cancer treatment (date of censoring was the last adequate response assessment before the starting of the new anti-cancer treatment), and death or progression after more than 1 missed assessment. Outcomes of progression included progression documented between scheduled response assessments (date of progression was the date of the first overall response assessment showing PD), death before first assessment of PD (date of progression was date of death), and death between adequate assessment visits (date of progression was date of death). No sensitivity analysis was planned for PFS.12
DOR was a secondary outcome in the FIGHT-202 trial and was assessed for cohorts A, B, and C, respectively. The number of patients who responded, who progressed or died, and who were censored were summarized. A KM plot of DOR was presented with its 95% CI; 95% CI was estimated using the Brookmeyer and Crowley method.46,12 Censoring of DOR was done in the same manner as the censoring of PFS (see details provided earlier for PFS). No sensitivity analysis was planned for DOR.12
DCR was a secondary outcome in the FIGHT-202 trial and was assessed for cohorts A, B, and C, respectively. The 95% CI for DCR was estimated using the exact method for binomial distribution.12 Patients with insufficient baseline or on-study response assessment data were considered nonresponders and were included in the denominators in the calculation of DCR.12 No sensitivity analysis for DCR was planned.12
OS was a secondary outcome in the FIGHT-202 trial and was assessed for cohorts A, B, and C, respectively. The number of patients who died and those who were censored were summarized. The KM plot of OS was presented with its 95% CI; 95% CI was estimated using the Brookmeyer and Crowley method.46,12 Reasons for censoring included lost to follow-up or still alive at the time of analysis (censoring occurred at the earlier of the date the patient was last known alive and the clinical data cut-off date for the analysis). The “last known alive” date was specified as the date of the last study visit or the date the patient was last known alive from the survival follow-up, whichever was later.12 No sensitivity analysis for OS was planned.12
All analyses performed on the HRQoL outcome (EORTC QLQ-C30) and symptom severity outcome (EORTC QLQ-BIL21) were done in the efficacy evaluable population (defined in the section that follows) for cohorts A, B, and C, respectively. Analyses were considered descriptive (i.e., non-inferential) in nature. No statistical comparisons were planned between cohorts. The standardized scores for each scale of the EORT QLQ-C30 and the EORTC QLQ-BIL21 were calculated as per the scoring guidance of the respective measure. Changes from baseline to each visit were presented for the March 22, 201916 and April 7, 202017 data cut-off dates. A raw score was considered as missing if the number of missing item values totalled 50% or more of the items that contributed to a scale.12 No MID was defined.12
In addition, the results of a post-hoc analysis were published in abstract presented at the 2021 Gastrointestinal Cancers Symposium.49 Additional information about this post-hoc analysis and its results was provided by the sponsor in its clinical summary document.12 Valle et al. (2021) performed subgroup analyses in cohort A whereby the changes from baseline to week 16 (cycle 6) for the EORTC QLQ-C30 and EORTC QLQ-BIL21 were summarized by the following subgroups: patients with a CR or PR, stable disease, and PD. Valle et al. (2021) noted that treatment-related changes in HRQoL would be expected to be apparent by week 16. Upon request, the sponsor explained that due to a rapid decline in the number of patients available to complete the questionnaires, a robust assessment of the data could only be conducted between baseline and cycle 6.17 Three key independent physicians consulted by the sponsor on how the drop-off in patients after cycle 6 might impact the interpretation of the data noted that changes in HRQoL would be apparent within 4 to 6 cycles after starting treatment.17 Post-hoc analyses were performed on the data from the March 22, 2019, data cut-off date and were considered descriptive (i.e., non-inferential) in nature. It was reported by the sponsor that nominal statistical significance was determined by a lack of overlap of the associated standard error bars around the point estimate.12 Graphs of the observed mean changes from baseline within each subgroup were generated and presented in the Clinical Summary Report.12
Subgroup Analyses
Subgroup analyses were planned a priori in the statistical analysis plan for the groups of patients listed subsequently. For each subgroup, a forest plot and the respective outcome’s 95% CI was provided. Subgroup analyses were conducted for the primary outcome, the ORR in cohort A (subgroup analyses were conducted for all patient groups listed subsequently), the PFS in cohort A (subgroup analyses were performed for all groups listed except for renal or hepatic impairment), and the DOR for cohort A (subgroup analyses were done only in patients with renal or hepatic impairment):
age category (< 65 years versus 65 to < 75 years versus ≥ 75 years)
sex (female versus male)
region (North America versus Western Europe versus rest of world)
baseline ECOG PS (0 versus 1 or 2)
metastatic disease present (yes versus no)
lines of prior therapy (1 line versus 2 lines versus ≥ 3 lines)
received previous platinum treatment (yes versus no)
renal impairment grade (normal versus mild versus moderate versus severe)
hepatic impairment grade (normal versus mild versus moderate versus severe).
Only the subgroup, baseline ECOG PS, which was identified in the CADTH review protocol, is reported in the efficacy section that follows.
Amendments12
The study protocol of the FIGHT-202 trial was amended 7 times (amendment 1: September 14, 2016, amendment 2: December 5, 2016, amendment 3: January 18, 2017, amendment 4: March 21, 2017, amendment 5: October 3, 2017, amendment 6: February 15, 2018, amendment 7: April 2, 2020).
Amendment 1 included changes to the patient eligibility criteria for completing the EORTC QLQ-BIL21 questionnaire. Only patients in the US, UK, Italy, Germany, and Korea were administered the QLQ-BIL21 instrument, given that this questionnaire was only translated and validated in the primary languages of those countries.
Amendment 2 included clarifications to the inclusion and exclusion criteria, guidance for dose reductions, the addition of updated clinical experience data, and minor administrative changes.
Amendment 3 included clarifications to requirements for HIV screening and enrolment parameters for cohort C (i.e., only patients from the US were allowed to enrol in cohort C).
Amendment 4 included revisions to permit patients to enrol based on local genomics testing results, with the final results to be determined by the central genomics laboratory. Statistical analyses of the primary and secondary outcomes and final cohort assignment were to be done based on the central genomics testing results. This amendment also included changes to the exclusion and screening criteria, the addition of updated clinical experience data, and minor administrative changes.
Amendment 5 implemented changes to increase the number of patients to be enrolled into the FIGHT-202 trial. The overall number of patients, including all 3 cohorts, was increased from 100 to 140; the number of patients to be enrolled into cohort A was increased from 60 to 100. A sample size of 100 patients was estimated to provide a greater than 95% probability (previously 80% probability with a sample size of 60) of having a 95% CI with a lower limit of greater than 15%. This change was done to ensure the most robust efficacy data to inform any future development decision. Following the European Medicines Agency’s Scientific Advice Working Party oral explanation, a sensitivity analysis was done at the time of the primary analysis showing that the proportion of the first 60 patients in cohort A (sample size as per original protocol) who achieved an objective response was consistent with the proportion of patients with an objective response among the 47 patients enrolled in cohort A after amendment 5.17,39 The European Medicines Agency assessment report noted that the increase in sample size could be considered acceptable.9 This amendment also included clarifications to study eligibility (a list of possible FGF and FGFR alterations that can be considered eligible for the study was added) and inclusion criteria, criteria for the futility analysis, and minor administrative changes.
Amendment 6 included clarifications to inclusion criteria, guidelines for dose reductions, ophthalmologic testing, and hyperphosphatemia grading. The comprehensive eye examination was revised to add fundoscopy with digital imaging.
Amendment 7 included guidelines for the management of serous retinal detachment and retinal pigmented epithelium detachment, inclusion of optical coherence tomography at the time of regular scheduled eye examinations and updates to the restricted medications (removal of CYP3A4 inducers and proton pump inhibitors and addition of organic cation transporter 2 substrates) and the prohibited medications (inclusion of moderate CYP3A4 inducers). This amendment also included other minor administrative changes.12
Table 12: Statistical Analysis of Efficacy End Points
End pointa | Statistical model | Sensitivity analyses |
|---|---|---|
OS Definition: Time from the start of the study drug (day 1) until the date of death due to any cause. | The KM method was used to estimate median OS and 95% CIs. The 95% CI for OS was calculated using the Brookmeyer and Crowley method.46 | None. |
PFS Definition: Time from the start of the study drug (day 1) to the date of progressive disease2 or death from any cause, whichever occurred first. | The KM method was used to estimate median PFS and 95% CIs. The 95% CI for PFS was calculated using the Brookmeyer and Crowley method.46 | None. |
ORR (primary end point) Definition: Best overall response is the best response recorded post baseline before and including the first PD, in the order of CR, PR, stable disease, PD, and NE.c A best overall response of CR or PR needs to be confirmed by IRC.12 | The 95% CI for ORR was calculated using the exact method for binomial distribution. | ORR analysis based on PPb population as a sensitivity analysis. |
DOR Definition: Time from the first overall response contributing to an objective responsed to the earlier of death from any cause or first overall response of PD occurring after the first overall response contributing to the objective response. | The KM method was used to estimate median DOR and 95% CIs. The 95% CI for DOR was calculated using the Brookmeyer and Crowley method.46 | None. |
DCR Definition: The proportion of patients with a best response of CR, PR, or stable disease based on RECIST 1.1 as assessed by the IRC. | The 95% CI for DCR was calculated using the exact method for binomial distribution. | None. |
EORTC QLQ-C30 | It was specified a priori in the statistical analysis plan12 that the scores for each scale would be calculated. No further analyses were specified. | None. |
EORTC QLQ-BIL21 | It was specified a priori in the statistical analysis plan12 that the scores for each scale would be calculated. No further analyses were specified. | None. |
CI = confidence interval; CR = complete response; DCR = disease control rate; EORTC = European Organisation for Research and Treatment of Cancer; HRQoL = health-related quality of life; IRC = independent review committee; KM = Kaplan–Meier; NE = not evaluable; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PP = per-protocol; PR = partial response; QLQ-BIL21 = Quality of Life Questionnaire Cholangiocarcinoma and Gallbladder Cancer Module 21; QLQ-C30 = Quality of Life Questionnaire Core 30; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
aOutcomes are presented in order of priority as identified in the CADTH review protocol.
bThe PP population comprises patients in the efficacy evaluable population who are considered to be sufficiently compliant with the protocol.
cSee Table 29 for definitions for the evaluation of the target lesions.
dPD based on RECIST 1.1 as assessed by IRC.
Source: Sponsor’s submission.12
Analysis Populations
All efficacy data were analyzed using the efficacy evaluable population, as defined in Table 13. In sensitivity analyses, ORR in cohort A was analyzed using the PP population as defined in Table 13. Analyses of safety were performed using the safety population (Table 13).
Table 13: Analysis Populations in the FIGHT-202 Trial
Analysis population | Description |
|---|---|
Efficacy evaluable population | All patients who have a known FGF/FGFR alteration confirmed by the central genomics laboratory and who received at least 1 dose of pemigatinib as well as all patients in the USa who have a negative result for FGF/ FGFR alteration from the central genomics laboratory and who received at least 1 dose of pemigatinib. |
PP population | Patients in the efficacy evaluable population who were considered to be sufficiently compliant with the protocol. The decision to exclude a patient from the PP population was done by the clinical team before database freeze. To identify potential patients for exclusion from the PP population, the following procedures were done:
|
Safety population | All enrolled patients who have received at least 1 dose of the study drug. |
FGF = fibroblast growth factor; FGFR = fibroblast growth factor receptor; PP = per-protocol.
aAs specified in protocol amendment 3 of the FIGHT-202 trial, only patients from the US were allowed to enrol in cohort C.
Source: Sponsor’s submission.12
Results
Patient Disposition
Details of the patient disposition in cohort A of the FIGHT-202 trial are summarized in Table 14. A total of 171 patients were screened and, of those, 146 patients were enrolled; 107 patients were enrolled into cohort A, 20 patients into cohort B, and 18 patients into cohort C. Patients (N = 25) who were not enrolled failed to meet the trial eligibility criteria. All patients enrolled into cohort A received pemigatinib. As of the primary (March 22, 2019) and updated (April 7, 2020) data cut-off dates, 76 (71.0%) and 98 (90.7%) patients in cohort A had discontinued treatment, respectively. The most common reason for treatment discontinuation was PD.16
As of the primary (March 22, 2019) and updated (April 7, 2020) data cut-off dates, a total of |||||||||||| and |||||||||||| patients in cohort A, respectively, had terminated the study. The main reason for study termination was death, and the percentage of patients who had died was |||||||||||| at the March 22, 2019, data cut-off date, and |||||||||||| at the April 7, 2020, data cut-off date. At the March 22, 2019, and April 7, 2020, data cut-off dates, respectively, 6 (5.6%) and |||||||||||| patients discontinued the study due to withdrawal of consent and 2 (1.9%) and |||||||||||| patients discontinued the study due to being lost to follow-up. There were 31 |||||||||| and ||||| patients (9.3%) participating in ongoing on-study treatment at the March 22, 2019, and April 7, 2020, data cut-off dates, respectively. A total of |||||||||||| and |||||||||||| patients remained in the study at the primary and updated data cut-off dates, respectively.16
Table 14: Patient Disposition, Safety Population of the FIGHT-202 Trial
Disposition | Outcome | |
|---|---|---|
Screened, n | 171 | |
Enrolled, na | 146 | |
Cohort assignment,b n | ||
Cohort A | 107 | |
Cohort B | 20 | |
Cohort C | 18 | |
Undetermined FGF or FGFR alterationc | 1 | |
Cohort A (safety population) | ||
Data cut-off date | March 22, 2019 | April 7, 2020 |
Treated, n | 107 | 108d |
With ongoing treatment, n (%) | |||||||||| | |||||||||| |
Discontinued from treatment phase, n (%) | 76 (71.0) | |||||||||| |
Reason for discontinuation from treatment phase, n (%): | ||
Progressive disease | 57 (53.3) | |||||||||| |
Withdrawal by participant | 5 (4.7) | |||||||||| |
Adverse event | 4 (3.7) | |||||||||| |
Physician decision | 4 (3.7) | |||||||||| |
Othere | 5 (4.7) | |||||||||| |
Death | 1 (0.9) | |||||||||| |
Lost to follow-up | 0 (0) | |||||||||| |
Protocol violation | 0 (0) | |||||||||| |
Patients still in study, n (%) | |||||||||| | |||||||||| |
Patients who discontinued from the study, n (%) | 48 (44.9) | |||||||||| |
Reason for discontinuation from the study n (%): | ||
Death | 38 (35.5) | |||||||||| |
Withdrew consent | 6 (5.6) | |||||||||| |
Progressive disease | 2 (1.9) | |||||||||| |
Lost to follow-up | 2 (1.9) | |||||||||| |
Efficacy evaluable populationf | 145 | |
Per-protocol populationf | 142 | |
HRQoL population | ||
EORTC QLQ-C30 population | ||
Cohort A assignment | 107 | |||||||||| |
Cohort B assignment | |||||||||| | |||||||||| |
Cohort C assignment | |||||||||| | |||||||||| |
Symptom severityg,h | ||
EORTC QLQ-BIL21 population | ||
Cohort A assignment | NRi | |||||||||| |
Cohort B assignment | NR | |||||||||| |
Cohort C assignment | NR | |||||||||| |
Safety populationj | 146 | |
ECOG = Eastern Cooperative Oncology Group; EORTC = European Organisation for Research and Treatment of Cancer; FGF = fibroblast growth factor; FGRF = fibroblast growth factor receptor; HRQoL = health-related quality of life; NR = not reported; QLQ-BIL21 = Quality of Life Questionnaire Cholangiocarcinoma and Gallbladder Cancer Module 21; QLQ-C30 = Quality of Life Questionnaire Core 30.
Note: Data cut-off dates of March 22, 2019, and April 7, 2020.
aEligibility criteria that were not met by patients were inclusion criteria (including primarily criterion 4 [documentation of FGF and FGFR gene alteration status through central laboratory], criterion 8 [ECOG Performance Status of 0 to 2], and criterion 7 [life expectancy ≥ 12 weeks]) followed by exclusion criteria (including primarily criterion 6 [abnormal laboratory parameters]). In addition, 2 patients were not enrolled into any of the cohorts because their FGFR gene alteration status could not be confirmed by the central laboratory.17
bCohort determination is based on the tumour FGF or FGFR status provided by the central genomics laboratory.
cOne participant from the safety population was assigned to a group labelled “undetermined” and excluded from the efficacy evaluable population because the local laboratory’s results for FGF or FGFR status could not be confirmed centrally due to technical issues with the tissue sample.16
dThe April 7, 2020, data cut-off date included 1 additional patient in cohort A who was enrolled after the August 30, 2019, data cut-off date.17
eOther reasons were consistent with progressive disease but were not considered “progressive disease.” They included clinical decline of patient without growth of tumour, patient was considered by the central radiology group to have progressive disease but not by the investigator, receipt of anti-cancer therapy due to brain metastasis, and patient withdrew consent after being taking off treatment due to a CT showing a suspicious lung lesion.17
fThree participants in the efficacy evaluable population were excluded from the per-protocol population due to protocol deviations16: 1 patient due to prior therapy with another FGFR inhibitor (for less than 11 days and ending 30 days before the first dose of pemigatinib), 1 patient due to trisegmentectomy (extended right hepatectomy) while on study, and 1 patient due to surgical removal of pulmonary malignant lesions while on study.16
gIn amendment 1 (September 14, 2016), language was added to denote that only patients in the US, UK, Italy, Germany, and Korea were administered the QLQ-BIL21 questionnaire because the questionnaire is translated and validated only in the primary languages of those countries.12
hHRQoL and symptom severity were assessed in the efficacy evaluable population. Patients who replied to at least 1 question of the patient-reported outcomes instruments were considered in the analyses.17
iUpon request, the number of patients who contributed to the results was provided for the April 7, 2020, data cut-off date but not for the March 22, 2019, data cut-off date.j The safety population included all 146 patients who were enrolled in the study as of the March 22, 2019, data cut-off date.16
Source: Clinical Study Reports,16 sponsor’s submission,12,17 and sponsor’s response.17
Protocol Violations
Protocol deviations in cohort A of the FIGHT-202 trial are summarized in Table 15. Missed assessments (most of which were missed laboratory assessments) were the most commonly reported protocol deviations, occurring in 95 patients (88.8%). The next most commonly reported deviations were related to non-compliance with study treatment, none of which were recorded as substantial deviations.16 Three patients in cohort A had study entry–criteria deviations: for 1 patient, the electrocardiogram displayed incomplete information at study entry (this patient achieved a PR with pemigatinib); 1 patient received treatment with an FGFR inhibitor for 11 days that ended 30 days before the first dose with pemigatinib (this patient did not achieve a response on pemigatinib); and 1 patient had missing serology results at screening (this patient did not achieve a response with pemigatinib).5 It was noted in the sponsor’s submission that none of the protocol deviations significantly affected the internal validity of the study data.16 This statement was echoed in the FDA report, which stated that none of the deviations were likely to impact the efficacy results in favour of pemigatinib.5
Table 15: Summary of Protocol Deviations in Cohort A of the FIGHT-202 Trial
Deviation category | Outcome (N = 107) |
|---|---|
Adverse event, n (%) | 2 (1.9) |
Informed consent, n (%) | 7 (6.5) |
Entry criteria, n (%) | 3 (2.8) |
Concomitant medications, n (%) | 0 (0.0) |
Non-compliance with study treatment, n (%) | 29 (27.1) |
Non-compliance with study procedure: Out-of-window assessment, n (%) | 53 (49.5) |
Non-compliance with study procedure: Missed assessment, n (%) | 95 (88.8) |
Other, n (%) | 19 (17.8) |
Note: Patients were counted once under each protocol deviation category.
Source: Clinical Study Report.16
Exposure to Study Treatments
Exposure to pemigatinib in cohort A at the March 22, 2019, and April 7, 2020, data cut-off dates is summarized in Table 16. The median duration of treatment with pemigatinib was 219 days (range, 7 to 730) and |||||||||||||||||||||||||||||||||||||| at the primary (March 22, 2019) and updated (April 7, 2020) data cut-off dates, respectively. The median overall compliance rates were high at both data cut-off dates (100%; mean = 100.37; SD = 3.296) and |||||||||||||||||||||||||||||||||||||||||||||||||||||| for the primary and updated data cut-off dates, respectively), indicating high treatment adherence in cohort A. Treatment compliance was evaluated by pill counts completed at the site.16
Table 16: Exposure to Pemigatinib (Cohort A), Safety Population
Detail | Outcome | |
|---|---|---|
Data cut-off date | March 22, 2019 | April 7, 2020 |
Treated, n | 107 | 108a |
Duration of treatment (days)b | ||
Mean (SD) | 247.4 (170.25) | ||||||||||||||||||||||||||| |
Median (range) | 219.0 (7 to 730) | ||||||||||||||||||||||||||| |
Number of treatment cycles | ||
Mean (SD) | 11.8 (7.93) | ||||||||||||||||||||||||||| |
Median | 10.0 | |||||||||||| |
Minimum, maximum | 1, 34 | |||||||||||| |
Participant exposure length, n (%) | ||
≤ 1 month | 3 (2.8) | |||||||||||| |
> 1 to 3 months | 18 (16.8) | |||||||||||| |
> 3 to 6 months | 21 (19.6) | |||||||||||| |
> 6 to 9 months | 26 (24.3) | |||||||||||| |
> 9 to 12 months | 18 (16.8) | |||||||||||| |
> 12 to 15 months | 8 (7.5) | |||||||||||| |
> 15 to 18 months | 5 (4.7) | |||||||||||| |
> 18 to 21 months | 5 (4.7) | |||||||||||| |
> 21 to 24 months | 3 (2.8) | |||||||||||| |
> 24 months | 0 (0.0) | |||||||||||| |
Overall compliance (%)c | ||
Mean (SD) | ||||||||||||||||||||||||||| | ||||||||||||||||||||||||||| |
Median | 100.00 | |||||||||||| |
Minimum, maximum | 90.0, 124.4 | ||||||||||||||||||||||||||| |
SD = standard deviation.
aThe April 7, 2020, data cut-off date included 1 additional patient in cohort A who had been enrolled after the August 30, 2019, data cut-off date.17
bTreatment duration in days is defined as date of last dose minus date of first dose plus 1.
cThe compliance rate (%) for each patient was computed as: (total actual dose [mg] divided by total prescribed dose [mg]) multiplied by 100.
Dose Modifications (Interruption, Reduction)
At the primary (March 22, 2019) and updated (April 7, 2020) data cut-off dates, respectively, 47 patients (43.9%) and |||||||||||||||||||||| of patients in cohort A (safety population) required a study treatment interruption due to a TEAE. The most common TEAE leading to dose interruption was stomatitis, occurring in 10 patients (9.3%) at the primary and updated data cut-off dates. Other commonly reported TEAEs leading to pemigatinib interruption included palmar-plantar erythrodysesthesia syndrome, arthralgia, and fatigue, occurring in 8 (7.5%), 5 (4.7%), and 4 (3.7%) patients at both data cut-off dates. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. The most commonly reported TEAEs leading to dose reductions were ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| of patients, respectively.16
Concomitant Medication
Concomitant medications were generally administered similarly across both data cut-off dates and reported for almost all patients in cohort A (safety population) (98.6% and |||||||||||||| of patients at the March 22, 2019, and April 7, 2020, data cut-off dates, respectively). As of the March 22, 2019, data cut-off date, the medications most commonly used (≥ 20%) included paracetamol (37.0%), ondansetron (|||||||), lorazepam (||||||||), sodium chloride (|||||||||), and omeprazole (21.9%). As of the April 7, 2020, data cut-off date, the medication most commonly used (≥ 20%) |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||. Concomitant phosphate binders were used by |||||||||||||| and ||||||||||| of patients at the March 22, 2019, and April 7, 2020, data cut-off dates, respectively; the most commonly used phosphate binder was sevelamer (March 22, 2019, data cut-off date: sevelamer = 8.2%, sevelamer carbonate = 4.1%, and sevelamer hydrochloride = 0.7%; April 7, 2020, data cut-off date: |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||).16
Subsequent Treatments
Information on any subsequent treatments received is available for only some patients.17 According to the sponsor, the study sites of the FIGHT-202 trial were not required (but were requested) to provide information on post-treatment therapies once pemigatinib was discontinued.17 For cohort A, information on subsequent treatments was available for ||||||||||||||, the majority of whom received FOLFIRI (|||||||||||||) as the first subsequent treatment.17 The next most commonly received subsequent treatments following pemigatinib included immunotherapies (||||||||||||||| received nivolumab and ||||||||||||||| received pembrolizumab) and targeted therapies (i.e., TAS-||||||||||||||||| and -||||||||||||||||||||||||], capecitabine [||||||||||||], and gemcitabine plus cisplatin [||||||||||||]). A range of chemotherapies was received by only | ||||||||||||||||||||||||||||||, including fluorouracil plus gemcitabine, irinotecan plus leucovorin, epirubicin plus cisplatin, and gemcitabine plus oxaliplatin).17
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported subsequently. This CADTH review focuses on cohort A, as cohorts B and C were not part of the requested reimbursement criteria submitted to CADTH and not approved in the Health Canada Notification of Compliance with conditions; therefore, cohorts B and C are beyond the scope of this review. Selected results for cohorts B and C have been included in Appendix 3.
Overall Survival
The OS results of the FIGHT-202 trial for cohort A at the March 22, 2019, and April 7, 2020, data cut-off dates are summarized in Table 17. With an additional 11 months of follow-up, OS at the April 7, 2020, data cut-off date was generally consistent with the OS results seen at the March 22, 2019, data cut-off date. As of the primary analysis (March 22, 2019), with a median follow-up time of 15.44 months, |||||||||||||||||||||| death events occurred in cohort A. There were 67 (62.6%) patients censored. Median OS was 21.06 months (95% CI, 14.82 to not evaluable). The KM curve is depicted in Figure 3 (panel A). The survival probabilities of patients surviving to 6 and 12 months were 88.6% (95% CI, 80.8 to 93.4) and 67.5% (95% CI, 56.4 to 76.3), respectively.16
As of the updated analysis (April 7, 2020), with a median follow-up time of 27.9 months,12 |||||||||||||||||||| death events occurred in cohort A. Forty-five patients (41.7%) were censored. Median OS was 17.48 months (95% CI, 14.42 to 22.93). The KM curve is depicted in Figure 3 (panel B). The survival probabilities of patients surviving to 6 and 12 months were ||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||) and 67.3% (95% CI, 57.4 to 75.4), respectively.16
Table 17: Summary of Primary and Secondary End Points for FIGHT-202
Variable | Pemigatinib, cohort A outcome | |
|---|---|---|
N = 107 | N = 108a | |
Efficacy outcomes, efficacy evaluable population | ||
Data cut-off date | March 22, 2019 | April 7, 2020 |
Median follow-up time,b months (range) | 15.44 (7.0 to 24.7) | 27.9 (|||||||||||||||||||||) |
Secondary outcome: OS | ||
Median OS, months (95% CI)c | 21.06 (14.82 to NE) | 17.48 (14.42 to 22.93) |
Events (death), n (%) | |||||||||||||||||||| | |||||||||||||||||||| |
Censored, n (%) | 67 (62.6) | |||||||||||||||||| |
KM estimates of OS at | ||
3 months (95% CI) | |||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||| |
6 months (95% CI) | 88.6 (80.8 to 93.4) | |||||||||||||||||||||||||||||||| |
9 months (95% CI) | |||||||||||||||||||||||||||||||| | 76.1 (66.7 to 83.2) |
12 months (95% CI) | 67.5 (56.4 to 76.3) | 67.3 (57.4 to 75.4) |
Secondary outcome: PFS (IRC assessment) | ||
Median PFS, months (95% CI)c | 6.93 (6.18 to 9.59) | 7.03 (6.08 to 10.48) |
Events (disease progression or death), n (%) | 71 (66.4) | |||||||||||||||||||| |
Disease progression, (%) | 63 (58.9) | |||||||||||||||||||| |
Death, n (%) | 8 (7.5) | |||||||||||||||||||| |
Censored, n (%) | 36 (33.6) | |||||||||||||||||||| |
KM estimates of PFS at: | ||
3 months (95% CI) | 78.9 (69.7 to 85.5) | |||||||||||||||||||||||||||||||| |
6 months (95% CI) | 61.7 (51.5 to 70.4) | |||||||||||||||||||||||||||||||| |
9 months (95% CI) | 45.3 (34.9 to 55.1) | |||||||||||||||||||||||||||||||| |
12 months (95% CI) | 29.2 (18.9 to 40.2) | |||||||||||||||||||||||||||||||| |
Primary outcome: ORR (IRC assessment) | ||
Objective response,d n (%) | 38 (35.5) | 40 (37.0) |
95% CIe | 26.50 to 45.35 | 27.94 to 46.86 |
Best overall response, n (%): | ||
Confirmed complete response | 3 (2.8) | 4 (3.7) |
Confirmed partial response | 35 (32.7) | 36 (33.3) |
Stable disease | 50 (46.7) | 49 (45.4) |
Progressive disease | 16 (15.0) | 16 (14.8) |
Not evaluablef | 3 (2.8) | 3 (2.8) |
Secondary outcome: DOR (IRC assessment)g | ||
Participants with confirmed objective responses, n (%) | 38 (35.5) | 40 (37.0) |
Participants with events, n (%) | 21 (55.3) | |||||||||||||||||||| |
Disease progression | 20 (52.6) | |||||||||||||||||||| |
Death | 1 (2.6) | |||||||||||||||||||| |
Participants censored, n (%) | 17 (44.7) | |||||||||||||||||||| |
Median DOR, months (95% CI)c | 7.49 (5.65 to 14.49) | 8.08 (5.65 to 13.14) |
KM estimates of DOR: | ||
3 months (95% CI) | 100.0 (100.0 to 100.0) | |||||||||||||||||||||||||||||||| |
6 months (95% CI) | 68.5 (49.0 to 81.8) | |||||||||||||||||||||||||||||||| |
9 months (95% CI) | 47.4 (27.6 to 64.9) | |||||||||||||||||||||||||||||||| |
12 months (95% CI) | 37.4 (18.6 to 56.2) | |||||||||||||||||||||||||||||||| |
Secondary outcome: DCR (IRC assessment) | ||
Disease control,h n (%) | 88 (82.2) | Not availablei |
95% CIe | 73.7 to 89.0 | Not availablei |
Best response, n (%): | Not availablei | |
Confirmed complete response | 3 (2.8) | Not availablei |
Confirmed partial response | 35 (32.7) | Not availablei |
Stable disease ≥ 39 days | 50 (46.7) | Not availablei |
CI = confidence interval; DCR = disease control rate; DOR = duration of response; IRC = independent review committee; ITT = intention to treat; KM = Kaplan–Meier; NA = not applicable; NE = not evaluable; NR = not reported; ORR = objective response rate; OS = overall survival; PFS = progression-free survival.
Note: Data cut-off dates are March 22, 2019, and April 7, 2020. Outcomes are presented in order of priority as identified in the CADTH review protocol.
aThe April 7, 2020, data cut-off date included 1 additional patient in cohort A who had been enrolled after the August 30, 2019, data cut-off date.17
bFollow-up time for all patients in the efficacy evaluable population in cohort A.
cThe 95% CI was calculated using the Brookmeyer and Crowley method (1982).
dParticipants who had a best overall response of complete response or partial response.
eThe CI was calculated based on the exact method for binomial distribution.
fA post-baseline tumour assessment was either not performed due to study discontinuation (2 participants) or was performed before the minimum interval of 39 days for an assessment of stable disease (1 participant).
gComplete and partial responses were confirmed.
hParticipants who have a best overall response of complete response, partial response or stable disease with measurements that meet the stable disease criteria after the date of first dose at a minimum interval of 39 days.
IThe DCR outcome was not generated for the April 7, 2020, data cut-off date since the primary focus of the analyses at that date was on the integrated safety summary for a new regulatory submission.17
Figure 3: Kaplan–Meier Estimates of Overall Survival — Cohort A, Efficacy Evaluable Population
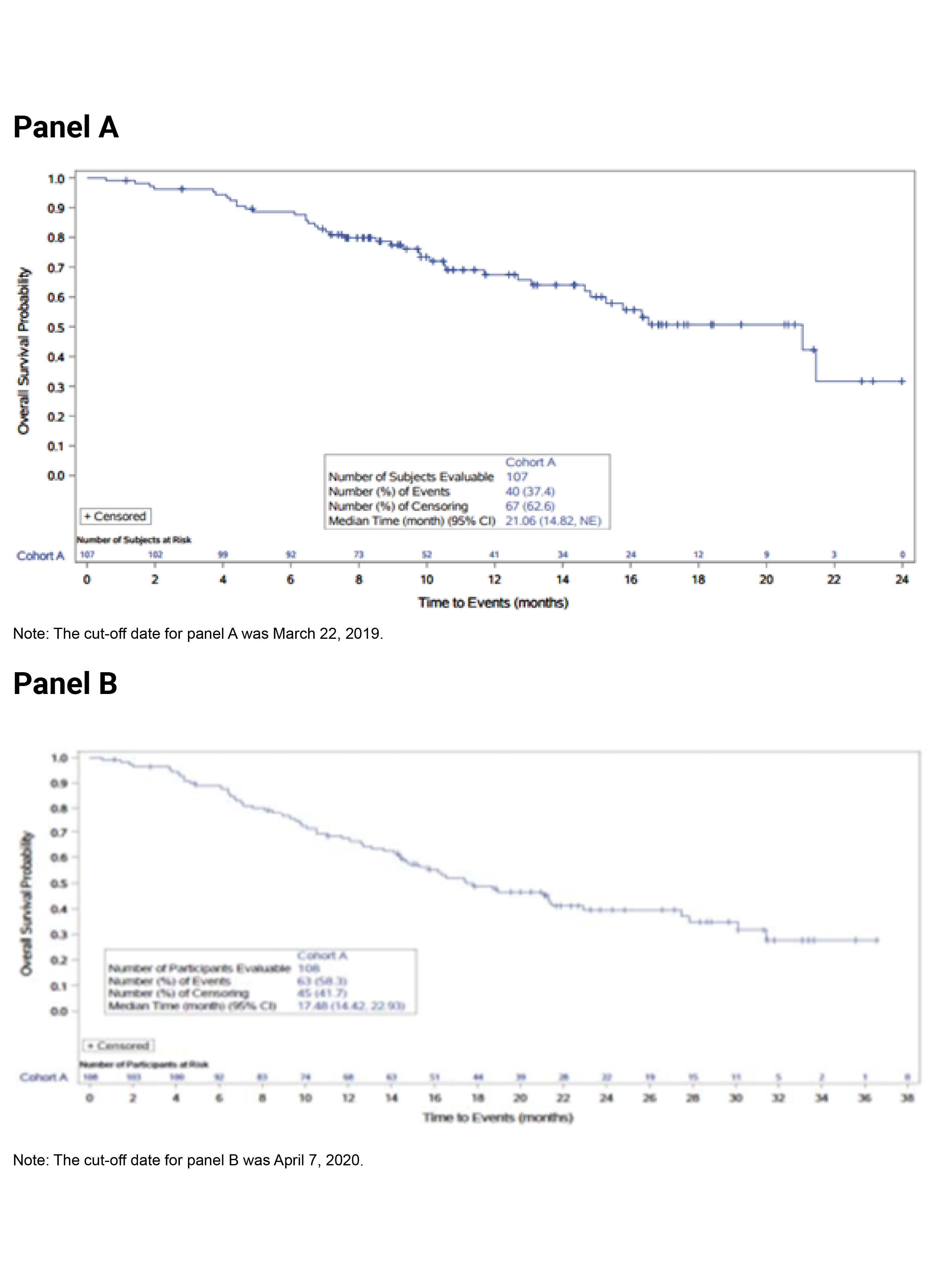
CI = confidence interval; NE = not evaluable.
Progression-Free Survival
The PFS results (based on IRC assessment) of the FIGHT-202 trial for cohort A at the March 22, 2019, and April 7, 2020, data cut-off dates are summarized in Table 17. With an additional 11 months of follow-up, PFS at the April 7, 2020, data cut-off date was generally consistent with the PFS results seen at the March 22, 2019, data cut-off date. As of the analysis of the data from the March 22, 2019, data cut-off date, with a median follow-up time of 15.44 months, 63 patients (58.9%) had experienced disease progression, 8 patients (7.5%) had died, and 36 patients (33.6%) were censored. Median PFS was 6.93 months (95% CI, 6.18 to 9.59). The KM curve is depicted in Figure 4 (panel A). The PFS probabilities at 6 and 12 months were 61.7 months (95% CI, 51.5 to 70.4) and 29.2 months (95% CI, 18.9 to 40.2), respectively. Among the censored patients (33.6%), ||||| patients had an ongoing response or stable disease (3 patients had a CR with a PFS duration of 6.24 to 22.57 months, 12 patients had a PR with a PFS duration of 6.87 to 19.32 months, and 13 patients had stable disease with a PFS duration of 2.73 to 19.32 months). The PFS results based on investigator assessment showed results consistent with those based on IRC assessment; median PFS was 7.03 months (95% CI, 6.87 to 9.63).16
As of the updated analysis (April 7, 2020), with a median follow-up time of 27.9 months, |||||||||||||||||| patients had experienced disease progression, 9 patients (8.3%) had died, and 27 patients (25.0%) were censored. Median PFS was 7.03 months (95% CI, 6.08 to 10.48). The KM curve is depicted in Figure 4 (panel B). The PFS probabilities at 6 and 12 months were |||||||||||||||||||||||||||||||||||||||| and ||||||||||||||||||||||||||||||||||||||||||, respectively. Among the censored patients, |||||||||||||||||||| patients had ongoing response or stable disease (||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||). Results for PFS based on investigator assessment were not reported for the April 7, 2020, data cut-off date in the sponsor’s submission.16
Figure 4: Kaplan–Meier Estimates of Progression-Free Survival — Cohort A, Efficacy Evaluable Population
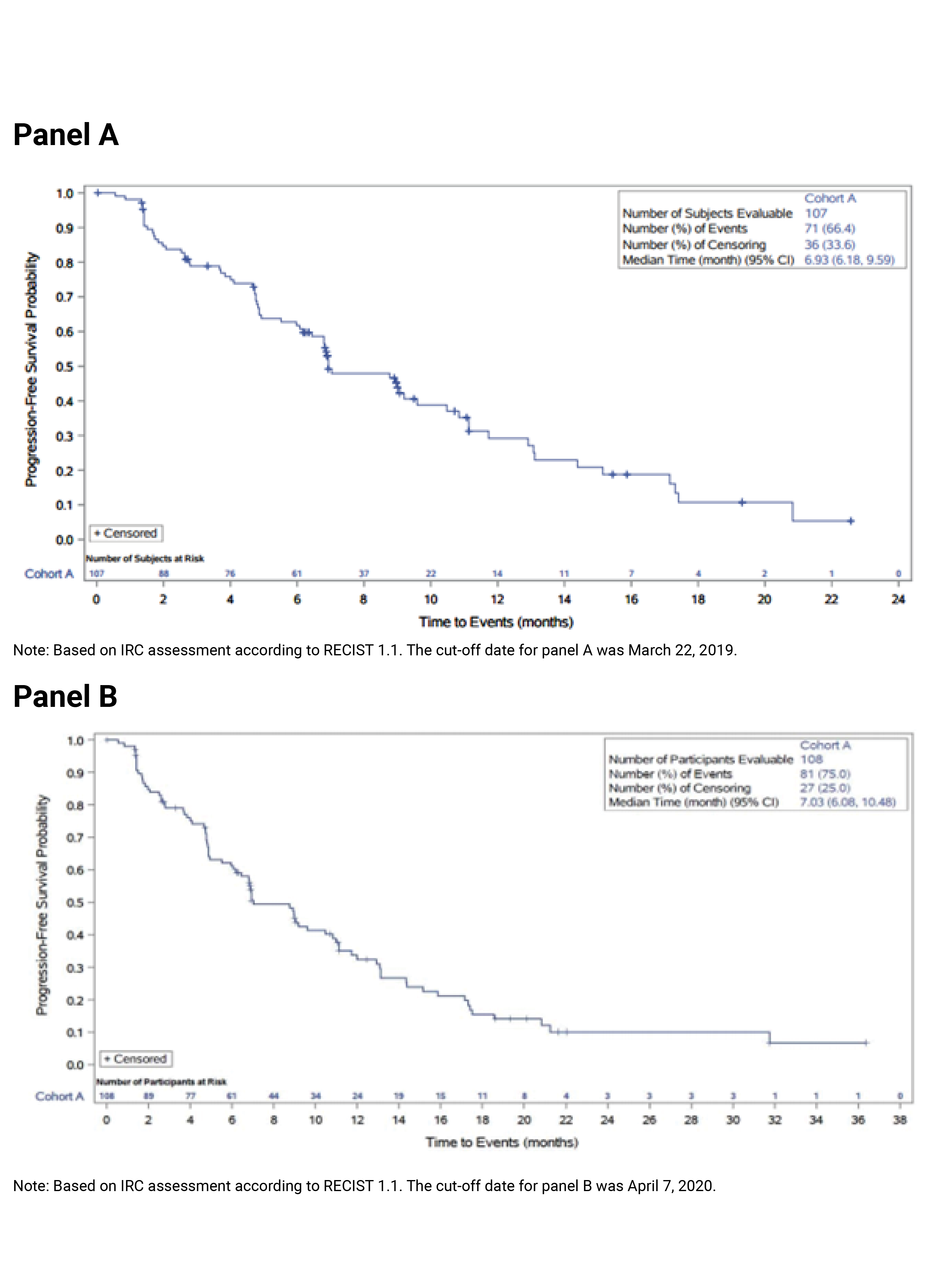
CI = confidence interval.
Source: Clinical Study Reports.16
The PFS results by subgroup of interest, as specified a priori in the protocol for this CADTH review, are summarized in Table 18. The treatment effect on PFS was consistent with the PFS analysis for all patients in cohort A across patients with ECOG PS of 0 and patients with ECOG PS of 1 or 2. Of note, the sample sizes of these subgroups were small (45 patients with ECOG PS of 0 and 62 patients with ECOG PS of 1 or 2) and relatively wide CIs in the subgroups reflected uncertainty in the effect estimates.
Table 18: PFS by ECOG PS, Cohort A, Efficacy Evaluable Population
ECOG PS subgroup | Pemigatinib cohort A outcome (N = 107) | Median PFS, months (95% CI) | |
|---|---|---|---|
Number of patients | Number of patients with PFS events | ||
ECOG PS, n | |||
0 | 45 | 25 | 9.59 (6.93 to 13.08) |
1 or 2 | 62 | 46 | 6.18 (4.73 to 9.00) |
CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group Performance Status; PFS = progression-free survival.
Note: Data cut-off date of March 22, 2019.
Source: Clinical Study Report.16
Objective Response Rate
The ORR results (based on IRC assessment) for the FIGHT-202 trial for cohort A at the March 22, 2019, and April 7, 2020, data cut-off dates are summarized in Table 17. The ORR results at the April 7, 2020, data cut-off date were generally consistent with the ORR results seen at the March 22, 2019, data cut-off date.
As of the analysis at the March 22, 2019, data cut-off date, the proportion of patients who achieved an objective response was 35.5% (N = 38) (95% CI, 26.50 to 45.35), including 3 patients (2.8%) with a CR and 35 patients (32.7%) with a PR. It was reported in the sponsor’s submission that since the lower limit of the 95% CI for ORR exceeded 15% (the predetermined threshold), the trial results would be considered positive. The ORR sensitivity analysis using the PP population showed results consistent with the ORR results for the efficacy evaluable population. The ORR results based on investigator assessment showed results that were generally consistent with those based on IRC; the proportion of patients with an objective response was 32.7% (N = 35) (95% CI, 23.95 to 42.45), including 4 patients (3.7%) with CRs and 31 patients (29.0%) with PRs.16
As of the updated analysis (April 7, 2020), the proportion of patients who achieved an objective response was 37.0% (N = 40) (95% CI, 27.94 to 46.86), including 4 patients (3.7%) with a CR and 36 patients (33.3%) with PRs. ||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||Results for ORR based on investigator assessment for the PP population were not reported for the April 7, 2020, data cut-off date in the sponsor’s submission.16
The ORR results by subgroup of interest, as specified a priori in the protocol for this CADTH review, are summarized in Table 19. The ORR results for the subgroup of interest suggest that the treatment effects on ORR for the subgroups of patients with an ECOG PS of 0 and 1 plus 2 were generally consistent with the overall population in cohort A. Of note, the sample sizes of these subgroups were small (45 patients with an ECOG PS of 0 and 62 patients with an ECOG PS of 1 or 2) and the relatively wide CIs in the subgroups reflected uncertainty in the effect estimates.
Table 19: Cohort A ORR by ECOG PS, Efficacy Evaluable Population
Subgroup | Pemigatinib cohort A (N = 107) | ORR, % (95% CI) |
|---|---|---|
ECOG PS, n | ||
0 | 45 | 48.9 (33.70 to 64.23) |
1 or 2 | 62 | 25.8 (15.53 to 38.50) |
CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ORR = overall response rate.
Note: Response assessed by independent reviewer and response was confirmed.
Note: Data cut-off date of March 22, 2019.
Source: Clinical Study Report.16
Duration of Response
The DOR results (based on IRC assessment) for the FIGHT-202 trial for cohort A at the March 22, 2019, and April 7, 2020, data cut-off dates are summarized in Table 17. In addition, results were presented for DOR at the August 12, 2019, data cut-off date. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The results for each data cut-off date are described in text that follows.
At the March 22, 2019, data cut-off date, among the 38 patients who achieved an objective response, median DOR was 7.49 months (95% CI, 5.65 to 14.49). The KM curve is depicted in Figure 5 (panel A). Of the 38 patients with an objective response, 35 patients (92%) had at least 6 months of follow-up from the time of initial response; the other 3 patients with an objective response had 5.2, 5.7, and 5.85 months of follow-up from the time of initial response. Median time-to-first response was |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||.39 There were 17 patients (44.7%) censored, of which 12 patients had an ongoing PR with a DOR ranging from 4.17 to 14.55 months, and 3 patients had an ongoing CR with a DOR of 4.83, 6.34, and 19.52 months, respectively. The probabilities of maintaining a response for at least 6 and 12 months were |||||||||||||||||||||||||||||||||||||||| and 37.4 (95% CI, 18.6 to 56.2), respectively. The DOR results that were based on investigator assessment were generally consistent with those based on IRC results; median DOR was ||||||||||||||||||||||||||||||||||||||||||||||||).16
As of the updated analysis at the August 12, 2019, data cut-off date, median DOR was ||||||||||||||||||||||||||||||||||||||||| months among the 38 respondents.16 All 38 patients with an objective response had at least ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. There were ||||||||||||||| patients censored. Among the 38 patients with an objective response, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||, respectively.
As of the updated analysis (April 7, 2020), among the 40 patients who achieved an objective response, median DOR was 8.08 months (95% CI, 5.65 to 13.14). The KM curve is depicted in Figure 5 (panel B). Among the 40 patients with an objective response, 23 patients (57.5%) had a DOR of at least 6 months, 15 patients (37.5%) had a DOR of at least 9 months, and 10 patients (25.0%) had a DOR of at least 12 months. There were ||||||||||||||| patients censored. The probabilities of maintaining a response for at least 6 and 12 months were ||||||||||||||||||||||||||||||||| and |||||||||||||||||||||||||||||||||||||||||||||, respectively. Results for DOR, based on investigator assessment, were not reported for the April 7, 2020, data cut-off date in the sponsor’s submission.16
Figure 5: Kaplan–Meier Estimate of Duration of Response Based on IRC Assessment, Cohort A, Efficacy Evaluable Population
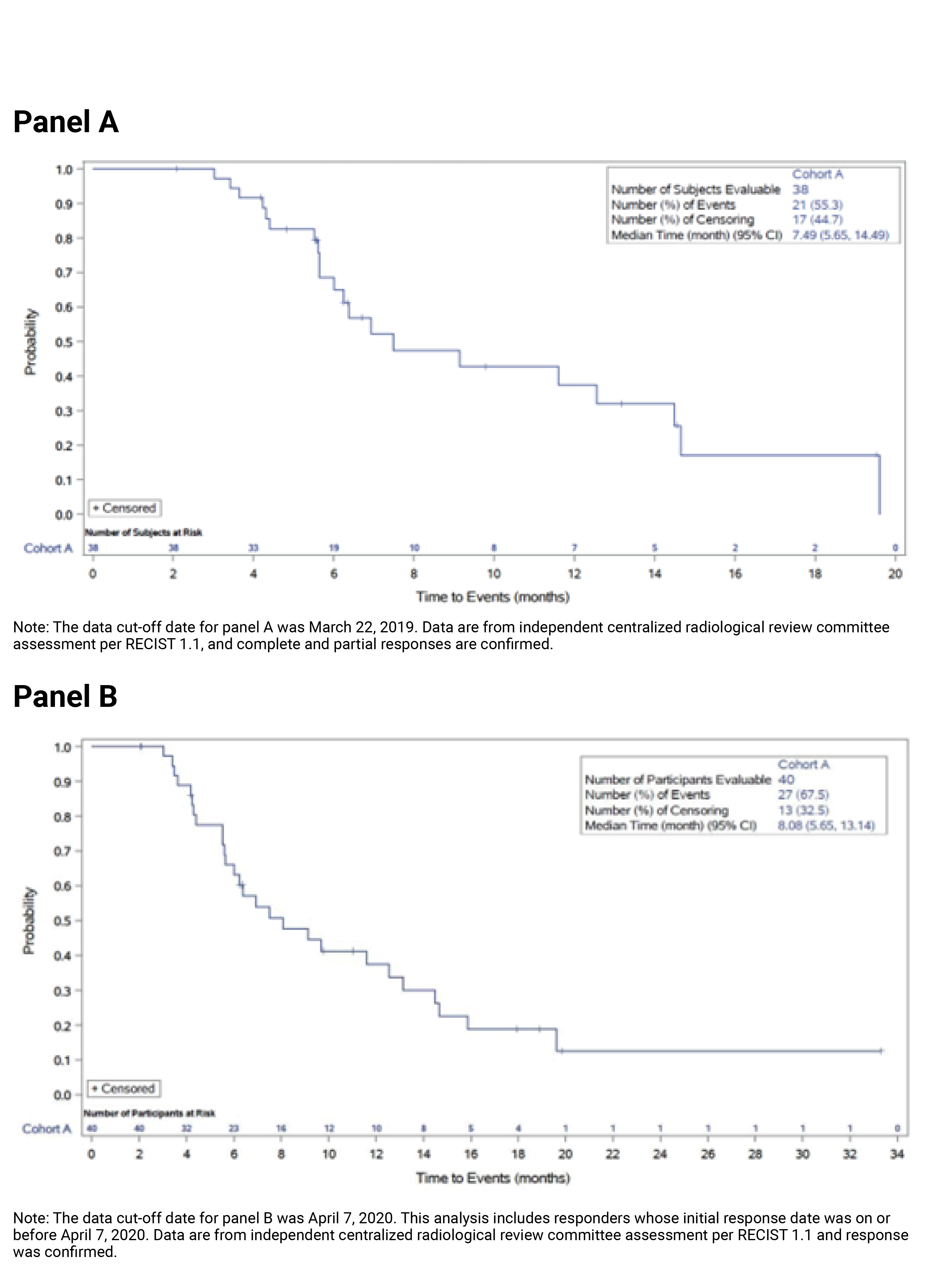
CI = confidence interval; IRC = independent review committee RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Source: Clinical Study Reports.16
Disease Control Rate
The DCR results (based on IRC assessment) for the FIGHT-202 trial for cohort A at the March 22, 2019, data cut-off date are summarized in Table 17. As of that date, the proportion of patients with a best response of CR, PR, or stable disease was 82.2% (n = 88) (95% CI, 73.7 to 89.0), including 3 patients (2.8%) with a CR, 35 patients (32.7%) with a PR, and 50 patients (46.7%) with stable disease for 39 or more days since the first pemigatinib dose. The DCR results based on investigator assessment (DCR = 86.9%; 95% CI, 79.0 to 92.7) were generally consistent with those based on IRC assessment.
The DCR outcome was not generated for the April 7, 2020, data cut-off date since the primary focus of the analyses at that date was on an integrated safety summary for a new regulatory submission.17
Patient-Reported Outcomes
Health-Related Quality of Life
EORTC Quality of Life Questionnaire Core 30
Completion rates for the EORT QLQ-C30 instrument declined over time. After week 16 (cycle 6) there were approximately ||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.17 The descriptive summary statistics of observed mean scores and mean changes from baseline at each assessment point for the EORT QLQ-C30 questionnaire (global health status [QoL] scale) at the April 2, 2020, data cut-off date are summarized in Figure 6 and Figure 7, respectively. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Overall observed scores from baseline to cycle 33 (March 22, 2019, data cut-off date) or to cycle 42 (April 7, 2020, data cut-off date) |||||||||||||||||||||||||||||||||||||||||||||||||||||||.16

