CADTH Reimbursement Review
Tralokinumab (Adtralza)
Sponsor: LEO Pharma Inc.
Therapeutic area: Atopic dermatitis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AAD
American Academy of Dermatology
AD
atopic dermatitis
AE
adverse event
AUC
area under the curve
CDA
Canadian Dermatology Association
CI
confidence interval
COVID-19
coronavirus disease 2019
CSPA
Canadian Skin Patient Alliance
DLQI
Dermatology Life Quality Index
EASI
Eczema Area and Severity Index
EASI-50
50% or greater reduction in Eczema Area and Severity Index from baseline
EASI-75
75% or greater reduction in Eczema Area and Severity Index from baseline
EASI-90
90% or greater reduction in Eczema Area and Severity Index from baseline
EQ-5D-5L
EQ-5D 5-Levels questionnaire
EQ VAS
EQ-5D Visual Analogue Scale
ESC
Eczema Society of Canada
FAS
full analysis set
HADS
Hospital Anxiety and Depression Scale
ICER
Institute for Clinical and Economic Review
IGA
Investigator’s Global Assessment
IL
interleukin
IMP
investigational medicinal product
ITC
indirect treatment comparison
JAK
Janus kinase
MAIC
matched adjusted indirect comparison
MAR
missing at random
MID
minimal important difference
NMA
network meta-analysis
NRS
numeric rating scale
OCS
oral corticosteroids
ODC
Origins Dermatology Centre
PGA
Physician’s Global Assessment
POEM
Patient-Oriented Eczema Measure
PP-NRS
peak pruritus numeric rating scale
RCT
randomized controlled trial
SAE
serious adverse event
SCORAD
Scoring of Atopic Dermatitis
SCORAD-50
50% decrease in Scoring Atopic Dermatitis
SCORAD-75
75% decrease in Scoring Atopic Dermatitis
SD
standard deviation
SF-36
Short Form (36) Health Survey
TCI
topical calcineurin inhibitor
TCS
topical corticosteroids
UVA
ultraviolet A
WPAI-GH
Work Productivity and Activity Impairment–General Health
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Tralokinumab (Adtralza), solution for subcutaneous injection, 150 mg single-use pre-filled syringe (150 mg per 1 mL) |
Indication | For the treatment of moderate to severe atopic dermatitis in adult patients whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable; Tralokinumab can be used with or without topical corticosteroids |
Reimbursement request | For the treatment of adult patients moderate to severe atopic dermatitis whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable and who had an inadequate trial or are ineligible for each of the following therapies: phototherapy (where available), methotrexate, and cyclosporine |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | October 13, 2021 |
Sponsor | LEO Pharma Inc. |
NOC = Notice of Compliance.
Introduction
Atopic dermatitis (AD) is the most common type of eczema. It is a chronic, relapsing, inflammatory skin condition characterized by severely itchy skin (pruritus) that results in red and swollen skin (rash). Lesions associated with AD may appear as fluid-filled vesicles that ooze, crack, and crust. Pruritus of the skin can cause frequent scratching and may result in lichenification (thickening of the skin) and secondary skin infections. Atopic dermatitis typically involves the skin folds behind the knees (popliteal areas) and in front of the elbows (antecubital areas). It may also appear on the face, neck, and hands. Individuals with AD have skin with impaired barrier function and reduced water-holding capacity, resulting in dry skin that requires treatment with specific bathing, cleansing, and moisturizing practices.
The goals of AD management are to prevent flares (episodes of worsening symptoms typically requiring escalation of treatment), and effectively manage flares when they occur by preventing disease progression. While there is no cure for AD, several therapeutic options are available to patients to manage the condition. The majority of patients treat AD by using general skin care methods, avoiding skin irritants, and applying topical anti-inflammatory treatments. If these common methods fail to improve AD, patients may use off-label systemic (i.e., immunosuppressant) therapy or other therapies such as phototherapy.
The most common pharmaceutical topical therapies include the use of topical corticosteroids (TCS) and topical calcineurin inhibitors (TCIs). The former act as anti-inflammatory therapies and are considered first-line treatment for AD.2 The latter are steroid-free, anti-inflammatory, immunosuppressant drugs that can be used long-term. In Canada, the 2 available second-line drugs are pimecrolimus and tacrolimus. Crisaborole, a topical phosphodiesterase type 4 inhibitor, is also available in Canada, although it is not recommended by CADTH for reimbursement.1,2 Phototherapy is another second-line therapy that is commonly used after failure of TCS, TCIs, and crisaborole.16
Systemic therapy for AD typically involves the use of antimicrobials, antihistamines, or immunomodulators.15-17 Immunomodulatory drugs, including methotrexate, cyclosporine, mycophenolate mofetil, azathioprine (listed in order of frequency of use in Canada), can be used in patients who are not responsive to other treatments.13,15,16 Dupilumab (Dupixent) is an interleukin (IL)-4 and IL-13 inhibitor indicated for use in adults and pediatric patients with moderate to severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. CADTH recommended that dupilumab be reimbursed with conditions and it is currently reimbursed by participating drug programs for patients whose AD is inadequately controlled with topical prescription therapies and who have demonstrated failure or intolerance to an adequate trial of phototherapy (where available), methotrexate, and cyclosporine.3
Tralokinumab is indicated for the treatment of moderate to severe AD in adult patients whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. It is administered by subcutaneous injection into the thigh or abdomen, and the recommended dosage for adult patients is an initial dose of 600 mg (4 injections of 150 mg) followed by 300 mg (2 injections of 150 mg each) administered every other week. At the prescriber’s discretion, dosing every fourth week may be considered for some patients who achieve clear or almost-clear skin after 16 weeks of treatment. Tralokinumab can be used with or without TCS. Tralokinumab may be used with TCIs.
The objective of this review was to perform a systematic review of the beneficial and harmful effects of tralokinumab for the treatment of moderate to severe AD in adult patients (> 18 years of age) whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from clinical expert(s) consulted by CADTH for the purpose of this review.
Patient Input
CADTH received 2 patient group submissions for the review of tralokinumab for AD: 1 from the Eczema Society of Canada (ESC) and a joint submission from the Canadian Skin Patient Alliance (CSPA) and Eczéma Québec. The ESC conducted a survey and interviews regarding how AD affects quality of life, experiences with symptoms and treatments, and the patient journey. The group received more than 3,000 responses from adults living with AD as well as their caregivers and family. The CSPA and Eczéma Québec created a web-based survey, to which 26 adults (85% patients and 8% caregivers) responded. The joint submission also included information from 56 Canadians with AD and caregivers who participated in health technology assessment surveys and interviews regarding Janus kinase (JAK) inhibitor treatments.
Patients who responded to the ESC survey described itching as the most burdensome symptom, with 72% and 95% of patients with moderate and severe AD, respectively, reporting feeling itchy multiple times a day. Moreover, 44% of respondents with severe disease were itchy all the time, and more than half of the respondents described being unable to control the urge to scratch their skin, and that it could be overwhelming and uncontrollable. Flares of worsening symptoms such as extreme itching and pain frequently led to loss of sleep. For instance, 63% and 86% of patients with moderate and severe AD, respectively, reported sleep disruptions and half of respondents with severe AD had lost sleep at least 8 nights per month. In the CSPA and Eczéma Québec submission, nearly all of those with AD experienced itching (98%), skin redness (91%), repeated rashes (87%), frequent scratching (87%), cracked skin (87%), and dry and rough skin (81%).
Patients expressed interest in new therapies that could reduce those symptoms. Those with moderate or severe AD noted the importance of having a medication that provided long-term relief and improved their quality of life. Respondents also indicated that new treatments should be covered by insurance or be affordable, allow them to stop using topical therapies, and be low-maintenance and not very time-consuming. Among the CSPA and Eczéma Québec respondents, 64% reported that it was important AD treatments did not require injections, while the other 34% were indifferent or reported it was not important.
In the ESC submission, patients reported accessing tralokinumab through a clinical trial, and many reported that it had significantly alleviated their pain, itching, discomfort, and the frequency of flares. Some patients reported experiencing improvements in 4 to 6 weeks, while others noted changes took a few months. According to patients who were interviewed by the ESC, an injectable medication is generally considered to be simpler and more convenient than other skin care routines and topicals, which can be messy and painstaking to administer. Some patients raised concerns over a fear of needles but anticipated they would be able to overcome this challenge.
Clinician Input
Input From Clinical Experts Consulted by CADTH
Two clinical experts with expertise in the diagnosis and management of AD consulted by CADTH indicated that some patients with moderate to severe AD respond to the current therapies; however, there is a need for additional therapies for patients with inadequate access to phototherapy or patients who experienced side effects with systemic therapies such as methotrexate and cyclosporine A. The clinical experts also stated that tralokinumab would complement other therapies and can be added to other treatments (excluding dupilumab, as both treatments act on similar receptors) such as TCS. The clinical experts indicated a trial of an appropriate topical therapy should be considered first before considering therapies such as tralokinumab, particularly because of the cost associated with tralokinumab. The clinical experts differed on the place in therapy of tralokinumab, with 1 clinical expert indicating that tralokinumab may offer a safer and more effective treatment option compared with the off-label systemic therapies currently available. The other clinical expert disagreed with that statement due to the lack of long-term evidence of safety and the fact that trials results were not encouraging. The clinical experts indicated that patients who would be best suited for treatment with tralokinumab are those with moderate to severe AD who have not responded to an adequate trial of topical therapies and an adequate trial of phototherapy. In terms of assessing response to treatment, the clinical experts were not aware of useful predictors of a good response to tralokinumab, but suggested that a clinically meaningful response would include improvements in quality-of-life scores, itch scores, and clinical scores from an Investigator’s Global Assessment (IGA) or the Eczema Area and Severity Index (EASI). One clinical expert indicated that the treatment response should be assessed monthly early on in treatment, and every 3 to 6 months later in the course of treatment, while the other noted that the response to tralokinumab should not be assessed earlier than 16 weeks, and that responders should be assessed every 6 months. According to both clinical experts, the factors to consider for discontinuation would be a lack of efficacy and adverse effects (e.g., severe conjunctivitis unresponsive to treatment measures). The clinical experts indicated that it would be reasonable to have a dermatologist diagnose, treat, and monitor patients receiving tralokinumab. The clinical experts indicated that tralokinumab is not expected to cause a dramatic shift in the current treatment paradigm but may present an additional option in the class of biologic therapies. Dupilumab has already established a precedent in this class of therapies.
Clinician Group Input
Input from 2 individual clinicians was received for the review of tralokinumab. One clinician, a dermatologist practising in British Columbia, provided input on behalf of the Canadian Dermatology Association (CDA) and the other was a dermatologist who practises at the Origins Dermatology Centre (ODC) in Saskatchewan. One clinician advised that tralokinumab would be used in first-line settings, while the other advised that topicals should be used as first-line therapy followed by phototherapy and then systemics, including biologics. According to the clinician input, tralokinumab would be relevant to clinical practice as 2/3 of patients who are treated with dupilumab do not achieve clear skin, creating a need for additional systemic medications with different mechanisms of action. Additionally, both clinicians emphasized that there is a need for treatments to be convenient and durable for patients. One clinician noted that this issue is of particular concern for Indigenous populations living in remote areas. These patients are often hard to reach virtually and have limited access to health care, which makes it extremely difficult monitor patient safety while they are on treatment with traditional systemic immunosuppressants. The clinician emphasized that traditional immunosuppressants can lead to side effects such as worsening of infection, cytopenias, and liver damage, for which many people on reserves and remote areas may not be able to receive adequate follow-up care. Both clinicians stated that patients with moderate to severe AD who do not respond to topicals and phototherapy have a high unmet need for this drug. Additionally, 1 clinician noted that women of childbearing age also have an unmet need as most off-label systemics are teratogenic.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review processes. The following were identified as key factors that could affect the implementation of a CADTH recommendation for tralokinumab:
Access to phototherapy may be limited in some areas of Canada. One clinical expert consulted by CADTH noted that phototherapy is typically accessible in urban areas, but access may be limited in rural areas. The expert noted that this barrier to phototherapy should be considered in the reimbursement review decision-making process.
Could tralokinumab be initiated in patients who have failed previous treatment with a biologic drug? One clinical expert noted that it is unlikely that they would initiate tralokinumab with patients who had failed a biologic, and they would more likely prescribe JAK inhibitors instead.
Should patients be required to have been enrolled in a previous trial of (or be ineligible for) cyclosporine, methotrexate, and phototherapy before initiating treatment with tralokinumab? One clinical expert consulted by CADTH noted that patients should follow the standard hierarchy of treatments, with biologics being the second-line treatment, and that a trial of 2 of the 4 immunomodulators (methotrexate, cyclosporine, mycophenolate mofetil, and azathioprine) should be considered before initiating tralokinumab.
Could the reimbursement criteria that were recommended for dupilumab (e.g., initiation and renewal criteria) be applicable to tralokinumab? One clinical expert consulted by CADTH noted that the criteria for dupilumab could be applicable for tralokinumab and could be implemented in clinical practice.
Should patients be required to undergo an adequate trial with dupilumab before being eligible for treatment with tralokinumab? One clinical expert consulted by CADTH noted that prior therapy with dupilumab should not be required for patients to be eligible for treatment with tralokinumab. Both experts agreed that tralokinumab should not be a rescue therapy for failed previous treatment with a biologic. It would be preferable to use tralokinumab first, and if it fails, then use dupilumab or JAK inhibitors.
On the question of whether tralokinumab should be used in combination with other JAK inhibitors, biologic disease-modifying antirheumatic drugs, phototherapy, or immunosuppressants, the clinical experts disagreed. One expert would not use tralokinumab with JAK inhibitors, but was not aware of any contraindications when combining tralokinumab with immunosuppressants or phototherapy. The other expert indicated that the only practical combination would be phototherapy and tralokinumab.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
The evidence for this review was derived from a systematic literature review of pivotal and phase III studies that was supplemented with additional studies to address important gaps in the evidence from randomized controlled trials (RCTs). The systematic review included 4 double-blind phase III RCTs.
Both the ECZTRA 1 (N = 802) and ECZTRA 2 (N = 794) trials were randomized, double-blind, placebo-controlled, identically designed, 52-week trials that evaluated the efficacy and safety of tralokinumab as a monotherapy compared to placebo in adults with moderate to severe AD. The studies had 3 key phases: an initial treatment phase (0 to 16 weeks), a maintenance treatment phase (16 to 52 weeks), and a safety follow-up (52 to 66 weeks). All patients used an emollient twice daily (or more often, as needed) for at least 14 days before randomization and were to continue this treatment throughout the trial. Patients were randomized in the initial treatment phase in a 3:1 ratio to either biweekly 300 mg tralokinumab injections (following the baseline 600 mg loading dose on day 0) or to a placebo administered every 2 weeks. At week 16, patients who achieved a clinical response (defined as an IGA score of 0 or 1, or at least 75% reduction in an EASI score from baseline [EASI-75]) and who were assigned to the tralokinumab group in the initial treatment phase were re-randomized in a 2:2:1 ratio to biweekly 300 mg tralokinumab injections, tralokinumab 300 mg every 4 weeks (alternating biweekly doses of placebo and 300 mg tralokinumab injections), or placebo. The primary outcomes were the percentage of patients achieving an IGA response of 0 (clear skin) or 1 (almost-clear skin) and the percentage of patients achieving an EASI-75 score at week 16, with secondary end points addressing symptom severity on the Scoring of Atopic Dermatitis (SCORAD), itch severity (worst daily pruritus numeric rating score [NRS]), and health-related quality of life (HRQoL) measures related to AD. Of the patients enrolled in the ECZTRA 1 trial, the overall mean age at baseline was 38.8 years, and 59.1% of the trial population were men. The mean body surface area involvement with AD was 53.1%, |||||||||||||||||||||||||||||||||||||||||||||| and the duration of AD was 28.3 years. Of the patients enrolled in the ECZTRA 2 trial, the mean age at baseline was 36.7 years, and 59.6% of the total trial population were men. At baseline, the mean body surface area involvement with AD was 52.7%, |||||||||||||||||||||||||||||||||| and the mean duration of AD was 28.1 years.
The ECZTRA 3 study (N = 380) was a randomized, double-blind, placebo-controlled, 32-week trial that evaluated the efficacy and safety of tralokinumab as a combination therapy with TCS compared to placebo plus TCS in adults with moderate to severe AD. All patients were to use an emollient twice daily (or more often, as needed) for at least 14 days before randomization and were to continue this treatment throughout the trial. The trial had a 16-week initial treatment period followed by an additional 16-week continuation period. On day 0 of the initial treatment period, patients received a loading dose of 600 mg of tralokinumab or placebo. In the initial treatment period, 380 patients were randomized in a 2:1 ratio to receive subcutaneous doses of tralokinumab or placebo every second week during the 16-week initial treatment period. At baseline, all patients were instructed to initiate treatment once daily with a supplied TCS (mometasone furoate 0.1% cream) on lesional skin and continue as needed throughout the trial. Patients randomized to tralokinumab in the initial treatment period who had a clinical response (defined as an IGA score of 0 or 1, or an EASI-75 score) at week 16 were re-randomized to the continuation treatment period in a 1:1 ratio, stratified by region (Europe and North America) and IGA response at week 16 (IGA 0 or 1, or IGA > 1): tralokinumab 300 mg every 2 weeks, or tralokinumab 300 mg every 4 weeks (alternating dose administrations of tralokinumab 300 mg and placebo). The trial evaluated the percentage of patients achieving an IGA response of 0 (clear) or 1 (almost clear) (IGA 0 or 1) and the percentage of patients achieving an EASI-75 score at week 16 (primary end points). The mean age at baseline was 39.1 years. In the group receiving tralokinumab every 2 weeks plus TCS, men and women were equally distributed. In the placebo plus TCS group, there was a higher proportion of men than women (66.1% versus 33.9%, respectively). Most patients (75.8%) were White. The mean body surface area involvement with AD was 48.1%, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| and the mean duration of AD was 28.2 years.
The ECZTRA 7 study (N = 277) was a randomized, double-blind, placebo-controlled, 26-week trial that evaluated the efficacy and safety of tralokinumab as a combination therapy with TCS compared to placebo plus TCS in adults with severe AD who were not adequately controlled with or have contraindications to oral cyclosporine A. Patients were randomized in a 1:1 ratio to receive tralokinumab 300 mg plus TCS or placebo plus TCS. The randomization was stratified by prior cyclosporine A use (yes or no), country (Germany: yes or no), and baseline disease severity (IGA = 3 or 4). All patients were instructed to use a supplied TCS (mometasone furoate 0.1% cream) once daily, as needed, on lesional skin during the treatment period. Each patient received a loading dose of 600 mg of tralokinumab or placebo. At subsequent visits in the treatment period, patients received either tralokinumab 300 mg every 2 weeks or placebo every 2 weeks. The trial evaluated the percentage of patients achieving an EASI-75 score at week 16 (the primary end point). |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| There was a higher proportion of men than women (59.6% versus 40.4% patients, respectively). Most patients (98.2%) were White. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Efficacy Results
Treatment with tralokinumab elicited a statistically significant improvement in markers of AD severity, such as IGA and EASI, at 16 weeks in adults with moderate to severe AD. For participants who achieved an IGA score of 0 or 1 at week 16, the difference between tralokinumab and placebo was 8.6% in the ECZTRA 1 trial (95% confidence interval [CI], 4.1 to 13.1; P = 0.002), 11.1% in the ECZTRA 2 trial (95% CI, 5.8 to 16.4; P < 0.001), and 12.4% in the ECZTRA 3 trial (95% CI, 2.9 to 21.9; P = 0.015), all favouring tralokinumab. In the ECZTRA 7 trial, for participants who achieved an IGA score of 0 or 1 at week 16, the difference between tralokinumab and placebo was ||||||||||||||||||||||||||||||||||; however, due to the insignificant difference between tralokinumab and placebo in the reduction of worst daily pruritus NRS outcome, which was first in the testing hierarchy, statistical testing was not conducted for this outcome.
For participants who achieved an EASI-75 score at week 16, the percent difference between tralokinumab and placebo was 12.1% in the ECZTRA 1 trial l(95% CI, 6.5 to 17.7; P < 0.001), 21.6% in the ECZTRA 2 trial (95% CI, 15.8 to 27.3; P < 0.001), 20.2% in the ECZTRA 3 trial (95% CI, 9.8 to 30.6; P < 0.001), and 14.1% in the ECZTRA 7 trial (95% CI, 2.5 to 25.7; P < 0.018). These differences were statistically significantly in favour of tralokinumab in all 4 trials.
The adjusted mean change from baseline in SCORAD was statistically significantly larger in the tralokinumab group compared with the placebo group at week 16 in the ECZTRA 1, ECZTRA 2, and ECZTRA 3 trials, with differences between tralokinumab and placebo of −10.4 in the ECZTRA 1 trial (95% CI, −14.4 to −6.5; P < 0.001), −14.0 in the ECZTRA 2 trial (95% CI, −18.0 to −10.1; P < 0.001), and −10.9 in the ECZTRA 3 trial (95% CI, −15.2 to −6.6; P < 0.001). The difference between tralokinumab and placebo in the ECZTRA 7 trial was −8.6 (95% CI, −13.0 to −4.2); however, due to the insignificant difference between tralokinumab and placebo in the reduction of worst daily pruritus NRS outcome, which was first in the testing hierarchy, statistical testing was not conducted for this outcome.
The adjusted mean change from baseline in Patient-Oriented Eczema Measure (POEM) scores also favoured tralokinumab when compared to placebo at week 16, with differences between tralokinumab and placebo of −4.6 in the ECZTRA 1 trial (95% CI, −6.0 to −3.1; P < 0.001), −5.1 in the ECZTRA 2 trial (95% CI, −6.5 to −3.6; P < 0.001), −4.0 in the ECZTRA 3 trial (95% CI, −5.6 to −2.4; P < 0.001), and −3.4 in the ECZTRA 7 trial (95% CI, −5.0 to −1.8; P < 0.001). However, this outcome was exploratory and was not adjusted for multiple testing in any of the included trials.
In terms of symptom reduction, for participants who achieved an improvement of at least 4 points in the weekly average of daily pruritus NRS scores at week 16, the difference between tralokinumab and placebo was 9.7% in the ECZTRA 1 trial (95% CI, 4.4 to 15.0; P = 0.002), 15.6% in the ECZTRA 2 trial (95% CI, 10.3 to 20.9; P < 0.001), 11.3% in the ECZTRA 3 trial (95% CI, 0.9 to 21.6; P = 0.037), and 9.7% in the ECZTRA 7 trial (95% CI, −2.0 to 21.4; P = 0.106). The between-group difference was statistically significantly in favour of tralokinumab in the ECZTRA 1, ECZTRA 2, and ECZTRA 3 trial, but not in the ECZTRA 7 trial.
Participants who received tralokinumab also experienced improvements in how much eczema interfered with sleep at week 16 based on the eczema-related sleep NRS, for which the difference between groups in the adjusted mean change from baseline at week 16 was −0.7 in the ECZTRA 1 trial (95% CI, −1.2 to −0.2; P = 0.007), −1.4% in the ECZTRA 2 trial (95% CI, −1.9 to −0.9; P < 0.001), −1.3% in the ECZTRA 3 trial (95% CI, −1.8 to −0.8; P < 0.001), and −0.8% in the ECZTRA 7 trial (95% CI, −1.3 to −0.2; P = 0.005) in favour of tralokinumab. The minimal important difference (MID) has not been identified for the eczema-related sleep NRS in populations with AD; this outcome was exploratory and was not adjusted for multiple testing in any of the included trials.
Treatment with tralokinumab also elicited a statistically significant improvement in HRQoL at week 16 based on the Dermatology Life Quality Index (DLQI) measure in the ECZTRA 1, ECZTRA 2, and ECZTRA 3 trials. For instance, the between-group difference in the adjusted mean change from baseline in the DLQI was statistically significantly larger in the tralokinumab group compared with the placebo group at week 16 in the ECZTRA 1 (−2.1; 95% CI, −3.4 to −0.8; P = 0.002), ECZTRA 2 (−3.9; 95% CI, −5.2 to −2.6; P < 0.001), and ECZTRA 3 (−2.9; 95% CI, −4.3 to −1.6; P < 0.001) trials. Treatment with tralokinumab also elicited a improvement in HRQoL at week 16 based on the DLQI measure in the ECZTRA 7 trial, in which the between-group difference in the adjusted mean change in the DLQI was larger in the tralokinumab group compared with the placebo group (−1.5; 95% CI, −2.6 to −0.4); however, as this outcome was ranked after the hierarchical analysis failed and was stopped, no appropriate statistical comparisons can be made. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. No MIDs have yet been identified in populations with AD for the DLQI, Short Form (36) Health Survey (SF-36), and EQ-5D 5-Levels questionnaire (EQ-5D-5L) outcome measures; the SF-36 and EQ-5D-5L outcomes were exploratory and were not adjusted for multiple testing in any of the included trials.
Harms Results
In the ECZTRA 1 trial, adverse events (AEs) were reported in 76.4% of patients (n = 460) treated with tralokinumab and 77.0% of patients (n = 151) treated with placebo at week 16, and serious adverse events (SAEs) were reported in 3.8% of patients (n = 23) treated with tralokinumab and 4.1% of patients (n = 8) treated with placebo. Treatment-emergent AEs leading to permanent discontinuation of the study drug were reported in 3.3% of patients (n = 20) treated with tralokinumab and 4.1% of patients (n = 8) treated with placebo at week 16. At week 52, AEs were reported in 79.4% of patients (n = 54) in the tralokinumab every 2 weeks group, 69.7% of patients (n = 53) in the tralokinumab every 4 weeks group, and 71.4% of patients (n = 25) in the placebo group. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In the ECZTRA 2 trial, AEs were reported in 61.5% of patients (n = 364) treated with tralokinumab and 66.0% of patients (n = 132) treated with placebo at week 16, while SAEs were reported in 1.7% of patients (n = 10) treated with tralokinumab and 2.5% of patients (n = 5) treated with placebo. Treatment-emergent AEs leading to permanent discontinuation of the study drug were reported in 1.5% of patients (n = 9) in the tralokinumab group and 1.5% of patients (n = 3) in the placebo group. At week 52, AEs were reported in 68.1% of patients (n = 62) in the tralokinumab every 2 weeks group, 62.9% of patients (n = 56) in the tralokinumab every 4 weeks group, and 69.6% of patients (n = 32) in the placebo group. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In the ECZTRA 3 trial, AEs were reported in 71.4% of patients (n = 180) treated with tralokinumab every 2 weeks plus TCS and 66.7% of patients (n = 84) treated with placebo plus TCS at week 16. SAEs were reported in 0.8% of patients (n = 2) treated with tralokinumab every 2 weeks plus TCS and 3.2% of patients (n = 4) treated with placebo plus TCS. Treatment-emergent AEs leading to permanent discontinuation of the study drug were reported in 2.4% of patients (n = 6) treated with tralokinumab every 2 weeks plus TCS and 0.8% of patients (n = 1) treated with placebo plus TCS at week 16. At week 32, AEs were reported in 69.6% of patients (n = 48) in the tralokinumab every 2 weeks plus TCS group and 59.4% of patients (n = 41) in the tralokinumab every 4 weeks plus TCS group. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In the ECZTRA 7 trial, AEs were reported in 77.5% of patients (n = 107) treated with tralokinumab every 2 weeks plus TCS and 78.8% of patients (n = 108) treated with placebo plus TCS at week 26; SAEs were reported in 0.7% of patients (n = 1) treated with tralokinumab every 2 weeks plus TCS and 3.6% patients (n = 5) treated with placebo plus TCS. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| No deaths were reported in the ECZTRA 7 trial.
Harms of special interest at week 16 included AD, which occurred in 25.9% of patients (n = 156) treated with tralokinumab and 38.3% of patients (n = 75) treated with placebo in the ECZTRA 1 trial and in 16.6% of patients (n = 98) treated with tralokinumab and 33.5% of patients (n = 67) treated with placebo in the ECZTRA 2 trial. Viral upper respiratory tract infection occurred in 23.1% of patients (n = 139) treated with tralokinumab and in 20.9% of patients (n = 41) treated with placebo in the ECZTRA 1 trial and in 8.3% of patients (n = 49) treated with tralokinumab and 18.5% of patients (n = 17) treated with placebo in the ECZTRA 2 trial. In the ECZTRA 3 and ECZTRA 7 trials, the most common AE was viral upper respiratory tract infection, which occurred in 19.4% of patients (n = 49) treated with tralokinumab plus TCS and 11.1% of patients (n = 14) treated with placebo plus TCS in the ECZTRA 3 trial and in 26.8% of patients (n = 37) treated with tralokinumab plus TCS and 25.5% of patients (n = 35) treated with placebo plus TCS in the ECZTRA 7 trial. Among notable harms at week 16, pruritus occurred in 5.3% of patients (n = 32) treated with tralokinumab and 5.1% of patients (n = 10) treated with placebo in the ECZTRA 1 trial; upper respiratory infraction occurred in 10.0% of patients (n = 59) treated with tralokinumab and 8.5% of patients (n = 17) treated with placebo in the ECZTRA 2 trial; conjunctivitis occurred in 11.1% of patients (n = 28) treated with tralokinumab plus TCS and 3.2% of patients (n = 4) treated with placebo plus TCS in the ECZTRA 3 trial; and headache occurred in 15.2% of patients (n = 21) treated with tralokinumab plus TCS and 9.5% of patients (n = 13) treated with placebo in the ECZTRA 7 trial.
Table 2: Summary of Key Results from the ECZTRA 1, ECZTRA 2, ECZTRA 3, and ECZTRA 7 Trials at Week 16
Result | ECZTRA 1 | ECZTRA 2 | ECZTRA 3 | ECZTRA 7 | ||||
|---|---|---|---|---|---|---|---|---|
Tralokinumab q.2.w. N = 601 | Placebo N = 197 | Tralokinumab q.2.w. N = 591 | Placebo N = 201 | Tralokinumab q.2.w. + TCS N = 252 | Placebo + TCS N = 126 | Tralokinumab q.2.w + TCS N = 138 | Placebo + TCS N = 137 | |
IGA score of 0 or 1 | ||||||||
IGA score of 0 or 1 at week 16, n (%) | 95 (15.8) | 14 (7.1) | 131 (22.2) | 22 (10.9) | 98 (38.9) | 33 (26.2) | |||||||||||||||| | |||||||||||||||| |
IGA score of 0 or 1 at week 16 difference vs. placebo, % (95% CI) | 8.6 (4.1 to 13.1; P = 0.002) | 11.1 (5.8 to 16.4; P < 0.001) | 12.4 (2.9 to 21.9; P = 0.015) | |||||||||||||||| | ||||
EASI-75 | ||||||||
Baseline EASI score mean (SD) | 32.2 |||||||||| | 32.9 |||||||||| | 32.1 |||||||||| | 32.6 |||||||||| | 28.8 |||||||||| | 30.4 |||||||||| | |||||||||| | |||||||||| |
EASI-75 score at week 16, n (%) | 150 (25.0) | 25 (12.7) | 196 (33.2) | 23 (11.4) | 141 (56.0) | 45 (35.7) | 88.6 (64.2) | 69.2 (50.5) |
EASI-75 week 16 difference vs. placebo, % (95% CI) | 12.1 (6.5 to 17.7; P < 0.001) | 21.6 (15.8 to 27.3; P < 0.001) | 20.2 (9.8 to 30.6; P < 0.001) | 14.1 (2.5 to 25.7; P = 0.018) | ||||
Harms, n (%) | ||||||||
AEs, n (%) | 460 (76.4) | 151 (77.0) | 364 (61.5) | 132 (66.0) | 180 (71.4) | 84 (66.7) | 107 (77.5) | 108 (78.8) |
SAEs, n (%) | 23 (3.8) | 8 (4.1) | 10 (1.7) | 5 (2.5) | 2 (0.8) | 4 (3.2) | 1 (0.7) | 5 (3.6) |
Any treatment-emergent AE leading to withdrawal from the trial, n (%) | |||||||||| | |||||||||| | |||||||||| | |||||||||| | |||||||||| | |||||||||| | |||||||||| | |||||||||| |
Deaths | |||||||||| | |||||||||| | |||||||||| | |||||||||| | |||||||||| | |||||||||| | 0 (0.0) | 0 (0.0) |
Notable harms | ||||||||
Dermatitis atopic, n (%) | 156 (25.9) | 75 (38.3) | 98 (16.6) | 67 (33.5) | 6 (2.4) | 10 (7.9) | 7 (5.1) | 16 (11.7) |
Viral upper respiratory tract infection, n (%) | 139 (23.1) | 41 (20.9) | 49 (8.3) | 17 (8.5) | 49 (19.4) | 14 (11.1) | 37 (26.8) | 35 (25.5) |
Conjunctivitis, n (%) | 43 (7.1) | 4 (2.0) | 18 (3.0) | 3 (1.5) | 28 (11.1) | 4 (3.2) | |||||||||| | |||||||||| |
AE = adverse event; CI = confidence interval; EASI-75 = 75% or greater reduction in Eczema Area and Severity Index from baseline; IGA = Investigator’s Global Assessment; q.2.w = every 2 weeks; SAE = serious adverse event; SD = standard deviation; TCS = topical corticosteroids; vs. = versus.
Source: Clinical Study Reports for ECZTRA 1,7 2,5 3,6 and 7.4
Table 3: Summary of Key Results from the ECZTRA 1 and ECZTRA 2 Trials at Week 52
Result | ECZTRA 1 | ECZTRA 2 | ||||
|---|---|---|---|---|---|---|
Tralokinumab q.2.w | Tralokinumab q.4.w | Placebo | Tralokinumab q.2.w | Tralokinumab q.4.w | Placebo | |
Primary end points | ||||||
IGA score of 0 or 1 at week 52, n/N (%) | 20/39 (51.3) | 14/36 (38.9) | 9/19 (47.4) | 32/54 (59.3) | 22/49 (44.9) | 7/28 (25.0) |
IGA score week 52 difference vs. placebo, % (95% CI)a | 6.0 (−21.8 to 33.7; P = 0.68) | −9.5 (−37.1 to 18.0; P = 0.50) | Reference | 34.1 (13.4 to 54.9; P = 0.004) | 19.9 (−1.2 to 40.9; P = 0.084) | Reference |
EASI-75 score at week 52, n/N (%) | 28/47 (59.6) | 28/57 (49.1) | 10/30 (33.3) | 43/77 (55.8) | 38/74 (51.4) | 9/42 (21.4) |
EASI-75 week 52 difference vs. placebo, % (95% CI)a | 21.2 (−0.2 to 42.6; P = 0.056) | 11.7 (−8.7 to 32.0; P = 0.27) | Reference | 33.7 (17.3 to 50.0; P < 0.001) | 30.0 (13.7 to 46.4; P = 0.001) | Reference |
Harms, n (%) | ||||||
AEs, n/N (%) | 54/68 (79.4) | 53/76 (69.7) | 25/35 (71.4) | 62/91 (68.1) | 56/89 (62.9) | 32/46 (69.6) |
SAEs, n/N (%) | 1/68 (1.5) | 3/76 (3.9) | NR | NR | 3/89 (3.4) | NR |
Any treatment-emergent AE leading to permanent discontinuation of study drug, n/N (%) | 1/68 (1.5) | 1/76 (1.3) | NR | 2/91 (2.2) | 1/89 (1.1) | NR |
Notable harmsb | ||||||
Viral upper respiratory tract infection, n/N (%) | 14/68 (20.6) | 18/76 (23.7) | 4/35 (11.4) | 9/91 (9.9) | 6/89 (6.7) | 7/46 (15.2) |
Bronchitis, n/N (%) | 3/68 (4.4) | 7/76 (9.2) | 2/35 (5.7) | 1/91 (1.1) | 3/89 (3.4) | NR |
Conjunctivitis, n/N (%) | 3/68 (4.4) | 4/76 (5.3) | NR | 5/91 (5.5) | 1/89 (1.1) | 2/46 (4.3) |
AE = adverse event; CI = confidence interval; EASI-75 = 75% or greater reduction in Eczema Area and Severity Index from baseline; IGA = Investigator’s Global Assessment; NR = not reported; q.2.w = every 2 weeks; q.4.w = every 4 weeks; SAE = serious adverse event; vs. = versus.
aMantel-Haenszel risk difference compared to placebo, stratified by region.
bData from maintenance safety analysis set.
Table 4: Summary of Key Results from the ECZTRA 3 Trial at Week 32
Results | Tralokinumab q.2.w. + TCS | Tralokinumab q.4.w + TCS |
|---|---|---|
Primary end points | ||
IGA score of 0 or 1 at week 32 achieved without rescue medication, n/N (%) | 43/48 (89.6) | 38/49 (77.6) |
EASI-75 score at week 32 achieved without rescue medication, n/N (%) | 62/67 (92.5) | 59/65 (90.8) |
Harms, n (%)a | ||
AEs, n/N (%) | 48/69 (69.6) | 41/69 (59.4) |
SAEs, n/N (%) | 3/69 (4.3) | 0/69 (0.0) |
Any treatment-emergent AE leading to permanent discontinuation of study drug, n/N (%) | 0/69 (0.0) | 1/69 (1.4) |
Notable harmsa | ||
Viral upper respiratory tract infection, n/N (%) | 12/69 (17.4) | 9/69 (13.0) |
Upper respiratory tract infection, n/N (%) | 7/69 (10.1) | 3/69 (4.3) |
Conjunctivitis, n/N (%) | 3/69 (4.3) | NR |
AE = adverse event; EASI-75 = 75% or greater reduction in Eczema Area and Severity Index from baseline; IGA = Investigator’s Global Assessment; NR = not reported; q.2.w = every 2 weeks; q.4.w = every 4 weeks; SAE = serious adverse event; TCS = topical corticosteroids.
aPatients in the continuation treatment safety analysis set who were tralokinumab responders at week 16.
Source: Clinical Study Report for ECZTRA 3.6
Critical Appraisal
Although the analyses were appropriate and investigators accounted for multiplicity, several limitations are associated with the design of the trials. First, the ECZTRA 1, ECZTRA 2, and ECZTRA 3 trials included a 2- to 6-week washout period during which no topical corticosteroid use was allowed. As noted by Wollenberg et al.,8 the patient population being studied has significant disease and high levels of prior medication use. The washout period therefore may have been long enough to exacerbate AD, leading to patients being labelled as “nonresponders” early in the studies. Second, the duration of the initial treatment period (16 weeks) in ECZTRA 1, 2, and 3 may not have been sufficient. Assessments of longer-term efficacy and safety in the ECZTRA 1, 2, and 3 trials were also limited by the fact that only patients who achieved a clinical response at week 16 were eligible to be re-randomized. As a result, the estimates of the effect in the maintenance phase are uncertain, and the analyses in the maintenance phase were not powered, which signifies that the long-term efficacy and safety of tralokinumab is uncertain. Another limitation is the absence of a comparator with a similar mechanism of action (e.g., dupilumab). Within the context of the trials, the performance of tralokinumab is therefore statistically significantly superior with respect to the primary and secondary end points in comparison to placebo. However, the intervention was not compared to another biologic currently available to patients. Last, pauses in dosing or the use of rescue medication in situations where the intervention was not available due to the coronavirus 2019 (COVID-19) pandemic during the ECZTRA 7 trial may have affected the results.
In terms of external validity, the ECZTRA 3 and ECZTRA 7 trials are more reflective of real-world practice because tralokinumab was combined with TCS as the intervention. In ECZTRA 1 and ECZTRA 2, patients who used rescue medication were considered “nonresponders,” which does not align with real-world use of biologics, which, according to the clinical experts consulted by CADTH for this review, are initiated as add-on therapy to TCS for active lesions.
Indirect Comparisons
Description of Studies
CADTH summarized and appraised 2 indirect treatment comparisons (ITCs): 1 matched adjusted indirect comparison (MAIC)9 submitted by the sponsor and a published network meta-analysis (NMA) by the Institute for Clinical and Economic Review (ICER).10 The ICER meta-analysis10 compared tralokinumab against dupilumab (the only drug approved for use in the treatment of AD at the time of this review), upadacitinib and abrocitinib (currently under review by Health Canada and CADTH for use in the treatment of AD), and several drugs that were not listed as under review by Health Canada or CADTH at the time of this review (e.g., nemolizumab, lebrikizumab, and baricitinib). The sponsor-submitted MAIC9 compared ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
Efficacy Results
Result from the NMA showed that tralokinumab was generally superior to placebo, while being inferior to upadacitinib (both 15 mg and 30 mg), abrocitinib 200 mg, and dupilumab 300 mg. These results were consistent when the treatments were used as monotherapy or in combination with topical therapies.
The sponsor-submitted MAIC reported that ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
Harms Results
Results from the sponsor-submitted MAIC indicate that ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
Critical Appraisal
Neither ITC included comments on the methods of study selection, data extraction, or quality assessment. The sponsor’s review was clearer on the methods for conducting the MAIC, referring to National Institute for Health and Care Excellence11 protocols, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
Conclusions regarding the long-term efficacy of tralokinumab compared to the active comparators relevant to this review cannot be drawn from the NMA, as it used results collected over a inappropriately short period of time for use in a study involving a chronic condition such as AD. There is also uncertainty due to the inherent heterogeneity across trials in the networks. The robustness of the comparative efficacy was further compromised by the lack of precision in some of the findings, and the results from the NMA must therefore be interpreted with caution.
Other Relevant Evidence
Description of Studies
One ongoing, open-label, single-arm, long-term, extension study (ECZTEND) has been summarized to provide additional evidence on the safety and efficacy of tralokinumab in patients with AD who have previously participated in clinical trials for tralokinumab (i.e., the ECZTRA 1 through 8 and TraSki trials). The ECZTEND study consists of a 2-week screening period (which is expected to overlap with the end of the parent trial for most patients), a 0.5- to 5-year treatment phase, and a 14-week follow-up beginning 2 weeks after the final dose. At the time of data cut-off, 1,174 patients were included in the ECZTEND trial. The primary outcome is safety or the number of AEs experienced during the study. The secondary outcomes apply to drug efficacy and include achieving an IGA score of 0 or 1 and an EASI-75 score at weeks 16, 56, 88, 104, 136, 152, 184, 216, and 248 during the treatment phase relative to baseline. Blinding of treatment allocation was maintained for patients who continued from a blinded parent trial and entered the open-label extension study.
Efficacy Results
Responders were defined as achieving an IGA score of 0 or 1 or an EASI-75 score. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Harms Results
Overall, 844 (71.9%) patients experienced 1 or more AEs, with the 3 most common AEs being viral upper respiratory tract infection (21.3%), dermatitis atopic (13.5%), and upper respiratory tract infection (7.1%). Other harms of special interest that were identified in CADTH’s systematic review protocol ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Nineteen patients (1.6%) withdrew due to an AE, and no deaths were reported.
Critical Appraisal
The ECZTEND trial lacked a comparator, which made it difficult to adjust for natural changes in the course of AD or the effects of potential confounders. Additionally, the open-label design may have influenced patient and clinician perceptions of improvement, which could affect the reporting of harms and efficacy measures. The number of patients screened from the parent trials was not reported, nor were the reasons for screening failures. Moreover, patients were recruited exclusively from the parent trials of tralokinumab, and only those who could tolerate the treatments were able to enrol in the ECZTEND study. No formal sample size or power calculations were performed, no control for multiplicity was described in the report, and there was no imputation of missing safety data. Most patients in the study were White (71.3%), which may be a product of the regions where the study took place (mainly Europe and North America). While the clinical experts CADTH consulted for this review were uncertain if race would bias the outcomes, this factor may limit how the results can be interpreted in the context of a broader patient population in Canada. Treatment history was not described in this report and whether the patients were treatment-naive and which medications they have had experience with (e.g., topical, systemic, or biologic) were unknown, which limits the generalizability of the results to other patients with AD and prevents comparisons with other treatments.
Conclusions
Three double-blind RCTs demonstrated that, compared with placebo, 16 weeks of treatment with tralokinumab was associated with statistically significant improvements in a range of outcomes that are important in the management of AD in adults, including overall severity of AD (EASI, IGA response, SCORAD), symptoms (pruritus NRS), and HRQoL (DLQI). These trials included the use of tralokinumab as monotherapy (ECZTRA 1 [N = 802] and ECZTRA 2 [N = 794]) and as combination therapy (ECZTRA 3 [N = 380]). The ECZTRA 7 trial, which evaluated the efficacy and safety of tralokinumab as a combination therapy with TCS compared to placebo plus TCS in adults with severe AD who are not adequately controlled with or have contraindications to oral cyclosporine A, demonstrated statistically significant improvement in EASI scores in the tralokinumab group compared to the placebo group. Tralokinumab was well-tolerated in the short term (16 weeks) and long-term (32 or 52 weeks) phase III studies. There were no direct comparisons between tralokinumab and other active AD treatments.
Results from a sponsor-submitted MAIC ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Results from the NMA conducted by ICER suggest that tralokinumab may be inferior to dupilumab in terms of most efficacy outcomes. However, the MAIC comparisons were |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. There is also uncertainty in the results from the NMA due to the inherent heterogeneity across trials in the networks. The robustness of the comparative efficacy was further compromised by the lack of precision in some of the findings, and the results from the indirect comparisons must be interpreted with caution.
Introduction
Disease Background
Atopic dermatitis is the most common type of eczema. It is a chronic, relapsing, inflammatory skin condition characterized by severely itchy skin (pruritus) that results in red and swollen skin (rash). Lesions associated with AD may appear as fluid-filled vesicles that ooze, crack, and crust. Pruritus of the skin can cause frequent scratching and may result in lichenification (thickening of the skin) and secondary skin infections. Atopic dermatitis typically involves the skin folds behind the knees (popliteal areas) and the skin folds in front of the elbows (antecubital areas). It may also appear on the face, neck, and hands. Individuals with AD have skin with impaired barrier function and reduced water-holding capacity, resulting in dry skin that requires treatment with specific bathing, cleansing, and moisturizing practices.
As a hereditary form of eczema, AD generally presents in infancy, with most cases beginning before the age of 5 years. The majority of these children will outgrow the condition by adolescence. It is common for children with AD to develop asthma and/or hay fever. This process is referred to as the “atopic march,” and AD is often the first step in the sequential development of these other atopic conditions. The clinical manifestations of AD vary with age, with infants showing AD on the extensor surfaces of the extremities, face, neck, scalp, and trunk. Children are typically affected on the flexural surfaces of the extremities, neck, wrists, and ankles, while adolescents and adults are generally affected on the flexural surfaces of the extremities and the hands and feet.
The CDA reports that the lifetime prevalence of AD approaches 17% in the Canadian population, and there is evidence to suggest that the prevalence has increased over the past 30 years. Patients often experience worsening itching symptoms throughout the night, and this may result in sleep loss, which may result in detrimental effects pertaining to school or work. Individuals with AD may also suffer from the social stigma of having a highly visible condition. Overall, these patient experiences describe a physically and mentally exhausting condition that can result in anxiety, depression, and decrease in quality of life.
The goals of AD management are to prevent flares (episodes of worsening of symptoms typically requiring escalation of treatment), and effectively manage flares when they occur by preventing progression of the disease. While there is no cure for AD, several therapeutic options are available to patients to manage the condition. The majority of patients treat AD by using general skin care methods, avoiding skin irritants, and applying topical anti-inflammatory therapy. If these common methods fail to improve AD, patients may use off-label systemic therapy (i.e., immunosuppressant therapy) or other therapies such as phototherapy.
Standards of Therapy
General Skin Care
General skin care practices for patients with AD include irritant avoidance and managing dry skin. The symptoms of AD may be reduced or prevented through the avoidance of known skin irritants or triggers.1,3 Some common irritants include temperature, humidity, dust, pets (animal dander), smoke, and grass. Using mild detergents to wash clothing, with no bleach or fabric softener and double-rinsing, has been recommended to those with AD. Dry skin associated with AD can be countered through specific bathing, cleansing, and moisturizing practices. Baths using lukewarm water and emulsifying oil followed by the use of moisturizers are recommended. Limiting the use of soap and fragranced products may also help reduce symptoms.1-3,14
Topical Therapy
While a number of nonpharmacological topical therapies exist for treating the symptoms of AD, the most common therapy is the use of moisturizers, which combat dry skin through hydration and the prevention of trans-epidermal water loss. Moisturizers are routinely used to provide some barrier protection for the skin from irritants or allergens and can act to soften skin, reduce itching, and minimize cracking, fissuring, and lichenification.3,14 Moisturizers are routinely used frequently throughout the day, preferably after bathing. Moisturizers can contain a combination of emollients, humectants, and occlusive drugs. Emollients such as glycol, glyceryl stearate, and soy sterols lubricate soften, and smooth out the surface of the skin by filling the spaces with droplets. Humectants (e.g., glycerol, lactic acid, and urea) attract water and increase the skin’s water-holding capacity. Humectants sting open skin and are not useful in children with AD. Occlusive drugs (e.g., petrolatum, dimethicone, and mineral oil) provide a layer of oil on the surface of the skin to slow trans-epidermal water loss, prevent water loss though evapouration, and increase the moisture content of the skin. The choice of moisturizer depends on the area of the body and the degree of dryness of the skin.3,14
The most common pharmaceutical topical therapies include the use of TCS and TCIs. Topical corticosteroids act as anti-inflammatory therapy and are considered to be first-line treatments for AD.2 There are more than 30 different types of TCS, which can take the form of lotions, creams, oily creams, ointments, or gels and be combined with other drugs such as antibiotics.15 The potency of TCS varies. In Canada, low-potency (1%) hydrocortisone is the most commonly prescribed type of TCS for the face.3 For the body, moderate-potency triamcinolone or betamethasone valerate are most commonly prescribed. All TCS varieties are applied directly to the area of affected skin before the use of emollients, and a response is typically seen within 10 to 14 days. Side effects associated with the long-term use of TCS include striae (stretch marks), petechiae (small red and/or purple spots), telangiectasia (small, dilated blood vessels on the surface of the skin), thinning of the skin, atrophy, and acne.2 Use of TCS is also recommended for children, according to the American Academy of Dermatology (AAD), with cautions regarding dosing, as children have a larger ratio of surface area to body mass and there are mixed results from various studies suggesting that systemic absorption may affect growth.
Topical calcineurin inhibitors are steroid-free, anti-inflammatory, immunosuppressant drugs that can be used long-term. In Canada, the 2 available second-line drugs are pimecrolimus and tacrolimus. Pimecrolimus 1% cream can be used for short-term and intermittent long-term therapy for mild to moderate AD and is effective in controlling pruritus.3 Topical tacrolimus is an ointment that can be used for short-term and intermittent long-term therapy of moderate to severe AD and demonstrates rapid and sustained AD symptom control.3,15 The most common AE associated with TCIs is application site–specific burning and irritation.2,3 Packaging for TCIs come with a black-box warning regarding lymphoma; however, long-term (10-year) surveillance studies have not found an increased risk of lymphoma over that of the general pediatric population.
Crisaborole, a topical phosphodiesterase type 4 inhibitor, is also available in Canada (although it is not recommended by CADTH for reimbursement).1,2 The advantage of the calcineurin inhibitors and crisaborole is that both can be safely applied to the face and creases, whereas TCS that are more potent than hydrocortisone 1% are inappropriate. Other topical therapies for AD include treatments with diluted bleach baths, which can help reduce the occurrence of secondary skin infections.3,16
Systemic Therapy
Systemic therapy for the treatment of AD typically involves the use of antimicrobials, antihistamines, or immunomodulators.15-17 Systemic antibiotic treatment can be used to counter widespread secondary bacterial infection. Many patients encounter infection with Staphylococcus aureus, and this may cause new inflammation and exacerbate AD symptoms. The choice of systemic antibiotic drug depends upon the skin culture and sensitivity profile. Sedating antihistamines have been used in cases in which patients are not achieving adequate sleep due to itching.1,15
Immunomodulatory drugs, including methotrexate, cyclosporine, mycophenolate mofetil, azathioprine (listed in order of frequency of use in Canada), can be used in patients who are not responsive to other treatments.13,15,16 However, these commonly used off-label treatments are administered at the lowest dose for the shortest duration possible due to side effects.16,17 According to the AAD, cyclosporine is an effective treatment in pediatric patients. The AAD notes that the evidence for the use of methotrexate in pediatric AD patients is limited; however, a recent 12-week study showed it to have a slower onset than low-dose cyclosporine but an increased time before relapse after discontinuation. Regarding azathioprine, the AAD noted there is evidence of efficacy in children, but recommended that its use should be reserved for recalcitrant AD, or for patients in whom AD is having a significant psychosocial impact. The AAD noted that mycophenolate mofetil was a relatively safe systemic therapy in pediatric AD patients, although its long-term (> 24 months) efficacy and safety in pediatric patients have not been studied. With respect to corticosteroids, there is a longstanding understanding that chronic use can affect growth in children. The AAD does not recommend corticosteroid use in children with AD unless they are given as part of a short-term transition to systemic immunomodulators.
Dupilumab (Dupixent) is an IL-4 and IL-13 inhibitor indicated for use in adult and pediatric patients with moderate to severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. CADTH recommended that dupilumab be reimbursed with conditions, and dupilumab is currently reimbursed by the participating drug programs for patients whose AD is inadequately controlled with topical prescription therapies and who have demonstrated failure or intolerance to an adequate trial of phototherapy (where available), methotrexate, and cyclosporine.3
Other Therapy
Phototherapy is another second-line therapy that is commonly used after failure of TCS, TCIs, and crisaborole. This therapy includes several sessions and is guided by a number of factors, including patient skin type and skin cancer history.16 According to AAD guidelines, phototherapy is considered to be a safe and effective treatment for AD in children. There are no studies of the long-term consequences of phototherapy use in pediatric AD patients, although an increased risk of nonmelanoma skin cancer has been reported in children receiving psoralen plus UV A (UVA) exposure to treat psoriasis.
Drug
Tralokinumab is indicated for the treatment of moderate to severe AD in adult patients whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. Tralokinumab can be used with TCIs. The recommended dosage of tralokinumab for adult patients is an initial dose of 600 mg (4 injections of 150 mg each) followed by 300 mg (2 injections of 150 mg each) administered every other week as subcutaneous injection. At the prescriber’s discretion, every-fourth-week dosing may be considered for some patients who achieve clear or almost-clear skin after 16 weeks of treatment. It is administered by subcutaneous injection into the thigh or abdomen. If someone other than the patient administers the injection, the upper arm can also be used. Tralokinumab is contraindicated in patients who are hypersensitive to this drug or to any ingredient in the formulation, including any nonmedicinal ingredient or component of the container. This drug has not been previously reviewed by CADTH.
Tralokinumab is a fully human immunoglobin G4 monoclonal antibody that specifically binds to the type 2 cytokine IL-13 and inhibits its interaction with the IL-13 receptor alpha-1 and alpha-2 subunits (of the type II receptor). Tralokinumab restores expression of skin barrier markers decreased by IL-13 in human keratinocytes.
The sponsor’s reimbursement request is for the treatment of adult patients with moderate to severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable and who had an adequate trial or are ineligible for each of the following therapies: phototherapy (where available), methotrexate, and cyclosporine.
The characteristics of tralokinumab and its most common comparators for the purpose of this review are presented in Table 5.
Table 5: Key Characteristics of Tralokinumab and Comparators
Characteristic | Tralokinumab | Dupilumab | Azathioprine | Mycophenolate mofetil | Cyclosporine | Methotrexate |
|---|---|---|---|---|---|---|
Mechanism of action | IL-13 cytokine inhibitor | Receptor antagonist that binds to the IL-4 receptor alpha subunit, shared by IL-4 and IL-13 receptor complexes | Immunosuppressive Antimetabolite — reduces proliferation of lymphocytes | Immunosuppressive Inhibits purine synthesis, reduces lymphocyte proliferation Reduces antibody formation by B lymphocytes | Immunosuppressive Inhibits IL-2 and T-cell activation | Immunosuppressive |
Indicationa | Treatment of moderate to severe AD in adult patients whose disease is not adequately controlled with topical prescription therapies or when therapies are not advisable | Treatment of patients aged 6 years and older with moderate to severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable | Rheumatoid arthritis Prevention of transplant rejection (renal) | Prevention of transplant rejection (renal) | Prevention of transplant rejection Psoriasis Rheumatoid arthritis Nephrotic syndrome | Various neoplasia Psoriasis Rheumatoid arthritis |
Route of administration | Subcutaneous | Subcutaneous | Oral | Oral or IV | Oral | Oral or SC |
Recommended dose | Initial dose of 600 mg (4 injections of 150 mg) followed by 300 mg (2 injections of 150 mg) administered every other week At prescriber’s discretion, q.4.w dosing may be considered for some patients who achieve clear or almost-clear skin after 16 weeks of treatment | ≥ 18 years: 600 mg, followed by 300 mg q.2.w 6 to 17 years:
| Renal transplant: initial dose 3 to 5 mg/kg daily. Then dose reduction maintenance level of 1 to 3 mg/kg daily. Rheumatoid arthritis: initial dose of 1 mg/kg (50 mg to 100 mg) as single dose or twice daily. Dose increments of 0.5 mg/kg daily up to a maximum of 2.5 mg/kg/day | 1 g orally twice a day 1 g IV twice a day | Psoriasis Initial: 2.5 mg/kg/day in 2 divided doses not to exceed 5 mg/kg/day | Varies with indication |
Serious adverse effects or safety issues | Upper respiratory tract infections Conjunctivitis Eosinophilia Conjunctivitis allergic | Conjunctivitis Keratitis Hypersensitivity Helminthic infections | Carcinogenic Leukopenia Thrombo-cytopenia Infection Hepatoxicity | Infection Lymphoma Progressive multifocal leuko-encephalopathy | Infection Malignancy Nephrotoxicity Hypertension Hepatotoxicity Neurotoxicity | Malignancy Serious rash Bone marrow suppression Vomiting and diarrhea Hepatotoxicity Pulmonary toxicity |
Other | Animal studies did not show any effects on male and female reproductive organs and on sperm; limited data on use during pregnancy | No evidence of fetal harm — however, with limited data | Fetal harm (mutagenic) | Fetal harm/pregnancy loss | Reports of fetal harm | Fetal harm (mutagenic) |
AD = atopic dermatitis; IL = interleukin; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; SC = subcutaneous.
aHealth Canada–approved indication.
Source: Product monographs for dupilumab,12 azathioprine,13 mycophenolate mofetil,14 cyclosporine,15 and methotrexate.16
Stakeholder Perspectives
Summary of Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
About the Patient Group(s) and Information Gathered
CADTH received 2 patient group submissions for the review of tralokinumab for AD. One was from the ESC and the other was a joint submission from the CSPA and Eczéma Québec.
The ESC is a registered Canadian charity working to improve the lives of those living with eczema through support, education, awareness, and research. For its submission, ESC conducted a survey and interviews covering how AD affects quality of life, experiences with symptoms and treatments, and the patient journey. The group received more than 3,000 responses from adults living with AD as well as their caregivers and family.
Both the CSPA and Eczéma Québec are nonprofit organizations. The CSPA is dedicated to advocating, educating, and supporting Canadians affected by skin, hair, and nail disorders. Eczéma Québec is a patient advisory committee created as a branch of the McGill University Hospital Network Centre of Excellence for Atopic Dermatitis and consists of adult patients with AD and health care practitioners in the field of AD. The 2 organizations created a web-based survey (available from April 26 to May 16, 2021) in English and French using the Survey Monkey platform asking about experiences with AD and tralokinumab. The survey was distributed via the groups’ newsletters and social media networks in addition to being sent to the clinical trial investigators, whose contact information was requested and received from the sponsor, to be shared directly with the clinical trial participants from the investigators. Twenty-six individuals responded to the survey (14 in French and 12 in English), with 81% of responses from Québec, 15% from Ontario, and 4% from France. Patients and caregivers made up the majority of respondents (85% and 8%, respectively) and all were adults. The groups’ submission (based on information gathered between March 29 and April 23, 2021) also included information from 56 Canadians with AD along with caregivers who participated in health technology assessment surveys and interviews regarding JAK inhibitor treatments. Most of the responses were from adults but 2 children younger than 12 years of age also contributed.
Disease Experience
Patients who responded to the ESC survey described itching as the most burdensome symptom, with 72% and 95% of those with moderate and severe AD, respectively, reporting feeling itchy multiple times a day. Moreover, 44% of respondents with severe disease were itchy all the time and more than half of the group described being unable to control the urge to scratch their skin and that it could be “overwhelming and uncontrollable.” Many patients reported having scars and marks from scratching as well as cracked and blistered skin that could bleed through clothing, while others had to vacuum daily to clean dead skin from the floors. Flares of worsening symptoms such as extreme itching and pain frequently led to loss of sleep. For example, 63% and 86% of patients with moderate and severe AD, respectively, noted sleep disruptions, and half of respondents with severe AD had lost sleep at least 8 nights per month. According to 1 patient, “It was so severe that the pain would interrupt my sleep, which would lead to more inflammation and pain, which would further interrupt my sleep and my ability to heal. I was trapped in a destructive cycle.”
Of the respondents to the CSPA and Eczéma Québec survey, 62% indicated having experienced symptoms for more than 10 years, with 15% and 39% of patients reporting having moderate and severe disease, respectively. Although individuals reported being affected by AD on all parts of their body, the most common areas were the backs of hands (68%), outsides of arms and/or elbows (57%), thighs and/or legs (57%), elbow folds (55%), neck (51%), and abdomen (51%). Nearly all of those with AD experienced itching (98%), skin redness (91%), repeated rashes (87%), frequent scratching (87%), cracked skin (87%), and dry and rough skin (81%). Disrupted sleep, pain, bleeding, skin flaking and thickening, swelling, and oozing were other issues that patients faced. One patient described what it has been like living with AD: “As I grew up, my disease got worse and worse, until it got to the point where I frequently had to miss school, and had trouble sleeping at night. On days when I could attend school, I was teased because of the way my skin looked, and people stared or made comments on my appearance…. I felt as though no one understood what it was like living in my skin.” Another patient recalled, “I used to be unable to do any sport without getting a huge debilitating rash. I also wouldn’t be able to shower or go swim if I didn't have unscented lotion with me. My eczema would be considered moderate to severe growing up and I was in constant pain.” Patients reported their average level of pain to be 4.95 out of 10 and the average level of itch to be 6.44 out of 10. More than half of the patients reported that their pain was controlled or that they were not experiencing pain, whereas most indicated that their itch was poorly controlled.
The symptoms of AD contribute negatively to stress and significantly affect mental health and relationships. The ESC also emphasized that the mental health impact of having AD is “a significant aspect of the condition and is often not understood by others, nor prioritized by health care providers.” Patients reported feelings of embarrassment, anxiety, depression, poor self-esteem, low energy, and suicidal thoughts, and described the symptoms as difficult for those without AD to fully appreciate and understand. One patient stated, “People don’t understand the reality of living with eczema. It affects how you operate as a human being. Tasks as simple as bathing can be excruciating. Falling asleep can be nearly impossible.” Beyond the psychological impacts, having AD could also make bathing and handwashing painful, which affects individuals’ hygiene and in turn causes other health and social issues. The ESC noted that poor hygiene could lead to a higher risk of infection and the need for systemic antibiotics and other treatments. Nearly a third of respondents with moderate or severe AD reported missing work events, and 30% changed their careers or gave up activities as a result of the condition. These challenges are captured in the following patient quotes: “I was in such bad shape that daily tasks became almost unbearable. I couldn’t work and wasn’t able to play with my children without breaking my skin” and “You are scared to move your body — even if it’s just walking or running errands — because a little drip of sweat can irritate you severely and make you stop everything to scratch.”
Living with AD also affects caregivers and family members, who reported loss of sleep and missed days of work or school, along with feelings of helplessness, guilt, and frustration. Caregivers of those with AD expressed needing “to be mindful of their comfort level going into public… especially if they're having a bad flare-up,” while another noted that caring for their family member could affect their sleep, productivity at work, and increase their overall stress. Family members also noted how AD affected patients’ behaviours, from influencing their clothing choices and wanting to cover their skin to not going out to see friends, and how the burden of treatment routines could cause tension between a patient and their immediate caregiver.
Experiences With Currently Available Treatments
The ESC noted that topical treatments can be effective at controlling AD for some patients, but there is still a small group of individuals who live with uncontrolled moderate and severe forms of AD for whom these medications are inadequate. Lack of symptom control from trying various drugs can be frustrating for patients, and dermatologists may recommend systemic treatments such as off-label immune-suppressing medications (e.g., methotrexate and cyclosporine), oral corticosteroids (e.g., prednisone), or phototherapy. The ESC acknowledged that a biologic drug was recently approved for AD, but despite this and the previously listed nontopical treatments, challenges and limitations to current treatments still exist for many patients. For example, a patient who received prednisone indicated that “it helped with flares but there were all kinds of side effects. I gained weight and couldn’t sleep because my body was racing all the time. It is not a long-term solution.” Other patients reported that phototherapy did not offer long-term control over the AD, and that clinics may not be accessible to all individuals. A patient who had experience with both topical treatments and phototherapy stated, “My dermatologist kept prescribing me harsher and harsher creams, but my skin just kept getting worse. I tried phototherapy, but it was more than 30 minutes away and my schedule couldn’t keep up. You have to go often for it to work and I just couldn’t.” Respondents in the CSPA and Eczéma Québec submission reported experiencing financial barriers to access, such as only being able to afford treatments due to their own employer benefits or spouse’s private insurance, which may be in addition to having to pay for medications for other conditions.
Three respondents in the CSPA and Eczéma Québec surveys reported experience with dupilumab, while none had used either TCIs or cyclosporine. There was a general sentiment that most treatments were not effective at managing AD, although the most efficacious were TCIs (60% of respondents), emollient creams or ointments (47%), and phototherapy (30%).
According to a recent survey conducted by the ESC, oral corticosteroids (OCS) may be used as “rescue medication” for AD flares and as systemic treatment. However, OCS were associated with the highest safety concerns among patients and may only be used for a short period of time, and patients expressed frustration that OCS were not a solution given the chronic nature of AD. Furthermore, flares that occurred after taking OCS were described by some patients as devastating.
The trial-and-error process of testing currently available treatments and still having uncontrolled disease for years has left patients with little hope and low expectations that medications will work. Patients expressed renewed optimism that, when new treatments come out, something might finally work. The patient group submissions emphasized the need for effective treatment options that are affordable, accessible, and easy to use; can control the most significant symptoms; and offer long-term relief for patients who do not respond to current medications.
Improved Outcomes
Patients expressed interest in new therapies that could reduce symptoms such as itching, flares, redness, inflammation pain, dryness, flaking, blistering, and cracked skin. Those with moderate or severe AD noted the importance of having a medication that provides long-term relief, allowing them to sleep better, heal, and avoid new flares, complications, and secondary infections. Respondents also felt that new treatments should be covered by insurance or be affordable, allow them to stop using topical therapies, be low-maintenance and not very time-consuming, and improve their quality of life. In the CSPA and Eczéma Québec surveys, 64% indicated it was important that AD treatments not require injections, while the other 34% were indifferent or reported that it was not important. When asked about preferred modes of administration, daily oral pills were ranked the highest, followed by daily topical treatments, and injections every other week.
Respondents appeared to be willing to accept serious side effects based on the severity of their AD and previous experiences with treatments. For example, according to the input from CSPA and Eczéma Québec, 1 patient answered, “Yes and no depending on the side effects. Enough for me would be stopping the itchiness… and break outs [flares].” Another stated, “No!! I only expect to reduce the intensity of the flares, not eliminate them,” and yet another shared, “I had injections for almost a year and I would not want them again.”
Based on the input from the ESC, patients also valued being able to carry out everyday activities such as bathing, being productive at work, exercising, and not feeling the psychological burden that comes with AD. Moreover, respondents reported that they simply want to feel comfortable in their skin and maintain social relationships without being self-conscious. As 1 patient put it, “You just want a medication that works. You want to be able to sleep, to fit in with your peers, and to not feel hopeless anymore.”
Experience With the Drug Under Review
From the ESC submission, patients had accessed tralokinumab through a clinical trial and many reported that it had significantly alleviated their pain, itching, discomfort, and the frequency of flares. Some patients experienced improvements in 4 to 6 weeks while others noted changes took a few months. One patient shared, “This drug changed my life. I have not had an open wound, infection, or even a skin eruption since about the first 6 months of this trial. I have only used topical ointment a handful of times since starting tralokinumab.” According to another, “Once I started experiencing an improvement from the medication, it has relieved my eczema symptoms ever since. I scratch minimally during sleep instead of all night long. Not having any open areas on me has improved my mindset and has made my life considerably better.” Furthermore, tralokinumab has allowed patients to resume daily routines and activities they were previously unable to enjoy, such as exercising, being outdoors, bathing, swimming, and playing with their children. The improvements were clear to 1 patient who stated, “Before my trial, I was existing, now I am a contributing member of my family and society. I hope that these drugs become covered by benefits for everyone (particularly when nothing else works).”
Side effects in the form of fatigue and temporary redness and irritation at the injection site have been reported by those who received tralokinumab. Nevertheless, patients reported that they would carefully consider the risks and benefits of a new medication and many would accept potential side effects if the treatment was able to provide symptom relief. According to patients who were interviewed by the ESC, an injectable medication is simpler and more convenient in general compared with other skin care routines and topicals that can be messy and painstaking to apply. Some patients raised concerns over a fear of needles, although they indicated they would be able to overcome this challenge.
Two patient respondents in the CSPA and Eczéma Québec submission had experience with tralokinumab. When considering previous medications, 1 patient stated that they “left many gaps, were ineffective. Tralokinumab greatly improved quality of life.” Overall, both individuals appeared satisfied with the drug, as illustrated in the following quotes:
This was the first time I used an injection treatment. It is much easier and cleaner than others. It was very easy to notice a difference for me as my face was the first area to start to improve and continued to improve. I don’t have to load up on creams before I get dressed or before bed. Itchy level has decreased noticeabl[y] and has increased my night sleep. I can use a moisturizing cream daily and it works to stop the cracking skin that makes day to day life so much more comfortable.
Tralokinumab greatly improved all symptoms including flaking, itching, redness, pain, sleep interruption etc. I have not noticed any negative side effects, or problems. Great improvement in quality of life.
The benefits also extended beyond symptom relief to other aspects of their lives: “[It] made everything easier, ability to do whatever I want and wear what I want. Not have to worry about what others are thinking when they see rashes on my face. Being able to do my job without having to worry about getting dirt on my skin or having to keep cool is a great thing” and “Tralokinumab greatly improved family dynamics, as I was less ill, and in pain, and able to engage in normal day to day activities.”
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of AD.
Unmet Needs
Some patients with moderate to severe AD respond to the current therapies. However, several issues suggest a need for additional therapies. For example, there may be inadequate access to phototherapy (patients need to travel to an office 3 times a week for at least 12 weeks), there are side effects with systemic therapies such as methotrexate and cyclosporine A, and, while subcutaneous dupilumab is offered in addition to therapy, treatment is hampered by cost and limited reimbursement (as well as side effects of conjunctivitis and facial dermatitis). Last, not all patients respond to the current therapies.
Place in Therapy
Tralokinumab’s mechanism of action appears to be through IL-13 inhibition. It would therefore complement other therapies and can be added to other treatments (excluding dupilumab, as both treatments act on similar receptors). It can complement the use of TCIs or be used after trying and failing topical therapies. Although 1 of the clinical experts indicated that there are no clinical reasons why it could not be used as a first-line treatment, both experts indicated that it makes sense to trial an appropriate topical therapy first before considering therapies such as tralokinumab. One clinical expert indicated that tralokinumab may offer a safer and more effective treatment option, as compared to the off-label systemic therapies that are currently available. The other clinical expert disagreed with that statement, referring to the lack of long-term evidence of safety and their own view that the efficacy data are not impressive. Overall, tralokinumab is not expected to cause a dramatic shift in the current treatment paradigm but may present an additional therapy in the class of small-molecule and biologic therapies. Dupilumab has already established a precedent in this class of therapies.
In terms of whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with tralokinumab, the clinical experts indicated that, for economic reasons, tralokinumab should be used after failing topical therapies. However, they were not aware of a clinical reason that could prevent tralokinumab from being a first-line treatment.
Patient Population
According to the clinical experts, it would make sense to treat patients with moderate to severe AD who have not responded to adequate topical therapies and an adequate trial of phototherapy if access was not an issue. Moderate to severe AD carries significant short-term and long-term effects on quality of life; all patients with uncontrolled moderate to severe AD would therefore benefit from intervention.
Patients with AD often have comorbid allergic disease. However, there is no strong evidence that tralokinumab helps reduce asthma exacerbations. Following the precedent set by the other biologic (dupilumab) assessments, such as the EASI, NRS, and IGA, quality-of-life scores such as the DLQI can be used. Severe AD is generally not difficult to diagnose. Pre-symptomatic patients should not be treated.
Patients who have conditions other than AD would be least suitable for treatment.
Assessing Response to Treatment
The clinical experts were not aware of good predictors of a good response to tralokinumab. The same assessments used for severity (NRS, IGA, and quality-of-life scores such as the DLQI and EASI) can also be used to assess response. A clinically meaningful response would include improvements in quality of life, itch and clinical scores (IGA or EASI). The clinical experts disagreed on when the treatment response should be assessed. One expert indicated that the treatment response should be assessed monthly early on in treatment, and every 3 to 6 months later on in the course of treatment. The other clinical expert would not typically assess response to tralokinumab earlier than 16 weeks and then every 6 months.
Discontinuing Treatment
According to the clinical experts, the factors to consider for discontinuation would be lack of efficacy and/or clinical response, and adverse effects (e.g., severe conjunctivitis unresponsive to treatment measures).
Prescribing Conditions
Hospital, specialty, and community clinics are appropriate places to initiate tralokinumab. The actual administration can be carried out at home with nursing support (subcutaneous injection). It would be reasonable to have a dermatologist diagnose, treat, and monitor patients receiving tralokinumab. No companion diagnostic testing is required.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups.
Two individual clinician inputs were received for the review of tralokinumab. One dermatologist practising in British Columbia provided input on behalf of the CDA and the other input was provided by a dermatologist who practises at the ODC in Saskatchewan. The CDA is a national medical specialty association that represents certified Canadian dermatologists. Its mandate is to promote the development of new medicine and surgery related to skin hair and nails; provide continuing professional education and development for its members; support patient care; and educate the public on sun protection and other aspects of skin, hair, and nail health. The ODC is an independent skin health centre that provides dermatology services to the general patient population in southern Saskatchewan. Additionally, the centre provides remote dermatology clinics in northern and southern Saskatchewan in the form of virtual care and teledermatology, and in-person clinics with a particular focus on providing culturally sensitive dermatology services to Indigenous populations.
Current Treatments
The clinician from the ODC informed CADTH that, other than dupilumab, there are currently no other FDA-approved systemic medications to treat moderate to severe AD. Traditional systemic immunosuppressants methotrexate and cyclosporin A are available but are associated with safety concerns.
The clinician from the CDA stated that most patients are initially treated with emollients, topical steroids, TCIs (pimecrolimus and tacrolimus) and/or phosphodiesterase inhibitors. The most commonly used of these treatments are topical steroids, which may take up to an hour or an hour and a half per application for moderate to severe disease. Newer antihistamines such as bilastine and rupatadine may be prescribed as adjunctive therapy to minimize itching, as they have a better safety profile compared with first-generation antihistamines, which can often cause dry mouth and diminish sleep quality and work productivity. The clinician explained that these antihistamines are often effective for approximately 90% of patients. Patients with more aggressive disease that does not respond to these treatments are treated with phototherapy (e.g., narrowband and broadband UV B, UVA and psoralen plus UVA), or systemic treatments in addition to topical therapy. However, psoralen plus UVA is now less commonly used due to an increase in skin cancer rates. The only FDA-approved systemic treatment, dupilumab, is administered subcutaneously every other week. Although this treatment has a good safety profile and has helped many patients, it has only been effective for approximately a third of patients with dermatitis. It is associated with side effects such as conjunctivitis and facial dermatitis, which can result in significant morbidity and lead the patient to terminate the treatment early. Off-label immunosuppressants such as methotrexate, cyclosporine, azathioprine, and mycophenolate mofetil can be used if there are no alternatives. However, the CDA clinician noted that the side effects of these immunosuppressants currently limit their use and that there is a lack of high-quality RCTs in dermatitis to advise the use of these immunosuppressants. These immunosuppressants require frequent laboratory monitoring, which can be troublesome for patients. The most commonly used immunosuppressants are methotrexate and cyclosporine. However, cyclosporine is considered a short-term (3- to 6-month) treatment due to adverse effects such as nephrotoxicity, structural renal damage, infections, and hypertension. Methotrexate is teratogenic and may cause bone marrow suppression, hepatic toxicity, malignancies, infections, and pulmonary fibrosis. Mycophenolate mofetil is also teratogenic and associated with serious infections and malignancies. Azathioprine is mutagenic and may cause bone marrow suppression and malignancies. The clinician added that immunosuppressants have put patients at an increased risk for these adverse effects during the COVID-19 pandemic. Systemic glucocorticoids can also be used in the short term but are associated with adverse effects and are therefore not considered for long-term therapy. Rebound is also common after systemic glucocorticoids are discontinued.
Unmet Needs
Both clinicians explained that the goals of treatment are to provide long-term relief of itching and clear the skin. The clinician from the CDA noted that 2/3 of patients who are treated with dupilumab do not achieve clear skin, and there is therefore a need for additional systemic medications with different mechanisms of action. Additionally, both clinicians emphasized that there is a need for treatments to be convenient and durable for patients. The clinician from the ODC specifically voiced this concern for Indigenous populations living in remote areas. These patients are often hard to reach virtually and have limited access to health care, which makes it extremely difficult to monitor patient safety while they are being treated with traditional systemic immunosuppressants. The clinician emphasized that traditional immunosuppressants can lead to side effects, such as worsening of infection, cytopenias, and liver damage, for which many people on reserves and remote areas may not be able to receive adequate follow-up care. There is therefore a significant need for treatments that not only have an improved safety and tolerability profile but are also easier to access. Both clinicians emphasized that the ultimate goal of treatment is to improve the overall quality of life of patients and enable them to regain their ability to function adequately in society.
Clinicians were asked to specify the patient groups that have the greatest unmet need for a drug such as tralokinumab. Both clinicians stated that patients with moderate to severe AD who do not respond to topicals and phototherapy have a high unmet need for this drug. Additionally, the clinician from the CDA noted that women of childbearing age also have an unmet need, as most off-label systemics are teratogenic. The clinician from the ODC re-emphasized the significant unmet need for Indigenous populations in Canada. The clinician stated that AD is a common skin disease among Indigenous populations, a situation that is often exacerbated by health barriers and disadvantages such as poverty, communicable disease, inadequate water supply, and difficulties accessing basic skin care products. Effective access to a treatment like tralokinumab can significantly help alleviate the symptoms of AD and promote a better quality of life.
Place in Therapy
The clinician from the CDA explained that tralokinumab is a biologic that has a different mechanism of action compared to dupilumab. Dupilumab is a monoclonal antibody directed against the alpha subunit of the IL-4 receptor, which is also a component of the IL-13 receptor. Tralokinumab is a monoclonal antibody against IL-13. The clinician explained that tralokinumab will be prescribed to patients with moderate to severe AD for whom topical therapy and phototherapy has failed. It would be prescribed as the first systemic drug, as well as after other systemics, including dupilumab, have failed. Furthermore, the clinician explained that tralokinumab would likely be used in addition to the topical therapies that the patient is using. However, once the AD improves, the topicals will most likely only be applied to any resistant areas.
Similarly, the clinician from the ODC advised that, in addition to tralokinumab, topical therapy would continue to be used for the remaining dermatitis. However, the clinician advised that chronic inflammation must be attended to and it is unsafe for patients to apply topical steroids daily on wide surface areas of the body.
Clinicians were asked to indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Both clinicians advised that it is unsafe to recommend the use of tralokinumab after systemics such as methotrexate and cyclosporine, as there is little evidence to support their use for AD. These systemics are also associated with significant toxicity concerns, such as renal damage, if used in the long term.
In terms of sequencing of therapies, the clinician from ODC advised that tralokinumab would be used in first-line settings. The clinician from CDA advised that topicals will continue to be used as a first-line therapy, followed by phototherapy and then systemics, including biologics. Currently, no data support the use of tralokinumab in patients who have failed dupilumab. It is possible that, because the mechanisms of action of tralokinumab and dupilumab are different, tralokinumab could work in patients in whom dupilumab did not work, and vice versa. It may also be possible for patients to be prescribed tralokinumab if they have conjunctivitis as a baseline atopic comorbidity. Additionally, tralokinumab may be used before other toxic systemic drugs. However, if tralokinumab and dupilumab do not work, patients will need to try another treatment, which could include immunosuppressives after the biologics.
Patient Population
Clinicians were asked to indicate which patients would be best suited for treatment with the drug under review. The clinician from CDA responded that patients with moderate to severe AD affecting more than 10% of their body surface area who experience intense itching, and patients who have failed to respond to topical and phototherapy, are best suited for this treatment. However, the clinician cautioned that there is no evidence to indicate which subtypes of patients with moderate to severe AD will respond better. Similarly, the clinician from the ODC noted that patients with moderate to severe AD who have failed traditional immunosuppressants would be best suited for tralokinumab. Both clinicians stated that these groups of patients are easily identifiable in clinical practice.
Clinicians were asked to identify which patients would be least suited to treatment. The clinician from the CDA explained that patients with mild AD or a different disease state would be least suited for this treatment. The clinician from ODC stated that patients with contraindications to tralokinumab such as hypersensitivity would be least suited to the treatment. In addition, the data are uncertain about the use of this treatment for pregnant women and children.
Assessing Response to Treatment
Clinicians were asked if it is possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review. The clinician from the CDA explained that a waterfall plot of patient responses has shown that most patients have some response to tralokinumab; however, it is unclear how nonresponders would be identified. The clinician from the ODC responded that the DLQI test can be administered easily to help identify patients who are most likely to exhibit a response. A virtual EASI and Psoriasis Area and Severity Index scores can also be used; however, they are challenging to conduct virtually.
The clinician from the CDA stated that itch NRS, body surface area, and the IGA scale are outcomes often used in clinical practice to determine whether a patient is responding to treatment. Furthermore, the clinician explained that, in clinical trials, SCORAD and the DQLI are often used in addition to the EASI; however, SCORAD is mainly a European instrument and is difficult to calculate. The clinician from the ODC also mentioned the utility of the EASI and DLQI, as well as the IGA scale and body surface area scoring.
The clinician from the ODC noted that a 4-point reduction in itching, a reduction in IGA to mild or better, and a reduction in the affected body surface area would indicate a clinically meaningful response to treatment. The clinician further noted that lichenification can take many months to resolve when patients are improving. The clinician from the CDA specified that a reduction of 50% or greater in the EASI from baseline (EASI-50) or an EASI-75 response, a 2-point drop in the IGA score, and in improvement in quality-of-life indices would be considered clinically meaningful responses.
The clinician from the CDA advised that responses to treatment should be assessed at 6-month intervals, and yearly thereafter. Many patients with moderate to severe AD do not show maximal improvements until after at least 6 months of treatment. The clinician from the ODC advised that patients should be assessed approximately every 3 months until they are relatively stable, after which they could be assessed annually, given the favourable safety profile of tralokinumab.
Discontinuing Treatment
Both clinicians noted that nonresponse to the treatment or disease progression would signal a patient to discontinue treatment. The clinician from the ODC also specified that AEs from tralokinumab would also lead to discontinuation; however, the clinician from the CDA noted that there are no major safety concerns with tralokinumab, and that injection-site reactions and conjunctivitis rarely lead to treatment discontinuation.
Prescribing Conditions
Both clinicians noted that the disease is diagnosed, treated, and monitored by a dermatologist. The clinician from the CDA also specified that allergists may also play a role in management of the disease. In terms of prescribing tralokinumab, the clinician from CDA advised that it is usually prescribed initially by an allergist or dermatologist and may be renewed by other physicians after 6 months of treatment.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 6.
Table 6: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Access to phototherapy seems to be limited across Canada. Is this factual or perceived among clinicians and dermatologists? | Phototherapy is accessible primarily in urban areas but not in rural areas. It is important to consider this barrier in the decision-making process. |
Would tralokinumab be initiated in patients who have failed previous treatment with a biologic? | No evidence indicates that it is acceptable to initiate tralokinumab in patients who have failed previous treatment with a biologic; however, the clinical experts agreed that it is unlikely that they would use tralokinumab with patients who have failed a biologic (e.g., dupilumab). Both dupilumab and tralokinumab have similar mechanisms; dupilumab seems to have higher efficacy, but tralokinumab may have fewer side effects. However, the experts are more likely to use JAK inhibitors following failed treatment with a biologic. |
Should patients be required to have tried an adequate trial or be ineligible for cyclosporine, methotrexate, and phototherapy before initiating tralokinumab? | Patients should follow the standard hierarchy of treatments. When assessing the paradigm of treatments, small-molecule treatments are effective. Methotrexate, for example, may work faster in comparison to tralokinumab. One clinical expert stated that, if economics was not an issue, it would make sense from a safety perspective to try small-molecule therapies before initiating immunosuppresants. Although phototherapy works quite well, access to this therapy is an issue. The other clinical expert stated that, from a pharmacoeconomic perspective, biologics should be second-line treatments, and that a trial of 2 of the 4 immunomodulators (methotrexate, cyclosporine, mycophenolate mofetil, and azathioprine) should be considered before initiating tralokinumab. |
The initiation criteria that were recommended by CDEC for dupilumab are:
Excluding the age of 12 years and older, could the initiation criteria align with dupilumab in the treatment of moderate to severe AD? | One clinical expert consulted by CADTH noted that the initiation criteria for dupilumab can be implemented in clinical practice and could be applied to tralokinumab. |
Will prior therapies required for eligibility include dupilumab (or biologics approved for AD)? | The experts agreed that tralokinumab should not be a rescue therapy for failed previous treatment with a biologic. The experts agreed that it would be possible to use tralokinumab first, and if tralokinumab failed, then use dupilumab or JAK inhibitors. A patient does not need to fail dupilumab, for example, to initiate tralokinumab. |
The CDEC renewal criteria for dupilumab are:
Should renewal criteria of tralokinumab be aligned with that of dupilumab EASI-75)? | The clinical experts consulted by CADTH noted that the renewal criteria can applied to tralokinumab. |
What should the approved initial duration of therapy be? | The approved initial duration of dupilumab is 6 months. It is reasonable to use that same duration for tralokinumab. During that time it is possible to capture patients who may not have been initial responders. |
The CDEC recommendation for dupilumab included the following 3 implementation considerations:
Should these 3 implementation considerations also be considered for tralokinumab? | One clinical expert consulted by CADTH noted that these implementation considerations are relevant for the reimbursement of tralokinumab and should be noted in the recommendation. |
Can tralokinumab be used in combination with other JAK inhibitors, biologic DMARDs, phototherapy or immunosuppressants? | The clinical experts had a difference of opinion. One clinical expert was not sure that tralokinumab should be used in combination with JAK inhibitors, but was not aware of any contraindications when combining tralokinumab with immunosuppressants or phototherapy. The expert would not use tralokinumab in combination with dupilumab. The other clinical expert agreed that there are no contraindications, but in practical terms, the only combination that would occur is phototherapy and tralokinumab, in part because of published studies that show that using phototherapy speeds up the dupilumab response. This clinical expert would only use tralokinumab in combination with phototherapy. |
Should tralokinumab be prescribed in consultation with a dermatologist and/or specialist? | A specialist would be required to diagnose, treat, and monitor patients taking tralokinumab. Appropriate specialists include dermatologists. |
With respect to “adequate trial” or “ineligible” for each of phototherapy (where available), methotrexate, and cyclosporine, can you define an “adequate trial” and what makes a patient “ineligible” for methotrexate or cyclosporine? | An adequate trial would be 3 months. A patient is ineligible for methotrexate or cyclosporine if they have intolerable side effects or a contraindication to those medications (e.g., renal disease). One clinical expert suggested using 2 immunosuppressants before initiating biologics. It is fairly easy for clinicians to come up with reasons to avoid using immunosuppressants (e.g., the patient has fatty liver disease or hypertension, both of which are common) or consider patients ineligible for immunosuppressants. |
AD = atopic dermatitis; CDEC = CADTH Canadian Drug Expert Committee; DMARDS = disease-modifying antirheumatic drugs; EASI = Eczema Area and Severity Index; EASI-75 = 75% reduction in Eczema Area and Severity Index from baseline; IGA = Investigator’s Global Assessment; JAK = Janus kinase; RCT = randomized controlled trial.
Clinical Evidence
The clinical evidence included in the review of tralokinumab is presented in 3 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of tralokinumab for the treatment of moderate to severe AD in adult patients (> 18 years of age) whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable.
Methods
Studies selected for inclusion in the systematic review include pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 7. Outcomes included in the CADTH review protocol reflect those considered to be important to patients, clinicians, and drug plans.
The systematic review protocol described in Table 7 was established before the granting of a Notice of Compliance by Health Canada.
Table 7: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adult patients (> 18 years of age) diagnosed with moderate to severe atopic dermatitis whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable Subgroups:
|
Intervention | Tralokinumab at an initial dose of 600 mg (4 injections of 150 mg each) followed by 300 mg (2 injections of 150 mg each) administered every other week as subcutaneous injections into the thigh or abdomen At prescriber’s discretion, every-fourth-week dosing may be considered for some patients who achieve clear or almost-clear skin after 16 weeks of treatment Tralokinumab can be used with or without topical corticosteroids; tralokinumab may be used with topical calcineurin inhibitors |
Comparator | When used alone or in combination with topical therapy:
|
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study designs | Published and unpublished phase III and IV RCTs |
AE = adverse event; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; NRS = numeric scale; POEM = Patient-Oriented Eczema Measure; RCT = randomized controlled trial; SAE = serious adverse event; SCORAD = Scoring Atopic Dermatitis; SF-36 = Short Form (36) Health Survey; UVA/B = UV A/B; WDAE = withdrawal due to adverse event.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.17
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946—) via Ovid and Embase (1974—) via Ovid. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were tralokinumab. Clinical trials registries searched included the US National Institutes of Health’s clinicaltrials.gov, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Appendix 1 provides detailed search strategies.
The initial search was completed on May 26, 2021. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee on September 22, 2021.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.18 Included in this search were the websites of regulatory agencies (US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Appendix 1 provides more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers and through contacts with appropriate experts. In addition, the sponsor of the drug was contacted for information regarding unpublished studies.
Findings from the Literature
Four studies were identified from the literature for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 8, Table 5 Table 9, and Table 10. A list of excluded studies is presented in Appendix 2.
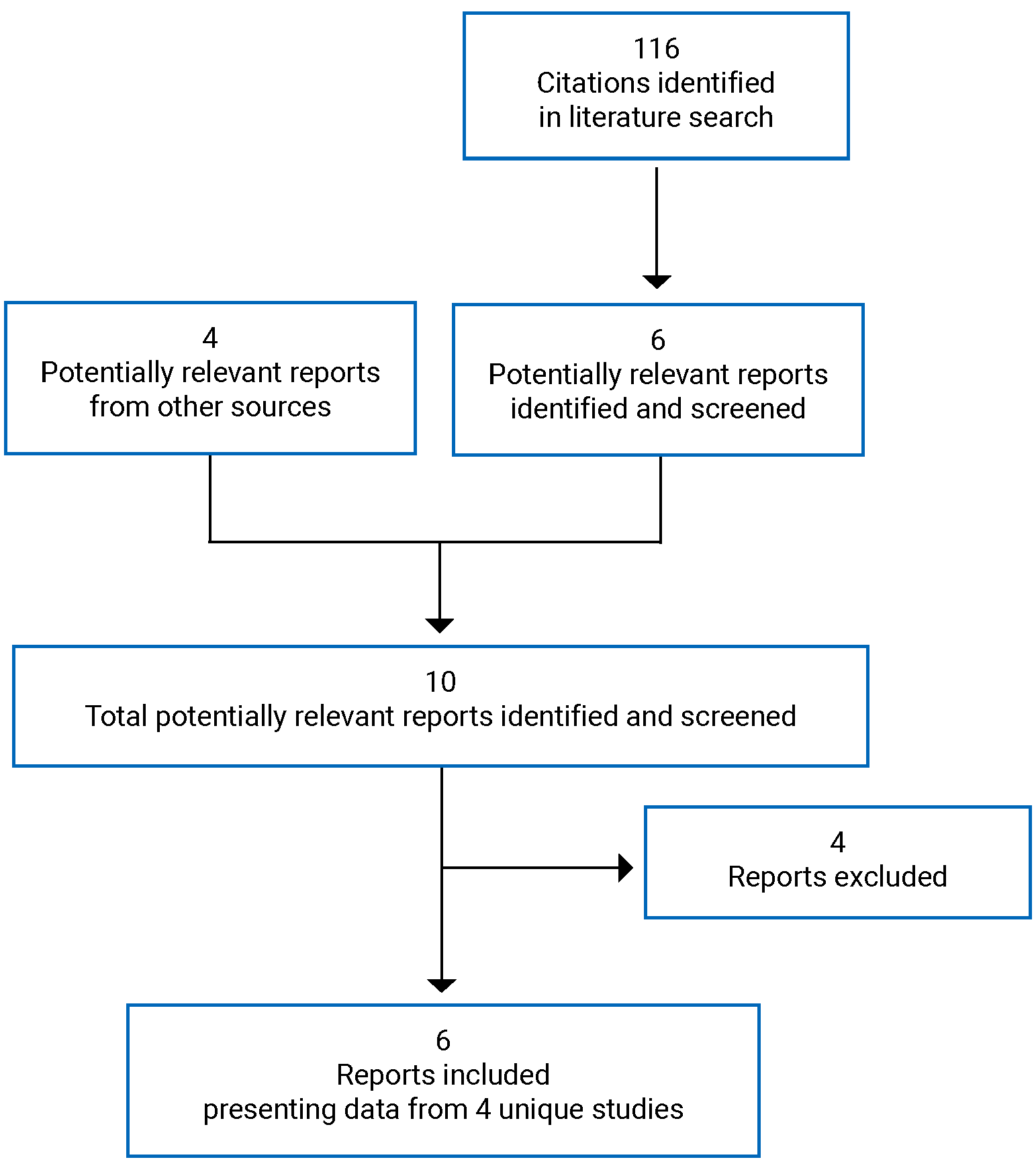
Table 8: Study Details for the ECZTRA 1 and ECZTRA 2 Trials
Study details | ECZTRA 1 and ECZTRA 2 |
|---|---|
Designs and populations | |
Study design | Double-blind RCT |
Locations | ECZTRA 1: US, Germany, France, Spain, Japan ECZTRA 2: US, Canada, Denmark, Great Britain, Italy, Poland, Russia, Australia, Korea |
Patient enrolment dates | ECZTRA 1: May 30, 2017, to March 5, 2018 ECZTRA 2: June 29, 2017, to April 26, 2018 |
Randomized (N) | ECZTRA 1: 802 ECZTRA 2: 794 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Initial treatment period (week 0 to 16):
Patients achieving a clinical response at week 16 (defined as an IGA of 0 or 1, or an EASI-75) continued into maintenance treatment until week 52 and who had been randomized to tralokinumab in the initial treatment period were re-randomized 2:2:1 to 1 of the following q.2.w. maintenance regimens (week 16 to 52):
|
Comparator(s) | Placebo (4 mL) at baseline, then placebo (2 mL) q.2.w. Patients randomized to placebo in the initial treatment period who achieved a clinical response at week 16 continued to receive placebo q.2.w. in the maintenance treatment period; patients who did not achieve a clinical response at week 16 as well as patients who did not maintain adequate clinical response during the maintenance treatment period were transferred to open-label tralokinumab 300 mg q.2.w. treatment with optional use of TCS up to week 52 |
Duration | |
Phase | |
Run-in | Up to 6 weeks |
Double-blind (initial treatment) | 16 weeks |
Double-blind (maintenance treatment) | 36 weeks |
Follow-up | 14 weeks |
Outcomes | |
Primary end points | Patients with IGA 0 or 1 (on a 5-point scale) and EASI-75 at week 16 |
Secondary and exploratory end points | Key secondary end points:
Other secondary end points:
Exploratory end points:
|
Notes | |
Publications | Wollenberg (2021)8 |
AD = atopic dermatitis; AE = adverse event; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI-50 = 50% or greater reduction in Eczema Area and Severity Index from baseline; EASI-75 = 75% or greater reduction in Eczema Area and Severity Index from baseline; EASI-90 = 90% or greater reduction in Eczema Area and Severity Index from baseline; IGA = Investigator’s Global Assessment; IMP = investigational medicinal product; NRS = numeric scale; PDE4 = phosphodiesterase type 4; POEM = Patient-Oriented Eczema Measure; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; RCT = randomized controlled trial; SC = subcutaneous; SAE = serious adverse event; SCORAD = Scoring Atopic Dermatitis; TCS = topical corticosteroids.
Table 9: Study Details of ECZTRA 3
Study details | ECZTRA 3 |
|---|---|
Designs and populations | |
Study design | Double-blind RCT |
Locations | US, Canada, Poland, Germany, Great Britain, Spain, Netherlands, Belgium |
Patient enrolment dates | March 19, 2018, to November 14, 2018 |
Randomized (N) | 380 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Initial treatment period (week 0 to 16) patients were randomized 2:1 to either the intervention (tralokinumab) or comparator group (placebo):
Patients randomized to tralokinumab in the initial treatment period who had a clinical response at week 16 (defined as IGA of 0 or 1, or at least 75% reduction in EASI score) were re-randomized into the continuation treatment period in a 1:1 ratio, stratified by region (Europe and North America) and IGA response at week 16 (IGA 0 or 1, or IGA > 1):
Patients randomized to placebo in the initial treatment period who had a clinical response at week 16 continued to receive placebo q.2.w. in the continuation treatment period via blinded treatment allocation. Patients randomized to tralokinumab or placebo in the initial treatment period who did not have a clinical response at week 16 received tralokinumab q.2.w. in the continuation treatment period. |
Comparator(s) | Placebo (4 mL) at baseline, then placebo (2 mL) q.2.w. |
Duration | |
Phase | |
Run-in | Up to 6 weeks |
Double-blind (initial treatment) | 16 weeks |
Double-blind (continuation treatment) | 16 weeks |
Follow-up | 14 weeks |
Outcomes | |
Primary end points | Patients with IGA 0 or 1 (on a 5-point scale) and EASI-75 at week 16 |
Secondary and exploratory end points | Key secondary end points:
Other secondary end points:
Exploratory end points:
|
Notes | |
Publications | Silvberg (2021)19 |
AD = atopic dermatitis; AE = adverse event; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI-50 = 50% or greater reduction in Eczema Area and Severity Index from baseline; EASI-90 = 90% or greater reduction in Eczema Area and Severity Index from baseline; IGA = Investigator’s Global Assessment; IMP = investigational medicinal product; NRS = numeric scale; PDE4 = phosphodiesterase type 4; POEM = Patient-Oriented Eczema Measure; q.2.w = every 2 weeks; q.4.w. = every 4 weeks; RCT = randomized controlled trial; SC = subcutaneous; SAE = serious adverse event; SCORAD = Scoring Atopic Dermatitis; TCI = topical calcineurin inhibitor; TCS = topical corticosteroids.
Source: Clinical Study Report for ECZTRA 3.6
Table 10: Study Details of ECZTRA 7
Study details | ECZTRA 7 |
|---|---|
Designs and populations | |
Study design | Double-blind RCT |
Locations | Poland, Germany, Great Britain, Spain, Belgium, Czech Republic, France |
Patient enrolment dates | December 13, 2018, to September 28 2020 |
Randomized (N) | 277 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | The first day of dosing was considered day 0 (visit 3, baseline). Each patient received 4 SC injections (1.0 mL each) of 150 mg tralokinumab or placebo to receive a loading dose of 600 mg tralokinumab or placebo (4.0 mL). At subsequent visits in the treatment period (week 0 to 26) patients were randomized 1:1 and stratified by prior cyclosporine A use (yes or no), country (Germany: yes or no), and baseline disease severity (IGA = 3 or 4):
Patients in both treatment groups applied a thin film of a supplied TCS once daily to areas with active lesions as needed; lower-potency TCS or TCI could be prescribed if needed for body areas where the supplied TCS was not advisable or for areas where continued treatment with TCS was considered unsafe |
Comparator(s) | Placebo (4 mL) at baseline, then placebo (2 mL) q.2.w. |
Duration | |
Phase | |
Run-in | Up to 6 weeks |
Double-blind | 26 weeks |
Follow-up | 14 weeks |
Outcomes | |
Primary end point | EASI-75 at week 16 |
Secondary and exploratory end points | Key secondary end points:
Other secondary end points:
Exploratory end points:
|
Notes | |
Publications | None |
AD = atopic dermatitis; AE = adverse event; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI-75 = 75% or greater reduction in Eczema Area and Severity Index from baseline; EASI-90 = 90% or greater reduction in Eczema Area and Severity Index from baseline; IGA = Investigator’s Global Assessment; IMP = investigational medicinal product; NRS = numeric rating scale; PDE4 = phosphodiesterase type 4; POEM = Patient-Oriented Eczema Measure; q.2.w. = every 2 weeks; RCT = randomized controlled trial; SC = subcutaneous; SCORAD = Scoring Atopic Dermatitis; TCI = topical calcineurin inhibitor; TCS = topical corticosteroids.
Source: Clinical Study Report for ECZTRA 7.4
Description of Studies
Four pivotal studies were included in this review:
ECZTRA 1 and 2
The ECZTRA 1 (N = 802) and ECZTRA 2 (N = 794) studies were 52-week, phase III, double-blind RCTs that aimed to evaluate the efficacy of tralokinumab compared with placebo in treating moderate to severe AD. The RCTs began with a screening period of 2 to 6 weeks, during which patients were assessed for study eligibility, and systemic and topical treatments for AD were washed out. All patients were to use an emollient twice daily (or more, as needed) for at least 2 weeks before randomization and were to continue this treatment throughout the trial. Following the screening period, the study had 3 key phases: an initial treatment phase (0 to 16 weeks), a maintenance treatment phase (16 to 52 weeks), and a safety follow-up (52 to 66 weeks) (Figure 2). At the start of the initial treatment phase, patients were randomized, stratified by region (North America, Japan, and Europe) and baseline disease severity (IGA of 3 or 4), in a 3:1 ratio to 1 of the following groups: a total loading dose of 600 mg of tralokinumab on day 0 followed by 300 mg of tralokinumab every 2 weeks or a total loading dose of 600 mg of placebo on day 0 followed by 300 mg of placebo every 2 weeks. Patients who achieved a clinical response at week 16 (defined as an IGA of 0 or 1, or an EASI-75 score) and who had been randomized to tralokinumab in the initial treatment phase were re-randomized 2:2:1 to 1 of the following groups: tralokinumab every 2 weeks, alternating dose administrations of placebo and tralokinumab every 2 weeks, or placebo every 2 weeks. Patients who did not achieve a clinical response at week 16, as well as those who did not maintain an adequate clinical response during the maintenance treatment period, were transferred to open-label tralokinumab every 2 weeks, with optional use of TCS. Patients randomized to the placebo group in the initial treatment period who achieved a clinical response at week 16 (defined as an IGA of 0 or 1, or an EASI-75 score) continued to receive placebo every 2 weeks in the maintenance treatment period. Randomization was conducted using a computer-generated randomization schedule stratified by region and baseline disease severity. The trials were unblinded once all randomized patients had completed the week 52 visit. At unblinding, all efficacy, safety, pharmacokinetic, and antidrug antibody data for the randomized treatment periods were collected and all end points used to test pre-specified hypotheses were final. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| (The Other Relevant Evidence section provides details).
Figure 2: ECZTRA 1 and ECZTRA 2 Trial Design
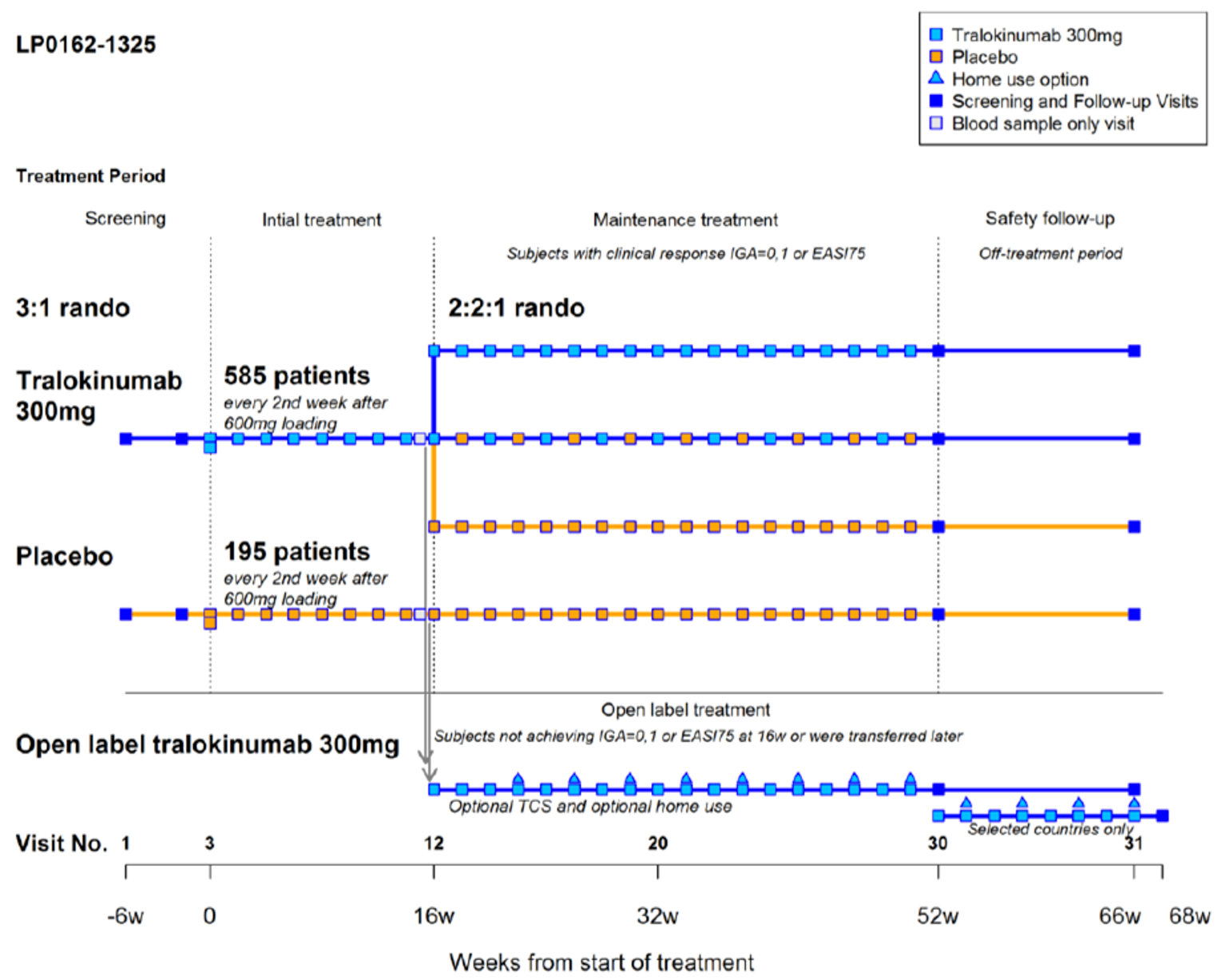
EASI75 = 75% or greater reduction in Eczema Area and Severity Index from baseline; IGA = Independent Investigator’s Assessment; rando = randomization; TCS = topical corticosteroids; w = week.