Note: This figure has been redacted at the sponsor’s request.
||||||||||||||||||||||||||||||||||||.17

Note: This figure has been redacted at the sponsor’s request.
|||||||||||||||||||||||||||||||||||||||||||||.17
Post-Hoc Analysis on HRQoL
Descriptive statistics of observed mean changes from baseline to week 16 (cycle 6) by subgroups of patients (i.e., patients with a CR or PR, stable disease, or PD) are summarized in Table 20 (baseline to cycle 6) and Figure 8 (baseline to cycle 39). The analysis population included 100 evaluable patients of the total 107 patients in cohort A. A definition of the evaluable population was not provided.49 The 3 subgroups included 36, 48, and 15 patients with a CR or PR, stable disease, and PD, respectively. The results suggested that the change in overall mean score for the overall health status scale appeared to be maintained in patients with a CR or PR (−0.3; SD = 1.3) and stable disease (−0.3; SD = 1.1), and declined in patients with PD (−1.2; SD = 0.8) (Table 20 and Figure 8).49 While similar results were observed for the emotional function scale, all patients appeared to show decline for the role and social functioning scales.49 The sponsor reported that the difference in mean change from baseline between patients with PD and those with either a CR or PR, or stable disease were driven by a reported increase in feelings of worry and tension in patients with PD at cycle 6.12 The sponsor additionally reported that differences in mean change from baseline were seen for the constipation scale between patients with PD and those with either a CR or PR, or stable disease; however, results were not reported and no definition of what constitutes a meaningful change was provided.12 Overall, Valle et al. (2021) concluded that changes in HRQoL appeared directionally more favourable in patients with a CR or PR, or stable disease than in patients with PD.49
Table 20: Mean Changes From Baseline to Week 16 by Best Overall Response for the EORTC QLQ-C30 and QLQ-BIL21, Cohort A, Evaluable Population
Mean (SD) score change | CR/PR | Stable disease | PD | |||
|---|---|---|---|---|---|---|
n | mean (SD) | n | mean (SD) | n | mean (SD) | |
QLQ-C30 | ||||||
Overall health status | 34 | −0.3 (1.3) | 35 | −0.3 (1.1) | 5 | −1.2 (0.8) |
Emotional functioning | 34 | 2.2 (17.0) | 36 | −0.6 (16.6) | 5 | −11.7 (9.5) |
Role functioning | 33 | −4.0 (24.0) | 36 | −12.0 (26.3) | 5 | −20.0 (29.8) |
Social functioning | 34 | −5.9 (25.6) | 36 | −11.1 (27.9) | 5 | −6.7 (14.9) |
QLQ-BIL21 | ||||||
Pain | 28 | −5.7 (20.0) | 27 | −3.5 (11.4) | 5 | 8.3 (5.9) |
Anxiety | 28 | −2.9 (13.4) | 26 | −5.4 (19.1) | 5 | 8.3 (11.8) |
Treatment side effects | 28 | 19.0 (36.8) | 24 | 13.9 (33.9) | 5 | 6.7 (49.4) |
CR = complete response; EORTC = European Organisation for Research and Treatment of Cancer; PD = progressive disease; PR = partial response; QLQ-BIL21 = Quality of Life Questionnaire Cholangiocarcinoma and Gallbladder Cancer Module 21; QLQ-C30 = Quality of Life Questionnaire Core 30; SD = standard deviation.
Note: The data cut-off date was March 22, 2019.
Source: Permission to reprint was granted by the publisher of Valle et al. (2021),49 https://ascopubs.org/doi/abs/10.1200/JCO.2021.39.3_suppl.276.

Note: This figure has been redacted at the sponsor’s request.
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.12
Symptom Severity
EORTC Quality of Life Questionnaire Cholangiocarcinomas and Gallbladder Cancer Module 21
Completion rates for the EORT QLQ-BIL21 instrument declined over time. After week 16 (cycle 6) there were approximately ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||.17 The descriptive summary statistics of observed mean scores and mean changes from baseline at each assessment point for the EORT QLQ-BIL21 questionnaire (eating and pain scales; other scales are found in Appendix 3, Figures 19 to 30) at the April 2, 2020, data cut-off date are summarized in Figure 9 (eating scale), Figure 10 (eating scale), Figure 11 (pain scale), and Figure 12 (pain scale), respectively. A definition for what constituted a clinically meaningful change from baseline in the present target population was not provided. Overall observed scores from baseline to cycle 33 (March 22, 2019, data cut-off date) or to cycle 42 (April 7, 2020, data cut-off date) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.16

Note: This figure has been redacted at the sponsor’s request.
|||||||||||||||||||||||||||||||||||||||||||.17

Note: This figure has been redacted at the sponsor’s request.
||||||||||||||||||||||||||||||||||||||||||||||||.17

Note: This figure has been redacted at the sponsor’s request.
||||||||||||||||||||||||||||||||||||||||||||||||.17

Note: This figure has been redacted at the sponsor’s request.
|||||||||||||||||||||||||||||||||||||||||||.17
Post-Hoc Analysis on HRQoL
Descriptive statistics of observed mean changes from baseline to week 16 (cycle 6) by subgroups of patients (i.e., patients with a CR or PR, stable disease, or PD) are summarized in Table 20 (baseline to cycle 6) and Figure 13 (baseline to cycle 39). The analysis population included 100 evaluable patients out of the 107 patients in cohort A; a definition of the evaluable population was not provided.49 The 3 subgroups included 36, 48, and 15 patients for patients with a CR or PR, stable disease, and PD, respectively.49 The results suggested that the overall mean score change for the pain scales appeared to decline in patients with a CR or PR (−5.7; SD = 20.0), and stable disease (−3.5; SD = 11.4) and increase in patients with PD (8.3; SD = 5.9) (Table 20 and Figure 13).49 The sponsor reported that the difference in mean change from baseline between patients with PD and those with either a CR or PR, or stable disease were driven by a reported increase in pain at night in patients with PD at cycle 6.12 Similar results were observed for the anxiety scale (Table 20), and the sponsor reported that the difference in mean change from baseline between patients with PD and those with either a CR or PR, or stable disease were driven by a reported increase in worry and decreased ability to enjoy oneself in patients with PD at cycle 6.12,49 All patients appeared to experience an increase in treatment side effects (Table 20).49 Overall, Valle et al. (2021) concluded that changes in HRQoL appeared directionally more favourable in patients with a CR or PD, or stable disease than in patients with PD.49