ECZTRA 3
The ECZTRA 3 (N = 380) study was a 46-week phase III, double-blind RCT that aimed to demonstrate that tralokinumab in combination with TCS is superior to placebo in combination with TCS in treating moderate to severe AD. The ECZTRA 3 trial began with a 2- to 6-week screening period during which patients were assessed for study eligibility, and when systemic and topical treatments for AD were washed out. During the screening period, all patients received an electronic diary to collect patient-reported outcome data from 2 weeks before randomization until week 32 (Figure 3). All patients were to use an emollient twice daily (or more, as needed) for at least 14 days before randomization and were to continue this treatment throughout the trial. Following the screening period, the RCT had 2 key phases: an initial treatment phase (0 to 16 weeks) and a continuation treatment phase (16 to 32 weeks). At the start of the initial treatment phase, patients were randomized in a 2:1 ratio into 1 of 2 groups, stratified by region (Europe and North America) and baseline disease severity (IGA = 3 or 4): a total loading dose of 600 mg of tralokinumab on day 0 followed by 300 mg of tralokinumab every 2 weeks or a total loading dose of 600 mg of placebo on day 0 followed by 300 mg of placebo every 2 weeks. From baseline, all patients were instructed to use a supplied TCS (mometasone furoate 0.1% cream; Europe: class 3 [potent], US: class 4 [mid-strength]) once daily, as needed, on lesional skin throughout the treatment periods. Patients who were randomized to tralokinumab in the initial treatment phase who had a clinical response at week 16 (defined as an IGA score of 0 or 1, or an EASI-75 score) were re-randomized into the continuation treatment period in a 1:1 ratio, stratified by region (Europe and North America) and IGA response at week 16 (IGA 0 or 1, or IGA > 1) to 1 of the following groups: tralokinumab every 2 weeks or alternating dose administrations of placebo and tralokinumab every 2 weeks. Patients randomized to placebo in the initial treatment phase who had a clinical response at week 16 continued to receive placebo every 2 weeks in the continuation treatment period by blinded treatment allocation. Patients randomized to tralokinumab or placebo in the initial treatment period who did not have a clinical response at week 16 received tralokinumab every 2 weeks in the continuation treatment period. All patients stayed on the TCS regimen during the continuation treatment phase. All patients who did not enter the ECZTEND trial continued in a 14-week off-treatment follow-up period for the assessment of safety at week 46. Randomization involved assigning patients the lowest available randomization number, and a central interactive voice response system was used to control the randomization and stratification factors.
The trial was unblinded once all randomized patients had completed the week 32 visit. At unblinding, all efficacy, safety, pharmacokinetic, and antidrug antibody data for the randomized treatment periods were collected and all end points used to test pre-specified hypotheses were final. The trial had 2 database locks: ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
Figure 3: ECZTRA 3 Trial Design
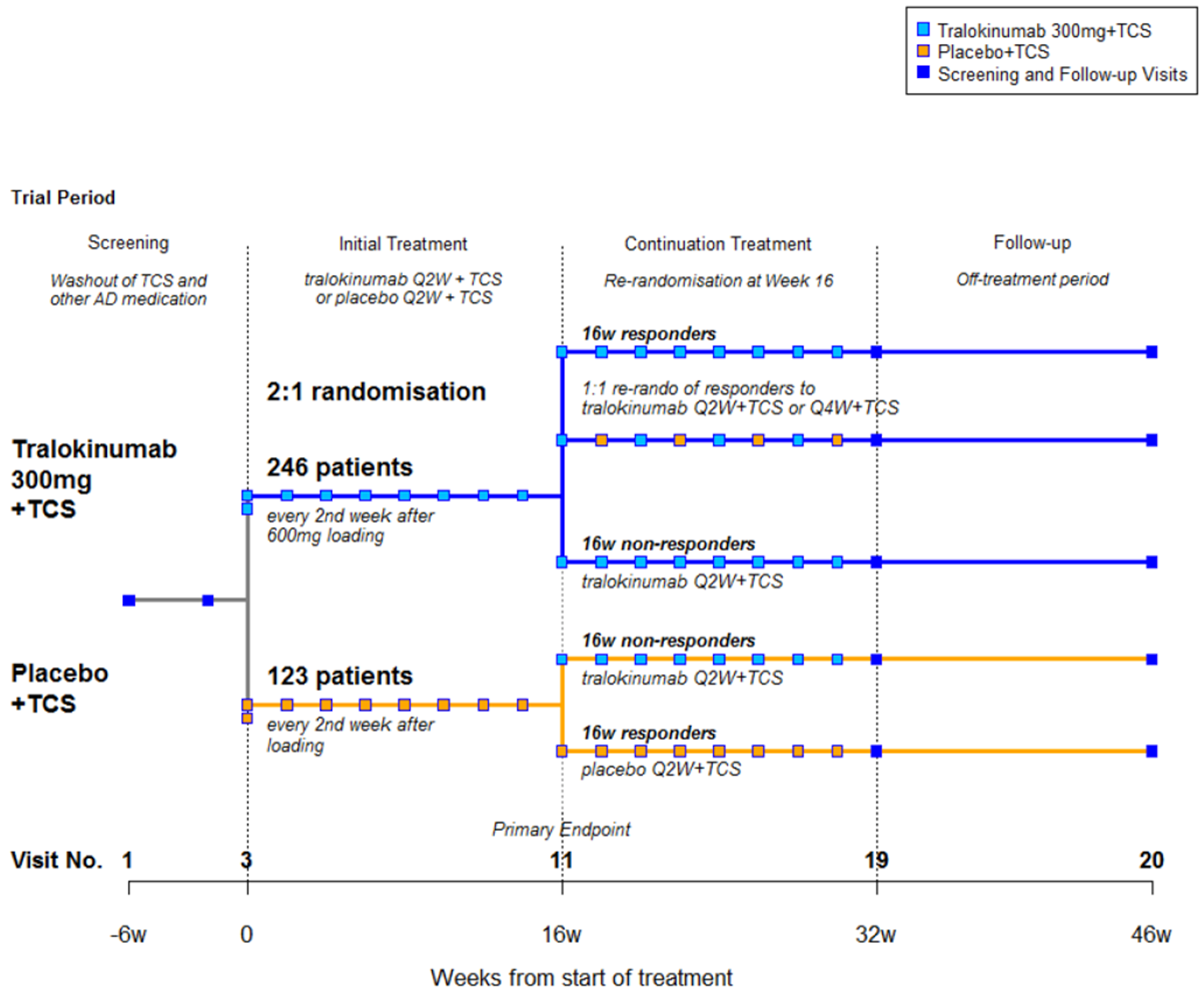
AD = atopic dermatitis; Q2W = every 2 weeks; Q4W = every 4 weeks; TCS = topical corticosteroids; w = week.
Source: Clinical Study Report for ECZTRA 3.6
ECZTRA 7
The ECZTRA 7 (N = 277) study was a 40-week phase III, double-blind RCT that aimed to demonstrate that tralokinumab in combination with TCS is superior to placebo in combination with TCS in treating severe AD in patients who are not adequately controlled with or have contraindications to oral cyclosporine A. The RCT began with a 2- to 6-week screening period during which patients were assessed for study eligibility and systemic and topical treatments for AD were washed out. During the screening period, all patients attended a screening visit 2 weeks before baseline, when they received electronic diary training and started completion of the patient-reported outcomes questionnaires in the diary (data entered into the diary were used to calculate baseline values of the patient-reported outcomes). All patients were to use an emollient twice daily (or more, as needed) for at least 14 days before randomization and were to continue this treatment throughout the trial. On lesional skin, the emollient was applied only when TCS was not applied; on TCS-untreated areas, the emollient was applied at all times. Following the screening period, patients were randomized in a 1:1 ratio to 1 of the following groups stratified by prior cyclosporine A use (yes or no), country (Germany: yes or no), and baseline disease severity (IGA = 3 or 4): a total loading dose of 600 mg of tralokinumab on day 0 followed by 300 mg of tralokinumab every 2 weeks or a total loading dose of 600 mg of placebo on day 0 followed by 300 mg of placebo every 2 weeks (Figure 4). Patients in both groups applied a thin film of a supplied TCS (mometasone furoate, 0.1% cream, Europe: class 3 [potent]) once daily to areas with active lesions as needed; lower-potency TCS or TCIs could be prescribed if needed for body areas where the supplied TCS are not advisable or for areas where continued treatment with TCS was considered unsafe. Topical therapy was discontinued when control was achieved. The safety and appropriateness of continued or repeated courses of TCS was monitored and supervised by the site staff. Patients who did not enter the ECZTEND trial after completion of the week 26 visit completed a 14-week off-treatment follow-up period for the assessment of safety. Interactive response technology was used to control randomization and stratification factors. The ECZTRA 7 trial had 1 primary outcome: the percentage of patients achieving an EASI-75 score at week 16. All staff involved in the conduct of the trial remained blinded to treatment allocation for the entire duration of the trial. This principle was applied to all investigator staff and to staff employed by the sponsor, except for sponsor staff who handled trial supply and who were directly involved in the execution of the analysis following double-blinding and unblinding. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Figure 4: ECZTRA 7 Trial Design
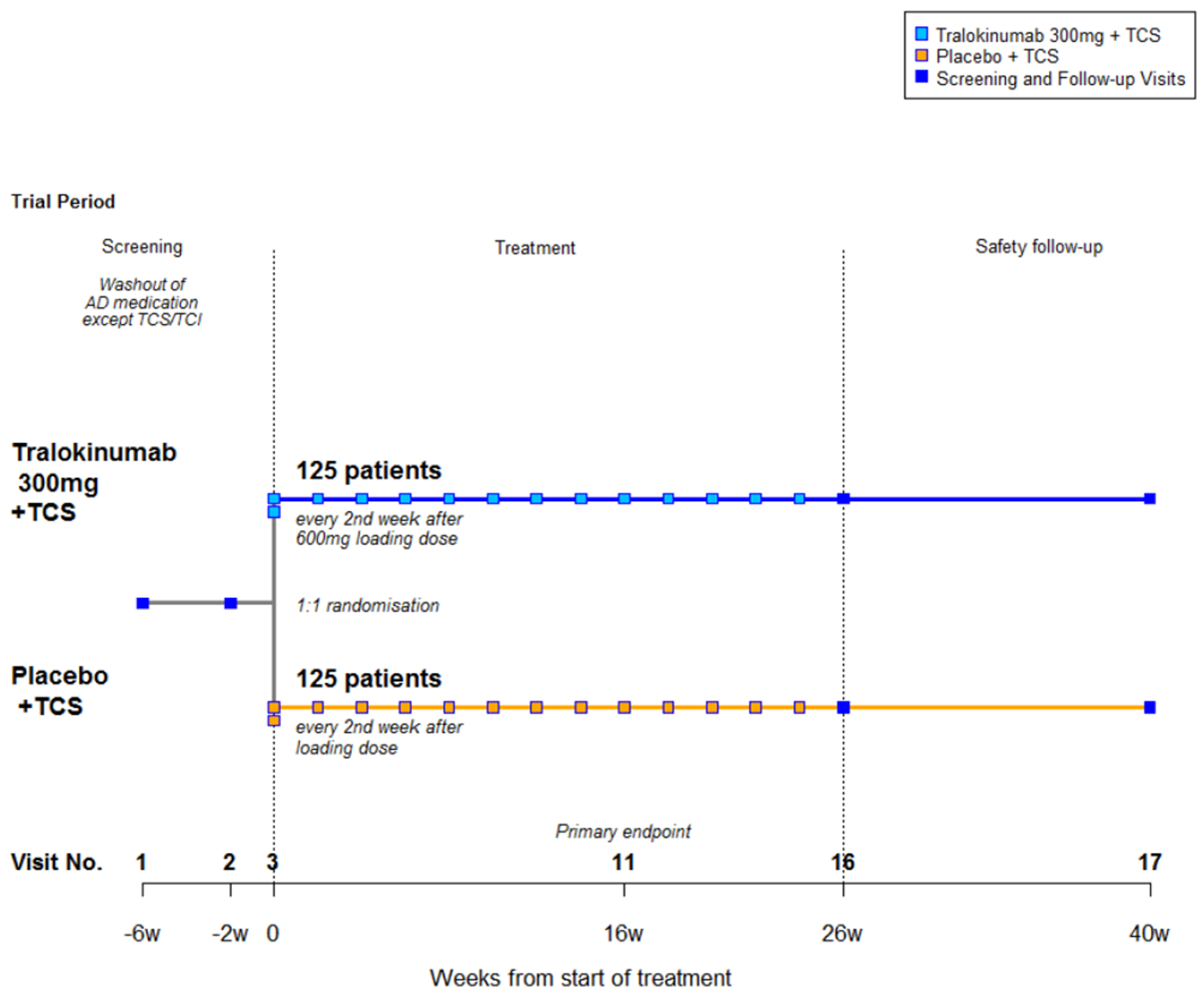
AD = atopic dermatitis; TCI = topical calcineurin inhibitors; TCS = topical corticosteroids; w = week.
Source: Clinical Study Report for ECZTRA 7.4
Populations
Inclusion and Exclusion Criteria
All RCTs included males and females 18 years of age or older, with a diagnosis of AD for at least 1 year who had inadequate response to treatment with topical medications or for whom topical medications were medically inadvisable. Inadequate response was defined as failure to achieve and maintain remission or a low disease activity state (comparable to IGA 0 = clear to 2 = mild) despite treatment with a daily regimen of TCS of medium to higher potency. Patients were required to have EASI of 12 or greater at screening and 16 or greater at baseline (with the exception of the ECZTRA 7 trial, in which where patients were required to have an EASI of at least 20 at screening and baseline), an IGA score of 3 or greater at screening and at baseline, AD involvement of at least 10% of body surface area at screening and baseline, and a worst daily pruritus NRS average score of 4 or greater during the week before baseline. Women of childbearing potential were required to use a highly effective form of birth control (confirmed by the investigator) throughout the trial and for at least for 16 weeks (5 half-lives) after last administration of treatment. Patients who had been treated with a topical phosphodiesterase type 4 inhibitor, TCS, or TCI within 2 weeks of randomization, or used systemic immunosuppressive or immunomodulating drugs, systemic corticosteroids, or 3 or more bleach baths within 4 weeks of baseline were excluded, as were those who used tanning beds or phototherapy within 6 weeks before randomization, received cell-depleting drugs within 6 months of randomization or other biologics within 5 half-lives or 12 weeks before randomization (whichever was longer).
Baseline Characteristics
Across the 4 trials, the overall mean age of patients at baseline was similar, ranging from 36.5 to 39.1 years. In the ECZTRA 1, ECZTRA 2, and ECZTRA 7 trials, more than 59% of the patients at baseline were men. The ECZTRA 3 trial had a slightly lower percentage of men at baseline (55%). The mean body surface area involvement with AD at baseline was approximately 53%, with the exception of the ECZTRA 3 trial, for which it was 48.1%. The median age of onset of AD was 3.0 (ECZTRA 1), 2.0 (ECZTRA 2), and 4.0 (ECZTRA 3); however, the median age of onset was much higher in the ECZTRA 7 trial (10.0). Compared to the ECZTRA 1, ECZTRA 2, and ECZTRA 3 trials, the ECZTRA 7 trial also had a slightly lower duration of AD (reported in years). The duration of AD was 26.2 years at baseline for the ECZTRA 7 trial, whereas the other 3 trials included patients with a duration of AD of 28 years. Greater than 99% of patients reported previous use of AD treatments across all 4 trials. The percentage of patients with severe disease (IGA = 4) was approximately 50% in all but the ECZTRA 3 trial, which included 46.3% of patients with severe AD. The details of the baseline characteristics of patients in each RCT are included in the following section.
ECZTRA 1
Of the patients enrolled in ECZTRA 1, the overall mean age at baseline was ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Patients were included from 3 regions: |||||||| of patients from North America (US), |||||||| of patients from Europe (|||||||| from Germany, |||||||| from France, and ||||| from Spain) and ||||| of patients from Asia (Japan). |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||, and the ||||||||||||||||||||||||||||||. For all randomized patients, |||||||||||||||||||| (moderate disease based on IGA) and 50.7% had IGA 4 (severe disease based on IGA), with comparable percentages for the tralokinumab every 2 weeks and placebo treatment groups, respectively. A total of 99.1% of patients reported use of any previous AD treatment, which included TCS (98.0% of patients), systemic steroids (59.4% of patients), topical and systemic calcineurin inhibitors (49.9% of patients), and phototherapy (48.1% of patients). No major differences in demographic and clinical characteristics were seen between-trial groups. Table 11 provides details.
ECZTRA 2
Of the patients enrolled in the ECZTRA 2 trial, the mean age at baseline was |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Patients were included from 4 regions: |||||| of patients from North America (|||||| from Canada and |||||| from the US), |||||| of patients from Europe (|||||| from Poland, |||||| from Great Britain, |||||| from Italy, |||||| from Russia, and |||||| from Denmark), |||||| of patients from Australia, and |||||| of patients from Asia (Korea). At baseline, the ||||||||||||||||||||||||||||||||||||||||||, the ||||||||||||||||||||||||||||||||||||, and the ||||||||||||||||||||||||||||||||||||||||||||||||. For all randomized patients, |||||||||||||||||||||||| (moderate disease based on IGA) and 48.7% had IGA 4 (severe disease based on IGA), and the percentages were comparable for the tralokinumab every 2 weeks and placebo treatment groups, respectively. A total of 99.7% of all randomized patients reported use of previous AD treatment, which included TCS (98.7% of patients), systemic steroids (67.4% of patients), |||||||||||||||||||||||||||||| calcineurin inhibitors (46.5% of patients), and phototherapy (43.7% of patients). No major differences in demographic and clinical characteristics were seen between-trial groups. Table 11 provides details.
ECZTRA 3
Of the patients enrolled in the ECZTRA 3 trial, |||||||||||||||||||||||||||||||||||||||||| and 55.0% of the trial population were men. In the placebo plus TCS group, there was a higher proportion of men than women (66.1% versus 33.9% patients, respectively). Most patients were White (75.8%) and ||||||||||||||||||||||||||||||||||||||||||||||||. There was a higher proportion of White patients (80.2% versus 66.9%) and a lower proportion of Asian patients (6.7% versus 18.9%) in the tralokinumab every 2 weeks plus TCS group than in the placebo plus TCS group, respectively. In both treatment groups, the distribution of ethnicity was comparable. Approximately ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. In both treatment groups, the distribution of patients between regions (North America and Europe) was comparable and the distribution of patients between countries did not differ markedly between the 2 treatment groups. At baseline, the ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. For all randomized patients, 53.2% had an IGA of 3 (moderate disease) and 46.3% had an IGA of 4 (severe disease). All patients reported previous use of AD treatments. Almost all of the patients (98.2%) had used TCS. The frequent use of TCS was in line with the target population of patients with a recent history (within 1 year before the screening visit) of inadequate response to treatment with TCS. High proportions of patients (≥ 40%) had also used systemic steroids (61.6%), ||||||||||||||||||||||||||||||||||||||||||||||||||||||, phototherapy (46.1%), and antibiotics (40.0%) in both treatment groups. In addition, previous use of cyclosporine (31.1% patients), methotrexate (15.5% patients), azathioprine (6.6% patients), and mycophenolate (3.2% patients) was also reported in both treatment groups. At baseline, previous use of methotrexate and azathioprine was less frequent in the tralokinumab every 2 weeks plus TCS group than in the placebo plus TCS group (11.5% versus 23.6% patients and 5.1% versus 9.4% patients, respectively). Table 12 provides details.
ECZTRA 7
Of the patients enrolled in the ECZTRA 7 trial, |||||||||||||||||||||||||||||||||||||||||||||||||||||| and 59.6% of the trial population were men. There was a higher proportion of men than women (59.6% versus 40.4% patients, respectively). Most patients (98.2%) were White and ||||||||||||||||||||||||||||||||||||||||||||||||. The patients were all located in Europe, mostly in Poland (27.8%). At baseline, the ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. For all randomized patients, 50.2% had IGA score of 3 (moderate disease) and 49.8% had IGA score of 4 (severe disease), and the ||||||||||||||||||||||||||||||||||||||||||||||||. All patients reported previous use of AD treatments. Almost all of the patients (99.6%) had used TCS. High proportions of patients (≥ 40%) had also used systemic steroids (68.2%), |||||||||||||||||||||||||||||||||||||||||| phototherapy (58.8%), |||||||||||||||||||||||||||||||||||||||||| in both treatment groups. In addition, previous use of cyclosporine (74.7% patients), methotrexate (17.7% patients), azathioprine (13.0% patients), and mycophenolate (2.9% patients) was also reported in both treatment groups. Table 13 provides details.
Table 11: Summary of Baseline Characteristics for the ECZTRA 1 and ECZTRA 2 Trials
Characteristic | ECZTRA 1 | ECZTRA 2 | ||
|---|---|---|---|---|
Tralokinumab q.2.w. N = 603 | Placebo N = 199 | Tralokinumab q.2.w. N = 593 | Placebo N = 201 | |
Mean (SD) age, years | 38.6 (13.7) | 39.4 (15.2) | 37.2 (14.7) | 35.1 (14.0) |
Male, n (%) | 351 (58.2) | 123 (61.8%) | 359 (60.5) | 114 (56.7) |
Race, n (%) | ||||
White | 426 (70.6) | 138 (69.3) | 374 (63.1) | 123 (61.2) |
African-American or African | 41 (6.8) | 18 (9) | 43 (7.3) | 17 (8.5) |
Asian | 120 (19.9) | 40 (20.1) | 154 (26.0) | 52 (25.9) |
Other or missing data | 16 (2.6) | 3 (1.5) | 22 (3.7) | 9 (4.5) |
Country, n (%) | ||||
US | 149 (24.7) | 49 (24.6) | 124 (20.9) | 47 (23.4) |
Germany | 200 (33.2) | 73 (36.7) | NA | NA |
France | 85 (14.1) | 23 (11.6) | NA | NA |
Spain | 73 (12.1) | 23 (11.6) | NA | NA |
Japan | 96 (15.9) | 31 (15.6) | NA | NA |
Australia | NA | NA | 90 (15.2) | 31 (15.4) |
Canada | NA | NA | 146 (24.6) | 44 (21.9) |
Denmark | NA | NA | 8 (1.3) | 2 (1.0) |
Great Britain | NA | NA | 55 (9.3) | 15 (7.5) |
Italy | NA | NA | 31 (5.2) | 10 (5.0) |
Korea | NA | NA | 58 (9.8) | 20 (10.0) |
Poland | NA | NA | 67 (11.3) | 27 (13.4) |
Russia | NA | NA | 14 (2.4) | 5 (2.5) |
Mean duration of atopic dermatitis, years (SD) | 27.9 (14.5) | 29.6 (15.1) | 28.3 (15.9) | 27.5 (14.7) |
EASI score mean (SD) | 32.2 (13.7) | 32.9 (13.9) | 32.1 (14.3) | 32.6 (13.9) |
Baseline BSA mean (SD) | 52.7 (24.1) | 54.2 (25.6) | 52.6 (25.6) | 53.0 (25.0) |
IGA score, n (%) | ||||
|||||||||||||||||||||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Severe disease | 305 (50.6) | 102 (51.3) | 286 (48.2) | 101 (50.2) |
SCORAD mean (SD) | 70.3 (13.0) | 71.7 (12.5) | 70.0 (13.4) | 70.5 (12.2) |
Weekly average worst daily pruritus NRS mean (SD) | 7.7 (1.4) | 7.7 (1.4) | 7.9 (1.5) | 8.0 (1.4) |
|||||||||||||||||||||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||||||||||||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Patients receiving prior systemic corticosteroids, n (%) | 357 (59.2) | 119 (59.8) | 410 (69.1) | 125 (62.2) |
Patients receiving prior systemic nonsteroidal immunosuppressants, n (%) | ||||
Azathioprine | 39 (6.5) | 7 (3.5) | 72 (12.1) | 25 (12.4) |
Cyclosporine | 227 (37.6) | 65 (32.7) | 204 (34.4) | 65 (32.3) |
Methotrexate | 77 (12.8) | 26 (13.1) | 127 (21.4) | 38 (18.9) |
Mycophenolate | 27 (4.5) | 9 (4.5) | 37 (6.2) | 14 (7.0) |
BSA = body surface area; EASI = Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; NA = not applicable; NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; q.2.w. = every 2 weeks; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation
Source: Clinical Study Reports for ECZTRA 17 and 2.5
Table 12: Summary of Baseline Characteristics for the ECZTRA 3 Trial
Characteristic | Tralokinumab q.2.w. + TCS N = 253 | Placebo N = 127 |
|---|---|---|
Mean (SD) age, years | |||||||||||| | |||||||||||| |
Male, n (%) | 125 (49.4) | 84 (66.1) |
Race, n (%) | ||
White | 203 (80.2) | 85 (66.9) |
African-American or African | 23 (9.1) | 12 (9.4) |
Asian | 17 (6.7) | 24 (18.9) |
Native Hawaiian or other Pacific Islander | 1 (0.4) | 1 (0.8) |
Other | 9 (3.6) | 5 (3.9) |
Country, n (%) | ||
US | 72 (28.5) | 28 (22.0) |
Canada | 34 (13.4) | 26 (20.5) |
Poland | 48 (19.0) | 19 (15.0) |
Germany | 42 (16.6) | 15 (11.8) |
Great Britain | 21 (8.3) | 13 (10.2) |
Spain | 12 (4.7) | 15 (11.8) |
Netherlands | 9 (3.6) | 7 (5.5) |
Belgium | 15 (5.9) | 4 (3.1) |
Mean duration of atopic dermatitis, years (SD) | 28.0 (16.5) | 28.7 (15.0) |
EASI score mean (SD) | 28.8 (12.0) | 30.4 (12.8) |
Baseline BSA mean (SD) | 47.6 (23.3) | 49.0 (25.9) |
IGA score, n (%) | ||
Moderate disease | 136 (53.8) | 66 (52.0) |
Severe disease | 116 (45.8) | 60 (47.2) |
SCORAD mean (SD) | 67.0 (13.3) | 68.9 (13.2) |
Worst pruritus NRS from electronic diary mean (SD) | 7.7 (1.5) | 7.9 (1.5) |
DLQI score mean (SD) | 17.6 (7.1) | 17.2 (7.2) |
Patients receiving prior systemic corticosteroids, n (%) | 148 (58.5) | 86 (67.7) |
Patients receiving prior systemic nonsteroidal immunosuppressants, n (%) | ||
Azathioprine | 13 (5.1) | 12 (9.4) |
Cyclosporine | 75 (29.6) | 43 (33.9) |
Methotrexate | 29 (11.5) | 30 (23.6) |
Mycophenolate | 7 (2.8) | 5 (3.9) |
BSA = body surface area; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; NRS = numeric rating scale; SCORAD = Scoring Atopic Dermatitis; q.2.w = every 2 weeks; SD = standard deviation; TCS = topical corticosteroids.
Source: Clinical Study Report for ECZTRA 3.6
Table 13: Summary of Baseline Characteristics for the ECZTRA 7 Trial
Characteristics | Tralokinumab q.2.w + TCS N = 140 | Placebo + TCS N = 137 |
|---|---|---|
|||||||||||||||||||||||||||||| | ||||||||||| | ||||||||||| |
Male, n (%) | 82 (58.6) | 83 (60.6) |
Race, n (%) | ||
White | 137 (97.9) | 135 (98.5) |
African-American or African | 0 (0) | 1 (0.7) |
Asian | 0 (0) | 1 (0.7) |
|||||||||||||||||||||||||||||||||||||||||||| | ||||||||||| | ||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||| | ||||||||||| | ||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||| | ||||||||||| | ||||||||||| |
Country, n (%) | ||
Belgium | 25 (17.9) | 27 (19.7) |
Czech Republic | 14 (10.0) | 12 (8.8) |
Germany | 22 (15.7) | 18 (13.1) |
Spain | 28 (20.0) | 21 (15.3) |
France | 12 (8.6) | 7 (5.1) |
||||||||||| | ||||||||||| | ||||||||||| |
Poland | 34 (24.3) | 43 (31.4) |
|||||||||||||||||||||||||||||||||||||||||||| | ||||||||||| | ||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||| | ||||||||||| | ||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||| | ||||||||||| | ||||||||||| |
IGA score, n (%) | ||
Moderate disease | 68 (49.3) | 70 (51.1) |
Severe disease | 70 (50.7) | 67 (48.9) |
|||||||||||||||||||||||||||||||||||||||||||| | ||||||||||| | ||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||| | ||||||||||| | ||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||| | ||||||||||| | ||||||||||| |
Patients receiving prior systemic corticosteroids, n (%) | 98 (70.0) | 91 (66.4) |
Patients receiving prior systemic nonsteroidal immunosuppressants, n (%) | ||
Azathioprine | 18 (12.9) | 18 (13.1) |
Cyclosporine | 105 (75.0) | 102 (74.5) |
Methotrexate | 23 (16.4) | 26 (19.0) |
Mycophenolate | 3 (2.1) | 5 (3.6) |
BSA = body surface area; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; NRS = numeric rating scale; SCORAD = Scoring Atopic Dermatitis; q.2.w. = every 2 weeks; SD = standard deviation; TCS = topical corticosteroids.
Source: Clinical Study Report for ECZTRA 7.4
Interventions
ECZTRA 1 and ECZTRA 2
Following the screening period, patients were randomized 3:1 to the intervention group or placebo for 16 weeks. In the intervention group, each patient received 4 subcutaneous injections (1 mL each) of 150 mg tralokinumab for a total loading dose of 600 mg tralokinumab on day 0. At subsequent visits in the initial treatment period (week 0 to 16), each patient received 2 injections (1 mL each) of 150 mg tralokinumab every 2 weeks. Patients randomized to the placebo group received an initial loading dose of 4 mL at day 0, and then 2 mL every second week. All injections were administered subcutaneously. Patients eligible for the maintenance period (week 16 to 52) were re-randomized 2:2:1 to receive either 300 mg tralokinumab every 2 weeks, alternating administrations of 300 mg tralokinumab and 2 mL placebo every 2 weeks, or 2 mL placebo every 2 weeks.
All patients were to use an additive-free, basic, bland emollient twice daily (or more often, as needed) for at least 14 days before randomization and were to continue this treatment throughout the trial. Rescue treatment for AD could be provided to trial patients at the discretion of the investigator. For analysis of the primary end points, patients who received rescue treatment during the initial treatment phase were considered nonresponders, but they continued tralokinumab or placebo treatment if the rescue treatment consisted of topical medications only. Investigators were encouraged to try topical treatments first and escalate to systemic medications only for patients who did not respond adequately after at least 14 days. Any WHO class of TCS could be used as topical rescue treatment, although it was unclear if any restrictions were imposed on frequency or dose of topical rescue treatment. A TCI could also be used but such use should be reserved for problem areas only (e.g., face, neck, intertriginous, and genitals). Systemic rescue treatment with corticosteroids or nonsteroidal immunosuppressive drugs (e.g., cyclosporine or methotrexate) required immediate discontinuation of tralokinumab. After the treatment with these drugs was completed, tralokinumab could be resumed if deemed appropriate by the investigator and the sponsor’s medical expert, but no sooner than 5 half-lives after the last dose of the systemic rescue treatment. Investigators could prescribe concomitant medications or treatments to provide adequate supportive care as deemed necessary, except for medications considered prohibited. The following concomitant medications related to AD treatment were permitted from screening through safety follow-up: oral antibiotics, antiviral, or antifungal therapy for skin infections as appropriate, stable doses of an emollient, and oral antihistamines.
ECZTRA 3
Following the screening period, patients were randomized 2:1 into the following groups, stratified by region (Europe and North America) and baseline disease severity (IGA = 3 or 4): a total loading dose of 600 mg tralokinumab or placebo on day 0 followed by tralokinumab 300 mg every 2 weeks (2 subcutaneous injections [1.0 mL each] of 150 mg tralokinumab) or placebo every 2 weeks (2 subcutaneous injections [1.0 mL each]). From baseline, all patients were instructed to initiate treatment once daily with a supplied TCS on lesional skin and continue as needed throughout the treatment period. At week 16, patients entered the continuation treatment period that lasted until week 32. The treatment assigned depended on the treatment in the initial treatment period and on the patient’s clinical response at week 16 (IGA of 0 or 1, or EASI-75). Patients randomized to tralokinumab in the initial treatment period who had a clinical response at week 16 were re-randomized to the continuation treatment period in a 1:1 ratio, stratified by region (Europe and North America) and IGA response at week 16 (IGA of 0 or 1, or IGA > 1) to receive either tralokinumab 300 mg every 2 weeks or tralokinumab 300 mg every 4 weeks (alternating dose administrations of tralokinumab 300 mg every 2 weeks and placebo every 2 weeks). Patients randomized to placebo in the initial treatment period who had a clinical response at week 16 continued to receive placebo every 2 weeks in the continuation treatment period via blinded treatment allocation. Patients randomized to tralokinumab or placebo in the initial treatment period who did not have a clinical response at week 16 were allocated to receive tralokinumab every 2 weeks in the continuation treatment period. All patients stayed on the TCS regimen during the continuation treatment period.
Rescue treatment for AD could be provided to patients at the discretion of the investigator. Investigators were encouraged to try topical treatments first and escalate to systemic medications only for patients who did not respond adequately after at least 14 days. Patients who received systemic rescue treatment with corticosteroids or nonsteroidal immunosuppressive drugs (cyclosporine, methotrexate, mycophenolate mofetil, or azathioprine) were required to immediately discontinue tralokinumab or placebo. After treatment with these medications was completed, treatment with the tralokinumab or placebo could be resumed if deemed appropriate by the investigator and the sponsor’s medical expert, but no sooner than 5 half-lives after the last dose of systemic rescue medication. Investigators could prescribe concomitant medications or treatments to provide adequate supportive care as deemed necessary, except for medications considered prohibited. The following concomitant medications related to AD treatment were permitted from screening through safety follow-up: oral antibiotics, antiviral, or antifungal therapy for skin infections as appropriate, and oral antihistamines.
ECZTRA 7
Following the screening period, patients were randomized 1:1 to 1 of the following groups stratified by prior cyclosporine A use (yes or no), country (Germany: yes or no), and baseline disease severity (IGA = 3 or 4): tralokinumab 600 mg loading dose at day 0 followed by tralokinumab 300 mg every 2 weeks or placebo 600 mg loading dose at day 0 followed by placebo every 2 weeks. Patients in both treatment groups applied a thin film of a supplied TCS once daily to areas with active lesions as needed; lower-potency TCS or a TCI could be prescribed if needed for body areas where the supplied TCS was not advisable or for areas where continued treatment with TCS was considered unsafe. Topical therapy was discontinued when control was achieved; discontinuation should preferably be gradual. The safety and appropriateness of continued or repeated courses of TCS therapy was monitored and supervised by the site staff. Patients, except those who entered the long-term extension trial after completion of the week-26 visit (ECZTEND), completed a 14-week off-treatment follow-up period for the assessment of safety, pharmacokinetics, and antidrug antibodies.
Rescue treatment for AD could be provided to trial patients at the discretion of the investigator. Investigators were encouraged to try topical treatments first and escalate to systemic medications only for patients who did not respond adequately after at least 14 days. Patients who received systemic rescue treatment with corticosteroids or nonsteroidal immunosuppressive drugs (e.g., methotrexate, mycophenolate mofetil, or azathioprine) were required to immediately discontinue tralokinumab or placebo. After treatment with these medications was completed, treatment with tralokinumab or placebo could be resumed if deemed appropriate by the investigator, but no sooner than 5 half-lives after the last dose of systemic rescue treatment. Investigators could prescribe concomitant medications or treatments to provide adequate supportive care as deemed necessary, except for medications considered prohibited. The following concomitant medications related to AD treatment were permitted from screening through safety follow-up: oral antibiotics, antiviral, or antifungal therapy for skin infections as appropriate, and oral antihistamines.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 14. These end points are then further summarized. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 3.
Table 14: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | ECZTRA 1 | ECZTRA 2 | ECZTRA 3 | ECZTRA 7 |
|---|---|---|---|---|
IGA score | Primary | Primary | Primary | Secondary |
EASI-75 | Primary | Primary | Primary | Primary |
SCORAD | Secondary | Secondary | Secondary | Secondary |
Worst daily pruritus NRS | Secondary | Secondary | Secondarya | Secondarya |
DLQI | Secondary | Secondary | Secondary | Secondary |
EASI-50 | Secondary | Secondary | Secondary | NA |
EASI-90 | Secondary | Secondary | Secondary | Exploratory |
SCORAD-75 | Secondary | Secondary | Secondary | NA |
SCORAD-50 | Secondary | Secondary | Secondary | NA |
Eczema-related sleep NRS | Exploratory | Exploratory | Exploratorya | Exploratorya |
||||||||||| | ||||||||||| | ||||||||||| | ||||||||||| | ||||||||||| |
POEM | Exploratory | Exploratory | Exploratory | Exploratory |
||||||||||| | ||||||||||| | ||||||||||| | ||||||||||| | ||||||||||| |
|||||||| | ||||||||||| | ||||||||||| | ||||||||||| | ||||||||||| |
||||||| | ||||||||||| | ||||||||||| | ||||||||||| | ||||||||||| |
DLQI = Dermatology Life Quality Index; EASI-50 = 50% or greater reduction in Eczema Area and Severity Index from baseline; EASI-75 = 75% or greater reduction in Eczema Area and Severity Index from baseline; EASI-90 = 90% or greater reduction in Eczema Area and Severity Index from baseline; IGA = Investigator’s Global Assessment; NA = not applicable; NRS = Numeric rating scale; POEM = Patient-Oriented Eczema Measure; SCORAD-50 = 50% decrease in Scoring Atopic Dermatitis; SCORAD-75 = 75% decrease in Scoring Atopic Dermatitis.
aPatient-reported outcomes collected in the electronic diaries on a daily basis in the morning. Patients attended a screening visit 2 weeks before baseline at which they received the diary training and started completion of the patient-reported outcomes questionnaires in the diary. Patients also recorded days of topical treatment use in their diary. Patients used the diary for 32 weeks and 26 weeks in the ECZTRA 3 and ECZTRA 7 trials, respectively.
Source: Clinical Study Report for ECZTRA 1,7 2,5 3,6 and 7.4
In the ECZTRA 1, ECZTRA 2, and ECZTRA 3 trials, the coprimary outcomes were patients with an IGA score of 0 or 1 at week 16 and patients who had achieved an EASI-75 score at week 16. In the ECZTRA 7 trial, the primary outcome was patients who had achieved an EASI-75 score at week 16.
Investigator’s Global Assessment
The IGA score is an investigator-reported assessment used to rate AD severity. It is based on a 5-point scale ranging from 0 to 4, in which “0” indicates clear, and “4” indicates severe AD.20 A decrease in score reflects improvement in signs and symptoms. The validity and reliability of the measure was determined to be adequate in patients with varying severities of AD based on a study by Bożek (2017),21 and reliability was also found to be adequate in a study conducted by Zhao et al. (2017),22 which compared patients with lighter and darker skin. No MID was identified in patients with AD.
Eczema Area and Severity Index
The EASI is a scale used in clinical trials to assess the severity and extent of AD.23 In the EASI, 4 disease characteristics of AD (erythema, infiltration or papulation, excoriations, and lichenification) are assessed for severity by the investigator on a scale of “0” (absent) to “3” (severe). The scores are added up for each of the 4 body regions (head, arms, trunk, and legs). The assigned percentages of body surface area for each section of the body are 10% for head, 20% for arms, 30% for trunk, and 40% for legs respectively. Each subtotal score is multiplied by the body surface area represented by that region. In addition, the affected area of AD assessed as a percentage by each body region is converted to a score of 0 to 6, in which the area is expressed as 0 (none), 1 (1% to 9%), 2 (10% to 29%), 3 (30% to 49%), 4 (50% to 69%), 5 (70% to 89%), or 6 (90% to 100%). Each of the body area scores is multiplied by the area affected. The total EASI score therefore ranges from 0 to 72 points, with higher scores indicating worse severity of AD.24 It is suggested that the severity of AD based on the EASI are categorized as follows: 0 = clear; 0.1 to 1.0 = almost clear; 1.1 to 7.0 = mild; 7.1 to 21.0 = moderate; 2l.1 to 50.0 = severe; and 50.1 to 72.0 = very severe.25 The EASI-50, EASI-75, EASI-90, and EASI-100 end points indicate improvements of 50% or greater, 75% or greater, 90% or greater, and 100% improvement from baseline, respectively.
The psychometric properties of the EASI have been examined in several studies of patients with AD.26-31 Validity of the EASI and SCORAD was adequate, with correlation coefficients ranging from 0.84 to 0.93.29 Intra- and inter-rater reliability were estimated to be adequate based on correlation coefficients ranging from 0.8 to 0.9, and responsiveness was adequate in populations of children and adults with AD.29 It was determined that the EASI is a validated scale and can be used reliably to assess the severity and extent of AD. Inter-rater reliability for the EASI was also assessed in a study that separated patients into 2 groups: those with lighter skin (melanin index ≤ 200; n = 11) and those with darker skin (melanin index > 200; n = 14), and was found to be adequate (intraclass correlation coefficients of 0.83 and 0.77, respectively).22 The overall MID is 6.6, based on results from 1 study that included patients with atopic eczema.32
Scoring Atopic Dermatitis
The SCORAD tool was developed to standardize the evaluation of the extent and severity of AD.33 It is considered a valid and reliable tool for the objective assessment of eczema clinical signs.34 The instrument assesses 3 components of AD: the extent of affected body surface area (0 to 100), severity (0 to 18), and symptoms (0 to 20). The extent of AD is assessed as a percentage of each defined body area and reported as the sum of all areas. The score ranges from 0 to 100. The severity of 6 specific signs of AD (redness, swelling, oozing and/or crusting, excoriation, skin thickening and/or lichenification, and dryness) is assessed using a 4-point scale (none = 0, mild = 1, moderate = 2, and severe = 3) with a minimum score of 0 and a maximum of 18. Relevant symptoms (itch and sleeplessness) are recorded by the patient or relative on a visual analogue scale, with scores ranging from 0 (no symptoms) to 10 (worst imaginable symptom) and a maximum possible score of 20. The SCORAD total is calculated based on the 3 components, with a maximum possible total score of 103, in which a higher score indicates poorer or a more severe condition.
The SCORAD tool has been found to be valid and reliable in patients with AD, with excellent agreement with global assessments of disease severity.23,35 Content validity has been deemed adequate, with good construct validity (Spearman R values range from 0.53 to 0.92) and internal consistency. Sensitivity to change and inter-observer reliability are also adequate; the latter with measurements of the intraclass coefficient from 0.84 to 0.99. However, intra-observer (test-retest) reliability was unclear.23 The MID has been estimated using mean change scores of SCORAD of patients that showed a relevant improvement based on IGA, defined as an “improvement” or “decline” of at least 1 point in the Physician’s Global Assessment (PGA) and IGA. A difference of 8.7 points in SCORAD was estimated as the MID for the patients with AD.32
Worst Pruritus Numeric Rating Score
The pruritus NRS is used to report the intensity of itches during a daily recall period. Patients rate their overall (average) and maximum intensity of itch experienced during the past 24 hours based on a scale of 0 to 10 (0 = “no itch” and 10 = “worst itch imaginable”).
The reliability of the NRS is adequate,36 with pooled intraclass coefficients in a range of 0.95 to 0.97.36 The NRS scores are stable over a period of time. The validity of the NRS has been shown using known-groups approaches with all results above the Cohen threshold of 0.80 for large effect sizes.36 Based on a study by Simpson et al., an estimated improvement on the pruritus NRS is at least a 3- to 4-point reduction.36 This estimate was calculated using anchor- and distribution-based methods and data from a phase IIb study of dupilumab used to treat patients with moderate to severe AD. Yosipovitch et al. 2019 also estimated the MID using both distribution-based methods as well as patient-reported outcome and clinician-reported outcome anchor-based methods.37 The MIDs were reported to range from 2.2. to 4.2 points for clinician-reported outcome anchors (i.e., EASI and IGA) but were generally lower at 0.76 and 2.6 points for the half–standard deviation (SD) distribution–based method and patient-reported outcome anchor (i.e., pruritus categorical scale), respectively. The investigators suggested that the pruritus categorical scale may be the more appropriate anchor as it had a stronger correlation with the NRS than with the EASI or IGA.
Dermatology Life Quality Index
The DLQI is a widely used HRQoL instrument. It is a 10-item questionnaire that assesses 6 different aspects that may affect quality of life.38,39 These aspects are symptoms and feelings, daily activities, leisure, work and school performance, personal relationships, and treatment. Each of the 10 questions is scored on a 4-point scale (0 = not at all or not relevant, 1 = a little, 2 = a lot, and 3 = very much), and the overall DLQI is calculated by summing the score of each question resulting in a numeric score between 0 and 30 (or a percentage of 30).38,39 The higher the score, the more quality of life is impaired. The meaning of the DLQI scores on a patient’s life is as follows40: 0 to 1 = no effect; 2 to 5 = small effect; 6 to 10 = moderate effect; 11 to 20 = very large effect; and 21 to 30 = extremely large effect.
The DLQI has previously been validated in patients with a variety of skin conditions, including eczema.41-44 Patel et al. (2019) aimed to assess the validity, responsiveness, and floor and ceiling effects of the DLQI in a group of 340 adults with AD, and determined that the DLQI has adequate content validity and moderate convergent validity when compared to the EASI (rS = 0.44), SCORAD (rS = 0.55), NRS-itch (rS = 0.59), and POEM (rS = 0.61).45 Convergent validity was also assessed in a 2021 study by Schwartzman et al. that included 994 adults with AD and showed similar outcomes for the EASI (rS = 0.48), SCORAD (rS = 0.48), NRS-itch (rS = 0.53 worst itch and rS = 0.49 average itch), and POEM (rS = 0.61) instruments.46 To assess discriminative validity, Patel et al. (2019) also determined that the DLQI has adequate discriminative validity, good internal consistency (Cronbach alpha = 0.89) and fair to moderate correlation among individual items (rho = 0.28 to 0.60). The DLQI was found to be less responsive for patients who showed either 1- or 3-point improvement and more responsive for those who showed at least 3-point worsening. A study by Holm et al. (2006) looked at 101 patients with AD (66 adults and 35 children) along with 30 healthy controls who completed the DLQI at 2 time points 6 months apart, and determined that the instrument is sensitive to differing severities of AD.47 Last, the Patel et al. study reported no floor or ceiling effects, defined as at least 15% of patients scoring in the lowest or highest values, for the overall DLQI scores, although they could be noted for individual items.45
A study by Basra et al. (2015) used an anchor-based approach to assess the responsiveness of the DLQI and estimate a MID.48 A total of 192 patients with different skin conditions ranging from acute to chronic were included (50.5% with psoriasis, 21.9% with acne, 12.5% with eczema, and 15.1% other). Details regarding the disease severity of patients were not reported. Using the Global Rating of Change Questionnaire as an anchor and considering a change in score of 2 or 3 points to be a “small,” an MID of 3.3 was estimated for the DLQI.48 There are a few limitations with the determination of the MID for the DLQI. The anchor-based approach used a subjective, global assessment of change that is subject to recall bias and not specific to AD.48 The authors acknowledged this and noted that the use of a global assessment was reasonable because the population comprised patients with a mix of diagnosed skin conditions; however, the lack of specificity to AD is a limitation that should be taken into consideration when applied to an AD-specific population.
Hospital Anxiety and Depression Scale
The Hospital Anxiety and Depression Scale (HADS) is a widely used patient-reported questionnaire designed to identify anxiety disorders and depression in patients at nonpsychiatric medical institutions. Repeated administration also provides information about changes in a patient’s emotional state.49-51 The HADS questionnaire contains 14 items that assess symptoms experienced in the previous week, including 7 items related to anxiety and 7 related to depression. Patients provided responses to each item based on a 4-point Likert scale. Each item is scored from 0 (the best) to 3 (the worst); a patient can therefore score between 0 and 21 for each subscale (anxiety and depression). A high score was indicative of a poor state. Scores of 11 or more on either subscale were considered to be a “definite case” of psychological morbidity, while scores of 8 to 10 represented a “probable case” and 0 to 7 “not a case.”51 One study52 indicated that the HADS has good construct validity when applied to adult AD patients, with no overall floor or ceiling effects. The author concluded that the HADS may be useful for the assessment of AD patients in clinical trials and practice, but added that additional research is needed to confirm the construct validity and to assess content validity and feasibility in research and clinical practice.52 No additional validity and MID information regarding application of the HADS to patients with AD was found in a literature search.
Patient-Oriented Eczema Measure
The POEM is a 7-item questionnaire used in clinical trials to assess disease symptoms in children and adults. Based on frequency of occurrence during the past week, the 7 items (dryness, itching, flaking, cracking, sleep loss, bleeding, and weeping) are assessed using a 5-point scale. The possible scores for each question are: “0” for no days, “1” for 1 to 2 days, “2” for 3 to 4 days, “3” for 5 to 6 days, and “4” for every day. The maximum total score is 28; a high score is indicative of a poor quality of life (0 to 2 indicates clear or almost clear; 3 to 7 mild eczema; 8 to 16 moderate eczema; 17 to 24 severe eczema; and 25 to 28 very severe eczema).53
The POEM has been tested and found to be adequate for validity, reliability, and responsiveness.30,54,55 Two 2020 studies by Silverberg et al. (N = 60256 and N = 29,157) assessed patient-reported outcomes in adults with AD. For convergent validity, Pearson (r) and Spearman (rS) correlation coefficients generally indicated a strong correlation between the POEM and the DLQI (r = 0.62,56 rS = 0.5957), NRS-itch (r = 0.58,56 rS = 0.45 and 0.5057 for worst itch and average itch, respectively), and the EASI (rS = 0.5257), but moderate correlation with the HADS anxiety and depression subscores (r = 0.33 and 0.31,56 respectively). The POEM also demonstrated adequate criterion validity through significant and stepwise increases with different levels of self-reported global AD severity (e.g., mild, moderate, and severe). Discriminant validity was generally adequate and the POEM showed varying ability to distinguish between mild and moderate AD (area under the curve [AUC] = 0.6757 to 0.75,56 poor to fair ability), mild and severe AD (AUC = 0.7057 to 0.89,56 fair to good ability), and moderate and severe AD (AUC = 0.5257 to 0.73,56 unable to fairly ability). Test-retest reliability, calculated using the intraclass correlation coefficient, was acceptable (0.86) when assessed in 189 patients.57 Changes in scores, a measure of responsiveness, were assessed, with closer correlation evident between the POEM and the DLQI (r = 0.67) compared to the EASI (r = 0.47) and itch NRS (r = 0.48 for both worst and average itch).57 Floor effects were noted for the overall POEM score and all individual items of the instrument for the study with 602 patients,56 while no floor effects were noted for the study with 291 patients.57 No ceiling effects were observed in either study.
Using data from 3 RCTs for AD treatment, Schram et al. (2012) reported an overall mean MID for the POEM to be 3.4 points (SD = 4.8), with an IGA score improvement of 1 points used as an anchor.31 A different study by Silverberg et al. (2020) estimated an MID of 5.0 points based on an anchor of at least a 1-point improvement for patient-reported global severity.57 The MID was smaller (3.7) for patients who reported baseline clear-to-mild AD or greater (6.1) for those who reported moderate to severe AD.
Short Form (36) Health Survey 36 (Acute Recall)
The SF-36 is a 36-item general health status assessment. Patients were asked to answer each question by selecting 1 of 3 to 6 categorical response options. The instrument instructions do not state a specific recall period; however, a recall period is defined for most items. The acute recall version, which asks patients about the last week, was used in this trial.58 This version of the survey (SF-36 Acute Recall) yields scores for 8 health domains (physical functioning, role physical, bodily pain, general health, vitality, social functioning, role emotional, and mental health) and 2 psychometrically derived summary scores (a physical component summary and a mental component summary). Although no literature was found that assessed the SF-36 Acute Recall for validity or reliability in patients with AD, the instrument has been validated in other disease areas such as psoriasis.59 A study by Holm et al. (2006) looked at 101 patients with AD (66 adults and 35 children) along with 30 healthy controls who completed the SF-36 at 2 time points with a 6-month interval.47 The physical component summary and mental component summaries showed little change between the assessments and between patient and control groups. Furthermore, using Wilcoxon rank scores, the investigators noted only small differences between patients with moderate and mild AD and those with mild AD and healthy controls. No MID was identified in patients with AD.
EQ-5D 5-Levels Questionnaire
The EQ-5D is a generic quality-of-life instrument that has been applied to a wide range of health conditions and treatments, including AD.60,61 The first of 2 parts of the EQ-5D is a descriptive system that classifies respondents (aged ≥ 12 years) into 1 of 243 distinct health states. The descriptive system consists of the following 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Each dimension has 3 possible levels (1, 2, or 3) representing “no problems,” “some problems,” and “extreme problems,” respectively. Respondents are asked to choose 1 level that reflects their own health state for each of the 5 dimensions. A scoring function can be used to assign a value (EQ-5D index score) to self-reported health states from a set of population-based preference weights.60,61 The second part is a 20 cm Visual Analogue Scale (EQ VAS) that has end points labelled 0 and 100, with respective anchors of “worst imaginable health state” and “best imaginable health state,” respectively. Respondents are asked to rate their own health by drawing a line from an anchor box to the point on the EQ VAS that best represents their own health on that day. The EQ-5D index and EQ VAS scores can be summarized and analyzed as continuous data.62,63 The EQ-5D-5L has been validated in terms of feasibility, ceiling effects, discriminatory power, and convergent validity in a diverse patient population from 6 countries with chronic conditions63; however, no literature was found that assessed the EQ-5D-5L for validity, reliability, or responsiveness in patients with AD.