Note: This figure has been redacted at the sponsor’s request.
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.12
Harms
Only those harms identified in the review protocol are reported subsequently. Refer to Table 21 for detailed harms data in cohort A at the March 22, 2019, and April 7, 2020, data cut-off dates. Results are similar at both data cut-off dates with no new safety concerns identified as of the latest data cut-off date; therefore, the safety results of the April 7, 2020, analyses are described in the text that follows.
Adverse Events
All patients in cohort A experienced at least 1 TEAE (100.0%). The most commonly reported TEAEs were alopecia (|||||||||), hyperphosphatemia (|||||||||), diarrhea (|||||||||), dysgeusia (||||||||||), fatigue (||||||||||), and nausea (||||||||||).16
Grade 3 or higher TEAEs occurred in ||||||||| of patients in cohort A (Table 21). The most commonly reported grade 3 or higher TEAE was hypophosphatemia. The percentage of patients experiencing |||||||||||||||||||||||||||||||||||| was ||||||||||. Other grade 3 or higher TEAEs included ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||.16
Serious Adverse Events
The percentage of patients experiencing serious TEAEs was ||||||||||| in cohort A. The most common serious TEAEs were pyrexia and cholangitis, each occurring in |||||||| of patients; abdominal pain, occurring in |||||||| of patients; and infective cholangitis, occurring in |||||||| of patients.16
Withdrawals Due to Adverse Events
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. AEs led to discontinuation of study treatment in ||||||||||||||||||||||||||||||| in cohort A. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.16
Mortality
TEAEs leading to death occurred relatively rarely in |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.16
Notable Harms
Notable harms as specified in the protocol included nail toxicity, serous retinal detachment, hyperphosphatemia, and hypophosphatemia.16
Nail Toxicity
The percentage of patients experiencing nail toxicity TEAEs was ||||||||||| in patients in cohort A. The most commonly reported ||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. The percentage of patients experiencing nail toxicity TEAEs of grade 3 or higher was |||||||, including nail discoloration, nail disorder, onychoclasis, and paronychia. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.16
Serous Retinal Detachment
The percentage of patients experiencing serous retinal detachment TEAEs in cohort A was ||||||||. The most commonly reported serous retinal detachment was |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.16
Hyperphosphatemia
The percentage of patients experiencing hyperphosphatemia TEAEs in cohort A was |||||||||. The most commonly reported hyperphosphatemia events were ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.16
Hypophosphatemia
The percentage of patients experiencing hypophosphatemia TEAEs in cohort A was |||||||||. The most commonly reported hypophosphatemia events were ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Hypophosphatemia TEAEs of grade 3 or higher occurred in |||||||| of patients, including only hypophosphatemia events. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||.16
Table 21: Summary of Harms, Safety Population
Harm | Pemigatinib cohort A outcome | |
|---|---|---|
N = 107 | N = 108 | |
Data cut-off date | March 22, 2019 | April 7, 2020a |
Patients with at least 1 TEAE | ||
n (%) | 107 (100.0) | |||||||||||||||||||||| |
Most common events,b n (%) | ||
Alopecia | |||| (58.9) | ||||||||||||| |
Hyperphosphatemia | |||| (55.1) | ||||||||||||| |
Diarrhea | |||| (52.3) | ||||||||||||| |
Dysgeusia | |||| (47.7) | ||||||||||||| |
Fatigue | |||| (44.9) | ||||||||||||| |
Nausea | |||| (40.2) | ||||||||||||| |
Constipation | ||||||||||||| | ||||||||||||| |
Stomatitis | ||||||||||||| | ||||||||||||| |
Dry mouth | ||||||||||||| | ||||||||||||| |
Dry eye | ||||||||||||| | ||||||||||||| |
Vomiting | ||||||||||||| | ||||||||||||| |
Decreased appetite | ||||||||||||| | ||||||||||||| |
Arthralgia | ||||||||||||| | ||||||||||||| |
Dry skin | ||||||||||||| | ||||||||||||| |
Hypophosphatemia | ||||||||||||| | ||||||||||||| |
Back pain | ||||||||||||| | ||||||||||||| |
Pain in extremity | ||||||||||||| | ||||||||||||| |
Abdominal pain | ||||||||||||| | ||||||||||||| |
Palmar-plantar erythrodysesthesia syndrome | ||||||||||||| | ||||||||||||| |
Urinary tract infection | ||||||||||||| | ||||||||||||| |
Weight decreased | ||||||||||||| | ||||||||||||| |
Headache | ||||||||||||| | ||||||||||||| |
Dizziness | ||||||||||||| | ||||||||||||| |
Epistaxis | ||||||||||||| | ||||||||||||| |
Hypercalcemia | ||||||||||||| | ||||||||||||| |
Dehydration | ||||||||||||| | ||||||||||||| |
Peripheral edema | ||||||||||||| | ||||||||||||| |
Anemia | ||||||||||||| | ||||||||||||| |
Pyrexia | ||||||||||||| | ||||||||||||| |
Asthenia | ||||||||||||| | ||||||||||||| |
Myalgia | ||||||||||||| | ||||||||||||| |
Gastroesophageal reflux disease | ||||||||||||| | ||||||||||||| |
Dyspepsia | ||||||||||||| | ||||||||||||| |
Upper abdominal pain | ||||||||||||| | ||||||||||||| |
Patients with at least 1 grade 3 or higher TEAE | ||
n (%) | 64 (59.8) | ||||||||||||| |
Most common events,c n (%) | ||
Hypophosphatemia | 13 (12.1) | ||||||||||||| |
Stomatitis | 8 (7.5) | ||||||||||||| |
Arthralgia | 7 (6.5) | ||||||||||||| |
Palmar-plantar erythrodysesthesia syndrome | 6 (5.6) | ||||||||||||| |
Abdominal pain | 5 (4.7) | ||||||||||||| |
Fatigue | 4 (3.7) | ||||||||||||| |
Diarrhea | 3 (2.8) | ||||||||||||| |
Hypotension | 4 (3.7) | ||||||||||||| |
Cholangitis | 3 (2.8) | ||||||||||||| |
||||||||||||||||||||||||||||||||||||||| | ||||||||||||| | ||||||||||||| |
Hyponatremia | 3 (2.8) | ||||||||||||| |
Anemia | 3 (2.8) | ||||||||||||| |
Blood alkaline phosphatase increased | 3 (2.8) | ||||||||||||| |
Dehydration | 3 (2.8) | ||||||||||||| |
Aspartate aminotransferase increased | 3 (2.8) | ||||||||||||| |
Hypertension | 3 (2.8) | ||||||||||||| |
Urinary tract infection | 3 (2.8) | ||||||||||||| |
Hyperbilirubinemia | 3 (2.8) | ||||||||||||| |
||||||||||||||||||||||| | ||||||||||||| | ||||||||||||| |
Nausea | 3 (2.8) | ||||||||||||| |
Patients with at least 1 serious TEAE | ||
n (%) | |||| (40.2) | ||||||||||||| |
Most common events,d n (%) | ||
Pyrexia | ||| (4.7) | ||||||||||||| |
Cholangitis | ||| (3.7) | ||||||||||||| |
Abdominal pain | ||| (3.7) | ||||||||||||| |
Infective cholangitis | ||| (2.8) | ||||||||||||| |
Small intestinal obstruction | 2 (1.9) | ||||||||||||| |
Chills | ||||||||||||| | ||||||||||||| |
Fatigue | ||||||||||||| | ||||||||||||| |
Bile duct obstruction | ||||||||||||| | ||||||||||||| |
Urinary tract infection | ||||||||||||| | ||||||||||||| |
Sepsis | ||||||||||||| | ||||||||||||| |
Bacteremia | ||||||||||||| | ||||||||||||| |
Blood bilirubin increased | ||||||||||||| | ||||||||||||| |
Failure to thrive | ||||||||||||| | ||||||||||||| |
Hypercalcemia | ||||||||||||| | ||||||||||||| |
Dehydration | ||||||||||||| | ||||||||||||| |
Device occlusion | ||||||||||||| | ||||||||||||| |
Acute kidney injury | ||||||||||||| | ||||||||||||| |
Pleural effusion | ||||||||||||| | ||||||||||||| |
Patients who stopped treatment due to TEAEs | ||
n (%) | 5 (4.7) | ||||||||||||| |
Most common events, n (%) | ||
Intestinal obstruction | 1 (0.9) | ||||||||||||| |
Gastrointestinal hemorrhage | 1 (0.9) | ||||||||||||| |
Bile duct obstruction | 1 (0.9) | ||||||||||||| |
Hyperbilirubinemia | 1 (0.9) | ||||||||||||| |
Biliary tract infection | ||||||||||||| | ||||||||||||| |
Sepsis | ||||||||||||| | ||||||||||||| |
Paraplegia | 1 (0.9) | ||||||||||||| |
Acute kidney injury | 1 (0.9) | ||||||||||||| |
Deaths | ||
Due to TEAEs,e n (%) | 3 (2.8) | ||||||||||||| |
Most common events, n (%) | ||
Bile duct obstruction | ||||||||||||| | ||||||||||||| |
Failure to thrive | ||||||||||||| | ||||||||||||| |
Notable harms | ||
Nail toxicity | ||
Nail toxicity (any-grade TEAEs), n (%) | 56 (52.3) | ||||||||||||| |
Onychomadesis | 13 (12.1) | ||||||||||||| |
Nail discoloration | 12 (11.2) | ||||||||||||| |
Nail dystrophy | 10 (9.3) | ||||||||||||| |
Onycholysis | 10 (9.3) | ||||||||||||| |
Paronychia | 9 (8.4) | ||||||||||||| |
Onychoclasis | 9 (8.4) | ||||||||||||| |
Nail disorder | 5 (4.7) | ||||||||||||| |
Onychomycosis | 4 (3.7) | ||||||||||||| |
Nail infection | 1 (0.9) | ||||||||||||| |
Nail ridging | 3 (2.8) | ||||||||||||| |
Nail toxicity | 3 (2.8) | ||||||||||||| |
Nail hypertrophy | 1 (0.9) | ||||||||||||| |
Onychalgia | 1 (0.9) | ||||||||||||| |
Nail toxicity (grade 3 or higher TEAEs), n (%) | ||||||||||||| | ||||||||||||| |
Nail toxicity (serious TEAEs), n (%) | ||||||||||||| | ||||||||||||| |
Serous retinal detachment | ||
Serous retinal detachment (any-grade TEAEs), n (%) | 4 (3.7) | 5 (4.6) |
Retinal detachment | 2 (||||) | 2 (1.9) |
Chorioretinal folds | ||||||||||||| | 1 (0.9) |
Detachment of retinal pigment epithelium | ||||||||||||| | ||||||||||||| |
Maculopathy | ||||||||||||| | ||||||||||||| |
Retinal thickening | ||||||||||||| | ||||||||||||| |
Serous retinal detachment (grade 3 or higher TEAEs), n (%) | ||||||||||||| | ||||||||||||| |
Serous retinal detachment (serious TEAEs), n (%) | ||||||||||||| | ||||||||||||| |
Hyperphosphatemia | ||
Hyperphosphatemia (any-grade TEAEs), n (%) | 62 (57.9) | ||||||||||||| |
Hyperphosphatemia | 59 (55.1) | ||||||||||||| |
Blood phosphorus increased | ||||||||||||| | ||||||||||||| |
Hyperphosphatemia (grade 3 or higher TEAEs), n (%) | ||||||||||||| | ||||||||||||| |
Hyperphosphatemia (serious TEAEs), n (%) | ||||||||||||| | ||||||||||||| |
Hypophosphatemia | ||
Hypophosphatemia (any-grade TEAEs), n (%) | |||| (25.2) | ||||||||||||| |
Hypophosphatemia | |||| (24.3) | ||||||||||||| |
Blood phosphorus decreased | ||||||||||||| | ||||||||||||| |
Hypophosphatemia (grade 3 or higher TEAEs), n (%) | 13 (12.1) | ||||||||||||| |
Hypophosphatemia (serious TEAEs), n (%) | ||||||||||||| | ||||||||||||| |
TEAE = treatment-emergent adverse event.
Note: Data cut-off dates were March 22, 2019, and April 7, 2020. A patient is counted only once for multiple events within a preferred term or system organ class.
aThe April 7, 2020, data cut-off date included 1 additional patient in cohort A who had been enrolled after the August 30, 2019, data cut-off date.17
bFrequency > 10% of patients at 1 or both of the 2 data cut-off dates.
cFrequency ≥ 2% of patients at 1 or both of the 2 data cut-off dates.
dFrequency ≥ 1% of patients at 1 or both of the 2 data cut-off dates.
eNone were considered treatment-related.
Critical Appraisal
Internal Validity
Primary objective of phase II design: The primary objective of phase II (randomized or non-randomized) trials is to document the safety outcomes and investigate whether the estimate of the effect for a new drug is large enough to use it in confirmatory phase III trials. Phase II trials may not accurately predict harm and/or effectiveness of treatments. There are numerous examples of phase III trials whose results did not support the phase II trial results.50 It is uncertain whether the results observed in this phase II trial would translate into positive phase III trials or into real-world clinical practice. There are currently no randomized phase III trials under way for this review’s target population. The clinical experts consulted by CADTH noted that, despite the high unmet need, conducting an RCT in this setting with a targeted therapy, such as pemigatinib, compared with the current therapies available in the second line in Canadian clinical practice, would likely not be feasible. According to the clinical experts, developing phase III RCTs is hindered by the overall low number of patients who meet the current indication and equipoise between pemigatinib and other chemotherapy drugs does not exist.
Limited interpretation of time-to-event end points: Interpretation of time‐to‐event end points such as OS or PFS is limited in single‐arm studies. While ORR may be directly attributable to the drug effect, the non-randomized design makes interpreting PFS and OS events attributable to pemigatinib challenging, since all patients in cohort A received the same treatment. The extent to which the observed survival is due to the natural history of the tumour or the intervention remains unclear.48 The FDA’s multi-discipline review for the assessment of pemigatinib in the present indication included the following reviewer comment: “In a single arm trial, FDA considers time-to-event end points to be uninterpretable, and the results will not be described in this review.”5 Consequently, the efficacy outcomes contributing to the FDA’s accelerated approval of pemigatinib in the current setting included response outcomes and no survival end points.5
Prognostic value of FGFR2: While there is strong genetic and functional evidence that FGFR genetic alterations can drive the formation of tumours,7 it is currently not known whether patients with an FGFR2 alteration represent a distinct prognostic subgroup.11 Retrospective studies19,21-23 in patients with CCA suggest that patients with FGFR alterations appear to have better prognoses compared with an unselected CCA population.9,19 Given the single‐arm design of the FIGHT-202 trial, it could be challenging to disentangle to what extent the observed results are due to a potential prognostic effect of FGFR2 fusions or rearrangements, or due to drug-associated effects. The clinical experts consulted on this issue by CADTH agreed that, although there is exploratory, preliminary data that FGFR2 genetic alterations may be associated with a more indolent disease progression,19 there is currently insufficient evidence to determine whether patients with FGFR2 fusion–positive CCA represent a distinct prognostic subgroup. The clinical experts were of the opinion that FGFR2 mutation status was likely not an important prognostic factor in the indicated patient population. The clinical experts considered that the median time from initial diagnosis to enrolment into cohort A of the FIGHT-202 trial was approximately 1 year, which is reflective of median OS with standard first-line therapy in this setting. The duration of first-line therapy for patients in cohort A of the FIGHT-202 trial was also reflective of clinical practice, according to the clinical experts (see Table 34 in Appendix 3). The clinical experts further agreed that progression on prior systematic therapy is a key prognostic factor in these patients, and they did not anticipate that patients would derive any substantial benefit from their underlying disease biology at the time they enrolled into the FIGHT-202 trial.
Open-label design: The FIGHT-202 trial had an open-label design whereby the investigator and the study participants are aware of their treatment status, which increases the risk of detection bias and performance bias. This has the potential to bias results and outcomes in favour of pemigatinib if the assessor (investigator or patient) believes the study drug is likely to provide a benefit. However, to mitigate the impact of this bias, the investigators used an IRC to evaluate responses using standardized criteria (i.e., based on RECIST 1.1 and assessed by an independent radiological review committee using central genomics laboratory results,12 with confirmation of a CR and PR at least 4 weeks after the initial assessment).12 Results for the investigators’ and IRC’s response assessments were generally consistent, suggesting that the potential of confounding effects on response outcomes due to the open-label design is likely not substantial. Furthermore, subjective outcomes (i.e., adverse outcomes and patient-reported outcomes) may be biased due to the open-label design. For example, if study personnel and patients knew that the treatment was pemigatinib (which is known to cause nail toxicity, hyperphosphatemia, and other AEs), this could have influenced the reporting of harms. Overall, the magnitude and direction of this bias remains unclear.
Statistical analyses: No formal statistical significance and hypotheses testing were performed and, thus, no P values were reported. Point estimates with 95% CIs were reported to estimate the magnitude of treatment effect. A greater than 95% probability of having a 95% CI for ORR in cohort A with a lower limit larger than 15% was the basis for the sample size determination and was regarded as the threshold for a positive study outcome. Results for ORR appeared consistent with the sample size assumptions, and the study recruited the intended number of patients.
Subgroup analysis: Methodological issues limited the ability to interpret the results from the subgroup analyses. The subgroup analyses were non-inferential, wide CIs reflected uncertainty in the effect estimates, and small sample sizes limited the generalizability to a broader population.
Small sample size: A limited number of patients were included in the efficacy evaluable dataset (n = 107) of cohort A. The magnitude of the treatment effect estimates observed in a small study sample may not be replicable in a larger study sample or generalizable to the target population in real-world clinical practice.
HRQoL and symptom severity assessments: The interpretation of results for the EORTC QLQ-C30 and EORTC QLQ-BIL21 questionnaire (i.e., the ability to assess trends over time) at later cycles is limited by the ||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. In addition, selection bias over time should be considered when interpreting results, as the long-term survivors tend to be the healthier patients. Furthermore, given the lack of pre-specified statistical analyses a priori in the statistical analysis plan for patient-reported outcomes, results from post-hoc analyses (i.e., changes from baseline to week 16 summarized by disease response subgroups) are considered exploratory in nature. As well, patient-reported outcomes in the post-hoc analysis were measured up to week 16, which may not represent an accurate picture of patients’ experiences with pemigatinib for a prolonged period of time. Given that the trial was non-randomized, the impact of pemigatinib on patient-reported outcomes in relation to other therapies is unknown.
The reliability of the EORTC QLQ-C30 instrument was evaluated in an international study in patients with BTC41 and showed that internal consistency was acceptable for most scales and that results for test–retest reliability were mixed.41 Estimates for MIDs in the literature were not found for the EORTC QLQ-BIL30 in patients with CCA or BTC.
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Therefore, it is unclear if the changes from baseline experienced by patients in the FIGHT-202 trial are reflective of a clinically meaningful change in patients with unresectable, locally advanced, or metastatic CCA with FGFR2 alterations.
The EORTC QLQ-BIL21 instrument was validated in an international study in patients with BTC that showed acceptable internal consistency for all multi-item scales and good test–retest reliability. Known group validity also showed an ability to distinguish between subgroups and there was some evidence of responsiveness. Estimates for MIDs in the literature were not found for the EORTC QLQ-BIL30 in patients with CCA or BTC| |||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Therefore, it is unclear if the changes from baseline experienced by patients in the FIGHT-202 trial are reflective of a clinically meaningful change in patients with unresectable, locally advanced, or metastatic CCA with FGFR2 alterations.
Overall, these methodological issues render results from the EORT QLQ-C30 and EORTC QLQ-BIL21 instruments inconclusive.
External Validity
Overall, the clinical experts consulted by CADTH agreed that the baseline patient characteristics of cohort A of the FIGHT-202 trial were reflective of patients they see in Canadian clinical practice for the present indication. Although the majority of patients in cohort A were enrolled in trial sites in the US and Europe, according to the clinical experts consulted by CADTH, the population enrolled in the trial was consistent with the population expected to be treated in Canadian clinical practice; furthermore, no different treatment effect would be expected based on different disease management practices across countries. According to the clinical expects’ opinion, as long as patients have the FGFR2 alteration, pemigatinib would be appropriate to administer after any of the prior therapies received by patients in the trial. However, the clinical experts agreed that patients should not have previously been treated with an FGFR2-targeted therapy. The majority of patients (61%) in cohort A of the FIGHT-202 trial had received 1 prior line of systemic therapy before trial enrolment, and 27% and 12% of patients had received 2, or 3 lines or more of prior systemic therapy, respectively. The clinical experts anticipated seeing the benefit of treatment with pemigatinib regardless of the number of previous lines of systemic therapy, as long as patients have the FGFR2 alteration. Furthermore, FGFR2 alterations occur rarely in eCCA and there was 1 patient in cohort A of the FIGHT-202 trial with FGFR2-positive eCCA, whereas all other patients in cohort A had iCCA. The clinical experts noted that patients with iCCA and eCCA are managed in a similar way in clinical practice and that the results observed in cohort A are generalizable to patients with FGFR2-positive eCCA based on the fact that FGFR2 is the target of the mechanism of action of pemigatinib and there is no biologic rationale to assume that pemigatinib’s safety profile would be different in patients with eCCA.
Concomitant medications received by patients in the trial appeared reflective of the medications patients would receive in Canadian clinical practice, according to the clinical experts consulted by CADTH.
Non-comparative design: The non-comparative design of the FIGHT-202 trial precludes the ability to assess the relative therapeutic benefit or safety of pemigatinib against currently available therapies in Canadian clinical practice. As noted previously, the clinical experts consulted by CADTH agreed that direct randomized comparisons between pemigatinib and currently used therapies are unlikely to take place in the setting of previously treated, unresectable, locally advanced, or metastatic CCA with an FGFR2 fusion or other rearrangement. In the absence of a direct comparison of pemigatinib with relevant treatment options, the sponsor submitted an ITC51 in the form of an unanchored MAIC comparing the efficacy of pemigatinib (cohort A of the FIGHT-202 trial) with each of the 2 treatment groups in the ABC-06 study. Please refer to the following section on indirect evidence for a detailed summary and critical appraisal of the sponsor’s submitted ITC. The results of the ITC favoured pemigatinib for PFS and OS in comparison with mFOLFOX plus ASC as well as with ASC alone. The clinical experts agreed that in the absence of robust comparative data on PFS and OS, no firm conclusions could be drawn on how pemigatinib compared with other relevant treatment options. The clinical experts consulted by CADTH anticipated, however, that based on the FIGHT-202 results and on poor results with existing treatment options in clinical practice, pemigatinib appeared to offer at least similar or improved clinical benefits compared with current therapies, with better tolerability.
Relevance of trial efficacy outcomes: The primary outcome in the FIGHT-202 trial was ORR in cohort A and secondary outcomes included DOR, DCR, PFS, and OS. According to the clinical experts consulted by CADTH, ORR, DOR, and DCR are clinically meaningful end points for patients with unresectable, locally advanced, or metastatic CCA that has progressed on prior therapy. Responses in this patient population are important because of accompanying delay in the worsening of symptoms and a slower decline in ECOG PS. According to the clinical experts, the majority of patients will have stable disease, followed by a PR and, rarely, a CR in response to first-line treatment; therefore, the clinical experts were not concerned about the low number of patients who achieved CR in the FIGHT-202 trial, but emphasized the clinical relevance and importance of maintaining stable disease in preventing an otherwise fast decline in patients in this setting. While the clinical experts agreed that, based on the available evidence, it was not possible to conclude whether the antitumour activity expressed as responses would translate into clinical benefits in terms of PFS and OS, they felt that durable responses could potentially delay tumour progression and result in prolonged survival benefit in this patient population.
Excluded patient subgroups: The FIGHT-202 trial excluded patients who would be intolerant to standard first-line therapy without experiencing PD. The clinical experts consulted by CADTH felt it would be reasonable to generalize the results from cohort A to patients with FGFR2 alterations who are intolerant to first-line therapy, given the favourable safety profile of oral pemigatinib. The clinical experts noted that as patients with an ECOG PS greater than 2 were excluded from the FIGHT-202 trial, there are no data to support the generalizability of treatment benefit in this patient population. The clinical experts were of the opinion that it would be reasonable to leave it up to the discretion of the treating physician to apply some flexibility in terms of using pemigatinib with slightly lower laboratory parameters than those outlined in the trial. It was agreed by the clinical experts that since patients with brain or central nervous system metastases that are untreated or have progressed were excluded from the trial, there are no data to support the generalizability of treatment benefit in this patient population.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
The FIGHT-202 study was a non-comparative study and the systematic review therefore does not provide any direct evidence for the relative efficacy of pemigatinib versus a relevant comparator. A focused literature search for network meta-analyses dealing with pemigatinib or CCA was run in MEDLINE All (1946–) through Ovid and Embase (1974–) through Ovid on July 16, 2021. No limits were applied, and conference abstracts were excluded from the search results. No relevant studies were identified comparing pemigatinib versus mFOLFOX, FOLFIRI, fluorouracil alone or in combination with cisplatin or oxaliplatin, capecitabine alone or in combination with cisplatin or oxaliplatin, or best supportive care in adults with previously treated, unresectable, locally advanced, or metastatic CCA with FGFR2 fusions or other rearrangements.
The sponsor submitted an ITC51 in the form of a MAIC between cohort A of the FIGHT-202 study and each of the 2 treatment groups in the ABC-06 study. The ABC-06 study compared an mFOLFOX regimen plus ASC versus ASC alone in patients with BTC. The results of the MAIC were used to inform the sponsor’s pharmacoeconomic submission.
Methods of Sponsor-Submitted MAIC
Objectives
Given the lack of an RCT comparing pemigatinib with a standard-of-care regimen, an ITC was conducted to provide evidence for the relative efficacy of pemigatinib versus relevant comparators.
Study Selection Methods
There were 2 literature searches, 1 performed on November 9, 2018 (original search) and a second performed on April 21, 2020, (updated search), to identify available clinical efficacy, safety, and tolerability evidence related to second-line treatment of advanced and/or metastatic CCA with FGFR2 fusions or rearrangements. Both searches used the same English-language search strategy in multiple databases (MEDLINE In-Process, Embase, MEDLINE, and the Cochrane Library) and the date ranges were from database inception to November 9, 2018 for the original search, and from October 1, 2018 to April 21, 2020, for the updated search. The following sources were also searched: conference proceedings from 6 oncology conferences starting from the year 2016; reference lists of relevant studies, systematic reviews within the previous 2 years, and meta-analyses; and reference lists from relevant articles from 7 health technology assessment agencies. For the original search, 2 independent reviewers screened abstracts, with a third reviewer assessing abstracts where there was disagreement or uncertainty. In those cases, the consensus of the majority was used to make a final decision. Full-text screening of relevant abstracts was performed in the same manner as for abstract screening. For the updated search, 2 independent reviewers screened abstracts and full-text articles, with a third reviewer independently resolving uncertainties regarding study inclusion at each screening stage.
Studies were eligible if they included adults with advanced and/or metastatic or surgically unresectable CCA with FGFR2 fusions or rearrangements that had failed to respond to at least 1 treatment. Studies were excluded if they reported on pediatric patients, patients that did not have metastatic or advanced cancer or FGFR2 fusions or rearrangements, patients who were treatment-naive, or patients with resectable CCA. Articles with unclear disease stage, FGFR2 status, or treatment line were included during abstract screening. Studies either had to be single-arm with a pharmacological intervention or had to compare a pharmacological intervention with placebo, best supportive care (as defined by the study author), or any other pharmacological intervention. Eligible study designs were RCTs, single-arm studies, observational studies, and systematic reviews, with the latter used only for bibliography searches. Preclinical studies, case reports, case series, pharmacokinetic studies, and economic studies were excluded. A list of relevant outcomes was provided, though it was unclear whether articles were screened based on available outcomes. These outcomes were: response rate; OS; PFS; time to treatment discontinuation; DOR; mortality; HRQoL; incidence of AEs; study or treatment discontinuation; and relationship between intermediate outcome (PFS and response rate) and OS, DCR, stable disease, time on treatment, time to response, overall response rate, and patient-reported outcomes.
In the original search, a total of 35 relevant results were identified reporting on 8 non-comparative studies, 1 retrospective observational study, and 11 ongoing studies with no results available. In addition, 111 articles were flagged: 32 were flagged as having a patient population with BTC and 79 were flagged as having a patient population with unclear FGFR2 mutation status. From the updated search, 829 new articles were included at the full-text screening stage; in addition, the 111 flagged articles from the original search were also included in the full-text screening. One of the most common reasons given for excluding full-text publications was “biliary tract cancer” (n = 117). In response52 to a request for clarification by the CADTH review team, the sponsor indicated that the following types of studies were excluded: studies that did not report a subgroup of patients with CCA, studies in which patients with CCA made up less than 80% of the study patients, and studies in which the percentages of patients with each type of BTC were not reported.
After full-text screening in the updated search, a total of 209 relevant publications were identified (of which 23 were identified from conference proceedings and 35 were the relevant results from the original search), reporting on 108 studies. Study quality for the 108 studies was assessed using the Downs and Black checklist, though it was unclear how or if these assessments were used.
For potential inclusion in the MAIC, additional criteria were applied to the 108 studies identified in the systematic literature searches. It was not clear at what point these criteria were established, and rationales were not provided for all of the criteria. KM plots of both OS and PFS were required so that pseudo patient-level data for these outcomes could be derived. The study had to include a treatment (if not pemigatinib) that was representative of standard of care, which seemed to be defined as chemotherapy. A minimum sample size of 20 patients was established, though a justification for this cut-off was not provided. After applying these additional criteria and eliminating a study in patients receiving a fourth-line or later-line therapy, 8 studies remained. Although the proportions of patients with an ECOG PS of 0 or 1 and patients with iCCA had to be high to match the patient population in the FIGHT-202 study, these criteria did not appear to be applied at this stage of the study selection process.
Of the 8 remaining studies, 2 were single-arm trials (including the FIGHT-202 study), 2 were RCTs, and 4 were retrospective studies. In the studies other than the FIGHT-202 study, publication dates ranged from 2012 to 2020, sample sizes for each treatment group ranged from 30 to 255, treatments included chemotherapy and ASC, median age ranged from 54 years to 65 years where reported, the percentage of male patients ranged from 43% to 66.7%, the percentage of patients with iCCA ranged from 16.7% to 94.6%, and the percentage of patients with an ECOG PS of 0 or 1 ranged from 64% to 100% where reported. FGFR2 mutation status was not reported for any of the studies, aside from the FIGHT-202 study.
Despite the sponsor’s explanation that studies were excluded during full-text screening if they did not report on the percentage of patients with CCA or if the percentage of patients with CCA was less than 80%, CADTH reviewers noted examples within the 8 considered studies that appeared to contradict this. The types of biliary cancers included in 1 study were not reported in the cited conference abstract53 and the percentage of patients with CCA was less than 80% in 3 studies.13,54,55
The ITC authors focused on 2 studies, Kim et al.55 and Lemarca et al.,13 based on sample size and recent date of publication (thought to more accurately reflect the current standard of care). The ITC was originally developed for the pemigatinib submission to the National Institute for Health and Care Excellence (NICE) and the study by Lemarca et al.,13 also known as the ABC-06 study, was chosen because it recruited patients in the UK.
MAIC Analysis Methods
The MAIC approach was selected due to the non-comparative nature of the FIGHT-202 study. The choice of the ABC-06 study for comparison with the FIGHT-202 study did not appear to be based on its specific interventions; however, the mFOLFOX regimen was considered by the clinical experts consulted by the ITC authors to be a relevant comparator in the second-line setting. Both ASC alone and mFOLFOX plus ASC from the ABC-06 study were compared with pemigatinib from the FIGHT-202 study.
According to the ITC authors, the following baseline characteristics were available for cohort A of the FIGHT-202 study and the patients in the ABC-06 study: median age, percentage of male patients, percentage of patients with iCCA, percentage of patients with an ECOG PS of 0 or 1, and percentage of patients with a serum albumin concentration of less than 35 g/L. Of these, the following covariates were chosen for adjustment: |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. The selection of covariates for adjustment did not appear to be preplanned and no rationale was provided for the covariates selected. Additionally, the following baseline characteristics were available for both studies and did not appear to be considered: disease stage, percentage of patients with prior surgery for cancer, and number of lines of prior systemic therapy for advanced or metastatic cancer. In the FIGHT-202 study, 9 patients from cohort A were excluded from the MAIC due to missing serum albumin values.
For each arm in the ASC-06 study, a logistic regression model was estimated using the method of moments based on individual patient data from the FIGHT-202 study and summary data from the relevant arm in the ABC-06 study. The model was used to approximate propensity scores for patients in the FIGHT-202 study, which reflected the odds for each patient being included in the ABC-06 study versus the FIGHT-202 study based on the distributions of the covariates included in the model. The baseline characteristics used to reweight the pemigatinib group, along with effective sample size, were presented for the pemigatinib group before weighting and after weighting to the mFOLFOX plus ASC and ASC only groups. The outcomes of PFS and OS were the only disease-related outcomes available for both studies and were generated on a pseudo patient level for the ABC-06 study by estimating times and survival probabilities from the published KM PFS and OS curves using a software tool. HRs for PFS and OS were determined using Cox proportional hazard models with bootstrapping used to estimate standard errors and CIs.
Results of Sponsor-Submitted MAIC
Summary of Included Studies
The ABC-06 study, the study selected for the MAIC with cohort A of the FIGHT-202 study (the pemigatinib group), was a phase III, open-label, multi-centre RCT conducted in the UK with patients enrolled from 2014 to 2018. Adult patients with locally advanced or metastatic BTC with disease progression in the previous 6 weeks on first-line cisplatin and gemcitabine chemotherapy were randomized (1:1) to receive ASC alone (ASC group) or mFOLFOX plus ASC (mFOLFOX plus ASC group). Randomization was stratified by platinum sensitivity (sensitive or refractory/resistant), serum albumin concentration (< 35 g/L or ≥ 35 g/L), and disease stage (locally advanced or metastatic).
Patients in the ABC-06 study had to have an ECOG PS of 0 to 1; a life expectancy of longer than 3 months; adequate hematological, renal, and hepatic function; and no evidence of ongoing infection, inadequate biliary drainage, metastatic disease to the brain, or clinically significant cardiovascular disease.
All patients received ASC, which consisted of early identification and treatment of biliary-related complications and cancer-related symptom management. Interventions included biliary drainage, antibiotics, analgesia, steroids, antiemetics, other palliative treatment for symptom control, palliative radiotherapy (e.g., for painful bone metastases), and transfusion of blood products. Patients had study visits every 4 weeks for ASC, which included physical examination, assessment of ECOG PS, symptom monitoring, review of concomitant medications, and assessment of liver and renal function with full blood count. Patients in the mFOLFOX plus ASC group also received chemotherapy every 2 weeks for a maximum of 12 cycles. At each cycle, patients received oxaliplatin 85 mg/m2 and L-folinic acid 175 mg (or folinic acid 350 mg) through IV infusion over 2 hours, and fluorouracil 400 mg/m2 through a 5- to 10-minute bolus on day 1. Fluorouracil 2,400 mg/m2 was started as a continuous IV infusion on day 1 and was finished on day 2. A maximum of 2 dose reductions for each drug was allowed, representing a 20% and 50% reduction from the initial dose. If treatment was delayed for more than 28 days due to toxicity, the patient permanently discontinued treatment. If oxaliplatin was discontinued due to toxicity, treatment with the other components of the regimen could continue with an increase in fluorouracil dose, according to local practice. Patients in the mFOLFOX plus ASC group with disease progression were subsequently treated at the clinician’s discretion. Following disease progression, patients in both the ASC alone group and mFOLFOX plus ASC group could receive treatment with experimental therapies in phase I trials.
Radiological assessment with CT (and, optionally, MRI) was performed in the mFOLFOX plus ASC group every 12 weeks until disease progression, and images were evaluated by investigators according to RECIST 1.1 criteria. Patients in the ASC alone group had radiological assessments only when clinically indicated. The primary end point of the ABC-06 study was OS in the intention-to-treat population. Secondary end points included PFS, radiological response, and QoL.
The key baseline characteristics of patients in cohort A of the FIGHT-202 study, and the ASC alone group and mFOLFOX plus ASC group in the ABC-06 study, are presented in Table 22. Compared with both treatment groups in the ABC-06 study, patients in cohort A of the FIGHT-202 study were younger (median age of 56 years versus 65 years) and less likely to be male (39% male versus 53% and 46% male). Almost all patients in cohort A of the FIGHT-202 study had iCCA, while 42% and 47% of patients in the mFOLFOX plus ASC and ASC alone groups, respectively, had iCCA. Almost all patients in both studies had an ECOG PS of 0 or 1, with a greater proportion of patients in cohort A of the FIGHT-202 study having an ECOG PS of 0 compared with the mFOLFOX plus ASC and ASC alone groups. FGFR2 mutation status was not reported in the ABC-06 study. Almost 40% of patients in cohort A of the FIGHT-202 study had more than 1 line of prior systemic therapy compared with none in the ABC-06 study.
Table 22: Summary of Baseline Characteristics
Characteristic | FIGHT-202 (cohort A) | ABC-06 | |
|---|---|---|---|
Pemigatinib (pre-weighting) N = 107 | mFOLFOX plus ASC N = 81 | ASC alone N = 81 | |
Median age,a years (range) | 56 (26 to 77) | 65 (26 to 84) | 65 (26 to 81) |
Male,a n (%) | 42 (39) | 43 (53) | 37 (46) |
Tumour site, n (%) | NA | NA | NA |
Intrahepatic | 105 (98) | 34 (42) | 38 (47) |
Extrahepatic | 1 (0.9) | 26 (32) | 19 (23) |
Gallbladder | ||| | 17 (21) | 17 (21) |
Ampulla | ||| | 4 (5) | 7 (9) |
Missing | 1 (0.9) | 0 | 0 |
Patients with FGFR2 fusions or rearrangements, n (%) | 107 (100) | NR | NR |
ECOG Performance Status,a n (%) | NA | NA | NA |
0 | 45 (42.1) | 25 (31) | 28 (35) |
1 | 57 (53.3) | 55 (68) | 52 (64) |
2 | 5 (4.7) | 0 | 0 |
Missing | 0 | 0 | 1 (1) |
Serum albumin ≥ 35 g/L,a n (%) | |||||||| | 62 (77) | 60 (74) |
Disease stage, n (%) | NA | NA | NA |
Locally advanced | 16 (15.0) | 14 (17) | 15 (19) |
Metastatic | 88 (82.2) | 67 (83) | 66 (81) |
Unknown or missing | 3 (2.8) | 0 | 0 |
Previous surgery, n (%) | 38 (35.5) | 34 (42) | 38 (47) |
Number of prior systemic therapies, n (%) | NA | NA | NA |
1 | 65 (60.7) | 81 (100) | 81 (100) |
2 | 29 (27.1) | 0 | 0 |
≥ 3 | 13 (12.1) | 0 | 0 |
ASC = active symptom control; ECOG = Eastern Cooperative Oncology Group; FGFR2 = fibroblast growth factor receptor 2; MAIC = matching-adjusted indirect comparison; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; NA = not applicable; NR = not reported.
aMean age and percentages of male patients, patients with an ECOG Performance Status ≤ 1, and patients with serum albumin ≥ 35 g/L were covariates in the logistic regression model used for adjustment.
Source: Sponsor-submitted MAIC report,51 FIGHT-202 Clinical Study Report,16 and Lamarca et al.13
Results
The regression model covariates are presented in Table 23 for the pemigatinib group before weighting and after weighting to match the mFOLFOX plus ASC group and the ASC alone group. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The effective sample size of the pemigatinib group was reduced to ||||||||||||||||||||||||| after weighting to match the mFOLFOX plus ASC and ASC alone groups, respectively. Other baseline characteristics after weighting were not presented in the sponsor-submitted ITC report.
Table 23: Summary of Model Covariates Before and After Weighting
Model covariate | Pemigatinib, before weighting |||||||||| | Pemigatinib, weighted to mFOLFOX plus ASC |||||||||| | Pemigatinib, weighted to ASC alone ||||||||||||||||||| |
|---|---|---|---|
|||||||||||||||||||||||||| | ||||||| | ||||||| | ||||||| |
|||||||||| | ||||||| | ||||||| | ||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||| | ||||||| | ||||||| |
||||||||||||||||||||||||||||||||||||||||||||||| | ||||||| | ||||||| | ||||||| |
ASC = active symptom control; ECOG = Eastern Cooperative Oncology Group; ESS = effective sample size; MAIC = matching-adjusted indirect comparison; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin.
Source: Sponsor-submitted MAIC report.51
Median follow-up duration in the ABC-06 study at data cut-off was 21.7 months (interquartile range, 17.2 months to 30.8 months). There were 2 patients and 1 patient lost to follow-up in the ASC alone and mFOLFOX plus ASC groups, respectively.
Overall Survival
Results for OS for pemigatinib versus mFOLFOX plus ASC are presented in Table 24. Following weighting of the pemigatinib group to match the mFOLFOX plus ASC group, median OS was |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| for the pemigatinib group versus |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| for the mFOLFOX plus ASC group, based on the March 22, 2019, data cut-off for the FIGHT-202 study. The corresponding HR was |||||||||||||||||||||||||||||||||||||||||||||||||||||||) and the HR using the results from the April 7, 2020, data cut-off was |||||||||||||||||||||||||||||||||||||||||||||||||||||||||. KM curves for OS are shown in Figure 14 for the unweighted pemigatinib group, weighted pemigatinib group, and mFOLFOX plus ASC group.
Table 24: Overall Survival, Pemigatinib vs. mFOLFOX Plus ASC
Outcome | Pemigatinib, before weighting |||||||| | Pemigatinib, weighted to mFOLFOX plus ASC |||||||||||||| | mFOLFOX plus ASC |||||||| |
|---|---|---|---|
OS events, n | |||| | |||| | |||| |
Median OS, months (95% CI),a | ||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||||||| |
HR (95% CI),b | ||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||||||| | ||||||| |
HR (95% CI),b April 7, 2020, data cut-off | ||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||||||| | ||||||| |
ASC = active symptom control; CI = confidence interval; ESS = effective sample size; MAIC = matching-adjusted indirect comparison; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; HR = hazard ratio; NA = not applicable; OS = overall survival; REF = reference group; vs. = versus.
NOTE: Results for the pemigatinib group are from the March 22, 2019, data cut-off unless otherwise indicated.
aKaplan–Meier estimates.
bHRs were determined using Cox proportional hazard models with bootstrapping to estimate CIs.
Source: Sponsor-submitted MAIC report.51

Note: This figure has been redacted at the sponsor’s request.
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
||||||||||||||||||||||||||.51
Results for OS for pemigatinib versus ASC are presented in Table 25. Following weighting of the pemigatinib group to match the ASC alone group, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||, based on the March 22, 2019, data cut-off for the FIGHT-202 study. The corresponding HR was |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. KM curves for OS are shown in Table 15 for the unweighted pemigatinib group, weighted pemigatinib group, and ASC alone group.
Table 25: Overall Survival, Pemigatinib Versus ASC
Outcome | Pemigatinib, before weighting ||||||||||||| | Pemigatinib, weighted to ASC ||||||||||||| | ASC ||||||||||||| |
|---|---|---|---|
OS events, n | |||| | |||| | |||| |
Median OS, months (95% CI)a | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| |
HR (95% CI),b | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||||||| |
HR (95% CI),b April 7, 2020, data cut-off | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||||||| |
ASC = active symptom control; CI = confidence interval; ESS = effective sample size; HR = hazard ratio; MAIC = matching-adjusted indirect comparison; NA = not applicable; OS = overall survival; Ref. = reference group.
NOTE: Results for the pemigatinib group are from the March 22, 2019, data cut-off unless otherwise indicated.
aKaplan–Meier estimates.
bHRs were determined using Cox proportional hazard models with bootstrapping to estimate CIs.
Source: Sponsor-submitted MAIC report.51
Figure 15: Kaplan–Meier Overall Survival Curves, Pemigatinib Versus ASC (Redacted)

Note: This figure has been redacted at the sponsor’s request.
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.51
Progression-Free Survival
Results for PFS for pemigatinib versus mFOLFOX plus ASC are presented in Table 26. Following weighting of the pemigatinib group to match the mFOLFOX plus ASC group, median PFS was ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| for the pemigatinib versus mFOLFOX plus ASC groups, based on the March 22, 2019, data cut-off for the FIGHT-202 study. The corresponding HR was |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. KM curves for PFS are shown in Figure 16 for the unweighted pemigatinib group, the weighted pemigatinib group, and the mFOLFOX plus ASC group.
Table 26: Progression-Free Survival, Pemigatinib vs. mFOLFOX Plus ASC
Outcome | Pemigatinib, before weighting ||||||||||| | Pemigatinib, weighted to mFOLFOX plus ASC ||||||||||||| | mFOLFOX plus ASC ||||||||||| |
|---|---|---|---|
PFS events, n | |||| | |||| | ||||| |
Median PFS, months (95% CI)a | ||||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||||||||||| |
HR (95% CI),b | ||||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||||||||||| | ||||| |
HR (95% CI),b April 7, 2020, data cut-off | ||||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||||||||||| | ||||| |
ASC = active symptom control; CI = confidence interval; ESS = effective sample size; MAIC = matching-adjusted indirect comparison; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; HR = hazard ratio; MAIC = matching-adjusted indirect comparison; NA = not applicable; PFS = progression-free survival; Ref. = reference group; vs. = versus.
Note: Results for the pemigatinib group are from the March 22, 2019, data cut-off, unless otherwise indicated.
aKaplan–Meier estimates.
bHRs were determined using Cox proportional hazard models with bootstrapping to estimate CIs.
Source: Sponsor-submitted MAIC report.51
Figure 16: Kaplan–Meier Progression-Free Survival Curves, Pemigatinib vs. mFOLFOX Plus ASC (Redacted)