A Canadian-specific estimate of a MID for the EQ-5D-5L was generated by simulating the effects of single-level transitions in each dimension.64 The results yielded MIDs with a summarized mean of 0.056 (SD = 0.011), and a summarized median of 0.056 (interquartile range = 0.049 to 0.063). No MID was identified in patients with AD.
Work Productivity and Activity Impairment–General Health
The impact of AD on the patient’s ability to perform both paid work and unpaid work and engage in regular activities was assessed by the Work Productivity and Activity Impairment–General Health (WPAI-GH) instrument.65 It consists of 6 items and measures absenteeism, presenteeism, and impairments in unpaid activity because of health problems during the past 7 days. The following 4 main outcomes can be generated from the WPAI-GH: percent of work time missed due to health for those who were currently employed, percent impairment while working due to health for those who were currently employed and actually worked in the past 7 days, percent of overall work impairment due to health for those who were currently employed, and percent activity impairment due to health for all respondents. No reports were found in the literature that assessed the instrument for validity, reliability, or responsiveness in patients with AD. No MID was identified in patients with AD.
Statistical Analysis
ECZTRA 1 and ECZTRA 2 Trials
Sample size: The investigators assumed a screening failure rate of 25%. Approximately 1,040 patients were expected to be screened and approximately 780 patients were planned to be randomized 3:1 to initial treatment (585 patients to tralokinumab and 195 patients to placebo). The sample sizes was chosen to ensure that sufficient safety information was collected and that a sufficient number of responders were re-randomized to maintenance treatment.
Database lock points: ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Multiplicity adjustment: To control the overall type I error rate, the primary analyses for the primary and secondary end points for the initial and maintenance treatment followed the testing. For the global (non-US) submission testing procedure, an IGA of 0 or 1 at week 16 between tralokinumab and placebo was evaluated at a 5% significance level. If the test was significant, an EASI-75 score at week 16 between tralokinumab and placebo was evaluated at a 5% significance level. If both these tests were significant, the significance level (alpha) was split between the analyses of the 3 secondary end points at week 16 and the analyses of the 2 maintenance end points at week 52. These groups of tests were tested in parallel with an alpha of 1% for the end points at week 16 and with an alpha of 4% for the maintenance end points at week 52. Evaluations of the 3 secondary end points at week 16 between tralokinumab and placebo used the Holm method66 for 3 ordered P values at a 1% significance level to adjust for multiplicity. The hypotheses for the maintenance treatment end points were tested sequentially in the specified order at the 4% significance level. The next hypothesis was only to be tested if the former was significant. If all P values for the 3 secondary end points at week 16 were significant, then the hypotheses for the maintenance treatment end points could be evaluated at a 5% significance level. Conversely, if all P values for the maintenance end points were significant, then the hypotheses for the 3 secondary end points at week 16 could be evaluated at a 5% significance level. The US submission testing procedure was executed as above, except that a reduction in the worst daily pruritus NRS weekly average of at least 4 at week 16 was tested after the sequential testing of an IGA of 0 or 1 and an EASI-75 score if these tests showed statistical significance.
Rescue medication: Rescue medication was summarized separately for the initial treatment period and the maintenance treatment period, and organized in a tabular format by type (topical and systemic) and group (corticosteroids, immunosuppressants, and other). For patients continuing with maintenance or open-label treatment at week 16, the summary tabulation of rescue medication during the initial treatment period included rescue medication taken between the first dose of initial treatment and the first dose of either maintenance or open-label treatment. For patients who did not continue with maintenance or open-label treatment, the summary tabulation of rescue medication during the initial treatment period included medications taken after the first dose and before the week 16 visit. The summary tabulation of rescue medication during the maintenance treatment period included rescue medication taken (but not necessarily initiated) after the first maintenance dose and excluded rescue medication initiated during the safety follow-up period.
Analysis: Categorical data were summarized using the number and percentage of patients in each category and treatment group. Continuous data were summarized using the mean, median, SD, and minimum and maximum values. Unless otherwise stated, all significance tests were 2-sided using the 5% significance level. All P values were nominal. All CIs were presented with a 95% degree of confidence.
For all analyses of the 2 primary outcomes at week 16, the difference in response rates between treatment groups were analyzed using the Cochran-Mantel-Haenszel test (single imputation analyses) or a combined inference from multiple Mantel-Haenszel risk differences and associated standard errors using the Rubin rule (multiple imputation analyses). The stratification factors included region (North America, Japan, and Europe) and baseline disease severity (IGA = 3 or 4). Treatment difference in response rates of IGA of 0 or 1 and an EASI-75 score after 16 weeks achieved without rescue medication were assessed, regardless of treatment discontinuation, and the primary estimand assessed the expected difference in response rates (defined as response obtained without initiation of any rescue medication) after 16 weeks, resulting from initiation of a treatment regimen with tralokinumab compared to a treatment regimen with placebo. For the primary analysis of primary end points at week 16, patients who had received rescue medication before the week 16 visit were considered nonresponders. Patients with missing data at week 16 and in whom rescue medication had not been used before week 16 were imputed as nonresponders. Three sensitivity analyses were conducted for the primary outcomes at week 16: a primary analysis in which all patients who permanently discontinued the investigational medicinal product (IMP) before week 16 were considered nonresponders, even if no rescue medication had been used; a primary analysis in which missing data at week 16 was imputed using last observation carried forward rather than using nonresponder imputation for patients who did not receive rescue medication and did not withdraw due to an AE or lack of efficacy; and a tipping-point analysis using multiple imputation in which patients who had received rescue medication before the week 16 visit were considered nonresponders. Missing week-16 responses were imputed from a Bernoulli distribution with a varying parameter for patients in the placebo group who did not use rescue medication. Patients in the tralokinumab group with missing week-16 data were imputed as nonresponders. Different percentages of placebo patients were considered responders for the different values of P. The tipping point is the value of P that changed the conclusion from significant to nonsignificant. Two main subgroup analyses were conducted at week 16 based on the primary outcomes: IGA of 0 or 1 by baseline IGA and IGA of 0 or 1 by region; and an EASI-75 score by baseline IGA and EASI-75 by region. For the maintenance treatment period (week 52) the 2 dichotomous maintenance end points were: an IGA of 0 or 1 at week 52 among patients with IGA of 0 or 1 at week 16 achieved without rescue medication after initial randomization to tralokinumab, and an EASI-75 score at week 52 among patients with an EASI-75 score at week 16 achieved without rescue medication after initial randomization to tralokinumab. All patients who had received rescue medication including TCS before the week-52 visit and/or had been transferred to open-label treatment with tralokinumab were considered nonresponders. Patients with missing data at week 52 were imputed as nonresponders. For each end point (IGA of 0 or 1 and an EASI-75 score at week 52) the difference in response rates between treatment groups was analyzed using the Cochran Mantel-Haenszel test stratified by region.
For the 3 secondary outcomes (reduction of worst daily pruritus NRS [weekly average] of 4 or greater from baseline to week 16, change in SCORAD from baseline to week 16, and change in DLQI score from baseline to week 16) all analyses were based on the full analysis set (FAS) at week 16. Data collected after permanent discontinuation of tralokinumab or placebo or after initiation of rescue medication were not included in the analysis. For patients who did not have any post-baseline data collected before initiation of rescue medication, the week-2 change was imputed as 0. In terms of the sensitivity analysis for the secondary outcomes at week 16, data collected after permanent discontinuation of tralokinumab or placebo or after initiation of rescue medication were not included. Multiple imputation of missing values was applied, based on regression models fitted on observed data from the placebo group.
No interim analyses were planned or conducted.
ECZTRA 3
Sample size: Assuming a screening failure rate of 25%, approximately 492 patients were planned to be screened and approximately 369 patients were planned to be randomized 2:1 to the initial treatment; 246 patients to tralokinumab q.2.w. plus TCS and 123 patients to placebo plus TCS. The sample size was chosen to ensure a sufficient combined power to demonstrate a statistically significant predefined difference between tralokinumab every 2 weeks plus TCS and placebo plus TCS for the primary end points.
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Multiplicity adjustment: The overall type I error rate for the primary analysis of the primary estimands for the primary and confirmatory secondary end points was protected by a combination of hierarchical testing and a Holm-Bonferroni multiplicity adjustment. The hypothesis relating to a specific end point could not be rejected unless all hypotheses relating to end points earlier in the hierarchy were also rejected. For the global (non-US) submission testing procedure an IGA of 0 or 1 at week 16 between tralokinumab every 2 weeks plus TCS and placebo plus TCS was evaluated at a 5% significance level. If the test was significant, an EASI-75 score at week 16 between tralokinumab every 2 weeks plus TCS and placebo plus TCS was evaluated at a 5% significance level. If both these tests were significant, the 5% significance level (alpha) was propagated to the 3 confirmatory secondary end points of pruritus, SCORAD, and DLQI at week 16. The evaluations of the 3 confirmatory secondary end points between tralokinumab every 2 weeks plus TCS and placebo plus TCS used the Holm method66 for 3 ordered P values at a 5% significance level to adjust for multiplicity. The US testing procedure was executed in a manner similar to the non-US submission except that pruritus at week 16 was tested after the sequential testing of an IGA of 0 or 1 and an EASI-75 score if these tests showed statistical significance. If the test of pruritus at week 16 was also significant at a 5% significance level, the 2 remaining confirmatory secondary end points of SCORAD and DLQI at week 16 were evaluated using the Holm method for 2 ordered P values at a 5% significance level to adjust for multiplicity.
Rescue medication: Rescue medication was summarized separately for the initial and continuation treatment periods. In addition, a summary tabulation of rescue medication by type (topical and systemic) and overall group (corticosteroids, immunosuppressants, and other) was made. For patients who continued with continuation treatment from week 16, the summary tabulation of rescue medication during the initial treatment period included rescue medication taken between the first dose of initial treatment and the first dose of continuation treatment. For patients who did not continue with continuation treatment, the summary tabulation of rescue medication during the initial treatment period included medication taken after the first dose and before the week-16 visit. The summary tabulation of rescue medication during the continuation treatment period included rescue medication taken (but not necessarily initiated) after the first continuation dose of tralokinumab and placebo and excluded rescue medication initiated during the safety follow-up period.
Analysis: Categorical data were summarized using the number and percentage of patients in each category and treatment group. Continuous data were summarized using the mean, median, SD, and minimum and maximum values. Unless otherwise stated, all significance tests were 2-sided using the 5% significance level. All P values were nominal. All CIs were presented with a 95% degree of confidence.
For all analyses of the 2 primary outcomes at week 16, the difference in response rates between treatment groups were analyzed using the Cochran-Mantel-Haenszel test (single imputation analyses) or a combined inference from multiple Mantel-Haenszel risk differences and associated standard errors using the Rubin rule (multiple imputation analyses). The stratification factors included: region (North America and Europe) and baseline disease severity (IGA = 3 or 4). Treatment difference in response rates of an IGA of 0 or 1 and an EASI-75 score after 16 weeks achieved without rescue medication were assessed, regardless of treatment discontinuation, and the primary estimand assessed the expected difference in response rates (defined as a response obtained without initiation of any rescue medication) after 16 weeks resulting from initiation of a treatment regimen with tralokinumab every 2 weeks plus TCS compared to a treatment regimen with placebo plus TCS. For the primary analysis of primary end points at week 16, patients who had received rescue medication before the week-16 visit were considered nonresponders. Patients with missing data at week 16 and in whom rescue medication had not been used before week 16 were imputed as nonresponders. The sensitivity and subgroup analyses at week 16 for the primary outcomes are identical to those conducted in the ECZTRA 1 and ECZTRA 2 trials, with the sole difference being that tralokinumab every 2 weeks + TCS was compared to placebo plus TCS group. The primary and sensitivity analyses on the secondary outcomes at week 16 were also identical to those of the ECZTRA 1 and ECZTRA 2 trials.
Descriptive statistics were used for end points in the continuation treatment period. For the continuation treatment period (week 32) the 2 dichotomous end points were an IGA of 0 or 1 at week 32 among patients with an IGA of 0 or 1 at week 16 achieved without rescue medication after initial randomization to tralokinumab and an EASI-75 score at week 32 among patients with an EASI-75 score at week 16 achieved without rescue medication after initial randomization to tralokinumab.
No interim analyses were planned or conducted.
ECZTRA 7
Sample size: With a significance level of 5%, a sample size of 250 patients would provide 99% power to detect a treatment difference for the primary end point, assuming an EASI-75 response rate at week 16 of 40% versus 15% for tralokinumab plus TCS and placebo plus TCS, respectively. Assuming a response rate of 30% versus 15% in a reduction of worst daily pruritus NRS (weekly average) score of at least 4 from baseline to week 16 for tralokinumab plus TCS and placebo plus TCS, respectively, the trial would provide at least 80% power to reject the hypotheses related to the primary end point and the secondary end point evaluating pruritus at week 16.
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Multiplicity adjustment: The primary and secondary end points were evaluated hierarchically in the order shown in Figure 5. The hypothesis relating to a specific end point could not be rejected unless all hypotheses relating to end points earlier in the hierarchy were also rejected at the 5% significance level. Hypothesis testing was based on the primary analysis of the primary estimand for each associated end point.
Figure 5: Testing Hierarchy for Primary and Secondary End Points in the ECZTRA 7 Trial

α = statistical significance level; DLQI = Dermatology Life Quality Index; EASI75 = 75% or greater reduction in Eczema Area and Severity Index from baseline; IGA = Investigator’s Global Assessment; NRS = numeric rating scale; SCORAD = Scoring Atopic Dermatitis.
Source: Clinical Study Report for ECZTRA 7.4
Rescue medication: Rescue medication was summarized for the entire treatment period as well as for the period up until the week 16 visit. Rescue treatment was also summarized for the periods before and after the start of the COVID-19 pandemic, within the treatment period. Additionally, rescue treatment was summarized for the entire treatment period by the variables of prior cyclosporine A use (yes or no), country (Germany: yes or no), and baseline disease severity (IGA = 3 or 4). A summary table of rescue treatment by type (topical and systemic) and by overall group (corticosteroids, immunosuppressants, and other) was made for the entire treatment period as well as for the period up until the week-16 visit.
Analysis: For the primary analysis for the primary end point (EASI-75 score at week 16) the difference in response rates between treatment groups was analyzed using the Cochran-Mantel-Haenszel test (single imputation analyses) or multiple Mantel-Haenszel risk differences and associated standard errors for a combined inference using the Rubin rule (multiple imputation analyses). The 2 stratification factors were prior cyclosporine A use (yes or no) and baseline disease severity (IGA = 3 or 4). Patients who received rescue treatment before the week-16 visit or permanently discontinued IMP, without prior patient onset of COVID-19, were considered nonresponders at all visits after the relevant event occurred. Any missing or collected data from patients who had COVID-19 as their first prior intercurrent event were handled differently and instead imputed assuming they were missing at random (MAR) following the start of patient onset of COVID-19. Data missing before any intercurrent event were handled as nonresponses, unless data were missing due to COVID-19, in which case they were imputed using the MAR assumption. The procedure for imputing values according to the rules described was implemented in 2 steps, with all missing or ignored data (irrespectively of reason) initially imputed using the MAR assumption based on available data from all patients. The nonresponder imputation of data not affected by COVID-19 was handled subsequently. For the sensitivity analysis, a tipping-point analysis tested the MAR assumption among patients in the tralokinumab plus TCS group who had missing data imputed at the week-16 visit due to patient onset of COVID-19 or data missing due to the COVID-19 pandemic. A quantity, delta, was added to all imputed week-16 values for patients in the tralokinumab plus TCS group who had patient onset of COVID-19 before week 16 or had data missing due to COVID-19 at week 16. Delta was defined as from the minimum to the maximum of the analyzed score (e.g., EASI). Each of the 100 modified imputed datasets was analyzed in the same manner as the primary analysis for the primary estimand. The tipping point was then considered the value of delta that changed the conclusion from significant to nonsignificant.
Analysis of the binary secondary end points was conducted as described for the primary end point. For the binary end points, subgroup analyses were applied for the primary analyses of the primary and tertiary estimands for the groups: sex, prior cyclosporine A use (yes or no), baseline disease severity (IGA = 3 or 4), country (Germany: yes or no), and baseline age (≤ 65 years or > 65 years). Interactions between subgroups and treatment effects were tested using a conditional logistic regression model. For the SCORAD and DLQI at the week-16 and week-26 continuous end points, the same subgroup analyses were performed for the primary and secondary continuous estimands. For the continuous secondary end points, data collected after permanent discontinuation of tralokinumab or placebo, after initiation of rescue treatment, or after patient onset of COVID-19 were not included in the analysis. Repeated measurements modelled the post-baseline responses up to week 16 and week 26 as change from baseline in SCORAD/DLQI = treatment × visit + baseline SCORAD/DLQI × visit + prior cyclosporine A use + country (Germany: yes or no) + baseline IGA. This model assumed that data were MAR within each treatment group.
No interim analyses were planned or conducted.
Analysis Populations
ECZTRA 1 and ECZTRA 2
In the FAS, all patients randomized to initial treatment who were exposed to tralokinumab or placebo were included and analyzed for efficacy up to week 16.
For the maintenance analysis set, all patients who received tralokinumab in the initial treatment period were re-randomized to maintenance treatment. Patients who were not exposed to maintenance treatment were excluded from the maintenance analysis set.
The safety analysis set comprised all patients randomized to initial treatment who were exposed to tralokinumab or placebo. The safety analysis set was therefore identical to the FAS.
The maintenance safety analysis set comprised all patients who were assigned to the maintenance treatment period and received at least 1 dose of maintenance treatment.
ECZTRA 3
In the FAS, patients randomized to initial treatment who were exposed to tralokinumab plus TCS or placebo plus TCS were analyzed for efficacy up to week 16.
The safety analysis set comprised all patients randomized to initial treatment who were exposed to treatment and for whom post-baseline safety data were available. Because all patients received the first dose of treatment at week 0 and were subsequently monitored for immediate drug reactions, all exposed patients were included in the safety analysis set. The safety analysis set was therefore identical to the FAS.
The continuation treatment analysis set comprised patients in the FAS who did not withdraw from the trial before or at the week-16 visit and who were exposed to at least 1 dose of treatment in the continuation treatment period.
The continuation treatment safety analysis set was defined in the same way as the continuation treatment analysis set. The safety follow-up analysis set comprised patients for whom the date of last contact was after the date of exposure end. Exposure end was defined as the week-32 visit for patients completing the treatment period or otherwise the date of permanent discontinuation of treatment for patients not completing the treatment period. For the analysis of efficacy, patients were included “as randomized.” For the analysis of safety, if patients were mistakenly given a treatment other than the treatment to which they were randomized, they were analyzed “as-treated.” They were therefore included in the group according to the treatment actually received. For the continuation treatment period, data were presented as planned treatment.
ECZTRA 7
All patients randomized to treatment and exposed to the tralokinumab plus TCS or placebo plus TCS were included in FAS and analyzed for efficacy.
Two safety analysis sets were defined: a safety analysis set for all patients exposed to tralokinumab plus TCS or placebo plus TCS within the treatment period, and a safety follow-up analysis set for all patients who had a date of last contact after the date of exposure end.
Decisions regarding inclusion and exclusion of patients from the trial analysis sets were documented in the analysis set definition document before breaking the randomization code.
Results
Patient Disposition
ECZTRA 1
Overall, 189 of 991 participants (19.1%) were screening failures. Among the screening failures, ||||||||| did not meet the inclusion criteria and ||||||||| met the exclusion criteria, 40 patients withdrew consent, 5 patients were lost to follow-up, and 16 patients reported other reasons for discontinuation before randomization. This left 802 patients to be randomized in the initial treatment period. Of the 802 randomized patients, 51 of 603 patients (8.5%) in the tralokinumab every 2 weeks treatment group and 18 of 199 patients (9.0%) in the placebo treatment group permanently discontinued treatment before week 16. In total, 729 patients (90.9% of all randomized patients) completed week 16 on treatment (Table 15). A total of 185 patients were week-16 tralokinumab responders assigned to maintenance treatment (71 in the tralokinumab every 2 weeks group, 78 in the tralokinumab every 4 weeks group, and 36 in the placebo group), and ||||||||| were week-16 placebo responders. Of the week-16 tralokinumab responders, 62.0% (n = 44) on tralokinumab every 2 weeks, 67.9% (n = 53) on tralokinumab every 4 weeks, and 58.3% (n = 21) on placebo completed the maintenance period, and 51.7% (n = 15) of week-16 placebo responders completed the maintenance period. Table 16 provides the disposition of patients assigned to maintenance treatment.
ECZTRA 2
Overall, 234 of 1,028 participants (22.8%) were screening failures. Among the screening failures, ||||||||| did not meet the inclusion criteria and ||||||||| met the exclusion criteria, 39 patients withdrew consent, 8 patients were lost to follow-up, and 14 patients reported other reasons for discontinuation before randomization. This left 794 patients to be randomized in the initial treatment period. Of the 794 randomized patients, 33 of 593 patients (5.6%) in the tralokinumab every 2 weeks treatment group and 22 of 201 patients (10.9%) in the placebo treatment group permanently discontinued treatment before week 16. In total, 737 patients (92.8% of all randomized patients) completed week 16 on treatment (Table 15). A total of 227 patients were week-16 tralokinumab responders assigned to maintenance treatment (91 in the tralokinumab every 2 weeks group, 90 in the tralokinumab every 4 weeks group, and 46 in the placebo group), and ||||||||| were week-16 placebo responders. Of the week-16 tralokinumab responders, 57.1% (n = 52) on tralokinumab every 2 weeks, 55.6% (n = 50) on tralokinumab every 4 weeks, 32.6% (n = 15) on placebo completed the maintenance period, and 51.6% (n = 16) of week-16 placebo responders completed the maintenance period. Table 16 provides the disposition of patients assigned to maintenance treatment.
ECZTRA 3
Overall, 127 of 507 participants (25.0%) were screening failures. Among the screening failures, 39 patients did not meet the inclusion criteria and 51 met the exclusion criteria, 25 patients withdrew consent, 6 patients were lost to follow-up, and 7 patients reported other reasons for discontinuation before randomization. This left 380 patients to be randomized in the initial treatment period. Of the 380 randomized patients, 235 of 253 patients (92.9%) in the tralokinumab every 2 weeks plus TCS group and 120 of 127 patients (94.5%) in the placebo plus TCS group completed the initial treatment period on treatment. Table 15 provides details. A total of 353 patients were assigned to continuation treatment, including 138 week-16 tralokinumab responders (evenly split between the tralokinumab every 2 weeks plus TCS and tralokinumab every 4 weeks plus TCS), 95 week-16 tralokinumab nonresponders, ||||||| week-16 placebo nonresponders, ||||||| week-16 placebo responders. In total, 98.6% (n = 68) and 94.2% (n = 65) assigned to the tralokinumab every 2 weeks plus TCS group and the tralokinumab every 4 weeks plus TCS group, respectively, completed the continuation period (week 32).
Table 17 provides the disposition of patients assigned to continuation treatment.
ECZTRA 7
Overall, 41 of 318 participants (12.9%) were screening failures. Among the screening failures, 29 patients did not meet eligibility criteria (inclusion and/or exclusion criteria), and 12 patients withdrew consent before randomization, leaving 277 patients to be randomized in the treatment period (week 0 to week 26). Of the 277 randomized patients, 125 of 140 patients (89.3%) in the tralokinumab plus TCS group and 120 of 137 patients (87.6%) in the placebo plus TCS group completed week 26 on treatment (Table 15).
Table 15: Patient Disposition for All Trials at Week 16
Disposition | ECZTRA 1 | ECZTRA 2 | ECZTRA 3 | ECZTRA 7 | ||||
|---|---|---|---|---|---|---|---|---|
Tralokinumab q.2.w. | Placebo | Tralokinumab q.2.w. | Placebo | Tralokinumab q.2.w. + TCS | Placebo + TCS | Tralokinumab + TCS | Placebo + TCS | |
Screened, n | 991 | 1,028 | 507 | 318 | ||||
Randomized, n (%) | 603 | 199 | 593 | 201 | 253 | 127 | 140 | 137 |
Discontinued from study, n (%) | 51 (9) | 18 (9) | 33 (6) | 22 (11) | 17 (7) | 6 (5) | |||||||||| | |||||||||| |
Reason for discontinuation, n (%) | ||||||||
|||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
|||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
|||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
|||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Full analysis set, n (%) | 601 (100) | 197 (99) | 591 (100) | 201 (100) | 252 (100) | 126 (99) | 138 (99) | 137 (100) |
Safety analysis set, n (%) | 602 (100) | 196 (99) | 592 (100) | 200 (100) | 252 (100) | 126 (99) | 138 (99) | 137 (100) |
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Any rescue medication, n (%) | ||||||||
Topical | 232 (39) | 103 (52) | 139 (23) | 85 (42) | 6 (2) | 10 (8) | 6 (4) | 16 (12) |
Systemic | 26 (4) | 17 (9) | 15 (3) | 33 (17) | 3 (1) | 6 (5) | 3 (2) | 8 (6) |
q.2.w. = every 2 weeks; TCS = topical corticosteroids.
Note: Patients who received rescue treatment during the initial treatment period were considered nonresponders, but they continued investigational medicinal product treatment if the rescue treatment consisted of topical medications only.
Source: Clinical Study Reports for ECZTRA 1,7 ECZTRA 2,5 ECZTRA 3,6 and ECZTRA 7.4
Table 16: Patient Disposition for the Maintenance Treatment phase in the ECZTRA 1 and ECZTRA 2 Studies
Disposition | ECZTRA 1 | ECZTRA 2 | ||||||
|---|---|---|---|---|---|---|---|---|
Week-16 tralokinumab responders | Week-16 placebo responders | Week-16 tralokinumab responders | Week-16 placebo responders | |||||
Tralokinumab q.2.w. | Tralokinumab q.4.w. | Placebo | Tralokinumab q.2.w | Tralokinumab q.2.w. | Placebo | |||
Assigned maintenance treatment, n | 71 | 78 | 36 | ||||||||| | 91 | 90 | 46 | ||||||||| |
Not dosed, n (%) | 3 (4.2) | 2 (2.6) | 1 (2.8) | ||||||||| | 0 (0.0) | 1 (1.1) | 0 (0.0) | ||||||||| |
Permanently discontinued IMP, n (%) | 4 (5.6) | 6 (7.7) | 4 (11.1) | ||||||||| | 9 (9.9) | 13 (14.4) | 5 (10.9) | ||||||||| |
Reason for discontinuation, n (%) | ||||||||
Adverse events | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Lost to follow-up | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Withdrawal by patient | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Lack of efficacy | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Other | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Transferred to open-label, n (%) | 23 (32.4) | 19 (24.4) | 10 (27.8) | ||||||||| | 29 (31.9) | 27 (30.0) | 26 (56.5) | ||||||||| |
With no use of rescue medication after re-randomization | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
With use of rescue medication after re-randomization | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Maintenance analysis set, n (%) | 68 (95.8) | 76 (97.4) | 35 (97.2) | ||||||||| | 91 (100.0) | 89 (98.9) | 46 (100.0) | ||||||||| |
Maintenance safety analysis set, n (%) | 68 (95.8) | 76 (97.4) | 35 (97.2) | ||||||||| | 91 (100.0) | 89 (98.9) | 46 (100.0) | ||||||||| |
Completed maintenance period, n (%) | 44 (62.0) | 53 (67.9) | 21 (58.3) | ||||||||| | 52 (57.1) | 50 (55.6) | 15 (32.6) | ||||||||| |
NR = not reported; q.2.w = every 2 weeks; q.4.w = every 4 weeks.
Table 17: Patient Disposition for the Continuation Treatment phase in the ECZTRA 3 Trial
Disposition | Week-16 tralokinumab responders | Week-16 tralokinumab nonresponders | Week-16 placebo nonresponders | Week-16 placebo responders | |
|---|---|---|---|---|---|
Tralokinumab q.2.w. + TCS | Tralokinumab q.2.w + TCS | Tralokinumab q.2.w + TCS | Tralokinumab q.2.w. + TCS | Placebo + TCS | |
Assigned continuation treatment, n | 69 | 69 | 95 | ||||||||| | ||||||||| |
Permanently discontinued IMP, n (%) | 1 (1.4) | 3 (4.3) | 7 (7.4) | ||||||||| | ||||||||| |
Reason for discontinuation, n (%) | |||||
Adverse events | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Withdrawal by patient | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Lack of efficacy | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Other | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Continuation treatment analysis set, n (%) | 69 (100.0) | 69 (100.0) | 95 (100.0) | ||||||||| | ||||||||| |
Continuation treatment safety analysis set, n (%) | 69 (100.0) | 69 (100.0) | 95 (100.0) | ||||||||| | ||||||||| |
Completed continuation period (week 32), n (%) | 68 (98.6) | 65 (94.2) | 87 (91.6) | ||||||||| | ||||||||| |
NR = not reported; q.2.w. = every 2 weeks; q.4.w = every 4 weeks; TCS = topical corticosteroids.
Source: Clinical Study Report for ECZTRA 3.6
Exposure to Study Treatments
ECZTRA 1
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
ECZTRA 2
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
ECZTRA 3
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
ECZTRA 7
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported in the following section.
Disease Severity
Investigator’s Global Assessment
One of the primary outcomes for the ECZTRA 1 and ECZTRA 2 trials was achieving an IGA score of 0 or 1 at week 16. In the ECZTRA 1 trial, 15.8% of patients in the intervention group achieved an IGA score of 0 or 1 at week 16 compared to 7.1% in the placebo group. The difference between tralokinumab and placebo (8.6%; 95% CI, 4.1 to 13.1; P = 0.002) was statistically significant, with the sensitivity analysis yielding the exact same result (Table 18). At week 52, of patients in the tralokinumab every 2 weeks group, 51.3% achieved an IGA score of 0 or 1 compared to 38.9% in the tralokinumab every 4 weeks group, and 47.4% in the placebo group (Table 19). The difference between tralokinumab every 2 weeks and placebo at week 52 (6.0%; 95% CI, −21.8 to 33.7; P = 0.68) was not statistically significant, nor was it statistically significant in the sensitivity analysis |||||||||||||||||||||||||||||||||||. The difference between tralokinumab every 4 weeks and placebo at week 52 (−9.5%; 95% CI, −37.1 to 18.0 P = 0.50) was also not statistically significant.
In the ECZTRA 2 trial, 22.2% of patients in the intervention group achieved an IGA score of 0 or 1 at week 16 compared to 10.9% in the placebo group. The difference between tralokinumab and placebo (11.1%; 95% CI, 5.8 to 16.4; P < 0.001) was statistically significant, and the sensitivity analysis yielded the exact same result (Table 20). At week 52, of patients in the tralokinumab every 2 weeks group, 59.3% achieved an IGA score of 0 or 1 compared to 44.9% in the tralokinumab every 4 weeks group, and 25.0% in the placebo group (Table 21). The difference between the tralokinumab every 2 weeks and placebo groups at week 52 (34.1%; 95% CI, 13.4 to 54.9; P = 0.004) was statistically significant, as was the result of the sensitivity analysis ||||||||||||||||||||||||||||||||||||||||||. The difference between the tralokinumab every 4 weeks and placebo groups at week 52 (19.9%; 95% CI, −1.2 to 40.9; P = 0.084) was not statistically significant, nor was the result of the sensitivity analysis ||||||||||||||||||||||||||||||||||||||||||.
In the ECZTRA 3 trial, 38.9% of patients in the intervention group achieved an IGA score of 0 or 1 at week 16 compared to 26.2% in the placebo group. The difference between tralokinumab and placebo (12.4%; 95% CI, 2.9 to 21.9; P = 0.015) was statistically significant, and the sensitivity analysis yielded the same result (Table 22). At week 32, of patients in the tralokinumab every 2 weeks plus TCS group, 89.6% achieved an IGA score of 0 or 1 without rescue medication compared to 77.6% in the tralokinumab every 4 weeks plus TCS group (Table 23). The IGA score of 0 or 1 was first in the testing hierarchy for the ECZTRA 3 trial.
In the ECZTRA 7 trial, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Due to the insignificant difference between tralokinumab plus TCS and placebo plus TCS in the reduction of worst daily pruritus NRS (weekly average) outcome, which is first in the testing hierarchy, P values are not reported for the IGA score at weeks 16 and 26 for the ECZTRA 7 trial.
Table 18: Summary of Efficacy Outcomes for the ECZTRA 1 Trial: Full Analysis Set
Outcomes | Tralokinumab q.2.w. N = 601 | Placebo N = 197 |
|---|---|---|
Primary end pointsa | ||
IGA score of 0 or 1 at week 16, n/N (%)a | 95/601 (15.8) | 14/197 (7.1) |
IGA score week 16 difference vs. placebo, % (95% CI)a,b | 8.6 (4.1 to 13.1; P = 0.002) | |
IGA score of 0 or 1 at week 16 sensitivity analysis, n/N (%) | 95/601 (15.8) | 14/197 (7.1) |
IGA score of 0 or 1 at week 16 sensitivity analysis difference vs. placebo, % (95% CI)b | 8.6 (4.1 to 13.1; P = 0.002) | |
EASI-75 score at week 16, n/N (%)a | 150/601 (25.0) | 25/197 (12.7) |
EASI-75 week 16 difference vs. placebo, % (95% CI)a,b | 12.1 (6.5 to 17.7; P < 0.001) | |
EASI-75 score at week 16 sensitivity analysis, n/N (%) | 148/601 (24.6) | 24/197 (12.2) |
EASI-75 score at week 16 sensitivity analysis difference vs. placebo, % (95% CI) | 12.3 (6.7 to 17.8; P < 0.001) | |
Secondary end pointsa | ||
Worst daily pruritus NRS (weekly average) reduction ≥ 4 at week 16, n/N (%)b | 119/594 (20.0) | 20/194 (10.3) |
Worst daily pruritus NRS difference vs. placebo, % (95% CI)a,b | 9.7 (4.4 to 15.0; P = 0.002) | |
Worst daily pruritus NRS (weekly average) reduction ≥ 4 at week 16 sensitivity analysis, n/N (%) | 119/594 (20.0) | 20/194 (10.3) |
Worst daily pruritus NRS (weekly average) reduction ≥ 4 at week 16 sensitivity analysis difference vs. placebo, % (95% CI) | 9.7 (4.4 to 15.0; P = 0.002) | |
Baseline worst daily pruritus NRS, mean (SD) | 7.7 (1.4) | 7.7 (1.4) |
Change from baseline to week 16 in SCORAD, adjusted mean change (SE)a | −25.2 (0.94) | −14.7 (1.80) |
SCORAD mean difference vs. placebo, (95% CI)a | −10.4 (−14.4 to −6.5; P < 0.001) | |
Change from baseline to week 16 SCORAD sensitivity analysis, adjusted mean change (SE) | −24.9 (1.23) | −17.2 (1.98) |
SCORAD at week 16 sensitivity analysis mean difference vs. placebo, (95% CI) | −7.7 (−11.4 to −3.9; P < 0.001) | |
Baseline SCORAD, mean (SD) | 70.3 (13.0) | 71.7 (12.5) |
Change from baseline to week 16 in DLQI, adjusted mean change (SE)a | −7.1 (0.31) | −5.0 (0.59) |
DLQI mean difference vs. placebo, (95% CI)a | −2.1 (−3.4 to −0.8; P = 0.002) | |
Change from baseline to week 16 DLQI sensitivity analysis, adjusted mean change (SE) | −7.5 (0.41) | −5.7 (0.63) |
DLQI at week 16 sensitivity analysis mean difference vs. placebo, (95% CI) | −1.8 (−3.0 to −0.6; P = 0.005) | |
Baseline DLQI, mean (SD) | 16.8 (7.1) | 17.0 (6.6) |
EASI-50 score at week 16, n (%) | 250 (41.6) | 42 (21.3) |
EASI-50 difference vs. placebo, % (95% CI) | 20.1 (13.3 to 26.8; P < 0.001) | |
EASI-90 score at week 16, n (%) | 87 (14.5) | 8 (4.1) |
EASI-90 difference vs. placebo, % (95% CI) | 10.3 (6.4 to 14.1; P < 0.001) | |
Worst daily pruritus NRS (weekly average) reduction ≥ 3 at week 16, n (%) | 177 (29.6) | 28 (14.4) |
Worst daily pruritus NRS reduction ≥ 3 difference vs. placebo, % (95% CI) | 15.2 (9.2 to 21.3; P < 0.001) | |
SCORAD-75 at week 16, n (%) | 53 (8.8) | 6 (3.0) |
SCORAD-75 difference vs. placebo, % (95% CI) | 5.7 (2.5 to 8.9; P = 0.007) | |
SCORAD-50 at week 16, n (%) | 156 (26.0) | 23 (11.7) |
SCORAD-50 difference vs. placebo, % (95% CI) | 14.1 (8.6 to 19.6; P < 0.001) | |
DLQI reduction ≥ 4 at week 16, n/N (%) | 258/578 (44.6) | 60/190 (31.6) |
DLQI reduction ≥ 4 at week 16, difference vs. placebo, % (95% CI) | 13.0 (5.4 to 20.5; P = 0.001) | |
Exploratory end points | ||
Eczema-related sleep NRS (weekly average) at week 16, adjusted mean change (95% CI) | −2.6 (−2.9 to −2.4) | −1.9 (−2.4 to −1.5) |
Eczema-related sleep NRS (weekly average) at week 16, mean difference vs. placebo, (95% CI) | −0.7 (−1.2 to −0.2; P = 0.007) | |
HADS anxiety score at week 16, adjusted mean change (95% CI) | |||||||||||||||||||| | |||||||||||||||||||| |
HADS anxiety score at week 16 difference vs. placebo, adjusted mean change (95% CI) | |||||||||||||||||||| | |
HADS depression score at week 16, adjusted mean change (95% CI) | |||||||||||||||||||| | |||||||||||||||||||| |
HADS depression score at week 16 difference vs. placebo, adjusted mean change (95% CI) | |||||||||||||||||||| | |
HADS anxiety and HADS depression scores < 8 at week 16, n/N (%) | |||||||||||||||||||| | |||||||||||||||||||| |
HADS anxiety and HADS depression scores < 8 at week 16 difference vs. placebo, % (95% CI) | |||||||||||||||||||| | |
POEM at week 16, adjusted mean change (95% CI) | |||||||||||||||||||| | |||||||||||||||||||| |
POEM at week 16 difference vs. placebo, adjusted mean change (95% CI) | |||||||||||||||||||| | |
POEM reduction ≥ 4 at week 16, n/N (%) | |||||||||||||||||||| | |||||||||||||||||||| |
POEM reduction ≥ 4 at week 16 difference vs. placebo, % (95% CI) | |||||||||||||||||||| | |
SF-36 physical component at week 16 | |||||||||||||||||||| | |||||||||||||||||||| |
SF-36 physical component at week 16 mean difference vs. placebo, (95% CI) | |||||||||||||||||||| | |
SF-36 mental component at week 16 | |||||||||||||||||||| | |||||||||||||||||||| |
SF-36 mental component at week 16, mean difference vs. placebo, (95% CI) | |||||||||||||||||||| | |
EQ-5D-5L at week 16, adjusted mean change (95% CI) | |||||||||||||||||||| | |||||||||||||||||||| |
EQ-5D-5L at week 16, mean difference vs. placebo (95% CI) | |||||||||||||||||||| | |
EQ VAS score week 16 adjusted mean change (95% CI) | |||||||||||||||||||| | |||||||||||||||||||| |
EQ VAS score week 16, mean difference vs. placebo (95% CI) | |||||||||||||||||||| | |
WPAI-GH work time missed at week 16 adjusted mean change (95% CI) | |||||||||||||||||||| | |||||||||||||||||||| |
WPAI-GH work time missed at week 16, mean difference vs. placebo (95% CI) | |||||||||||||||||||| | |
WPAI-GH impairment while working at week 16 adjusted mean change (95% CI) | |||||||||||||||||||| | |||||||||||||||||||| |
WPAI-GH impairment while working at week 16, mean difference vs. placebo (95% CI) | |||||||||||||||||||| | |
WPAI-GH overall work impairment at week 16 adjusted mean change (95% CI) | |||||||||||||||||||| | |||||||||||||||||||| |
WPAI-GH overall work impairment at week 16, mean difference vs. placebo (95% CI) | |||||||||||||||||||| | |
WPAI-GH activity impairment at week 16 adjusted mean change (95% CI) | |||||||||||||||||||| | |||||||||||||||||||| |
WPAI-GH activity impairment at week 16, mean difference vs. placebo (95% CI) | |||||||||||||||||||| | |
CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI-50 = 50% or greater reduction in Eczema Area and Severity Index from baseline EASI-75 = 75% or greater reduction in Eczema Area and Severity Index from baseline; EQ-5D-5L = EQ-5D 5-Levels questionnaire; EQ VAS = EQ-5D Visual Analogue Scale; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; SCORAD = Scoring Atopic Dermatitis; SCORAD-50 = 50% decrease in Scoring Atopic Dermatitis; SCORAD-75 = 75% decrease in Scoring Atopic Dermatitis; SD = standard deviation; SE = standard error; SF-36 = Short Form (36) Health Survey; vs. = versus; WPAI-GH = Work Productivity and Activity Impairment–General Health.
aThe primary and secondary end points were tested sequentially under the hierarchical testing strategy in the following order: IGA score of 0 or 1 at week 16 (primary end point), EASI-75 at week 16 (primary end point), pruritus NRS scores at week 16 (secondary end point), SCORAD at week 16 (secondary end point), and DLQI at week 16 (secondary end point). These secondary end points were tested in parallel with an alpha of 1% for the end points at week 16 and with an alpha of 4% for the maintenance end points at week 52. The maintenance end points were tested sequentially in the following order: IGA 0 or 1 at week 52 between q.2.w. and placebo, EASI-75 at week 52 between q.2.w. and placebo, IGA 0 or 1 at week 52 between q.4.w. and placebo, and EASI-75 at week 52 between q.4.w. and placebo.
bMantel-Haenszel risk difference, stratified by region and baseline IGA.
Source: Clinical Study Report for ECZTRA 1.7
Table 19: Outcomes — ECZTRA 1 Trial Maintenance End Points
Outcomes | Tralokinumab q.2.w. | Tralokinumab q.4.w. | Placebo |
|---|---|---|---|
IGA score of 0 or 1 at week 52, n/N (%)a | 20/39 (51.3) | 14/36 (38.9) | 9/19 (47.4) |
IGA score week 52 difference vs. placebo, % (95% CI)a,b | 6.0 (−21.8 to 33.7; P = 0.68) | −9.5 (−37.1 to 18.0; P = 0.50) | Reference |
IGA score of 0 or 1 at week 52 sensitivity analysis, n/N (%)b | |||||||||||||||||||| | |||||||||||||||||||| | |||||||||||||||||||| |
IGA score week 52 sensitivity analysis difference vs. placebo, % (95% CI)a,b | |||||||||||||||||||| | |||||||||||||||||||| | |||||||||||||||||||| |
EASI-75 score at week 52, n/N (%)a | 28/47 (59.6) | 28/57 (49.1) | 10/30 (33.3) |
EASI-75 week 52 difference vs. placebo, % (95% CI)a,b | 21.2 (−0.2 to 42.6; P = 0.056) | 11.7 (−8.7 to 32.0; P = 0.27) | Reference |
EASI-75 score at week 52 sensitivity analysis, n/N (%)a | |||||||||||||||||||| | |||||||||||||||||||| | |||||||||||||||||||| |
EASI-75 week 52 sensitivity analysis difference vs. placebo, % (95% CI)a,b | |||||||||||||||||||| | |||||||||||||||||||| | |||||||||||||||||||| |
IGA score of 0 or 1 at week 52: patients in maintenance analysis set with EASI-75 and IGA ≥ 2 at week 16, n/N (%) | |||||||||||||||||||| | |||||||||||||||||||| | |||||||||||||||||||| |
IGA score of 0 or 1 at week 52: patients in maintenance analysis set with EASI-75 and IGA ≥ 2 at week 16, difference vs. placebo, % (95% CI)b | |||||||||||||||||||| | |||||||||||||||||||| | |||||||||||||||||||| |
IGA 0 or 1 or EASI-75 at week 52: patients in maintenance analysis set with IGA 0 or 1 or EASI-75 at week 16 achieved without rescue medication, n/N (%) | |||||||||||||||||||| | |||||||||||||||||||| | |||||||||||||||||||| |
IGA 0 or 1 or EASI-75 at week 52: patients in maintenance analysis set with IGA 0 or 1 or EASI-75 at week 16 achieved without rescue medication, % (95% CI)b | |||||||||||||||||||| | |||||||||||||||||||| | |||||||||||||||||||| |
SCORAD at week 16, n/N (mean [SD]) | |||||||||||||||||||| | |||||||||||||||||||| | |||||||||||||||||||| |
SCORAD at week 52, n/N (mean [SD]) | |||||||||||||||||||| | |||||||||||||||||||| | |||||||||||||||||||| |
Worst daily pruritus NRS at week 16, n/N (mean [SD]) | |||||||||||||||||||| | |||||||||||||||||||| | |||||||||||||||||||| |
||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||| | |||||||||||||||||||| | |||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||| | |||||||||||||||||||| | |||||||||||||||||||| |
CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI-75 = 75% or greater reduction in Eczema Area and Severity Index from baseline; EQ-5D-5L = EQ-5D 5-Levels questionnaire; EQ VAS = EQ-5D Visual Analogue Scale; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; q.2.w. = every 2 weeks; q.4.w = every 4 weeks; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation; SF-36 = Short Form (36) Health Survey; vs. = versus; WPAI-GH = Work Productivity and Activity Impairment–General Health.
Note: Patients who achieved a clinical response at week 16 were eligible to continue maintenance treatment and were included in this dataset, and the outcomes reported were achieved without rescue medication.
aThe primary and secondary end points were tested sequentially under the hierarchical testing strategy in the following order: IGA score of 0 or 1 at week 16 (primary end point), EASI-75 at week 16 (primary end point), pruritus at week 16 (secondary end point), SCORAD at week 16 (secondary end point), and DLQI at week 16 (secondary end point). These secondary end points were tested in parallel with an alpha of 1% for the end points at week 16 and with an alpha of 4% for the maintenance end points at week 52. The maintenance end points were tested sequentially in the following order: IGA 0 or 1 at week 52 between q.2.w. and placebo, EASI-75 at week 52 between q.2.w. and placebo, IGA 0 or 1 at week 52 between q.4.w. and placebo, and EASI-75 at week 52 between q.4.w. and placebo.
bMantel-Haenszel risk difference compared to placebo, stratified by region.
Note: Redacted rows have been deleted.
Source: Clinical Study Report for ECZTRA 1.7
Table 20: Summary of Efficacy Outcomes for the ECZTRA 2 Trial: Full Analysis Set
Outcomes | Tralokinumab q.2.w. N = 591 | Placebo N = 201 |
|---|---|---|
Primary end pointsa | ||
Patients with IGA score of 0 or 1 at week 16, n/N (%)a | 131/591 (22.2) | 22/201 (10.9) |
IGA score difference vs. placebo, % (95% CI)a,b | 11.1 (5.8 to 16.4; P < 0.001) | |
IGA score of 0 or 1 at week 16 sensitivity analysis, n/N (%) | 131/591 (22.2) | 22/201 (10.9) |
IGA score of 0 or 1 at week 16 sensitivity analysis difference vs. placebo, % (95% CI)b | 11.1 (5.8 to 16.4; P < 0.001) | |
EASI-75 score at week 16, n/N (%)a | 196/591 (33.2) | 23/201 (11.4) |
EASI-75 difference vs. placebo, % (95% CI)a,b | 21.6 (15.8 to 27.3; P < 0.001) | |
EASI-75 score at week 16 sensitivity analysis, n/N (%) | 196/591 (33.2) | 23/201 (11.4) |
EASI-75 score at week 16 sensitivity analysis difference vs. placebo, % (95% CI) | 21.6 (15.8 to 27.3; P < 0.001) | |
Secondary end pointsa | ||
Reduction of worst daily pruritus NRS (weekly average) ≥ 4 at week 16, % (95% CI)a | 25.0 (21.7 to 28.7) | 9.5 (6.2 to 14.4) |
Reduction of worst daily pruritus NRS (weekly average) ≥ 4 at week 16 difference vs. placebo, % (95% CI)a | 15.6 (10.3 to 20.9; P < 0.001) | |
Worst daily pruritus NRS (weekly average) reduction ≥ 4 at week 16 sensitivity analysis, n/N (%) | 144/575 (25.0) | 19/200 (9.5) |
Worst daily pruritus NRS (weekly average) reduction ≥ 4 at week 16 sensitivity analysis difference vs. placebo, % (95% CI) | 15.6 (10.3 to 20.9; P < 0.001) | |
Change from baseline to week 16 in SCORAD, adjusted mean change (SE)a | −28.1 (0.92) | −14.0 (1.79) |
SCORAD, mean difference vs. placebo, (95% CI)a | −14.0 (−18.0 to −10.1; P < 0.001) | |
Baseline SCORAD, mean (SD) | 70.0 (13.4) | 70.5 (12.2) |
Change from baseline to week 16 SCORAD sensitivity analysis, adjusted mean change (SE) | −26.9 (1.06) | −13.8 (2.00) |
SCORAD at week 16 sensitivity analysis, mean difference vs. placebo, (95% CI) | −13.0 (−17.1 to −9.0; P < 0.001) | |
Change from baseline to week 16 in DLQI, adjusted mean change (SE)a | −8.8 (0.30) | −4.9 (0.60) |
DLQI, mean difference vs. placebo, (95% CI)a | −3.9 (−5.2 to −2.6; P < 0.001) | |
Change from baseline to week 16 DLQI sensitivity analysis, adjusted mean change (SE) | −8.6 (0.36) | −5.2 (0.68) |
DLQI at week 16 sensitivity analysis, mean difference vs. placebo, (95% CI) | −3.4 (−4.8 to −2.0; P < 0.001) | |
Baseline DLQI, mean (SD) | 17.7 (7.1) | 17.8 (7.3) |
EASI-50 at week 16, % (95% CI) | 49.9 (45.9 to 53.9) | 20.4 (15.4 to 26.5) |
EASI-50 at week 16 difference vs. placebo, % (95% CI) | 29.3 (22.5 to 36.1; P < 0.001) | |
EASI-90 at week 16, % (95% CI) | 18.3 (15.4 to 21.6) | 5.5 (3.1 to 9.5) |
EASI-90 at week 16 difference vs. placebo, % (95% CI) | 12.7 (8.3 to 17.0; P < 0.001) | |
Reduction of worst daily pruritus NRS (weekly average) ≥ 3 at week 16, % (95% CI) | 34.1 (30.4 to 38.1) | 14.0 (9.9 to 19.5) |
Reduction of worst daily pruritus NRS (weekly average) ≥ 3 at week 16 difference vs. placebo, % (95% CI) | 20.1 (13.9 to 26.2; P < 0.001) | |
SCORAD-75 at week 16, % (95% CI) | 11.5 (9.2 to 14.3) | 3.5 (1.7 to 7.0) |
SCORAD-75 at week 16, difference vs. placebo, % (95% CI) | 8.0 (4.4 to 11.6; P < 0.001) | |
SCORAD-50 at week 16, % (95% CI) | 33.5 (29.8 to 37.4) | 14.4 (10.2 to 20.0) |
SCORAD-50 at week 16, difference vs. placebo, % (95% CI) | 18.9 (12.8 to 25.1; P < 0.001) | |
DLQI reduction ≥ 4 at week 16, n/N (%) | 325/577 (56.3) | 54/198 (27.3) |
DLQI reduction ≥ 4 at week 16 difference vs. placebo, % (95% CI) | 28.9 (21.4 to 36.3; P < 0.001) | |
Exploratory end points | ||
Eczema-related sleep NRS (weekly average) at week 16, adjusted mean change (95% CI) | −2.9 (−3.1 to −2.7) | −1.5 (−1.9 to −1.1) |
Eczema-related sleep NRS (weekly average) at week 16, mean difference vs. placebo, (95% CI) | −1.4 (−1.9 to −0.9; P < 0.001) | |
||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |
CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI-50 = 50% or greater reduction in Eczema Area and Severity Index from baseline; EASI-75 = 75% or greater reduction in Eczema Area and Severity Index from baseline; EASI-90 = 90% or greater reduction in Eczema Area and Severity Index from baseline; EQ-5D-5L = EQ-5D 5-Levels questionnaire; EQ VAS = EQ-5D Visual Analogue Scale; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; SCORAD = Scoring Atopic Dermatitis; SCORAD-50 = 50% decrease in Scoring Atopic Dermatitis; SCORAD-75 = 75% decrease in Scoring Atopic Dermatitis; SD = standard deviation; SE = standard error; SF-36 = Short Form Health Survey; vs. = versus; WPAI-GH = Work Productivity and Activity Impairment–General Health.
aThe primary and secondary end points were tested sequentially under the hierarchical testing strategy in the following order: IGA score of 0 or 1 at week 16 (primary end point), EASI-75 at week 16 (primary end point), pruritus NRS scores at week 16 (secondary end point), SCORAD at week 16 (secondary end point), and DLQI at week 16 (secondary end point). These secondary end points were tested in parallel with an alpha of 1% for the end points at week 16 and with an alpha of 4% for the maintenance end points at week 52. The maintenance end points were tested sequentially in the following order: IGA 0 or 1 at week 52 between q.2.w. and placebo, EASI-75 at week 52 between q.2.w. and placebo, IGA 0 or 1 at week 52 between q.4.w. and placebo, and EASI-75 at week 52 between q.4.w. and placebo.
bMantel-Haenszel risk difference, stratified by region and baseline IGA.