Note: This figure has been redacted at the sponsor’s request.
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.51
Critical Appraisal of Sponsor-Submitted MAIC
Due to the non-comparative design of the FIGHT-202 study, the approach of using a MAIC to compare pemigatinib with a relevant comparator was appropriate. The following limitations of the systematic literature search methods were identified: the final stage of study selection for the MAIC did not appear to use preplanned criteria and the criteria for excluding publications during full-text screening for the reason “biliary tract cancer” may not have been consistently applied. However, the clinical experts consulted by CADTH for this review agreed that the ABC-06 study was likely the most relevant trial in this setting, given that it represents the only RCT evidence for treating CCA beyond the first-line setting. The selection of an RCT rather than a retrospective study was an appropriate choice, since patients who enrol in a clinical trial likely differ overall compared with those included in retrospective studies.
Although the statistical methods used for reweighting the pemigatinib group (cohort A of the FIGHT-202 study) and for estimating the 95% CIs were appropriate, there were potentially important underlying differences between the FIGHT-202 and ABC-06 studies. In particular, all patients in cohort A of the FIGHT-202 study had FGFR2 fusions or rearrangements, while patients in the ABC-06 study were not selected based on FGFR2 mutation status, and FGFR2 mutation status was not reported. Given that FGFR2 fusions and rearrangements occur almost exclusively in iCCA and that the prevalence of FGFR2 fusions and rearrangements is less than 20%56 in patients with iCCA, there is likely a large disparity in FGFR2 mutation status between the study populations. While the FIGHT-202 study included only patients with CCA, the ABC-06 study included patients with BTC, which encompasses gallbladder cancer and ampullary cancer in addition to CCA. Due to this difference and the distribution of FGFR2 fusions and rearrangements between the types of CCA, 98.1% of patients in cohort A of the FIGHT-202 study had iCCA compared with 42% and 47% in the mFOLFOX plus ASC and ASC alone groups, respectively. Since disease type and FGFR2 status were more restricted in the FIGHT-202 study, these differences could not be addressed through the weighting of patients in the pemigatinib group. The natural history among bile duct cancer subtypes appears variable with different prognosis.57
The covariates chosen for adjustment were based on |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Although the ITC authors claimed that this selection was based on the availability of this information for both the FIGHT-202 and ABC-06 studies, the following baseline characteristics were also available for both studies and did not appear to be considered: disease stage, percentage of patients with prior surgery for cancer, and number of lines of prior systemic therapy for advanced or metastatic cancer. It was not clear if the selection of covariates for adjustment was based on the need to adjust for predetermined prognostic factors and effect modifiers. Baseline characteristics other than the chosen covariates were not reported for the pemigatinib group after weighting. The clinical experts consulted by CADTH for this review were of the opinion that the number of lines of previous therapy was of key importance in terms of prognosis. The clinical experts were not aware of any additional prognostic factors and/or effect modifiers that were not reported in both studies and that should have been considered.
Another difference was noted related to the median duration of follow-up; which was 21.7 months (interquartile range = 17.2 to 30.8) in the ABC-06 trial and 15.44 months (minimum = 7.0; maximum = 24.7) and 27.9 months (||||||||||||||||||||||||||||||||||), respectively, at the March 22, 2019, and April 7, 2020, data cut-off dates in cohort A of the FIGHT-202 trial. The differing lengths of follow-up between the trials were not adjusted in the MAIC and may also contribute to heterogeneity, especially for the survival analyses.
While there are retrospective studies suggesting that the presence of FGFR2 mutations in CCA may be associated with a better prognosis,18,19 the clinical experts consulted by CADTH were of the opinion that FGFR2 mutation status was not an important prognostic factor in the indicated patient population. The clinical experts considered the fact that patients in both the FIGHT-202 and ABC-06 trials had progressed on prior systemic therapy to be of greater importance in terms of prognosis. The clinical experts expected patients in the FIGHT-202 study to have more advanced disease than patients in the ABC-06 study because the FIGHT-202 study population was more heavily pre-treated overall. It is unclear whether the pemigatinib group was more or less similar to the ABC-06 groups in this respect following weighting, as the weighting process did not take the number of prior lines of systemic therapy into account. If substantial differences remained, these differences could have led to bias against pemigatinib in all of the comparisons.
The limitations with regard to the external validity identified for cohort A in the FIGHT-202 study also apply to the MAIC results. In addition, the effective sample size of the pemigatinib group was reduced by approximately ||||||| after weighting to the mFOLFOX plus ASC and ASC alone groups, and it is unclear how representative the post-weighting pemigatinib groups are of cohort A of the FIGHT-202 study. It is apparent that patients from cohort A of the FIGHT-202 study with an ECOG PS of 2 were effectively excluded from the pemigatinib group during the weighting process. Other changes noted in the pemigatinib group following weighting were that mean age increased by approximately ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||| for comparison with mFOLFOX plus ASC and ASC alone, respectively. While both male and female patients would have been adequately represented in the post-weighting pemigatinib groups, it is not clear how the age distribution was affected.
After adjustment based on the 4 chosen covariates, there was substantial loss in precision for the efficacy estimates, as the effective sample size was reduced by approximately |||||||. Adding more covariates for adjustment may have further reduced the available precision.
Comparisons of pemigatinib with other relevant comparators (FOLFIRI, fluorouracil alone or in combination with cisplatin or oxaliplatin, and capecitabine alone or in combination with cisplatin or oxaliplatin) were not available. Given that mFOLFOX plus ASC is the only therapy beyond the first-line setting with RCT evidence of an OS benefit, the clinical experts consulted by CADTH expected that mFOLFOX plus ASC would have the greatest efficacy out of all the relevant comparators.
Summary
For the unanchored MAIC to produce unbiased treatment effect estimates, both effect modifiers and prognostic variables need to be adjusted for in the analysis. Residual confounding remains the major limitation of the MAIC despite adjusting for age, sex, ECOG PS, and serum albumin in the comparisons of pemigatinib with mFOLFOX plus ASC and ASC alone. While any bias introduced by the differences between the FIGHT-202 and ABC-06 studies in the number of prior lines of systemic therapy may have been against pemigatinib, the substantial differences in FGFR2 mutation status and tumour site between trials introduce a high degree of uncertainty in the OS and PFS results. Furthermore, MAICs cannot account for unknown cross-trial differences; thus, the MAIC estimates are susceptible to bias from unknown confounding. An evaluation of potential bias from residual confounding was not reported; therefore, the magnitude of this bias in the relative treatment effect estimates is unclear. Overall, it remains uncertain whether pemigatinib provides additional OS or PFS benefit versus mFOLFOX plus ASC or ASC alone.
Other Relevant Evidence
This section includes 1 additional relevant report included in the sponsor’s submission to CADTH that was suggested to be a study for the indication for this CADTH review. FIGHT-101 is an ongoing, open-label phase I/II dose-escalation and expansion study of pemigatinib among participants with previously treated advanced malignancies with and without an FGF/FGFR alteration. As of February 2019, FIGHT-101 enrolled 160 participants from 14 study sites in the US and Denmark, 116 of which received at least 1 dose of pemigatinib monotherapy. Sixteen participants who were treated with pemigatinib monotherapy had CCA, ||||| of whom had FGFR2 rearrangements or fusions and received pemigatinib 13.5 mg orally once daily on a schedule of 2 weeks on, 1 week off, for each 21-day cycle.
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. The best overall response of a PR was observed in ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. In terms of safety outcomes, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. However, due to the open-label design and the limited data on the efficacy of pemigatinib on CCA within the report, the ability to interpret these results is considerably limited.
Discussion
Summary of Available Evidence
The CADTH systematic review included 1 phase II trial of pemigatinib that evaluated the efficacy and safety of pemigatinib in patients with advanced, metastatic, or surgically unresectable CCA with FGFR2 alterations, other FGF or FGFR alterations, or no FGF or FGFR alterations, whose disease did not respond to previous therapy. The FIGHT-202 trial (N = 146) is an ||||||||||||||||, multi-centre, open-label, single-arm phase II trial that assigned patients to 3 cohorts, depending on the patient’s FGF/FGFR status (cohort A: FGFR2 fusions or rearrangements; cohort B: FGF/FGFR alterations other than FGFR2 fusions or rearrangements; or cohort C: negative for FGF/FGFR alterations). This CADTH review focuses on cohort A, as cohorts B and C were not part of the requested reimbursement criteria submitted to CADTH and not approved in the Health Canada Notice of Compliance with conditions. All enrolled participants received oral pemigatinib (13.5 mg orally once daily on a schedule of 2 weeks on, 1 week off for each 21-day cycle). The primary outcome was ORR in cohort A, and secondary outcomes included ORR in cohort B, cohort A plus B, and cohort C; PFS, DOR, DCR, OS, and safety were assessed in all 3 cohorts. Exploratory end points included HRQoL and symptom severity.
Adults diagnosed with advanced, metastatic, or surgically unresectable CCA with FGFR2-positive disease who had documented disease progression after at least 1 line of prior systemic therapy, were enrolled into cohort A of the Fight-202 trial. The majority of patients had iCCA, an ECOG PS of 0 or 1, had received 1 or 2 previous lines of systemic therapies for advanced or metastatic disease, and were 56 years old (range, 26 to 77 years).
In addition to the systematic review, 1 sponsor-submitted ITC was summarized and appraised for this review. The other relevant evidence section included the summary of an open-label phase I/II trial (FIGHT-101) of pemigatinib in patients with advanced malignancies.
Interpretation of Results
Efficacy
The FIGHT-202 trial achieved the predetermined threshold for a positive outcome (lower limit of the 95% CI for ORR > 15%) in cohort A. As of the March 22, 2019, data cut-off date, after a median follow-up time of 15.44 months, the proportion of patients with an objective response, (the primary end point in cohort A) was 35.5% (95% CI, 26.50 to 45.35). A total of 3 patients had achieved a CR, 35 patients had a PR, and 50 patients had stable disease as the best response; the DCR was 82.2%. The results at the April 7, 2020, data cut-off date, with 27.9 months of follow-up, were consistent with the previous data cut-off date, with an ORR of 37% (95% CI, 27.94 to 46.86) and 4, 36, and 49 patients achieving a CR, PR, and stable disease as best response, respectively; DCR was not reported for the April 7, 2020, data cut-off date. Median DOR was 7.49 months (95% CI, 5.65 to 14.49) and 8.08 months (95% CI, 5.65 to 13.14) at the March 22, 2019, and April 7, 2020, data cut-off dates, respectively. The FIGHT-202 trial included no formal statistical significance and hypotheses testing, and point estimates with 95% CIs were reported to estimate the magnitude of treatment effect. A greater than 95% probability to have a 95% CI for ORR in cohort A with a lower limit larger than 15% was the basis for the sample size determination and was regarded as the threshold for a positive study outcome. Results for the subgroup of interest, as pre-specified in the protocol for this CADTH systemic literature review, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. However, given that the trial was not designed to detect differences in treatment effects across subgroups, no conclusions can be made on the basis of subgroup results. OS and PFS were assessed as secondary outcomes in the FIGHT-202 trial; median OS and PFS, respectively, were 21.06 months (95% CI, 14.82 to not evaluable) and 6.93 months (95% CI, 6.18 to 9.59) at the March 22, 2019, data cut-off date. Consistent results were observed at the April 7, 2020, data cut-off date, with median OS and PFS being 17.48 months (95% CI, 14.42 to 22.93) and 7.03 months (95% CI, 6.08 to 10.48), respectively. Interpretation of time‐to‐event end points such as OS or PFS is limited in single‐arm studies; since all patients in cohort A received the same treatment, the extent to which the observed survival is due to the natural history of the tumour or the intervention remains unclear.48 The primary objective of phase II (randomized or non-randomized) trials is to document the safety outcomes and investigate whether the estimate of effect for a new drug is large enough to use it in confirmatory phase III trials. A phase II trial may not accurately predict harm and/or effectiveness of treatments. The clinical experts consulted by CADTH noted that, despite the high unmet need, conducting an RCT in this setting with a targeted therapy, such as pemigatinib, compared with the available therapies currently used in the second line in Canadian clinical practice would likely not be feasible. According to the clinical experts, developing phase III RCTs is hindered by the overall low number of patients who meet the current indication and equipoise between pemigatinib and other chemotherapy drugs does not exist.
According to the clinical experts consulted by CADTH and the registered clinician groups providing input for this submission, the responses achieved with pemigatinib were clinically relevant, important to patients in this setting, and clearly higher in comparison with what could be observed with the therapies currently used in this setting. The clinically experts consulted by CADTH noted that durable responses in this patient population are important because of accompanying delay in the worsening of symptoms and a slower decline in ECOG PS. The clinical experts emphasized the clinical relevance and importance of even maintaining stable disease in preventing an otherwise fast decline in patients in this oftentimes last line of treatment. This view was echoed by the input provided by the patient advocacy group, which highlighted tumour response, maintenance of response, delay in disease progression, and QoL as important treatment goals for patients. While the clinical experts agreed that, based on the available evidence, it was not possible to conclude whether the antitumour activity expressed as responses would translate into clinical benefits in terms of PFS and OS, they felt that the preliminary survival results from the trial were very encouraging and that durable responses could potentially delay tumour progression and result in prolonged survival benefit in this patient population.
In the absence of a direct comparison of pemigatinib with relevant treatment options, the sponsor submitted an ITC51 in the form of an unanchored MAIC comparing the efficacy of pemigatinib (cohort A of the FIGHT-202 trial) with each of the 2 treatment groups in the ABC-06 study. The results of the ITC favoured pemigatinib for PFS and OS in comparison with mFOLFOX plus ASC as well as compared with ASC alone. The CADTH critical assessment identified several limitations with the sponsor’s submitted MAIC, including heterogeneity across study designs and populations and the inability to adjust for all potential confounders and prognostic variables in the MAIC. The clinical experts agreed with the CADTH clinical review team that, given the absence of robust comparative data on PFS and OS, the ability to interpret the relative treatment effects observed between pemigatinib and FOLFOX plus ASC and ASC alone was limited, and no firm conclusions could be drawn on how pemigatinib compared with other relevant treatment options. The clinical experts consulted by CADTH anticipated, however, that based on the FIGHT-202 results and on poor results with existing treatment options in clinical practice, pemigatinib appeared to offer at least similar or improved clinical benefits compared with current therapies, with better tolerability. The clinical experts consulted by CADTH noted there is currently insufficient evidence to determine whether patients whose cancer is positive for an FGFR2 fusion represent a distinct prognostic subgroup. The clinical experts agreed that progression on prior systematic therapy is a key prognostic factor in these patients, and they did not anticipate that patients would derive any substantial benefit from their underlying disease biology at the time they enrolled into the FIGHT-202 trial.
Input received by the patient advocacy group, the registered clinicians, and the clinical experts consulted by CADTH highlighted HRQoL as an important outcome and treatment goal for patients. The overall observed scores from baseline to cycle 42 were |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. However, given the non-comparative, open-label design of the trial, the lack of a pre-specified analysis of the patient-reported outcomes data, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. The clinical experts consulted by CADTH noted that HRQoL in this setting is low and unstable. They anticipated that, given the observed responses in the FIGHT-202 trial, pemigatinib would likely improve or at least maintain patients’ HRQoL.
While patients recruited in the FIGHT-202 trial were considered representative of patients in Canadian clinical practice, the clinical experts consulted by CADTH noted that it would be reasonable to generalize the results of cohort A of the FIGHT-202 trial to patients who are intolerant to first-line therapy, which was a group of patients excluded from the trial. As well, given the acceptable safety profile of pemigatinib, the clinical experts felt it would be reasonable to leave it up to the discretion of the treating physician to apply some flexibility in terms of using pemigatinib in patients with slightly lower laboratory parameters compared with those outlined in the trial. The clinical experts anticipated seeing the benefit of treatment with pemigatinib regardless of the number of previous lines of systemic therapy, as long as patients have the FGFR2 alteration. However, the clinical experts agreed that patients should not have been treated previously with an FGFR2-targeted therapy. Furthermore, the clinical experts noted that patients with iCCA and eCCA are managed in a similar way in clinical practice and that results observed in cohort A are generalizable to patients with FGFR2-positive eCCA, based on the fact that FGFR2 is the target of the mechanism of action of pemigatinib and there is no biologic rationale to assume that pemigatinib’s safety profile would be different in patients with eCCA.
Harms
The single-arm, non-randomized design of the FIGHT-202 trial made interpreting the safety events attributable to pemigatinib challenging, since all patients in cohort A received the same treatment. This should be considered when reviewing the incidence of TEAEs. All patients in cohort A experienced at least 1 TEAE. The most commonly reported TEAEs included alopecia, hyperphosphatemia, diarrhea, and dysgeusia. The most commonly reported grade 3 or higher and serious TEAEs, respectively, were hypophosphatemia and pyrexia. The clinical experts consulted by CADTH noted that most TEAEs associated with pemigatinib could be managed with dose modifications, and that treatment discontinuation due to TEAEs was relatively rare. From the review of notable harms, it appeared that toxicities from pemigatinib were mostly seen as hyperphosphatemia and nail toxicities. Overall, deaths were few and no TEAE leading to death was considered treatment-related. Overall, the clinical experts consulted by CADTH agreed with the registered clinicians who provided input to this submission that the TEAEs observed with pemigatinib were generally acceptable and could be adequately managed in clinical practice. This was reflective of the patient experience with pemigatinib reported in the patient input received, which stated that, overall, patients had little challenge dealing with the side effects of pemigatinib. Furthermore, it was emphasized by the clinical experts consulted by CADTH that the toxicity with pemigatinib appeared favourable compared with currently available chemotherapy options. Examples of side effects from chemotherapy that may be avoided with pemigatinib were neuropathy and neutropenia, according to the clinical experts.
Conclusions
One phase II, singe-arm, open-label trial (FIGHT-202) provided evidence regarding the efficacy and safety of pemigatinib in patients with advanced, metastatic, or surgically unresectable CCA with FGFR2 alterations (cohort A) whose disease did not respond to a previous therapy. The FIGHT-202 trial achieved the predetermined threshold for a positive outcome (lower limit of the 95% CI for ORR > 15%) in cohort A. The clinical experts consulted by CADTH felt that the achieved ORR of 37% (April, 7, 2020, data cut-off date) was clinically meaningful for the target population and durable (median of 8.08 months; range, 5.65 to 13.14). In the opinion of the clinical experts, the observed responses appeared higher than what is seen with the therapies currently used in the second line in this setting. There was uncertainty around the magnitude of the clinical benefit, given the limitations in the evidence from this non-comparative phase II clinical trial. The clinical experts consulted by CADTH noted that, despite the high unmet need, conducting an RCT with a targeted therapy, such as pemigatinib, compared with the currently available therapies used in the second line in Canadian clinical practice would likely not be feasible. While the secondary efficacy outcomes, OS and PFS, appeared supportive of the observed ORR achievements, the non-randomized design of the FIGHT-202 trial made interpreting the PFS and OS events attributable to pemigatinib challenging. In the absence of a direct comparison of pemigatinib with relevant treatment options, the sponsor submitted an ITC. However, the CADTH critical assessment identified limitations with the sponsor’s submitted unanchored MAIC (including heterogeneity across study designs and populations and the inability to adjust for all potential confounders and prognostic variables), which limited the ability to interpret the relative treatment effects observed between pemigatinib and other treatments. The results for the HRQoL and symptom severity exploratory outcomes remained inconclusive due to a number of important limitations. The toxicity profile of pemigatinib was considered manageable by the clinical experts consulted by CADTH and appeared favourable compared with currently available chemotherapy options. However, the non-comparative design of the FIGHT-202 trial made interpreting the safety events attributable to pemigatinib challenging, since all patients in cohort A received the same treatment.
References
1.Zhou X, Wang J, Tang M, et al. Hepatocellular carcinoma with hilar bile duct tumor thrombus versus hilar Cholangiocarcinoma on enhanced computed tomography: a diagnostic challenge. BMC Cancer. 2020;20(1):54. PubMed
2.Valle JW, Borbath I, Khan SA, Huguet F, Gruenberger T, Arnold D. Biliary cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up †. Ann Oncol. 2016;27:v28-v37. PubMed
3.NCCN clinical practice guidelines in Oncology (NCCN Guidelines). Hepatobiliary cancers. Version 3.2021, June 15, 2021. ACCESSED: August 10, 2021. 2021: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1438.
4.Meza-Junco J, Montano-Loza AJ, Ma M, Wong W, Sawyer MB, Bain VG. Cholangiocarcinoma: has there been any progress? Canadian journal of gastroenterology = Journal canadien de gastroenterologie. 2010;24(1):52-57.
5.Center for Drug Evaluation R. Multidiscipline review(s). Pemazyre (pemigatinib) administration. Company: Incyte Corporation. Application No.:213736Orig1s000. Approval date: 04/17/2020 (FDA approval package). Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2020: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/213736Orig1s000MultidisciplineR.pdf. Accessed 2021 Jul 8.
6.American Cancer Society. Survival Rates for Bile Duct Cancer ACCESSED: July 20, 2021. https://www.cancer.org/cancer/bile-duct-cancer/detection-diagnosis-staging/survival-by-stage.html.
7.NICE. National Institute for Health and Care Excellence. Single Technology Appraisal. Pemigatinib for treating relapsed or refractory advanced cholangiocarcinoma with FGFR2 alterations [ID3740]. Committee Papers. 2021: https://www.nice.org.uk/guidance/ta722/documents/committee-papers-3.
8.Amerian Cancer Society. Signs and Symptoms of Bile Duct Cancer. Accessed July 25, 2021. https://www.cancer.org/cancer/bile-duct-cancer/detection-diagnosis-staging/signs-symptoms.html.
9.European Medicines Agency. Assessment report Pemazyre International non-proprietary name: pemigatinib. Accessed August 5, 2021. 2021: https://www.ema.europa.eu/en/medicines/human/EPAR/pemazyre.
10.Lamarca A, Barriuso J, McNamara MG, Valle JW. Molecular targeted therapies: Ready for “prime time” in biliary tract cancer. J Hepatol. 2020;73(1):170-185. PubMed
11.Saborowski A, Lehmann U, Vogel A. FGFR inhibitors in cholangiocarcinoma: what's now and what's next? Ther Adv Med Oncol. 2020;12:1758835920953293-1758835920953293. PubMed
12.Drug Reimbursement Review sponsor submission: pemigatinib, 4.5 mg, 9 mg, and 13.5 mg, oral tablets [internal sponsor's package]. Pointe Claire (QC): Incyte Biosciences Canada Corporation; 2021 Jun 23.
13.Lamarca A, Palmer DH, Wasan HS, et al. Second-line FOLFOX chemotherapy versus active symptom control for advanced biliary tract cancer (ABC-06): a phase 3, open-label, randomised, controlled trial. Lancet Oncol. 2021;22(5):690-701. PubMed
14.National Library of Medicine. PubChem. Pemigatinib. Accessed September 8, 2021. https://pubchem.ncbi.nlm.nih.gov/compound/Pemigatinib.
15.PemazyreTM (pemigatinib tablets): 4.5 mg, 9 mg, and 13.5 mg, oral [product monograph]. Wilmington (DE): Incyte Corporation; 2021 Sep 8.
16.Clinical Study Report: INCB 54828-202. A phase 2, open-label, single-arm, multicenter study to evaluate the efficacy and safety of INCB054828 in subjects with advanced/metastatic or surgically unresectable cholangiocarcinoma including FGFR2 translocations who failed previous therapy (FIGHT-202) [internal sponsor's report]. Pointe Claire (QC): Incyte Corporation; 2019 Aug 12.
17.Incyte response to September 1, 2021 DRR request for additional information regarding pemigatinib DRR review [internal additional sponsor's information]. Pointe Claire (QC): Incyte Biosciences Canada; 2021 Sep 10.
18.De Luca A, Esposito Abate R, Rachiglio AM, et al. FGFR Fusions in Cancer: From Diagnostic Approaches to Therapeutic Intervention. Int J Mol Sci. 2020;21(18). PubMed
19.Jain A, Borad MJ, Kelley RK, et al. Cholangiocarcinoma With FGFR Genetic Aberrations: A Unique Clinical Phenotype. JCO Precision Oncology. 2018(2):1-12. PubMed
20.NIH. National Cancer Institute. Bile Duct Cancer (Cholangiocarcinoma) Treatment (PDQ®)–Patient Version. ACCESS July 10, 2021. https://www.cancer.gov/types/liver/patient/bile-duct-treatment-pdq#_1.
21.Bibeau K, Féliz L, Barrett S, Na L, Lihou CF, Asatiani E. Progression-free survival in patients with cholangiocarcinoma with FGFR2 fusions or rearrangements: An exploration of response to systemic therapy. J Clin Oncol. 2020;38(4_suppl):588-588.
22.Churi CR, Shroff R, Wang Y, et al. Mutation profiling in cholangiocarcinoma: prognostic and therapeutic implications. PLoS One. 2014;9(12):e115383. PubMed
23.Graham RP, Barr Fritcher EG, Pestova E, et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum Pathol. 2014;45(8):1630-1638. PubMed
24.FDA U.S. Food & Drug Administration. FDA grants accelerated approval to pemigatinib for cholangiocarcinoma with an FGFR2 rearrangement or fusion Accessed September 3, 2021. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pemigatinib-cholangiocarcinoma-fgfr2-rearrangement-or-fusion.
25.European Medicines Agency. EU/3/18/2066: pemigatinib. Accessed August 8. https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu3182066.
26.European Medicines Agency. ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS. Accessed: June 28, 2021. https://www.ema.europa.eu/en/documents/product-information/pemazyre-epar-product-information_en.pdf.
27.FDS U.S. Food & Drug Administration. FDA grants accelerated approval to infigratinib for metastatic cholangiocarcinoma. Accessed September 3, 2021.
28.ClinicalTrials.gov. A Study to Evaluate the Efficacy and Safety of Pemigatinib Versus Chemotherapy in Unresectable or Metastatic Cholangiocarcinoma (FIGHT-302).NCT03656536. Accessed August 20. 2021. https://www.clinicaltrials.gov/ct2/show/NCT03656536.
29.ClinicalTrials.gov. Phase 3 Study of BGJ398 (Oral Infigratinib) in First Line Cholangiocarcinoma With FGFR2 Gene Fusions/Translocations. NCT03773302. Accessed September 3, 2021. https://clinicaltrials.gov/ct2/show/NCT03773302.
30.ClinicalTrials.gov. Futibatinib Versus Gemcitabine-Cisplatin Chemotherapy as First-Line Treatment of Patients With Advanced Cholangiocarcinoma Harboring FGFR2 Gene Rearrangements (FOENIX-CCA3).NCT04093362. Accessed September 3, 2021. https://www.clinicaltrials.gov/ct2/show/NCT04093362.
31.Lamarca A, Hubner RA, David Ryder W, Valle JW. Second-line chemotherapy in advanced biliary cancer: a systematic review. Ann Oncol. 2014;25(12):2328-2338. PubMed
32.ClinicalTrials.gov.Study of AG-120 in Previously Treated Advanced Cholangiocarcinoma With IDH1 Mutations (ClarIDHy) (ClarIDHy).NCT02989857. Accessed September 3, 2021. https://clinicaltrials.gov/ct2/show/NCT02989857.
33.OneLive. FDA Grants Priority Review to Ivosidenib in IDH1-Mutant Cholangiocarcinoma Accessed September 8, 2021. 2021: https://www.onclive.com/view/fda-grants-priority-review-to-ivosidenib-in-idh1-mutant-cholangiocarcinoma.
34.Businesswire. Incyte Announces Approval of Pemazyre® (pemigatinib) in Japan for the Treatment of Patients with Unresectable Biliary Tract Cancer (BTC) with a Fibroblast Growth Factor Receptor 2 (FGFR2) Fusion Gene, Worsening After Cancer Chemotherapy. https://www.businesswire.com/news/home/20210323005530/en/Incyte-Announces-Approval-of-Pemazyre%C2%AE-pemigatinib-in-Japan-for-the-Treatment-of-Patients-with-Unresectable-Biliary-Tract-Cancer-BTC-with-a-Fibroblast-Growth-Factor-Receptor-2-FGFR2-Fusion-Gene-Worsening-After-Cancer-Chemotherapy. Accessed October 12, 2021.
35.GoDRUGBANK.Pemigatinib. Accessed: September 9, 2021. https://go.drugbank.com/drugs/DB15102.
36.Bekaii-Saab TS, Valle JW, Van Cutsem E, et al. FIGHT-302: phase III study of firstline (1L) pemigatinib (PEM) versus gemcitabine (GEM) plus cisplatin (CIS) for cholangiocarcinoma (CCA) with FGFR2 fusions or rearrangements. J Clin Oncol. 2020;38(4):TPS592-TPS592.
37.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 Guideline Statement. J Clin Epidemiol. 2016;75:40-46. PubMed
38.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: Accessed 2021 August 16.: https://www.cadth.ca/grey-matters.
39.Abou-Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 2020;21(5):671-684. PubMed
40.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228-247. PubMed
41.Kaupp-Roberts SD, Yadegarfar G, Friend E, et al. Validation of the EORTC QLQ-BIL21 questionnaire for measuring quality of life in patients with cholangiocarcinoma and cancer of the gallbladder. Br J Cancer. 2016;115(9):1032-1038. PubMed
42.Friend E, Yadegarfar G, Byrne C, et al. Development of a questionnaire (EORTC module) to measure quality of life in patients with cholangiocarcinoma and gallbladder cancer, the EORTC QLQ-BIL21. Br J Cancer. 2011;104(4):587-592. PubMed
43.Lamarca A, Palmer DH, Wasan HS, et al. ABC-06 | A randomised phase III, multi-centre, open-label study of active symptom control (ASC) alone or ASC with oxaliplatin / 5-FU chemotherapy (ASC+mFOLFOX) for patients (pts) with locally advanced / metastatic biliary tract cancers (ABC) previously-treated with cisplatin/gemcitabine (CisGem) chemotherapy. J Clin Oncol. 2019;37(15_suppl):4003-4003.
44.Lowery MA, Goff LW, Keenan BP, et al. Second-line chemotherapy in advanced biliary cancers: A retrospective, multicenter analysis of outcomes. Cancer. 2019;125(24):4426-4434. PubMed
45.Ying J, Chen J. Combination versus mono-therapy as salvage treatment for advanced biliary tract cancer: A comprehensive meta-analysis of published data. Crit Rev Oncol Hematol. 2019;139:134-142. PubMed
46.Brookmeyer RaC, J. A Confidence Interval for the Median Survival Time. Accessed August 10, 2021. International Biometric Society; 1982: https://www.jstor.org/stable/2530286.
47.Food and Drug Administration (FDA). Guidance for Industry: Clinical Trial Endpoints for the Approval of Non-Small Cell Lung Cancer Drugs and Biologics. Accessed: September 3, 2021 2015: https://www.fda.gov/downloads/drugs/guidances/ucm259421.pdf. Accessed April 12, 2019.
48.Food and Drug Administration (FDA). Guidance for Industry: Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics. 2018. Accessed September 10, 2019. 2018: https://www.fda.gov/downloads/Drugs/Guidances/ucm071590.pdf.
49.Valle JW, Bibeau K, Cho Y, et al. Longitudinal evaluation of quality of life (QoL) in patients (Pts) with FGFR2-driven cholangiocarcinoma (CCA) treated with pemigatinib. J Clin Oncol. 2021;39(3_suppl):276-276.
50.FDA. U.S. Food & Drug Administration. 22 case studies where phase 2 and phase 3 trials had divergent results. 2017: https://www.fda.gov/media/102332/download.
51.Pemigatinib 4.5 mg, 9 mg, 13.5 mg, oral tablets: Indirect and Mixed Treatment Comparison Report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: pemigatinib, 4.5 mg, 9 mg, 13.5 mg, oral tablets. Pointe Claire (QC): Incyte Biosciences Canada Corporation; 2021 Jun 23.
52.Incyte response to August 4, 2021 DRR request for additional information regarding pemigatinib DRR review [internal additional sponsor's information]. Pointe Claire (QC): Incyte Biosciences Canada; 2021 Aug 10.
53.Westin GFM, Alsidawi S, Chandrasekharan C, et al. Outcomes of second line treatment in patients with advanced and metastatic biliary cancers. J Clin Oncol. 2017;35(4_suppl):420-420.
54.Belkouz A, de Vos-Geelen J, Mathôt RAA, et al. Efficacy and safety of FOLFIRINOX as salvage treatment in advanced biliary tract cancer: an open-label, single arm, phase 2 trial. Br J Cancer. 2020;122(5):634-639. PubMed
55.Kim BJ, Yoo C, Kim KP, et al. Efficacy of fluoropyrimidine-based chemotherapy in patients with advanced biliary tract cancer after failure of gemcitabine plus cisplatin: retrospective analysis of 321 patients. Br J Cancer. 2017;116(5):561-567. PubMed
56.Bekaii-Saab TS, Bridgewater J, Normanno N. Practical considerations in screening for genetic alterations in cholangiocarcinoma. Ann Oncol. 2021;28:28. PubMed
57.Ji JH, Song HN, Kim RB, et al. Natural history of metastatic biliary tract cancer (BTC) patients with good performance status (PS) who were treated with only best supportive care (BSC). Jpn J Clin Oncol. 2015;45(3):256-260. PubMed
58.Reeve BB, Wyrwich KW, Wu AW, et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centered outcomes and comparative effectiveness research. Qual Life Res. 2013;22(8):1889-1905. PubMed
59.Giesinger JM, Kieffer JM, Fayers PM, et al. Replication and validation of higher order models demonstrated that a summary score for the EORTC QLQ-C30 is robust. J Clin Epidemiol. 2016;69:79-88. PubMed
60.Fayers PM, Aaronson NK, Bjordal K, et al. The EORTC QLC-C30 scoring manual (3rd Edition). Brussels (BE): European Organisation for Research and Treatment of Cancer (EORTC); 2001: https://www.eortc.org/app/uploads/sites/2/2018/02/SCmanual.pdf. Accessed 2021 Aug 12.
61.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-144. PubMed
62.King MT. The interpretation of scores from the EORTC quality of life questionnaire QLQ-C30. Qual Life Res. 1996;5(6):555-567. PubMed
63.Snyder CF, Blackford AL, Sussman J, et al. Identifying changes in scores on the EORTC-QLQ-C30 representing a change in patients' supportive care needs. Qual Life Res. 2015;24(5):1207-1216. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: July 19, 2021
Alerts: Biweekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type
Limits: Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ab | Abstract |
.dq | Candidate term word (Embase) |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase) |
.nm | Name of substance word (MEDLINE) |
.ot | Original title |
.pt | Publication type |
.rn | Registry number |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(pemigatinib* or Pemazyre* or INCB054828 or INCB-054828 or INCB54828 or INCB-54828 or ibi375 or ibi-375 or Y6BX7BL23K).ti,ab,kf,ot,hw,rn,nm.
(FIGHT-202* or FIGHT202* or FIGHT-302 or FIGHT302*).ti,ab,kf.
1 or 2
use medall
*pemigatinib/
(pemigatinib* or Pemazyre* or INCB054828 or INCB-054828 or INCB54828 or INCB-54828 or ibi375 or ibi-375).ti,ab,kw,dq.
(FIGHT-202* or FIGHT202* or FIGHT-302 or FIGHT302*).ti,ab,kw,dq.
5 or 6 or 7
use oemezd
not conference abstract.pt.
4 or 10
remove duplicates from 11
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search terms – pemigatinib, Pemazyre, or INCB054828]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
[Search terms – pemigatinib, Pemazyre, or INCB054828]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms – pemigatinib, Pemazyre, or INCB054828]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms – pemigatinib, Pemazyre, or INCB054828]
Grey Literature
Search dates: July 2 – 16, 2021
Keywords: pemigatinib, Pemazyre, INCB054828, INCB-054828, cholangiocarcinoma, bile duct cancer, biliary tract cancer
Limits: None
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search
Appendix 2: Excluded Studies
Reference | Reason for exclusion |
|---|---|
Bekaii-Saab TS, Valle JW, Van Cutsem E, et al. FIGHT-302: phase III study of firstline (1L) pemigatinib (PEM) versus gemcitabine (GEM) plus cisplatin (CIS) for cholangiocarcinoma (CCA) with FGFR2 fusions or rearrangements. J Clin Oncol. 2020;38(4):TPS592-TPS592.36 | Not relevant population. |
Note: This table has not been copy-edited.
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 29: Evaluation of Target Lesions
Response outcomes | Description |
|---|---|
Complete response | Disappearance of all non-nodal target lesions and any pathological lymph nodes must have become normal (i.e., decreases in short axis to < 10 mm). |
Partial response | At least a 30% decrease in sum of the diameters of target lesions, taking as preference the baseline SOD. Additionally, progression of target lesions must not be present (see Section 7.7.4). |
Stable disease | Neither sufficient shrinkage of target lesions to qualify for PR, nor sufficient increase to qualify for PD. |
Progressive disease | At least a 20% increase in the sum of diameters of target lesions, taking as reference the nadir sum of diameters (or the baseline, if the baseline is the nadir value). In addition to the relative increase of 20% in sum of diameters, the sum of diameters must also demonstrate an absolute increase of ≥ 5 mm. |
Not evaluable | If ≥ one target lesions is classified as NE for a particular time point, the sum of diameters and percent change cannot be accurately determined, and the target response will be NE for that time point. The only exception is if the sum of diameters of the evaluable target lesions show a ≥ 20% increase from the nadir sum of diameters and an absolute increase of ≥ 5 mm. In this case, the target response will be PD. |
Note: These definitions should be used to evaluate response based on target lesions at each time point after screening. A radiographic target response will be determined based on the radiographic target lesions identified during the radiology review.
Source: Sponsor’s response.17
Key Results for Cohorts B and C of the FIGHT-202 Trial
Cohort B and C were not part of the sponsor’s reimbursement request to CADTH.
Table 30: Summary of Overall Survival, Efficacy Evaluable Population (March 22, 2019, Data Cut-Off Date)
Variable | FIGHT-202 Trial | ||
|---|---|---|---|
Cohort A (N = 107) | Cohort B (N = 20) | Cohort C (N = 18) | |
Number (%) of subjects who died | 40 (37.4) | 16 (80.0) | 14 (77.8) |
Number (%) of subjects censored | 67 (62.6) | 4 (20.0) | 4 (22.2) |
Median overall survival (months) (95% CI)a | 21.06 (14.82 to NE) | 6.70 (2.10 to 10.55) | 4.02 (2.33 to 6.47) |
Kaplan-Meier estimates (95% CI) of overall survival of: | |||
3 months | 96.2 (90.3 to 98.6) | 67.4 (41.2 to 83.9) | 68.8 (40.5 to 85.6) |
6 months | 88.6 (80.8 to 93.4) | 50.5 (26.4 to 70.5) | 31.3 (11.4 to 53.6) |
9 months | 77.4 (68.0 to 84.4) | 33.7 (13.9 to 54.9) | 25.0 (7.8 to 47.2) |
12 months | 67.5 (56.4 to 76.3) | 22.5 (7.0 to 43.2) | 12.5 (2.1 to 32.8) |
CI = confidence interval; NE = not evaluable.
aThe 95% CI is calculated using the Brookmeyer and Crowley method (1982).
Source: Clinical Study Report.16
Figure 17: Kaplan-Meier Estimates of Overall Survival, Efficacy Evaluable Population (March 22, 2019, Data Cut-Off Date)
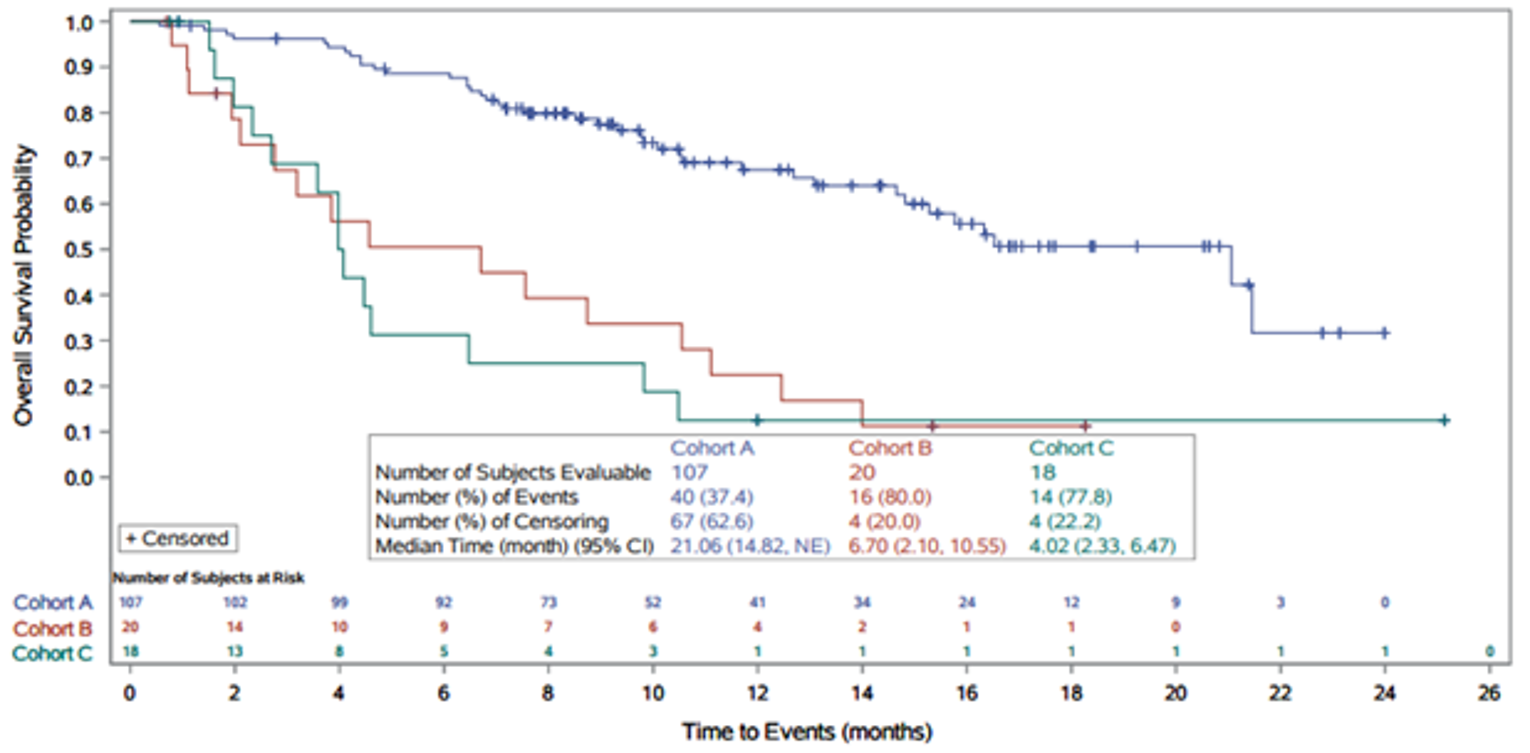
Source: Clinical Study Report.16
Table 31: Summary of PFS Based on IRC Assessment According to RECIST 1.1, Efficacy Evaluable Population (March 22, 2019, Data Cut-Off Date)
Variable | FIGHT-202 Trial | ||
|---|---|---|---|
Cohort A (N = 107) | Cohort B (N = 20) | Cohort C (N = 18) | |
Number (%) of participants with events | 71 (66.4) | 17 (85.0) | 16 (88.9) |
Disease progression | 63 (58.9) | 13 (65.0) | 12 (66.7) |
Death | 8 (7.5) | 4 (20.0) | 4 (22.2) |
Number (%) of participants censored | 36 (33.6) | 3 (15.0) | 2 (11.1) |
Median PFS (months) (95% CI) a | 6.93 (6.18 to 9.59) | 2.10 (1.18 to 4.86) | 1.68 (1.25 to 1.84) |
Kaplan-Meier estimates (95% CI) of PFS at: | |||
3 months | 78.9 (69.7 to 85.5) | 37.9 (16.3 to 59.5) | 12.7 (2.1 to 33.3) |
6 months | 61.7 (51.5 to 70.4) | 25.3 (8.1 to 47.1) | 6.4 (0.4 to 25.1) |
9 months | 45.3 (34.9 to 55.1) | 12.6 (2.1 to 32.9) | 0.0 (NE to NE) |
12 months | 29.2 (18.9 to 40.2) | 0.0 (NE to NE) | 0.0 (NE to NE) |
CI = confidence interval. IRC = independent review committee: NE = not evaluable; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
aThe 95% CI was calculated using the Brookmeyer and Crowley method (1982).
Source: Clinical Study Report.16
Figure 18: Kaplan-Meier Estimates of Progression-Free Survival Based on IRC Assessment According to RECIST 1.1, Efficacy Evaluable Population (March 22, 2019, Data Cut-Off Date)
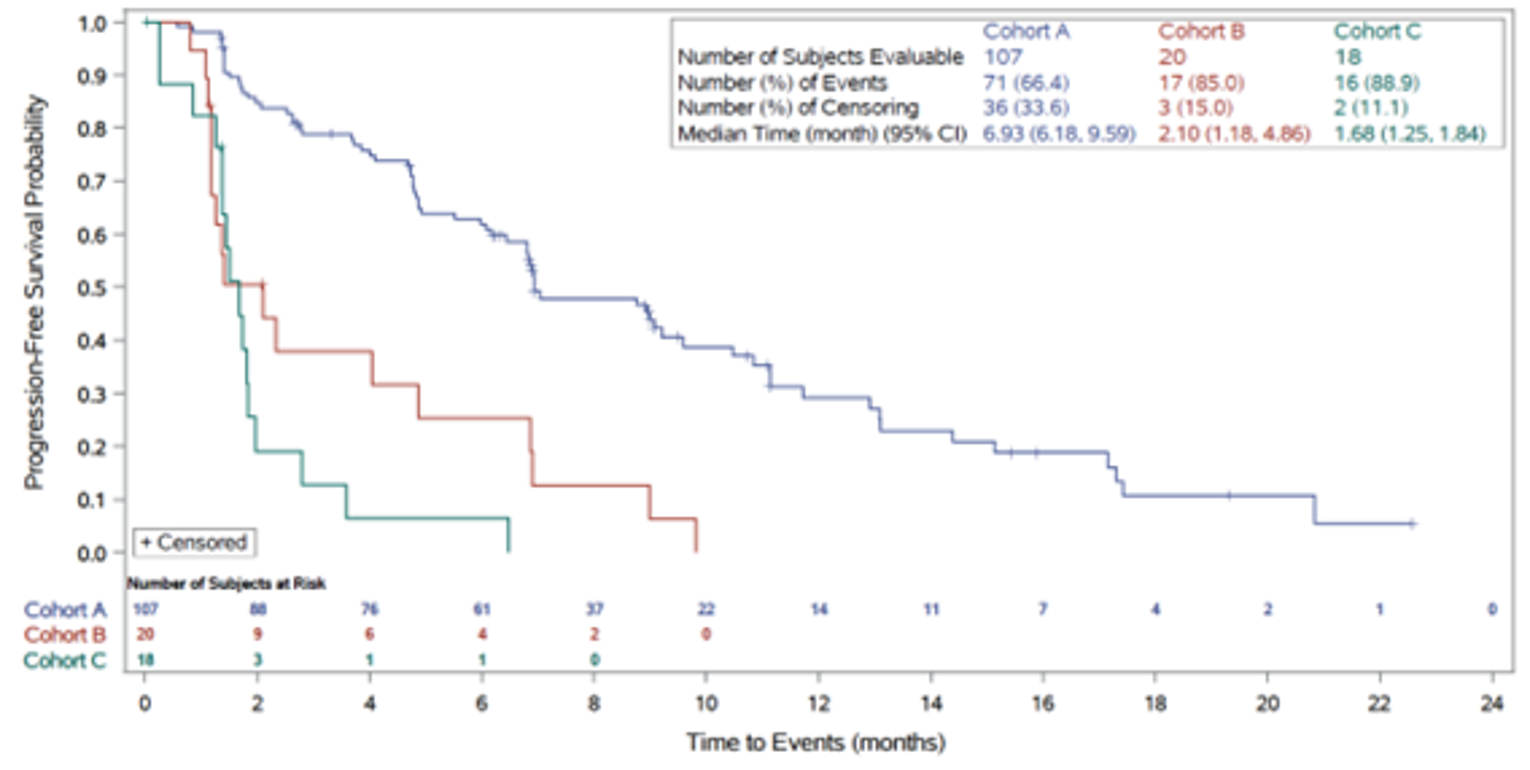
CI = confidence interval; IRC = independent review committee; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Source: Clinical Study Report.16
Table 32: Summary of Best Overall Response and Objective Response Rate Based on IRC Assessment According to RECIST 1.1, Efficacy Evaluable Population (March 22, 2019, Data Cut-Off Date)
Variable | FIGHT-202 Trial | ||
|---|---|---|---|
Cohort A + B (N = 127) | Cohort B (N = 20) | Cohort C (N = 18) | |
Objective responsea, n (%) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
95% CIb | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Best overall response, n (%) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Confirmed complete response | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Confirmed partial response | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Stable disease | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Progressive disease | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Not evaluablec | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
aParticipants who have a best overall response of complete response or partial response.
bThe CI was calculated based on the exact method for binomial distribution.
cPost-baseline tumour assessment was not performed due to study discontinuation (2 participants in cohort A, 4 participants in cohort B, 3 participants in cohort C) or was performed prior to the minimum interval of 39 days for an assessment of stable disease (one participant in cohort A, one participant in cohort B).
Source: Clinical Study Report.16
Duration of Response
Because there were no IRC-assessed confirmed tumour responses in cohorts B or C, DOR was not evaluable in those cohorts.
Table 33: Summary of Disease Control Rate Based on IRC Assessment According to RECIST 1.1, Efficacy Evaluable Population (March 22, 2019, Data Cut-Off Date)
Variable | FIGHT-202 Trial | ||
|---|---|---|---|
Cohort A (N = 107) | Cohort B (N = 20) | Cohort C (N = 18) | |
Disease control, n (%)a | 88 (82.2) | 8 (40.0) | 4 (22.2) |
95% CIb | 73.7, 89.0 | 19.1, 63.9 | 6.4, 47.6 |
Best response, n (%) | 8 (7.5) | 4 (20.0) | 4 (22.2) |
Confirmed complete | 3 (2.8) | 0 | 0 |
Confirmed partial | 35 (32.7) | 0 | 0 |
Stable disease ≥ 39 days | 50 (46.7) | 8 (40.0) | 4 (22.2) |
aParticipants who have a best overall response of complete response, partial response or stable disease with measurements that meet the stable disease criteria after the date of first dose at a minimum interval of 39 days.
b95% CI was calculated based on the exact method for binomial distribution.
Source: Clinical Study Report.16
Table 34: Treatment Pattern — Duration of Therapy by Treatment Line, All Enrolled Patients (April 7, 2020, Data Cut-Off Date)
Variable | FIGHT-202 Trial | |||
|---|---|---|---|---|
Any (N = 145) | Cohort A (N = 108) | Cohort B (N = 20) | Cohort C (N = 17) | |
Duration of the first line (days) | ||||
n | |||||||||| | |||||||||| | |||||||||| | |||||||||| |
Q1 | |||||||||| | |||||||||| | |||||||||| | |||||||||| |
Median | |||||||||| | |||||||||| | |||||||||| | |||||||||| |
Q3 | |||||||||| | |||||||||| | |||||||||| | |||||||||| |
Minimum–maximum | |||||||||| | |||||||||| | |||||||||| | |||||||||| |
Gemcitabine plus | ||||
n | 97 | 72 | 12 | 13 |
Q1 | 64.0 | 75.5 | 56.5 | 50.0 |
Median | 152.0 | 155.0 | 109.5 | 62.0 |
Q3 | 260.0 | 279.0 | 178.5 | 314.0 |
Minimum, maximum | 24, 1,161 | 29, 1,161 | 29, 196 | 24, 832 |
Not gemcitabine plus | ||||
n | 41 | 32 | 7 | 2 |
Q1 | 43.0 | 43.0 | 93.0 | 41.0 |
Median | 88.0 | 73.0 | 124.0 | 91.0 |
Q3 | 126.0 | 123.0 | 218.0 | 141.0 |
Minimum, maximum | 28, 658 | 28, 658 | 31, 400 | 41, 141 |
Note: treatment duration was calculated as (treatment end date-treatment start date +1). Missing treatment dates were imputed based on the rules described in the SAP. Treatment lines with dates unable to be imputed were removed from the calculation.
Sponsor’s response.17
Figure 19: Mean Curve of EORTC QLQ-BIL21 Over Time — Jaundice, Efficacy Evaluable Population, Cohorts A, B, and C (April 7, 2020, Data Cut-Off Date) (Redacted)