Note: Redacted rows have been deleted.
Source: Clinical Study Report for ECZTRA 2.5
Table 21: Outcomes — ECZTRA 2 Trial Maintenance End Points
Outcomes | Tralokinumab q.2.w. | Tralokinumab q.4.w. | Placebo |
|---|---|---|---|
IGA score of 0 or 1 at week 52, n/N (%)a | 32/54 (59.3) | 22/49 (44.9) | 7/28 (25.0) |
IGA score week 52 difference vs. placebo, % (95% CI)a,b | 34.1 (13.4 to 54.9; P = 0.004) | 19.9 (−1.2 to 40.9; P = 0.084) | Reference |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
EASI-75 score at week 52, n/N (%)a | 43/77 (55.8) | 38/74 (51.4) | 9/42 (21.4) |
EASI-75 week 52 difference vs. placebo, % (95% CI)a,b | 33.7 (17.3 to 50.0; P < 0.001) | 30.0 (13.7 to 46.4; P = 0.001) | Reference |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI-75 = 75% or greater reduction in Eczema Area and Severity Index from baseline; EQ-5D-5L = EQ-5D 5-Levels questionnaire; EQ VAS = EQ-5D Visual Analogue Scale; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation; SF-36 = Short Form (36) Health Survey; vs. = versus; WPAI-GH = Work Productivity and Activity Impairment–General Health.
Note: Patients who achieved a clinical response at week 16 were eligible to continue maintenance treatment and were included in this dataset, and the outcomes reported were achieved without rescue medication.
aThe primary and secondary end points were tested sequentially under the hierarchical testing strategy in the following order: IGA score of 0 or 1 at week 16 (primary end point), EASI-75 at week 16 (primary end point), pruritus NRS scores at week 16 (secondary end point), SCORAD at week 16 (secondary end point), and DLQI at week 16 (secondary end point). These secondary end points were tested in parallel with an alpha of 1% for the end points at week 16 and an alpha of 4% for the maintenance end points at week 52. The maintenance end points were tested sequentially in the following order: IGA 0 or 1 at week 52 between q.2.w. and placebo, EASI-75 at week 52 between q.2.w. and placebo, IGA 0 or 1 at week 52 between q.4.w. and placebo, and EASI-75 at week 52 between q.4.w. and placebo.
bMantel-Haenszel risk difference compared to placebo, stratified by region.
Note: Redacted rows have been deleted.
Source: Clinical Study Report for ECZTRA 2.5
Table 22: Summary of Efficacy Outcomes for the ECZTRA 3 Trial: Full Analysis Set
Outcomes | Tralokinumab q.2.w. + TCS N = 252 | Placebo + TCS N = 126 |
|---|---|---|
Primary end pointsa | ||
Patients with IGA score of 0 or 1 at week 16, n/N (%)a | 98/252 (38.9) | 33/126 (26.2) |
IGA score at week 16 difference vs. placebo, % (95% CI)a,b | 12.4 (2.9 to 21.9; P = 0.015) | |
IGA score of 0 or 1 at week 16 sensitivity analysis, n/N (%) | 98/252 (38.9) | 33/126 (26.2) |
IGA score of 0 or 1 at week 16 sensitivity analysis difference vs. placebo, % (95% CI)b | 12.4 (2.9 to 21.9; P = 0.015) | |
EASI-75 score at week 16, n/N (%)a | 141/252 (56.0) | 45/126 (35.7) |
EASI-75 difference vs. placebo, % (95% CI)a,b | 20.2 (9.8 to 30.6; P < 0.001) | |
EASI-75 score at week 16 sensitivity analysis, n/N (%) | 140/252 (55.6) | 44/126 (34.9) |
EASI-75 score at week 16 sensitivity analysis difference vs. placebo, % (95% CI) | 20.6 (10.2 to 30.9; P < 0.001) | |
Secondary end pointsb | ||
Reduction of worst daily pruritus NRS (weekly average) ≥ 4 at week 16, % (95% CI)a | 45.4 (39.3 to 51.6) | 34.1 (26.4 to 42.8) |
Reduction of worst daily pruritus NRS (weekly average) ≥ 4 at week 16 difference vs. placebo, % (95% CI)a | 11.3 (0.9 to 21.6; P = 0.037) | |
Worst daily pruritus NRS (weekly average) reduction ≥ 4 at week 16 sensitivity analysis, n/N (%) | 45.4 (39.3 to 51.6) | 34.1 (26.4 to 42.8) |
Worst daily pruritus NRS (weekly average) reduction ≥ 4 at week 16 sensitivity analysis difference vs. placebo, % (95% CI) | 11.3 (0.9 to 21.6; P = 0.037) | |
Change from baseline to week 16 in SCORAD, adjusted mean change (SE)a | −37.7 (1.25) | −26.7 (1.83) |
SCORAD mean difference vs. placebo, (95% CI)a | −10.8 (−15.2 to −6.5; P < 0.001) | |
Change from baseline to week 16 SCORAD sensitivity analysis, adjusted mean change (SE) | −37.5 (1.27) | −26.8 (1.80) |
SCORAD at week 16 sensitivity analysis mean difference vs. placebo, (95% CI) | −10.9 (-−5.2 to −6.6; P < 0.001) | |
Baseline SCORAD, mean (SD) | 67.0 (13.3) | 68.9 (13.2) |
Change from baseline to week 16 in DLQI, adjusted mean change (SE)b | −11.7 (0.39) | −8.8 (0.56) |
DLQI mean difference vs. placebo, (95% CI)a | −2.9 (−4.3 to −1.6; P < 0.001) | |
Change from baseline to week 16 DLQI sensitivity analysis, adjusted mean change (SE) | −11.6 (0.40) | −8.8 (0.57) |
DLQI at week 16 sensitivity analysis, mean difference vs. placebo, (95% CI) | −2.8 (−4.2 to −1.5; P < 0.001) | |
Baseline DLQI, mean (SD) | 17.6 (7.1) | 17.2 (7.2) |
EASI-50 at week 16, % (95% CI) | 79.4 (73.9 to 83.9) | 57.9 (49.2 to 66.2) |
EASI-50 at week 16 difference vs. placebo, % (95% CI) | 21.3 (11.3 to 31.3; P < 0.001) | |
EASI-90 at week 16, % (95% CI) | 32.9 (27.4 to 39.0) | 21.4 (15.2 to 29.4) |
EASI-90 at week 16 difference vs. placebo, % (95% CI) | 11.4 (2.1 to 20.7; P = 0.022) | |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||| | |||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||| | |
DLQI reduction ≥ 4 at week 16, n/N (%) | 207/278 (83.5) | 81/123 (65.9) |
DLQI reduction ≥ 4 at week 16 difference vs. placebo, % (95% CI) | 17.6 (8.0 to 27.1; P < 0.001) | |
Exploratory end points | ||
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||| | |||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||| | |
CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI-50 = 50% or greater reduction in Eczema Area and Severity Index from baseline; EASI-90 = 90% or greater reduction in Eczema Area and Severity Index from baseline; EQ-5D-5L = EQ-5D 5-Levels questionnaire; EQ VAS = Visual Analogue Scale; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; q.2.w. = every 2 weeks; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation; SE = standard error; TCS = topical corticosteroids.
aThe primary and secondary end points were tested sequentially under the hierarchical testing strategy in the following order: IGA score of 0 or 1 at week 16 (primary end point), EASI-75 at week 16 (primary end point), pruritus NRS scores at week 16 (secondary end point), SCORAD at week 16 (secondary end point), and DLQI at week 16 (secondary end point).
bMantel-Haenszel risk difference, stratified by region and baseline IGA.
Note: Redacted rows have been deleted.
Source: Clinical Study Report for ECZTRA 3.6
Table 23: Outcomes — ECZTRA 3 Trial Continuation Treatment Period End Points
Outcomes | Tralokinumab q.2.w. + TCS | Tralokinumab q.4.w. + TCS |
|---|---|---|
IGA score of 0 or 1 at week 32 achieved without rescue medication, n/N (%)a | 43/48 (89.6) | 38/49 (77.6) |
EASI-75 score at week 32 achieved without rescue medication, n/N (%)a | 62/67 (92.5) | 59/65 (90.8) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||| | ||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||| | ||||||||||||||||||||| |
EASI-50 response at week 32, n/N (%) | 68/69 (98.6) | 63/69 (91.3) |
EASI-75 response at week 32, n/N (%) | 64/69 (92.8) | 60/69 (87.0) |
EASI-90 response at week 32, n/N (%) | 50/69 (72.5) | 44/69 (63.8) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||| | ||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||| | ||||||||||||||||||||| |
CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI-50 = 50% or greater reduction in Eczema Area and Severity Index from baseline; EASI-75 = 75% or greater reduction in Eczema Area and Severity Index from baseline; EASI-90 = 90% or greater reduction in Eczema Area and Severity Index from baseline; EQ-5D-5L = EQ-5D 5-Levels questionnaire; EQ VAS = EQ-5D Visual Analogue Scale; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation; TCS = topical corticosteroids.
Note: Patients treated with tralokinumab q.2.w. plus TCS in the initial treatment period who had a clinical response at week 16 were re-randomized in a 1:1 ratio to tralokinumab 300 mg q.2.w. or tralokinumab 300 mg q.4.w. stratified by region (Europe and North America) and IGA response at week 16 (IGA 0 or 1, or IGA > 1). Patients re-randomized to tralokinumab 300 mg every 4 weeks were treated with alternating dose administration of tralokinumab 300 mg and placebo every second week.
aMantel-Haenszel risk difference compared to placebo, stratified by region.
Note: Redacted rows have been deleted.
Source: Clinical Study Report for ECZTRA 3.6
Table 24: Summary of Efficacy Outcomes for the ECZTRA 7 Trial: Full Analysis Set
Efficacy outcomes | Tralokinumab q.2.w. + TCS N = 138 | Placebo + TCS N = 137 | |
|---|---|---|---|
Primary end pointsa | |||
EASI-75 score at week 16, n (%)b | 88.6 (64.2) | 69.2 (50.5) | |
EASI-75 difference vs. placebo, % (95% CI)a,b | 14.1 (2.5 to 25.7; P = 0.018) | ||
Secondary end points | |||
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||| | ||||||||||||||||||||| | |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||| | ||
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||| | ||||||||||||||||||||| | |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||| | ||
Percent change from baseline in EASI at week 16, adjusted mean change (SE) | −79.2 (2.6) | −65.8 (2.7) | |
Percent change from baseline in EASI at week 16, difference vs. placebo, (95% CI) | −13.3 (−20.7 to −6.0) | ||
EASI-75 score at week 26, n (%)a | 95.0 (68.8) | 75.7 (55.3) | |
EASI-75 at week 26 difference vs. placebo, % (95% CI)a,b | 14.1 (2.9 to 25.3) | ||
Reduction of worst daily pruritus NRS (weekly average) reduction ≥ 4 to week 16, n/N (%)a | 61/134 (45.5) | 48/135 (35.6) | |
Worst daily pruritus NRS week 16 difference vs. placebo, % (95% CI)a,b | 9.7 (−2.0 to 21.4; P = 0.106) | ||
Worst daily pruritus NRS (weekly average) reduction ≥ 4 to week 26, n/N (%)a | 63/134 (47.2) | 54/135 (39.7) | |
Worst daily pruritus NRS week 26 difference vs. placebo, % (95% CI)a,b | 7.3 (−4.6 to 19.2) | ||
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||| | ||||||||||||||||||||| | |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||| | ||
Change from baseline to week 16 in DLQI, adjusted mean change (SE)a | −11.2 (0.40) | −9.6 (0.40) | |
DLQI week 16, mean difference vs. placebo, (95% CI)b | −1.5 (−2.6 to −0.4) | ||
Change from baseline to week 26 in DLQI, adjusted mean change (SE)a | −11.5 (0.40) | −9.9 (0.40) | |
DLQI week 2, difference vs. placebo, (95% CI)b | −1.6 (−2.7 to −0.5) | ||
Exploratory end points | |||
EASI-50 at week 16, n (%) | 110.4 (80.0) | 95.3 (69.5) | |
EASI-50 at week 16, difference vs. placebo, % (95% CI) | 10.6 (0.3 to 20.8; P = 0.043) | ||
EASI-50 at week 26, n (%) | 111.2 (80.5) | 91.9 (67.1) | |
EASI-50 at week 26, difference vs. placebo, % (95% CI) | 13.7 (3.5 to 23.9; P = 0.008) | ||
EASI-90 at week 16, n (%) | 56.7 (41.1) | 40.2 (29.3) | |
EASI-90 at week 16, difference vs. placebo, % (95% CI) | 12.3 (1.1 to 23.6; P = 0.032) | ||
EASI-90 at week 26, n (%) | 67.1 (48.6) | 49.8 (36.4) | |
EASI-90 at week 26, difference vs. placebo, % (95% CI) | 12.9 (1.4 to 24.4; P = 0.027) | ||
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||| | ||||||||||||||||||||| | |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||| | ||
Change from baseline in eczema-related sleep NRS (weekly average) at week 16, adjusted mean change (SE) | −4.1 (0.2) | −3.4 (0.2) | |
Change from baseline in eczema-related sleep NRS (weekly average) at week 16, difference vs. placebo, (95% CI) | −0.8 (−1.3 to −0.2; P = 0.005) | ||
Change from baseline in eczema-related sleep NRS (weekly average) at week 26, adjusted mean change (SE) | −4.3 (0.2) | −3.7 (0.2) | |
Change from baseline in eczema-related sleep NRS (weekly average) at week 26, difference vs. placebo, (95% CI) | −0.6 (−1.1 to −0.0; P = 0.037) | ||
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||| | ||||||||||||||||||||| | |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||| | ||
CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI-50 = 50% or greater reduction in Eczema Area and Severity Index from baseline; EASI-75 = 75% or greater reduction in Eczema Area and Severity Index from baseline; EASI-90 = 90% or greater reduction in Eczema Area and Severity Index from baseline; EQ-5D-5L = EQ-5D 5-Levels questionnaire; EQ VAS = EQ-5D Visual Analogue Scale; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation; SE = standard error; SF-36 = Short Form (36) Health Survey; TCS = topical corticosteroid; vs. = versus.
aThe primary and secondary end points that were tested sequentially under the hierarchical testing strategy in the following order: EASI-75 at week 16 (primary end point), and then the following secondary end points were tested sequentially: reduction of worst daily pruritus NRS (weekly average) of at least 4 from baseline to week 16, change in SCORAD from baseline to week 16, change in DLQI score from baseline to week 16, IGA score of 0 or 1 at week 16, EASI-75 at week 26, reduction of worst daily pruritus NRS (weekly average) scores of at least 4 from baseline to week 26, change in SCORAD from baseline to week 26, change in DLQI score from baseline to week 26, IGA score of 0 or 1 at week 26.
bMantel-Haenszel risk difference, stratified by prior cyclosporine A use and baseline IGA.
Note: Redacted rows have been deleted.
Source: Clinical Study Report for ECZTRA 7.4
Sleep Disturbance
In the ECZTRA 1 trial, there was significantly greater improvement in how much eczema interfered with sleep, based on the eczema-related sleep NRS (weekly average) score, in the tralokinumab every 2 weeks group compared to the placebo group at week 16 (−0.7%; 95% CI, −1.2 to −0.2; P = 0.007) (Table 18). |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| In the ECZTRA 2 trial, there was also greater improvement in how much eczema interfered with sleep in the tralokinumab every 2 weeks group compared to the placebo group at week 16 (−1.4%; 95% CI, −1.9 to −0.9; P < 0.001) (Table 20). ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| In the ECZTRA 3 trial, there was greater improvement in how much the eczema interfered with the sleep in the tralokinumab every 2 weeks plus TCS group compared to the placebo plus TCS at week 16 group (−1.3%; 95% CI, −1.8 to −0.8; P < 0.001) (Table 22). At week 32, the mean eczema-related sleep NRS (weekly average) score was 1.3 (SD = 1.7) compared to 1.7 (SD = 1.9) for placebo. In the ECZTRA 7 trial, there was greater improvement in how much eczema interfered with sleep at week 16 (−0.8%; 95% CI, −1.3 to −0.2; P = 0.005) and week 26 (−0.6%; 95% CI, −1.1 to −0.0; P = 0.037) (Table 24). Eczema-related sleep NRS (weekly average) scores were not part of the testing hierarchy in any RCT.
Health-Related Quality of Life
Dermatology Life Quality Index
In the ECZTRA 1 trial, the adjusted mean change from baseline in the DLQI was statistically significantly larger in the tralokinumab group compared with the placebo group at week 16 (−2.1; 95% CI, −3.4 to −0.8; P = 0.002), and the sensitivity analysis also yielded a statistically significant result. (Table 18). The DLQI at week 16 fell within the testing hierarchy for ECZTRA 1. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The proportion of patients with a reduction in the DLQI of at least 4 points from baseline, among patients with a DLQI score of at least 4 at baseline, was higher in the tralokinumab group compared with the placebo group at week 16 (13.0%; 95% CI, 5.4 to 20.5; P = 0.001).
In the ECZTRA 2 trial, the adjusted mean change from baseline in the DLQI was statistically significantly larger in the tralokinumab group compared with the placebo group at week 16 (−3.9; 95% CI, −5.2 to −2.6; P < 0.001), and the sensitivity analysis also yielded a significant difference (−3.4; 95% CI, −4.8 to −2.0; P < 0.001) (Table 20). The DLQI at week 16 fell within the testing hierarchy for the ECZTRA 2 trial. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The proportion of patients with a reduction in the DLQI of at least 4 points from baseline, among patients with a DLQI of at least 4 at baseline, was higher in the tralokinumab group compared with the placebo group at week 16 (28.9%; 95% CI, 21.4 to 36.3; P < 0.001).
In the ECZTRA 3 trial, the adjusted mean change from baseline in the DLQI was statistically significantly larger in the tralokinumab every 2 weeks plus TCS group compared with the placebo plus TCS group at week 16 (−2.9; 95% CI, −4.3 to −1.6; P < 0.001), and the sensitivity analysis also yielded a significant result (Table 22). The proportion of patients with a reduction in the DLQI of at least 4 points from baseline, among patients with a DLQI of at least 4 at baseline, was statistically significantly higher in the tralokinumab every 2 weeks plus TCS group compared with the placebo plus TCS group at week 16 (17.6%; 95% CI, 8.0 to 27.1; P < 0.001). The DLQI outcome fell within the testing hierarchy for the ECZTRA 3 trial.
In the ECZTRA 7 trial, the adjusted mean change from baseline in the DLQI was larger in the tralokinumab every 2 weeks plus TCS group compared with the placebo plus TCS group at week 16 (−1.5; 95% CI, −2.6 to −0.4), ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| At week 26, the adjusted mean change from baseline in the DLQI was larger in the tralokinumab every 2 weeks plus TCS group compared with the placebo plus TCS group (−1.6; 95 CI, −2.7 to −0.5). The proportion of patients with a reduction in the DLQI of at least 4 points from baseline, among patients with a DLQI of at least 4 at baseline, was not higher in the tralokinumab every 2 weeks plus TCS group compared with the placebo plus TCS group at week 16 (4.9%; 95% CI, −4.8 to 14.6). |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Due to the insignificant difference between tralokinumab plus TCS and placebo plus TCS in the reduction of worst daily pruritus NRS (weekly average) scores outcome, which is first in the testing hierarchy, P values are not reported for the DLQI at weeks 16 and 26 for the ECZTRA 7 trial.
Short Form (36) Health Survey
In the ECZTRA 1 trial, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The SF-36 was not part of the testing hierarchy in the ECZTRA 1 trial.
In the ECZTRA 2 trial, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The SF-36 was not part of the testing hierarchy in the ECZTRA 2 trial.
In the ECZTRA 7 trial, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The SF-36 was not part of the testing hierarchy in the ECZTRA 7 trial.
EQ-5D 5-Levels Questionnaire
In the ECZTRA 1 trial, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||The EQ-5D-5L was not part of the testing hierarchy in the ECZTRA 1 trial.
In the ECZTRA 2 trial, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The EQ-5D-5L was not part of the testing hierarchy in the ECZTRA 2 trial.
In the ECZTRA 3 trial, 3 trial, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The EQ-5D-5L was not part of the testing hierarchy in the ECZTRA 3 trial.
In the ECZTRA 7 trial ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The EQ-5D-5L was not part of the testing hierarchy in the ECZTRA 7 trial.
Mood
Hospital Anxiety and Depression Scale
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The HADS anxiety and depression scores were not part of the testing hierarchy in the ECZTRA 1 trial.
In ECZTRA 2, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The HADS anxiety and depression scores were not part of the testing hierarchy in the ECZTRA 2 trial.
In the ECZTRA 3 trial, the mean improvement (reduction) in HADS anxiety scores from baseline to week 16 was larger in the tralokinumab group compared to placebo (−1.3; 95% CI, −2.1 to −0.6; P < 0.001) (Table 22). The mean improvement (reduction) in HADS depression scores from baseline to week 16 was also larger in the tralokinumab group compared to placebo (−0.8; 95% CI. −1.5 to −0.2; P = 0.015) (Table 22). |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The HADS anxiety and depression scores were not part of the testing hierarchy in the ECZTRA 3 trial.
In ECZTRA 7, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The HADS anxiety and depression scores were not part of the testing hierarchy in the ECZTRA 7 trial.
Productivity
Work Productivity and Activity Impairment–General Health
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The WPAI-GH scores were not part of the testing hierarchy for the ECZTRA 1 trial.
In the ECZTRA 2 trial, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The WPAI-GH scores were not part of the testing hierarchy for the ECZTRA 2 trial.
Subgroup Analysis
In the ECZTRA 1, ECZTRA 2, and ECZTRA 3 trials, a higher number of patients who had moderate disease severity at baseline achieved an IGA score of 0 or 1 and an EASI-75 score at week 16 compared to those who had severe disease severity at baseline in both treatment groups. Irrespective of disease severity at baseline, a higher proportion of patients achieved an IGA score of 0 or 1 and an EASI-75 score at week 16 in the tralokinumab group compared with the placebo group. In the ECZTRA 7 trial a higher proportion of patients achieved an EASI-75 score in the tralokinumab plus TCS group compared with the placebo plus TCS group. A notable difference between ECZTRA 7 and the other trials is that a higher proportion of patients with severe disease at baseline achieved an EASI-75 score at week 16 compared to patients with moderate disease at baseline. Baseline disease severity is the only subgroup of interest based on this report’s protocol; failure to respond, contraindications, or intolerance to 1 or more systemic therapies or the presence of other comorbid conditions such as asthma are not reported as subgroups in the clinical trial reports.
Table 25: Subgroup Analyses of the Primary End Points at Week 16 with Regard to Disease Severity
Subgroup | ECZTRA 1 | ECZTRA 2 | ECZTRA 3 | ECZTRA 7 | ||||
|---|---|---|---|---|---|---|---|---|
Tralokinumab q.2.w. N = 601 | Placebo N = 197 | Tralokinumab q.2.w. N = 591 | Placebo N = 201 | Tralokinumab q.2.w. plus TCS N = 252 | Placebo plus TCS N = 126 | Tralokinumab q.2.w. plus TCS N = 138 | Placebo plus TCS N = 137 | |
Primary end points | ||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||||
|||||||||||||||||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||||
||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
COVID-19 = coronavirus 2019; EASI = Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; IMP = investigational medicinal product; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; TCS = topical corticosteroids.
Source: Clinical Study Reports for ECZTRA 1,7 ECZTRA 2,5 ECZTRA 3,6 and ECZTRA 7.4
Harms
Only those harms identified in the review protocol are reported. In all trials, exposure start was defined as the date and time of first dose of tralokinumab or placebo, and exposure end was defined as the date of the week 52 (ECZTRA 1 and ECZTRA 2) or 32 (ECZTRA 3) visit. Otherwise, exposure end was defined as the date of permanent discontinuation of tralokinumab or placebo or, if missing, the date of last treatment administration. Table 26 and Table 27 provide detailed harms data.
Adverse Events
In the ECZTRA 1 trials, AEs were reported in 76.4% (n = 460) of tralokinumab and 77.0% (n = 151) of placebo patients, and in the ECZTRA 2 trial, AEs were reported in 61.5% (n = 364) of tralokinumab patients and 66.0% (n = 132) of placebo patients during the initial treatment period (Table 26). In the ECZTRA 3 trial, AEs were reported in 71.4% (n = 180) of tralokinumab every 2 weeks plus TCS patients and 66.7% (n = 84) of placebo plus TCS patients, and in the ECZTRA 7 trial, AEs were reported in 77.5% (n = 107) of tralokinumab every 2 weeks plus TCS patients and 78.8% (n = 108) of placebo plus TCS patients in the initial treatment period (Table 27).
During the maintenance treatment period, AEs were reported in 79.4% (n = 54) of the tralokinumab every 2 weeks group, 69.7% (n = 53) of the tralokinumab every 4 weeks group, and 71.4% (n = 25) of the placebo group in the ECZTRA 1 trial (Table 28), and 68.1% (n = 62) of the tralokinumab every 2 weeks group, 62.9% (n = 56) tralokinumab every 4 weeks group, and 69.6% (n = 32) of the placebo group in the ECZTRA 2 trial (Table 29). In the continuation treatment period of the ECZTRA 3 trial, AEs occurred in 69.6% (n = 48) of the tralokinumab every 2 weeks plus TCS group, 59.4% (n = 41) of the tralokinumab every 4 weeks plus TCS group for those in the week-16 tralokinumab responders group (Table 30).
Serious Adverse Events
In the ECZTRA 1 trial, SAEs were reported in 3.8% (n = 23) of tralokinumab patients and 4.1% (n = 8) of placebo patients, and in the ECZTRA 2 trial, SAEs were reported in 1.7% (n = 10) of tralokinumab patients and 2.5% (n = 5) of placebo patients at week 16 (Table 26). In the maintenance treatment period, SAEs were reported in 1.5% (n = 1) of the tralokinumab every 2 weeks group, 3.9% (n = 3) of the tralokinumab every 4 weeks group among the week-16 tralokinumab responders in the ECZTRA 1 trial (Table 28), and 3.4% (n = 3) of the tralokinumab every 4 weeks group in the ECZTRA 2 trial (Table 29).
In the ECZTRA 3 trials, SAEs were reported in 0.8% (n = 2) of tralokinumab every 2 weeks plus TCS patients and 3.2% (n = 4) of placebo plus TCS patients, and in the ECZTRA 7 trial, SAEs were reported in 0.7% (n = 1) of tralokinumab every 2 weeks plus TCS patients and 3.6% (n = 5) of placebo plus TCS patients (Table 27). Serious adverse events were reported in 4.3% (n = 3) of the tralokinumab every 2 weeks plus TCS group (week-16 tralokinumab responders) in the ECZTRA 3 trial (Table 30).
Withdrawal Due to Adverse Events
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| During the maintenance treatment phase, 2 patients withdrew from the ECZTRA 1 trial (Table 28), 3 withdrew from the ECZTRA 2 trial (Table 29), and 10 withdrew from the ECZTRA 3 trial due to AEs (Table 30).
Mortality
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| No deaths were reported in the ECZTRA trial ||||||||| 7.
Notable Harms
Harms of special interest included dermatitis atopic, which occurred in 25.9% (n = 156) of tralokinumab patients and 38.3% (n = 75) of placebo patients in the ECZTRA 1 trial, and 16.6% (n = 98) of tralokinumab patients and 33.5% (n = 67) of placebo patients in the ECZTRA 2 trial. Viral upper respiratory tract infection occurred in 23.1% (n = 139) of tralokinumab patients and 20.9% (n = 41) placebo patients in the ECZTRA 1 trial, and in 8.3% (n = 49) of the tralokinumab patients and 18.5% (n = 17) of placebo patients in the ECZTRA 2 trial (Table 26). In the ECZTRA 3 and ECZTRA 7 trials, the most common AE was viral upper respiratory tract infection, which occurred in 19.4% (n = 49) of tralokinumab plus TCS patients and 11.1% (n = 14) of placebo plus TCS patients (ECZTRA 3) and 26.8% (n = 37) of tralokinumab plus TCS patients and 25.5% (n = 35) of placebo plus TCS patients (ECZTRA 7). Among notable harms, pruritus occurred in 5.3% (n = 32) of tralokinumab patients and 5.1% (n = 10) of placebo patients in the ECZTRA 1 trial; upper respiratory infraction occurred in 10.0% (n = 59) of tralokinumab patients and 8.5% (n = 17) of placebo patients in the ECZTRA 2 trial; conjunctivitis occurred in 11.1% (n = 28) of tralokinumab plus TCS patients and 3.2% (n = 4) of placebo plus TCS patients in the ECZTRA 3 trial; and headache occurred in 15.2% (n = 21) of tralokinumab plus TCS patients and 9.5% (n = 13) of placebo patients in the ECZTRA 7 trial (Table 27). Table 28, Table 29, and Table 30 detail the notable harms in the maintenance treatment and continuation treatment periods.
Table 26: Summary of Harms for the ECZTRA 1 and ECZTRA 2 Trials in the Initial Treatment Period: Safety Analysis Set
Harms | ECZTRA 1 | ECZTRA 2 | ||
|---|---|---|---|---|
Tralokinumab q.2.w. N = 602 | Placebo N = 196 | Tralokinumab q.2.w. N = 592 | Placebo N = 200 | |
Patients with ≥ 1 adverse event | ||||
Patients with any treatment-emergent AE, n (%) | 460 (76.4) | 151 (77.0) | 364 (61.5) | 132 (66.0) |
Most common events, 8% in any group, n (%) | ||||
Dermatitis atopic | 156 (25.9) | 75 (38.3) | 98 (16.6) | 67 (33.5) |
Viral upper respiratory tract infection | 139 (23.1) | 41 (20.9) | 49 (8.3) | 17 (8.5) |
Serious adverse event | ||||
Patients with a serious adverse event, n (%) | 23 (3.8) | 8 (4.1) | 10 (1.7) | 5 (2.5) |
Patients who stopped treatment due to adverse events | ||||
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Deaths | ||||
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Harms of special interest | ||||
Skin and subcutaneous tissue disorders, n (%) | ||||
Pruritus | 32 (5.3) | 10 (5.1) | 12 (2.0) | 5 (2.5) |
Infections and infestations, n (%) | ||||
Conjunctivitis | 43 (7.1) | 4 (2.0) | 18 (3.0) | 3 (1.5) |
Upper respiratory tract infection | NR | NR | 59 (10.0) | 17 (8.5) |
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Skin infection | NR | NR | 12 (2.0) | 11 (5.5) |
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Eczema herpaticum | NR | NR | 2 (0.3) | 5 (2.5) |
General, n (%) | ||||
|||||||||||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Nervous system disorders, n (%) | ||||
Headache | 28 (4.7) | 10 (5.1) | 16 (2.7) | 6 (3.0) |
Gastrointestinal disorders, n (%) | ||||
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
|||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Psychiatric disorders, n (%) | ||||
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Eye disorders, n (%) | ||||
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Vascular disorders, n (%) | ||||
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Blood and lymphatic system disorders, n (%) | ||||
|||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Respiratory, thoracic, and mediastinal disorders, n (%) | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
AE = adverse event; NR = not reported; q.2.w. = every 2 weeks.
Source: Clinical Study Reports for ECZTRA 17 and ECZTRA 2.5
Table 27: Summary of Harms for the ECZTRA 3 and ECZTRA 7 Trials in the Initial Treatment Period: Safety Analysis Set
Harms | ECZTRA 3 | ECZTRA 7 | ||
|---|---|---|---|---|
Tralokinumab q.2.w. + TCS N = 252 | Placebo + TCS N = 126 | Tralokinumab q.2.w. + TCS N = 138 | Placebo + TCS N = 137 | |
Patients with ≥ 1 adverse event | ||||
Patients with any treatment-emergent AE, n (%) | 180 (71.4) | 84 (66.7) | 107 (77.5) | 108 (78.8) |
Most common events, 9% in any group, n (%) | ||||
Viral upper respiratory tract infection | 49 (19.4) | 14 (11.1) | 37 (26.8) | 35 (25.5) |
Conjunctivitis | 28 (11.1) | 4 (3.2) | ||||||||| | ||||||||| |
Headache | 22 (8.7) | 6 (4.8) | 21 (15.2) | 13 (9.5) |
Serious adverse event | ||||
Patients with a serious adverse event, n (%) | 2 (0.8) | 4 (3.2) | 1 (0.7) | 5 (3.6) |
Patients who stopped treatment due to adverse events | ||||
||||||||||||||||||||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Deaths | ||||
|||||||||||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Harms of special interest | ||||
Skin and subcutaneous tissue disorders, n (%) | ||||
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Dermatitis atopic | 6 (2.4) | 10 (7.9) | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Infections and infestations, n (%) | ||||
Upper respiratory tract infection | 19 (7.5) | 6 (4.8) | 10 (7.2) | 10 (7.3) |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
General, n (%) | ||||
Injection-site reactions | 17 (6.7) | 0 (0.0) | NR | NR |
||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Gastrointestinal disorders, n (%) | ||||
|||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
|||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | 8 (5.8) |
Eye disorders, n (%) | ||||
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
|||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Vascular disorders, n (%) | ||||
||||||||| | ||||||||| | ||||||||| | 3 (2.2) | 7 (5.1) |
Respiratory, thoracic, and mediastinal disorders, n (%) | ||||
||||||||| | ||||||||| | ||||||||| | 4 (2.9) | 8 (5.8) |
AE = adverse event; NR = not reported; q.2.w. = every 2 weeks; TCS = topical corticosteroids.
Source: Clinical Study Reports for ECZTRA 36 and ECZTRA 7.4
Table 28: Summary of Harms for the ECZTRA 1 Trial in the Maintenance Treatment Period: Safety Analysis Set
Harms | Week-16 tralokinumab responders | Week-16 placebo responders | ||
|---|---|---|---|---|
Tralokinumab q.2.w. N = 68 | Tralokinumab q.4.w. N = 76 | Placebo N = 35 | Placebo N = 29 | |
Patients with ≥ 1 adverse event | ||||
Patients with any treatment-emergent AE, n (%) | 54 (79.4) | 53 (69.7) | 25 (71.4) | ||||||||| |
Most common events, n (%) | ||||
|||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Viral upper respiratory tract infection | 14 (20.6) | 18 (23.7) | 4 (11.4) | ||||||||| |
Serious adverse event | ||||
Patients with a serious adverse event, n (%) | 1 (1.5) | 3 (3.9) | NR | ||||||||| |
Patients who stopped treatment due to adverse events | ||||
Any treatment-emergent AE leading to permanent discontinuation of study drug, n (%) | 1 (1.5) | 1 (1.3) | NR | ||||||||| |
Deaths (reported in Table 24) | ||||
Harms of special interest | ||||
Skin and subcutaneous tissue disorders, n (%) | ||||
Pruritus | 2 (2.9) | 4 (5.3) | 1 (2.9) | ||||||||| |
Infections and infestations, n (%) | ||||
Conjunctivitis | 3 (4.4) | 4 (5.3) | NR | ||||||||| |
Upper respiratory tract infection | 1 (1.5) | 2 (2.6) | 1 (2.9) | ||||||||| |
Influenza | 4 (5.9) | 3 (3.9) | 1 (2.9) | ||||||||| |
Bronchitis | 3 (4.4) | 7 (9.2) | 2 (5.7) | ||||||||| |
Nasopharyngitis | NR | 3 (3.9) | 2 (5.7) | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
General, n (%) | ||||
Injection-site reactions | 5 (7.4) | 7 (9.2) | 1 (2.9) | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Nervous system disorders, n (%) | ||||
Headache | 6 (8.8) | 2 (2.6) | 3 (8.6) | ||||||||| |
Gastrointestinal disorders, n (%) | ||||
|||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Eye disorders, n (%) | ||||
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Vascular disorders, n (%) | ||||
Hypertension | 1 (1.5) | 2 (2.6) | NR | || |
Blood and lymphatic system disorders, n (%) | ||||
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Respiratory, thoracic, and mediastinal disorders, n (%) | ||||
Asthma | 4 (5.9) | 1 (1.3) | NR | ||||||||| |
AE = adverse event; NR = not reported; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks.
Source: Clinical Study Report for ECZTRA 1.7
Table 29: Summary of Harms for the ECZTRA 2 Trial in the Maintenance Treatment Period: Safety Analysis Set
Harms | Week-16 tralokinumab responders | Week-16 placebo responders | ||
|---|---|---|---|---|
Tralokinumab q.2.w. N = 91 | Tralokinumab q.4.w. N = 89 | Placebo N = 46 | Placebo N = 31 | |
Patients with ≥ 1 adverse event | ||||
Patients with any treatment-emergent AE, n (%) | 62 (68.1) | 56 (62.9) | 32 (69.6) | ||||||||| |
Most common events, n (%) | ||||
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Upper respiratory tract infection | 14 (5.4) | 9 (10.1) | 3 (6.5) | ||||||||| |
Viral upper respiratory tract infection | 9 (9.9) | 6 (6.7) | 7 (15.2) | ||||||||| |
Serious adverse event | ||||
Patients with a serious adverse event, n (%) | NR | 3 (3.4) | NR | ||||||||| |
Patients who stopped treatment due to adverse events | ||||
Any treatment-emergent AE leading to permanent discontinuation of study drug, n (%) | 2 (2.2) | 1 (1.1) | NR | ||||||||| |
Deaths (reported in Table 24) | ||||
Harms of special interest | ||||
Skin and subcutaneous tissue disorders, n (%) | ||||
Pruritus | 2 (2.2) | 2 (2.2) | 2 (4.3) | ||||||||| |
Infections and infestations, n (%) | ||||
Conjunctivitis | 5 (5.5) | 1 (1.1) | 2 (4.3) | ||||||||| |
Influenza | 2 (2.2) | 1 (1.1) | 1 (2.2) | ||||||||| |
Bronchitis | 1 (1.1) | 3 (3.4) | NR | ||||||||| |
|||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
|||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
General, n (%) | ||||
Injection-site reaction | 4 (4.4) | 4 (4.5) | NR | ||||||||| |
|||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Nervous system disorders, n (%) | ||||
Headache | 2 (2.2) | 2 (2.2) | NR | ||||||||| |
Gastrointestinal disorders, n (%) | ||||
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Eye disorders, n (%) | ||||
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Vascular disorders, n (%) | ||||
Hypertension | 1 (1.1) | 1 (1.1) | 3 (6.5) | ||||||||| |
Blood and lymphatic system disorders, n (%) | ||||
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Respiratory, thoracic, and mediastinal disorders, n (%) | ||||
Asthma | 2 (2.2) | 3 (3.4) | 3 (6.5) | ||||||||| |
AE = adverse event; NR = not reported; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks.
Source: Clinical Study Report for ECZTRA 2.5
Table 30: Summary of Harms for the ECZTRA 3 Trial in the Continuation Treatment Period: Safety Analysis Set
Harms | Week-16 tralokinumab responders | Week-16 tralokinumab nonresponders | |
|---|---|---|---|
Tralokinumab q.2.w. plus TCS N = 69 | Tralokinumab q.4.w. plus TCS N = 69 | Tralokinumab q.2.w. plus TCS N = 95 | |
Patients with ≥ 1 adverse event | |||
Patients with any treatment-emergent AE, n (%) | 48 (69.6) | 41 (59.4) | 62 (65.3) |
Most common events, n (%) | |||
Dermatitis atopic | 1 (1.4) | 1 (1.4) | 8 (8.4) |
Upper respiratory tract infection | 7 (10.1) | 3 (4.3) | 6 (6.3) |
Viral upper respiratory tract infection | 12 (17.4) | 9 (13.0) | 20 (21.1) |
Serious adverse event | |||
Patients with a serious adverse event, n (%) | 3 (4.3) | NR | 2 (2.1) |
Patients who stopped treatment due to adverse events | |||
Any treatment-emergent AE leading to permanent discontinuation of study drug, n (%) | 0 (0.0) | 1 (1.4) | 0 (0.0) |
Deaths (reported in Table 25) | |||
Harms of special interest | |||
Skin and subcutaneous tissue disorders, n (%) | |||
||||||||| | ||||||||| | ||||||||| | ||||||||| |
Infections and infestations, n (%) | |||
|||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| |
General, n (%) | |||
Injection-site reactions | 5 (7.2) | 4 (5.8) | 5 (5.3) |
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| |
Nervous system disorders, n (%) | |||
Headache | 2 (2.9) | 5 (7.2) | 7 (7.4) |
Gastrointestinal disorders, n (%) | |||
||||||||| | ||||||||| | ||||||||| | ||||||||| |
Eye disorders, n (%) | |||
||||||||||||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| |
Vascular disorders, n (%) | |||
|||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| |
Blood and lymphatic system disorders, n (%) | |||
|||||||||||||||||| | ||||||||| | ||||||||| | ||||||||| |
Respiratory, thoracic, and mediastinal disorders, n (%) | |||
||||||||| | ||||||||| | ||||||||| | ||||||||| |
AE = adverse event; NR = not reported; q.2.w. = every 2 weeks; q.4.w = every 4 weeks; TCS = topical corticosteroids.
Source: Clinical Study Report for ECZTRA 3.6
Critical Appraisal
Internal Validity
All the included trials were randomized, double-blind, and placebo-controlled. Each trial was clearly described with specific objectives, end points, and interventions. The randomization method for the initial randomization and re-randomization across trials consisted of a central interactive web response system and an interactive voice response system was used to control the randomization and stratification factors. The baseline demographics and disease characteristics were generally similar between treatment groups in each trial, suggesting adequate randomization. However, there is an imbalance in the ECZTRA 3 trial’s demographic data with regard to race and gender. In the placebo group, 84 of 127 patients (66.1%) were male and 85 of 127 (66.9%) were White. That imbalance was also present in the ECZTRA 7 trial, in which 135 of 137 (98.5%) of patients were White, and slightly more than 60% (n = 83) were male. It is unclear if these imbalances biased the data in any way, but some impact on the generalizability to a diverse Canadian population is likely. The randomization method allowed for allocation concealment. Neither the patients nor any of the investigators involved in the treatment or clinical evaluation and monitoring of the patients were aware of the treatment received. The packaging and labelling of the intervention contained no evidence of its identity, and the intervention was handled and administered by a qualified, unblinded health care professional at the site who was not involved in the management of trial patients and who did not perform any of the assessments. At LEO Pharma (the sponsor), selected staff remained blinded to treatment allocation for the entire duration of the trial. The sponsor’s medical experts and safety advisors continued monitoring and cleaning data after being unblinded. Due to the identical packaging and labelling, patients in the ECZTRA 7 trial who self-injected the intervention were not unblinded. There was the potential for unblinding for patient safety reasons. While 24-hour emergency unblinding was allowed, only health care professionals who were not part of the trial staff could make emergency requests.
The primary outcomes assessed in the trial were based on IGA and EASI scores. The EASI has been shown to be both reliable and valid for the assessment of severity and extent of AD.24 The MID for both outcomes was not identified in the clinical trial reports, but previous research has found that the MID for the EASI is 6.6 points.31 Despite the identification of an MID for the EASI in AD populations, it is unclear if the 75% improvement cut-off of the EASI-75 score, which was the primary outcome across trials, is clinically meaningful. More work is needed to validate the uses of EASI-75 in populations with AD and determine a MID. The lack of an MID for the IGA score limits the ability to determine clinical relevance. The fact that the IGA assessments were based on the patient’s condition at the time of observation only and not compared to the condition at an earlier visit can be considered a limitation. It may have been useful to compare IGA scores across visits to determine if statistically significant changes were occurring over time. The clinical experts consulted by CADTH expected IGA and EASI measures to be used to assess AD severity, as a clinically meaningful response would include improvements in quality of life and itch scores. Other outcome measures such as SCORAD, pruritus NRS, DLQI, and POEM have adequate reliability and validity in patients with AD.
Subgroup analyses were specified a priori and conducted across the trials. The stratification by region was appropriate considering the recruitment in various countries and, for stratification by disease severity, it made sense to differentiate patients with mild to severe disease from patients with clear or almost-clear AD. The protocol of this report also listed other subgroups, such as the presence of other comorbid conditions (e.g., asthma) or the failure of patients to respond to 1 or more systemic therapies before initiating tralokinumab. These subgroups were not part of the analysis plan, but perhaps should have been, given that the majority of AD patients have a history of prior medication use.
Across trials, for all analyses at week 16, the differences in response rates between treatment groups were analyzed using the Cochran-Mantel-Haenszel test (single imputation analyses) or a combined inference from multiple Mantel-Haenszel risk differences and associated standard errors using the Rubin rule (multiple imputation analyses). There were no concerns related to the data lock points or protocol amendments made in the trials. The decision to impute those who used rescue medication and patients with missing data as nonresponders was appropriate.
Although the analyses were appropriate and investigators accounted for multiplicity, several limitations are associated with the design of the trials. First, the ECZTRA 1, ECZTRA 2, and ECZTRA 3 trials included a 2-week washout period during which no topical corticosteroid use was allowed. As noted by Wollenberg et al.,8 the patient population being studied has significant disease and high levels of prior medication use. The washout period therefore may have been long enough to exacerbate AD, leading to patients being labelled as “nonresponders” early in the studies. Second, the duration of the initial treatment period (16 weeks) in the ECZTRA 1, ECZTRA 2, and ECZTRA 3 trials may not have been sufficient. A further limitation affecting assessments of longer-term efficacy and safety in the ECZTRA 1, ECZTRA 2, and ECZTRA 3 trials is that only patients who achieved a clinical response at week 16 were eligible to be re-randomized. As a result, the estimates of effects in the maintenance phase are uncertain, and the analyses in the maintenance phase were not powered, which signifies that the long-term efficacy and safety of tralokinumab is uncertain. In a related issue, both the ECZTRA 3 and ECZTRA 7 trials used TCS in combination with tralokinumab and placebo, and the additive impact of the TCS can be seen in the results. For example, EASI-75 responses occurred in 50.5% of patients in the placebo group in the ECZTRA 7 trial (a percentage that is relatively high for a placebo group). Also, in the ECZTRA 3 and ECZTRA 7 trials, the cumulative amount of TCS used in the placebo group was higher than in the tralokinumab group, and results could therefore have been biased against tralokinumab; however, it is unclear how much this additional use of TCS biased the results. Last, pauses in dosing or the use of rescue medication in situations where the intervention was not available due to COVID-19 during the ECZTRA 7 trial may have affected the results.
External Validity
Two of the 4 trials (ECZTRA 2 and ECZTRA 3) included patients from Canada. In the ECZTRA 2 trial, 146 patients (24.6%) in the tralokinumab group and 44 placebo patients (21.9%) were from Canada. In the ECZTRA 3 trial, the numbers were lower: 34 patients (13.4%) in the tralokinumab plus TCS group and 26 placebo plus TCS patients (20.5%) were from Canada.
The inclusion and exclusion criteria for each study were clearly described and almost identical across trials. The clinical experts consulted for this review did not identify any issues with the inclusion or exclusion criteria or with the outcomes used to assess AD severity, symptoms, and HRQoL. The experts also stated that they considered the attrition rate to be low across studies because treatment was administered in the context of a clinical trial, and more patients would be expected to drop out in clinical practice in comparison to the numbers seen in these studies. Further, the ECZTRA 3 trial is more reflective of real-world practice because tralokinumab was combined with TCS as the intervention. In the ECZTRA 1 and ECZTRA 2 trials, patients who used rescue medication (including TCS) were considered “nonresponders,” which does not align with real-world use of biologics, which are initiated as add-on therapy to TCS for active lesions in 80% to 90% of patients.67,68 Another limitation is the absence of a comparator with a similar mechanism of action (e.g., dupilumab). Within the context of the trials, tralokinumab is associated with statistically significantly superior outcomes on the primary and secondary end points in comparison to placebo. However, the intervention is not compared to another biologic that is currently available to patients.
Generalizability of results is also limited because the design led to small sample sizes in the maintenance phases of the trials, and conclusions cannot be drawn regarding the long-term efficacy of tralokinumab. One clinical expert indicated that more injections are required with tralokinumab compared to dupilumab. The noticeable improvements in outcomes in the ECZTRA 3 and ECZTRA 7 trials, even in placebo groups, shows the additive effect of TCS.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
Several treatments are under development for moderate to severe AD. Three JAK inhibitors (abrocitinib, upadacitinib, and baricitinib) and monoclonal antibodies such as tralokinumab are all being developed. Because no head-to-head evidence comparing tralokinumab against other relevant treatments used to treat AD was available for this review, a review of indirect evidence was undertaken. The aim of this section is to summarize and critically appraise any ITCs that compare tralokinumab with other treatments for the management of moderate to severe AD in both monotherapy and combination regimes.
Patients with moderate to severe AD were evaluated in this review. Two ITCs, a MAIC submitted by the sponsor9 and an NMA published by the ICER10 that was identified in a separate literature search, were summarized and critically appraised.
Description of Identified Indirect Treatment Comparisons
The ICER performed an NMA10 to compare the efficacy and safety of abrocitinib, baricitinib, tralokinumab, and upadacitinib to each other, dupilumab, and placebo in patients with moderate to severe AD in both monotherapy and combination therapy regimes with TCS. While a second population of mild to moderate AD was also analyzed, it will not be summarized in this section as this target population is not of interest to the indication under review. While many outcomes were planned for inclusion, only EASI-50, EASI-75, EASI-90, IGAs of 0 or 1, and peak pruritus numeric rating scale (PP-NRS) of at least 4 were reported in the NMA.
The sponsor-submitted MAIC9 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
An overview of both ITCs is provided in Table 31.
Table 31: Overview of Included Indirect Treatment Comparisons
Characteristics | ICER network meta-analysis10 | Sponsor-submitted MAIC9 |
|---|---|---|
Population | Adults with moderate to severe AD | ||||||||||||||||||||||||||||||||||||||||||||| |
Intervention |
| ||||||||||||||||||||||||| |
Comparators |
| |||||||||||||||||||||||||||||||||||||||||||||||||| |
Outcomes |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||| |
Study design | RCTs | ||||||||| |
Publication | English only; included abstracts from conference proceedings | ||||||||| |
Exclusion criteria | Articles indexed as guidelines, letters, editorials, narrative reviews, case reports, or news items. | ||||||||| |
Databases searched | MEDLINE, EMBASE, Cochrane Library (Cochrane Database of Systematic Reviews and CENTRAL); searches were conducted from 1996 to present | ||||||||| |
Selection process | Full-text articles screened by a single reviewer, providing justification for exclusions | ||||||||| |
Data extraction process | Not specified | ||||||||| |
Quality assessment | Criteria published by the US Preventive Services Task Forces | ||||||||| |
AD = atopic dermatitis; CSA = cyclosporine A; DLQI = Dermatology Life Quality Index; EASI-50 = 50% or greater reduction in Eczema Area Severity Index from baseline; EASI-75 = 75% or greater reduction in Eczema Area Severity Index from baseline; EASI-90 = 90% or greater reduction in Eczema Area Severity Index from baseline; ICER = Institute for Clinical and Economic Review; IGA = Investigator's Global Assessment, MAIC = matched adjusted indirect comparison; NA = not applicable; POEM = Patient-Oriented Eczema Measure; PP-NRS = peak pruritus numeric rating scale; RCT = randomized control trial; SCORAD = Scoring Atopic Dermatitis; TCS = topical corticosteroids.
Source: Sponsor-submitted MAIC9 and ICER network meta-analysis.10
Network Meta-Analysis Conducted by the Institute for Clinical and Economic Review
Objectives and Rationale
The objectives of the ITC were to assess the relative efficacy and safety of abrocitinib, baricitinib, tralokinumab, and upadacitinib, as compared to each other, dupilumab, and placebo in populations with moderate to severe AD. A separate analysis was conducted examining mild to moderate AD, but will not be considered for the purposes of this evaluation.
Study Eligibility, Selection Process, and Data Extraction
The MEDLINE and EMBASE databases and the Cochrane Library (both the Cochrane Database of Systematic Reviews and CENTRAL) were searched. Specific study design filters to identify RCTs for each database were applied.
Studies were included if they were RCTs that reported the outcomes of interest, included a treatment of interest, involved the population of interest, were published since 1996, and were reported in English. Abstracts and conference proceedings were included. Articles indexed as guidelines, letters, editorials, narrative reviews, case reports, or news items were excluded. Screening was performed by a single investigator, as was inclusion and exclusion. It is also not reported how data extraction was conducted and whether more than 1 reviewer was involved.
Comparators
The comparators of interest were abrocitinib, baricitinib, tralokinumab, and upadacitinib compared to each other, topical therapies, and dupilumab (Table 31).
Outcomes
Efficacy outcomes identified to be assessed were: patient-reported pruritus or itching, EASI-50, EASI-75, and EASI-90 or relative change from baseline, IGA, sleep, SCORAD, POEM, DLQI, Children's Dermatology Life Quality Index, anxiety and depression (e.g., HADS), EQ-5D, measures of productivity (e.g., WPAI-GH), and other patient-reported symptom and quality-of-life measures. Safety outcomes intended to be assessed included AEs, treatment-emergent AEs, SAEs, discontinuation due to AEs, thrombotic events, infections, hematological abnormalities, malignancy, and all-cause mortality.
After data collection, NMAs were performed only on EASI, IGA, and PP-NRS (Table 31). Other outcomes were not reported due to inconsistent or limited data reporting and only described narratively.
Quality Assessment of Included Studies
Quality assessment was performed using criteria published by the US Preventive Services Task Force, rating each study as good, fair, or poor. Criteria used in the ratings were comparability of groups, reliability and validity of measurement instruments, intervention clarity, outcomes, attention to confounders, and performance of intention-to-treat analysis. Publication bias for the review was assessed by searching the clinicaltrials.gov database of trials to identify trials completed more than 2 years ago that would have met the inclusion criteria for which no findings have been published.
Evidence Networks
The evidence networks for the outcome of EASI-75 for both monotherapy and combination therapies are shown in this section, as well as that for pooling the 2 together in 1 analysis (Figure 6 and Figure 7). Networks for other outcomes were similar and based on reporting of the outcomes in each trial.
Figure 6: Evidence Network Diagram for Included Studies for Monotherapy Trials
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; EASI-75 = 75% or greater reduction in Eczema Area Severity Index from baseline; PBO = placebo; Q2W = every 2 weeks; TRA = tralokinumab; UPA = upadacitinib.
Note: Numbers in nodes are doses in milligrams.
Source: Institute for Clinical and Economic Review network meta-analysis,10 reprinted with permission.
Figure 7: Evidence Network Diagram for Included Studies for Combination Therapy Trials
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; PBO = placebo; Q2W = every 2 weeks; TRA = tralokinumab; UPA = upadacitinib.
Note: Numbers in nodes are doses in milligrams.
Source: Institute for Clinical and Economic Review network meta-analysis,10 reprinted with permission.
Indirect Treatment Comparison Methods
The NMA was conducted using a Bayesian framework. The IGA and PP-NRS variables were analyzed as dichotomous outcomes using a binomial likelihood and log link. The EASI outcomes were analyzed as ordinal data with 4 distinct groups (EASI < 50, EASI-50, EASI-75, and EASI-90). Mutually exclusive groups were created by reclassifying the data as ranges (< 50, 50 to 74, 75 to 89, and ≥ 90). A multinomial likelihood model with a probit link used methods from the National Institute for Health and Clinical Excellence Decision Support Unit.69 Unspecified noninformative priors were used for all model parameters. A Markov chain Monte Carlo simulation was performed by discarding the first 50,000 iterations as burn-in and basing inferences on an additional 50,000 iterations using 3 chains. Convergence was determined through visual examinations of the Brook-Gelman-Rubin diagnostic and historical plots. Models were run for both fixed and random effects, and they were run both adjusted and unadjusted for differences in placebo response. Models with the lowest deviance information criterion were considered to have the best fit between fixed and random effects. The model with placebo adjustment was considered a better fit if the regression coefficient was statistically significant and there was a reduction in between-trial heterogeneity. Only the analysis for the primary model was reported.
No information was given on details of the assessment of statistical heterogeneity, statistical consistency, or transitivity. Many trials had multiple arms with multiple doses of the same drug — these were treated as separate nodes within the NMA — and there was no pooling of arms to create nodes.
The models performed are summarized in Table 32.
Table 32: Network Meta-Analysis Reported by the ICER
Outcome | Trial type | Model | Number of trials |
|---|---|---|---|
EASI | a) Monotherapy only b) Combination only | Multinomial with probit link | a) 15 b) 6 |
IGA | a) Monotherapy only b) Combination only | Binomial with log link | a) 14 b) 6 |
PP-NRS4 | a) Monotherapy only b) Combination only | Binomial with log link | a) 14 b) 5 |
EASI = Eczema Area and Severity Index; ICER = Institute for Clinical and Economic Review; IGA = Investigator’s Global Assessment; PP-NRS4 = severity of peak pruritus numeric scale response.
Source: ICER network meta-analysis.10
Table 33: Indirect Treatment Comparison Analysis Methods for the ICER Report
Indirect treatment comparison methods | Network meta-analysis |
|---|---|
Priors | Noninformative |
Assessment of model fit | Deviance information criterion |
Assessment of consistency | Not reported |
Assessment of convergence | Brook-Gelman-Rubin diagnostic and historical plots |
Follow-up time points | 12 to 16 weeks |
Sensitivity analyses | Not reported in the network meta-analysis (only cost-effectiveness analysis) |
Subgroup analyses (descriptive only; not included in network meta-analysis) | Age (children, adolescents, and adults) Disease severity (moderate and severe) |
ICER = Institute for Clinical and Economic Review,
Source: ICER network meta-analysis.10
Results
Out of 58 trials identified for potential inclusion in the NMA, 21 were eventually included: 15 in the monotherapy analysis and 6 in the combination therapy analysis. Reasons for exclusion were not specified. All 21 trials enrolled adults and only 2 trials (Heads Up and JADE COMPARE) included active comparator groups (i.e., dupilumab 300 mg every 2 weeks). All trials were conducted over 12 to 16 weeks, used stable doses, and were similar in terms of age, duration of AD, and disease diversity. Although dupilumab was tested at different doses, only the FDA-approved dose of 300 mg once every 2 weeks was included in the NMA. All studies were of parallel design and assessed to be of “good” quality according to the US Preventive Services Task Force rating scale. All studies used some form of imputation, but the methods varied across studies. Multiple imputation, last observation carried forward, and nonresponse imputation were used in various combinations to account for missing data. Table 34 provides the important characteristics and baseline demographics of the included studies.
Table 34: Baseline Characteristics of Included Studies
Trial | Monotherapy or combination therapy | Doses | Sample size (N) | EASI (mean) | Mean age, years | Mean disease duration, years | IGA score of 4 (%) |
|---|---|---|---|---|---|---|---|
Abrocitinib trials | |||||||
JADE MONO-1a | Monotherapy | 100 mg 200 mg | 387 | 30.2 | 32.4 | 23.4 | 40.7 |
JADE MONO-2a | Monotherapy | 100 mg 200 mg | 391 | 28.5 | 35.1 | 21.0 | 32.2 |
JADE COMPARE | Combination | 100 mg 200 mg DUP 300 mg | 837 | 30.9 | 37.7 | 22.7 | 35.4 |
Gooderham (2019) | Monotherapy | 100 mg 200 mg | 167 | 25.6 | 40.8 | 23.0 | 40.8 |
Baricitinib trials | |||||||
BREEZE-AD 1 | Monotherapy | 1 mg 2 mg 4 mg | 624 | 31.0 | 35.7 | 25.7 | 41.8 |
BREEZE-AD 2 | Monotherapy | 1 mg 2 mg 4 mg | 615 | 33.5 | 34.5 | 24.0 | 50.5 |
BREEZE-AD 5 | Monotherapy | 1 mg 2 mg | 440 | 27.1 | 39.7 | 23.7 | 41.7 |
BREEZE-AD 7 | Combination | 2 mg | 329 | 29.57 | 33.8 | 24.03 | 45.0 |
Guttman-Yassky (2018) | Combination | 2 mg 4 mg | 104 | 21.23 | 36.5 | 22.03 | NR |
Tralokinumab trials | |||||||
ECZTRA 1 | Monotherapy | 300 mg | 802 | 29.3 | 37.0 | 27.5 | 50.9 |
ECZTRA 2 | Monotherapy | 300 mg | 794 | 28.9 | 32.0 | 25.3 | 49.2 |
ECZTRA 3 | Combination | 300 mg | 380 | 25.5 | 36.0 | 26.0 | 46.3 |
Upadacitinib trials | |||||||
MEASURE UP 1a | Monotherapy | 15 mg 30 mg | 847 | 29.5 | 34.0 | NR | 45.2 |
MEASURE UP 2a | Monotherapy | 15 mg 30 mg | 836 | 29.1 | 33.6 | NR | 54.9 |
AD-UP | Combination | 15 mg 30 mg | 907 | 29.6 | 34.1 | NR | 52.9 |
Heads Up | Monotherapy | UPA 30 mg DUP 300 mg | 692 | NR | NR | NR | NR |
Guttman-Yassky (2018) | Monotherapy | 7.5 mg 15 mg 30 mg | 167 | 25.6 | 40.8 | 23.0 | 40.8 |
Dupilumab trials | |||||||
LIBERTY AD SOLO 1 | Monotherapy | 300 mg | 671 | 30.7 | 38.7 | 26.7 | 48.3 |
LIBERTY AD SOLO 2 | Monotherapy | 300 mg | 708 | 29.4 | 34.7 | 24.8 | 48.3 |
LIBERTY AD CHRONOS | Combination | 300 mg | 740 | 29.8 | 31.2 | 26.7 | 47.7 |
Thaci (2016) | Monotherapy | 100 mg 200 mg 300 mg | 379 | 31.9 | 37.0 | 28.0 | 47.3 |
DUP = dupilumab; EASI = Eczema Area and Severity Index; NR = not reported; UPA = upadacitinib.
Note: All time points at 16 weeks except JADE MONO-1, JADE MONO-2, (12 weeks) and COMPARE (12 and 16 weeks).
aPooled estimates from this trial were in patients 12 and older.
Source: Institute for Clinical and Economic Review network meta-analysis,10 modified with permission.
Monotherapy
Table 35: Network Meta-Analysis Inputs for Monotherapy Outcomes
Trial | Arm | IGA | PP-NRS4 | EASI scores | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Response | Response | 50 | 75 | 90 | |||||||
N | n | N | n | N | n | N | n | N | n | ||
JADE MONO-1 | ABRO 200 mg | 120 | 58 | 121 | 68 | RD | RD | 120 | 78 | RD | RD |
ABRO 100 mg | 122 | 28 | 122 | 44 | RD | RD | 122 | 47 | RD | RD | |
PBO | 60 | 4 | 60 | 11 | RD | RD | 60 | 7 | RD | RD | |
JADE MONO-2 | ABRO 200 mg | 140 | 53 | 140 | 75 | RD | RD | 139 | 85 | RD | RD |
ABRO 100 mg | 139 | 42 | 141 | 67 | RD | RD | 139 | 62 | RD | RD | |
PBO | 70 | 7 | 70 | 8 | RD | RD | 70 | 8 | RD | RD | |
Goderham (2019) | ABRO 200 mg | 48 | 21 | 44 | 28 | 48 | 38 | 48 | 31 | 48 | 21 |
ABRO 100 mg | 54 | 16 | 50 | 25 | 54 | 30 | 54 | 22 | 54 | 14 | |
PBO | 52 | 3 | 51 | 13 | 52 | 14 | 52 | 8 | 52 | 5 | |
ECZTRA 1 | TRA 300 mg | 601 | 95 | 594 | 119 | 601 | 250 | 601 | 150 | 601 | 87 |
PBO | 197 | 14 | 194 | 20 | 197 | 42 | 197 | 25 | 197 | 8 | |
ECZTRA 2 | TRA 300 mg | 591 | 131 | 575 | 144 | 591 | 295 | 591 | 196 | 591 | 108 |
PBO | 201 | 22 | 200 | 19 | 201 | 41 | 201 | 23 | 201 | 11 | |
MEASURE UP 1 | UPA 30 mg | 243 | 148 | 238 | 145 | RD | RD | 243 | 192 | RD | RD |
UPA 15 mg | 239 | 119 | 234 | 125 | RD | RD | 239 | 166 | RD | RD | |
PBO | 241 | 21 | 233 | 26 | RD | RD | 241 | 43 | RD | RD | |
MEASURE UP 2 | UPA 30 mg | 247 | 125 | 246 | 150 | RD | RD | 247 | 180 | RD | RD |
UPA 15 mg | 243 | 93 | 240 | 103 | RD | RD | 243 | 144 | RD | RD | |
PBO | 242 | 12 | 238 | 24 | RD | RD | 242 | 32 | RD | RD | |
Heads Up | UPA 30 mg | NR | NR | 336 | 120 | RD | RD | 348 | 247 | 348 | 211 |
DUP 300 mg q.2.w. | NR | NR | 340 | 188 | RD | RD | 344 | 210 | 344 | 133 | |
Guttman-Yassky (2020) | UPA 30 mg | 42 | 21 | 36 | 19 | 42 | 35 | 42 | 29 | 42 | 21 |
UPA 15 mg | 42 | 13 | 32 | 19 | 42 | 30 | 42 | 22 | 42 | 11 | |
PBO | 41 | 1 | 35 | 2 | 41 | 9 | 41 | 4 | 41 | 1 | |
BREEZE-AD 1 | BARI 2 mg | 123 | 14 | 100 | 12 | 123 | 37 | 123 | 23 | 123 | 13 |
BARI 1 mg | 127 | 15 | 105 | 11 | 127 | 32 | 127 | 22 | 127 | 11 | |
PBO | 249 | 12 | 222 | 16 | 249 | 38 | 249 | 22 | 249 | 12 | |
BREEZE-AD 2 | BARI 2 mg | 123 | 13 | 106 | 16 | 123 | 34 | 123 | 22 | 123 | 11 |
BARI 1 mg | 125 | 11 | 100 | 6 | 125 | 23 | 125 | 16 | 128 | 8 | |
PBO | 244 | 11 | 213 | 10 | 244 | 30 | 244 | 15 | 244 | 6 | |
BREEZE-AD 5 | BARI 2 mg | 146 | 35 | 131 | 33 | 146 | 51 | 146 | 43 | 146 | 30 |
BARI 1 mg | 147 | 19 | 132 | 21 | 147 | 29 | 147 | 19 | 147 | 11 | |
PBO | 147 | 8 | 123 | 7 | 147 | 19 | 147 | 12 | 147 | 5 | |
SOLO 1 | DUP 300 mg q.2.w. | 244 | 85 | 213 | 87 | 224 | 154 | 224 | 115 | 224 | 80 |
PBO | 224 | 23 | 212 | 26 | 224 | 55 | 224 | 33 | 224 | 17 | |
SOLO 2 | DUP 300 mg q.2.w. | 233 | 84 | 225 | 81 | 233 | 152 | 233 | 103 | 233 | 70 |
PBO | 236 | 20 | 221 | 21 | 236 | 52 | 236 | 28 | 236 | 17 | |
THACI (2016) | DUP 300 mg q.2.w. | 64 | 19 | NR | NR | 64 | 50 | 64 | 34 | 64 | 1R9 |
PBO | 61 | 1 | NR | NR | 61 | 18 | 61 | 7 | 61 | 2 | |
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; PBO = placebo; N = total number; n = number responding; NR = not reported; q.2.w. = every 2 weeks; RD = data redacted; TRA = tralokinumab; UPA = upadacitinib.
Source: Institute for Clinical and Economic Review network meta-analysis,10 modified with permission.
Results from the NMA for monotherapy in all 3 of the EASI responses show that tralokinumab was superior to placebo, comparable to abrocitinib 100 mg, and inferior to upadicitinib (30 mg and 15 mg), abrocitinib 200 mg, and dupilumab 300 mg. For IGA response, tralokinumab was superior only to placebo, while being comparable to abrocitinib 100 mg, and inferior to upadacitinib (30 mg and 15 mg), abrocitinib 200 mg, and dupilumab 300 mg. For the PP-NRS response, tralokinumab was superior to placebo, comparable to abrocitinib (100 mg and 200 mg), and inferior to upadacitinib (30 mg and 15 mg) and dupilumab 300 mg. Results from the NMA for monotherapy are presented in Table 36.
Table 36: Results From the Network Meta-Analysis of the Median Relative Risk (95% Credible Interval) of EASI-50, EASI-75, EASI-90, IGA, and PP-NRS of at Least 4 Points Responses in Monotherapy Trials in Adult patients
Comparator | EASI-50 (RR, 95% CrI) | EASI-75 (RR, 95% CrI) | EASI-90 (RR, 95% CrI) | IGA (RR, 95% CrI) | PP-NRS of at Least 4 Points (RR, 95% CrI) |
|---|---|---|---|---|---|
Tralokinumab vs. | |||||
Placebo | 2.14 (1.80 to 2.47) | 2.61 (2.09 to 3.18) | 3.32 (2.5 to 4.27) | 2.2 (1.47 to 3.3) | 2.29 (1.17 to 4.08) |
Upadacitinib 15 mg | 0.65 (0.54 to 0.78) | 0.56 (0.44 to 0.7) | 0.46 (0.33 to 0.63) | 0.33 (0.2 to 0.53) | 0.51 (0.23 to 0.99) |
Upadacitinib 30 mg | 0.57 (0.48 to 0.67) | 0.46 (0.36 to 0.56) | 0.35 (0.25 to 0.46) | 0.25 (0.16 to 0.39) | 0.46 (0.22 to 0.88) |
Abrocitinib 100 mg | 0.83 (0.65 to 1.05) | 0.78 (0.57 to 1.08) | 0.72 (0.48 to 1.1) | 0.59 (0.3 to 1.12) | 0.78 (0.34 to 1.72) |
Abrocitinib 200 mg | 0.63 (0.51 to 0.76) | 0.53 (0.4 to 0.69) | 0.42 (0.29 to 0.61) | 0.36 (0.2 to 0.65) | 0.55 (0.25 to 1.15) |
Dupilumab 300mg Q2W | 0.71 (0.59 to 0.85) | 0.63 (0.49 to 0.8) | 0.55 (0.39 to 0.75) | 0.47 (0.28 to 0.76) | 0.47 (0.23 to 0.94) |
CrI = credible interval; EASI-50 = 50% or greater reduction in Eczema Area and Severity Index from baseline; EASI-75 = 75% or greater reduction in Eczema Area and Severity Index from baseline; EASI-90 = 90% or greater reduction in Eczema Area and Severity Index from baseline; IGA = Investigator’s Global Assessment; PP-NRS = peak pruritus numeric rating scale; Q2W = every 2 weeks.
Note: All analyses were carried out using the random effects, unadjusted models. Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between Tralokinumab and comparators. Estimates in bold signify that the 95% credible interval does not contain a risk ratio of 1. Values greater than 1 favour tralokinumab treatment.
Source: Institute for Clinical and Economic Review network meta-analysis,10 modified with permission.
Combination Therapy
Table 37: Network Meta-Analysis Inputs for Combination Therapy Outcomes
Trial | Arm | IGA | PP-NRS4 | EASI scores | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Response | Response | 50 | 75 | 90 | |||||||
N | n | N | n | N | n | N | n | N | n | ||
JADE COMPARE | ABRO 200 mg | 221 | 105 | 172 | 108 | 221 | 193 | 221 | 157 | 221 | 108 |
ABRO 100 mg | 230 | 80 | 168 | 79 | 229 | 186 | 229 | 138 | 229 | 87 | |
DUP 300 mg q.2.w. | 232 | 90 | 189 | 108 | 232 | 195 | 232 | 152 | 232 | 90 | |
PBO | 124 | 16 | 94 | 27 | 124 | 71 | 124 | 38 | 124 | 14 | |
ECZTRA 3 | TRA 300 mg | 252 | 98 | 249 | 113 | 252 | 200 | 252 | 141 | 252 | 83 |
PBO | 126 | 33 | 126 | 43 | 126 | 73 | 126 | 45 | 126 | 27 | |
AD-UP | UPA 30 mg | 260 | 150 | 258 | 168 | RD | RD | 260 | 201 | RD | RD |
UPA 15 mg | 261 | 107 | 252 | 134 | RD | RD | 261 | 172 | RD | RD | |
PBO | 264 | 30 | 256 | 39 | RD | RD | 264 | 68 | RD | RD | |
BREEZE-AD7 | BARI 2 mg | 109 | 26 | 97 | 37 | 109 | 70 | 109 | 47 | 109 | 18 |
PBO | 109 | 16 | 104 | 21 | 109 | 45 | 109 | 25 | 109 | 15 | |
Guttman-Yassky (2018) | BARI 2 mg | 37 | 8 | NR | NR | 37 | 21 | 37 | 11 | 37 | 7 |
PBO | 49 | 4 | NR | NR | 49 | 18 | 49 | 10 | 49 | 3 | |
LIBERTY AD CHRONOS | DUP 300 mg q.2.w. | 106 | 41 | 102 | 60 | 106 | 85 | 106 | 73 | 106 | 42 |
PBO | 315 | 39 | 299 | 59 | 315 | 118 | 315 | 73 | 315 | 35 | |
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; EASI = Eczema Area and Severity Index; N = total number; n = number responding; NR = not reported; PBO = placebo; PP-NRS4 = severity of peak pruritus numeric scale response; q.2.w. = every 2 weeks; RD = data redacted; TRA = tralokinumab; UPA = upadacitinib.
Note: All interventions (including placebo) were administered with topical corticosteroids.
Source: Institute for Clinical and Economic Review network meta-analysis,10 reprinted with permission.
Results from the NMA for combination therapy show that for the EASI-50, EASI-75, EASI-90, and IGA responses, tralokinumab was shown to be superior to placebo, and inferior to upadacitinib (30 mg and 15 mg), abrocitinib (100 mg and 200 mg), and dupilumab 300 mg. For the PP-NRS responses tralokinumab was superior to placebo, comparable to abrocitinib 100 mg and inferior to upadacitinib (30 mg and 15 mg), abrocitinib 200 mg and dupilumab 300 mg. Results from the NMA for monotherapy are presented in Table 38.
Table 38: Results From the Network Meta-Analysis of the Median Relative Risk (95% Credible Interval) of EASI-50, EASI-75, EASI-90, IGA, and PP-NRS of at Least 4 Points Responses in Combination Trials in Adult patients
Comparator | EASI-50 (RR, 95% CrI) | EASI-75 (RR, 95% CrI) | EASI-90 (RR, 95% CrI) | IGA (RR, 95% CrI) | PP-NRS of at Least 4 Points (RR, 95% CrI) |
|---|---|---|---|---|---|
Tralokinumab vs. | |||||
Placebo | 1.44 (1.23 to 1.64) | 2.30 (1.94 to 2.68) | 2.13 (1.51 to 2.88) | 1.63 (1.11 to 2.35) | 1.42 (1.03 to 1.91) |
Upadacitinib 15 mg | 0.76 (0.64 to 0.87) | 0.63 (0.48 to 0.79) | 0.5 (0.34 to 0.7) | 0.48 (0.31 to 0.74) | 0.52 (0.36 to 0.75) |
Upadacitinib 30 mg | 0.69 (0.58 to 0.79) | 0.53 (0.41 to 0.65) | 0.36 (0.25 to 0.51) | 0.35 (0.23 to 0.53) | 0.42 (0.3 to 0.57) |
Abrocitinib 100 mg | 0.83 (0.7 to 0.98) | 0.75 (0.57 to 0.97) | 0.65 (0.43 to 0.95) | 0.6 (0.37 to 0.98) | 0.71 (0.48 to 1.08) |
Abrocitinib 200 mg | 0.76 (0.64 to 0.88) | 0.63 (0.48 to 0.8) | 0.5 (0.34 to 0.71) | 0.45 (0.29 to 0.69) | 0.49 (0.35 to 0.68) |
Dupilumab 300mg Q2W | 0.79 (0.67 to 0.92) | 0.68 (0.53 to 0.87) | 0.57 (0.39 to 0.81) | 0.54 (0.35 to 0.83) | 0.56 (0.39 to 0.78) |
CrI = credible interval; EASI-50 = 50% or greater reduction in Eczema Area and Severity Index from baseline; EASI-75 = 75% or greater reduction in Eczema Area and Severity Index from baseline; EASI-90 = 90% or greater reduction in Eczema Area and Severity Index from baseline; IGA = Investigator’s Global Assessment; PP-NRS = peak pruritus numeric rating scale; Q2W = every 2 weeks.
Note: All analyses were carried out using the random effects, unadjusted models. Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between Tralokinumab and comparators. Estimates in bold signify that the 95% credible interval does not contain a risk ratio of 1. Values greater than 1 favour tralokinumab treatment.
Source: Institute for Clinical and Economic Review network meta-analysis,10 modified with permission.
Harms
An NMA of harms data was not reported. Narrative summaries of the reported safety data indicated that nausea, conjunctivitis, and herpetic infection were more common in treatments than in placebo. Treatment-emergent AEs, SAEs, and discontinuations due to AEs were low and generally similar among treatments.
While no subgroup analyses were performed within the NMA, age and disease severity were examined using in-confidence data provided to ICER by manufacturers. Results in adolescents were deemed to be similar to those in adults in abrocitinib, while abrocitinib showed greater efficacy among patients with severe disease compared to those with moderate disease.
Critical Appraisal of ICER Network Meta-Analysis
The eligibility criteria, PICOT terms (patient or population, intervention, comparison, outcome and time), and search strategy were comprehensive and the authors reported that they followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines in the conduct of the review. Quality assessment of included studies was performed using a referenced tool. End point data were collected at 16 weeks for all trials except for the monotherapy abrocitinib trials, in which data were collected at 12 weeks. The pivotal trials for abrocitinib and upadacitinib enrolled adolescents, but the majority of patients were still at least 18 years old, and the reviewers attempted to stratify by age. The trials were similar in terms of key baseline characteristics age (31 to 41 years), duration of disease (21 to 28 years), and disease severity (32% to 55% IGA of 4). No unpublished studies meeting the inclusion criteria were identified in the clincaltrials.gov database, indicating no evidence of publication bias.
There are many critical appraisal points to be made regarding the NMA conducted by the ICER:
Study screening was not verified by a second party, as all studies were screened by a single reviewer. The recommended practice is for screening by 2 or more independent reviewers to reduce the risk of selection bias.
For data extraction, there is no mention of duplicate data extraction or data validation methods.
While most of the trials had an end point of 16 weeks, all the monotherapy abrocitinib trials had an end point of 12 weeks. The true effect this point of clinical heterogeneity has on the final results is uncertain.
The results from the combination therapy were based on only 6 trials; despite this, credible intervals were precise. In fact, they were in general more precise than the results of monotherapy (which was the subject of 15 trials). The reason for this apparent discrepancy is unclear.
The authors made no mention of the transitivity issue and the testing for consistency. While the lack of head-to-head comparisons among active treatments would make tests for consistency difficult, there were closed loops within the network that could have been tested.
No sensitivity analysis appears to have been conducted within the NMA to explore any possible assumptions made by the reviewers. There is also no indication of an adjustment made in the model to account for the correlation in the 3 arm trials.
The NMA results are presented only for EASI, IGA, and PP-NRS outcomes; other planned outcomes were not assessed due to inconsistent or limited data reporting. Tables are presented giving narrative information on safety data. The lack of reporting for these additional outcomes of interest increases the likelihood of reporting bias.
All trials included in the review used imputation to adjust for missing data (combinations of multiple imputation, nonresponder imputation, or last observation carried forward), although there was no systematic difference in imputation methods across end points. It is unknown what kind of effect this may have had on final results.
Sponsor-Submitted Matched Adjusted Indirect Comparison
This section was redacted based on the sponsor request.
Summary of Indirect Evidence
Each of the ITCs examined for this review had different populations, methods, and focus. The ICER performed an NMA to compare the JAK inhibitors (abrocitinib, upadicitinib, and baricitinb) with tralokinumab and dupilumab in patients with moderate to severe AD. The sponsors compared ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The ITCs can be considered independent from each other as the 2 trials included in the sponsor's MAIC9 were omitted from the NMA conducted by the ICER.10
The ICER review used Bayesian methods to perform the NMA, while the sponsor review involved an ITC using a MAIC, referring to National Institute for Health and Care Excellence protocols,11 however, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| While they are difficult to compare, as the ITCs used different metrics of analysis (the ICER used a risk ratio |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||) the EASI-50, EASI-75, and EASI-90 results from the 2 analyses for tralokinumab versus dupilumab are presented side by side in Table 39. Both monotherapy and combination therapy results are shown for the ICER review.
Table 39: Tralokinumab Versus Dupilumab for EASI Outcomes in the ICER and Sponsor Reviews
Outcome | ICER network meta-analysis: monotherapy (risk ratio) | ICER network meta-analysis: combination therapy (risk ratio) | Sponsor MAIC (risk difference) |
|---|---|---|---|
EASI-50 | 0.71 (0.59 to 0.85) | 0.79 (0.67 to 0.92) | ||||||||| |
EASI-75 | 0.63 (0.49 to 0.80) | 0.68 (0.53 to 0.87) | ||||||||| |
EASI-90 | 0.55 (0.39 to 0.75) | 0.57 (0.39 to 0.81) | ||||||||| |
IGA 0 or 1 | 0.47 (0.28 to 0.76) | 0.54 (0.35 to 0.83) | ||||||||| |
EASI-50 = 50% or greater reduction in Eczema Area and Severity Index from baseline; EASI-75 = 75% or greater reduction in Eczema Area and Severity Index from baseline; EASI-90 = 90% or greater reduction in Eczema Area and Severity Index from baseline; ICER = Institute for Clinical and Economic Review; IGA = Investigator’s Global Assessment; MAIC = matched adjusted indirect comparison.
Note: Risk ratio values smaller than 1 favour dupilumab. Risk difference values smaller than 0 favour dupilumab.
Source: Sponsor-submitted MAIC9 and ICER network meta-analysis.10
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| the ICER’s NMA showed the smallest difference at this threshold, for both monotherapy and combination therapy. Credible intervals in the ICER review were much more precise than|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| For the IGA outcome, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||; however, the NMA conducted by the ICER found that dupilumab had twice the efficacy of tralokinumab (in both monotherapy and combination therapy) with credible intervals that completely favoured dupilumab.
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
It is evident from both ITCs that some uncertainty remains regarding the efficacy and safety of tralokinumab compared to dupilumab and JAK inhibitors (abrocitinib, baricitinib, and upadacitinib). ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. However, in the ICER review, dupilumab was clearly superiority in all outcomes. Compared to the JAK inhibitors in the NMA, tralokinumab was generally inferior in efficacy compared to upadicitinib and abrocitinib. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Conclusion
Two ITCs were identified, reviewed, and critically appraised. One was submitted by the sponsor (||||||||||||||||||) and the other was published by the ICER (as a network meta-analysis). Both reviews failed to report how studies were selected and how data were extracted. The sponsors were forthcoming about their method of analysis, but the ICER omitted many important details about its methodology, making it difficult to assess the quality of the NMA.
The NMA was mostly concerned with comparing abrocitinib, baricitinib, tralokinumab, and upadacitinib with dupilumab in monotherapies and combination therapies. The authors concluded that upadacitinib and abrocitinib had more favourable outcomes compared with baricitinib and tralokinumab. Overall, all 4 treatments were rated by the ICER as promising but inconclusive compared to topical therapy alone. Evidence on upadicitinib and abrocitinib compared to dupilumab was considered to be insufficient, while evidence on baricitinib and tralokinumab compared to dupilumab was considered to be comparable to inferior. The comparison of the 4 treatments to each other was rated as inconclusive.
Focusing on tralokinumab, the NMA showed tralokinumab (at a 300 mg dose) was generally superior to placebo and generally inferior to upadacitinib (both 15 mg and 30 mg), abrocitinib 200 mg, and dupilumab 300 mg. It was similar to baricitinib (1 mg and 2 mg) and abrocitinib 100 mg. This applied to treatments taken alone and in combination with topical therapies.
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Focusing on the comparison between tralokinumab and dupilumab, The NMA conducted by the ICER suggested that tralokinumab may be inferior to dupilumab in several efficacy outcomes, while the results of the sponsor-submitted |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Other Relevant Evidence
This section includes submitted long-term extension studies and additional relevant studies included in the sponsor’s submission to CADTH that were considered to address important gaps in the evidence included in the systematic review.
Long-Term Extension Studies
One ongoing, long-term extension study (ECZTEND) has been summarized to provide additional evidence on the safety and efficacy of tralokinumab in patients with AD. Data presented in this summary were collected until April 30, 2020, and presented in the intermediate clinical trial report.
Methods
The ECZTEND study is an ongoing, open-label, single-arm, long-term extension study of the safety and efficacy of tralokinumab in patients with AD who have previously participated in clinical trials for tralokinumab (i.e., ECZTRA 1 through 8 and TraSki). The study is taking place at |||||||||, including centres in Canada, Belgium, the Czech Republic, France, Germany, Italy, Japan, Poland, Spain, UK, and the US.71 The ECZTEND study consists of a 2-week screening period (which is expected to overlap with the end of the parent trial for most patients), a 0.5- to 5-year treatment phase, and a 14-week follow-up beginning 2 weeks after the final dose (Figure ). At the time of data cut-off, 1,174 patients were included in the ECZTEND trial. The primary outcome is safety or the number of AEs experienced during the study. The secondary outcomes are drug efficacy and include achieving an IGA score of 0 or 1 and an EASI-75 score at weeks 16, 56, 88, 104, 136, 152, 184, 216, and 248 during the treatment phase relative to baseline. Blinding of treatment allocation was maintained for patients who continued from a blinded parent trial and entered the open-label extension.
Figure 8: Study Flow Diagram for the ECZTEND Open-Label Extension Study
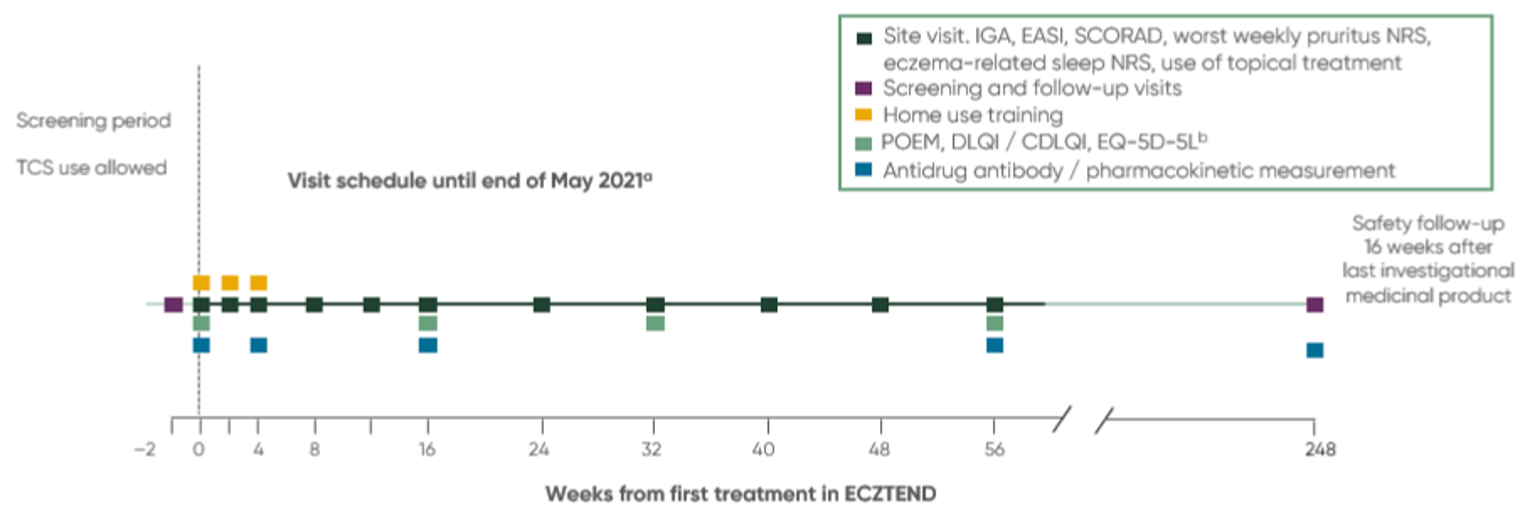
CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EQ-5D-5L = EQ-5D 5-Levels questionnaire; IGA = Investigator’s Global Assessment; NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; SCORAD = Scoring Atopic Dermatitis; TCS = topical corticosteroids.
a After May 2021, some site visits switched to telephone visits.
b Patients from the ECZTRA 6 parent trial will not perform the EQ-5D-5L.
Source: Clinical Study Report of ECZTEND.73
Populations
Individuals were eligible to participate in ECZTEND if they met the following inclusion criteria:
Completed a parent trial for tralokinumab including ECZTRA 1, ECZTRA 2, ECZTRA 3, ECZTRA 4, ECZTRA 5, ECZTRA 6, ECZTRA 7, ECZTRA 8, or TraSki (ECZTRA 1, ECZTRA 2, ECZTRA 3, and ECZTRA 7 were pivotal trials reviewed and described earlier in this report).
Able and willing to self-administer or have a caregiver administer treatment at home after training during the first 3 site visits (with exceptions).
Use a stable emollient at least twice per day for a minimum of 14 days before baseline and continue to use it throughout the study and follow-up.
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||Individuals were excluded from participating in ECZTEND if they met the following exclusion criteria:
More than 26 weeks elapsed since the patient received their last dose in the parent trial.
Any condition leading to permanent discontinuation during the parent trial; AE leading to temporary discontinuation or SAE during the parent trial related to tralokinumab that may indicate continued treatment would pose an unreasonable safety risk.
Treatment with any of the following: systemic immunosuppressant or immunomodulating drugs and/or systemic corticosteroids (within 5 half-lives before baseline of ECZTEND), topical phosphodiesterase type 4 inhibitors (2 weeks before baseline), any marketed biologic therapy (3 to 6 months before baseline).
Clinically significant infection (4 weeks before baseline), helminth parasitic infection (6 months before informed consent), tuberculosis requiring treatment (within 12 months before screening).
Any of the following health conditions: ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||, tuberculosis, known primary immunodeficiency disorder ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||Baseline characteristics of patients in the ECZTEND trial are summarized in Table 40. Briefly, the |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| More than half of the patients were male (57%) and most individuals were White (71.3%), Asian (17.2%), or Black (8.6%). Nearly equal proportions of patients were from Europe and North America (46.5% and 46.2%, respectively) followed by Japan (7.3%); ||||||||||||||||||||||||||||||||||||||||||||||||||. Patients transitioned from 1 of 4 parent trials: ECZTRA 1 (38.3%), ECZTRA 2 (25.0%), ECZTRA 3 (24.0%), and ECZTRA 5 (12.7%). ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In terms of patient disease history, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| measured at parent study baseline. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| measured at ECZTEND baseline. At parent study baseline, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| which correspond to moderate or severe disease. When assessed at baseline of the open-label extension, most patients had |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Additional baseline measures of disease were captured using the SCORAD, POEM, DLQI, pruritus NRS, and eczema-related sleep NRS.
Table 40: Baseline Characteristics of ECZTEND Open-Label Extension Study — FAS
Characteristics | Tralokinumab, all patients (N = 1,174) |
|---|---|
Demographic | 1,174 (100) |
|||||||||||||||||||||||||||||| | |||||||||| |
Age (years), median (range) | 38.0 (18.0 to 87.0) |
Male, n (%) | 675 (57.5) |
Race, n (%) | 1,172 (99.8) |
White | 836 (71.3) |
Asian | 201 (17.2) |
Black | 101 (8.6) |
Native Hawaiian or other Pacific Islander | 3 (0.3) |
American Indian or Alaska Native | 2 (0.2) |
Other | 29 (2.5) |
Region, n (%) | 1,174 (100) |
Europe | 546 (46.5) |
North America | 542 (46.2) |
|||||||||| | |||||||||| |
|||||||||| | |||||||||| (7.3) |
Parent trial, n (%) | |
ECZTRA 1 | 450 (38.3) |
ECZTRA 2 | 293 (25.0) |
ECZTRA 3 | 282 (24.0) |
ECZTRA 5 | 149 (12.7) |
Time (days) from last dose in parent trial, n (%) | 1,174 (100) |
|||||||||||||||||||| | |||||||||| |
Median (range) | 36.0 (7.0, 148.0) |
Disease historya | |
|||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||| |
IGA score | |
|||||||||| | |
|||||||||| | |||||||||| |
|||||||||| | |||||||||| |
AD = atopic dermatitis; BSA = body surface area; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; FAS = full analysis set; IGA = Investigator’s Global Assessment; NR = not reported; NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation.
aSafety analysis set used.
bMeasured at parent study baseline.
cMeasured at ECZTEND study baseline.
Note: Redacted rows have been deleted.
Source: Clinical Study Report for ECZTEND.73
Interventions
Adult patients received a loading dose of tralokinumab 600 mg, administered as 4 separate 1.0 mL subcutaneous injections of tralokinumab 150 mg, followed by tralokinumab 300 mg every 2 weeks thereafter. A minimum of 7 days was required between any 2 doses.
During the first 3 treatment visits (weeks 0, 2, and 4), patients were trained by site staff to self-inject tralokinumab, or have a caregiver administer the drug, to continue treatment at home afterwards. Patients from open-label arms of parent trials (ECZTRA 1 and ECZTRA 2) who had experience with self-administration of tralokinumab could self-inject from home starting at baseline without additional training. Patients were monitored for immediate post-dose drug reactions during the first 3 treatment visits as some patients were treatment-naive (i.e., they received placebo) during the parent trials. Similarly, patients from open-label arms or trials (ECZTRA 1, ECZTRA 2, ECZTRA 4, and ECZTRA 6) who had received at least 3 doses of medication did or will not require additional monitoring.
Use of TCS (US class ≥ 4 or Europe class ≤ 3) or TCIs was optional in some of the parent trials and continued to be optional during the open-label extension. This aligns with the intended commercial use of tralokinumab, which is with or without concurrent use of TCS. Other permitted concomitant medications included oral antibiotics, antiviral, antifungal therapies for skin infections, oral antihistamines, and stable doses of emollients.
Rescue medication was allowed, at the investigator’s discretion, starting with topical treatments and escalating to systemic treatments if the patient had an inadequate response after at least 14 days using the topical treatment. Topical treatments could include TCS with a classification of less than 4 in the US and greater than 3 in Europe and be used concurrently with tralokinumab. Patients were to be monitored for local or systemic TCS toxicity. Systemic treatments could include corticosteroids or nonsteroidal systemic immunosuppressive drugs (e.g., cyclosporine, methotrexate, mycophenolate mofetil, and azathioprine) and tralokinumab was to be immediately discontinued. Once systemic treatment was completed, tralokinumab could resume at the investigator’s discretion after at least 5 half-lives of the last dose of systemic treatment.
Prohibited concomitant therapies, in addition to those listed in the exclusion criteria and as rescue medications, were phototherapy and 3 or more bleach baths per week.
Outcomes
The primary outcome of the ECZTEND trial was the safety of subcutaneous administration of tralokinumab every 2 weeks in patients with AD and was summarized as AEs, SAEs, harms of special interest, withdrawals due to AEs, and deaths. Secondary outcomes were for drug efficacy and include achieving an IGA score of 0 or 1 and an EASI-75 score relative to the parent study baseline at weeks 16 and 56 of the open-label extension. Exploratory outcomes that were summarized include the IGA, EASI, worst weekly pruritus NRS, and eczema-related weekly sleep NRS scores.
Statistical Analysis
At the time of data cut-off, 1,174 patients were included in the ECZTEND trial. All patients who received tralokinumab were included in the FAS, which was used to assess efficacy. The safety analysis set was identical to the FAS and used to assess the safety of tralokinumab. Demographic and baseline characteristics were presented as descriptive statistics. For the primary safety end point, data were presented as the number of AEs during the treatment period. Results for the secondary end points (IGA and EASI) and exploratory end points were presented as descriptive statistics (e.g., number of observations, mean, SD, median, and range) by visit. An observed-cases method was used to tabulate data by visit (i.e., only patients who attended a specific visit). No formal sample size or power calculations were performed for this study and missing data were not imputed for safety results.
Patient Disposition
Patient disposition for the ECZTEND trial is summarized in Table 41. The number of patients screened was not reported. Of the 1,174 patients who were enrolled, 1,035 (88.2%) were still participating at the time of the data cut-off for the intermediate report and 139 (11.8%) had discontinued. Reasons for permanent discontinuation included patients lost to follow-up (2.5%), lack of efficacy (2.0%), AEs (1.6%), patient withdrawal (1.4%), and other reasons (e.g., pregnancy, protocol deviation, physician decision, or administrative reasons) (4.3%).
Table 41: Patient Disposition of ECZTEND Open-Label Extension Study
Disposition | Tralokinumab |
|---|---|
Screened | NR |
Randomized | 1,174 |
Ongoing, n (%) | 1,035 |
Discontinued study, n (%) | 139 (11.8) |
Lost to follow-up | 29 (2.5) |
Lack of efficacy | 24 (2.0) |
AE | 19 (1.6) |
Patient withdrawal | 16 (1.4) |
Othera | 51 (4.3) |
FAS | 1,174 (100) |
SAS | 1,174 (100) |
AE = adverse event; FAS = full analysis set; NR = not reported; SAS = safety analysis set.
aOther reasons include pregnancy, protocol deviation (concomitant medication and eligibility), physician decision, or administrative reasons (patient moved, relocated, busy, transportation issues, and personal reasons).
Source: Clinical Study Report for ECZTEND.73
Exposure to Study Treatments
Of the 1,174 patients in ECZTEND, 1,023 (87.1%) were recently treated with tralokinumab and were exposed during the parent studies to various treatment regimens, including a mix of placebo, tralokinumab every 2 weeks with or without TCS, and tralokinumab every 4 weeks with or without TCS. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Patient exposure to tralokinumab was reported as the time from the first dose of study drug to the last visit or permanent discontinuation before |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| or a median of 58.1 weeks (range = 0.0 to 82.3). By week 56, |||||||||||| of patients had discontinued from the study.
Efficacy
Secondary and exploratory outcomes for efficacy are summarized in Table 42. Responders were defined as achieving an IGA score of 0 or 1 an EASI-75 score. At weeks 16 and 56, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
At total of 345 patients from the ECZTRA 1 and ECZTRA 2 trials enrolled in the ECZTEND trial at least 60 weeks before the data cut-off of April 30, 2020. Forty (11.6%) of these patients permanently discontinued the trial. Patients from the ECZTRA 1 and ECZTRA 2 parent trials who enrolled in ECZTEND at least 60 weeks before the data cut-off received continued tralokinumab treatment for 2 years: 52 weeks in the ECZTRA 1 and ECZTRA 2 trials followed by 56 weeks in the ECZTEND trial. Of the patients receiving continued tralokinumab treatment for 2 years, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. In addition, analysis in which missing data and discontinuations were imputed as nonresponses, |||||||||||||||||||||||||||||||||||||||||||. In a second additional analysis |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| responders at week 56. At that point, the proportion of EASI-75 responders among patients from the ECZTRA 1 and ECZTRA 2 parent trials was 82.5%. In addition, analysis ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. In a second additional analysis ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. At week 56, the proportion of EASI-90 responders among patients from the ECZTRA 1 and ECZTRA 2 parent trials (i.e., patients receiving continued tralokinumab treatment for 2 years) was 59.8%. In addition, analysis |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| In a second additional analysis ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 42: Efficacy Outcomes in ECZTEND Open-Label Extension Study — FAS
Outcomes | Tralokinumab, all patients (N = 1,174) |
|---|---|
Secondary outcomes: respondersa | |
IGA score 0 or 1, n/N (%) | |
Week 16 | |||||||||||| |
Week 56 | |||||||||||| |
EASI-75, n/N (%) | |
Week 16 | |||||||||||| |
Week 56 | |||||||||||| |
Exploratory outcomes | |
EASI score, week 56 |||||||||||| | |
Absolute score, mean (SD) | |||||||||||| |
Worst weekly pruritus NRS score, week 56 |||||||||||| | |
Absolute score, mean (SD) | |||||||||||| |
Eczema-related weekly sleep NRS score, week 56 |||||||||||| | |
Absolute score, mean (SD) | |||||||||||| |
EASI = Eczema Area and Severity Index; EASI-75 = 75% or greater reduction in Eczema Area and Severity Index from baseline; FAS = full analysis set; IGA = Investigator’s Global Assessment; NR = not reported; NRS = numeric scale; SD = standard deviation.
aDefined as achieving an IGA score of 0 or 1 or an EASI-75 score.
Source: Clinical Study Report for ECZTEND.73
Harms
Table 43 summarizes the harms outcomes in the ECZTEND trial. Overall, 844 patients (71.9%) experienced at least 1 treatment-emergent AE, with the 3 most common AEs being viral upper respiratory tract infection (21.3%), dermatitis atopic (13.5%), and upper respiratory tract infection (7.1%). Other harms of special interest that were identified in CADTH’s systematic review protocol include conjunctivitis (3.8%), injection-site reactions (2.4%), conjunctivitis allergic (2.0%), ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Nineteen patients (1.6%) withdrew due to an AE, and no deaths were reported.
Table 43: Harms Outcomes in ECZTEND Open-Label Extension Study — SAS
Harm | Tralokinumab, all patients (N = 1,174) |
|---|---|
Patients with ≥ 1 AE, n (%) | 844 (71.9) |
AE,a n (%) | |
Infections and infestations | |||||||||||| |
Viral upper respiratory tract infection | 250 (21.3) |
Upper respiratory tract infection | 83 (7.1) |
Conjunctivitis | 45 (3.8) |
Influenza | 43 (3.7) |
Gastroenteritis | 30 (2.6) |
Bronchitis | 27 (2.3) |
Sinusitis | 27 (2.3) |
Rhinitis | 26 (2.2) |
Herpes simplex | 25 (2.1) |
Oral herpes | 24 (2.0) |
Skin and subcutaneous disorders | |||||||||||| |
Dermatitis atopic | 158 (13.5) |
Pruritus | 28 (2.4) |
Gastrointestinal disorders | |||||||||||| |
Diarrhea | 40 (3.4) |
Musculoskeletal and connective tissue disorders | |||||||||||| |
Back pain | 33 (2.8) |
General disorders and administration site conditions | |||||||||||| |
Injection-site reactions | 28 (2.4) |
Respiratory, thoracic, and mediastinal disorders | |||||||||||| |
Cough | 37 (3.2) |
Eye disorders | |||||||||||| |
Conjunctivitis allergic | 24 (2.0) |
Nervous system disorders | |||||||||||| |
Headache | 49 (4.2) |
Harms of special interest, n (%) | |
Upper respiratory tract infections | 83 (7.1) |
Conjunctivitis | 45 (3.8) |
Injection-site reactions | 28 (2.4) |
Eosinophilia | |||||||||||| |
Hypersensitivity reactions | |||||||||||| |
Facial dermatitis | |||||||||||| |
Patients with ≥ 1 SAE, n (%) | 55 (4.7) |
SAE,b n (%) | |
Infections and infestations | |||||||||||| |
Tonsillitis | |||||||||||| |
Neoplasms benign, malignant, and unspecified (includes cysts and polyps) | |||||||||||| |
Breast cancer | |||||||||||| |
Invasive ductal breast carcinoma | |||||||||||| |
Respiratory, thoracic, and mediastinal disorders | |||||||||||| |
Asthma | |||||||||||| |
Skin and subcutaneous tissue disorders | |||||||||||| |
Dermatitis atopic | |||||||||||| |
Patients with ≥ 1 WDAE, n (%) | 19 (1.6) |
Deaths, n (%) | NR |
AE = adverse event; NR = not reported; SAE = serious adverse event; SAS = safety analysis set; WDAE = withdrawal due to adverse event.
aAt least 2% of patients in ECZTEND.
bAt least 2% of patients in ECZTEND.
Source: Clinical Study Report for ECZTEND.73
Critical Appraisal
Internal Validity
The ECZTEND trial is a single-arm study and the lack of a comparator makes it difficult to adjust for natural changes in the course of AD or the effects of potential confounders. Additionally, the open-label design may have influenced the perception of improvement by patients and clinicians, which could affect the reporting of harms and efficacy measures. The number of patients screened from the parent trials was not reported, nor were the reasons for screening failures. Moreover, patients were recruited exclusively from the parent trials of tralokinumab and only those who could tolerate the treatments were able to enrol in the ECZTEND open-label extension. The number of patient discontinuations and some reasons for withdrawal were reported. No formal sample size or power calculations were performed and no control for multiplicity was described in the report. Blinding of treatment allocation in the parent study (tralokinumab versus placebo and dose assignment) was maintained and post-dose monitoring took place during the first 3 treatment visits as patients who received placebo in the parent trials were tralokinumab-naive. The study also used both clinician- and patient-reported outcome measures, although secondary end points included only clinician-reported outcomes. Furthermore, there is a lack of data available for patient-reported outcomes, and consequently information from the patient perspective, in the intermediate clinical study report, limiting any conclusions that can be drawn regarding the long-term use of tralokinumab. The lack of efficacy outcomes data for later time points was acknowledged by the sponsor and is largely due to patients enrolling at different times and not reaching the assessments yet. An MID of 8.7 to 24.2 points74 on the EASI for patients with moderate to severe AD was noted from the literature and the mean decrease of |||||||||||| points from the parent study baseline to week 56 of the ECZTEND trial exceeded that difference. Rescue medication was used only when deemed necessary by the investigator, although the study report did not describe if and how frequently it was used and which patients (treatment-naive or -experienced) required it during the trial. The limitations of the study design make it challenging to interpret the results and form conclusions with certainty.
External Validity
The ECZTEND study is a large, multi-centre study that has included 1,174 patients with AD so far, of whom ||||||||||||||| are Canadians. Patients range widely in age |||||||||||||||||||||||| and treatment has been for a mean duration of |||||||||||| weeks, which is expected to be an acceptable duration to assess treatment response, although it is uncertain if this is sufficient to assess the long-term safety and maintained efficacy of tralokinumab. Most patients in the study were White (71.3%), which may be a product of the regions where the study took place (mainly Europe and North America). While the clinical experts CADTH consulted on this review were uncertain if race would bias the outcomes, this factor may limit how the results can be applied to a broader patient population in Canada. Treatment history was not described in this report and it is unknown if patients were treatment-naive or which medications they have had experience with (e.g., topical, systemic, or biologic), which limits the generalizability of the results to other patients with AD. It was noted that patients and/or caregivers received training for the first 3 site visits to continue with at-home administration. No data on adherence were reported and it is unknown if tralokinumab administered subcutaneously is acceptable to patients. Input from patient groups submitted to CADTH for this review was mixed. Some patients indicated they would prefer an injectable medication while others preferred noninjectable treatments. The clinical experts consulted for this review noted a similar mix of opinions, and they expected that some patients would prefer a less-frequent subcutaneous medication to a daily, oral pill, while other patients who fear needles would not. It was noted that patients would be under the supervision of a dermatologist or allergist at least every 8 weeks during the treatment phase, which the sponsor acknowledged would be more frequent than standard clinical practice. It is possible that increased access and attention from health care professionals could affect the measurement of treatment outcomes, particularly subjective outcomes. The secondary outcomes for efficacy were IGA and EASI assessments which, in combination with the patient-reported outcomes, appear to reflect patient interests. The use of TCS or TCIs was optional, which, according to the sponsor, aligns with the intended commercial use of tralokinumab. The limitations noted here are important considerations when generalizing the study results to a broader Canadian population with AD.