Note: This figure has been redacted at the sponsor’s request.
||||||||||||||||||||||||||||||||||||||||17
Figure 20: Mean Percent Change From Baseline of EORTC QLQ-BIL21 Over Time — Jaundice, Efficacy Evaluable Population, Cohorts A, B, and C (April 7, 2020, Data Cut-Off Date) (Redacted)

Note: This figure has been redacted at the sponsor’s request.
||||||||||||||||||||||||||||||||||||||||17
Figure 21: Mean Curve of EORTC QLQ-BIL21 Over Time — Tiredness, Efficacy Evaluable Population, Cohorts A, B, and C (April 7, 2020, Data Cut-Off Date) (Redacted)

Note: This figure has been redacted at the sponsor’s request.
||||||||||||||||||||||||||||||||||||||||17
Figure 22: Mean Percent Change From Baseline of EORTC QLQ-BIL21 Over Time — Tiredness, Efficacy Evaluable Population, Cohorts A, B, and C (April 7, 2020, Data Cut-Off Date) (Redacted)

Note: This figure has been redacted at the sponsor’s request.
||||||||||||||||||||||||||||||||||||||||17
Figure 23: Mean Curve of EORTC QLQ-BIL21 Over Time ‑— Anxiety, Efficacy Evaluable Population, Cohorts A, B, and C (April 7, 2020, Data Cut-Off Date) (Redacted)

Note: This figure has been redacted at the sponsor’s request.
||||||||||||||||||||||||||||||||||||||||17
Figure 24: Mean Percent Change from Baseline of EORTC QLQ-BIL21 Over Time ‑— Anxiety, Efficacy Evaluable Population, Cohorts A, B, and C (April 7, 2020, Data Cut-Off Date) (Redacted)

Note: This figure has been redacted at the sponsor’s request.
||||||||||||||||||||||||||||||||||||||||17
Figure 25: Mean Curve of EORTC QLQ-BIL21 Over Time — Treatment Side Effects, Efficacy Evaluable Population, Cohorts A, B, and C (April 7, 2020, Data Cut-Off Date) (Redacted)

Note: This figure has been redacted at the sponsor’s request.
||||||||||||||||||||||||||||||||||||||||17
Figure 26: Mean Percent Change From Baseline of EORTC QLQ-BIL21 Over Time – Treatment Side Effects, Efficacy Evaluable Population, Cohorts A, B, and C (April 7, 2020, Data Cut-Off Date) (Redacted)

Note: This figure has been redacted at the sponsor’s request.
||||||||||||||||||||||||||||||||||||||||17
Figure 27: Mean Curve of EORTC QLQ-BIL21 Over Time — Drains, Efficacy Evaluable Population, Cohorts A, B, and C (April 7, 2020, Data Cut-Off Date) (Redacted)

Note: This figure has been redacted at the sponsor’s request.
||||||||||||||||||||||||||||||||||||||||17
Figure 28: Mean Percent Change From Baseline of EORTC QLQ-BIL21 Over Time — Drains, Efficacy Evaluable Population, Cohorts A, B, and C (April 7, 2020, Data Cut-Off Date) (Redacted)

Note: This figure has been redacted at the sponsor’s request.
||||||||||||||||||||||||||||||||||||||||17


Appendix 4: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
EORTC QLQ-BIL21, an exploratory efficacy outcome in the FIGHT-202 study
EORTC QLQ-C30, an exploratory efficacy outcome in the FIGHT-202 study
Findings
Table 35: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-BIL21 | A 21-item, patient-reported, cholangiocarcinoma and gallbladder cancer–specific, quality of life questionnaire using 4-point Likert scales. It includes 5 multi-item scales (eating symptoms, jaundice symptoms, tiredness, pain symptoms, and anxiety symptoms) and 3 single-item scales (treatment side effects, difficulties with drainage bags/tubes, and concerns about weight loss). | Validity: Evidence for construct validity was found for all scales except for the jaundice and weight loss scales. Reliability: All multi-item scales have been shown to have acceptable internal consistency. All scales have been shown to have acceptable test–retest reliability. Responsiveness: There is some evidence for responsiveness to change in the eating, jaundice, tiredness, pain, treatment side effects, and anxiety scales. | Not identified |
EORTC QLQ-C30 | A 30-item, patient-reported, cancer-specific, quality of life questionnaire using 4- and 7-point Likert scales. It consists of 5 multi-item functional scales (physical, role, emotional, cognitive, and social), 3 multi-item symptom scales (fatigue, nausea-vomiting, and pain), 6 single-item symptom scales (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial impact), and a 2-item global quality of life scale. | Validity: No evidence for validity was found in patients with cholangiocarcinoma or biliary tract cancer. Reliability: Acceptable internal consistency was found for all scales except for the physical function, cognitive function, and nausea/vomiting scales, which had mixed results. Results for test–retest reliability were also mixed, with intraclass correlation coefficients for the scales ranging from 0.52 to 0.92. Responsiveness: No evidence for responsiveness to change was found in patients with cholangiocarcinoma or biliary tract cancer. | Not identified for patients with cholangiocarcinoma or biliary tract cancer. Estimates of MIDs for the global quality of life scale in patients with breast, colorectal, and small-cell lung cancer ranged from 10 to 19 points for improvement. |
EORTC = European Organisation for Research and Treatment of Cancer; MID = minimal important difference; QLQ-BIL21 = Quality of Life Questionnaire Cholangiocarcinoma and Gallbladder Cancer Module 21; QLQ-C30 = Quality of Life Questionnaire Core 30.
EORTC QLQ-BIL21
The EORTC QLQ-BIL21 is a disease-specific module to be used in addition to the EORTC QLQ-C30 to assess HRQoL in patients with CCA and gallbladder cancer.42 It consists of 21 questions, with 18 of the items grouped into 5 scales: eating symptoms (4 items), jaundice symptoms (3 items), tiredness (3 items), pain symptoms (4 items), and anxiety symptoms (4 items).42 The remaining 3 items are single-item assessments of treatment side effects, difficulties with drainage bags/tubes, and concerns about weight loss.42 Patients complete the questionnaire based on a 1-week recall period by rating each item on a 4-point Likert scale (1 = not at all; 2 = a little; 3 = quite a bit; 4 = very much).42 The scores are then transformed linearly to a 0 to 100 scale to yield scale scores using EORTC guidelines, with higher scores indicating more severe symptoms.41,42 The questions have been translated according to QoL group guidelines into Mandarin Chinese, Italian, German, Dutch, Spanish, and Hindi.41,42
During development of the EORTC QLQ-BIL21,42 52 patients rated relevance of each item in a provisional version of the test. Final item selection was based on these results and interviews with the patients. An international study was conducted to validate the EORTC QLQ-BIL21 in patients with BTC.41 The study included 172 adult patients with CCA and 91 patients with gallbladder cancer who had an expected minimum survival of 3 months and were undergoing treatment.41 Patients completed the EORTC QLQ-C30 and QLQ-BIL21 and KPS was recorded 1 month or less before treatment and again 2 months later. The total number of questionnaires available was 478.41 Patients were assigned to 1 of 3 groups based on which of the following treatments they were to receive: surgical treatment; chemotherapy, radiotherapy, photodynamic therapy, or laser therapy; or supportive care only. Prior stents or drains and prior active treatment (provided residual effects had resolved) were permitted for all treatment groups. Patients who received IV chemotherapy at the 2-month assessment were excluded from the test–retest reliability assessment.
Internal consistency was acceptable (alpha ≥ 0.7058) for all multi-item scales of the EORTC QLQ-BIL21 at each time point and when the baseline and 2-month time point results were pooled.41 Test–retest reliability was assessed for 67 patients who completed the questionnaire again 2 weeks after the 2-month assessment using the ICC and was found to be acceptable (ICC ≥ 0.7058) for all scales.41
Construct validity was assessed using the known groups approach by comparing baseline EORTC QLQ-BIL21 scores for patients with a KPS of less than 70 and greater than 70 at baseline (N = 238 to 256 for each scale).41 Patients with a KPS of less than 70 had a significantly greater EORTC QLQ-BIL21 score than patients with a KPS of greater than 70 (P < 0.05) for all scales, except for the jaundice and weight loss scales.41
Responsiveness of the scales over time was assessed by comparing scores between the baseline and 2-month follow-up assessments (N = 154 to 178).41 However, responsiveness was not assessed using an anchor, definition of response, or responsiveness statistics. With the 3 treatment groups pooled together, P value was less than 0.05 for the mean change in the eating, jaundice, tiredness, pain, and treatment side effects scales.41 There was worsening in the eating, tiredness, and treatment side effects scales and improvement in the jaundice and pain scales.41 Patients with surgical treatment had improvement (P < 0.05) in the jaundice scale and worsening in the tiredness scale, patients with medical therapies had improvement in the jaundice, pain, and anxiety scales and worsening in treatment side effects scale, and patients with supportive care only had worsening in the tiredness scale.41
The single-item assessment of difficulties with drainage bags and tubes was considered irrelevant by 29 patients.41 The study authors noted that not all patients experience drains during their treatment and that perhaps there should be a “not applicable” option for responding to that item.41
Estimates of MIDs were not found for the EORTC QLQ-BIL21.
EORTC QLQ-C30
The QLQ-C30 is one of the most commonly used patient-reported outcomes measures in oncology clinical trials. It is a multi-dimensional, cancer-specific, evaluative measure of HRQoL. It was designed specifically for the purpose of assessing changes in participants’ HRQoL in clinical trials, in response to treatment.59 The core questionnaire of the EORTC QLQ-C30 consists of 30 questions that are scored to create 5 multi-item functional scales (physical [5 items], role [2 items], emotional [4 items], cognitive [2 items], and social [2 items] functioning), 3 multi-item symptom scales (fatigue [3 items], nausea/vomiting [2 items], and pain [2 items]), 6 single-item symptom scales (dyspnea, insomnia, appetite loss, constipation, diarrhea and financial impact), and a 2-item QoL scale.
The EORTC QLQ-C30 uses a 1-week recall period in assessing function and symptoms. Most questions have 4 response options (1 = not at all, 2 = a little, 3 = quite a bit, 4 = very much). For the 2 items that form the global QoL scale, the response format is a 7-point Likert-type scale, with anchors between 1 (very poor) and 7 (excellent).60
Raw scores for each scale are computed as the average of the items that contribute to a particular scale. This scaling approach is based upon the assumption that it is appropriate to provide equal weighting to each item that comprises a scale. There is also an assumption that, for each item, the interval between response options is equal (for example, the difference in score between “not at all” and “a little” is the same as “a little” and “quite a bit,” at a value of 1 unit). Each raw scale score is converted to a standardized score that ranges from 0 to 100 using a linear transformation, with a higher score reflecting better function on the function scales, higher symptoms on the symptom scales, and better QoL (i.e., higher scores simply reflect higher levels of response on that scale). Thus, a decline in score on the symptom scale would reflect an improvement, whereas an increase in score on the function and QoL scale would reflect an improvement.60 According to the EORTC QLQ-C30 scoring algorithm, if there are missing items for a scale (i.e., the participant did not provide a response), the score for the scale can still be computed if there are responses for at least one-half of the items. In calculating the scale score, the missing items are simply ignored, an approach that assumes that the missing items have values equal to the average of those items for what the respondent completed.
Reliability of the EORTC QLQ-C30 was evaluated in the validation study for the EORTC QLQ-BIL21.41 Patients completed the EORTC QLQ-C30 ≤ 1 month before treatment and again 2 months later. Internal consistency was acceptable (alpha ≥ 0.7058) for all scales of the EORTC QLQ-C30 at each time point and when the baseline and 2-month time point results were pooled, except for the physical function (alpha = 0.47), cognitive function (alpha = 0.65), and nausea/vomiting (alpha = 0.67) scales at baseline.41 Test–retest reliability was assessed for 67 patients who completed the questionnaire again 2 weeks after the 2-month assessment and ICCs ranged from 0.52 to 0.92 with no further detail provided.41 Construct validity and responsiveness were not assessed for the QLQ-C30.41 Estimates for MIDs in the literature were not found for the EORTC QLQ-C30 in patients with CCA or BTC.
MIDs for the EORTC QLQ-C30 have been estimated in patients with other types of cancer. One study by Osoba et al.61 was based on a sample of patients with breast and small-cell lung cancer. An anchor-based approach was taken, using global ratings of change measured by a subjective significance questionnaire as an anchor. The mean change in scores for patients who indicated a small difference, either positive or negative, was 5 to 10 points.61 A “moderate” change reported by patients had corresponding changes of about 10 to 20.61 Similar findings were reported in a study by King, et al.62 In the study by Osoba et al.,61 mean change in the global QoL scale in patients who felt “a little better” following the initiation of chemotherapy was approximately 12 (as estimated from a column graph) for patients with breast cancer and 18.1 for patients with small-cell lung cancer. Results in patients who felt “a little worse” were inconsistent as there was improvement in the global QoL scale for small-cell lung cancer and worsening for breast cancer.61 Of note, these MID estimates may not be appropriate for all cancer patients and are based on previous versions of the questionnaire.
More recently in 2015, a Canadian study estimated the MIDs of EORTC QLQ-C30 scales using data from 193 newly diagnosed breast and colorectal cancer patients undergoing surgery.63 The Supportive Care Needs Survey-Short Form-34 (SCNS-SF34) and the EORTC QLQ-C30 were collected at baseline (2 to 7 days post discharge from surgery), 3 weeks, and 8 weeks.63 Individual items determined by the study authors to be relevant to the EORTC QLQ-C30 physical function, role function, emotional function, global health, pain, and fatigue scales were used as anchors.63 Mean changes in these 6 EORTC QLQ-C30 scales associated with improvement, worsening, and no change in the corresponding SCNS-SF34 items were then calculated.63 For improvement, the following statistically significant mean changes were found in patients with improvement from “some unmet need” (low, moderate, and high need responses pooled together) to “not applicable” (patient not experiencing the issue): 17.3 for physical function, 32.3 for role function, 16.7 for emotional function, 20.1 for global health, −31.0 for pain, and −25.9 for fatigue.63 The following statistically significant mean changes were found in patients with improvement from “Some Unmet Need” to “Satisfied” (issue applies but is being adequately addressed): 15.2 for physical function, 18.1 for role function, 9.8 for global health, −28.6 for pain, and −19.0 for fatigue.63 Results for worsening were mixed as some scales (particularly physical function) did not change in the hypothesized direction and most changes were not statistically significant. For worsening, the following statistically significant mean changes were found in patients with worsening from “not applicable” to “some unmet need”: −21.2 for emotional function, −13.4 for global health, and 20.8 for fatigue.63 For worsening, patients with worsening from “Satisfied” to “Some Unmet Need” had a statistically significant mean change of −9.3 for emotional function.63 Mean change was statistically significant and in the direction opposite to the hypothesized direction for role function.63 The results should be interpreted with caution, given the limited sample sizes (ranging from 8 to 58) for each estimate, the unknown validity of using single items of the SCNS-SF34 as anchors, and the patient population being composed of patients with breast or colorectal cancer only.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
ASC
active symptom control
CCA
cholangiocarcinoma
FGFR2
fibroblast growth factor receptor 2
FOLFIRI
folinic acid, fluorouracil, and irinotecan hydrochloride
FOLFOX
folinic acid, fluorouracil, and oxaliplatin
HR
hazard ratio
ICER
incremental cost-effectiveness ratio
MAIC
matching-adjusted indirect comparison
mFOLFOX
modified folinic acid, fluorouracil, and oxaliplatin
NGS
next-generation sequencing
OS
overall survival
PFS
progression-free survival
RDI
relative dose intensity
QALY
quality-adjusted life-year
ToT
time on treatment
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Pemigatinib (Pemazyre), tablet |
Submitted price | Pemigatinib, $830.30 per 4.5 mg, 9 mg, or 13.5 mg tablets |
Indication | Proposed: For the treatment of adults with previously treated, unresectable, locally advanced, or metastatic cholangiocarcinoma with a fibroblast growth factor 2 (FGFR2) fusion or other rearrangement |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | September 8, 2021 |
Reimbursement request | As per indication |
Sponsor | Incyte Biosciences Canada Corporation |
Submission history | No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-effectiveness analysis Partitioned survival model |
Target population | Adult patients with previously treated, unresectable, locally advanced, or metastatic CCA with an FGFR2 fusion or rearrangement, aligned with the proposed Health Canada indication |
Treatment | Pemigatinib |
Comparators | ASC alone (consisting of treatments that include biliary drainage, antibiotics, analgesia, steroids, and antiemetics, as well as palliative radiotherapy and blood transfusions) mFOLFOX plus ASC |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (20 years) |
Key data sources | FIGHT-202 trial, a phase II, open-label, single-arm, multinational trial (pemigatinib) and sponsor-conducted MAIC (mFOLFOX plus ASC and ASC alone) |
Submitted results | Sequential analyses:
|
Key limitations |
|
CADTH reanalysis results |
|
ASC = active symptom control; CCA = cholangiocarcinoma; FGFR2 = fibroblast growth factor receptor 2; ICER = incremental cost-effectiveness ratio; LY = life-year; MAIC = matching-adjusted indirect comparison; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; OS = overall survival; PFS = progression-free survival; PSM = partitioned survival model; QALY = quality-adjusted life-year.
Conclusions
The CADTH clinical review found that, given the absence of robust comparative data on progression-free survival (PFS) and overall survival (OS), the ability to interpret the relative treatment effects observed between pemigatinib and folinic acid, fluorouracil, and oxaliplatin (FOLFOX) plus active symptom control (ASC) versus ASC alone was limited, and no firm conclusions could be drawn on how pemigatinib compared with other relevant treatment options.
Given the high degree of uncertainty concerning the magnitude of clinical benefit, CADTH was unable to perform a base-case analysis. The reanalysis performed by CADTH utilizes more appropriate assumptions, but these estimates are highly uncertain.
CADTH undertook reanalyses to address limitations relating to: the incorporation of MAIC-derived comparative efficacy estimates into the sponsor’s analysis, long-term extrapolations for pemigatinib PFS and OS, selecting comparator extrapolations for PFS and OS, the assumption that utility values vary by whether patients are on or off treatment, genetic testing costs, RDI, and mFOLFOX costs.
Based on the CADTH reanalysis, the incremental cost-effectiveness ratio (ICER) for pemigatinib relative to ASC and mFOLFOX was estimated to be $252,718 and $261,226 per quality-adjusted life-year (QALY) gained, respectively. A sequential analysis could not be performed due to the efficacy of pemigatinib being contingent on whether data were matched to the ASC arm or ASC plus mFOLFOX arm of the ABC-06 trial. At these ICERs, at least a 95% to 100% reduction in the price of pemigatinib is required for pemigatinib to achieve an ICER of $50,000 per QALY gained compared with mFOLFOX and ASC, respectively. The reason a price reduction of 100% would be required is due to the high cost of testing, estimated to be $38,000 to identify a single patient eligible for treatment with pemigatinib. If testing costs were $0 then, to be cost-effective relative to ASC, a 77% price reduction is needed, or a 72% price reduction versus FOLFOX.
The uncertainty in the comparative efficacy data for pemigatinib meant that the magnitude of benefit associated with pemigatinib compared with ASC and FOLFOX could not be reliably determined. Consequently, CADTH was unable to determine a base-case estimate regarding pemigatinib’s cost-effectiveness. Instead, CADTH conducted an exploratory reanalysis on the sponsor’s base case. According to the clinical experts consulted by CADTH for this review, pemigatinib could be equal to or better than alternative treatments currently received by patients. The price reductions noted by CADTH assume substantially improved efficacy with pemigatinib, which is highly uncertain.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Three groups collaborated for a single patient input submission: 2 Canadian patient groups, the Canadian Liver Foundation and Canadian Organization for Rare Disorders, and 1 international organization, the Cholangiocarcinoma Foundation. Patient input was gathered through an online survey and through a virtual focus group with 3 Canadian participants that included patients and caregivers affected by bile duct cancers, including those with fibroblast growth factor receptor 2 (FGFR2) gene fusions or rearrangements. Twenty-seven respondents completed the entire survey, 12 of whom identified as Canadian. A total of 15% of respondents had been diagnosed with FGFR2 fusions. Patients reported that the experience of cholangiocarcinoma (CCA) influenced their overall quality of life, with fatigue being noted as the most problematic and common symptom, followed by anxiety. Other concerning symptoms reported included unintended weight loss, insomnia, and gastrointestinal problems. Among the 74% of patients who had received treatment, all had received chemotherapy and most indicated that the side effects were worth the benefits, which were noted to include reduced pain and increased OS. In terms of hopes for improved outcomes, patients noted there is a lack of treatment options, and that quality of life is valued as much or more than quantity. One Canadian survey respondent and 2 focus group participants had received pemigatinib. Patients who received that drug noted that dose adjustments were required due to side effects (including hair loss, headaches, diarrhea, and sore joints) but found that with these changes, the drug was tolerable. Patients receiving pemigatinib hoped for a reduction in nodule size, no new growth and stability in the tumour, and remission.
Registered clinician input was received from 2 groups, the Ontario Health Gastrointestinal Cancer Drug Advisory Committee and the Canadian Gastrointestinal Oncology Evidence Network (CGOEN) plus other CCA-treating physicians. The clinician input noted that the current care pathway for patients with unresectable disease includes cisplatin plus gemcitabine for first-line therapy. Second-line treatments include mFOLFOX; folinic acid, fluorouracil, and irinotecan hydrochloride (FOLFIRI); and capecitabine. Clinicians reported that prolonging survival, delaying disease progression and maintaining quality of life, reducing symptom severity, and minimizing AEs are desired outcomes for a new treatment. Clinicians noted that patients would first need to be treated with a standard of care first-line therapy before receiving pemigatinib, in line with pemigatinib’s position in the FIGHT-202 trial, meaning that pemigatinib would be used as a later or last line of treatment. CGOEN noted that FGFR2 testing is required to identify eligible patients but that there is no current publicly funded mechanism for this testing in Canada and such testing is not currently routine.
The drug plan input noted there is no standard of care for patients upon progression after first-line therapy, but that second-line options included mFOLFOX or FOLFIRI, capecitabine, or best supportive care. The drug plan input considered whether patients who were currently receiving second-line treatment (e.g., mFOLFOX) should switch to pemigatinib, or whether pemigatinib should be used for subsequent therapy. The drug plans also noted that genetic testing for FGFR2 is not routinely available and asked what the best timing would be for testing of mutation status.
Several of the drug plans’ concerns were addressed in the sponsor’s model:
OS and the health-state utilities capturing symptoms were included
AEs associated with pemigatinib and comparators were included in the pharmacoeconomic analysis
genetic testing was included in the sponsor’s analysis.
In addition, CADTH addressed some of these concerns as follows:
Genetic testing was assumed not to occur in patients who are not receiving pemigatinib, given that genetic testing is not covered in a world without pemigatinib.
Economic Review
The current review is for pemigatinib for adult patients with previously treated, unresectable, locally advanced, or metastatic CCA with an FGFR2 fusion or rearrangement.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of pemigatinib compared with ASC alone and mFOLFOX plus ASC. The model population comprised adult patients with previously treated, unresectable, locally advanced, or metastatic CCA with an FGFR2 fusion or rearrangement, which was aligned with the proposed Health Canada indication.
Pemigatinib is available as a 4.5 mg, 9 mg, or 13.5 mg tablet. The recommended dose of pemigatinib is 13.5 mg orally once daily for 14 consecutive days followed by 7 days off therapy, in 21-day cycles. At the sponsor’s submitted price of $830.2987 per 13.5 mg tablet, the 28-day cycle cost of pemigatinib is $15,499, or $202,039 annually if patients remained on therapy for a full year (assuming a total of 17.4 21-day cycles annually). No drug-acquisition costs were modelled for ASC, which could consist of biliary drainage, antibiotics, analgesia, steroids, and antiemetics as well as palliative radiotherapy and blood transfusions.1 The cost of mFOLFOX used by the sponsor in the model was $3,333 per 28-day cycle. A 24-week stopping rule was applied for mFOLFOX, in line with its use in the ABC-06 study. No wastage was assumed in the model.
The clinical outcomes of interest were QALYs and life-years. The economic analysis was undertaken over a lifetime (20-year) time horizon from the perspective of a Canadian public health care payer. Discounting (1.5% per annum) was applied to both costs and outcomes.
Model Structure
The sponsor submitted a partitioned survival model with 5 health states: progression-free on treatment, progression-free off treatment, progressed disease on treatment, progressed disease off treatment, and death (Figure 1). All patients entered the model in the progression-free on-treatment state. Because patients enter the model progression-free after having received at least 1 line of prior therapy, “progression-free” in the model refers to disease progression during or after receiving pemigatinib or a comparator. The proportion of people with progression-free and progressed disease was first determined by fitting survival curves to unadjusted PFS and OS data from cohort A of the FIGHT-202 study. The proportion of pemigatinib patients who remained on therapy over time was determined by fitting survival curves to time on treatment (ToT) data from cohort A of the FIGHT-202 trial. As ToT was always less than PFS, the default in the model was such that the proportion of patients with progressed disease on treatment was always 0.
Model Inputs
The model’s baseline population characteristics and clinical efficacy parameters for pemigatinib were characterized by the planned subgroup (cohort A) of the FIGHT-202 study. The FIGHT-202 study was a phase II, open-label, single-arm study to evaluate the efficacy and safety of pemigatinib in patients with previously treated, locally advanced, or metastatic CCA with and without FGFR2 fusions or rearrangements. Cohort A of the FIGHT-202 trial consisted of patients with FGFR2 fusions or rearrangements. The sponsor assumed that the baseline patient characteristics of cohort A of FIGHT-202 (mean age = 55 years; proportion males = 39%; body surface area = 1.88 m2) reflected the Canadian population. Mean age and gender distribution were used to adjust the general population mortality data, sourced from Statistics Canada, to match the demographics of cohort A.
PFS, OS, and ToT curves for pemigatinib were generated using unadjusted data from cohort A of the FIGHT-202 study. Extrapolation curves were selected based on clinical plausibility and visual and statistical fit to the trial’s Kaplan–Meier data. Figure 2 and Figure 4 present the observed and predicted OS and PFS for pemigatinib, respectively. Comparator survival data were informed by relative treatment effects derived from the sponsor-conducted MAIC study. The MAIC considered patient-level data from FIGHT-202 for pemigatinib matched to aggregate data from ABC-06, a randomized, phase III, open-label study comparing mFOLFOX plus ASC with ASC alone in patients with all types of biliary tract cancers. The resulting weighted hazard ratios (HRs) for OS for pemigatinib compared with mFOLFOX plus ASC and ASC alone were applied to the sponsor’s selected survival curve for pemigatinib OS to derive comparator OS (Figure 3). As no PFS data for ASC alone were reported in ABC-06, an HR for PFS for ASC alone was not derived. Instead, it was assumed that PFS for ASC alone was equal to that of mFOLFOX plus ASC. Therefore, the MAIC-derived HR for PFS for mFOLFOX plus ASC was applied to the sponsor’s selected survival curve for pemigatinib and used to derive PFS for both mFOLFOX plus ASC and ASC alone (Figure 5). While ToT was modelled using a survival curve for pemigatinib (Figure 6), for comparators, ToT was assumed to be equal to PFS. Grade 3 or greater adverse events (AEs) were included if they occurred in 5% or more of patients for any comparator, and were naively derived from FIGHT-202 for pemigatinib and ABC-06 for mFOLFOX plus ASC and ASC alone.
Health-state utility values were derived from cohort A of the FIGHT-202 study. European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30 data were mapped to EuroQol 5-Dimensions 3-Levels Questionnaire utilities, with a UK tariff applied.2 Utilities were age-adjusted by applying a multiplier to the health-state utility value. An IV medication administration disutility of 0.025 was applied to patients receiving mFOLFOX plus ASC while patients were on treatment.3 Disutility and durations of AEs sourced from the literature were applied based on their frequency. In the absence of data to inform AE disutility, assumptions based on clinical expert opinion were used to estimate the disutility associated with the AE.
Costs in the model included the cost of treatment acquisition, drug administration, health care resource use, and AE costs. Dose interruptions for pemigatinib were adjusted by calculating the percentage of doses received as a proportion of the expected number of doses without any interruptions. To adjust for patients taking pemigatinib for 2 weeks then not taking it for a week, in the model, pemigatinib weekly costs were adjusted by averaging the number of days per week they would be taking medication (4.67 days). Costs of subsequent therapies upon progression were not included in the model. Pain medication was included for patients in the progressed-disease health states. A cost per administration of mFOLFOX was derived based on chair time and a nurse visit to discontinue infusion. Health care resource use included medical oncologist visits, CT scans, and blood tests using costs from the Ontario Schedule of Benefits and Fees and the Schedule of Benefits for Laboratory Services.4,5 Frequency of health care resource use by health state is presented in Table 12. End-of-life costs were approximated based on the costs associated with pancreatic cancer from de Oliveira et al. (2016) and inflated to 2021 values.6 AE costs were sourced from the Ontario Case Costing Initiative database ambulatory and inpatient care codes.7 Costs for FGFR genetic testing were applied to 100% of pemigatinib patients and 75% of mFOLFOX plus ASC and ASC alone patients. These costs incorporated the costs of testing patients who would test negative by adjusting the cost of the test according to the prevalence of the mutation. The cost of the genetic test was based on a previous CADTH review where the cost of adding a gene to a panel was $750.8
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (2,500 iterations for the base-case and scenario analyses). The probabilistic findings are presented subsequently.
Base-Case Results
Pemigatinib was associated with a QALY gain of 1.23 at an additional cost of $177,324, resulting in an ICER of $143,604 compared with ASC alone. Compared with ASC alone, mFOLFOX plus ASC was dominated (i.e., less effective and more costly). In a pairwise comparison with pemigatinib, the ICERs for ASC and mFOLFOX versus pemigatinib were $143,604 and $127,359 per QALY gained, respectively. At the end of the 20-year time horizon, 1% of pemigatinib patients remained alive. Of the 1.65 QALYs accrued for pemigatinib, 0.92 (59%) occurred during the first 2 years of the model time horizon.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total LYs | Total QALYs | Pairwise ICER ($/QALY) (pemigatinib vs. comparator) | Sequential ICER ($/QALY) |
|---|---|---|---|---|---|
ASC | 69,907 | 0.61 | 0.42 | 143,604 | Reference |
mFOLFOX + ASC | 89,316 | 0.67 | 0.41 | 127,359 | Dominated |
Pemigatinib | 247,231 | 2.56 | 1.65 | NA | 143,604 |
ASC = active symptom control; ICER = incremental cost-effectiveness ratio; LY = life-year; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; NA = not applicable; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted extensive probabilistic scenario analyses. When a shorter (10-year) time horizon was used, the ICER for pemigatinib compared with ASC increased to $159,040. Results were also sensitive to using the MAIC-adjusted survival analysis (rather than the sponsor’s unadjusted base case), increasing the ICER to $160,408 and $166,411 when mFOLFOX and ASC were adjusted, respectively. When the Weibull and generalized gamma curves were used to extrapolate pemigatinib OS, the ICER increased to $207,363 and $160,554 per QALY compared with ASC. Using the sponsor’s societal perspective, the ICER for pemigatinib increased to $179,274 compared with ASC.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
Comparative efficacy estimates derived from the MAIC are uncertain. As FIGHT-202 was a single-arm study and because no head-to-head studies were conducted comparing pemigatinib with ASC or mFOLFOX, the sponsor conducted an unanchored MAIC to derive comparator efficacy rates to be used in the model. The CADTH Clinical Review Report identified several limitations with the sponsor’s MAIC, including heterogeneity across study designs and populations and the inability to adjust for all potential confounders and prognostic variables in the MAIC. Not conducting a head-to-head trial means there are potentially known and unknown confounders that could influence the comparative efficacy estimates derived from the MAIC. Consequently, residual confounding remains the major limitation of the MAIC, despite adjusting for some baseline characteristics.
As the sponsor’s MAIC created 2 different patient populations — 1 that made cohort A of FIGHT-202 look similar to the ASC arm of ABC-06 and a second population that made cohort A of FIGHT-202 look similar to the mFOLFOX arm of ABC-06 — the model produced 2 different results for pemigatinib. One that should only be interpreted alongside the ASC arm of the ABC-06 trial and 1 that should only be interpreted alongside the mFOLFOX arm. Therefore, only pairwise comparisons between pemigatinib and each of the comparators are appropriate.
Additionally, the sponsor’s approach to deriving PFS and OS for comparators was to take unadjusted extrapolations from FIGHT-202 and apply MAIC-derived HRs to get ASC and mFOLFOX PFS and OS estimates. First, it is inappropriate to use the unadjusted FIGHT-202 Kaplan–Meier data because it assumes that the MAIC HRs derived from an adjusted FIGHT-202 population will apply to the entire FIGHT-202 trial, which would negate the need for matching in the first place. Second, the approach of applying HRs to pemigatinib extrapolations assumes there are proportional hazards between pemigatinib and comparators. As there are no head-to-head data, there is no evidence that a proportional hazards assumption will hold; therefore, applying constant HRs is inappropriate.
Finally, AE rates were derived from a naive comparison rather than the MAIC, which further assumes that the baseline characteristics between FIGHT-202 and ABC-06 were the same which, based on the sponsor’s MAIC, was not found to be the case.
In the CADTH reanalyses, data from the FIGHT-202 trial were matched to the ASC arm of the ABC-06 trial, as per the sponsor’s MAIC. Rather than deriving comparative efficacy from weighted HRs, CADTH fitted independent survival curves to the ASC data and the ASC-matched pemigatinib data. A pairwise analysis was then performed between ASC and pemigatinib. Data from the FIGHT-202 trial was then matched to the FOLFOX arm of the ABC-06 trial, as per the sponsor’s MAIC. CADTH fitted independent survival curves to the FOLFOX data and the FOLFOX-matched pemigatinib data. A pairwise analysis was then performed between FOLFOX and pemigatinib.
CADTH was unable to address the limitation regarding naively derived AE rates; however, it is not expected that this would have a large influence on cost-effectiveness estimates.
Sponsor’s selected parametric functions for long-term outcomes did not meet face validity. In the FIGHT-202 trial, after 36 months of exposure, approximately 5% of patients on pemigatinib remained progression-free. For OS, after 22 months, approximately 41% of patients remained alive. To extrapolate pemigatinib outcomes to the model time horizon, the sponsor used parametric survival functions. The sponsor selected a log-logistic distribution to extrapolate pemigatinib OS, which resulted in a sustained post-progression survival benefit. This meant that patients who progressed on pemigatinib would have substantially improved OS outcomes relative to those who progressed on another therapy. According to the clinical experts consulted by CADTH for this review, this is neither expected nor proven. In the sponsor’s report, they also note: “One clinician consulted in the development of the economic model noted that they would expect approximately 10% of patients to be progression-free at 2 years.”1 The sponsor’s chosen log-logistic curve predicts 13% progression-free at 2 years, which is the furthest away from 10% compared with all other options. Although the sponsor argues the log-logistic has better statistical fit, this only relates to a curve’s ability to fit the known data and has little weight in determining long-term outcomes. For ASC and mFOLFOX, the data from the ABC-06 trial were very mature and most events, either progression or death, had occurred within the trial period.9 This meant that model outcomes for ASC and mFOLFOX were more influenced by the survival curve's ability to interpolate, rather than extrapolate, the data.
The CADTH reanalyses selected alternative pemigatinib survival functions such that the post-progression survival benefit associated with pemigatinib was aligned with clinical expert expectations.
For FOLFOX and ASC, CADTH chose curves that best interpolated the data while ensuring that post-progression survival outcomes were not greater than for pemigatinib.
The sponsor’s approach to modelling ToT was uncertain. In the sponsor’s submission, ToT was assumed to equal PFS for mFOLFOX. The clinical experts consulted for this review deemed this to be inappropriate, as mFOLFOX has cumulative neurotoxicity such that some patients may not be able to tolerate sustained treatment until progression. Instead, ToT for some mFOLFOX patients may be less than PFS, as they could discontinue before progression. In the sponsor’s submission, as there is no ToT data available from ABC-06, this assumption could not be changed.
For pemigatinib, a separate ToT curve was fitted to the FIGHT-202 trial data to estimate pemigatinib treatment costs. The experts consulted by CADTH for this review stated that while ToT for pemigatinib could be less than PFS, they would expect this to be rare in clinical practice and, if it were to occur, would expect progression in 1 to 2 months. The sponsor assumed that time to treatment discontinuation followed an exponential function, meaning that the rate of discontinuation was constant over time. However, they assumed a log-normal distribution for PFS, meaning that a considerable proportion of patients would discontinue therapy but remain progression-free for an extended period. Examining the pemigatinib PFS and ToT Kaplan–Meier data demonstrated that these curves closely followed each other, indicating that PFS and ToT were similar for pemigatinib in FIGHT-202.
Finally, ToT was based on the full patient population from the FIGHT-202 trial, whereas the PFS data were based on an adjusted population to match the ASC and FOLFOX arms of the ABC-06 trial. Given that ToT is inherently linked to PFS, any changes to PFS through matching should lead to a change in ToT.
To reflect the clinical experts’ expectations regarding ToT for pemigatinib and to ensure that modelled costs for pemigatinib reflected the approach for estimating costs for mFOLFOX, in the CADTH reanalyses, ToT was assumed to be equal to PFS for pemigatinib.
Costs of genetic testing are uncertain. According to clinician feedback and the sponsor, genetic testing for FGFR2 fusions or rearrangements is not currently publicly funded.1 To estimate genetic testing costs, the sponsor applied a cost of $750, which was sourced from a previous CADTH review and was the cost associated with adding a genetic test onto an existing next-generation sequencing (NGS) panel.10 This approach is inappropriate, as the cost of the initial NGS panel is not accounted for, and NGS panels are not currently publicly funded. As a scenario analysis, the sponsor explored the costs associated with NGS panel testing from FoundationOne, a private laboratory, which was US$5,800, which was deemed to be a more appropriate approximation of NGS costs but was not converted to Canadian dollars. The sponsor’s base case also appropriately applied the prevalence of FGFR2 fusions and rearrangements to calculate testing costs, such that the cost of testing patients found to be negative was incorporated into the model.
In addition to 100% of pemigatinib patients receiving genetic testing, the sponsor assumed that 75% of patients receiving mFOLFOX and ASC would also undergo testing. While some patients in the current context may receive genetic testing either privately through out-of-pocket payments or through research studies, given that testing is not currently funded and will not inform treatment decisions if pemigatinib is not available, it is inappropriate to apply genetic testing costs to comparator arms.
In the CADTH reanalyses, the cost of genetic testing was the cost associated with the NGS panel from FoundationOne converted to Canadian dollars using the average Bank of Canada exchange rate from August 30, 2020 to August 30, 2021 (1.2690).11 Additionally, CADTH assumed 0% of ASC and mFOLFOX patients will receive publicly funded genetic testing.
Health-state utility values do not meet face validity. The sponsor derived utility values from FIGHT-202 for the following health states: progression-free on treatment, progression-free off treatment, progressed disease on treatment, progressed disease off treatment (Table 20). Patients with progression-free disease who were off treatment had a lower utility value (||||||||||||||||||) applied than that of patients in any of the progressed-disease health states. According to the clinical experts consulted for this review, there is no reason for a patient with progression-free disease who is off treatment to have worse quality of life than someone with progressed disease. Additionally, results of the European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30 questionnaire was assessed at baseline and then every 3 cycles starting with cycle 3 until discontinuation of the study treatment and at the end-of-treatment visit, meaning that the progression-free off-treatment utility value is taken at the time of discontinuation. If patients discontinue treatment due to intolerable AEs, their health-related quality of life will likely improve upon discontinuation. Finally, applying these utility values led to results that are not clinically expected; mFOLFOX plus ASC was found to predict greater life-years than ASC alone, but because no ASC patients fell into the progression-free off-treatment health state, treatment with ASC led to greater QALYs (Table 3).
To address this limitation, utilities in the CADTH reanalyses did not differ depending on whether patients were on or off treatment.
The analysis did not include the costs of subsequent therapies. The sponsor’s model incorporated costs and outcomes of pemigatinib, ASC, and FOLFOX; however, upon progression, no additional treatments are incorporated. This is inappropriate as, according to the clinical experts consulted for this review, if patients progress on second-line treatment and have a good Eastern Cooperative Oncology Group Performance Status, they can receive subsequent chemotherapy. Treatment with subsequent therapies was observed in FIGHT-202, as discussed in the CADTH Clinical Review Report. Further, subsequent therapy options are expected to differ depending on second-line treatments according to clinical experts; if patients received pemigatinib as second-line therapy, they would be expected to receive mFOLFOX upon progression, but if patients received second-line mFOLFOX, they would receive FOLFIRI or other drugs upon progression. As subsequent treatments differ by second-line therapy and, given there are differences in the costs of third-line regimens, this introduces some uncertainty in the cost-effectiveness estimates.
CADTH was unable to address this limitation. The direction and magnitude of not incorporating subsequent therapies is unknown.
The following limitations were identified but were not deemed key limitations:
The incorporation of relative dose intensity (RDI) is inappropriate. The sponsor incorporated an RDI of ||||||, which was calculated from FIGHT-202 based on doses received as a proportion of the expected number of doses without interruptions. CADTH was unable to validate this value as a means of accounting for dose interruptions. RDI was not reported in the sponsor’s clinical study report; however, treatment compliance was reported and found to be high, as concluded in the CADTH Clinical Review Report. Additionally, though patients may miss a dose of pemigatinib, this might not influence overall costs to public drug plans, as full drug claims will be dispensed. Finally, RDI was not applied to mFOLFOX therapy.
CADTH reanalyses assumed an RDI of 1 for pemigatinib.
Costs used for the mFOLFOX regimen were uncertain. Costs for the components for the mFOLFOX regimen were sourced from a previous CADTH review.12 CADTH found wholesale prices for mFOLFOX components from the IQVIA Delta PA database13 that were deemed to be more appropriate.
CADTH reanalysis used wholesale costs from IQVIA Delta PA.
Health care resource use estimates are uncertain. The sponsor incorporated health care resource use by assuming that the estimated clinician visits and bloodwork would occur once every 3 months for those with progression-free and progressed disease, and the CT scan would occur once every 3 and 12 months for those with progression-free and progressed disease, respectively.1 According to the clinical experts consulted for this review, patients are expected to see clinicians and have bloodwork monitoring done more frequently when in the progressed-disease state versus the progression-free disease state. Additionally, while the study protocol for pemigatinib specifies that patients receiving pemigatinib will need to see an eye specialist and undergo optic coherence tomography imaging before initiation and at regular intervals (every 3 cycles), or as clinically indicated thereafter, to monitor for serous retinal detachment, the costs of these exams for pemigatinib patients were not included.14
Given the relatively low overall costs of resource use compared with other model costs, assumptions regarding visit frequency are unlikely to change conclusions regarding the cost-effectiveness of pemigatinib. Not incorporating eye exam or optical coherence tomography costs for pemigatinib favours pemigatinib, as only patients receiving pemigatinib would require eye exams.
ASC costing assumption estimates are uncertain. According to the clinical experts consulted for this review, ASC may differ depending on a patient’s second-line therapy; however, the magnitude and direction are uncertain. Likewise, given that pemigatinib improves OS, this would lead to more ASC costs being incurred in the pemigatinib arm.
Exclusion of ASC costs is likely to favour pemigatinib but the magnitude of impact on cost-effectiveness is unlikely to be large.
The analysis does not include all relevant comparators. The sponsor’s analysis compared pemigatinib with ASC and mFOLFOX, as these were the only comparators indicated for CCA treatment. In addition to ASC and mFOLFOX, there are several other off-label medications for second-line treatment of CCA that are used in Canadian clinical practice, based on feedback from the clinical experts consulted for this review (per Appendix 1 and the comparator table provided by the sponsor).14 According to CADTH economic guidelines, all interventions currently used and potentially displaced should be identified, and those that decision-makers are currently funding or that are commonly used should be included.15 While not indicated for CCA, some off-label treatments are listed as a full benefit in jurisdictions, meaning that clinicians can prescribe medications for any indication, including those that are off-label.14 Additionally, CADTH economic guidelines note that comparator selection should not be limited by the availability of data.15 The exclusion of comparators that may be displaced if pemigatinib is publicly reimbursed may favour pemigatinib, as these comparators are associated with lower annual costs (see Appendix 1), though the comparative benefits are unknown.
CADTH was unable to address this limitation and, as such, the cost-effectiveness of pemigatinib compared with off-label therapies that are currently reimbursed is unknown.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
The CADTH reanalyses addressed several limitations within the economic model, summarized in Table 4. CADTH was unable to address limitations regarding the uncertainty in comparative efficacy estimates due to FIGHT-202 being a single-arm trial and uncertainties arising in the MAIC, and not including costs of subsequent therapies. As such, the changes shown in Table 4 reflect a CADTH reanalysis rather than a base-case estimate of the cost-effectiveness of pemigatinib compared with ASC and mFOLFOX. The CADTH reanalysis was derived by making changes in model parameter values and assumptions, in consultation with clinical experts.
Table 4: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption | ||
|---|---|---|---|---|
Correctionsa to sponsor’s base case | ||||
None | — | — | ||
Changes to derive the CADTH base case | ||||
1. Comparative efficacy | Weighted hazard ratios | Extrapolated curves | ||
2. MAIC-adjusted survival analysis | Pemigatinib OS and PFS were unadjusted | Pemigatinib OS and PFS were adjusted to match the ASC and FOLFOX arms from the ABC-06 trial: a) ABC-06 ASC adjusted b) ABC-06 mFOLFOX adjusted | ||
3. Time on treatment for pemigatinib | Not equal to PFS | Equal to PFS | ||
4. Pemigatinib PFS extrapolation | Log-normal | Weibull | ||
5. Pemigatinib OS extrapolation | Log-logistic | Weibull | ||
6. Comparator PFS extrapolation | Not applicable (based on HRs) | Log-normal (PFS assumed equal for ACS and FOLFOX due to absence of evidence for ASC PFS) | ||
7. Comparator OS extrapolation | Not applicable (based on HRs) | ASC: Log-normal mFOLFOX: Weibull | ||
8. Utility values | Assume treatment status effect (see Table 13). | Do not assume treatment status effect (|||||||| and |||||||| for progression-free and progressed disease, respectively) | ||
9. Genetic testing frequency and costs | $750 per test:
| $7,360 per test:
| ||
10. Relative dose intensity | ||||||||||||% | 100% | ||
11. mFOLFOX costs | Total per week = $833.20:
| Total per week = $307.13: | ||
CADTH reanalysis: ASC vs. pemigatinib | — | 1 + 2a + 3 + 4 + 5 + 6 + 7a + 8 + 9 + 10 | ||
CADTH reanalysis: mFOLFOX vs. pemigatinib | — | 1 + 2b + 3 + 4 + 5 + 6 + 7b + 8 + 9 + 10 + 11 | ||
ASC = active symptom control; HR = hazard ratio; MAIC = matching-adjusted indirect comparison; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; OS = overall survival; PFS = progression-free survival.
aCorrections are minor errors (e.g., transcription errors between report and model, misapplication of distributions, or standard errors in probabilistic analyses) that are not identified as limitations.
The results of CADTH’s stepped analysis are presented in Table 14 and Table 15 for pemigatinib versus ASC and mFOLFOX, respectively. The efficacy of pemigatinib in the sponsor’s analysis is dependent on whether the population in FIGHT-202 is adjusted to the ASC cohort or mFOLFOX cohort from the ABC-06 trial. CADTH notes that outcomes for pemigatinib were not substantially different when the population was adjusted to the ASC cohort relative to the FOLFOX population. Compared with ASC, pemigatinib was $209,585 more expensive and yielded 0.83 greater QALYs, leading to an ICER of $252,718 per QALY gained (Table 5). Compared with FOLFOX, pemigatinib was $198,154 more expensive and yielded 0.76 greater QALYs, leading to an ICER of $261,226 per QALY gained (Table 6). Changing the survival curve to extrapolate pemigatinib OS resulted in the largest change to the sponsor’s base case. At a willingness-to-pay threshold of $50,000, pemigatinib is 0% likely to be cost-effective compared with either ASC or mFOLFOX. Of the total costs for pemigatinib, 61% were drug costs (Table 16). The majority of costs for ASC and mFOLFOX came from terminal care costs. Of the 1.24 total QALYs for pemigatinib when compared with ASC, 0.62 are accrued during the first 2 years of the model’s time horizon. Of the 1.25 total QALYs for pemigatinib when compared with mFOLFOX, 0.53 are accrued during the first 2 years of the model’s time horizon.
Table 5: Summary of the CADTH Reanalysis Results for Pemigatinib vs. ASC
Drug | Total costs ($) | Total LYs | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
ASC | 66,895 | 0.58 | 0.41 | Reference |
Pemigatinib (ASC adjusted) | 276,480 | 1.79 | 1.24 | 252,718 |
ASC = active symptom control; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus.
Table 6: Summary of the CADTH Reanalysis Results for Pemigatinib vs. mFOLFOX
Drug | Total costs ($) | Total LYs | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
mFOLFOX | 77,945 | 0.72 | 0.49 | Reference |
Pemigatinib (mFOLFOX adjusted) | 276,099 | 1.82 | 1.25 | 261,226 |
ICER = incremental cost-effectiveness ratio; LY = life-year; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; QALY = quality-adjusted life-year; vs. = versus.
Scenario Analysis Results
CADTH undertook price-reduction analyses in its reanalysis (Table 7). These analyses demonstrated that a price reduction of 95% would be required for pemigatinib to be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY compared with mFOLFOX. For ASC, a price reduction approaching 100% is required for pemigatinib to be considered cost-effective when compared with ASC. When testing costs were set to $0, the price reduction required for pemigatinib to be cost-effective at a willingness-to-pay threshold of $50,000 per QALY compared with ASC and mFOLFOX was 77% and 72%, respectively.
Table 7: CADTH Price-Reduction Analyses
Analysis | ICERs for pemigatinib vs. ASC and mFOLFOX | |||
|---|---|---|---|---|
Sponsor base case ($) | CADTH reanalysis ($) | |||
Price reduction | ASC | mFOLFOX | ASC | mFOLFOX |
No price reduction | 143,604 | 127,359 | 252,718 | 261,226 |
10% | 128,897 | 112,792 | 232,758 | 238,427 |
20% | 115,506 | 99,314 | 211,560 | 214,927 |
30% | 101,650 | 85,533 | 190,917 | 194,114 |
40% | 87,167 | 71,079 | 171,020 | 170,806 |
50% | 72,816 | 56,824 | 149,908 | 148,989 |
60% | 58,475 | 42,566 | 129,915 | 126,879 |
70% | 44,407 | NA | 109,882 | 104,537 |
80% | NA | NA | 88,930 | 82,937 |
90% | NA | NA | 68,379 | 59,688 |
100% | NA | NA | 48,908 | 38,348 |
ASC = active symptom control; ICER = incremental cost-effectiveness ratio; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; NA = not applicable; vs. = versus.
Issues for Consideration
The clinical experts consulted for this review would generalize the results from the FIGHT-202 trial to patients with CCA in the first line of treatment who do not tolerate standard of care for the first line, not just to patients whose disease has progressed. Because the inclusion criteria for the FIGHT-202 trial specified that patients’ disease must have progressed after at least 1 line of prior systemic therapy, the cost-effectiveness of pemigatinib for patients whose cancer does not progress is not known. Use of pemigatinib in the population with non–progressed disease would also lead to a higher budget impact than that estimated by the sponsor and CADTH.
Treatment with pemigatinib will require testing to determine eligibility. Genetic testing for FGFR2 fusions or rearrangements to determine pemigatinib eligibility is not routinely available or funded by the public health care payer.
Overall Conclusions
The CADTH clinical review found that given the absence of robust comparative data on PFS and OS, the ability to interpret the relative treatment effects observed between pemigatinib and FOLFOX plus ASC and ASC alone was limited and no firm conclusions could be drawn on how pemigatinib compared with other relevant treatment options.
Given the high degree of uncertainty concerning the magnitude of clinical benefit, CADTH was unable to perform a base-case analysis. The reanalysis performed by CADTH utilizes more appropriate assumptions but notes that these estimates are highly uncertain.
CADTH undertook reanalyses to address limitations relating to: the incorporation of MAIC-derived comparative efficacy estimates into the sponsor’s analysis, long-term extrapolations for pemigatinib PFS and OS, selecting comparator extrapolations for PFS and OS, the assumption that utility values vary by whether patients are on or off treatment, genetic testing costs, RDI, and mFOLFOX costs.
Based on the CADTH reanalysis, the ICER for pemigatinib relative to ASC and mFOLFOX was estimated to be $252,718 and $261,226 per QALY gained, respectively. A sequential analysis could not be performed due to the efficacy of pemigatinib being contingent on whether the MAIC matched the data from the ASC arm or mFOLFOX arm of the ABC-06 trial. At these ICERs, at least a 95% to 100% reduction in the price of pemigatinib is required for pemigatinib to achieve an ICER of $50,000 per QALY gained compared with mFOLFOX and ASC, respectively. The reason price reductions approach 100% is due to the high cost of testing, estimated to be $38,000, to identify a single patient eligible for treatment with pemigatinib. If testing costs were $0, then to be cost-effective relative to ASC, a 77% price reduction is needed, or 72% versus FOLFOX.
The uncertainty in the comparative efficacy data for pemigatinib meant that the magnitude of benefit associated with pemigatinib compared with ASC and FOLFOX could not be reliably determined. Consequently, CADTH was unable to determine a base-case estimate regarding pemigatinib’s cost-effectiveness. Instead, CADTH conducted an exploratory reanalysis on the sponsor’s base case. According to the clinical experts consulted by CADTH for this review, pemigatinib could be equal to or better than alternative treatments currently received by patients. The price reductions noted by CADTH assume substantially improved efficacy with pemigatinib, which is highly uncertain.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: pemigatinib, 4.5 mg, 9 mg, and 13.5 mg, oral tablets. Pointe Claire (QC): Incyte Biosciences Canada Corporation; 2021 Jun 23.
2.Longworth L, Yang Y, Young T, et al. Use of generic and condition-specific measures of health-related quality of life in NICE decision-making: a systematic review, statistical modelling and survey. Health Technology Assessment, no. 18.9. Southampton (UK): National Institute for Health Research (NIHR); 2014: https://www.ncbi.nlm.nih.gov/books/NBK261616/?report=classic. Accessed 2021 Oct 18.
3.NICE. Single technology appraisal: Pomalidomide with dexamethasone for treating relapsed and refractory multiple myeloma after at least two regimens including lenalidomide and bortezomib (review of TA338) [ID985] Committee papers. National Institute for Health and Care Excellence (NICE); 2016: https://www.nice.org.uk/guidance/ta427/documents/committee-papers. Accessed 2021 Aug 11.
4.Schedule of benefits for physician services under the Health Insurance Act: effective April 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: http://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master20200306.pdf. Accessed 2021 Sept 23.
5.Schedule of benefits for laboratory services: effective July 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: http://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf. Accessed 2021 Oct 18.
6.de Oliveira C, Pataky R, Bremner KE, et al. Phase-specific and lifetime costs of cancer care in Ontario, Canada. BMC Cancer. 2016;16(1):809. PubMed
7.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2021 Oct 18.
8.CADTH. Larotrectinib for neurotrophic tyrosine receptor kinase (NTRK) locally advanced or metastatic solid tumours – details. 2019; https://cadth.ca/larotrectinib-neurotrophic-tyrosine-receptor-kinase-ntrk-locally-advanced-or-metastatic-solid. Accessed 2021 Aug 11.
9.Lamarca A, Palmer DH, Wasan HS, et al. Second-line FOLFOX chemotherapy versus active symptom control for advanced biliary tract cancer (ABC-06): a phase 3, open-label, randomised, controlled trial. The Lancet Oncology. 2021;22(5):690-701. PubMed
10.CADTH pan-Canadian Oncology Drug Review. Final economic guidance report: larotrectinib (Vitrakvi) for neurotrophic tyrosine receptor kinase (NTRK) positive solid tumours. Ottawa (ON): CADTH; 2019: https://cadth.ca/sites/default/files/pcodr/Reviews2019/10159LarotrectinibNTRK%2BSolidTumours_fnEGR_NOREDACT-ABBREV_Post_31Oct2019_final.pdf. Accessed Sept 3 2021.
11.Bank of Canada. Daily exchange rates lookup: Currency converter. 2021; https://www.bankofcanada.ca/rates/exchange/daily-exchange-rates-lookup/. Accessed 2021 Sep 9.
12.CADTH pan-Canadian Oncology Drug Review. Final economic guidance report: panitmumab (Vectibix) for left-sided metastatic colorectal cancer. Ottawa (ON): CADTH; 2018: https://cadth.ca/sites/default/files/pcodr/pcodr_panitumumab_vectibix_ls_mcrc_fn_egr%20.pdf. Accessed 2021 Sep 14.
13.DeltaPA. Danbury (CT): IQVIA; 2021: https://www.iqvia.com/. Accessed 2021 Oct 18.
14.Drug Reimbursement Review sponsor submission: pemigatinib, 4.5 mg, 9 mg, and 13.5 mg, oral tablets [internal sponsor's package]. Pointe Claire (QC): Incyte Biosciences Canada Corporation; 2021 Jun 23.
15.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/dv/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2021 Oct 18.
16.Cancer Care Ontario. FOLFIRI - regimen monograph. 2019; https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/49306. Accessed 2021 Aug 26.
17.Nehls O, Oettle H, Hartmann JT, et al. Capecitabine plus oxaliplatin as first-line treatment in patients with advanced biliary system adenocarcinoma: a prospective multicentre phase II trial. Br J Cancer. 2008;98(2):309-315. PubMed
18.Cancer Care Ontario. CAPECISP - regimen monograph. 2019; https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/46811. Accessed 2021 Sept 14.
19.Flemming JA, Zhang‐Salomons J, Nanji S, Booth CM. Increased incidence but improved median overall survival for biliary tract cancers diagnosed in Ontario from 1994 through 2012: a population‐based study. Cancer. 2016;122(16):2534-2543. PubMed
20.Mukkamalla SKR, Naseri HM, Kim BM, Katz SC, Armenio VA. Trends in incidence and factors affecting survival of patients with cholangiocarcinoma in the United States. J Natl Compr Canc Netw. 2018;16(4):370-376. PubMed
21.Bridgewater J, Lopes A, Wasan H, et al. Prognostic factors for progression-free and overall survival in advanced biliary tract cancer. Ann Oncol. 2016;27(1):134-140. PubMed
22.Jain A, Borad MJ, Kelley RK, et al. Cholangiocarcinoma with FGFR genetic aberrations: a unique clinical phenotype. JCO Precision Oncology. 2018;2:1-12. PubMed
23.Lowery MA, Goff LW, Jordan E, et al. Second-line chemotherapy (CTx) outcomes in advanced biliary cancers (ABC): a retrospective multicenter analysis. American Society of Clinical Oncology; 2016.
24.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: pemigatinib, 4.5 mg, 9 mg, and 13.5 mg, oral tablets. Pointe Claire (QC): Incyte Biosciences Canada Corporation; 2021 Jun 23.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from the clinical expert(s) and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost-Comparison Table for Previously Treated, Unresectable, Locally Advanced, or Metastatic Cholangiocarcinoma
Treatment | Strength/ concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | 28-day cycle cost ($) |
|---|---|---|---|---|---|---|
Pemigatinib | 4.5 mg 9 mg 13.5 mg | Tablet | 830.2987a | 13.5 mg orally once daily for 14 consecutive days followed by 7 days off therapy, in 21-day cycles | 830.30 | 15,499b |
mFOLFOX | ||||||
Oxaliplatin (generic) | 5 mg/mL | Solution for injection 50 mg/10 mL 100 mg/20 mL 200 mg/40 mL | 36.2700c 72.5400c 145.0800c | 85 mg/m2 every 14 days | 10.36 | 290 |
Leucovorin (generic) | 10 mg/mL | Vial for injection 50 mg/5 mL 500 mg/50 mL | 68.9400c | 350 mg/m2 every 14 days | 64.02 | 1,792 |
Fluorouracil (generic) | 50 mg/mL | Solution for injection 0.5 g/10 mL 5 g/100 mL | 16.0900c 160.9000c | 400 mg/m2 then 2,400 mg/m2 every 14 days | 2.30 11.49 | 64 322 |
mFOLFOX | 88.17 | 2,469 | ||||
mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin.
aSponsor’s submitted price.
bThis cost represents the average 28-day cost. In a given 28-day period, due to patients being on therapy for 14 days followed by being off therapy for 7 days, patients may incur a minimum of 14 days of cost up to a maximum of 21 days depending on when the patient starts and pauses treatment. This was calculated by taking the average cost per day for a 21-day course ($830.2987 × 14/21 = $553.53) and then calculating the average cost per 28 days ($553.53 × 28 = $15,499).
cIQVIA Delta PA.13
Table 9: CADTH Cost-Comparison Table for Off-Label Treatments Used in Previously Treated, Unresectable, Locally Advanced, or Metastatic Cholangiocarcinoma
Treatment | Strength/ concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | 28-day cycle cost ($) |
|---|---|---|---|---|---|---|
FOLFIRIb | ||||||
Irinotecan (generic) | 20 mg/mL | Solution for injection 40 mg/2 mL 100 mg/5 mL 300 mg/15 mL 500 mg/25 mL | 3.2400a 8.1000a 24.3000a 40.5000a | 180 mg/m2 every 14 days | 1.97 | 55 |
Leucovorin (generic) | 10 mg/mL | Vial for injection 50 mg/5 mL 500 mg/50 mL | 68.9400a | 350 mg/m2 every 14 days | 64.02 | 1,792 |
Fluorouracil (generic) | 50 mg/mL | Solution for injection 0.5 g/10 mL 5 g/100 mL | 16.0900a 160.9000a | 400 mg/m2 then 2,400 mg/m2 every 14 days | 2.30 11.49 | 64 322 |
FOLFIRI | 79.78 | 2,234 | ||||
Capecitabine (may be used along or in combination with cisplatin or oxaliplatin)d | ||||||
Capecitabine alone | ||||||
Capecitabine | 150 mg 500 mg | Tablet | 0.4575c 1.5250c | 1,000 mg/m2 twice daily for 14 consecutive days followed by 7 days off therapy, in 21-day cyclesd | 11.59 | 216e |
XELOX (capecitabine plus oxaliplatin) | ||||||
Capecitabine | 150 mg 500 mg | Tablet | 0.4575c 1.5250c | 1,000 mg/m2 twice daily for 14 consecutive days followed by 7 days off therapy, in 21-day cyclesd | 11.59 | 216e |
Oxaliplatin | 5 mg/mL | Solution for injection 50 mg/10 mL 100 mg/20 mL 200 mg/40 mL | 36.2700a 72.5400a 145.0800a | 130 mg/m2 on day 1 in 21 day cycle d | 8.64 | 242 |
XELOX (capecitabine plus oxaliplatin) | 20.23 | 458 | ||||
Capecitabine plus cisplatin | ||||||
Capecitabine | 150 mg 500 mg | Tablet | 0.4575c 1.5250c | 1,000 mg/m2 twice daily for 14 consecutive days followed by 7 days off therapy, in 21-day cyclesf | 11.59 | 216e |
Cisplatin | 1 mg/mL | Solution for injection 10 mg/10 mL 50 mg/50 mL 100 mg/100 mL | 27.0000 135.0000 270.0000 | 60 mg/m2 on day 1 in 21 day cyclef | 15.43 | 432 |
Capecitabine plus cisplatin | 27.02 | 648 | ||||
Paclitaxel | ||||||
Paclitaxel | 6 mg/mL | Solution for injection 30 mg/ 5 mL 100 mg/ 16.7 mL 300 mg/ 50 mL | 300.0000 1,002.0000 3,000.0000 | 180 mg/m2 every 21 daysg | 157.14 | 4,400 |
Note: All surface area–based dosing assumed a body surface area of 1.88 m2.
aIQVIA Delta PA.13
bDose obtained from Cancer Care Ontario and confirmed to be appropriate by the clinical experts consulted for this review.16
cOntario Drug Benefit formulary (accessed August 2021).
dDose obtained from Nehls O, Oettle H, Hartmann JT, et al.17 and confirmed to be appropriate by the clinical experts consulted for this review.
eThis cost represents the average 28-day cost. In a given 28-day period, due to patients being on therapy for 14 days followed by being off therapy for 7 days, patients may incur a minimum of 14 days of cost up to a maximum of 21 days depending on when the patient starts and pauses treatment. This was calculated by taking the average cost per day for a 21-day course ($11.59 × 14/21 = $7.73) and then calculating the average cost per 28 days ($7.73 × 28 = $216).
fDose obtained from Cancer Care Ontario18 and confirmed to be appropriate by the clinical experts consulted for this review.
gDose informed by clinical expert feedback.
Appendix 2: Submission Quality
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | As the clinical information was derived from a single-arm trial the population had to be restricted to match trial populations of other comparators. This restriction of trial data, although necessary to avoid an even more flawed naive comparison, means that the data being used in the economic model is not reflective of the full Health Canada population. |
Model has been adequately programmed and has sufficient face validity | Yes | Not applicable. |
Model structure is adequate for decision problem | No | Partition survival models assume no explicit relationship between progression-free survival and overall survival. In the review, this leads to the perverse conclusion that patients who progress can live substantially longer than those who do not progress. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | Not applicable. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | See limitation regarding comparative efficacy assumptions. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | Not applicable. |
Note: This table has not been copy-edited.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
PD = progressed disease; PF = progression free.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 11: Sponsor’s Base-Case Distribution Choices and Survival Estimates for Pemigatinib
Extrapolation | Sponsor’s selected survival distribution | Survival estimates |
|---|---|---|
Overall survival | Log-logistic | 12% alive at 5 years |
Progression-free survival | Log-normal | 13% progression-free at 2 years |
Time on treatment | Exponential | 11% on treatment at 2 years |
Figure 2: Observed and Predicted Overall Survival Data for Pemigatinib
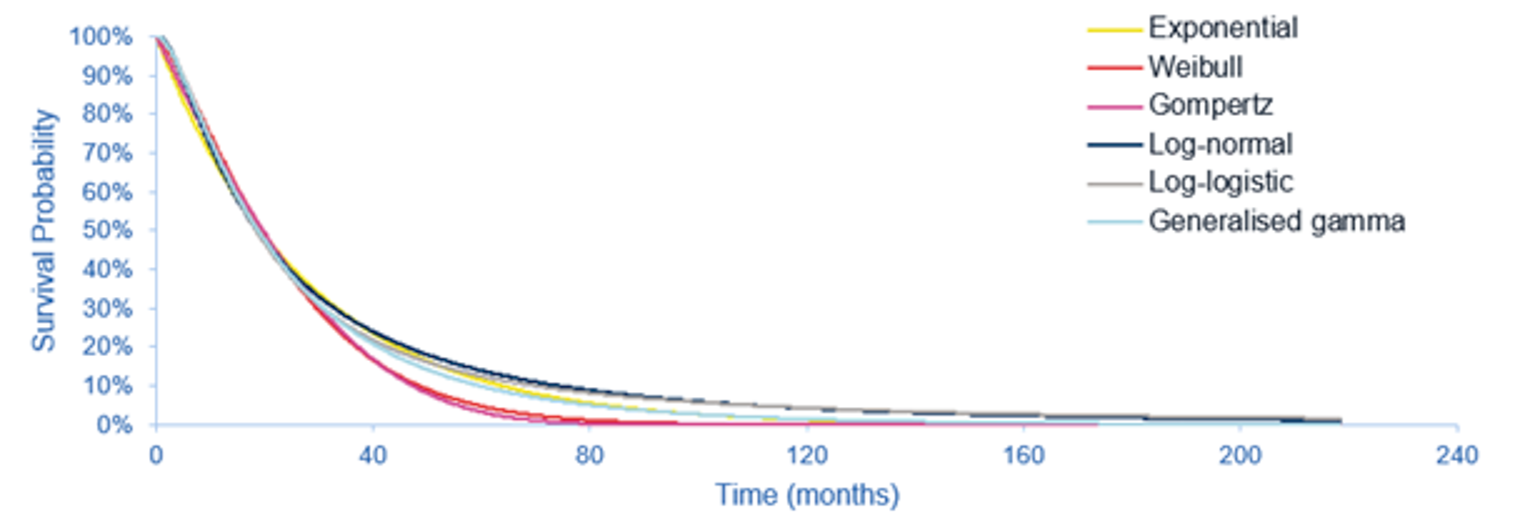
KM = Kaplan-Meier.
Source: Sponsor’s pharmacoeconomic submission.1
Figure 3: ASC Alone OS-Informed MAIC HR Compared With Pemigatinib OS
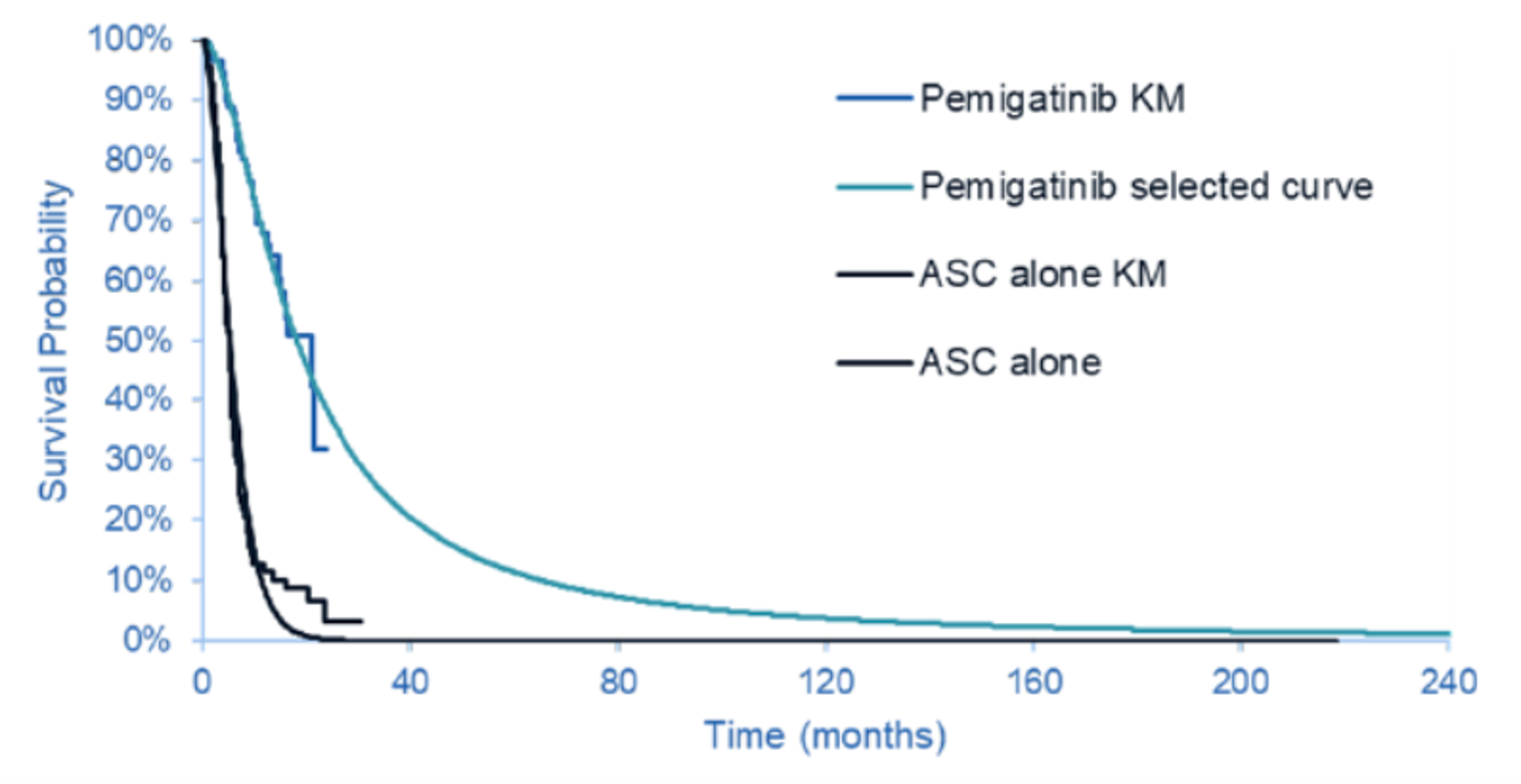
ASC = active symptom control; HR = hazard ratio; KM = Kaplan-Meier; MAIC = matching-adjusted indirect comparison; OS = overall survival.
Source: Sponsor’s pharmacoeconomic submission.1
Figure 4: Observed and Predicted Progression-Free Survival Data for Pemigatinib
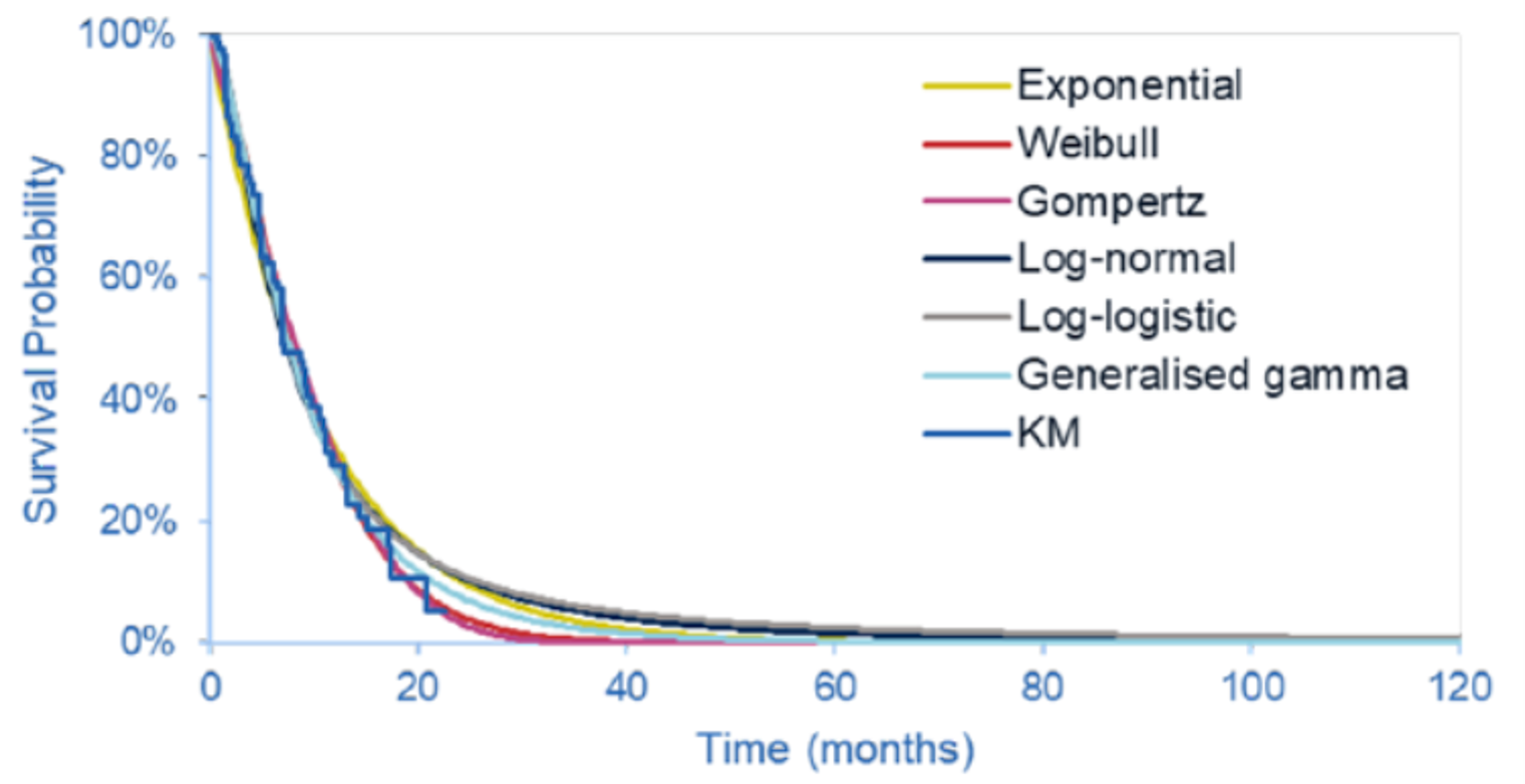
KM = Kaplan-Meier.
Source: Sponsor’s pharmacoeconomic submission.1
Figure 5: mFOLFOX Plus ASC PFS-Informed MAIC HR, Compared With Pemigatinib PFS
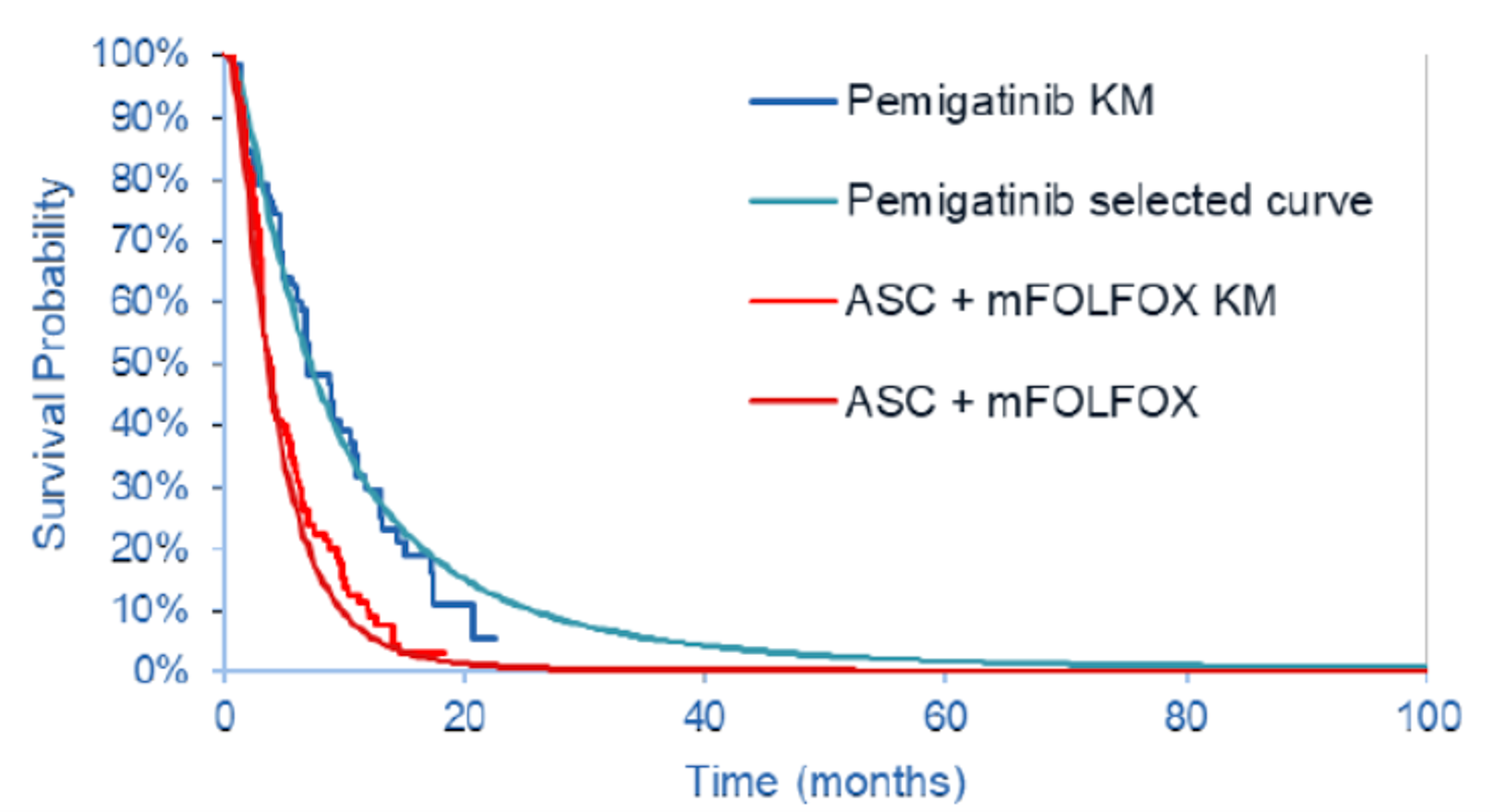
ASC = active symptom control; HR = hazard ratio; KM = Kaplan-Meier; MAIC = matching-adjusted indirect comparison; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; PFS = progression-free survival.
Source: Sponsor’s pharmacoeconomic submission.1
Figure 6: Predicted Time-on-Treatment Data for Pemigatinib