Discussion
Summary of Available Evidence
Four sponsor-funded, phase III, double-blind, placebo-controlled RCTs (ECZTRA 1, ECZTRA 2, ECZTRA 3, and ECZTRA 7), which featured adult populations with moderate to severe AD whose disease is not adequately controlled with topical prescription therapies or for whom those therapies are not advisable, were included in this review. Each study had a screening and/or washout phase of 2 to 6 weeks, during which patients were expected to use an emollient twice daily (or more often, as needed) for at least 14 days before randomization and were to continue this treatment throughout the trial. The randomization method across trials consisted of assigning patients the lowest randomization number (a 5-digit number), and a central interactive web response system and interactive voice response system was used to control the randomization and stratification factors. The primary end points for each study were the number of patients who achieved an IGA score of 0 or 1 and an EASI-75 score, with the exception of the ECZTRA 7 study, which only assessed the number of patients achieving an EASI-75 score as the primary end point. The following is a brief description of each trial:
ECZTRA 1 (N = 802) and ECZTRA 2 (N = 794) were randomized, double-blind, placebo-controlled, identically designed 52-week trials that evaluated the efficacy and safety of tralokinumab as a monotherapy compared to placebo in adults with moderate to severe AD. Patients were randomized in the initial treatment phase in a 3:1 ratio to either the biweekly 300 mg tralokinumab injections (following a baseline 600 mg loading dose on day 0) or to placebo administered every 2 weeks. At week 16, patients who achieved a clinical response (defined as an IGA score of 0 or 1 or an EASI-75 score) and were assigned to the tralokinumab group in the initial treatment phase were re-randomized in a 2:2:1 ratio to biweekly 300 mg tralokinumab injections, alternating biweekly doses of placebo and 300 mg tralokinumab injections, or placebo.
ECZTRA 3 (N = 380) was a randomized, double-blind, placebo-controlled, 32-week trial that evaluated the efficacy and safety of tralokinumab as a combination therapy with TCS compared to placebo plus TCS in adults with moderate to severe AD. All patients were to use an emollient twice daily (or more often, as needed) for at least 14 days before randomization and were to continue this treatment throughout the trial. The trial had a 16-week initial treatment period followed by an additional 16-week continuation period. On day 0 of the initial treatment period, patients received a loading dose of 600 mg tralokinumab or placebo. In the initial treatment period, 380 patients were randomized in a 2:1 ratio to receive subcutaneous doses of tralokinumab or placebo every second week during the 16-week initial treatment period. Patients randomized to tralokinumab in the initial treatment period who had a clinical response (defined as an IGA score of 0 or 1 or an EASI-75 score) at week 16 were re-randomized into the continuation treatment period in a 1:1 ratio to tralokinumab 300 mg every 2 weeks, or tralokinumab 300 mg every 4 weeks (alternating dose administrations of tralokinumab 300 mg and placebo).
ECZTRA 7 (N = 277) was a randomized, double-blind, placebo-controlled, 26-week trial that evaluated the efficacy and safety of tralokinumab as a combination therapy with TCS compared to placebo plus TCS in adults with severe AD who were not adequately controlled with or have contraindications to oral cyclosporine A. Patients were randomized in a 1:1 ratio to receive tralokinumab 300 mg plus TCS or placebo plus TCS.
For 3 studies (ECZTRA 1, ECZTRA 2, and ECZTRA 3) the 2- to 6-week washout period may have been long enough to exacerbate AD, leading to patients being labelled as “nonresponders” early in the studies, particularly as the patient population has significant disease and high levels of prior medication use. Because the 3 studies only allowed patients who achieved a clinical response at week 16 to continue into the maintenance treatment phase, the outcomes in the maintenance phase need to be interpreted with caution. In a related issue, the sample sizes in the maintenance phase were too small to make any conclusions regarding the long-term efficacy and safety of tralokinumab. The lack of a study that compared tralokinumab with a drug with a similar mechanism of action means there is no direct comparison regarding the safety and efficacy of tralokinumab versus a similar drug currently available to patients.
Two ITCs, a MAIC9 submitted by the sponsor and an NMA published by the ICER,10 were summarized and critically appraised. The ICER performed an NMA to compare JAK inhibitors (abrocitinib, upadicitinib, and baricitinb) with tralokinumab and dupilumab in patients with moderate to severe AD. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
There is 1 ongoing, open-label, single-arm, long-term extension study (ECZTEND) of the safety and efficacy of tralokinumab in patients with AD who have previously participated in clinical trials for tralokinumab. Overall, ||||||| patients were still participating at the time of the data cut-off.
Interpretation of Results
Efficacy
In the ECZTRA 1, ECZTRA 2, and ECZTRA 3 trials, tralokinumab elicited a statistically significant improvement in markers of AD severity, such as IGA, EASI, and SCORAD, at 16 weeks in adults with moderate to severe AD. However, it is important to note that, for the coprimary end points, the percentage of responders achieving an IGA score of 0 or 1 and an EASI-75 score is higher at week 16 when tralokinumab is combined with TCS, as was the case in the ECZTRA 3 trial. After 52 weeks in the ECZTRA 1 and ECZTRA 2 trials, the percentage of responders achieving an IGA score of 0 or 1 is higher when tralokinumab is used every 2 weeks compared to every 4 weeks, and the same is true after 32 weeks (continuation treatment phase) in the ECZTRA 3 trial. The same pattern is true for EASI-75, which suggests that tralokinumab may be a slower-acting drug that requires more than a 16-week period to significantly improve AD severity from a clinical standpoint. Furthermore, based on ECZTRA 3 trial data, tralokinumab used in combination with TCS may provide superior results compared to using tralokinumab as a monotherapy. The measurements used in the testing to assess the severity and extent of AD (IGA and EASI-75) do not have an MID that has been identified in AD patients. This is important, considering the 75% cut-off for EASI-75 is rather subjective.
The sponsor’s reimbursement request is for the treatment of adult patients with moderate to severe AD whose disease is not adequately controlled with topical prescription therapies or for whom those therapies are not advisable and who had an adequate trial or were ineligible for each of the following therapies: phototherapy (where available), methotrexate, and cyclosporine. The ECZTRA 7 trial evaluated the efficacy and safety of tralokinumab as a combination therapy with TCS compared to placebo plus TCS in adults with severe AD who are not adequately controlled with or have contraindications to oral cyclosporine A. Results from the ECZTRA 7 trial demonstrated that statistically significantly more patients in the tralokinumab treatment group achieved an EASI-75 in comparison with the placebo group. However, when the worst daily pruritus NRS outcome was assessed, no statistical significance was reported and, given that the worst daily pruritus NRS outcome was tested first in the hierarchy after EASI-75, it is unknown whether tralokinumab would demonstrate a statistically significant result in comparison to placebo for other efficacy outcomes such as an IGA score of 0 or 1.
The sponsor-submitted MAIC was ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
A summary and critical appraisal of an NMA conducted by ICER was also undertaken. Results from the NMA showed that tralokinumab was generally superior to placebo and inferior to upadacitinib (both 15 mg and 30 mg), abrocitinib 200 mg, and dupilumab 300 mg. These results were consistent when these treatments were used as monotherapies or in combination with topical therapies.
The ongoing, long-term extension study (ECZTEND) to provide additional evidence on the safety and efficacy of tralokinumab in patients with AD who have previously participated in clinical trials for tralokinumab (i.e., ECZTRA 1 to 8 and TraSki) has shown |||||||||||| of responders achieving an IGA score of 0 or 1, and |||||||| achieving an EASI-75 score at week 16. These percentages are higher than in the parent trials, but they do not improve at week 56 (|||||| of responders achieved an IGA score of 0 or 1 and ||||||| of responders achieve an EASI-75 score). However, these percentages may be a result of patients being recruited exclusively from the participants who could tolerate the treatments in parent trials of tralokinumab. Results were not controlled for multiplicity and there was no imputation of missing safety data. It also unknown if patients in the ECZTEND trial had a history with other AD treatments, such as TCS or biologics.
Harms
The most common AE was dermatitis atopic and viral upper respiratory tract infection. For example, 25.9% (n = 156) and 16.6% (n = 98) of patients in the tralokinumab group in ECZTRA 1 and ECZTRA 2, respectively, had dermatitis atopic. Viral upper respiratory tract infections were reported in 23.1% (n = 139), 19.4% (n = 49), and 26.8% (n = 37) of patients in the tralokinumab group in the ECZTRA 1, ECZTRA 3, and ECZTRA 7 trials, respectively.
Serious adverse events at week 16 were reported in 3.8% (n = 23), 1.7% (n = 10), 0.8% (n = 2), and 0.7% (n = 1) of patients receiving tralokinumab in ECZTRA 1, ECZTRA 2, ECZTRA 3, and ECZTRA 7, respectively. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In the ECZTEND trial, 844 patients (71.9%) experienced at least 1 AE, with the 3 most common AEs being viral upper respiratory tract infection (21.3%), dermatitis atopic (13.5%), and upper respiratory tract infection (7.1%). Other harms of special interest that were identified in CADTH’s systematic review protocol include conjunctivitis (3.8%), injection-site reactions (2.4%), conjunctivitis allergic (2.0%),|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Nineteen patients (1.6%) withdrew due to AEs and no deaths were reported. Overall, conclusions cannot be drawn regarding the long-term safety of tralokinumab due to the limitations of the design of the ECZTRA trials, which allowed only patients with a clinical response into the maintenance phase of the studies, leading to sample sizes that were too small to draw conclusions about safety with confidence.
Conclusions
Three double-blind RCTs demonstrated that, compared with placebo, 16 weeks of treatment with tralokinumab was associated with statistically significant improvements in a range of outcomes that are important in the management of AD in adults, including overall severity of AD (EASI, IGA response, and SCORAD), symptoms (pruritus), and HRQoL (DLQI). These trials included the use of tralokinumab as monotherapy (ECZTRA 1 [N = 802] and ECZTRA 2 [N = 794]) and as combination therapy (ECZTRA 3 [N = 380]). The ECZTRA 7 trial, which evaluated the efficacy and safety of tralokinumab as a combination therapy with TCS compared to placebo plus TCS in adults with severe AD who are not adequately controlled with or have contraindications to oral cyclosporine A, only demonstrated statistically significant improvement in EASI scores in the tralokinumab group compared to the placebo group. Tralokinumab was well-tolerated in the short term (16 weeks) and long-term (32 or 52 weeks) phase III studies. There were no direct comparisons between tralokinumab and other active AD treatments.
Results from a sponsor-submitted MAIC ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Results from the NMA conducted by the ICER suggest that tralokinumab may be inferior to dupilumab in terms of most efficacy outcomes. However, the MAIC comparisons were ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. There is also uncertainty regarding the results from the NMA conducted by the ICER due to the inherent heterogeneity across trials in the networks. The robustness of the comparative efficacy was further compromised by a lack of precision in some of the findings, and results from the indirect comparisons must be interpreted with caution.
References
1.Pfizer. Eucrisa (crisaborole) ointment, 2% for topical use [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2021: https://pdf.hres.ca/dpd_pm/00060960.PDF.
2.CADTH Drug Reimbursement Expert Review Committee final recommendation: crisaborole (eucrisa); Pfizer Canada Inc. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/cdr/complete/SR0570-Eucrisa-cdec-rec-april-2-2019.pdf.
3.CADTH Drug Reimbursement Expert Review Committee final recommendation: dupilumab (Dupixent); Sanofi Genzyme, a division of sanofi-aventis Canada Inc. Ottawa (ON): CADTH; 2020: https://www.cadth.ca/sites/default/files/cdr/complete/SR0636%20Dupixent%20-%20CDEC%20Final%20%20Recommendation%20April%2024%2C%202020%20for%20posting.pdf.
4.Clinical Study Report: LP0162-1346 Tralokinumab in combination with topical corticosteroids in subjects with severe atopic dermatitis who are not adequately controlled with or have contraindications to oral cyclosporine A ECZTRA 7 (ECZema TRAlokinumab trial no. 7) [internal sponsor's report]. City (PROV): Leo Pharma Inc.; 2021.
5.Clinical Study Report: LP0162-1326 Tralokinumab monotherapy for moderate to severe atopic dermatitis ECZTRA 2 (ECZema TRAlokinumab trial no. 2) [internal sponsor's report]. City (PROV): LEO Pharma Inc.; 2020.
6.Clinical Study Report: LP0162-1339 Tralokinumab in combination with topical corticosteroids for moderate to severe atopic dermatitis ECZTRA 3 (ECZema TRAlokinumab trial no. 3) [internal sponsor's report]. City (PROV): Leo Pharma Inc.; 2020.
7.Clinical Study Report: LP0162-1325 Tralokinumab monotherapy for moderate to severe atopic dermatitis ECZTRA 1 (ECZema TRAlokinumab trial no. 1) [internal sponsor's report]. City (PROV): LEO Pharma Inc.; 2020.
8.Wollenberg A, Blauvelt A, Guttman-Yassky E, et al. Tralokinumab for moderate to severe atopic dermatitis: results from two 52-week, randomized, double-blind, multicentre, placebo-controlled phase III trials (ECZTRA 1 and ECZTRA 2). Br J Dermatol. 2021;184(3):437-449. PubMed
9.Leo Pharma. Matching-Adjusted Indirect Comparisons of Tralokinumab vs. Dupilumab based on ECZTRA 7 and LIBERTY AD CAFÉ data for Atopic Dermatitis. 2021.
10.Atlas SJ, Brouwer E, Fox G, Carlson JJ, Campbell JD, Agboola F, Hansen RN, Pearson SD, Rind DM. JAK Inhibitors and Monoclonal Antibodies for the Treatment of Atopic Dermatitis: Effectiveness and Value; Evidence Report. Institute for Clinical and Economic Review, July 9, 2021. https://icer.org/assessment/atopic-dermatitis-2021/#timeline.
11.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. Methods for Population-Adjusted Indirect Comparisons in Health Technology Appraisal. Med Decis Making. 2018;38(2):200-211. PubMed
12.Dupixent (dupilumab): Solution for subcutaneous injection, 300 mg single-use syringe, 300 mg single-use pen, 200 mg single-use syringe [product monograph]. Laval (QC): Sanofi-aventis Canada Inc.; 2021.
13.Imuran (Azathioprine): Tablets USP, 50 mg, oral [product monograph]. Oakville (ON): Aspen Pharmacare Canada Inc; 2020.
14.CellCept (mycophenolate mofetil): Capsules 250 mg, Film-coated tablets 500 mg, oral [product monograph]. Mississauga (ON): Hoffmann-La Roche Ltd; 2021.
15.Cyclosporine Capsules (cyclosporine) Soft gelatin capsules, 25 mg, 50 mg, 100 mg, oral [product monograph]. Varennes (QC): Strides Pharma Canada Inc; 2020.
16.Methotrexate tablets (methotrexate): Tablets USP, 10 mg, oral [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2019.
17.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
18.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 May 11.
19.Silverberg JI, Toth D, Bieber T, et al. Tralokinumab plus topical corticosteroids for the treatment of moderate to severe atopic dermatitis: results from the double-blind, randomized, multicentre, placebo-controlled phase III ECZTRA 3 trial. Br J Dermatol. 2021;184(3):450-463. PubMed
20.Futamura M, Leshem YA, Thomas KS, Nankervis H, Williams HC, Simpson EL. A systematic review of Investigator Global Assessment (IGA) in atopic dermatitis (AD) trials: Many options, no standards. J Am Acad Dermatol. 2016;74(2):288-294. PubMed
21.Bozek A, Reich A. Assessment of Intra- and Inter-Rater Reliability of Three Methods for Measuring Atopic Dermatitis Severity: EASI, Objective SCORAD, and IGA. Dermatology. 2017;233(1):16-22. PubMed
22.Zhao CY, Hao EY, Oh DD, et al. A comparison study of clinician-rated atopic dermatitis outcome measures for intermediate- to dark-skinned patients. Br J Dermatol. 2017;176(4):985-992. PubMed
23.Schmitt J, Langan S, Deckert S, et al. Assessment of clinical signs of atopic dermatitis: a systematic review and recommendation. J Allergy Clin Immunol. 2013;132(6):1337-1347. PubMed
24.Hanifin JM, Thurston M, Omoto M, Cherill R, Tofte SJ, Graeber M. The eczema area and severity index (EASI): assessment of reliability in atopic dermatitis. EASI Evaluator Group. Exp Dermatol. 2001;10(1):11-18. PubMed
25.Leshem YA, Hajar T, Hanifin JM, Simpson EL. What the Eczema Area and Severity Index score tells us about the severity of atopic dermatitis: an interpretability study. Br J Dermatol. 2015;172(5):1353-1357. PubMed
26.Badia-Tahull M, Cobo-Sacristan S, Leiva-Badosa E, et al. Use of Subjective Global Assessment, Patient-Generated Subjective Global Assessment and Nutritional Risk Screening 2002 to evaluate the nutritional status of non-critically ill patients on parenteral nutrition. Nutr Hosp. 2014;29(2):411-419. PubMed
27.Barbier N, Paul C, Luger T, et al. Validation of the Eczema Area and Severity Index for atopic dermatitis in a cohort of 1550 patients from the pimecrolimus cream 1% randomized controlled clinical trials programme. Br J Dermatol. 2004;150(1):96-102. PubMed
28.Hanifin J, Thurston M, Omoto M, Cherill R, Tofte S, Graeber M. The eczema area and severity index (EASI): assessment of reliability in atopic dermatitis. EASI Evaluator Group. Exp Dermatol. 2001;10(1):11-18. PubMed
29.Schmitt J, Langan S, Deckert S, et al. Assessment of clinical signs of atopic dermatitis: a systematic review and recommendation J Allergy Clin Immunol. 2013;132(6):1337-1347. PubMed
30.Schmitt J, Langan S, Williams HC, European Dermato-Epidemiology N. What are the best outcome measurements for atopic eczema? A systematic review. J Allergy Clin Immunol. 2007;120(6):1389-1398. PubMed
31.Schram M, Spuls P, Leeflang M, Lindeboom R, Bos J, Schmitt J. EASI, (objective) SCORAD and POEM for atopic eczema: responsiveness and minimal clinically important difference Allergy. 2012;67(1):99-106. PubMed
32.Schram ME, Spuls PI, Leeflang MM, Lindeboom R, Bos JD, Schmitt J. EASI, (objective) SCORAD and POEM for atopic eczema: responsiveness and minimal clinically important difference. Allergy. 2012;67(1):99-106. PubMed
33.Alexander H, Patton T, Jabbar-Lopez ZK, Manca A, Flohr C. Novel systemic therapies in atopic dermatitis: what do we need to fulfil the promise of a treatment revolution? F1000Research. 1000;8. PubMed
34.Chalmers JR, Schmitt J, Apfelbacher C, et al. Report from the third international consensus meeting to harmonise core outcome measures for atopic eczema/dermatitis clinical trials (HOME). Br J Dermatol. 2014;171(6):1318-1325. PubMed
35.Rehal B, Armstrong AW. Health outcome measures in atopic dermatitis: a systematic review of trends in disease severity and quality-of-life instruments 1985-2010. PLoS ONE [Electronic Resource]. 2011;6(4):e17520. PubMed
36.Simpson E, Beck L, Gadkari A, Eckert L, Reaney M. Defining a responder on the Peak Pruritus Numerical Rating Scale (NRS) in patients with moderate to severe atopic dermatitis: detailed analysis from randomized trials of dupilumab J Am Acad Dermatol. 2017;76(6 Suppl 1):AB93.
37.Yosipovitch G, Reaney M, Mastey V, et al. Peak Pruritus Numerical Rating Scale: psychometric validation and responder definition for assessing itch in moderate to severe atopic dermatitis. Br J Dermatol. 2019;181(4):761-769. PubMed
38.Shikiar R, Harding G, Leahy M, Lennox RD. Minimal important difference (MID) of the Dermatology Life Quality Index (DLQI): results from patients with chronic idiopathic urticaria. Health Qual Life Outcomes. 2005;3:36. PubMed
39.Finlay AY, Khan GK. Dermatology Life Quality Index (DLQI)--a simple practical measure for routine clinical use. Clin Exp Dermatol. 1994;19(3):210-216. PubMed
40.Basra MK, Salek MS, Camilleri L, Sturkey R, Finlay AY. Determining the minimal clinically important difference and responsiveness of the Dermatology Life Quality Index (DLQI): further data. Dermatology. 2015;230(1):27-33. PubMed
41.Lewis V, Finlay A. 10 years experience of the Dermatology Life Quality Index (DLQI). J Investig Dermatol Symp Proc. 2004;9(2):169-180. PubMed
42.Rehal B, Armstrong A. Health outcome measures in atopic dermatitis: a systematic review of trends in disease severity and quality-of-life instruments 1985-2010. PLoS One. 2011;6(4):e17520. PubMed
43.Reilly M, Lavin P, Kahler K, Pariser D. Validation of the Dermatology Life Quality Index and the Work Productivity and Activity Impairment-Chronic Hand Dermatitis questionnaire in chronic hand dermatitis. J Am Acad Dermatol. 2003;48(1):128-130. PubMed
44.Wallenhammar L, Nyfjall M, Lindberg M, Meding B. Health-related quality of life and hand eczema--a comparison of two instruments, including factor analysis. J Invest Dermatol. 2004;122(6):1381-1389. PubMed
45.Patel KR, Singam V, Vakharia PP, et al. Measurement properties of three assessments of burden used in atopic dermatitis in adults. Br J Dermatol. 2019;180(5):1083-1089. PubMed
46.Schwartzman G, Lei D, Yousaf M, et al. Validity and reliability of Patient-Reported Outcomes Measurement Information System Global Health scale in adults with atopic dermatitis. J Am Acad Dermatol. 2021;20:20. PubMed
47.Holm EA, Wulf HC, Stegmann H, Jemec GB. Life quality assessment among patients with atopic eczema. Br J Dermatol. 2006;154(4):719-725. PubMed
48.Basra M, Salek M, Camilleri L, Sturkey R, Finlay A. Determining the minimal clinically important difference and responsiveness of the Dermatology Life Quality Index (DLQI): further data. Dermatology. 2015;230(1):27-33. PubMed
49.Bjelland I, Dahl AA, Haug TT, Neckelmann D. The validity of the Hospital Anxiety and Depression Scale. An updated literature review. J Psychosom Res. 2002;52(2):69-77. PubMed
50.Herrmann C. International experiences with the Hospital Anxiety and Depression Scale--a review of validation data and clinical results. J Psychosom Res. 1997;42(1):17-41. PubMed
51.Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand. 1983;67(6):361-370. PubMed
52.Silverberg JI, Gelfand JM, Margolis DJ, et al. Measurement Properties of the Hospital Anxiety and Depression Scale Used in Atopic Dermatitis in Adults. J Invest Dermatol. 2019;139(6):1388-1391. PubMed
53.Charman CR, Venn AJ, Williams HC. The patient-oriented eczema measure: development and initial validation of a new tool for measuring atopic eczema severity from the patients' perspective. Arch Dermatol. 2004;140(12):1513-1519. PubMed
54.Eichenfield L, Tom W, Chamlin S, et al. Guidelines of care for the management of atopic dermatitis: section 1. Diagnosis and assessment of atopic dermatitis J Am Acad Dermatol. 2014;70(2):338-351. PubMed
55.Schmitt J, Spuls PI, Thomas KS, et al. The Harmonising Outcome Measures for Eczema (HOME) statement to assess clinical signs of atopic eczema in trials. J Allergy Clin Immunol. 2014;134(4):800-807. PubMed
56.Silverberg JI, Margolis DJ, Boguniewicz M, et al. Validation of five patient-reported outcomes for atopic dermatitis severity in adults. Br J Dermatol. 2020;182(1):104-111. PubMed
57.Silverberg JI, Lei D, Yousaf M, et al. Comparison of Patient-Oriented Eczema Measure and Patient-Oriented Scoring Atopic Dermatitis vs. Eczema Area and Severity Index and other measures of atopic dermatitis: A validation study. Ann Allergy Asthma Immunol. 2020;125(1):78-83. PubMed
58.Ware JE, Jr., Sherbourne CD. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Med Care. 1992;30(6):473-483. PubMed
59.Frendl DM, Ware JE, Jr. Patient-reported functional health and well-being outcomes with drug therapy: a systematic review of randomized trials using the SF-36 health survey. Med Care. 2014;52(5):439-445. PubMed
60.Brooks R. EuroQol: the current state of play. Health Policy. 1996;37(1):53-72. PubMed
61.EuroQol G. EuroQol--a new facility for the measurement of health-related quality of life. Health Policy. 1990;16(3):199-208. PubMed
62.Alberta HQCo. 2014 Alberta population norms for EQ-5D-5L 2014; https://d10k7k7mywg42z.cloudfront.net/assets/542f01f2edb2f37083002e54/2014_EQ_5D_5L_report_FINALFINAL.pdf.
63.M van Reenen BJ. EQ-5D-5L user guide: basic information on how to use the EQ-5D-5L instrument. 2015; https://euroqol.org/wp-content/uploads/2016/09/EQ-5D-5L_UserGuide_2015.pdf.
64.McClure NS, Sayah FA, Xie F, Luo N, Johnson JA. Instrument-Defined Estimates of the Minimally Important Difference for EQ-5D-5L Index Scores. Value Health. 2017;20(4):644-650. PubMed
65.Reilly MC, Zbrozek AS, Dukes EM. The validity and reproducibility of a work productivity and activity impairment instrument. Pharmacoeconomics. 1993;4(5):353-365. PubMed
66.Holm S. A Simple Sequentially Rejective Multiple Test Procedure. Scandinavian Journal of Statistics. 1979;6(2):65-70.
67.Wang C, Kraus CN, Patel KG, Ganesan AK, Grando SA. Real-world experience of dupilumab treatment for atopic dermatitis in adults: a retrospective analysis of patients' records. Int J Dermatol. 2020;59(2):253-256. PubMed
68.Abraham S, Haufe E, Harder I, et al. Implementation of dupilumab in routine care of atopic eczema: results from the German national registry TREATgermany. Br J Dermatol. 2020;183(2):382-384. PubMed
69.Dias S, Welton NJ, Sutton AJ, Ades AE. NICE Decision Support Unit Technical Support Documents. NICE DSU Technical Support Document 2: A Generalised Linear Modelling Framework for Pairwise and Network Meta-Analysis of Randomised Controlled Trials. London: National Institute for Health and Care Excellence (NICE). Copyright © 2014 National Institute for Health and Clinical Excellence, unless otherwise stated. All rights reserved.; 2014.
70.Signorovitch JE, Wu EQ, Yu AP, et al. Comparative effectiveness without head-to-head trials: a method for matching-adjusted indirect comparisons applied to psoriasis treatment with adalimumab or etanercept. Pharmacoeconomics. 2010;28(10):935-945. PubMed
71.clinicaltrials.gov. Long-term Extension Trial in Subjects With Atopic Dermatitis Who Participated in Previous Tralokinumab Trials - ECZTEND. https://clinicaltrials.gov/ct2/show/NCT03587805.
72.Drug Reimbursement Review sponsor submission: tralokinumab, 150 mg pre-filled syringes (150 mg/1 mL) for subcutaneous injection [internal sponsor's package]. Thornhill (ON): LEO Pharma Inc; 2021 Apr 27.
73.Clinical Study Report: LP0162-1337 Long-term extension trial in subjects with atopic dermatitis who participated in previous tralokinumab trials – ECZTEND [internal sponsor's report]. City (PROV): Leo Pharma Inc.; 2021.
74.Silverberg JI, Lei D, Yousaf M, et al. What are the best endpoints for Eczema Area and Severity Index and Scoring Atopic Dermatitis in clinical practice? A prospective observational study. Br J Dermatol. 2021;184(5):888-895. PubMed
75.Chalmers J, Schmitt J, Apfelbacher C, et al. Report from the third international consensus meeting to harmonise core outcome measures for atopic eczema/dermatitis clinical trials (HOME). Br J Dermatol. 2014;171:1318-1325. PubMed
76.Leshem Y, Hajar T, Hanifin J, Simpson E. What the Eczema Area and Severity Index score tells us about the severity of atopic dermatitis: an interpretability study. Br J Dermatol. 2015;172(5):1353-1357. PubMed
77.Severity scoring of atopic dermatitis: the SCORAD index. Consensus report of the European Task Force on Atopic Dermatitis. Dermatology. 1993;186(1):26-31. PubMed
78.Eczema H-HOMf. Core Outcomes for trials. http://www.homeforeczema.org/research/index.aspx.
79.Charman CR, Venn AJ, Williams HC. The patient-oriented eczema measure: development and initial validation of a new tool for measuring atopic eczema severity from the patients' perspective. Arch Dermatol. 2004;140(12):1513-1519. PubMed
80.Spuls PI, Gerbens LAA, Simpson E, et al. Patient-Oriented Eczema Measure (POEM), a core instrument to measure symptoms in clinical trials: a Harmonising Outcome Measures for Eczema (HOME) statement. Br J Dermatol. 2017;176(4):979-984. PubMed
81.Apfelbacher CJ, Heinl D, Prinsen CA, et al. Measurement properties of adult quality-of-life measurement instruments for eczema: protocol for a systematic review. Syst Rev. 2015;4:48. PubMed
82.EuroQol--a new facility for the measurement of health-related quality of life. Health Policy. 1990;16(3):199-208. PubMed
83.Corporation R. 36-Item Short Form Survey (SF-36). https://www.rand.org/health-care/surveys_tools/mos/36-item-short-form.html.
84.Mease PJ, Menter MA. Quality-of-life issues in psoriasis and psoriatic arthritis: outcome measures and therapies from a dermatological perspective. J Am Acad Dermatol. 2006;54(4):685-704. PubMed
85.Shikiar R, Bresnahan BW, Stone SP, Thompson C, Koo J, Revicki DA. Validity and reliability of patient reported outcomes used in psoriasis: results from two randomized clinical trials. Health Qual Life Outcomes. 2003;1:53. PubMed
86.ASSOCIATES R. WPAI General Information. 2019; http://www.reillyassociates.net/WPAI_General.html.
87.Bjelland I, Dahl A, Haug T, Neckelmann D. The validity of the Hospital Anxiety and Depression Scale. An updated literature review. J Psychosom Res. 2002;52(2):69-77. PubMed
88.Herrmann C. International experiences with the Hospital Anxiety and Depression Scale--a review of validation data and clinical results. J Psychosom Res. 1997;42(1):17-41. PubMed
89.Zigmond A, Snaith R. The hospital anxiety and depression scale Acta Psychiatr Scand. 1983;67(6):361-370. PubMed
90.Silverberg JI, Gelfand JM, Margolis DJ, et al. Measurement properties of the Hospital Anxiety and Depression Scale used in atopic dermatitis in adults. J Invest Dermatol. 2019;139(6):1388-1391. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946–)
Embase (1974–)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: May 26, 2021
Alerts: Biweekly search updates until project completion
Study types: No filters were applied to limit the retrieval by study type.
Limits:
No date or language limits were used
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase) |
.dq | Candidate term word (Embase) |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
((tralokinumab* or Adtralza* or cat 354 or cat354 or GK1LYB375A).ti,ab,kf,ot,hw,rn,nm
1 use medall
*tralokinumab/
(tralokinumab* or Adtralza* or cat 354 or cat354).ti,ab,kw,dq.
3 or 4
5 use oemezd
6 not (conference review or conference abstract).pt.
2 or 7
remove duplicates from 8
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- Studies with results | tralokinumab OR Adtralza]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms -- tralokinumab OR Adtralza]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- tralokinumab OR Adtralza]
Grey Literature
Search dates: May 14 to May 19, 2021
Keywords: tralokinumab, Adtralza, atopic dermatitis
Limits: None
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search.
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
Gonçalves F, Freitas E, Torres T. Selective IL-13 inhibitors for the treatment of atopic dermatitis. Drugs in Context. 2021;10. | Study design: narrative review |
Panettieri Jr RA, Sjöbring U, Péterffy A, Wessman P, Bowen K, Piper E, Colice G, Brightling CE. Tralokinumab for severe, uncontrolled asthma (STRATOS 1 and STRATOS 2): two randomised, double-blind, placebo-controlled, phase 3 clinical trials. The Lancet Respiratory medicine. 2018 Jul 1;6(7):511-25. | Study population: participants aged 12–75 years with severe asthma that was inadequately controlled despite use of inhaled corticosteroids |
Sawangjit R, Dilokthornsakul P, Lloyd-Lavery A, Lai NM, Dellavalle R, Chaiyakunapruk N. Systemic treatments for eczema: a network meta‐analysis. Cochrane Database of Systematic Reviews. 2020(9). | Study design: systematic review |
Zhang Y, Cheng J, Li Y, He R, Pan P, Su X, Hu C. The safety and efficacy of anti–IL-13 treatment with tralokinumab (CAT-354) in moderate to severe asthma: a systematic review and meta-analysis. The Journal of Allergy and Clinical Immunology: In Practice. 2019 Nov 1;7(8):2661-71. | Study population: patients with moderate to severe asthma |
Appendix 3: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
Investigator’s Global Assessment (IGA)
Eczema Area and Severity Index (EASI)
Scoring Atopic Dermatitis (SCORAD)
Numeric rating scale (NRS)
Pruritus NRS (e.g., worst daily and average daily)
Patient Global Impression of Severity (PGI-S)
Patient-Oriented Eczema Measure (POEM)
Dermatology Life Quality Index (DLQI)
EG-5D 5-Levels questionnaire (EQ-5D-5L)
Short Form (36) Health Survey version 2 (SF-36 v2)
Work Productivity and Activity Impairment–General Health questionnaire (WPAI-GH)
Hospital Anxiety and Depression Scale (HADS).
Findings
Table 14 lists the outcome measures that were used in each of the pivotal studies and Table 46 summarizes the outcome measures, measurement properties, and MIDs of those that were included in the CADTH systematic review protocol. Instrument versions and method of information capture appeared to be consistent across all relevant studies.
Table 46: Summary of Outcome Measures and Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
IGA | A 5-point scale that provides a global clinical assessment of AD severity by the investigator. A decrease in score indicates an improvement in symptoms. | Validity: Adequate in patients with AD. Reliability: Adequate in patients with AD. Responsiveness: No evidence found for patients with AD. | Not identified in populations with AD. |
EASI | A scale used in clinical trials to assess the severity and extent of AD for 4 disease characteristics on 4 regions of the body. A decrease in score indicates an improvement in symptoms. | Validity: Adequate in patients with AD. Reliability: Adequate in patients with AD. Responsiveness: Adequate in patients with AD. | 6.6 points, but can range from 1.1 to 24.6 points depending on the anchor used, how improvement is defined, and severity of AD. |
SCORAD | A tool used in clinical research to evaluate the extent and intensity of AD by assessing 6 investigator-reported symptoms and 2 patient-reported symptoms. A decrease in score indicates an improvement in symptoms. | Validity: Adequate in patients with AD. Reliability: Adequate in patients with AD. Responsiveness: Adequate in patients with AD. | 8.7 points, but can range from 2.7 to 29.2 points depending on the anchor used, how improvement is defined, and severity of AD. |
Eczema-related sleep NRS | A single-item, 11-point scale allowing patients to rate how AD has affected their sleep. A decrease in score indicates an improvement in symptoms. | Validity: No evidence found for patients with AD. Reliability: No evidence found for patients with AD. Responsiveness: No evidence found for patients with AD. | Not identified in populations with AD. |
Pruritus NRS | A single-item, 11-point scale allowing patients to rate the severity of itch due to AD. A decrease in score indicates an improvement in symptoms. | Validity: Adequate in patients with AD. Reliability: Adequate in patients with AD. Responsiveness: Moderate to adequate in patients with AD. | 2.2 to 4 points, may depend on anchor used. |
PGI-S | A single-item, 4-point scale allowing patients to rate their perception of their overall AD symptom severity during the last 24 hours. A decrease in score indicates an improvement in symptoms. | Validity: No evidence found for patients with AD. Reliability: No evidence found for patients with AD. Responsiveness: No evidence found for patients with AD. | Not identified in populations with AD. |
POEM | A 7-item questionnaire used in clinical trials to assess disease symptoms in patients with AD by assessing 7 symptoms and how frequently each was experienced in the past week. A decrease in score indicates an improvement in symptoms. | Validity: Adequate in patients with AD. Reliability: Adequate in patients with AD. Responsiveness: Adequate in patients with AD. | 3.4 to 6.1 points, may depend on anchor used and severity of AD. |
DLQI | A 10-item questionnaire allowing patients to rate their perception of how their dermatological condition has affected their QoL. A decrease in score indicates an improvement in symptoms. | Validity: Moderate to adequate in patients with AD. Reliability: Moderate to adequate in patients with AD. Responsiveness: Moderate to adequate in patients with AD. | 2 to 7 points in populations with a variety of skin condition. Not identified in populations with AD. |
EQ-5D-5L | A patient-reported, generic, QoL instrument that has been applied to a wide range of health conditions and treatments. A decrease in score indicates a decrease in QoL. | Validity: Adequate in diverse patient populations; no evidence found for patients with AD. Reliability: No evidence found for patients with AD. Responsiveness: No evidence found for patients with AD. | 0.056 points for the Canadian population. Not identified in populations with AD. |
SF-36 v2 | A patient-reported, 36-item, generic QoL questionnaire that has been applied to a wide range of health conditions and treatments. A decrease in score indicates a decrease in QoL. | Validity: Adequate in patients with psoriasis; no evidence found for patients with AD. Reliability: No evidence found for patients with AD. Responsiveness: Not sensitive in patients with AD. | 2.6 to 3.9 points for the PCS and 3.9 to 6.0 points for the MCS in populations with moderate to severe plaque psoriasis. Not identified in populations with AD. |
WPAI-GH | A patient-reported questionnaire measuring impairments on work productivity and daily activities due to generic health. A decrease in score indicates an improvement. | Validity: Adequate in general, employed populations; no evidence found for patients with AD. Reliability: Adequate in general, employed populations; no evidence found for patients with AD. Responsiveness: No evidence found for patients with AD. | Not identified in populations with AD. |
HADS | A patient-reported, 14-item questionnaire designed to identify anxiety disorders and depression in patients at nonpsychiatric medical institutions. A decrease in score indicates an improvement in symptoms. | Validity: Adequate in diverse patient populations; no evidence found for patients with AD. Reliability: Assessed in diverse patient populations; no evidence found for patients with AD. Responsiveness: Assessed in diverse patient populations; no evidence found for patients with AD. | Not identified in populations with AD. |
AD = atopic dermatitis; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EQ-5D-5L = EuroQol 5-dimension 5-level; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; MCS = mental component summary; MID = minimal important difference; NRS = numeric scale; PCS = physical component summary; PGI-S = Patient Global Impression of Severity; POEM = Patient-Oriented Eczema Measure; QoL = quality of life; SCORAD = Scoring Atopic Dermatitis; SF-36 v2 = Short Form (36) Health Survey version 2; WPAI-GH = Work Productivity and Activity Impairment–General Health.
Investigator’s Global Assessment
The IGA is a 5-point scale used to rate a patient’s global AD severity ranging from 0 to 4 as outlined in Table 47 where a decrease in score indicates an improvement in signs and symptoms.4-7,54 In the ECZTRA studies, assessments were based on the patient’s condition at the time of observation only and not compared to the condition at an earlier visit.4-7
Table 47: Investigator's Global Assessment Scale
Score | Grade | Standard IGA scale | IGA morphological descriptors |
|---|---|---|---|
0 | Clear | No inflammatory signs of atopic dermatitis | No erythema and no elevation (papulation/infiltration). |
1 | Almost clear | Just perceptible erythema and just perceptible papulation/infiltration | Barely perceptible erythema and/or minimal lesion elevation (papulation/infiltration) that is not widespread. |
2 | Mild disease | Mild erythema and mild papulation/infiltration | Visibly detectable, light pink erythema and very slight elevation (papulation/infiltration). |
3 | Moderate disease | Moderate erythema and moderate papulation/infiltration | Dull red, clearly distinguishable erythema and clearly perceptible but not extensive elevation (papulation/infiltration). |
4 | Severe disease | Severe erythema and severe papulation/infiltration | Deep, dark red erythema, marked and extensive elevation (papulation/infiltration). |
IGA = Investigator’s Global Assessment.
Source: Clinical Study Report for ECZTRA 1.7
A study by Bożek et al. (2016) investigated the validity and reliability of the IGA, SCORAD, and EASI.21 Ten dermatologists assessed 10 adult patients with varying severities of AD twice in the same day. Pearson correlation coefficients were calculated for the first and second assessments of different instruments and all were found to be between 0.66 and 0.87 indicating strong correlations and adequate validity. The weakest correlation was between the first IGA and second EASI measurements while the strongest was between the first EASI and second SCORAD measurements. Overall, reliability was adequate for the IGA measurements with the exception of one patient who had a statistically significant difference between the 2 assessments. The results were similar with the SCORAD: generally reliable with a statistically significant difference for one patient though this was a different individual from the IGA. The EASI scores showed no significant differences between the 2 assessments and demonstrated adequate reliability. Intra-rater reliability, measured by the intraclass correlation coefficient, was fair (0.54 ± 0.28) for the IGA and good for the overall SCORAD (0.66 ± 0.2) and EASI (0.71 ± 0.21) scores.21 The inter-rater reliability variability, measured by the coefficient of variation, was moderate for the IGA (33.0 ± 12.3) and overall SCORAD score (28.1 ± 9.4) and high for the overall EASI score (66.5 ± 26.9).21 Inter-rater reliability of the IGA was also assessed in a study that separated patients into 2 groups, those with lighter skin (melanin index ≤200; n = 11) and darker skin (melanin index >200; n = 14), and was found to be adequate (intraclass correlation coefficient = 0.80 and 0.70, respectively). No literature was found that assessed the IGA for responsiveness in patients with AD.
No MID was identified in patients with AD.
Eczema Area and Severity Index
The EASI is a physician-administered scale used in clinical trials and clinical practice to assess the severity and extent of AD.4-7 In the ECZTRA studies, assessments were based on the patient’s condition at the time of observation only and not compared to the condition at an earlier visit. The EASI has been recommended by the Harmonising Outcome Measures for Eczema as the core outcome measure for the clinical signs of eczema in clinical trials.55,75
Table 48 outlines how scoring is performed with the EASI. Essentially, 4 disease characteristics of AD (erythema, infiltration/papulation, excoriation, and lichenification) are given a severity score (SS) by the investigator on a scale from 0 (none/absent) to 3 (severe) where half points (i.e., 0.5, 1.5, 2.5) may also be used.4-7 The SS are added up for each of the 4 body regions (head/neck, trunk, upper extremities, and lower extremities). The summed SS for each region is multiplied by an area score (AS) that represents the amount of body surface area that is affected by AD and ranges from 0 to 6 (0 = 0% affected area, 1 = 1% to 9%, 2 = 10% to 29%, 3 = 30% to 49%, 4 = 50% to 69%, 5 = 70% to 89%, and 6 = 90% to 100%). A weighting factor is assigned to each region of the body (10% for head/neck, 30% for trunk, 20% for upper extremities, and 40% for lower extremities) and the summed SS for each region is multiplied by the corresponding weighting factor. The final EASI score is the sum of the scores for the 4 body regions and ranges from 0 to 72 where a higher number indicates more severe AD. It has been suggested that the severity of AD be based on EASI scores as follows: 0 = clear; 0.1 to 1.0 = almost clear; 1.1 to 7.0 = mild; 7.1 to 21.0 = moderate; 21.1 to 50.0 = severe; 50.1 to 72.0 = very severe.76
Table 48: Eczema Area and Severity Index Calculation
Body region | Erythema | Induration/ papulation | Excoriation | Lichenification | Area score | Weighting factor | Score |
|---|---|---|---|---|---|---|---|
Head/neck | (SS + | SS + | SS + | SS) | x AS | x 0.1 | — |
Trunk | (SS + | SS + | SS + | SS) | x AS | x 0.3 | — |
Upper extremities | (SS + | SS + | SS + | SS) | x AS | x 0.2 | — |
Lower extremities | (SS + | SS + | SS + | SS) | x AS | x 0.4 | — |
Sum of scores for each of the 4 body regions (out of 72) | — | ||||||
AS = area score (see text for allowable values); SS = severity score (see text for allowable values).
Source: Clinical Study Report for ECZTRA 1.7
The psychometric properties of the EASI have been examined in several studies of patients with AD.26-31 Validity of the EASI and SCORAD was adequate with correlation coefficients ranging from 0.84 to 0.93.29 Intra- and inter-rater reliability were estimated to be adequate based on correlation coefficients ranging from 0.8 to 0.9, and responsiveness was adequate in populations of children and adults with AD.29 It was determined that the EASI is a validated scale and can be used reliably in the assessment of severity and extent of AD. Inter-rater reliability for the EASI was also assessed in a study that separated patients into 2 groups, those with lighter skin (melanin index ≤200; n = 11) and those with darker skin (melanin index >200; n = 14), and was found to be adequate (intraclass correlation coefficient = 0.83 and 0.77, respectively).22
An estimated MID of 6.6 points improvement has been reported in the literature.31 In a separate study, Silverberg et al. (2021) used an anchor-based approach to estimate a MID for the EASI from a sample of 826 children and adults with AD.74 The anchors included the patient-reported global assessment (PtGA), PGA, and validated investigator global assessment scale for AD (vIGA-AD). Absolute MIDs ranged from 1.1 to 24.6 points based on the anchor used, how improvement was defined for each anchor (i.e., 1-grade improvement and 2-grades improvement), and baseline AD severity. For instance, 1-grade improvements were 10.9, 14.0, and 14.9 points when using the PtGA, PGA, and vIGA-AD as anchors, respectively, and MIDs for 2- or at least 3-grade improvements were larger. Furthermore, when distinguishing by baseline disease severity, MIDs ranged from 1.1 to 7.9 for patients with mild AD, 8.7 to 13.5 for moderate AD, and 15.4 to 24.2 for severe AD.
Scoring Atopic Dermatitis (SCORAD)
The Scoring Atopic Dermatitis (SCORAD) tool is used in clinical research and was developed to standardize the evaluation of the extent, intensity, and subjective symptoms of AD.77 In the ECZTRA studies, assessments were based on the patient’s condition at the time of observation only and not compared to the condition at an earlier visit.4-7
The SCORAD assesses 3 components of AD: extent, intensity, and subjective symptoms.4-7 The extent of AD is assessed as a percentage of each defined body area and reported as the sum of all areas with a maximum score of 100% (extent score). The intensity of 6 specific symptoms of AD (erythema, edema/papulation, oozing/crusting, excoriation, lichenification, and dryness) is assessed by the investigator on an average representative area using a 4-point scale (0 = none, 1 = mild, 2 = moderate, and 3 = severe). The scores of the 6 symptoms are summed with a maximum overall score of 18 points (symptom score). The patient-reported symptoms, average itch and sleeplessness over the past 3 days and nights, are recorded on a visual analogue scale for each symptom (0 = no symptom and 10 = worst imaginable symptom) with a maximum possible score of 20 (subjective score). The final score is calculated as (extent score / 5) + (7 * sum of specific symptom scores / 2) + (total subjective score). The maximum possible total SCORAD score is 103, where a higher value indicates more severe AD.
The SCORAD has been adequately tested for validity, reliability, and responsiveness in patients with AD.30,54,55,75 Schmitt et al. conducted a review of the literature for instruments used to assess atopic eczema and found that convergent and divergent construct validity were adequate (correlation coefficient >0.70 or <0.70, respectively) and overall internal consistency was acceptable (correlation 0.64 to 0.86).30,55 Furthermore, inter-observer reliability was adequate (e.g., correlation coefficient >0.80) as was sensitivity to change (correlation >0.80 and area under the curve = 0.73). The intraclass correlation coefficient, assessing intra-rater reliability, was calculated to be 0.66 indicating fair to good reliability in patients with AD.26
A MID was estimated from the data of 3 trials in patients with atopic eczema. The mean change in SCORAD score of patients who showed a relevant change, defined as an ‘improvement’ or ‘decline’ of at least 1 point on the PGA or IGA, suggested a MID of 8.7 points.31 In a separate study, Silverberg et al. (2021) used an anchor-based approach to estimate a MID from a sample of 826 children and adults with AD.74 The anchors included the patient-reported global assessment (PtGA), PGA, and validated investigator global assessment scale for AD (vIGA-AD). Absolute MIDs ranged from 2.7 to 29.2 points based on the anchor used, how improvement was defined for each anchor (i.e., 1-grade improvement and 2-grades improvement), and baseline AD severity. For instance, 1-grade improvements were 16.6, 19.9, and >20 points when using the PtGA, PGA, and vIGA-AD as anchors, respectively, and MIDs for 2- or at least 3-grade improvements were larger. Furthermore, when distinguishing by baseline disease severity, MIDs ranged from 2.7 to 15.8 for patients with mild AD, 17.5 to 23.3 for moderate AD, and 22.3 to 29.2 for severe AD.
Eczema-Related Sleep Numerical Rating Scale
The Eczema-Related Sleep Numerical Rating Scale (NRS) is a single-item question where patients are asked to rate how AD affected their sleep each night using an 11-point NRS where 0 = did not interfere and 10 = completely interfered.4-7 This assessment was completed using the electronic diary each morning based on the night before.
No literature was found that assessed the eczema-related sleep NRS for validity, reliability, or responsiveness.
No MID was identified in patients with AD.
Pruritus NRS
The pruritus NRS is a single-item question where patients are asked to rate their itch using an 11-point NRS where 0 = no itch and 10 = worst itch imaginable. The ECZTRA trials assessed both the worst severity of itch and average itch a patient experienced over the past 24 hours.4-7 These assessments were completed using the electronic diary each morning based on the day before. The peak pruritus NRS (for worst itch over the last 24 hours) has been recommended by the Harmonising Outcome Measures for Eczema to be used as one of 2 main instruments for patient-reported symptoms in clinical trials, the other being the POEM.37,78
The instrument’s validity, reliability, and responsiveness have been assessed in studies of adult patients with AD by Yosipovitch et al. (2019)37 (N = 379 and N = 1,379) and Silverberg et al. (2020)56 (N = 602). Yosipovitch et al. examined the content validity of the NRS for both worst itch and average itch in the last 24 hours by conducting interviews with 14 adults with moderate to severe AD.37 Data from dupilumab trials for moderate to severe AD were used to assess psychometric properties and compared the peak pruritus NRS to both patient- and clinician-reported outcomes such as the SCORAD itch visual analogue scale, DLQI itch item, pruritus categorical scale, Patient Global Assessment of Disease Status (PGADS), EASI, and IGA.37 Construct validity was adequate (Pearson correlation coefficient [r] = 0.61 to 0.77) between the NRS and instruments measuring similar concepts (i.e., itch) compared to more general instruments (i.e., EASI and IGA) (r = 0.09 to 0.24) which was expected.37 Convergent validity was also strong between the NRS-itch and POEM (r = 0.58), but moderate with the DLQI (r = 0.48) and HADS anxiety and depression subscores (0.33 and 0.31, respectively).56 Known-groups validity was demonstrated for patients who reported milder itch compared to those who reported more severe itch on the NRS.37 The NRS-itch also demonstrated adequate criterion validity through significant and stepwise increases with different levels of self-reported global AD severity (e.g., mild, moderate, and severe).56 Discriminant validity was adequate (based on the area under the curve [AUC]) and showed good or excellent ability to distinguish between mild and moderate (AUC = 0.71), mild and severe (AUC = 0.88), and moderate and severe (AUC = 0.78) forms of AD.56 Test-retest reliability was adequate based on strong intraclass correlation coefficients (0.95 to 0.96) for weeks 15 and 16 of the dupilumab RCT which was when symptoms were expected to have stabilized.37 Similar to construct validity, when measuring responsiveness, instruments examining similar concepts were expected to show greater association and larger effect size. Responsiveness to change was strong for patient-reported outcomes (r = 0.64 to 0.77) and moderate for clinician-reported outcomes (r = 0.46 to 0.50) and standardized effect size estimates were strong (-1.1 and -1.3 for the different dupilumab trials).37 Silverberg et al. did not note any floor or ceiling effects for the instrument.56
Simpson et al. estimated an improvement on the pruritus NRS to be at least 3 to 4 points reduction.36 This estimate was calculated using anchor- and distribution-based methods on data from a phase IIb study of dupilumab used to treat patients with moderate to severe AD. Yosipovitch et al. also estimated the MID using both distribution-based methods as well as patient-reported outcome and clinician-reported outcome anchor-based methods.37 MIDs were reported to range from 2.2. to 4.2 points for clinician-reported outcome anchors (i.e., EASI and IGA) but were generally lower at 0.76 and 2.6 points for the half-SD distribution-based method and patient-reported outcome anchor (i.e., pruritus categorical scale), respectively. The investigators suggested that the pruritus categorical scale may be the more appropriate anchor since it had a stronger correlation with the NRS than the EASI or IGA.
Patient Global Impression of Severity
The Patient Global Impression of Severity is a single-item question assessing the patient’s perception of their overall AD symptom severity during the last 24 hours and rated on a 4-point scale from no symptoms to severe symptoms.5-7 These assessments were completed using the electronic diary each day from week 2 to week 52 for the ECZTRA 1, ECZTRA 2, and ECZTRA 3 studies.
No literature was found that assessed the Patient Global Impression of Severity for validity, reliability, or responsiveness in patients with AD.
No MID was identified in patients with AD.
Patient-Oriented Eczema Measure
The POEM is a 7-item questionnaire used in clinical trials and clinical practice to assess disease symptoms in patients with AD.4-7 Seven symptoms (itching, sleep, bleeding, weeping, cracking, flaking, and dryness) are assessed for how often each was experienced over the past week based on a 4-point scale (0 = no days, 1 = 1 to 2 days, 2 = 3 to 4 days, 3 = 5 to 6 days, 4 = every day). The total score is the sum of the 7 items and ranges from 0 to 28 where a higher score indicates more severe disease. It has been suggested that a score from 0 to 2 indicates clear or almost-clear skin, 3 to 7 is mild eczema, 8 to 16 is moderate eczema, 17 to 24 is severe eczema, and 25 to 28 is very severe eczema.79 The POEM has been recommended by the Harmonising Outcome Measures for Eczema to be used as one of 2 main instruments for patient-reported symptoms in clinical trials, the other being the peak pruritus NRS.78,80
The POEM has been tested and found to be adequate for validity, reliability, and responsiveness.30,54,55 Two 2020 studies by Silverberg et al. assessed patient-reported outcomes in adults with AD (N = 60256 and N = 29157). For convergent validity, Pearson (r) and Spearman (rS) correlation coefficients generally indicated a strong correlation between the POEM and the DLQI (r = 0.62,56 rS = 0.5957), NRS-itch (r = 0.58,56 rS = 0.45 and 0.5057 for worst itch and average itch, respectively), and the EASI (rS = 0.5257), but moderate correlation with the HADS anxiety and depression subscores (r = 0.33 and 0.31,56 respectively). The POEM also demonstrated adequate criterion validity through significant and stepwise increases with different levels of self-reported global AD severity (e.g., mild, moderate, and severe). Discriminant validity was generally adequate (based on the area under the curve [AUC]) and the POEM showed varying ability to distinguish between mild and moderate AD (AUC = 0.6757 to 0.75,56 poor to fair ability), mild and severe AD (AUC = 0.7057 to 0.89,56 fair to good ability), and moderate and severe AD (AUC = 0.5257 to 0.73,56 unable to fair ability). Test-retest reliability, calculated using the intraclass correlation coefficient, was acceptable (0.86) when assessed in 189 patients.57 Changes in scores, a measure of responsiveness, was assessed and correlated closest between the POEM and the DLQI (r = 0.67) compared to the EASI (r = 0.47) and NRS-itch (r = 0.48 for both worst and average itch).57 Floor effects were noted for the overall POEM score and all individual items of the instrument for the study with 602 patients56 while no floor effects were noted for the study with 291 patients.57 Furthermore, no ceiling effects were observed in either study.
Using data from 3 trials for AD treatment, Schram et al. (2012) reported an overall mean MID of the POEM to be 3.4 points (SD = 4.8) where an IGA score improvement of 1 point was used as an anchor.31 A different study by Silverberg et al. (2020) estimated a MID of 5.0 points based on an anchor of at least 1 point improvement for patient-reported global severity.57 The MID was smaller (3.7) for patients who reported baseline clear-to-mild AD or larger (6.1) for those who reported moderate to severe AD.
Dermatology Life Quality Index
The DLQI is a self-reported, 10-item questionnaire used to assess the patient’s perception of the impact that their skin condition has on their HRQoL during the past week.4-7 The instrument covers items such as dermatology-related symptoms and feelings, daily activities, leisure, work or school, personal relationships, and the treatment received. Each item is scored on a 4-point scale (0 = not at all/not relevant, 1 = a little, 2 = a lot, and 3 = very much). The overall score is the sum of the 10 item scores and ranges from 0 to 30 where a higher score indicates lower HRQoL. The overall score can be interpreted as follows: 0 to 1 = no effect, 2 to 5 = small effect, 6 to 10 = moderate effect, 11 to 20 = very large effect, and 21 to 30 = extremely large effect.48 The DLQI has been recommended by the Harmonising Outcome Measures for Eczema as the QoL measure in clinical trials for eczema.81
The DLQI has previously been validated in patients with a variety of skin conditions, including eczema.41-44 Patel et al. (2019) aimed to assess the validity, responsiveness, and floor and ceiling effects of the DLQI in a group of 340 adults with AD.45 Content validity was assessed in 50 patients, 92% of whom agreed that the questionnaire addressed the issues related to their AD suggesting adequate content validity while those who disagreed responded that it did not address their sleep disturbances. Using Spearman correlation (rS) methods, the DLQI showed moderate convergent validity when compared to the EASI (rS = 0.44), SCORAD (rS = 0.55), NRS-itch (rS = 0.59), and POEM (rS = 0.61). Convergent validity was also assessed in a 2021 study by Schwartzman et al. that included 994 adults with AD and showed similar outcomes for the EASI (rS = 0.48), SCORAD (rS = 0.48), NRS-itch (rS = 0.53 worst itch and rS = 0.49 average itch), and POEM (rS = 0.61) instruments.46 To assess discriminative validity, Patel et al. (2019) used the Wilcoxon rank sum test which generally showed significant and clear stepwise increases for increasing severity when comparing the DLQI to the EASI, SCORAD, NRS-itch, and POEM.45 The questionnaire also demonstrated good internal consistency (Cronbach alpha = 0.89) and fair to moderate correlation among individual items (rho = 0.28 to 0.60). Responsiveness was assessed using Cohen’s D between baseline and the next visit (time interval not indicated) for patients who showed at least a 1-point change and at least a 3- to 4-point change on the POEM (3 to 4 points was taken to be the MID of the POEM). The DLQI was found to be less responsive for patients who showed either one- or 3-point improvement and more responsive for those who showed at least 3-point worsening. A study by Holm et al. (2006) looked at 101 patients with AD (66 adults and 35 children) along with 30 healthy controls who completed the DLQI at 2 time points with a 6-month interval between.47 Using Wilcoxon rank scores, the investigators noted a statistically significant difference (P < 0.0001) in scores could be observed between patients with moderate and mild AD and those with mild AD and health controls suggesting the instrument is sensitive to differing severities of AD. Lastly, the Patel et al. study showed no floor or ceiling effects, defined as at least 15% of patients scoring in the lowest or highest values, for the overall DLQI scores though they could be noted for individual items.45
A study by Basra et al. (2015) used an anchor-based approach to assess the responsiveness of the DLQI and estimate a MID.48 A total of 192 patients with different skin conditions ranging from acute to chronic were included (50.5% with psoriasis, 21.9% with acne, 12.5% with eczema, and 15.1% other). Detail regarding the disease severity of patients was not reported. Using the Global Rating of Change Questionnaire as an anchor and a change in score of 2 or 3 points to be a “small change,” a MID was estimated to be 3.3 for the DLQI.48 There are a few limitations with the determination of the MID for the DLQI. The anchor-based approach was based on a subjective, global assessment of change that is subject to recall bias and it is not specific to AD.48 The authors acknowledged this and noted that the use of a global assessment was reasonable because the population was comprised of patients with a mix of diagnosed skin conditions; however, the lack of specificity to AD is a limitation that should be taken into consideration when applied to an AD-specific population.
Concerns have been identified regarding unidimensionality and the behaviour of items of the DLQI in different psoriatic patient populations with respect to their cross-cultural equivalence, age, and gender; however, these concerns were only identified in 2 of the 12 international studies identified.48 Furthermore, a patient’s emotional aspects may be underrepresented which may be a reason for unexpectedly low DLQI scores in patients with more emotionally disabling conditions such as vitiligo. To overcome this, it has been suggested that the DLQI be combined with more emotionally-oriented measures such as the mental component of the SF-36 or hospital anxiety and depression scales.48 Another limitation is the absence of benchmarks for the MID of DLQI scores in general dermatological conditions, although there have been some attempts to determine these differences for specific conditions.48
EQ-5D 5-Levels Questionnaire
The EQ-5D is a generic, self-reported, QoL instrument developed by the EuroQol Group that is applicable to a wide range of health conditions and treatments, including AD.60,63,82 As a generic measure of HRQoL that can capture the net effect of treatment benefits and harms, the EQ-5D provides valuable information from the patient perspective. The original 3-level version, EQ-5D-3L, was introduced in 1990 and composed of 5 dimensions pertaining to HRQoL. Respondents indicated their health status in terms of 5 HRQoL dimensions based on 3 levels of severity. To improve sensitivity and reduce ceiling effects, the EQ-5D-3L was updated to have 5 levels in 2005 resulting in the EQ-5D-5L which was used in the studies of this review.
The EQ-5D-5L consists of a descriptive system and the EQ VAS.63 The descriptive system comprises 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Patients respond to each dimension using 5 levels where 1 = no problems, 2 = slight problems, 3 = moderate problems, 4 = severe problems, and 5 = extreme problems or unable to perform. Respondents are asked to choose the level that reflects their health state for each of the 5 dimensions. In total, there are 3,125 possible unique health states defined by the EQ-5D-5L, with 11111 and 55555 representing the best and worst health states, respectively, for each of the 5 domains. The numerical values assigned to levels 1 to 5 for each dimension reflect rank order categories of function. In terms of measurement properties, these are ordinal data; they do not have interval properties and therefore, are not used to produce an individual dimension score. Results from the EQ-5D-5L descriptive system can be converted into a single index score using a scoring algorithm taking the local patient and population preferences into account. Therefore, the index score is a country-specific value and a major feature of the instrument.62 The range of index scores will differ according to the scoring algorithm used; however, in all scoring algorithms of the EQ-5D-5L, a score of 0 represents the health state dead and 1.0 reflects perfect health. Negative scores are also possible for health states that society, not the patient, considers to be worse than dead. Index scores in the pivotal studies of this review were based on the UK country-specific value set.4-7
The EQ VAS records the respondent’s self-rated health on a vertical VAS where the end points are labelled 0 (the worst health you can imagine) and 100 (the best health you can imagine). The respondents are asked to mark an X on the point of the VAS that best represents their health on that day. The EQ-5D index and VAS scores can be summarized and analyzed as continuous data.62,63 Hence, the EQ-5D produces 3 types of data for each respondent:
a profile indicating the extent of problems on each of the 5 dimensions represented by a 5-digit descriptor, such as 11121 or 21143,
a population preference-weighted health index score based on the descriptive system, and
a self-reported assessment of health status based on the EQ VAS.
The EQ-5D-5L has been validated in terms of feasibility, ceiling effects, discriminatory power, and convergent validity in a diverse patient population from 6 countries with chronic conditions63; however, no literature was found that assessed the EQ-5D-5L for validity, reliability, or responsiveness in patients with AD.
A Canadian-specific estimate of a MID for the EQ-5D-5L was generated by simulating the effects of single-level transitions in each dimension.64 The results yielded MIDs with a summarized mean of 0.056 (SD = 0.011), and a summarized median of 0.056 (interquartile range = 0.049, 0.063). No MID was identified in patients with AD.
Short Form (36) Health Survey Version 2
The 36-Item SF-36 v2 is a general health status instrument that has been used extensively in clinical trials in many disease areas.59 The questionnaire consists of 36 items representing 8 subscales: physical functioning (PF; 10 items), role physical (RP; 4 items), bodily pain (BP; 2 items), general health (GH; 5 items), vitality (VT; 4 items), social functioning (SF; 2 items), role emotional (RE; 3 items), and mental health (MH; 5 items). The second question of the survey is a single item not used for scoring the 8 scales, but instead used to estimate the general health from a cross-sectional standpoint.59,83 For each of the 8 domains, a subscale score can be calculated.59 The SF-36 also provides 2 component summaries, the physical component summary (PCS; made up of PF, RP, BP, and GH) and the mental component summary (MCS; made up of VT, SF, RE, and MH), derived from aggregating the 8 domains according to a scoring algorithm. The PCS and MCS and 8 subscales are each measured from 0 to 100, which are t-scores (mean of 50 and SD of 10) that have been standardized to the US general population. Thus, a score of 50 on any scale would be mean score of the general US population, while a score 10 points lower (i.e., 40) would be one SD below the mean. On any of the scales, an increase in score indicates improvement in health status.
Although no literature was found that assessed the SF-36 v2 for validity or reliability in patients with AD, the instrument has been validated in other disease areas such as psoriasis.59 A study by Holm et al. (2006) looked at 101 patients with AD (66 adults and 35 children) along with 30 healthy controls who completed the SF-36 at 2 time points with a 6-month interval between.47 PCS and MCS scores showed little change between the assessments and between patient and control groups. Furthermore, using Wilcoxon rank scores, the investigators noted only small differences between patients with moderate and mild AD and those with mild AD and healthy controls. The results were statistically significant (P < 0.05) for the MCS but not statistically significant for the PCS suggesting the instrument is not sensitive or responsive to differing severities of AD.
Based on anchor data, the developer of the SF-36 proposed the following minimal mean group differences for the individual domain scores: physical functioning, 3; role physical, 3; bodily pain, 3; general health, 2; vitality, 2; social functioning, 3; role emotional, 4; and mental health.43,84 It should be noted that these MID values were determined to be appropriate for groups with mean t score ranges of 30 to 40; for higher t score ranges, MID values may be higher. As these MID values were based on clinical and other non-patient-reported outcomes, they do not necessarily identify the smallest difference that patients would consider important. The MIDs of the PCS and MCS were also estimated in a study of patients with moderate to severe plaque psoriasis to range from 2.57 to 3.91 and from 3.89 to 6.05, respectively.85 No MID was identified in patients with AD.
Work Productivity and Activity Impairment–General Health
The Work Productivity and Activity Impairment (WPAI) is a questionnaire that measures impairments on work productivity and daily activities due to generic health (WPAI-GH) or specific health problems (WPAI-SHP).86 The 2 versions were created using the same template and respondents answer based on general health status or a specific health problem, disease, or condition. The instrument consists of 6 items and measures impairments on both paid and unpaid work over the last 7 days. A patient’s employment status is determined first after which they answer 3 questions related to: work hours missed due to health issues, work hours missed for other reasons, and hours worked. There are 2 final questions asking how health issues have impacted productivity at work and activities outside of work rated on an 11-point scale from 0 = no impairment to 10 = complete impairment. Four main outcomes are assessed including percent work time missed, percent impairment for those who worked in the last 7 days, percent overall work impairment, and percent activity impairment.5,7 A higher number indicates greater impairment at work and during non-work activities.
The instrument has been validated in a sample of 106 employed individuals who had some symptom or health problem in the 7 days prior to completing the questionnaire.65 Respondents completed a baseline, self-administered questionnaire then were randomly assigned to either complete a second self-administered version or an interviewer-administered version via telephone call at least 4 hours later. The WPAI was also compared to 3 items from the SF-36 (role function physical, role function emotional, and pain). Overall, highest correlations were observed among work productivity and non-work activities with the SF-36 as well as with the interviewer-administered questionnaires compared to self-administered formats. For reproducibility, there were no statistically significant changes from the first assessment to the second and all were done the same day.
No literature was found that assessed the instrument for validity, reliability, or responsiveness in patients with AD.
No MID was identified in patients with AD.
Hospital Anxiety and Depression Scale (HADS)
The Hospital Anxiety and Depression Scale (HADS) is a widely used, patient-reported questionnaire designed to identify anxiety disorders and depression in patients at nonpsychiatric medical institutions. Repeated administration also provides information about changes in a patient’s emotional state.87-89 The HADS questionnaire contains 14 items that assess symptoms experienced in the previous week, among which, 7 items are related to anxiety and 7 items are related to depression. A patient responds to each item based on a 4-point Likert scale ranging from 0 to 3 where a higher score indicates a worse health state. Scores can range from 0 to 21 for either subscale (i.e., anxiety and depression). Scores 11 or greater on either subscale are considered to be a 'definite case' of psychological morbidity, while scores from 8 to 10 represent 'probable case’ and 0 to 7 'not a case'.89
The psychometric properties of HADS have been assessed in various conditions; however, no literature was found that assessed the instrument for validity, reliability, or responsiveness in patients with AD.87 One study indicated that while the HADS may be useful for the assessment of patients with AD in clinical trials and practice, the authors concluded that additional research is needed to confirm the construct validity and to assess content validity and feasibility in either setting.90
No MID was identified in patients with AD.
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
CDEC
CADTH Canadian Drug Expert Committee
CMA
cost-minimization analysis
MAIC
matching adjusted indirect comparison
NMA
network meta-analysis
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Tralokinumab (Adtralza), 150 mg single-use pre-filled syringe |
Submitted price | Tralokinumab, 150 mg subcutaneous injection: $422.26 per syringe |
Indication | For the treatment of moderate to severe atopic dermatitis in adult patients whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. Tralokinumab can be used with or without topical corticosteroids |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | October 13, 2021 |
Reimbursement request | For the treatment of adult patients with moderate to severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable and who had an adequate trial or were ineligible for each of the following therapies: phototherapy (where available), methotrexate, and cyclosporine |
Sponsor | LEO Pharma Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Information
Component | Description |
|---|---|
Type of economic evaluation | Cost-minimization analysis |
Target population | Adult patients with moderate to severe atopic dermatitis whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable and who had an adequate trial or were ineligible for each of the following therapies: phototherapy (where available), methotrexate, and cyclosporine. |
Treatment | Tralokinumab |
Comparator | Dupilumab |
Perspective | Canadian publicly funded health care payer |
Key data source | Sponsor-submitted matching adjusted indirect comparison |
Costs considered | Drug acquisition costs |
Time horizon | 2 years (induction period and 1 maintenance year) |
Submitted results | Cost savings of $14,427 for tralokinumab vs. dupilumab |
Key limitations | The assumption of similar clinical efficacy for tralokinumab and dupilumab to support the conduct of a cost-minimization analysis is highly uncertain, as the ITCs appraised by the CADTH clinical review team |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Limitations identified in the ITCs introduce uncertainty to the findings. The use of an alternative maintenance dosing schedule applied at week 16 onward for a proportion of the target population on tralokinumab is not reflective of likely Canadian clinical practice and underestimates total costs associated with tralokinumab. A prior CDEC recommendation for dupilumab included a submitted price for dupilumab lower than the current publicly available list price for dupilumab used in the sponsor’s analysis. Additionally, CDEC advised that a significant price reduction was necessary for dupilumab to be cost-effective. As CADTH is not aware of the confidentially negotiated prices, the price of dupilumab is uncertain and significant reductions in its price may limit or eliminate the cost savings associated with tralokinumab. |
CADTH reanalysis results | CADTH conducted a reanalysis using the standard maintenance dose suggested in the product monograph for all patients to reflect the maintenance dosing schedule expected in Canadian clinical practice. Based on the CADTH reanalysis, tralokinumab was associated with a per-patient savings of $7,060 over a 2-year time horizon. CADTH considered scenario analyses exploring the cost of dupilumab. Should a 54% reduction in price, as per the CADTH pharmacoeconomic report for a prior dupilumab submission, be negotiated, tralokinumab would be associated with an incremental per-patient cost of $21,201 over the 2-year time horizon. CADTH was unable to address the uncertainty associated with the comparative efficacy of tralokinumab compared to dupilumab. Should tralokinumab be considered clinically inferior to dupilumab, a cost-minimization analysis is not appropriate to assess the cost-effectiveness of tralokinumab, and the cost-effectiveness of tralokinumab would be unknown. |
CDEC = CADTH Canadian Drug Expert Committee; ITC = indirect treatment comparison.
Conclusions
Assuming equal efficacy and safety for tralokinumab and dupilumab, the sponsor conducted a cost-minimization analysis (CMA) over a 2-year time horizon comparing drug costs alone. In the CADTH reanalysis, at the submitted price for tralokinumab and the publicly available price for dupilumab, tralokinumab is associated with a per-patient cost savings of $7,060 over a 2-year period. In a scenario analysis in which a price reduction for dupilumab in line with previous CADTH recommendations for this indication was considered, tralokinumab was no longer a cost-saving treatment. This cost comparison assumes clinical similarity between tralokinumab and dupilumab. Evidence from indirect treatment comparisons appraised by CADTH suggests that, for most efficacy analyses, the true difference between dupilumab and tralokinumab is unknown and ranges from |||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||. A CMA is insufficient to explore the clinical uncertainty associated with these findings. Should tralokinumab be found to be clinically inferior to dupilumab, the cost-effectiveness of tralokinumab would be unknown.
CADTH assessed tralokinumab within the context of the sponsor’s reimbursement request, which was specific to being listed in the same manner as dupilumab. The cost-effectiveness of tralokinumab according to its broader Health Canada indication, or in situations where dupilumab is not an available treatment option, is unknown.
Economic Review
The current review is for tralokinumab (Adtralza) for adult patients with moderate to severe atopic dermatitis.
Economic Information
Summary of Sponsor’s Economic Information
The sponsor submitted a CMA for tralokinumab compared with dupilumab. The target population of the analysis was adult patients with moderate to severe atopic dermatitis whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable, and who had an adequate trial or were ineligible for each of the following therapies: phototherapy (where available), methotrexate, and cyclosporine. The target population for this review is aligned with the sponsor’s reimbursement request, which is the same as the CADTH Canadian Drug Expert Committee (CDEC) recommendation for dupilumab (Dupixent). The target population and reimbursement request are narrower than those of the Health Canada indication, which states tralokinumab is indicated for the treatment of moderate to severe atopic dermatitis in adult patients whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. The sponsor’s request for a deviation to focus its analysis on the reimbursement request rather than the Health Canada–indicated population was granted by CADTH. The recommended dosages of both tralokinumab and dupilumab are identical; both are administered using a 600 mg loading dose, followed by 300 mg every 2 weeks.1,2
The sponsor assumed no differences in efficacy or safety between tralokinumab and dupilumab based on a sponsor-submitted matching adjusted indirect treatment comparison (MAIC).3 All clinical benefits and resource use beyond drug acquisition costs were assumed to be equivalent, and the sponsor’s base case considered only drug acquisition costs. The analysis was conducted from the perspective of the public health care payer over a 2-year time horizon to capture differences in costs in the first and second years due to differences in the loading and maintenance doses in the first year and second year. In the sponsor’s base case, |||||| of patients on tralokinumab were assumed to switch to a dosing schedule of every 4 weeks after 16 weeks of treatment with tralokinumab, while the remainder were assumed to continue on every 2 weeks. Tralokinumab was associated with savings of $6,663 in its first year of use and $7,764 in the second year, for a total savings of $14,427 over the 2-year time horizon in comparison with dupilumab.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total year 1 costs ($) | Total year 2 costs ($) | 2-year costs | |
|---|---|---|---|---|
Total costs | Incremental costs vs. dupilumab ($)a | |||
Dupilumab | 26,425 | 25,070 | 51,495 | Reference |
Tralokinumab | 19,762 | 17,306 | 37,068 | −14,427 |
vs. = versus.
aIncremental costs are the costs of tralokinumab minus those of the comparator. Negative incremental costs indicate tralokinumab is less expensive.
Source: Sponsor’s pharmacoeconomic submission.3
The sponsor also conducted scenario analyses testing alternative assumptions over the 2-year time horizon, including: incorporating dispensing fees and markups; assuming that |||||| of patients would continue treatment every 4 weeks after the first 6 months; assuming that |||||| and |||| of patients would adopt a dosing schedule of every 4 weeks after the first 16 weeks, respectively; and varying the discount rate by 0%, 3%, and 5%. Results remained robust to these changes, with tralokinumab remaining a cost-saving drug in all scenarios.
CADTH Appraisal of the Sponsor’s Economic Information
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis.
The assumption of similar clinical efficacy and safety for tralokinumab and dupilumab is likely inappropriate: In the absence of a head-to-head comparison between tralokinumab and dupilumab, the sponsor conducted an anchored MAIC of the efficacy and safety outcomes in patients with moderate to severe atopic dermatitis using individual patient data from the ECZTRA-7 and LIBERTY AD CAFÉ trials to indirectly compare the relative effects of tralokinumab to dupilumab. (The Indirect Evidence section of the CADTH Clinical Review Report provides details.) These trials were selected for the MAIC as their patient populations were relevant to the reimbursement request. The sponsor’s MAIC analysis |||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. However, CADTH’s clinical review of the sponsor’s MAIC indicated that, in addition to several methodological limitations associated with the MAIC (e.g., differences in patient populations, methods, and focus), the differences between tralokinumab and dupilumab in the primary efficacy outcome (a 75% or greater improvement in the Eczema Area and Severity Index score from baseline) ranged from |||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. The clinical experts consulted by CADTH for this review indicated that the observed range of risk differences included values that would be considered clinically meaningful. CADTH’s clinical review further appraised a network meta-analysis (NMA) conducted by the Institute for Clinical and Economic Review that compared the relative effects of abrocitinib, baricitinib, tralokinumab, and upadacitinib with dupilumab in monotherapies and combination therapies. The results of this NMA, along with |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| suggested tralokinumab may be inferior to dupilumab in most efficacy outcomes |||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||. The evidence from the appraised ITCs highlights substantial uncertainty regarding the assumption of clinical similarity, which is required for a CMA or cost comparison to be considered appropriate. A cost-utility analysis likely would have been more appropriate to determine the cost-effectiveness of tralokinumab in comparison with dupilumab. The true cost-effectiveness of tralokinumab in comparison with dupilumab is therefore unknown.
CADTH was unable to account for this limitation in reanalysis.
The maintenance dosing schedule (week 16 onward) for tralokinumab is not reflective of likely clinical practice: The sponsor assumed that, following the 16-week induction period on a dosing schedule of every 2 weeks, |||||| of patients who received tralokinumab would continue with dosing every 2 weeks while |||||||||||||| would switch to an dosing schedule of every 4 weeks. The clinical experts consulted by CADTH indicated that, in Canadian clinical practice, tralokinumab would typically be administered every 2 weeks, and the majority of patients are more likely to remain on such a schedule than to switch to every 4 weeks after 16 weeks of induction because every 2 weeks is the schedule they would have initially responded to. The sponsor’s assumption that |||||| of patients on tralokinumab would switch to every 4 weeks at week 16 nearly doubles the cost difference between dupilumab and tralokinumab each year over the 2-year time horizon.
CADTH addressed this limitation by revising the dosing schedule to reflect that all patients (100%) would receive tralokinumab every 2 weeks throughout the time horizon.
The price of dupilumab is uncertain: The submitted price for dupilumab ($959.94 per pre-filled syringe) as part of a recent CADTH Common Drug Review submission is lower than the list price for dupilumab set by the Ontario Exceptional Access Program ($978.70 per pre-filled syringe). Additionally, the confidentially negotiated price for dupilumab is unknown. According to the 2020 CDEC reimbursement recommendation for dupilumab, a price reduction was required to improve the cost-effectiveness of dupilumab relative to standard of care at its submitted price to reach a willingness-to-pay threshold of $50,000 per quality-adjusted life-year.4 As CADTH is not aware of the confidentially negotiated prices, the true price of dupilumab is uncertain, which affects the anticipated cost savings associated with tralokinumab. Should the price paid for dupilumab be lower than the list price, the incremental cost savings associated with dupilumab are likely to be overestimated, while cost savings with tralokinumab may not be realized should the price of dupilumab be substantially lower than its list price.
CADTH conducted scenario analyses to explore the impact of the lower submitted price of dupilumab ($959.94 per pre-filled syringe) and the impact of the suggested 54% price reduction on the submitted price for dupilumab as part of the submission in support of the April 24, 2020, CDEC recommendation. The price reduction listed in the April 24, 2020, recommendation is for an analysis separate from the reduction applicable to the final CDEC reimbursement population and is used to explore the impact of a significantly lower price for dupilumab on the incremental costs of tralokinumab.
CADTH Reanalyses of the Economic Information
The CADTH reanalysis of the sponsor’s base case included 1 change, which was the assumption that all patients would remain on a dosing schedule of every 2 weeks, rather than having a proportion of patients switch to every 4 weeks after a period of time. The CADTH base-case reanalysis was derived in consultation with clinical experts. Results of the reanalysis are provided in Table 4. Tralokinumab was associated with a cost savings of $7,060 over the 2-year time horizon.
Table 4: Summary of the CADTH Reanalysis Results
Drug | Drug costs only | 2-year costs | ||
|---|---|---|---|---|
Total year 1 costs ($) | Total year 2 costs ($) | Total costs ($) | Incremental costs vs. dupilumab ($)a | |
Dupilumab | 26,425 | 25,070 | 51,495 | Reference |
Tralokinumab | 22,802 | 21,633 | 44,435 | −7,060 |
vs. = versus.
Note: Reanalyses are based on publicly available prices of the comparator treatments, and the results presented are deterministic.
aIncremental costs are tralokinumab minus the comparator. Negative incremental costs indicated tralokinumab is less expensive.
CADTH conducted 2 additional scenario analyses testing alternative prices for dupilumab (Table 7). When considering the submitted price from the prior CADTH Common Drug Review submission for dupilumab, tralokinumab was associated with a cost savings of $6,073 over a 2-year time horizon. In the scenario considering a substantial price reduction in the submitted price for dupilumab from the previous scenario analysis in alignment with the April 24, 2020, CDEC recommendation for dupilumab, tralokinumab was associated with incremental costs of $21,201 over the 2-year time horizon.
Issues for Consideration
The sponsor’s reimbursement request and analysis population are narrower than those of the Health Canada indication: The sponsor submitted a request for a deviation to focus its submission on its reimbursement request, which was aligned with the CDEC recommendation for dupilumab, rather than the broader Health Canada indication for tralokinumab. This request for deviation was approved by CADTH. The cost-effectiveness of tralokinumab for its broader Health Canada indication was not assessed by CADTH and is therefore unknown.
The listing status of dupilumab across jurisdictions varies: Dupilumab may not be accessible in all jurisdictions and as a result may not be a relevant comparator in all jurisdictions for the reimbursement request of interest. Tralokinumab is more expensive than immunosuppressants, and retinoids are used off-label for the systemic treatment of atopic dermatitis, although the cost-effectiveness of tralokinumab in comparison with these interventions in situations where dupilumab is not available is unknown.
Conclusions
Based on the CADTH clinical review report, the assumption of similar clinical efficacy for tralokinumab and dupilumab is likely inappropriate. Results from a sponsor-submitted MAIC |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Results from an NMA conducted by the Institute for Clinical and Economic Review suggest that tralokinumab may be inferior to dupilumab in terms of most efficacy outcomes. However, these results are associated with a high level of imprecision and uncertainty.
CADTH identified an additional issue with the proportion of patients receiving an alternative maintenance dose to the recommended standard maintenance dose within the sponsor’s submitted CMA. Addressing this limitation resulted in cost savings for tralokinumab of $7,060 over a 2-year time horizon in comparison with dupilumab. In a scenario analysis in which a price reduction with dupilumab in line with previous CADTH recommendations for this indication was considered, tralokinumab was no longer associated with cost savings. However, given the conclusions of the CADTH clinical review, which |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||, a CMA is insufficient to explore the clinical uncertainty associated with these findings. Should tralokinumab be clinically inferior to dupilumab, the cost-effectiveness of tralokinumab is unknown.
CADTH assessed tralokinumab within the context of the sponsor’s reimbursement request, which was specific to being listed in the same manner as dupilumab. The cost-effectiveness of tralokinumab according to its broader Health Canada indication, or in situations where dupilumab is not an available treatment option, is unknown.
References
1.tralokinumab: 150 mg pre-filled syringes (150 mg/1 mL) for subcutaneous injection [product monograph]. Thornhill (ON): LEO Pharma Inc; 2020 Oct 4.
2.Dupixent (dupilumab) solution for subcutaneous injection 150 mg/mL [product monograph]. Laval (QC): Sanofi-aventis Canada, Inc; 2021: https://pdf.hres.ca/dpd_pm/00042394.PDF. Accessed 2021 Jul 28.
3.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: tralokinumab, 150 mg pre-filled syringes (150 mg/1 mL) for subcutaneous injection. Thornhill (ON): LEO Pharma Inc; 2021 Apr 27.
4.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: dupilumab (Dupixent-Sanofi-Aventis Canada Inc.). Ottawa (ON): CADTH; 2020: https://cadth.ca/sites/default/files/cdr/complete/SR0636%20Dupixent%20-%20CDEC%20Final%20%20Recommendation%20April%2024%2C%202020%20for%20posting.pdf. Accessed 2021 July 27.
5.Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2. Management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71(1):116-132. PubMed
6.Sidbury R, Davis DM, Cohen DE, et al. Guidelines of care for the management of atopic dermatitis: section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71(2):327-349. PubMed
7.CADTH drug reimbursement reivew: pharmacoeconomic report dupiliumab (Dupixent). Ottawa (ON): CADTH; 2020: https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/sr0636-dupixent-pharmacoeconomic-review-report.pdf. Accessed 2021 Jul 28.
8.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: tralokinumab, 150 mg pre-filled syringes (150 mg/1 mL) for subcutaneous injection. Thornhill (ON): LEO Pharma Inc; 2021 Apr 27.
9.Barbarot S, Auziere S, Gadkari A, et al. Epidemiology of atopic dermatitis in adults: results from an international survey. Allergy. 2018;73(6):1284-1293. PubMed
Appendix 1: Additional Economic Information
Note that this appendix has not been copy-edited.
Additional Details on the CADTH Reanalyses and Additional Analyses
Table 5: CADTH Cost Comparison Table for Systemic Therapy of Moderate to Severe Atopic Dermatitis — Biologics
Drug/comparator | Concentration | Dosage form | Price ($) | Recommended dosage | Average daily drug costs | Annual cost ($) |
|---|---|---|---|---|---|---|
Tralokinumab (Adtralza) | 150 mg/mL | Pre-filled syringe | 422.2600a | 600 mg as an initial dose, followed by 300 mg every 2 weeks Note: At prescriber’s discretion, every fourth week dosing may be considered for some patients who achieve clear or almost clear skin after 16 weeks of treatment. | Year 1: 62.47 Year 2+:59.27 | Year 1: 22,802 Year 2+: 21,633 |
Dupilumab (Dupixent) | 200 mg/ 1.14 mL 300 mg/ 2mL | Pre-filled syringe | 978.7000b | 600 mg as an initial dose, followed by 300 mg every 2 weeksb | Year 1: 72.40 Year 2+: 69.72 | Year 1: 26,424 Year 2+: 25,446 |
Note: Annual period assumes 52 weeks or 365 days for all comparators.
aSponsor’s submitted price for each dosage; price per smallest dispensable unit; cost per pack = $1,689.0400.
bOntario Exceptional Access Program.
Table 6: CADTH Cost Comparison Table for Systemic Therapy of Moderate to Severe Atopic Dermatitis (not Indicated for AD)
Treatment | Strength | Dosage form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost per treatment course ($) |
|---|---|---|---|---|---|---|
Immunosuppressants | ||||||
Azathioprine (generic) | 50 mg | Tablet | 0.2405 | 1 to 3 mg/kg per daya | 0.48 to 1.20 | 176 to 439 |
Cyclosporine (generic) | 10 mg 25 mg 50 mg 100 mg | Capsule | 0.6700 0.9952 1.9400 3.8815 | 150 to 300 mg per daya | 5.82 to 11.64 | 2,125 to 4,250 |
Methotrexate (generic) | 2.5 mg | Tablet | 0.6325 | 7.5 to 25 mg per weeka | 1.90 to 6.33 per week | 99 to 329 |
Mycophenolate mofetil | 250 mg 500 mg | Capsule | 0.3712 0.7423 | 500 mg to 3,000 mg dailya | 0.37 to 2.22 | 135 to 813 |
Retinoidsb | ||||||
Acitretin (Soriatane) | 10 mg 25 mg | Capsule | 1.2965 2.2770 | 10 to 50 mg dailyb | 1.30 to 2.59 | 473 to 967 |
Alitretinoin (Toctino) | 10 mg 30 mg | Capsule | 22.6490 | 30 mg dailyb | 22.65 | 8,266 |
Note: Unit prices of medications are taken from the Ontario Drug Benefit Formulary (accessed July 2021), unless otherwise indicated, and do not include dispensing fees. Unless otherwise indicated, treatment course for all systemic therapies is based on an annual period of 52 weeks (or 365 days) as per the CADTH dupilumab (Dupixent) review. Cost calculations assume an adult weight of 70 kg.
aRecommended dosage based on the American Atopic Dermatology Guidelines.5,6
bRecommended dosage aligned with the previous CADTH Pharmacoeconomic Review for Dupilumab (Dupixent).7
Scenario Analyses
Table 7: Scenario Analyses for the Uncertainty in the Price of Dupilumab
Scenario analysis | Drug | Total year 1 costs ($) | Total year 2 costs ($) | 2-Year costs | |
|---|---|---|---|---|---|
Total costs | Incremental costs vs. dupilumab ($) | ||||
Submitted price of dupilumab as per recent dupilumab submission | Dupilumab | 25,918 | 24,590 | 50,508 | Reference |
Tralokinumab | 22,802 | 21,633 | 44,435 | −6,073 | |
A 54% reduction in the price of dupilumab as per the recent dupilumab submission | Dupilumab | 11,922 | 11,311 | 23,234 | Reference |
Tralokinumab | 22,802 | 21,633 | 44,435 | 21,201 | |
aIncremental costs are the costs of tralokinumab minus the costs of the comparator. Negative incremental costs indicate tralokinumab is less expensive.
Appendix 2: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Key Takeaways of the Budget Impact Analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
In the submitted budget impact analysis (BIA), the sponsor assessed the introduction of tralokinumab for the treatment of adults with moderate to severe atopic dermatitis whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable, and who had an adequate trial or would be ineligible for each of the following therapies: phototherapy (where available), methotrexate, and cyclosporine. The target population is aligned with the reimbursement request, which is narrower than the Health Canada indication, which states tralokinumab is indicated for the treatment of moderate to severe atopic dermatitis in adult patients whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. The sponsor submitted a request for deviation to CADTH before submission to focus their analysis on the reimbursement population, which was approved by CADTH. The BIA was undertaken from the perspective of the Canadian public drug plans, over a 3-year time horizon.8 The sponsor’s base case only considered drug acquisition costs, along with mark-up and dispensing fees. In the reference scenario, the sponsor assumed that patients would be eligible to receive dupilumab. In the new drug scenario, tralokinumab was assumed to displace market shares of dupilumab.8
The sponsor undertook an epidemiological approach to estimate the total population size eligible for tralokinumab following a multi-step approach.8 This required using Canadian population data to estimate the total pan-Canadian adult population by province, estimating the adult population by province and incorporating an average province-specific annual population growth rate. This was followed by using IQVIA Pharmastat data to estimate the proportion of patients who would receive public coverage, and a clinician’s analysis to estimate that approximately |||| of the pan-Canadian adult population was diagnosed with moderate to severe atopic dermatitis, |||||| of whom were assumed to have had an adequate trial of 2 off-label immunosuppressants.8 Of these eligible patients, the sponsor assumed that |||||| would be treated with biologics/targeted therapy and assumed that this proportion would increase over the 3-year period.8 Key inputs to the BIA are presented in Table 8.
While the sponsor presented pan-Canadian BIA results, an option to modify key model parameters and inputs based on the entire pan-Canadian adult population was unavailable, and, therefore, the budget impact results could only be adjusted by province. The market share and cost inputs remained the same across jurisdictions.
Table 8: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as baseline / year 1 / year 2 / year 3, if appropriate) |
|---|---|
Target population | |
Total population in Canada | 31,012,688 |
Proportion of adults a | 80.1% |
Proportion of public patients a | 63.3% |
Annual population increase a | 1.1% |
Prevalence of atopic dermatitisb | ||||| |
Proportion with moderate to severe diseaseb | |||| |
Patients with adequate trial of phototherapy and/or at least 2 off-label immunosuppressantsb | |||| |
Treated populationa | |||||| / |||| / |||| / |||| |
Number of patients eligible for drug under review | |||||| / |||| / |||| / |||| |
Market uptake (3 years) | |
Uptake (reference scenario) Dupilumab | |||| / |||| / |||| |
Uptake (new drug scenario) Tralokinumab Dupilumab | |||| / |||| / |||| |||| / |||| / |||| |
Cost of treatment (per-patient) | |
Cost of treatment, per year Tralokinumab Dupilumab | $20,984 (year 1); $18,652 (subsequent years) $28,016 (year 1); 26,979 (subsequent years) |
aInputs are based on pan-Canadian averages calculated by CADTH using jurisdiction-specific values.
bDefault pan-Canadian values as per sponsor’s submitted model.
Summary of the Sponsor’s Budget Impact Analysis Results
Results of the sponsor’s submitted base case suggested that the introduction of tralokinumab for the treatment of moderate to severe atopic dermatitis in adult patients as per the reimbursement request would result in a budget savings of $6,702,691 in year 1, $12,255,132 in year 2, and $15,539,538 in year 3, for a total budget savings of $34,497,361 over the 3-year time horizon.
CADTH Appraisal of the Sponsor’s Budget Impact Analysis
CADTH identified the following key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Uncertainty regarding the number of patients eligible to receive tralokinumab: The sponsor used an epidemiological approach to identify the patient population eligible to receive tralokinumab. The clinical experts consulted by CADTH noted several areas of uncertainty with the estimates and assumptions used to derive the target population size. First, the sponsor only considered the prevalence of atopic dermatitis among Canadian adults and did not consider the incidence of atopic dermatitis among adults as part of their methodological approach. This is inappropriate because the sponsor omits new cases of atopic dermatitis from the population, thereby underestimating the total number of adult patients eligible for treatment with tralokinumab. The sponsor assumed that the prevalence of atopic dermatitis among adults in Canada was ||||||, with approximately |||| having moderate to severe disease. CADTH’s clinical experts indicated that while the prevalence of atopic dermatitis among adults was reasonable, the proportion of adult patients with moderate to severe disease likely ranged between 3 and ||||. Among adult atopic dermatitis patients with moderate to severe disease, the sponsor assumed |||||| would be eligible for biologics/targeted therapy, and approximately |||||| of these patients would be treated. In the sponsor’s base case, this value was assumed to increase year over year up to |||| in 3 years’ time. CADTH’s clinical experts noted that the proportion eligible for biologics/targeted therapy was expected to be higher, at approximately |||| to 70%, and there is uncertainty associated with the proportion of patients who receive biologics/targeted therapy across Canada, though the estimated treated population size appeared to align with their expectations. Lastly, the sponsor considered that a proportion of patients receive public coverage for tralokinumab based on Pharmastat data, however, the actual proportion of patients who receive public coverage by jurisdiction remains unknown and adds further uncertainty to the estimated eligible population for tralokinumab. Altogether, the clinical experts’ feedback suggests that the estimated population size derived from the sponsor’s assumptions and inputs is associated with some uncertainty.
CADTH addressed this limitation by changing the proportion of patients eligible for biologics to 70%. To address the uncertainty in the other estimates, CADTH undertook scenario analyses to explore the impact of (a) decreasing the proportion of patients with moderate to severe disease to 3% (b) revising the proportion of patients eligible for biologics/targeted therapy to 60%; (c) assuming that the proportion of the treated population remained constant across the 3-year time horizon; (d) assuming that all patients (100%) would receive public coverage.
Maintenance dosing schedule (week 16 onwards) for tralokinumab is not reflective of clinical practice: In the submitted CMA and budget impact analysis, the sponsor assumed that following the 16-week induction period on a Q2W (i.e., every 2 weeks) dosing schedule, |||||| of patients who received tralokinumab would continue with Q2W dosing while |||||| switched to a Q4W (i.e., every 4 weeks) dosing schedule. However, the clinical experts consulted by CADTH indicated that in Canadian clinical practice, tralokinumab would typically be administered on a Q2W dosing schedule and that the vast majority of patients are likely to remain on the Q2W dosing schedule after 16 weeks.
CADTH addressed this limitation by revising the dosing schedule to reflect that all patients (100%) would receive tralokinumab every 2 weeks, to align with the CADTH base case for the CMA.
Price of dupilumab is uncertain: The submitted price for dupilumab ($959.94 per pre-filled syringe) as part of a recent CDR submission is lower than the list price for dupilumab as per the Ontario Exceptional Access Program ($978.70 per pre-filled syringe). Additionally, the confidential negotiated price for dupilumab is unknown. As per the April 2020 CDEC reimbursement recommendation for dupilumab, a price reduction was required to improve the cost-effectiveness of dupilumab relative to standard of care at its submitted price to reach a willingness-to-pay threshold of $50,000 per quality-adjusted life-year.4 As CADTH is not aware of the confidential negotiated prices, there is uncertainty in the true price of dupilumab, which affects the anticipated budgetary savings associated with tralokinumab. Should the price paid for dupilumab be lower than the list price, the incremental budgetary savings associated with dupilumab are likely to be overestimated, while budgetary savings with tralokinumab may not be realized should the price of dupilumab be substantially lower than its list price.
CADTH conducted scenario analyses to explore the impact of the lower submitted price of dupilumab ($959.94 per pre-filled syringe); and the impact of the suggested 54% price reduction on the submitted price for dupilumab as part of the submission in support of the April 24,2020 CDEC recommendation. CADTH notes that the price reduction listed in the recommendation is for an analysis separate from the 1 applicable to the final CDEC reimbursement population and is used to explore the impact of a significantly lower price of dupilumab on the incremental costs of tralokinumab.
An additional limitation was identified but was not considered to be a key limitation:
Limitations with the programming of the submitted BIA model: The submitted model was non-modifiable at the pan-Canadian level, as changes to inputs could only be made for individual jurisdictions rather than at an aggregate level for the pan-Canadian population. Due to limitations with the programming of the submitted BIA model, CADTH attempted to address this issue by summing jurisdictional inputs, or determining the average, such as the total Canadian population size, proportion of Canadian adults, proportion of Canadian adults with public coverage, change in annual population growth, and proportion treated with biologics/targeted therapy, as appropriate to derive an approximate pan-Canadian result. While CADTH was unable to reproduce the sponsor’s base case results, similar results were produced. The 3-year total budget savings of the aggregated pan-Canadian analysis as reported by the sponsor was approximately $34,397,361 while the approximated base case parameters resulted in a 3-year total budget savings of $39,279,563. There is some uncertainty introduced by the approach CADTH has taken, but this approach allowed CADTH to determine the impact of alternative assumptions when addressing other limitations with the BIA.
CADTH addressed this limitation by modifying inputs and assumptions to derive an approximation of the sponsor’s base case results (Corrections to the sponsor’s base case are noted in Table 9).
CADTH Reanalyses of the Budget Impact Analysis
Changes made to the sponsor’s BIA as part of the CADTH reanalysis are provided in Table 9. All analyses are from the public drug plan perspective.
Table 9: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Pan-Canadian results were fixed and unamenable to changes. | Province- specific parameters: a. Total Canadian population b. Percentage of adults c. Percent annual population increase d. Percentage of adults assumed to receive public coverage across provinces e. Proportion of the treated population across provinces | Pan-Canadian parameters approximated: a. Summed the total Canadian population b. Estimated the proportion of adults based on the average across provinces c. Estimated percent annual population increase based on averages across provinces d. Estimated the proportion of adult patients eligible for coverage based on an average of those assumed to receive public coverage across provinces e. Estimated the proportion of the population treated year-by-year based on an average across provinces |
Changes to derive the CADTH base case | ||
1. Proportion of patients eligible for biologic/targeted therapy (i.e., those with an adequate trial of phototherapy and/or at least 2 off-label immunosuppressants) | |||||| | 70% |
2. Maintenance dosing schedule | |||||| of patients on Q2W and |||||| of patients on Q4W after the initial 16 weeks of treatment | All patients (100%) remain on a Q2W dosing schedule, following induction period |
CADTH base case | Reanalyses 1 + 2 | |
The sponsor’s 3-year costs savings under the drug plan perspective was $39,279,563 when considering the results of CADTH’s approximation of the sponsor’s base case that was required due to limitations with the sponsor’s BIA programming. Applying the changes in Table 9 resulted in a decrease in budget savings to $25,621,769 over the 3 years. The results of the CADTH stepwise reanalyses are presented in summary format in Table 10 and a more detailed breakdown is presented in Table 11.
Table 10: Summary of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total |
|---|---|
Sponsor’s submitted base case | −$34,497,361 |
CADTH’s approximation of sponsor base casea | −$39,279,563 |
CADTH reanalysis 1 | −$57,282,697 |
CADTH reanalysis 2 | −$16,753,854 |
CADTH base caseb | −$25,621,769 |
aThe sponsor’s approximated base case results reported here was approximated by using an average of all relevant inputs as noted in Table 7.
bThe CADTH base case reanalysis is based on the CADTH approximation of the sponsor’s base case.
Table 11: Detailed Breakdown of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Sponsor-submitted base case | Reference | $55,337,868 | $70,287,028 | $96,613,598 | $115,475,443 | $282,376,069 |
New drug | $55,337,868 | $63,584,337 | $84,358,466 | $99,935,905 | $247,878,708 | |
Budget impact | $0 | -$6,702,691 | −$12,255,132 | −$15,539,538 | −$34,497,361 | |
CADTH’s approximation of sponsor base case | Reference | $62,928,241 | $80,622,518 | $109,388,263 | $130,986,194 | $320,996,975 |
New drug | $62,928,241 | $72,919,322 | $95,454,424 | $113,343,665 | $281,717,411 | |
Budget impact | $0 | -$7,703,196 | −$13,933,839 | −$17,642,529 | −$39,279,563 | |
CADTH base case | Reference | $91,770,351 | $117,574,505 | $159,524,550 | $191,021,532 | $468,120,588 |
New drug | $91,770,351 | $112,390,402 | $150,483,152 | $179,625,264 | $442,498,819 | |
Budget impact | $0 | -$5,184,103 | −$9,041,398 | −$11,396,269 | −$25,621,769 |
CADTH conducted the following additional scenario analyses:
Applying the assumption that the prevalence of adult atopic dermatitis is (a) 2.1%; and (b) 4.9% based on published literature.9
Applying the assumption that 3% of adult patients with atopic dermatitis have moderate to severe disease.
Applying the assumption that approximately 60% of adult patients with moderate to severe atopic dermatitis will be eligible for biologics/targeted therapy.
Keeping the proportion of patients who are treated with biologics/targeted therapy (||||||) constant each year over the 3-year time horizon.
Applying the assumption that 100% of patients would receive public coverage.
Applying the submitted price of dupilumab ($959.94) based on the CDR submission associated with the April 24, 2020, CDEC reimbursement recommendation for dupilumab.
Applying a 54% reduction to the submitted price of dupilumab based on the April 24, 2020, CDEC reimbursement recommendation for dupilumab.4
The scenario analyses indicate that decreasing the estimated population size leads to fewer cost savings with introduction of tralokinumab, whereas increasing the eligible population size leads to greater anticipated cost savings. Lowering the price of dupilumab leads to a decrease in the anticipated budgetary savings with tralokinumab.
Table 12: CADTH Scenario Analyses
Stepped analysis | Budget impact | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
CADTH scenario analysis 1a | Reference | $27,143,343 | $34,775,558 | $47,183,318 | $56,499,326 | $138,458,202 |
New drug | $27,143,343 | $33,242,232 | $44,509,101 | $53,128,599 | $130,879,932 | |
Budget impact | $0 | −$1,533,326 | −$2,674,216 | −$3,370,727 | -$7,578,270 | |
CADTH scenario analysis 1b | Reference | $63,334,468 | $81,142,968 | $110,094,408 | $131,831,762 | $323,069,138 |
New drug | $63,334,468 | $77,565,207 | $103,854,570 | $123,966,731 | $305,386,509 | |
Budget impact | $0 | −$3,577,761 | −$6,239,838 | −$7,865,030 | −$17,682,630 | |
CADTH scenario analysis 2 | Reference | $55,062,211 | $70,544,703 | $95,714,730 | $114,612,919 | $280,872,353 |
New drug | $55,062,211 | $67,434,241 | $90,289,891 | $107,775,158 | $265,499,291 | |
Budget impact | $0 | −$3,110,462 | −$5,424,839 | −$6,837,761 | −$15,373,062 | |
CADTH scenario analysis 3 | Reference | $78,660,301 | $100,778,147 | $136,735,329 | $163,732,742 | $401,246,218 |
New drug | $78,660,301 | $96,334,631 | $128,985,559 | $153,964,512 | $379,284,702 | |
Budget impact | $0 | −$4,443,517 | −$7,749,770 | −$9,768,230 | −$21,961,517 | |
CADTH scenario analysis 4 | Reference | $79,893,229 | $77,772,189 | $78,619,285 | $79,475,608 | $235,867,083 |
New drug | $79,893,229 | $74,343,053 | $73,879,978 | $74,550,921 | $222,773,951 | |
Budget impact | $0 | −$3,429,136 | −$4,739,308 | −$4,924,687 | −$13,093,132 | |
CADTH scenario analysis 5 | Reference | $144,976,858 | $185,741,714 | $252,013,508 | $301,771,773 | $739,526,995 |
New drug | $144,976,858 | $177,551,979 | $237,730,099 | $283,768,189 | $699,050,266 | |
Budget impact | $0 | −$8,189,736 | −$14,283,409 | −$18,003,584 | −$40,476,729 | |
CADTH scenario analysis 6 | Reference | $90,028,224 | $115,342,523 | $156,496,208 | $187,395,265 | $459,233,995 |
New drug | $90,028,224 | $111,118,173 | $148,817,564 | $177,667,079 | $437,602,816 | |
Budget impact | $0 | −$4,224,351 | −$7,678,644 | −$9,728,185 | −$21,631,180 | |
CADTH scenario analysis 7 | Reference | $41,890,569 | $53,669,435 | $72,818,444 | $87,195,925 | $213,683,804 |
New drug | $41,890,569 | $75,964,512 | $102,794,794 | $123,559,436 | $302,318,742 | |
Budget impact | $0 | $22,295,077 | $29,976,350 | $36,363,511 | $88,634,938 |
Note: All scenario analyses are conducted based on the CADTH base case undertaken from the drug program plan perspective.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.