KM = Kaplan-Meier.
Source: Sponsor’s pharmacoeconomic submission.1
Table 12: Frequency of Health Care Resource Use
Resource | Monthly visit frequency | |
|---|---|---|
Progression-free | Progressed disease | |
Clinical exam | 0.333 | 0.333 |
CT scan | 0.333 | 0.083 |
Blood tests | 0.333 | 0.333 |
Source: Sponsor’s pharmacoeconomic submission.1
Table 13: Sponsor’s Health-State Utility Values
Health state | Mean utility value | Source |
|---|---|---|
Progression-free on treatment | |||||||||||||||||||| | FIGHT-202 utility analysis |
Progression-free off treatment | |||||||||||||||||||| | FIGHT-202 utility analysis |
Progressive disease on treatment | |||||||||||||||||||| | FIGHT-202 utility analysis |
Progressive disease off treatment | |||||||||||||||||||| | FIGHT-202 utility analysis |
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Table 14: Summary of the Stepped Analysis of the CADTH Reanalysis Results for Pemigatinib vs. ASC
Stepped analysis | Drug | Total costs ($) | Total LYs | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|---|
Sponsor’s base case (deterministic) | ASC | 69,776 | 0.60 | 0.42 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.65 | 148,488 | |
CADTH reanalysis 1: Sponsor fitted survival curves to PFS and OS for patients receiving ASC | ASC | 69,281 | 0.57 | 0.40 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.65 | 146,327 | |
CADTH reanalysis 2a: ASC adjusted survival analysis | ASC | 69,882 | 0.79 | 0.54 | Ref. |
Pemigatinib | 252,616 | 2.40 | 1.59 | 175,467 | |
CADTH reanalysis 3: Pemigatinib ToT | ASC | 69,776 | 0.60 | 0.42 | Ref. |
Pemigatinib | 278,023 | 2.53 | 1.69 | 163,867 | |
CADTH reanalysis 4: Pemigatinib PFS | ASC | 69,794 | 0.60 | 0.42 | Ref. |
Pemigatinib | 244,091 | 2.53 | 1.67 | 139,321 | |
CADTH reanalysis 5: Pemigatinib OS | ASC | 69,768 | 0.60 | 0.42 | Ref. |
Pemigatinib | 253,093 | 1.94 | 1.28 | 212,835 | |
CADTH reanalysis 6: Reanalysis 1 + Sponsor fitted survival curve to PFS for patients receiving ASC | ASC | 69,821 | 0.57 | 0.4 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.65 | 146,327 | |
CADTH reanalysis 7a: Reanalysis 1 + CADTH fitted survival curve to OS for patients receiving ASC | ASC | 69,828 | 0.58 | 0.40 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.65 | 147,024 | |
CADTH reanalysis 8: Utility values | ASC | 69,776 | 0.60 | 0.42 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.73 | 139,844 | |
CADTH reanalysis 9: Genetic testing | ASC | 66,853 | 0.60 | 0.42 | Ref. |
Pemigatinib | 286,890 | 2.53 | 1.65 | 178,771 | |
CADTH reanalysis 10: Relative dose intensity | ASC | 69,776 | 0.60 | 0.42 | Ref. |
Pemigatinib | 256,835 | 2.53 | 1.65 | 151,978 | |
CADTH reanalysis 1+2a+3+4+5+6+7a+ 8+9+10 (deterministic) | ASC | 66,905 | 0.58 | 0.41 | Ref. |
Pemigatinib | 274,857 | 1.77 | 1.22 | 255,631 | |
CADTH reanalysis 1+2a+3+4+5+6+7a+ 8+9+10 (probabilistic) | ASC | 66,895 | 0.58 | 0.41 | Ref. |
Pemigatinib | 276,480 | 1.79 | 1.24 | 252,718 |
ASC = active symptom control; ICER = incremental cost-effectiveness ratio; OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-year; Ref. = reference; ToT = time on treatment; vs. = versus.
Note: Results of all steps are presented deterministically. The cumulative CADTH base case is presented probabilistically, as well.
Table 15: Summary of the Stepped Analysis of the CADTH Reanalysis Results for Pemigatinib vs. mFOLFOX
Stepped analysis | Drug | Total costs ($) | Total LYs | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|---|
Sponsor’s base case (deterministic) | mFOLFOX | 89,282 | 0.66 | 0.41 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.65 | 132,099 | |
CADTH reanalysis 1: Sponsor fitted survival curves to PFS and OS for patients receiving ASC | mFOLFOX | 90,613 | 0.82 | 0.50 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.65 | 141,451 | |
CADTH reanalysis 2b: FOLFOX adjusted survival analysis | mFOLFOX | 92,121 | 0.85 | 0.53 | Ref. |
Pemigatinib | 252,589 | 2.45 | 1.62 | 148,078 | |
CADTH reanalysis 3: Pemigatinib ToT | mFOLFOX | 89,282 | 0.66 | 0.41 | Ref. |
Pemigatinib | 278,023 | 2.53 | 1.69 | 147,931 | |
CADTH reanalysis 4: Pemigatinib PFS | mFOLFOX | 89,276 | 0.66 | 0.41 | Ref. |
Pemigatinib | 244,091 | 2.53 | 1.67 | 122,779 | |
CADTH reanalysis 5: Pemigatinib OS | mFOLFOX | 89,070 | 0.67 | 0.41 | Ref. |
Pemigatinib | 253,093 | 1.94 | 1.28 | 189,873 | |
CADTH reanalysis 6: Reanalysis 1+ CADTH fitted survival curve to PFS for patients receiving mFOLFOX | mFOLFOX | 90,613 | 0.82 | 0.50 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.65 | 141,451 | |
CADTH reanalysis 7b: Reanalysis 1 + CADTH fitted survival curve to OS for patients receiving mFOLFOX | mFOLFOX | 90,230 | 0.72 | 0.44 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.65 | 134,484 | |
CADTH reanalysis 8: Utility values | mFOLFOX | 89,282 | 0.66 | 0.46 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.73 | 128,237 | |
CADTH reanalysis 9: Genetic testing | mFOLFOX | 86,359 | 0.66 | 0.41 | Ref. |
Pemigatinib | 286,890 | 2.53 | 1.65 | 162,259 | |
CADTH reanalysis 10: Relative dose intensity | mFOLFOX | 89,282 | 0.66 | 0.41 | Ref. |
Pemigatinib | 256,835 | 2.53 | 1.65 | 135,575 | |
CADTH reanalysis 11: mFOLFOX costs | mFOLFOX | 80,516 | 0.66 | 0.41 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.65 | 139,192 | |
CADTH reanalysis 1+2b+3+4+5+6+7b+ 8+9+10+11 (deterministic) | mFOLFOX | 78,735 | 0.72 | 0.49 | Ref. |
Pemigatinib | 274,862 | 1.79 | 1.24 | 264,674 | |
CADTH reanalysis 1+2b+3+4+5+6+7b+ 8+9+10+11 (probabilistic) | mFOLFOX | 77,945 | 0.72 | 0.49 | Ref. |
Pemigatinib | 276,099 | 1.82 | 1.25 | 261,226 |
ICER = incremental cost-effectiveness ratio; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-year; Ref. = reference; ToT = time on treatment; vs. = versus.
Note: results of all steps are presented deterministically. The cumulative CADTH base case is presented probabilistically, as well.
Table 16: Disaggregated Summary of CADTH’s Reanalysis Results (Pemigatinib vs. ASC)
Parameter | Pemigatinib | ASC | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 1.79 | 0.58 | 1.21 |
Progression-free | 0.82 | 0.48 | 0.34 |
Progressed disease | 0.97 | 0.10 | 0.87 |
Discounted QALYs | |||
Total | 1.24 | 0.41 | 0.83 |
Progression-free, on treatment | 0.58 | 0.34 | 0.24 |
Progressed disease, off treatment | 0.66 | 0.07 | 0.59 |
Discounted costs ($) | |||
Total | 276,480 | 66,895 | 209,585 |
Acquisition | 169,439 | 0 | 169,439 |
Administration | 0 | 0 | 0 |
Adverse events | 1,360 | 270 | 1,090 |
Resource use | 41,231 | 982 | 40,249 |
Terminal care | 64,451 | 65,643 | -1,192 |
ICER ($/QALY) | 252,718 | ||
ASC = active symptom control; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus.
Table 17: Disaggregated Summary of CADTH’s Reanalysis Results (Pemigatinib vs. mFOLFOX)
Parameter | Pemigatinib | mFOLFOX | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 1.82 | 0.72 | 1.09 |
Progression-free, on treatment | 0.82 | 0.33 | 0.48 |
Progression-free, off treatmenta | 0 | 0.14 | −0.14 |
Progressed disease | 1 | 0.25 | 0.75 |
Discounted QALYs | |||
Total | 1.25 | 0.49 | 0.76 |
Progression-free, on treatment | 0.58 | 0.23 | 0.35 |
Progression-free, off treatmenta | 0 | 0.10 | −0.10 |
Progressed disease | 0.68 | 0.17 | 0.51 |
Discounted costs ($) | |||
Total | 276,099 | 77,945 | 198,154 |
Acquisition | 169,439 | 5,339 | 163,868 |
Administration | 0 | 5,138 | −5,138 |
Adverse events | 1,359 | 953 | 406 |
Resource use | 41,237 | 1,143 | 40,095 |
Terminal care | 64,296 | 65,373 | −1,077 |
ICER ($/QALY) | 261,226 | ||
ICER = incremental cost-effectiveness ratio; LY = life-year; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; QALY = quality-adjusted life-year; vs. = versus.
aCADTH notes that in the sponsor’s model, the maximum time on treatment for mFOLFOX is set to be 24 weeks or so; after that, patients can remain progression-free but they all switch to the “progression-free without treatment” state at 25 weeks.

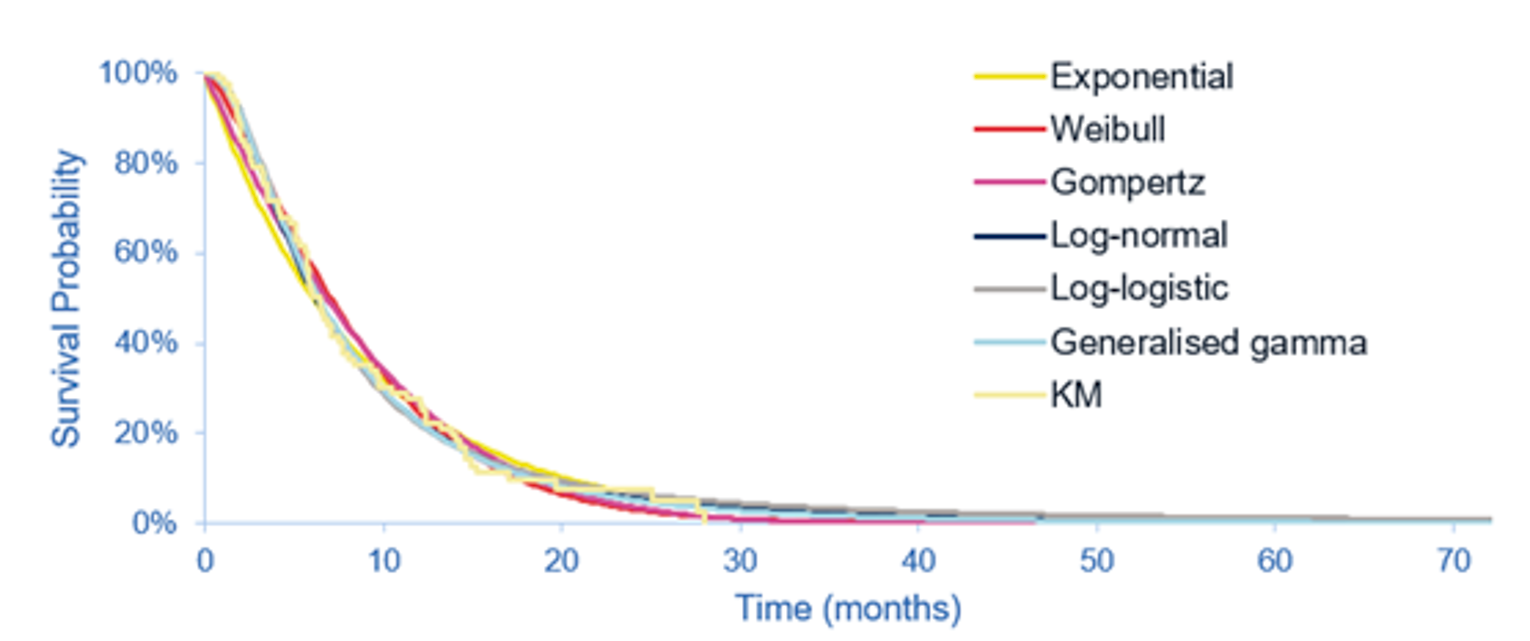
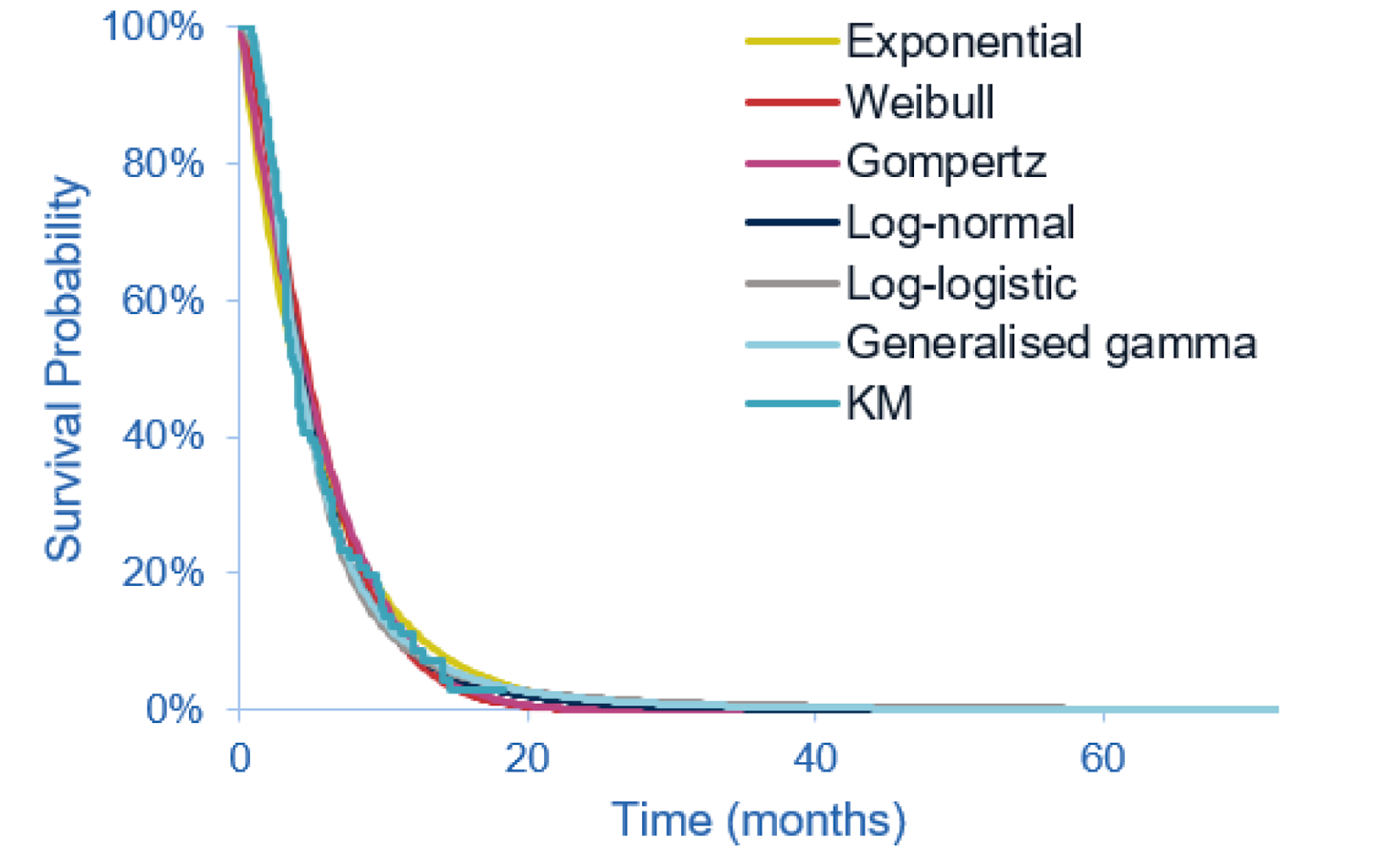
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 18: Summary of Key Takeaways
Key Takeaways of the Budget Impact Analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a budget impact analysis (BIA) estimating the budget impact of reimbursing pemigatinib for the treatment of previously treated, unresectable, locally advanced, or metastatic CCA patients with an FGFR2 fusion or rearrangement. The BIA base case was undertaken from a publicly funded drug plan perspective considering only drug costs over a 3-year time horizon. Pemigatinib costs were calculated by incorporating an RDI of ff observed in FIGHT-202. Stopping rules were applied for comparator treatments according to their respective clinical trials. Costs of subsequent therapies upon progression on second-line therapy were not included.
The analytic framework, which used an epidemiology-based approach, leveraged data from multiple sources in the literature and assumptions based on clinical expert input to determine the estimated population size (see Table 19). New patients were added to the BIA each year using a population growth rate of 4.4%.19 The sponsor compared a reference scenario where pemigatinib is not reimbursed as adjuvant therapy, with a new-drug scenario, where pemigatinib is funded as adjuvant therapy as per the Health Canada indication. Treatments available in the reference included mFOLFOX plus ASC, ASC alone, and clinical trial drugs. As it was assumed that all therapies will be taken with ASC, and ASC was assumed to not differ between comparators, costs for ASC were not assigned. A scenario analysis was conducted exploring drug costs along with costs of genetic testing and drug administration. Key inputs to the BIA are documented in Table 19.
Table 19: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3, if appropriate) |
|---|---|
Target population | |
Incidence of cholangiocarcinoma | 2.8 per 100,00020 |
Patients who are unresectable and eligible for first-line treatment with chemotherapy | 70%21 |
Percentage of patients completing genetic testing (including FGFR2) | 100%-Assumption |
Percentage FGFR2-positive | 19.24%22 (CCAs only) |
Percentage of patients treated with first line moving to second line | 100%-Assumption |
Percentage of eligible patients who move to third line | 52%23 |
Percentage of patients with public drug program coverage | 80%-Assumption |
Number of patients eligible for the drug under review | 143 / 149 / 154 |
Market uptake (3 years) | |
Uptake (reference scenario) ASC mFOLFOX + ASC Clinical trials | 45% / 45% / 45% 45% / 45% / 45% 10% / 10% / 10% |
Uptake (new-drug scenario) Pemigatinib ASC mFOLFOX + ASC Clinical trials | |||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||| 10% / 10% / 10% |
Cost of treatment (per patient) | |
Cost of treatment over 1 week Pemigatinib ASC mFOLFOX + ASC Clinical trials | $3,784.00 $0 $833.20 $0 |
ASC = active symptom control; FGFR2 = fibroblast growth factor receptor 2; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin.
Summary of the Sponsor’s BIA Results
The sponsor’s base case estimated the net budget impact of introducing pemigatinib for the treatment of previously treated, unresectable, locally advanced, or metastatic CCA patients with an FGFR2 fusion or rearrangement to be $5,241,637 in year 1, $10,944,538 in year 2, and $14,282,622 in year 3, for a total budget impact over 3 years of $30,468,797.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the results of the BIA:
The incidence-based approach to estimating market size may underestimate pemigatinib costs. The sponsor used an incidence approach to estimate the number of patients eligible for pemigatinib, along with applying an annual incidence growth rate. The sponsor’s approach only captures the costs associated with newly diagnosed patients and does not capture pemigatinib costs incurred by patients remaining on pemigatinib beyond the first year of treatment. This is inappropriate as in both the sponsor’s and CADTH’s pharmacoeconomic analysis, approximately 30% of patients receiving pemigatinib do so for more than 1 year.
As CADTH was unable to address this limitation, the CADTH base case likely underestimates the budget impact associated with reimbursing pemigatinib as costs are only captured for the first year of treatment with pemigatinib.
The uptake of pemigatinib is not aligned with clinical expert expectations. In the sponsor’s base case, it was assumed that ||| | ||| | ||| of eligible patients would uptake pemigatinib, should it become available. According to the clinical experts consulted by CADTH for this review, approximately 80% of patients are expected to initiate treatment upon pemigatinib becoming available, and this is expected to reach 90% to 100% by year 3.
In the CADTH reanalysis, the proportion of eligible patients who will use pemigatinib in year 1, 2, and 3 was changed to 80%, 85%, and 90%, respectively.
RDI implementation was inappropriate. The sponsor calculated an RDI of xx% based on the FIGHT-202 trial.24 CADTH was unable to validate this value as a means of accounting for dose interruptions. RDI was not reported in the sponsor’s clinical study report, however, treatment compliance was reported and found to be high, as concluded in the CADTH Clinical Review Report. Additionally, though patients may miss a dose of pemigatinib, this might not influence overall costs to public drug plans, as full drug claims will be dispensed. Finally, an RDI was not applied to mFOLFOX therapy.
CADTH reanalyses assumed an RDI of 100%.
The rate of growth of CCA is uncertain. The sponsor incorporated population growth in the model based on the growth in incidence of CCA.24 Two CCA growth rates were sourced from the literature: 1 for intrahepatic CCA (iCCA) (7%) and 1 for extrahepatic CCA (eCCA) (1.80%).19 The growth rate used in the sponsor’s base case was the mean of the iCCA and eCCA growth rates (4.40%).24 According to the CADTH Clinical Review Report, 98% of cohort A in FIGHT-202 had intrahepatic CCA.
To align with the expected patient population receiving pemigatinib, in the CADTH reanalyses, CCA growth rates were changed to those associated with intrahepatic CCA (7%).
Assigning clinical trials market shares is inappropriate. In the sponsor’s reference and new-drug scenario, 10% of eligible patients were assumed to be in clinical trials, which has no associated drug costs. This is inappropriate because it reduces the market size for the number of patients eligible for pemigatinib. Should pemigatinib become available and be deemed to be clinically effective, it is less likely that patients with FGFR2 fusions would enrol in clinical trials.
In CADTH reanalysis, 0% market share was assigned to clinical trials in the reference and new-drug scenario.
The percentage of patients diagnosed who are unresectable and eligible for first-line treatment with chemotherapy is not aligned with Canadian clinical practice. In the sponsor’s base case, 70% of diagnosed patients are unresectable and eligible for 1L treatment with chemotherapy.21 This value was derived from a study examining prognostic factors in advanced biliary tract cancers in the UK, which reported that 30% and 70% of patients had locally advanced and metastatic disease, respectively.21 According to clinical experts consulted for this review, 80% to 90% of patients will be unresectable and eligible for first-line chemotherapy at diagnosis.
CADTH reanalyses assumed that 85% of diagnosed patients are eligible for first-line chemotherapy to align with Canadian clinical practice.
Costs used for the mFOLFOX regimen were uncertain. Costs for the components for the mFOLFOX regimen were sourced from a previous CADTH review.12 CADTH found wholesale prices for mFOLFOX components from the IQVIA Delta PA database, which were deemed to be more appropriate. Additionally, the dose used for Leucovorin was inconsistent with that used in the ABC-06 trial.
The CADTH reanalysis used wholesale costs from IQVIA.
Genetic testing costs are not transparently programmed into the sponsor’s BIA model. While the sponsor’s BIA model allows for exploration of broader health care system costs like administration and genetic testing, this is conducted through a visual basics programmed scenario analysis. As such, it is unclear whether the scenario considers genetic testing for just pemigatinib patients or if patients receiving mFOLFOX and ASC also have genetic testing costs applied. As the costs of FGFR2 testing are not currently publicly covered, it is not expected that genetic testing costs will be covered for patients in the reference scenario or for patients who receive ASC and mFOLFOX in the new-drug scenario.
CADTH was unable to resolve this issue but notes the cost of additional testing may be substantial.
The proportion of patients eligible for public coverage is uncertain. The sponsor’s base-case analysis assumed that 80% of eligible patients in each jurisdiction would have public coverage.24 Intravenous oncology drugs are likely to be fully covered. Depending on the jurisdiction, oral oncology drugs may be fully reimbursed or may only be reimbursed by regular public drug plans. Neither of these scenarios is reflected in the sponsor’s base case.
To address uncertainty regarding the proportion eligible for public drug plan coverage, CADTH conducted a scenario analysis exploring 100% coverage across jurisdictions.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s base case by: increasing uptake of pemigatinib, changing the RDI to 100%, using the growth rate associated with iCCA, removing clinical trials market share from the reference and new-drug scenarios, assuming 85% of patients are diagnosed and unresectable, and changing mFOLFOX costs. Table 20 notes the assumptions used by the sponsor in comparison to those used by CADTH in its reanalysis.
Table 20: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case (none) | ||
Changes to derive the CADTH base case | ||
1. Uptake | |||||||||||||||||| | 80% / 85% / 90%a |
2. Relative dose intensity | ||||||||||||% | 100% |
3. Growth rate | 4.40% | 7% |
4. Clinical trials market share | 10% | 0% |
5. Percent diagnosed and unresectable | 70% | 85% |
6. mFOLFOX prices | Fluorouracil: $0.003/mg Oxaliplatin: $10.20/mg Calcium folinate: $0.05/mg | Fluorouracil: $0.03218/mg13 Oxaliplatin: $0.7254/mg13 Calcium folinate $1.378/mg13 |
CADTH base case | 1 + 2 + 3 + 4 + 5 + 6 | |
ASC = active symptom control; BIA = budget impact analysis; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin.
aThe market share for clinical trials was set to 0% in this step. The remaining market share was evenly distributed across ASC and mFOLFOX.
Applying these changes increased the total 3-year budget impact to $63,606,331. The results of the CADTH stepwise reanalysis are presented in summary format in Table 21 and a more detailed breakdown is presented in Table 22.
Table 21: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $30,468,797 |
CADTH reanalysis 1: Uptake 80% / 85% / 90% | $47,063,969 |
CADTH reanalysis 2: RDI 100% | $31,252,365 |
CADTH reanalysis 3: Growth rate 7% | $32,245,142 |
CADTH reanalysis 4: Removing clinical trials | $30,026,435 |
CADTH reanalysis 5: 85% unresectable | $36,997,825 |
CADTH reanalysis 6: mFOLFOX prices | $31,602,144 |
CADTH base case | $63,606,331 |
BIA = budget impact analysis; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; RDI = relative dose intensity.
CADTH also conducted additional scenario analyses to address remaining uncertainty:
Reduced the price of pemigatinib to the value in which it would be cost-effective at a $50,000 per QALY threshold (95 and 100% compared with mFOLFOX and ASC, respectively)
100% of the population is eligible for public coverage
Table 22: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|
Submitted base case | Reference | $1,270,369 | $1,326,265 | $1,384,621 | $3,981,255 |
New drug | $6,512,006 | $12,270,803 | $15,667,243 | $34,450,052 | |
Budget impact | $5,241,637 | $10,944,538 | $14,282,622 | $30,468,797 | |
CADTH base case | Reference | $845,650 | $904,845 | $968,184 | $2,718,679 |
New drug | $19,417,451 | $22,018,662 | $24,888,897 | $66,325,010 | |
Budget impact | $18,571,801 | $21,113,817 | $23,920,712 | $63,606,331 | |
CADTH scenario analysis 1a: 95% pemigatinib price reduction | Reference | $845,650 | $904,845 | $968,184 | $2,718,679 |
New drug | $1,131,546 | $1,229,874 | $1,336,422 | $3,697,842 | |
Budget impact | $285,896 | $325,028 | $368,238 | $979,163 | |
CADTH scenario analysis 1b: 100% pemigatinib price reductiona | Reference | $845,650 | $904,845 | $968,184 | $2,718,679 |
New drug | $169,130 | $135,727 | $96,818 | $401,675 | |
Budget impact | -$676,520 | -$769,118 | -$871,366 | -$2,317,004 | |
CADTH scenario analysis 2: 100% public drug coverage | Reference | $1,057,062 | $1,131,056 | $1,210,230 | $3,398,349 |
New drug | $24,271,814 | $27,523,328 | $31,111,121 | $82,906,263 | |
Budget impact | $23,214,752 | $26,392,271 | $29,900,891 | $79,507,913 |
BIA = budget impact analysis
aAlthough cost saving from a drug budget perspective there would still be incremental costs to the health system due to testing costs which are not included here.
Stakeholder Input
Patient Group Input
Authors of the submission: Durhane Wong-Rieger, Nem Maksimovic, Isabel Gardett
Canadian Liver Foundation, Canadian Organization for Rare Disorders, and the Cholangiocarcinoma Foundation
About the Patient Groups
Founded in 1969, the Canadian Liver Foundation (CLF) was the first organization in the world dedicated to supporting education and research into all forms of liver disease. Today, the CLF continues to be the only national health charity committed to reducing the incidence and impact for Canadians of all ages living with or at risk for liver disease. The CLF is the only registered charity in Canada directing funds specifically for liver disease research in all its forms and has invested more than $37 million in the scientific search for causes, preventative measures and potential treatments for liver disease. The CLF reaches millions of Canadians through our public and professional education programs, patient support programs and other awareness, fundraising and outreach efforts. Website: www.liver.ca
The Canadian Organization for Rare Disorders (CORD) is Canada’s national network for organizations representing all those with rare disorders. CORD provides a strong common voice to advocate for health policy and a healthcare system that works for those with rare disorders. CORD works with governments, researchers, clinicians and industry to promote research, diagnosis, treatment and services for all rare disorders in Canada. Website: www.raredisorders.ca
Founded in 2006 by a family who lost a loved one to cholangiocarcinoma, the Cholangiocarcinoma Foundation’s (CCF) mission is to find a cure and improve the quality of life for those affected by cholangiocarcinoma (bile duct cancer). CCF has grown to become the leading global resource in research, education, and public awareness. The CCF’s objective is finding a cure which relies on . research that provides essential resources and knowledge for the field and innovative research that opens new pathways for diagnosis and drug discovery. Website: www.cholangiocarcinoma.org
Information Gathering
Recruitment: Responses reflect direct patient input from two sources: online survey and a virtual focus group. Recruitment for the survey was targeted specifically to patients and caregivers affected by bile duct cancer (cholangiocarcinoma), especially those with FGFR2 gene infusions or rearrangements” by the Canadian Organization for Rare Disorders (CORD), the Canadian Liver Foundation (CLF), and Cholangiocarcinoma Foundation (based in the USA) through their patient databases and social media. The Cholangiocarcinoma Foundation (CCF) recruited and facilitated the virtual focus group of three Canadian patients diagnosed with cholangiocarcinoma with FGFR2 infusions.
Responses: Patients provided input through survey available on Survey Monkey from 24 June to 11 July 2021. The introduction specified that the purpose of the survey was to provide patient input to the Canadian Agency for Drugs and Technologies in Health (CADTH); however both Canadians and non-Canadians were invited to take part. There were 32 respondents, with 27 who completed the entire survey, and the feedback reported here reflects those 27 complete responses. Among these, 12 (44%) identified as Canadian; 13 (48%) as American, and 2 (7%) as “Other.” Canadian respondents reported home provinces as British Columbia, Alberta, Ontario, Quebec, and New Brunswick.
Among the 27 survey respondents, 15 (56%) identified as “diagnosed with bile duct cancer”; 10 (37%) as caregivers or family members; and two (7%) as having symptoms of bile duct cancer but not diagnosis). Additionally, 18 (67%) patients reported they had intrahepatic bile duct cancer; four (15%) had extrahepatic bile duct cancer; two (7%) had both forms; and three (11%) were unsure or preferred not to answer.
In terms of stage at time of diagnosis, survey respondents were somewhat “evenly” split across Stage IIA/B (n=7); Stage IIIA/B (n=6), and Stage IV (n=6), with two diagnosed at Stage I and seven who did not know. Additionally, 10 respondents (37%) reported their cancer was diagnosed as resectable and 11 (41%) said it was not; importantly, however, 12 (44%) said they did not know.
In terms of time since diagnosis, respondents were distributed relatively evenly across timeframes, with 26% diagnosed between 2 to 5 years ago, 4% more than 5 years ago, and 22% each for “less than 6 months” ago, “6 to 12 months” ago, and “1 to 2 years” ago.
When asked whether they had a diagnosis of tumour gene mutations, four (15%) responded that they had been diagnosed with FGFR2 fusions; none had been diagnosed with either NTRK fusions or IDH1 mutations; four (15%) said they had been diagnosed with other gene mutations; four reported they had no diagnosis of gene mutations; and the largest grouping, 15 (56%) did not know.
Age at time of diagnosis was somewhat evenly distributed across four groupings, with 30% over 65 years old, 26% under 45 years of age, 22% between 45 and 54 years old, and 19% between 55 and 64 years old. Overall, 58% of patients were female and 42% male.
While the number of respondents is small, we thought it worthwhile to note some of the key differences between Canadian and USA respondents in terms of age at diagnosis, time since diagnosis, and stage at time of diagnosis (see Table 1). Notably, Canadian patients were both younger and older in terms of age at diagnosis. The “time since diagnosis” seems to be about the same across borders, with more Americans diagnosed in the last six months. However, Canadians seem to be diagnosed at a later stage of cancer development, with most at Stages III and IV while Americans somewhat more likely to be at Stage II. The terms cholangiocarcinoma and bile duct cancer will be used interchangeably throughout this submission.
Table 1: Characteristics of Respondents in Canada cf. USA
Respondents | Age @ time of diagnosis | Time since diagnosis | Stage @ diagnosis | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
< 45 | 45- 54 | 55- 64 | > 65 | < 6 mos | 6-12 mos | 1-2 yrs | 2-5 yrs | > 5 yrs | Stage I | Stage IIA/B | Stage IIIA/B | Stage IV | Don’t know | |
Patients residing in Canada (n=12) | 25% | 8% | 17% | 43% | 17% | 25% | 33% | 25% | 0% | 8% | 17% | 33% | 25% | 17% |
Patients residing in USA (n=13) | 23% | 31% | 23% | 23% | 31% | 23% | 15% | 23% | 8% | 8% | 38% | 15% | 15% | 23% |
All respondents (n=25) | 24% | 20% | 20% | 32% | 24% | 24% | 24% | 24% | 4% | 8% | 28% | 24% | 20% | 20% |
To supplement direct patient feedback, some information from an abstract presented at the Gastrointestinal Cancers Symposium in 2020 on the diagnostic journey and life impacts of cholangiocarcinoma is included.
Disease Experience
Disease experience was elicited in the survey through (1) an open-ended question asking respondents to describe the experience of the patient and caregivers and (2) ratings along a predefined matrix of “problems or issues experienced by persons due to bile duct cancer.” Focus group members were also asked to describe their experience.
Overall, it was clear that cholangiocarcinoma has a major impact on the patients’ quality of life, including daily activities and relationships, as well as their mental well-being. The experiences of Canadian and American patients were highly similarly so the combined results are reported here. Based on the matrix of options, the problems that were rated as having the most impact (“very much” and “much”) were those related to “overall quality of life”, further articulated as participation in activities or relationships, including “family, social, work, and school” and “intimacy or sexual desire.” In terms of physical symptoms, “fatigue” was most problematic, rated as having “very much” or “much” impact by more than 80% of respondents. Likewise, about 80% reported experiencing “anxiety” “very much” to “somewhat” problematic. Other physical symptoms experienced by about two-thirds of respondents (67% to 72%) were “unintended weight loss” and “insomnia.” Gastrointestinal problems, “abdominal pain” and “constipation” were reported by a similar percentage. About two-thirds experienced “somewhat” to “very much” issues with “depression” and about 50% had experiences of “neuropathy.”
There findings echo those reported in the 2020 GCS abstract presentation, where a survey of cholangiocarcinoma patients found they experienced considerable or serious impact on daily lives, work productivity, quality of life, mental health, and sexual functioning. Additionally, patients reported having experience symptoms on average about two years prior to a diagnosis.
Importantly, the qualitative responses from the focus group and survey participants provide invaluable insights on the emotional and psychological toll of a “rare cancer” on patients and families. As is the case with many rare cancers, patients experienced delayed diagnosis and misdiagnosis which then delayed or eliminated some treatment options and contributed significantly to the stress and anxiety experienced.
“Months of misdiagnosis, centered on colo-rectal investigation (scans & scopes) due to symptoms being diarrhea and inability to digest food properly (progressive food intolerance's), after becoming rapidly jaundice, focus was re-directed and bile duct cancer diagnosed.”
“Doctor I first saw was extremely judgmental and focused on my weight and not on the cholangio thankfully I knew I had to get to a second opinion and maybe a third. That is critical and thankfully my family doc helped me secure appts with several leaders in cholangiocarcinoma. I was glad I got cc in the USA where we don’t have to fight for second opinions and then be declined.”
“Brutal. My dad went to the emergency room twice for abdominal pain. Both times he was told it was muscular and take something for acid. After 3 months of pain, he finally found a doctor that touched his liver and felt a mass.”
“The assumption that my husband is an alcoholic over and over again is annoying and disrespectful.”
“Had I not asked to have my cysts in my liver looked at closer with an ultrasound my Cholangiocarcinoma would not have been diagnosed.”
With cholangiocarcinoma, like many rare cancers, a diagnosis may often exacerbate rather than alleviate fears and anxieties, since there is little known about the condition and its prognosis, few specialists, few or no approved treatments, and often short life expectancy. Many patients do their own research but will then face barriers accessing promising options.
“Being diagnosed with a rare cancer is frightening and leads you down an uncertain path. There is very little information about the cancer itself, treatment options, and prognosis. It is unsettling to know that little research is being conducted for your particular cancer.”
“Then 6 weeks of test to finally learn he had 1 year to live and we were told he could get chemo if wanted to try to maybe slow progression.”
“Twin sister diagnosed with cat allergy by her GP. Intense itching and feeling unwell, vomiting. Stage 4 on diagnosis via A&E. Discovered it was cancer when an oncology secretary phoned to make an appt. Told terminal, emotional support zero. … No surgery or immunotherapy offered. No trials.”
“Up front patient experience was horrible. The disease is rare and therefore Drs give death sentences and minimal information on trials and or immunotherapy available. Drs do not talk about biopsies for mutations. All this is needed for trials to potentially prolong our lives.”
Respondents also reported having to do their own research to get a second opinion, access to appropriate tests, clinical trials, and treatment alternatives. It is not clear from such a small sample whether Canada lags other countries (USA in this case) in diagnosis and treatment but it is notable that the Canadian respondents report diagnosis at a later stage (III or IV) compared to Americans (Stage II).
“I went to my GP with a reoccurring pain in my upper right abdomen and after 3 months of tests I was diagnosed and giving 8 months to live. Sought second opinion and had surgery within 4 days of that meeting.”
“We are in Canada. We had to do research about gene mutation test cause no one mentioned that. We research protocols to find some hope. Started one with maybe keytruda (or placebo) with gem-cis at McGill. We investigated another protocol at CHUM and were told to forget about it and go home try to enjoy the rest.”
Finally, respondents spoke about the mental health impact on themselves and their concerns for their family and the lack of resources to address these.
“I feel alone, and scared, and hopeful.”
“That’s a game play every day. It’s hard. Some days I can go on and some days it’s just too hard. It is a struggle and my husband looks at me helplessly. Some days I am weak and some days I am good. It is a really tough game.”
“I try to leave it at the back of my mind at all times. I just try to live life daily without thinking about it. I talked to a psychiatrist for a bit but it did not help a lot. It was hard with my daughter, I am afraid to leave her. She will be alone. That is the worst thing that is on my mind is leaving her behind.”
“I’ve dealt with depression for a long time but I’ve been okay for a long time too. My concerns have been about my family. I am worried about my husband’s mental health. I have my late night cry, many nights. One of the most difficult parts is that I really feel fine and look fine, and it just seems very strange. You get through one day at a time. You keep going.”
“My father's diagnosis was very grim, but we have seen more hope from Facebook support groups for people with CC. We are all very emotional but positive.”
Experiences With Currently Available Treatments
Among the 27 survey respondents, 20 (74%) had or were currently receiving treatment, while seven (26%) had not received any treatment. Interestingly, only 67% of Canadians had received treatment while 85% of Americans had done so. Among the 20 who had received treatment, 60% had received surgery, 100% had or were currently receiving chemotherapy, and 20% had or were currently receiving radiation therapy. There was little or no different between Canadian and American responses. When presented with other potential treatments, only one patient (American) indicated receiving intra-arterial embolism and none of the Canadians or Americans had received any therapies targeted at specific gene mutations, with the exception of one Canadian who reported receiving pemigatinib (Pemazyre). In addition, two of the focus group participants (Canadian) were also receiving pemigatinib, all through special (compassionate) access from the manufacturer.
Given that bile duct cancer is very rare, highly aggressive, often diagnosed late, imminently life-threatening, and treatable with only a very few options, respondents were simply asked to describe in their own words the effectiveness of treatments experienced, tolerability of side effects, and their opinion as to whether treatment was “worthwhile”
There are two overarching themes regarding the experience of treatment that emerged from a content analysis of the responses. First, surgery, which is considered the 1st line option, was not always feasible or effective. Second, there are no chemotherapies specific for cholangiocarcinoma so patients experience considerable anxiety and uncertainty when undertaking course of treatment. There is limited reliable information about accessible treatment options on-line and many are highly reliant on their specialist to recommend and sometimes to source the best treatment option.
1. Resection (surgery) is worthwhile, if applicable, because it could “get rid” of the cancer. But it doesn’t always work.
“Surgery worthwhile as I was told by oncologist only way to cure this cancer is to cut it out"
“Not effective. Resection done in august 2020 with good margins, not found anywhere else. Chemo done to “just be sure”. May 2021 reoccurrence.”
2. Given the short life expectancy and the few treatment options, chemotherapy can be worth the side effects if it can reduce symptoms and extend life.
“The oncologist advised that 50% of patients diagnosed with this type of cancer, live up to 4 months; 10% up to 6 months and 5% up to a year.”
“The start of treatment (cisplatin and gemcitabine) with keytruda or placebo) really improved. Almost no more pain. No more need to take dilaudid.”
“Side effects are worth it if it will add years to your life. It's just hard to make plans around chemo because of side effects.”
“The treatments my husband received have been effective to date. The chemo was difficult to tolerate, but worth it since he is coming up on 3 years since diagnosis.”
“…the side effects on xeloda were tough...and I have developed trigger fingers in right hand quite bad requiring OT...and steroid injection...neuropathy in feet is bad...but I am alive and NED ...so thankful
“Somewhat effective. It's been over 12 years since diagnosis. I will be starting Folfox in 2 days, so I believe it's worth trying.”
Improved Outcomes
Responses on the effectiveness, side effectiveness, and value of available therapies point to the need for improved therapies and indeed improved outcomes but also convey the somewhat “resigned” and even “fatalistic” attitude of patients with rare conditions with limited research and development investment, few treatment options, and poor access to promising therapies. We identified the following themes on unmet needs.
1. There are not enough treatment options.
“Chemo has shrunk my tumour however I am developing toxicity so have to discontinue soon. Surgery is not an option and I have no mutations radiation is next attempt but beyond that not sure what options I have.”
“There seems to be a lot more options in the USA then here in Canada, not sure how effective they are…”
“Appalling options - death inevitable. It was worthwhile having some treatment as my sister was with us for a little longer, but it’s an evil disease.”
2. Quality of life is as important or more that quantity.
“I am definitely into quality of life over quantity. So far I’ve done ok managing symptoms.
”The oncologist also stated that the life expectancy might not change with treatment. The only reason that mom had undergone radiation, is the radiologist advised it might actually control the pain. The life experience remained the same.”
3. Given the ability to diagnosis gene mutations, there needs to be more research to develop more targeted therapies.
“I wish we had more options and it seems like now everyone is wanting to target FGFR which is only about 10% I believe of all patients...wish companies would move on and attempt to target other mutations that don't have a lot of meds ”
“From our experiences, chemo might not be a first option when there are more treatment options than before, especially when chemo alone doesn't seem very effective for bile duct cancer. Yet, taking oral drugs such as FGFR2 inhibitors or TKI secure much better quality of life without travelling to hospitals according to busy schedule.”
“The treatments take a toll on the physical capabilities but they are worthwhile. Since it is a genetic mutation we remain hopeful that the right targeted therapy will be developed.”
Experience With Drug Under Review
Given that a second targeted therapy for FGFR2 gene fusions (infigratinib or Truseltiq) was also being reviewed at this time by Health Canada (on the Project ORBIS collaborative framework), it made sense to ask about both of these drugs in this survey. Overall, about 12 (44%) of survey respondents did not know or were unsure they had been informed about targeted therapies for (any) gene mutations. About one-third (n=9) reported they had heard of either pemigatinib or infigratinib, or both, with a slightly higher proportion of Americans claiming knowledge that did Canadians. Two of the survey respondents (one Canadian and one American) and two of the focus group participants (both Canadian) reported direct experience with pemigatinib; none had experience with infigratinib. The focus group participants seemed to have accessed pemigatinib through compassionate access from the company.
“I had the FGFR2 mutation, so my doctor did what he could – I am on Pemazyre now – I don’t know what he had to jump through or do to get it here, but it took 3-4 months before he could give me the drug.”
Overall, respondents indicated they had to go through a period of adjustment (to get the right dosage) but overall had very little challenge dealing with the side effects of pemigatinib.
“First cycle was tough at 13.5 mg with multiple side effects, lowered to 9 for 2nd and 3rd cycles - this reduced side effects somewhat but not all. I am currently trying alternating 9/4.5 and all side effects have stopped except hair loss, while it is still falling out some places have started to grow back (weird). Feeling a lot better daily not having to deal with certain side effects, outlook on life hasn’t changed much as I know my time is limited. As I only have 1 daughter it is hard on her as she doesn’t have a sibling to talk with, she already had anxiety and depression before I was diagnosed.”
“I was okay for the most part with Pemazyre – I lost most of my hair, the first dosage was headaches, diarrhea, sore knuckles in fingers. I’ve tolerated most of it.”
Those survey respondents who indicated some knowledge about the targeted therapies were asked to discuss their expectations for the medication and what they believed or hoped it would do for them. Several also indicated that their expectations were based on feedback and discussions in cholangiocarcinoma (CC) support groups.
1. The first overriding theme that emerged was the “realistic but hopeful” expectation that the therapy would stabilize or reduce tumour size and disease progression.
“I am currently on Pemazyre … I am hoping for reduction in size of nodules and no new growth or at least stability.” “My expectations are that they will be as effective as possible at shrinking or stopping the growth of tumours.”
“I have seen great success with patients on Pemazyre. I know someone who was on the trial and has done exceptionally well on it.”
“Hope they would give remission and/or stability.”
“Just help and maybe control disease for a certain amount of time, which would be a lot!”
2. The second emergent theme was the hope that these therapies would stimulate development of more targeted therapies.
“I follow several CC support groups and read about folks who take these meds. Those who participate in these studies strongly encourage all CC patients to get genomic testing and apply for appropriate programs. Pemazyre seems especially well accepted. I have the IDH2 mutation and strongly hope for a targeted medication to treat it.”
“I was hoping for a second generation of FGFR2 inhibitors for when patients develop resistance to the current FGFR2.”
“I believe of all patients. wish companies would move on and attempt to target other mutations that don't have a lot of meds”
3. Third, not exactly a theme, but it would be unconscionable not include the hope that these therapies, because they are targeted, could have long-lasting benefits and perhaps even a cure.
“Hopefully they will prove to be effective in treatment and cure for CC.”
Companion Diagnostic Test
Access to targeted therapies requires diagnostic testing for specific genomic mutations to ensure the appropriate patients have access to the right therapy.
Anything Else?
These respondents are typical of the bile duct cancer community, where diagnosis is often incidental and delayed, referral to the right specialists is by chance rather than directed, treatment options are few and mostly ineffectual, access is made worse by the lack of knowledge among treaters and their unavailability in many settings, and little hope is offered. The identification of gene mutations and the development of therapies that target these are the best news possible at this time.
Survey respondents were provided a brief overview about the FGFR2 gene mutation and how it works in bile duct cancer, a brief description of the two targeted therapies for FGFR2 mutations and the clinical trial results with each.
They were asked to rate “…how important is it for bile duct cancer patients with FGFR2 fusions to have an option to access targeted therapies, if it is appropriate for them?”
All (100%) respondents indicated it was “very important” to have targeted therapies available. Some of the supporting comments were as follows.
“We need many options as everyone reacts differently with each treatment.”
“These meds can be a huge game changer and have proven success in trials. Patients with FGFR MUST be given access to these meds.”
“All cancer patients live on hope and these medications provide a substantial dose of that.”
Finally, all respondents were given the opportunity to provide any additional recommendations or comments.
“We need to educate more about this cancer in Canada, there are very few oncologists that specialize in the treatment as it is rare.”
“I am so glad that research on this cancer being done. This cancer is devastating lives, and young lives at that. At 70 years old, I feel I still have living to do and would happily try something that would help prolong my life if it doesn't mean quality of life is ruined. Targeted therapy sounds so much less invasive and more helpful in curing this cancer.”
“No question, if it works on some people, it’s still worth a try. Our drug approval systems are slow, that is the real problem. None of us can wait. This is an issue with time. We don’t have time to waste.”
“We need options. Options are what get us through. If I can get another 8 months of therapy that is doing some harm to the active cells, I will take it and hopefully during that time, a miracle would come along.”
“It would be awesome. I benefited somewhat. Definitely, we need options. Hopefully it will be all available to us at some point. We should all be able to get it.
“There needs to be more research done to find a cure. Money from the government should be used as it is used in other research for different cancers.”
“Canadians should have more options. It’s a shame, I read posts about people in India complaining about their limited options and it’s the same for us in Canada. I wish I was in the US.”
“These targeted therapies will help other people with cancer who currently do not have treatment or whose treatment does not work. The more research we do on targeted therapies and the more access people have to these therapies the more we will be able to advance in this field.”
Patient Groups’ Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
This submission was completed by the staff and volunteers of CORD, the CLF, and the CCF. Outside input for this submission came from the patients and caregivers who participated in interviews and those who responded to the online survey.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
Data collection and analysis was completed by the staff and volunteers of CORD, the CLF, and the CCF.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
The Canadian Liver Foundation (CLF) is committed to bringing liver research to life for all Canadians through liver research, education, patient support and advocacy. The CLF receives funding from a variety of sources with the majority coming from donations from individuals across the country. We use these funds to support CLF liver awareness, education, patient support and research grant programs.
The CLF receives some program funding in the form of unrestricted educational grants from pharmaceutical companies. Grant agreements are established in support of activities initiated by the CLF and prohibit the funder from having any input or influence in program objectives or deliverables.
Table 2: Conflict of Interest Declaration for the Canadian Liver Foundation
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None | — | — | — | — |
Table 3: Conflict of Interest Declaration for the Canadian Organization for Rare Disorders
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None | — | — | — | — |
Table 4: Conflict of Interest Declaration for the Cholangiocarcinoma Foundation
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Incyte Corporation | — | — | — | X |
Taiho Oncology | — | — | — | X |
QED Therapeutics | — | — | — | X |
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Name: Durhane Wong-Rieger
Position: President & CEO
Patient Group: Canadian Organization for Rare Disorders
Date: July 16, 2021
Clinician Group Input
Ontario Health (Cancer Care Ontario) Gastrointestinal Cancer Drug Advisory Committee (GI DAC)
Authors of the submission: Dr. Erin Kennedy, Dr. Jim Biagi, Dr. Tim Asmis, Dr. Christine Brezden-Masley
About Ontario Health (Cancer Care Ontario) Gastrointestinal Cancer Drug Advisory Committee (GI DAC)
Please describe the purpose of your organization. Include a link to your website (if applicable).
OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug- related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
Please describe how you gathered the information included in the submission.
This input was jointly discussed at a DAC meeting
Current Treatments
Describe the current treatment paradigm for the disease
Focus on the Canadian context. Please include drug and non-drug treatments. Drugs without Health Canada approval for use in the management of the indication of interest may be relevant if they are routinely used in Canadian clinical practice. Are such treatments supported by clinical practice guidelines? Treatments available through special access programs are relevant. Do current treatments modify the underlying disease mechanism? Target symptoms?
Response: 1L: Cis-Gem; 2L: FOLFOX; best supportive care or clinical trial if patients still suitable for treatment
Treatment Goals
What are the most important goals that an ideal treatment would address?
Examples: Prolong life, delay disease progression, improve lung function, prevent the need for organ transplant, prevent infection or transmission of disease, reduce loss of cognition, reduce the severity of symptoms, minimize adverse effects, improve health-related quality of life, increase the ability to maintain employment, maintain independence, reduce burden on caregivers.
Response: Prolong life, delay disease progression reduce the severity of symptoms minimize adverse effects, improve health-related quality of life
Treatment Gaps (Unmet Needs)
Considering the treatment goals in Section 4, please describe goals (needs) that are not being met by currently available treatments.
Examples: Not all patients respond to available treatments. Patients become refractory to current treatment options. No treatments are available to reverse the course of disease. No treatments are available to address key outcomes. Treatments are needed that are better tolerated. Treatment are needed to improve compliance. Formulations are needed to improve convenience.
Response: No treatment options available beyond 2L
Which patients have the greatest unmet need for an intervention such as the drug under review?
Would these patients be considered a subpopulation or niche population? Describe characteristics of this patient population. Would the drug under review address the unmet need in this patient population?
Response: FGFR2 fusions or other rearrangement
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Is there a mechanism of action that would complement other available treatments, and would it be added to other treatments? Is the drug under review the first treatment approved that will address the underlying disease process rather than being a symptomatic management therapy? Would the drug under review be used as a first-line treatment, in combination with other treatments, or as a later (or last) line of treatment? Is the drug under review expected to cause a shift in the current treatment paradigm?
Response: Pemigatinib will be a later or last line of treatment.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
If so, please describe which treatments should be tried, in what order, and include a brief rationale.
Response: Patients should have at least one line of systemic therapy previously.
How would this drug affect the sequencing of therapies for the target condition?
If appropriate for this condition, please indicate which treatments would be given after the therapy has failed and specify whether this is a significant departure from the sequence employed in current practice. Would there be opportunity to treat patients with this same drug in a subsequent line of therapy? If so, according to what parameters?
Response: NA
Which patients would be best suited for treatment with the drug under review?
Which patients are most likely to respond to treatment with the drug under review? Which patients are most in need of an intervention? Would this differ based on any disease characteristics (e.g., presence or absence of certain symptoms, stage of disease)?
Response: FGFR2 fusions or rearrangement population who received previous systemic therapy
How would patients best suited for treatment with the drug under review be identified?
Examples: Clinician examination or judgement, laboratory tests (specify), diagnostic tools (specify) Is the condition challenging to diagnose in routine clinical practice? Are there any issues related to diagnosis? (e.g., tests may not be widely available, tests may be available at a cost, uncertainty in testing, unclear whether a scale is accurate or the scale may be subjective, variability in expert opinion.) Is it likely that misdiagnosis occurs in clinical practice (e.g., underdiagnosis)? Should patients who are pre-symptomatic be treated considering the mechanism of action of the drug under review?
Response: Molecular sequencing (e.g., NGS or equivalent) will be needed to identify the FGFR2 alterations. Currently there is no routine testing for FGFR2 fusions or rearrangements.
Which patients would be least suitable for treatment with the drug under review?
Response: NA
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
If so, how would these patients be identified?
Response: NA
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Are the outcomes used in clinical practice aligned with the outcomes typically used in clinical trials?
Response: Clinical and radiographic assessment
What would be considered a clinically meaningful response to treatment?
Examples: Reduction in the frequency or severity of symptoms (provide specifics regarding changes in frequency, severity, and so forth). Attainment of major motor milestones. Ability to perform activities of daily living. Improvement in symptoms. Stabilization (no deterioration) of symptoms.
Consider the magnitude of the response to treatment. Is this likely to vary across physicians?
Response: Reduction in the frequency or severity of symptoms. Improvement in symptoms. Stabilization (no deterioration) of symptoms.
How often should treatment response be assessed?
Response: Routine clinical and radiographic evaluation
What factors should be considered when deciding to discontinue treatment?
Examples: Disease progression (specify; e.g., loss of lower limb mobility). Certain adverse events occur (specify type, frequency, and severity). Additional treatment becomes necessary (specify)
Response: disease progression or adverse events
What settings are appropriate for treatment with the drug under review?
Examples: Community setting, hospital (outpatient clinic), specialty clinic
Response: Community setting – take home cancer drug
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
If so, which specialties would be relevant?
Response: NA
Additional Information
Is there any additional information you feel is pertinent to this review?
Response: Companion diagnostics testing for FGFR2 fusions or rearrangements are not routinely done currently or expected in the near future. FIGHT-202 is a phase 2 study without a direct comparator. However, the results are clinically meaningful in a disease with limited treatment options.
Conflict of Interest Declarations for Ontario Health (Cancer Care Ontario) Gastrointestinal Cancer Drug Advisory Committee
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
OH-CCO provided secretariat support to the DAC in completing this input.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dr. Erin Kennedy
Position: Ontario Cancer Lead; surgeon
Date: 9-July-2021
Table 5: Declaration for OH-CCO Gastrointestinal Cancer Drug Advisory Committee Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None Declared | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Jim Biagi
Position: Medical Oncologist
Date: 9-July-2021
Table 6: Declaration for OH-CCO Gastrointestinal Cancer Drug Advisory Committee Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None Declared | — | — | — | — |
Declaration for Clinician 3
Name: Dr. Tim Asmis
Position: Medical Oncologist
Date: 9-July-2021
Table 7: Declaration for OH-CCO Gastrointestinal Cancer Drug Advisory Committee Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None Declared | — | — | — | — |
Declaration for Clinician 4
Name: Dr. Christine Brezden-Masley
Position: Medical Oncologist, Mount Sinai Hospital; Medical Director, Cancer Program for Sinai Health; Director, Marvelle Koffler Breast Centre, Mount Sinai Hospital; Senior Clinical Scientist, Lunenfeld-Tanenbaum Research Institute at Mount Sinai Hospital; Associate Professor of Medicine, University of Toronto
Date: 9-July-2021
Table 8: Declaration for OH-CCO Gastrointestinal Cancer Drug Advisory Committee Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None Declared | — | — | — | — |
The Canadian Gastrointestinal Oncology Evidence Network (CGOEN) and Other Cholangiocarcinoma-Treating Physicians
Authors of the Submission:
Dr. Vincent Tam, Medical Oncologist, Tom Baker Cancer Centre, Calgary. Disease site specialty: Gastrointestinal cancers.
Dr. Jennifer Knox, Medical Oncologist, Princess Margaret Cancer Centre, Toronto. Disease site Specialty: Gastrointestinal cancers.
Dr. Howard Lim, Medical Oncologist, BC Cancer Agency, Vancouver. Disease site specialty: gastrointestinal cancers.
Dr. Brandon Meyers, Medical Oncologist, Juravinski Cancer Centre, Hamilton, Disease site specialty(s): gastrointestinal cancers, head & neck cancers.
Dr. Ravi Ramjeesingh, Medical Oncologist, Dalhousie University, Halifax. Disease site Specialty: Gastrointestinal cancers, particular focus on hepatobiliary cancers
Dr. Eric Chen, Medical Oncologist, Princess Margaret Cancer Centre, Toronto. Disease site specialty: gastrointestinal cancers.
Dr. Sharlene Gill, Medical Oncologist, BC Cancer Agency, Vancouver. Specialty: Gastrointestinal (GI) malignancies.
Dr. Petr Kavan, Medical Oncologist, McGill University Health Centre. Disease site specialty: gastrointestinal (GI) cancers and neuroendocrine tumors (NETs).
Dr. Yoo-Joung Ko, Medical Oncologist, Sunnybrook Odette Cancer Centre, Toronto. Disease site specialty: gastrointestinal cancers.
Dr. Jennifer Spratlin, Medical Oncologist, Cross Cancer Institute, Edmonton. Specialty: GI malignancies.
About CGOEN and Other Cholangiocarcinoma-Treating Physicians
The Canadian GI Oncology Evidence Network (CGOEN) is a virtual and inclusive network of Canadian GI Oncology clinicians who contribute to the knowledge of GI cancer and its treatments, including participating in clinical trials, conducting observational research, and involvement in local/provincial and national clinical guideline development and health technology assessment. Some members of the clinician group participating in this submission are members of the International Cholangiocarcinoma Research Network (ICRN), a global collaboration of researchers and research centres working to improve knowledge about cholangiocarcinoma etiology, prevention, early detection, treatment, and prognosis. https://cholangiocarcinoma.org/international-cholangiocarcinoma-research-network/
Information Gathering
Please describe how you gathered the information included in the submission.
Information gathered for this submission was based on relevant data from the FIGHT-202 trial and expert evidence- based review by Canadian gastrointestinal cancer specialists.
Current Treatments
Describe the current treatment paradigm for the disease
Response: Cholangiocarcinoma (CCA) is a heterogeneous group of uncommon, fatal malignancies arising from the biliary tract with limited treatment options. The true incidence of CCA is not known because of difficulties in establishing diagnosis. The prognosis of CCA patients with metastatic disease is poor, with a median overall survival of less than a year, and a five year survival rate of < 20%
While surgical resection with negative margin offers the only potentially curative option, the majority of patients present at locally advanced or metastatic stages when surgical resection is not feasible. The incidence of CCA has increased globally over the past few decades, and the mortality rate remains high due to the aggressiveness of the disease and resistance to medical treatment.
With traditional chemotherapy regimens demonstrating limited effectiveness in CCA, research has focused on targeted treatments.
Standard treatment options for unresectable (including metastatic and recurrent) bile duct cancer include palliative therapy, chemotherapy, immunotherapy and targeted therapy. Palliative therapy can include relief of biliary obstruction through placement of bile duct stents. Palliative radiation may be beneficial for some patients.
Systemic chemotherapy is appropriate for select patients with the combination of cisplatin plus gemcitabine (CisGem) being the current standard of care first-line therapy. However, patients with poor performance status may not derive benefit from the doublet and the use of single-agent gemcitabine may be appropriate in these patients. Cisplatin may also be substituted by oxaliplatin in cases of renal impairment.
Second-line treatment usually consists of a fluoropyrimidine-based chemotherapy regimen such as FOLFOX, capecitabine or FOLFIRI. There is evidence of a small survival benefit with second-line FOLFOX compared to supportive care only.
The moderate survival benefit provided by CisGem has motivated much research aimed at identifying more effective treatments in this setting.
Currently, agents targeting FGFR2 fusion and IDH1/2 mutations hold great promise for improving the management of CCA. Pemigatinib, a fibroblast growth factor receptor (FGFR) 2 inhibitor, received (accelerated) approval in April 2020 by the US Food and Drug Administration (FDA) in CCA patients harboring FGFR2 gene fusions or other rearrangements and is the first targeted therapy to be approved for the treatment of CCA.
Treatment Goals
What are the most important goals that an ideal treatment would address?
Response: Extending survival, delaying disease progression and maintaining quality of life while on therapy are goals of current research into new treatments for the management of CCA.
Treatment Gaps (Unmet Needs)
Considering the treatment goals in Section 4, please describe goals (needs) that are not being met by currently available treatments.
Response: While CisGem is the standard of care first-line therapy for CCA it offers only moderate survival benefit with most of patients reporting a median survival of less than one year. Second-line treatment with FOLFOX improved survival to only 6·2 months compared to 5·3 months with supportive care alone.
Treatments for metastatic biliary cancer are needed to prolong survival for a larger period of time. Second-line treatments are required which have a meaningful survival benefit.
Which patients have the greatest unmet need for an intervention such as the drug under review?
The drug under review (pemigatinib) would benefit CCA patients harboring FGFR2 gene fusions or other rearrangements. FGFR2 fusions and rearrangements are found almost exclusively in intrahepatic cholangiocarcinoma occurring in 10–16% of patients (Abou-Alfa GK, Sahai V, Hollebecque A,Vaccaro G, Melisi D, Al-Rajabi R, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. The Lancet2020).
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Response: Currently 2nd line systemic treatments for patients with cholangiocarcinoma and FGFR2 fusions or rearrangements offer inadequate efficacy. Pemigatinib would offer these patients improved efficacy along with improved quality of life while on treatment.
As an oral drug, pemigatinib would also contribute to improved quality of life because it would require fewer visits by the patient to the cancer center, and less chair time for the patient.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
Response: The FIGHT-202 study was a phase 2 study to evaluate efficacy and safety of Pemigatinib in patients who had failed previous study. As such, it would continue to be appropriate to first treat patients with the standard of care front-line therapy before prescribing pemigatinib to CCA patients harboring FGFR2 gene fusions or other rearrangements.
It would also be reasonable to consider pemigatinib upfront for patients deemed unsuitable for cisplatin/gemcitabine as 1L therapy.
How would this drug affect the sequencing of therapies for the target condition?
Response: Refer to preceding response.
Which patients would be best suited for treatment with the drug under review?
Response: The drug under review (pemigatinib) would be best suited to CCA patients harboring FGFR2 gene fusions or other rearrangements, with an ECOG performance status of 0-2.
How would patients best suited for treatment with the drug under review be identified?
Response: Patients best suited for treatment should be prescreened for FGF/FGFR status using DNA sequencing.
Which patients would be least suitable for treatment with the drug under review?
Response: Biliary cancer patients without FGFR2 fusions or rearrangements and patients with compromised hepatic function or significant hyperbilirubinemia would be least suitable for treatment with pemigatinib.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
Response: Intrahepatic cholangiocarcinoma (iCCA) has highly actionable genomic targets including mutations in Fibroblast Growth Factor Receptor (FGFR), particularly FGFR2. Comprehensive genomic profiling (CGP), the backbone for precision oncology, opens the opportunity for tailored therapies such as pemigatinib.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Response: In clinical practice, the patient’s clinical condition and CT imaging are used to determine whether a patient is responding to treatment. If a patient is symptomatic from their cancer and a treatment results in improvement in the symptom then this may be an indication of response. The most objective measurement is CT imaging to compare the sizes of the primary cancer and metastases. CT imaging response is frequently used to assess outcomes in clinical trials.
What would be considered a clinically meaningful response to treatment?
Response: A clinically meaningful response to treatment would be maintenance or improvement in quality of life and prolongation of survival.
How often should treatment response be assessed?
Response: Treatment response and tolerance of the treatment should be assessed clinically every 3 weeks and response should be assessed radiographically with CT imaging every 2-3 months.
What factors should be considered when deciding to discontinue treatment?
Response: Patients would discontinue treatment if there is clear evidence of cancer progression on imaging, poor tolerance of the treatment which cannot be improved with dose delays or reductions, or patient preference to stop treatment.
What settings are appropriate for treatment with the drug under review?
Examples: Community setting, hospital (outpatient clinic), specialty clinic
Response: Pemigatinib can be taken at home as prescribed by a medical oncologist.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
Response: None
Additional Information
Is there any additional information you feel is pertinent to this review?
Response: We acknowledge that the FIGHT-202 study which supports the use of pemigatinib in the second-line setting is a phase 2 study. A phase 3 study in this particular setting will most likely never be done due to the rarity of this indication.
FGFR2 mutation status will need to be assessed in cholangiocarcinoma patients being considered for pemigatinib and there is current no funded mechanism for this in Canadian provinces. With respect to comprehensive genomic profiling (CGP), over 20 cancer therapies linked to over 15 genomic biomarkers have been approved in Canada, with many more quickly emerging. This emphasizes the growing value of precision medicine in cancer care and reinforces the need for all Canadians to have access to these therapies and the molecular tests needed to prescribe them. A plan for publicly funding CGP in patients with cancer is crucial for enabling our healthcare system to keep pace with rapidly evolving molecular testing needs (Impact Mediocom Inc with Lim, H., Sheffield, B., Karachiwala, H.,Leighl, N., Doherty, M., Sehdev, S., Slater, J. Determining Priority Access To Comprehensive Genomic Profiling For Canadian Patients With Cancer. https://www.impactmedicom.com/publications/report).
Conflict of Interest Declarations for Canadian Gastrointestinal Oncology Evidence Network (CGOEN) and Other Cholangiocarcinoma-Treating Physicians
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
None.
Declaration for Clinician 1
Name: Dr. Jennifer Knox
Position: Medical Oncologist, Princess Margaret Cancer Centre
Date: 13-07-2021
Table 9: Declaration for Canadian Gastrointestinal Oncology Evidence Network and Other Cholangiocarcinoma-Treating Physicians Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None Declared | — | — | — | — |
Declaration for Clinician 2
Name: Howard Lim
Position: Medical Oncologist, BC Cancer Agency
Date: 13-07-2021
Table 10: Declaration for Canadian Gastrointestinal Oncology Evidence Network and Other Cholangiocarcinoma-Treating Physicians Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None Declared | — | — | — | — |
Declaration for Clinician 3
Name: Vincent Tam
Position: Medical Oncologist, Tom Baker Cancer Centre
Date: 13-07-2021
Table 11: Declaration for Canadian Gastrointestinal Oncology Evidence Network and Other Cholangiocarcinoma-Treating Physicians Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Incyte Biosciences Canada | X | — | — | — |
Declaration for Clinician 4
Name: Brandon Meyers
Position: Medical Oncologist, Juravinski Cancer Centre
Date: 13-07-2021
Table 12: Declaration for Canadian Gastrointestinal Oncology Evidence Network and Other Cholangiocarcinoma-Treating Physicians Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None Declared | — | — | — | — |
Declaration for Clinician 5
Name: Eric Chen
Position: Medical Oncologist, Princess Margaret Cancer Center
Date: July 14, 2021
Table 13: Declaration for Canadian Gastrointestinal Oncology Evidence Network and Other Cholangiocarcinoma-Treating Physicians Clinician 5
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Taiho | X | — | — | — |
Eisai | X | — | — | — |
Roche | X | — | — | — |
Declaration for Clinician 6
Name: Petr Kavan MD, PhD
Position: Medical Oncologist, co-chairmen GI oncology tumor site McGill
Date: 13-07-2021
Table 14: Declaration for Canadian Gastrointestinal Oncology Evidence Network and Other Cholangiocarcinoma-Treating Physicians Clinician 6
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 7
Name: Yoo-Joung Ko
Position: Medical Oncologist, Sunnybrook Odette Cancer Centre
Date: 13-07-2021
Table 15: Declaration for Canadian Gastrointestinal Oncology Evidence Network and Other Cholangiocarcinoma-Treating Physicians Clinician 7
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None Declared | — | — | — | — |
Declaration for Clinician 8
Name: Sharlene Gill
Position: Medical Oncologist, BC Cancer - Vancouver
Date: 13-07-2021
Table 16: Declaration for Canadian Gastrointestinal Oncology Evidence Network and Other Cholangiocarcinoma-Treating Physicians Clinician 8
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No relevant disclosures | — | — | — | — |
Declaration for Clinician 9
Name: Ravi Ramjeesingh
Position: Medical Oncologist, Department of Medicine, Dalhousie University
Date: 07/14/2021
Table 17: Declaration for Canadian Gastrointestinal Oncology Evidence Network and Other Cholangiocarcinoma-Treating Physicians Clinician 9
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Roche | X | — | — | — |
Novartis | X | — | — | — |
Ipsen | X | — | — | — |
Amgen | X | — | — | — |
Eisai | X | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.